Embed Size (px)
Citation preview

Instrumentelle Analytik Atom- und Molekülspektren Seite N032_Wechselwirkung_b_BAneu.doc - 1/24
2.2.2 Moleküle und Molekülspektren
Definition: Ein Molekül ist eine Verbindung von zwei oder mehreren Atomen zu einem stabilen Verband mit definierten chemischen Eigenschaften und definierter Struktur.
Ursachen: - Anziehende Kräfte zwischen Atomen - Gemeinsames Verwenden von Elektronen - Coulombanziehung zwischen Katíon und Anion
Kräfte zwischen Atomen - Bindungstypen
Kovalente oder homöopolare Bindung: Bindungsenergie nur quantenmechanisch zu verstehen. Großer Einfluss von Pauli-Prinzip und Austausch-Symmetrien.
Ionische Bindung: Die Elektronenstruktur der beteiligten Atome bewirkt einen (teilweisen) Elektronentransfer zu einem Atom. Es entstehen Anion und Kation, die sich mit der Coulombkraft anziehen.
Van-der-Waals-Bindung: Anziehende, elektrostatische Dipol-Dipol-Wechselwirkung von induzierten oder permanenten Dipolmomenten.
Metallische Bindung: kovalent, wobei die Bindungselektronen über den gesamten Kristall verteilt sind (delokalisiert).
Wasserstoffbrückenbindung: ähnlich van-der-Waals, aber stärker. Verantwortlich für die Sekundärstruktur in biologischen Komplexen.
Molekülorbital MO (LCAO-Methode)
Ebenso wie in Atomen befinden sich in Molekülen die Elektronen in genau festgelegten Zuständen, hier Molekülorbitale genannt. Die Molekülorbitale werden in der Quantenmechanik durch Linearkombination der Atomorbitale gebildet (LCAO-Methode).
Wesentlich dabei ist, dass bei dem Überlapp der Wellenfunktionen (additiv oder subtraktiv) immer bindende und antibindende Orbitale entstehen.
Modell für das H2-Molekülion:
bag ss 11 : gs1
bindendes MO mit Elektronendichte zwischen den Kernen Anziehung
bau ss 11 : *1 us
antibindendes MO ohne Elektronen- dichte (Knoten) zwischen den Kernen Abstoßung
H-Atom H2-Molekülion

Instrumentelle Analytik Atom- und Molekülspektren Seite N032_Wechselwirkung_b_BAneu.doc - 2/24
Typen von Molekülorbitalen
core-Orbitale enthalten die inneren Elektronen. Diese Elektronen tragen in erster Näherung nichts zu den chemischen Bindungen bei. Ihre Anregung benötigt energiereiche Röntgenstrahlung.
n-Orbitale enthalten Elektronen der freien, nicht bindenden Elektronenpaare eines Atoms/Moleküls.
Für die Bindung bedeutsam sind die Molekülorbitale, die durch Kombinieren von s-, p-, und d-Atom-orbitalen entstehen. Hinsichtlich der Symmetrie zur Bindungsachse unterscheidet man drei Typen.
-Orbitale Die -Orbitale entstehen durch Überlappung von Atom-Orbitalen, die symmetrisch zur Molekülachse (Bindungsachse) liegen. Jedes bindende -Orbital, das mit 2 Elektronen besetzt ist, ergibt eine Einfachbindung (-Bindung). Durch die Überlappung entsteht neben dem bindenden -Orbital immer auch ein antibindendes *-Orbital. Das antibindende Orbital ist stets energiereicher und ist im Grundzustand meist auch nicht besetzt. Jedes -Orbital kann von maximal 2 Elektronen (Elektronenpaar) besetzt werden.
Zwei s-Atomorbitale kombinieren zu einem bindenden s- und zu einem antibindenden s*-Molekülorbital.
Zwei pz-Atomorbitale kombinieren zu einem bindenden p- und zu einem antibindenden p*-Molekülorbital.
-Orbitale entstehen durch Überlappung von Atomorbitalen, die senkrecht zu den Bindungen angeordnet sind. Sie sind für die Doppel- und Mehrfachbindungen im Molekül verantwortlich. Da die Bindungsstärke von Mehrfachbindungen im Verhältnis kleiner ist als die von Einfachbindungen, liegen die -Orbitale energetisch höher als die -Orbitale. Jedes -Orbital kann von maximal 2 Elektronen (Elektronenpaar) besetzt werden.
Zwei px- oder zwei py- Atomorbitale ergeben ein (ungerades) bindendes pu-Molekülorbital und ein (gerades) antibindendes pu*-Molekülorbital.
-Orbitale sind gekennzeichnet durch zwei, zueinander senkrecht stehende, die Verbindungsachse enthaltene Knotenflächen.
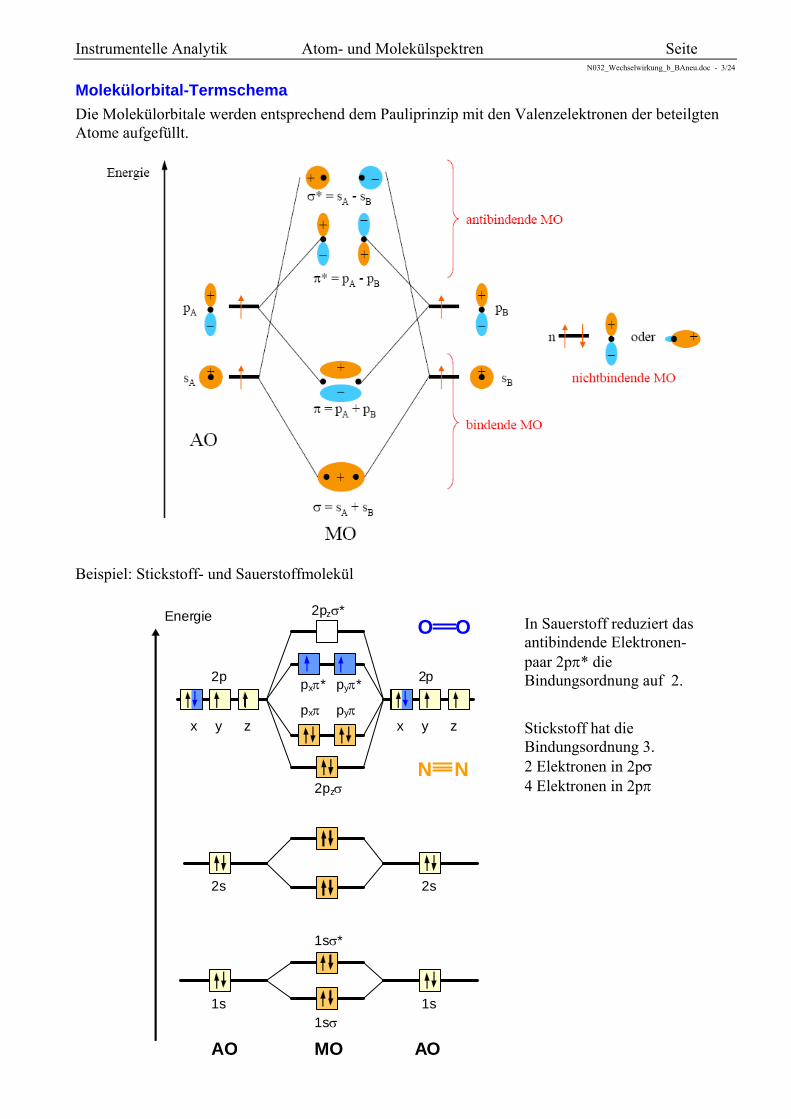
Instrumentelle Analytik Atom- und Molekülspektren Seite N032_Wechselwirkung_b_BAneu.doc - 3/24
Molekülorbital-Termschema
Die Molekülorbitale werden entsprechend dem Pauliprinzip mit den Valenzelektronen der beteilgten Atome aufgefüllt.
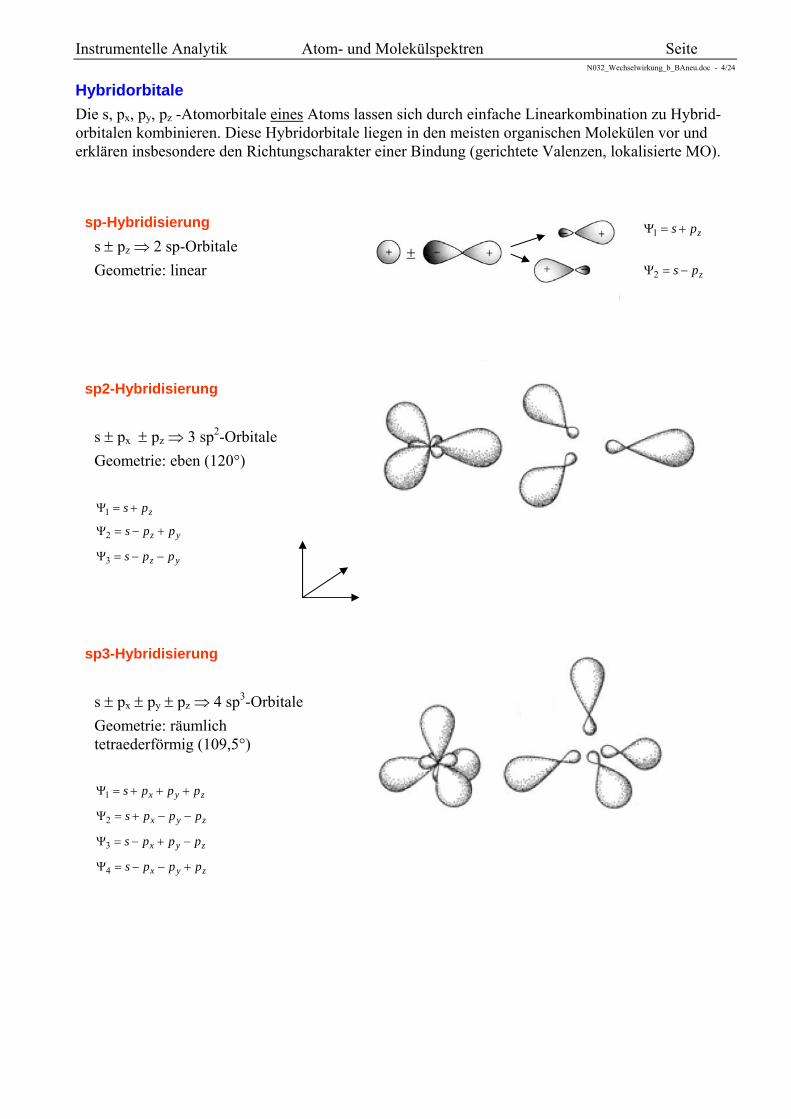
Beispiel: Stickstoff- und Sauerstoffmolekül
In Sauerstoff reduziert das antibindende Elektronen-paar 2p* die Bindungsordnung auf 2.
Stickstoff hat die Bindungsordnung 3. 2 Elektronen in 2p 4 Elektronen in 2p
AO AOMO
2s 2s
1s 1s
2p 2p
x y z x y zpx py
1s*
1s
2pz
px* py*
2pz* O O
N N
Energie
Instrumentelle Analytik Atom- und Molekülspektren Seite N032_Wechselwirkung_b_BAneu.doc - 4/24
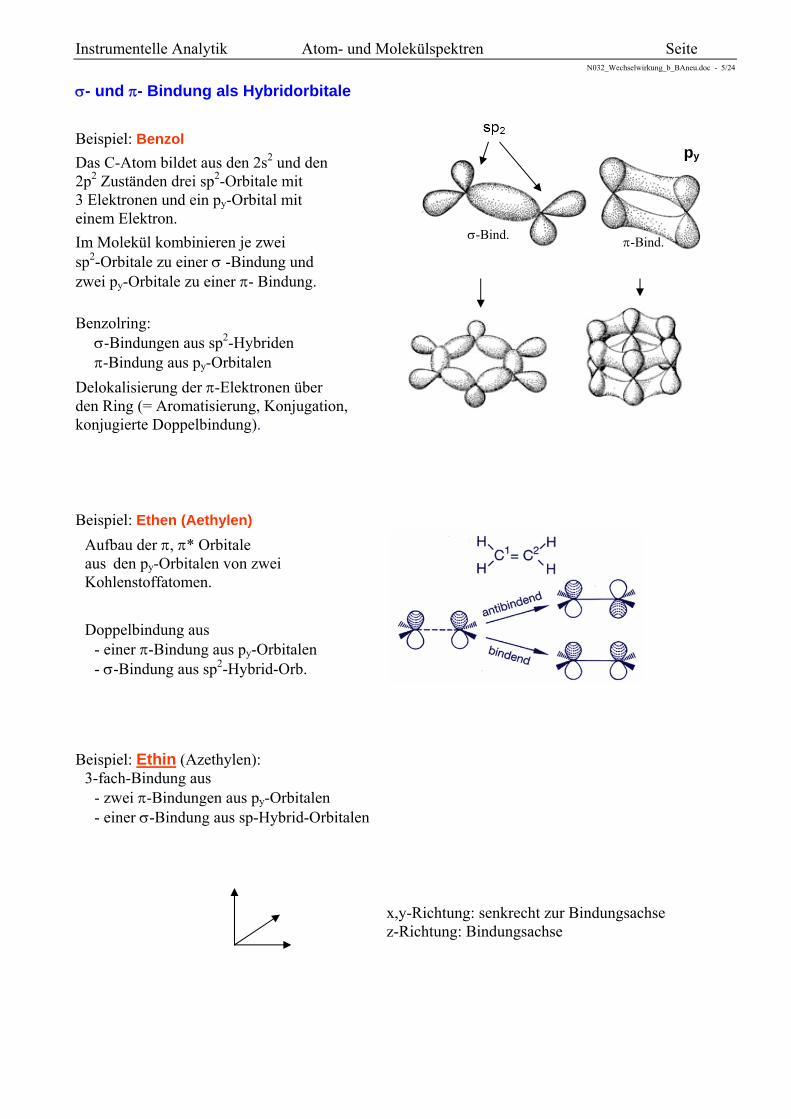
Hybridorbitale
Die s, px, py, pz -Atomorbitale eines Atoms lassen sich durch einfache Linearkombination zu Hybrid-orbitalen kombinieren. Diese Hybridorbitale liegen in den meisten organischen Molekülen vor und erklären insbesondere den Richtungscharakter einer Bindung (gerichtete Valenzen, lokalisierte MO).
sp-Hybridisierung
s pz 2 sp-Orbitale
Geometrie: linear
sp2-Hybridisierung
s px pz 3 sp2-Orbitale
Geometrie: eben (120°)
zps 1
yz pps 2
yz pps 3
sp3-Hybridisierung
s px py pz 4 sp3-Orbitale
Geometrie: räumlich tetraederförmig (109,5°)
zyx ppps 1
zyx ppps 2
zyx ppps 3
zyx ppps 4
zps 1
zps 2
Instrumentelle Analytik Atom- und Molekülspektren Seite N032_Wechselwirkung_b_BAneu.doc - 5/24
- und - Bindung als Hybridorbitale
Beispiel: Benzol
Das C-Atom bildet aus den 2s2 und den 2p2 Zuständen drei sp2-Orbitale mit 3 Elektronen und ein py-Orbital mit einem Elektron.
Im Molekül kombinieren je zwei sp2-Orbitale zu einer -Bindung und zwei py-Orbitale zu einer - Bindung.
Benzolring: -Bindungen aus sp2-Hybriden -Bindung aus py-Orbitalen
Delokalisierung der -Elektronen über den Ring (= Aromatisierung, Konjugation, konjugierte Doppelbindung).
Beispiel: Ethen (Aethylen)
Aufbau der , * Orbitale aus den py-Orbitalen von zwei Kohlenstoffatomen.
Doppelbindung aus - einer -Bindung aus py-Orbitalen - -Bindung aus sp2-Hybrid-Orb.
Beispiel: Ethin (Azethylen): 3-fach-Bindung aus - zwei -Bindungen aus py-Orbitalen - einer -Bindung aus sp-Hybrid-Orbitalen
x,y-Richtung: senkrecht zur Bindungsachse z-Richtung: Bindungsachse
-Bind. -Bind.
py

Instrumentelle Analytik Atom- und Molekülspektren Seite N032_Wechselwirkung_b_BAneu.doc - 6/24
2.2.2.1 Rotation von Molekülen, Rotationsspektren (starrer Rotator)
Die Gesamtenergie von Molekülen ist durch die schnelle Bewegung der Elektronen (elektronische Zustände) und die relativ langsame Bewegung der Kerne bestimmt.
Die Kernbewegung lässt sich wiederum in eine relativ schnelle Schwingungsbewegung, eine langsamere Rotation und die Translation zerlegen.
Für ein zweiatomiges Molekül (Atommassen M1 und M2), reduzierte Masse M und Massenträgheitsmoment I ergibt sich die Rotationsenergie Erot zu
I
JIErot 22
1 22 22
22211 MRRMRMI
21
21
MMMMM
Wegen der Quantisierung des Drehimpulses J erhält man für die Gleichgewichtslage der Atome R = Re die Rotationsenergie
)1(2
)1()(
2
2
JJhcBMR
JJJE ee
rot
mit der “Rotationskonstante“ Be
24 e
e cMRB
[Be] = cm-1
Die Energieabstände benachbarter Rotationsniveaus wachsen mit J an. Bei Wechselwirkung mit elektromagnetischer Strahlung gilt für die Übergänge:
J = 1:
2
2)1()()1(
erotrotrot MR
JJEJEE
)1(2~,1
JB
hcE
erot
JJ
Übergänge treten aber nur auf, wenn die Moleküle ein Dipolmoment besitzen, daher gibt es keine reinen Rotationsspektren bei homonuklearen Molekülen.
Übergangsenergien Erot 10-3 eV
Reine Rotationsübergänge sind für die Laboranalytik aber uninteressant, da sie bei Zimmertemperatur schon thermisch angeregt sind. Absorption und Emission werden von thermischer Strahlung überdeckt (Etherm 0,026 eV).
Beispiel: H2 - Molekül M = MH/2 = 8,410-28 kg, Re = 0,7410-10 m,
I = MR2e = 4,610-48 kgm2
Erot = 1,210-21 J(J+1) Nm = 7 meV J(J+1) Be = 60,8 cm-1
)13(2)34(~ eB
Instrumentelle Analytik Atom- und Molekülspektren Seite N032_Wechselwirkung_b_BAneu.doc - 7/24
2.2.2.2 Molekülschwingungen, Schwingungsspektren
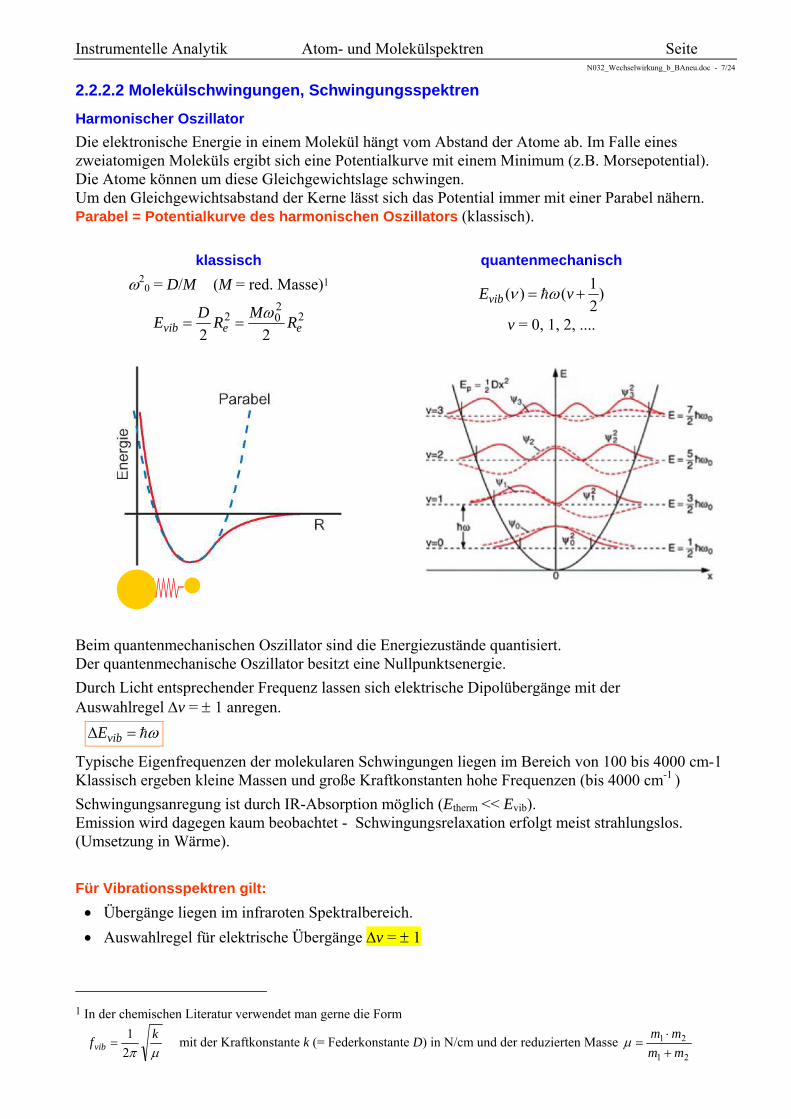
Harmonischer Oszillator
Die elektronische Energie in einem Molekül hängt vom Abstand der Atome ab. Im Falle eines zweiatomigen Moleküls ergibt sich eine Potentialkurve mit einem Minimum (z.B. Morsepotential). Die Atome können um diese Gleichgewichtslage schwingen. Um den Gleichgewichtsabstand der Kerne lässt sich das Potential immer mit einer Parabel nähern. Parabel = Potentialkurve des harmonischen Oszillators (klassisch).
klassisch
20 = D/M (M = red. Masse)1
2202
22 eevib RMRDE
quantenmechanisch
)2
1()( vEvib
v = 0, 1, 2, ....
Beim quantenmechanischen Oszillator sind die Energiezustände quantisiert. Der quantenmechanische Oszillator besitzt eine Nullpunktsenergie.
Durch Licht entsprechender Frequenz lassen sich elektrische Dipolübergänge mit der Auswahlregel v = 1 anregen.
vibE
Typische Eigenfrequenzen der molekularen Schwingungen liegen im Bereich von 100 bis 4000 cm-1 Klassisch ergeben kleine Massen und große Kraftkonstanten hohe Frequenzen (bis 4000 cm-1 )
Schwingungsanregung ist durch IR-Absorption möglich (Etherm << Evib). Emission wird dagegen kaum beobachtet - Schwingungsrelaxation erfolgt meist strahlungslos. (Umsetzung in Wärme).
Für Vibrationsspektren gilt:
Übergänge liegen im infraroten Spektralbereich.
Auswahlregel für elektrische Übergänge v = 1
1 In der chemischen Literatur verwendet man gerne die Form
kfvib 2
1 mit der Kraftkonstante k (= Federkonstante D) in N/cm und der reduzierten Masse
21
21
mmmm

Instrumentelle Analytik Atom- und Molekülspektren Seite N032_Wechselwirkung_b_BAneu.doc - 8/24
Schwingungsanregung durch IR-Absorption erfolgt nur bei Vorhandensein eines dynamischen Dipolmomentes, d.h. ein Schwingungszustand ist IR-aktiv, wenn sich bei der Schwingung das Dipolmoment ändert. (dµ/dr 0). Ein statisches Dipolmomemt muss nicht vorhanden sein, es reicht, wenn sich durch die Schwingungsanregung das Dipolmoment ändert.
intensive Banden im IR-Spektrum bei polaren Molekülen mit Heteroatomen. unpolare Molekülteile ergeben schwache IR-Absorptionsbanden. vollständig symmetrische Schwingungen sind nicht IR-aktiv.
Anharmonischer Oszillator
In Wirklichkeit ist das Potential nicht harmonisch. Bei hohen Anregungsenergien und großer Schwingungsamplitude wird das Potential aufgeweicht und die Energieabstände werden kleiner.
An der Dissoziationsgrenze gehen die Zustände in das Translationskontinuum über.
Übergänge auch zu Oberschwingungen mit v 1 möglich.
Normalschwingungen
Ein mehratomiges Molekül mit N Atomen hat 3N Bewegungsfreiheitsgrade. Drei dieser Freiheitsgrade werden durch die Translation und drei (zwei bei linearen Molekülen) durch die Rotation festgelegt.
Damit verbleiben für die Schwingungsbewegung im molekülfesten Koordinatensystem:
f = 3N - 5 Freiheitsgrade für lineare (gestreckte) Moleküle f = 3N - 6 Freiheitsgrade für alle anderen
Man beobachtet, dass die Atome in einem Molekül nicht völlig irregulär schwingen, sondern genau (3N - 5) bzw. (3N - 6) Normalschwingungen existieren, bei denen sich alle Atome mit derselben Frequenz und fester Phasenlage bewegen (gekoppelte Schwingungen). Durch Überlagerung dieser voneinander unabhängigen Eigenschwingungen kann das gesamte Schwingungsverhalten eines Moleküls erklärt werden.
Beispiel: Normalschwingungen von H2O
H O H f = 3N-6 = 3
Instrumentelle Analytik Atom- und Molekülspektren Seite N032_Wechselwirkung_b_BAneu.doc - 9/24
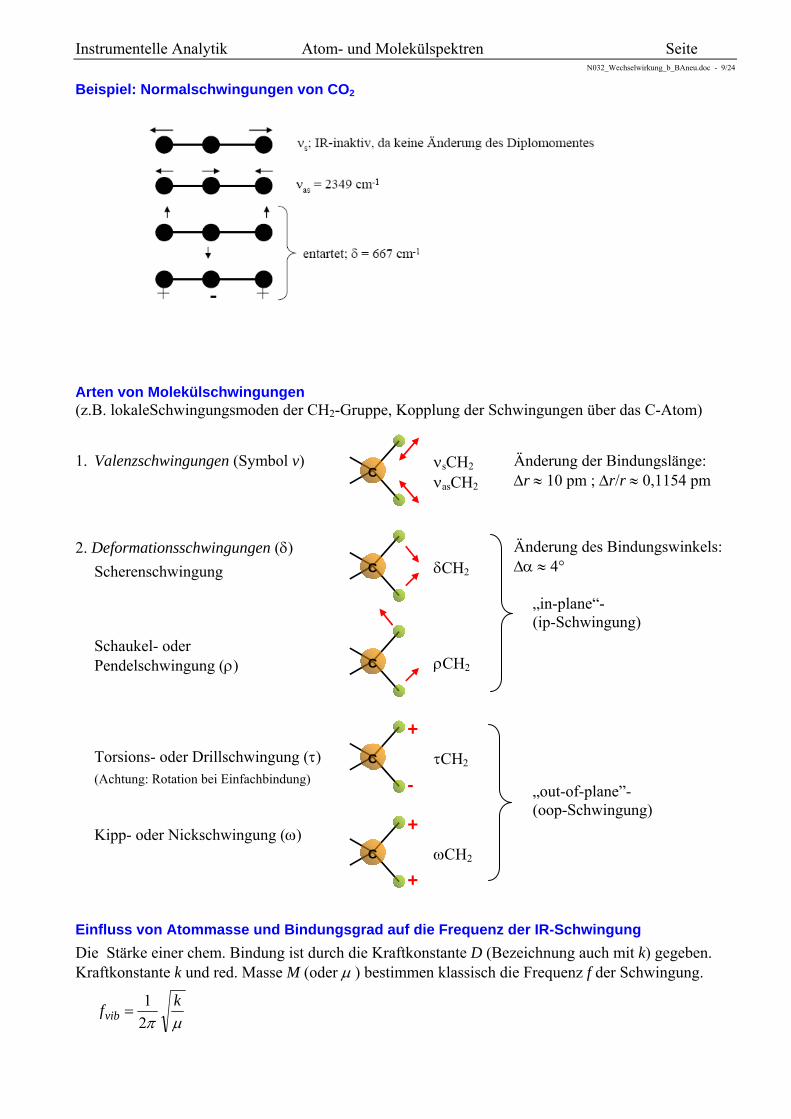
Beispiel: Normalschwingungen von CO2
Arten von Molekülschwingungen (z.B. lokaleSchwingungsmoden der CH2-Gruppe, Kopplung der Schwingungen über das C-Atom)
1. Valenzschwingungen (Symbol v)
Änderung der Bindungslänge: r 10 pm ; r/r 0,1154 pm
2. Deformationsschwingungen ()
Scherenschwingung
Änderung des Bindungswinkels: 4° „in-plane“- (ip-Schwingung)
Schaukel- oder Pendelschwingung ()
Torsions- oder Drillschwingung () (Achtung: Rotation bei Einfachbindung)
„out-of-plane”- (oop-Schwingung)
Kipp- oder Nickschwingung ()
Einfluss von Atommasse und Bindungsgrad auf die Frequenz der IR-Schwingung
Die Stärke einer chem. Bindung ist durch die Kraftkonstante D (Bezeichnung auch mit k) gegeben. Kraftkonstante k und red. Masse M (oder ) bestimmen klassisch die Frequenz f der Schwingung.
kfvib 2
1
CsCH2 asCH2
C CH2
C CH2
C CH2
C CH2
+
-
+
+

Instrumentelle Analytik Atom- und Molekülspektren Seite N032_Wechselwirkung_b_BAneu.doc - 10/24
Schematischer Aufbau eines IR-Spektrums
Schwingungsbanden funktioneller Gruppen und lokalisierte Schwingungen (Charakteristische Banden)
Schwingungen betreffen streng genommen immer ein Molekül als Ganzes. In guter Näherung lassen sich jedoch viele Schwingungen eines Moleküls isoliert betrachten.
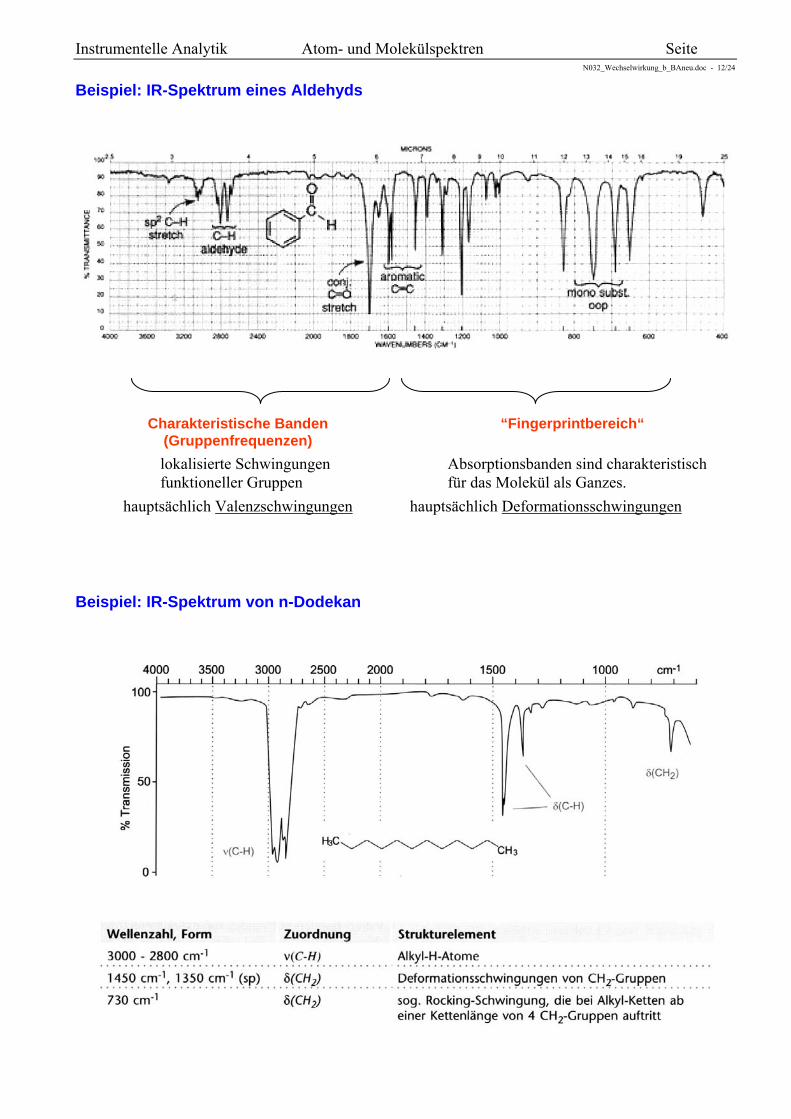
Die Methylengruppe CH2 eines Moleküls schwingt dann so, dass der Schwerpunkt aller drei Atome in Ruhe bleibt, d.h., bewegen sich die beiden H-Atome auf das C zu, so bewegt sich auch das C ein wenig auf die H-Atome zu. Da die H-Atome aber wesentlich leichter sind, als die C-Atome, bewegt sich das C-Atom viel weniger. Es befindet sich annähernd in Ruhe (siehe Beispiel n-Dodekan).
Für die Interpretation von Spektren sind besonders diese lokalisierten Schwingungen nützlich, die sich auf eine funktionelle Gruppe oder Einzelbindungen beschränken. Sie besitzen charakteristische Absorptionsfrequenzen in einem typischen, schmalen Bereich und sind damit charakteristisch für die funktionelle Gruppe. Die Frequenzen sind annähernd konstant und nur wenig von ihrer chemischen Umgebung im Molekül abhängig.
mit zunehmender Atommasse nimmt die Frequenz ab. (2 ~ D/M)
mit zunehmendem Bindungsgrad nimmt die Kraftkonstante und damit die Frequenz zu.
D [N/cm]
4,1
9,6
15,6
Instrumentelle Analytik Atom- und Molekülspektren Seite N032_Wechselwirkung_b_BAneu.doc - 11/24
Kennzeichnung von lokalisierten Schwingungen und benutzte Symbole: vs (CH2) symmetrische (C-H)-Valenzschwingung einer CH2-Gruppe vas (CH2) asymmetrische (C-H)-Valenzschwingung einer CH2-Gruppe δs (CH3) symmetrische (C-H)-Deformationsschwingung einer CH3-Gruppe δas
(CH3) asymmetrische (C-H)-Deformationsschwingung einer CH3-Gruppe
Die lokalisierten Schwingungen funktioneller Gruppen werden in geringem Maße von den restlichen Molekülteilen beeinflusst (Kopplung) und führen zu Verschiebungen im Spektrum, die wiederum Rückschlüsse auf die Struktur des Molekülrestes zulässt.
Beispiel: Die Absorption der C=O Doppelbindung einer Carboxylgruppe (-COOH) liegt je nach Umgebung im Bereich von 1680...1760 cm-1.
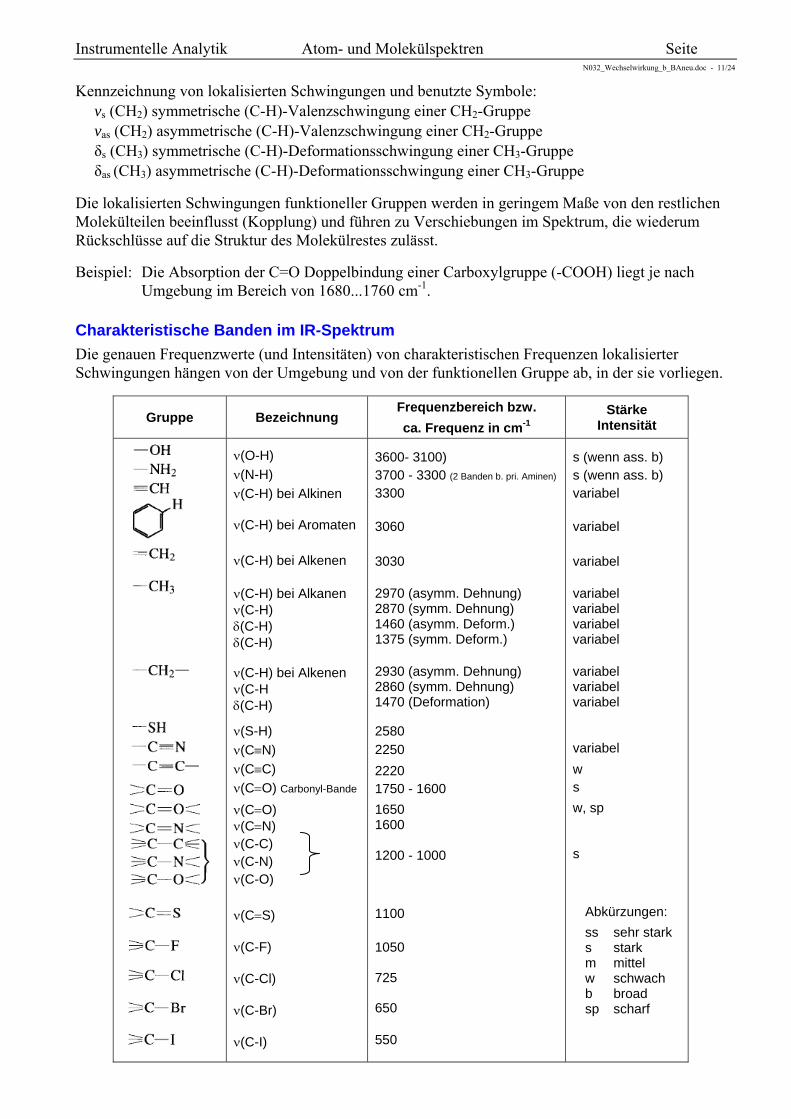
Charakteristische Banden im IR-Spektrum
Die genauen Frequenzwerte (und Intensitäten) von charakteristischen Frequenzen lokalisierter Schwingungen hängen von der Umgebung und von der funktionellen Gruppe ab, in der sie vorliegen.
Gruppe Bezeichnung
Frequenzbereich bzw.
ca. Frequenz in cm-1 Stärke
Intensität
(O-H) (N-H) (C-H) bei Alkinen (C-H) bei Aromaten (C-H) bei Alkenen (C-H) bei Alkanen (C-H) (C-H) (C-H) (C-H) bei Alkenen (C-H (C-H)
(S-H) (CN) (CC) (CO) Carbonyl-Bande (CO) (CN) (C-C) (C-N) (C-O) (CS) (C-F) (C-Cl) (C-Br) (C-I)
3600- 3100) 3700 - 3300 (2 Banden b. pri. Aminen) 3300 3060 3030 2970 (asymm. Dehnung) 2870 (symm. Dehnung) 1460 (asymm. Deform.) 1375 (symm. Deform.) 2930 (asymm. Dehnung) 2860 (symm. Dehnung) 1470 (Deformation) 2580 2250 2220 1750 - 1600 1650 1600 1200 - 1000 1100 1050 725 650 550
s (wenn ass. b) s (wenn ass. b) variabel variabel variabel variabel variabel variabel variabel variabel variabel variabel variabel w s w, sp s
Abkürzungen: ss sehr stark s stark m mittel w schwach b broad sp scharf
Instrumentelle Analytik Atom- und Molekülspektren Seite N032_Wechselwirkung_b_BAneu.doc - 12/24
Beispiel: IR-Spektrum eines Aldehyds
Charakteristische Banden (Gruppenfrequenzen)
“Fingerprintbereich“
lokalisierte Schwingungen funktioneller Gruppen
hauptsächlich Valenzschwingungen
Absorptionsbanden sind charakteristisch für das Molekül als Ganzes.
hauptsächlich Deformationsschwingungen
Beispiel: IR-Spektrum von n-Dodekan

Instrumentelle Analytik Atom- und Molekülspektren Seite N032_Wechselwirkung_b_BAneu.doc - 13/24
Beispiel: IR-Spektrum von Lidocain
Instrumentelle Analytik Atom- und Molekülspektren Seite N032_Wechselwirkung_b_BAneu.doc - 14/24
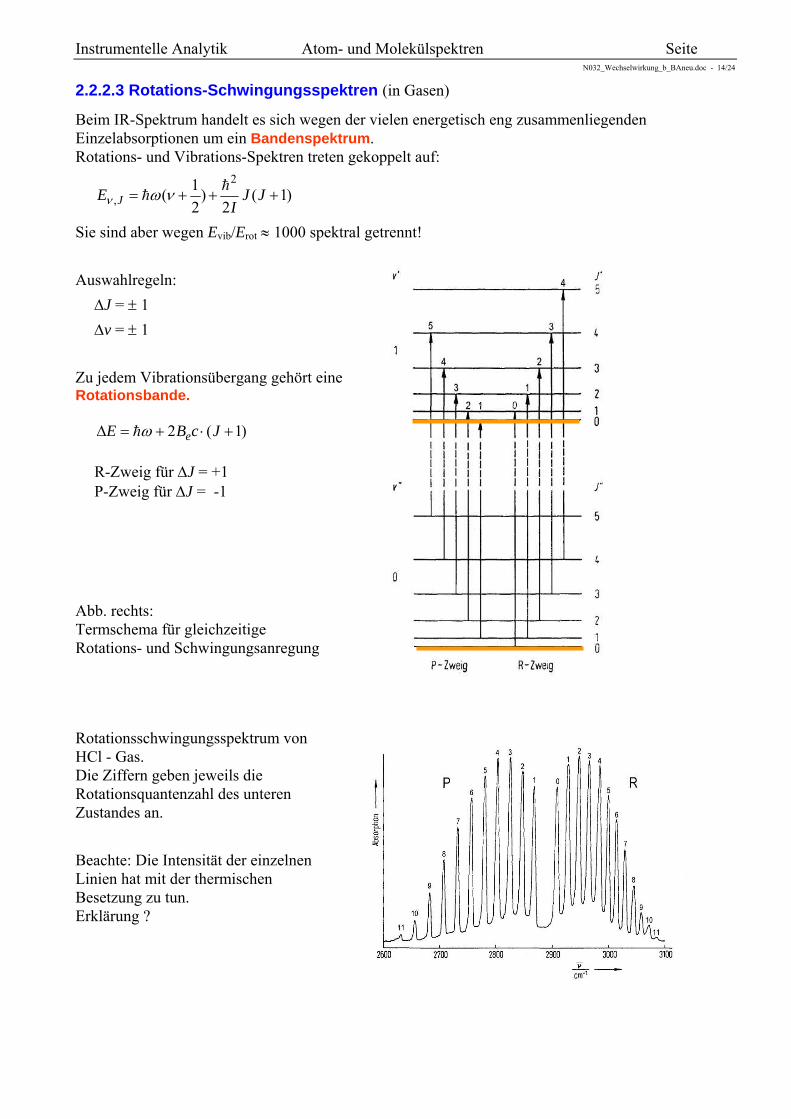
2.2.2.3 Rotations-Schwingungsspektren (in Gasen)
Beim IR-Spektrum handelt es sich wegen der vielen energetisch eng zusammenliegenden Einzelabsorptionen um ein Bandenspektrum. Rotations- und Vibrations-Spektren treten gekoppelt auf:
)1(2
)2
1(
2
, JJI
E J
Sie sind aber wegen Evib/Erot 1000 spektral getrennt!
Auswahlregeln:
J = 1
v = 1
Zu jedem Vibrationsübergang gehört eine Rotationsbande.
)1(2 JcBE e
R-Zweig für J = +1 P-Zweig für J = -1
Abb. rechts: Termschema für gleichzeitige Rotations- und Schwingungsanregung
Rotationsschwingungsspektrum von HCl - Gas. Die Ziffern geben jeweils die Rotationsquantenzahl des unteren Zustandes an.
Beachte: Die Intensität der einzelnen Linien hat mit der thermischen Besetzung zu tun. Erklärung ?

Instrumentelle Analytik Atom- und Molekülspektren Seite N032_Wechselwirkung_b_BAneu.doc - 15/24
2.2.2.3 Ramanstreuung und Rayleighstreuung
Rayleighstreuung Das elektrische Feld der Lichtwelle regt die Elektronen in Atomen oder Molekülen zu erzwungenen Schwingungen an. Sie schwingen gegen den viel schwereren Kern als Ladungswolke hin und her und bilden einen schwingenden Dipol.
Für das induzierte Dipolmoment gilt:
tEE e sinˆ
Der Dipol strahlt mit der gleichen Frequenz eine Welle ab. Für die Intensität gilt:
4StreuI
Ramanstreuung
Bei intensiver Bestrahlung (Laser) einer molekularen Probe, wird neben der eingestrahlten Frequenz auch eine Reihe von verschobenen Frequenzen im Streulicht gefunden. Diese inelastische Streuung lässt sich als Energieaustausch mit den Schwingungszuständen des Moleküls erklären.
Wenn die Atomkerne Schwingungen ausführen, werden die Bindungen gestaucht und gedehnt. Damit ändert sich die Polarisierbarkeit der Elektronen, d.h. sie sind bei den Extrempositionen der Kerne unterschiedlich leicht verschiebbar.
Die Polarisierbarkeit ändert sich periodisch mit den Schwingungsbewegungen im Molekül.
tvib sin10
])cos()[cos(ˆ2
sinˆsinˆ)sin( 1010 ttEEtEt vibevibeeevib
vibeRaman fff
Die Polarisation P = n strahlt zwei zusätzlichen Frequenzen ab
unterschiedliche Polarisierbarkeit

Instrumentelle Analytik Atom- und Molekülspektren Seite N032_Wechselwirkung_b_BAneu.doc - 16/24
Ein Schwingungszustand eines Moleküls ist Raman-aktiv, wenn sich bei der Schwingung die Polarisierbarkeit ändert. (d/dr 0)
Quantenmechanische Deutung des Ramaneffektes
Absorption in beliebige virtuelle Niveaus zwischen dem elektronischen Grundzustand Ek und dem 1. angeregten Zustand Ej. Die mittlere Aufenthaltsdauer in den virtuellen Niveaus beträgt nur etwa 10-13 s. Emission bei gleichzeitiger Erzeugung/Vernichtung eines Schwingungszustandes.
Zusammenfassung
Rotationsspektroskopie spielt in der Instrumentellen Analytik keine Rolle: Kann in der Regel nur in gasförmigem Zustand durchgeführt werden und erfordert einem hohen apparativen Aufwand (Kryotechnik).
IR- Spektroskopie (MIR) ist in der Regel eine absorptionsspektroskopische Methode. Erfasst Molekülschwingungen (Schwingungsrelaxation strahlungslos).
Ein Molekül mit N Atomen besitzt f = 3N-6 (3N-5) verschiedene Eigenschwingungen.
Eine Schwingung ist dann IR-aktiv, wenn sich im Verlauf der Schwingung das Dipolmoment des Moleküls ändert. Polare funktionelle Gruppen mit Heteroatomen führen deshalb zu intensiven Banden. Unpolare Moleküle ergeben schwache Banden.
Ramanspektroskopie ist eine emissionsspektroskopische Methode.
Eine Schwingung ist dann raman-aktiv, wenn sich im Verlauf der Schwingung die Polarisierbarkeit des Moleküls ändert.
Ramanstreuung beruht auf einem 2-Photonenprozess.
Die Intensität des Ramanstreulichtes ist extrem schwach.
IR-Spektroskopie und Ramanspektroskopie ergänzen sich gegenseitig. Häufig sind IR-aktive Schwingungen ramanverboten und umgekehrt (Komplementarität).
IR-Spektrometrie Information über funktionelle Gruppen Raman-Spektrometrie Information über Gerüstaufbau, Elektronenverteilung der C-Atome
Ej Ej
Ek Ek
Ei Ei
hS hAS
Stokes Strahlung
Anti-Stokes Strahlung

Instrumentelle Analytik Atom- und Molekülspektren Seite N032_Wechselwirkung_b_BAneu.doc - 17/24
IR-Spektrometrie Raman-Spektrometrie
IR-Aktiv Raman-Aktiv
IR-Absorption wird beobachtet, wenn während einer Schwingung eine Änderung des Dipolmomentes im Molekül eintritt.
Raman-Streuung wird beobachtet, wenn sich während einer Schwingung die Polarisierbarkeit des Moleküls ändert.
Die Intenstät der IR-Schwingungsbande ist proportional zum Quadrat der Änderung des Dipolmomentes mit der Normalkoordinate q.
2
0
q
I IR
Die Intenstät der Raman-Bande ist proportional zum Quadrat der Änderung der Polarisierbarkeit mit der Normalkoordinate q.
2
0
q
IRaman
Auswahlregeln:
Allgemein gilt für ein Molekül mit (Punkt-)Symmetriezentrum das sog. Alternativverbot.
Alle Schwingungen symmetrisch zum Symmetriezentrum sind im IR-Spektrum verboten.
Alle Schwingungen antisymmetrisch zum Symmetriezentrum sind im Raman-Spektrum verboten.
Ist kein Symmetriezentrum vorhanden, so können einige (aber nicht unbedingt alle) Schwingungen sowohl Raman-, wie auch IR-aktiv sein.
Generell kann gesagt werden, dass die Infrarot-Spektrometrie mehr über funktionelle Gruppen aussagt und die Raman-Spektrometrie hilfreich für die Charakterisierung des Kohlenstoffgerüsts ist (warum?).
Molekül (z.B. CO2)
Schwingungsmode
Variation von
als Funktion der Normalkoordinate q
(= Auslenkung)
q
q
q
Ableitung von Raman-aktiv ?
0 Ja
= 0 Nein
= 0 Nein
Variation von als Funktion der
Normalkoordinate q
q
q
q
Ableitung von IR-aktiv ?
= 0 Nein
0 Ja
0 Ja
-
+ - -
+ - - + - - +-

Instrumentelle Analytik Atom- und Molekülspektren Seite N032_Wechselwirkung_b_BAneu.doc - 18/24
2.2.2.4 Molekülspektren (elektronische Spektroskopie)
Bei der optischen Anregung eines Moleküls mit sichtbarem oder UV-Licht werden im allgemeinen viele Freiheitsgrade (Zustände) gleichzeitig verändert.
Die Änderung des elektronischen Zustandes ergibt analog zu den Atomen die größten Energiedifferenzen und bestimmt die Erscheinung des Spektrums.
Die Änderung der Schwingungsbewegung (Zustand) beim elektronischen Übergang ergibt für jeden elektronischen Übergang eine stark strukturierte Bande aus Übergängen zwischen vibronischen Zuständen.
Zusätzlich kann durch eine Änderung der Rotation beim Übergang eine Bande von rovibronischen Übergängen beobachtet werden. Dies tritt aber nur in der Gasphase auf. In Flüssigkeiten sind Rotationen stark behindert.
Elektronische Übergänge in vielatomigen Molekülen
Für die Spektroskopie (UV/VIS) sind nur die am weitesten von den Atomkernen entfernten Elektronen/Orbitale und deren Zustände von Interesse, da nur sie angeregt werden können . Für die tiefer liegenden Elektronen ist mehr Energie erforderlich.
Absorptionen können durch n-, -, und -Elektronen hervorgerufen werden.
Übergänge im UV/VIS-Breich (200 - 800 nm) erfolgen von * oder von n *
(n * oder * Übergänge sind i.a. schwach oder bereits im VUV.)
Schema der vier möglichen Elektronenübergänge. Die Dicke der Pfeile deutet die Intensität des Übergangs an.
Übergänge sind dann besonders intensiv, wenn die Orbitale von Anfangs- und Endzustand stark überlappen und die Auswahlregeln erfüllt sind.
Instrumentelle Analytik Atom- und Molekülspektren Seite N032_Wechselwirkung_b_BAneu.doc - 19/24
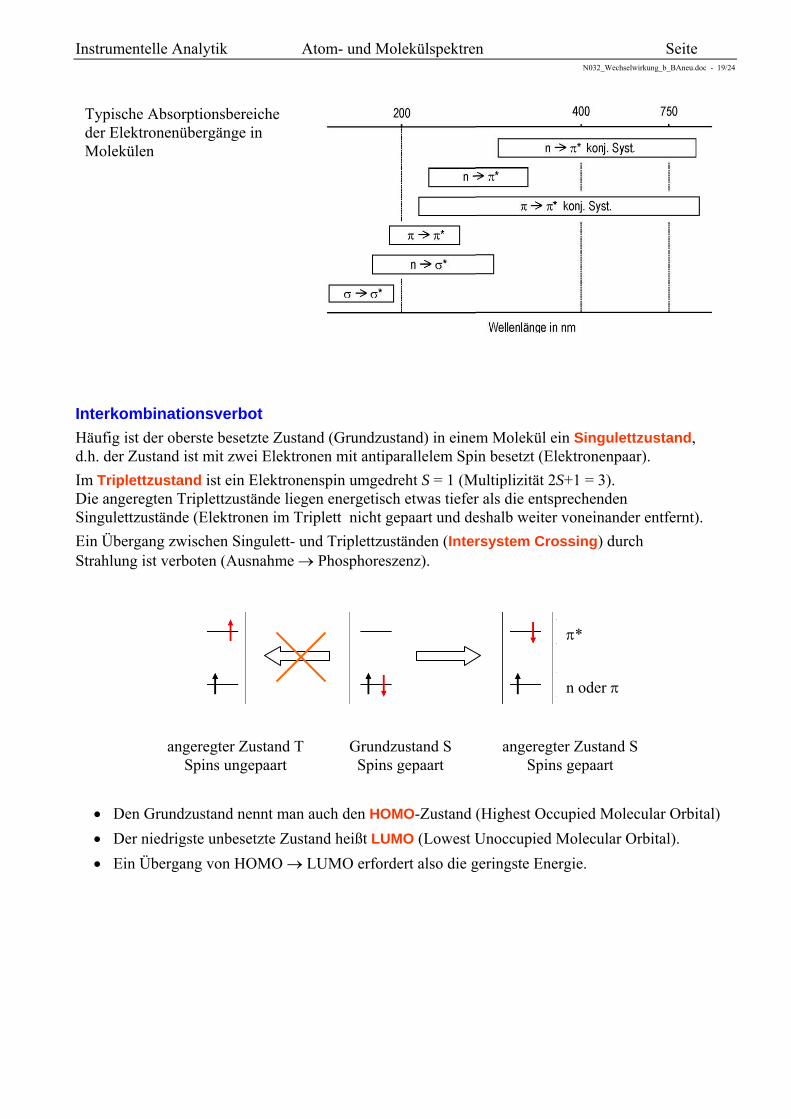
Typische Absorptionsbereiche der Elektronenübergänge in Molekülen
Interkombinationsverbot
Häufig ist der oberste besetzte Zustand (Grundzustand) in einem Molekül ein Singulettzustand, d.h. der Zustand ist mit zwei Elektronen mit antiparallelem Spin besetzt (Elektronenpaar).
Im Triplettzustand ist ein Elektronenspin umgedreht S = 1 (Multiplizität 2S+1 = 3). Die angeregten Triplettzustände liegen energetisch etwas tiefer als die entsprechenden Singulettzustände (Elektronen im Triplett nicht gepaart und deshalb weiter voneinander entfernt).
Ein Übergang zwischen Singulett- und Triplettzuständen (Intersystem Crossing) durch Strahlung ist verboten (Ausnahme Phosphoreszenz).
angeregter Zustand T Spins ungepaart
Grundzustand S Spins gepaart
angeregter Zustand S Spins gepaart
Den Grundzustand nennt man auch den HOMO-Zustand (Highest Occupied Molecular Orbital)
Der niedrigste unbesetzte Zustand heißt LUMO (Lowest Unoccupied Molecular Orbital).
Ein Übergang von HOMO LUMO erfordert also die geringste Energie.
*
n oder

Instrumentelle Analytik Atom- und Molekülspektren Seite N032_Wechselwirkung_b_BAneu.doc - 20/24
Systematik der Molekülspektren Jablonski-Schema: Schematische Darstellung der Energieniveaus (und möglichen Übergänge) in einem Molekül. Unter Beachtung der geltenden Auswahlregeln können alle möglichen Anregungsübergänge und Emissionsübergänge dem Jablonski-Schema entnommen werden.
S0
S1
S2
T1
T2
Moleküle können nach dem Jablonski-Schema auf unterschiedliche Art angeregt werden. Je nachdem ob zusätzlich zu den Elektronen auch noch Schwingungen und Rotationen angeregt werden, ist die Energie der Absorption unterschiedlich. Dies führt im Spektrum zu den breiten Banden.
)1()(2
2
2
1 JJEEEEEIerotvibeges
Für die Übergänge gilt: rotibe EEEE
mit: 12
1122
12 )()( vibE
)1()1( 112
2
222
2
12
JJJJEIIrot
Dabei gelten die Auswahlregeln
J = 1 (Rotationsquantenzahl ! nicht Gesamtdrehimpuls) S = 0 (Interkombinationsverbot) v nach Franck-Condon-Prinzip
*
n bzw A =
R =
IC =
Fl =
ISC =
Ph =
Instrumentelle Analytik Atom- und Molekülspektren Seite N032_Wechselwirkung_b_BAneu.doc - 21/24
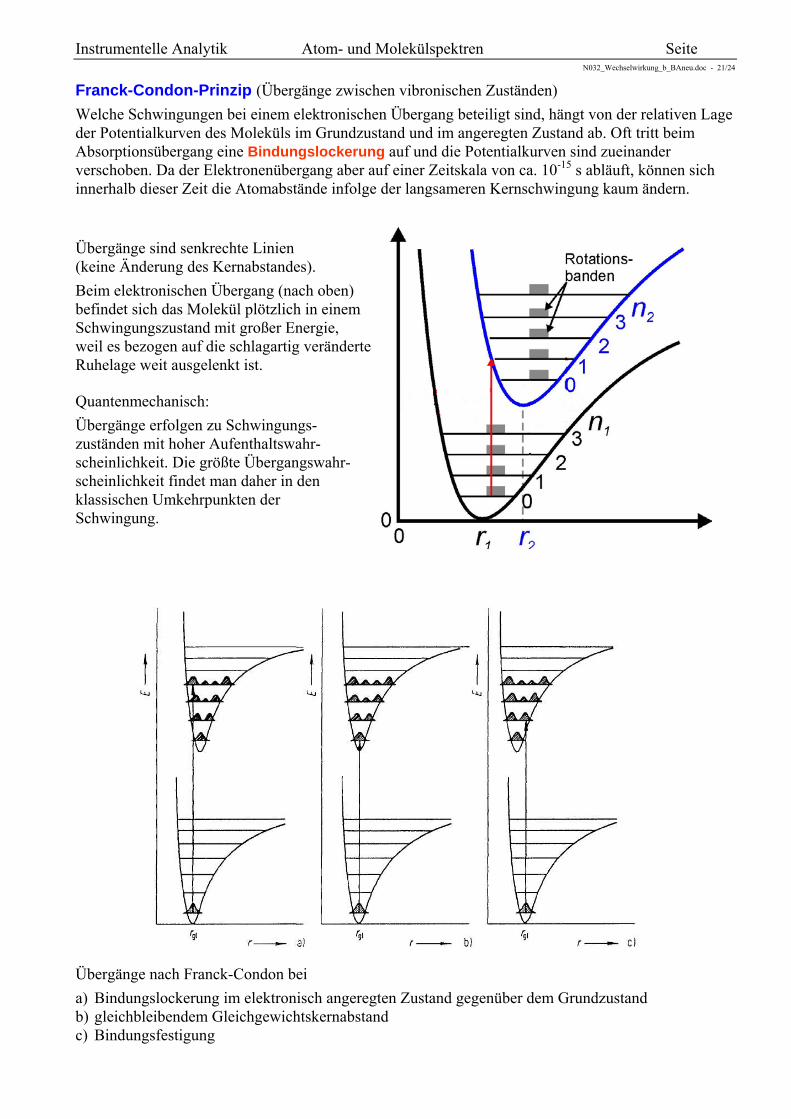
Franck-Condon-Prinzip (Übergänge zwischen vibronischen Zuständen)
Welche Schwingungen bei einem elektronischen Übergang beteiligt sind, hängt von der relativen Lage der Potentialkurven des Moleküls im Grundzustand und im angeregten Zustand ab. Oft tritt beim Absorptionsübergang eine Bindungslockerung auf und die Potentialkurven sind zueinander verschoben. Da der Elektronenübergang aber auf einer Zeitskala von ca. 10-15 s abläuft, können sich innerhalb dieser Zeit die Atomabstände infolge der langsameren Kernschwingung kaum ändern.
Übergänge sind senkrechte Linien (keine Änderung des Kernabstandes).
Beim elektronischen Übergang (nach oben) befindet sich das Molekül plötzlich in einem Schwingungszustand mit großer Energie, weil es bezogen auf die schlagartig veränderte Ruhelage weit ausgelenkt ist.
Quantenmechanisch:
Übergänge erfolgen zu Schwingungs- zuständen mit hoher Aufenthaltswahr- scheinlichkeit. Die größte Übergangswahr- scheinlichkeit findet man daher in den klassischen Umkehrpunkten der Schwingung.
Übergänge nach Franck-Condon bei
a) Bindungslockerung im elektronisch angeregten Zustand gegenüber dem Grundzustand b) gleichbleibendem Gleichgewichtskernabstand c) Bindungsfestigung
Instrumentelle Analytik Atom- und Molekülspektren Seite N032_Wechselwirkung_b_BAneu.doc - 22/24
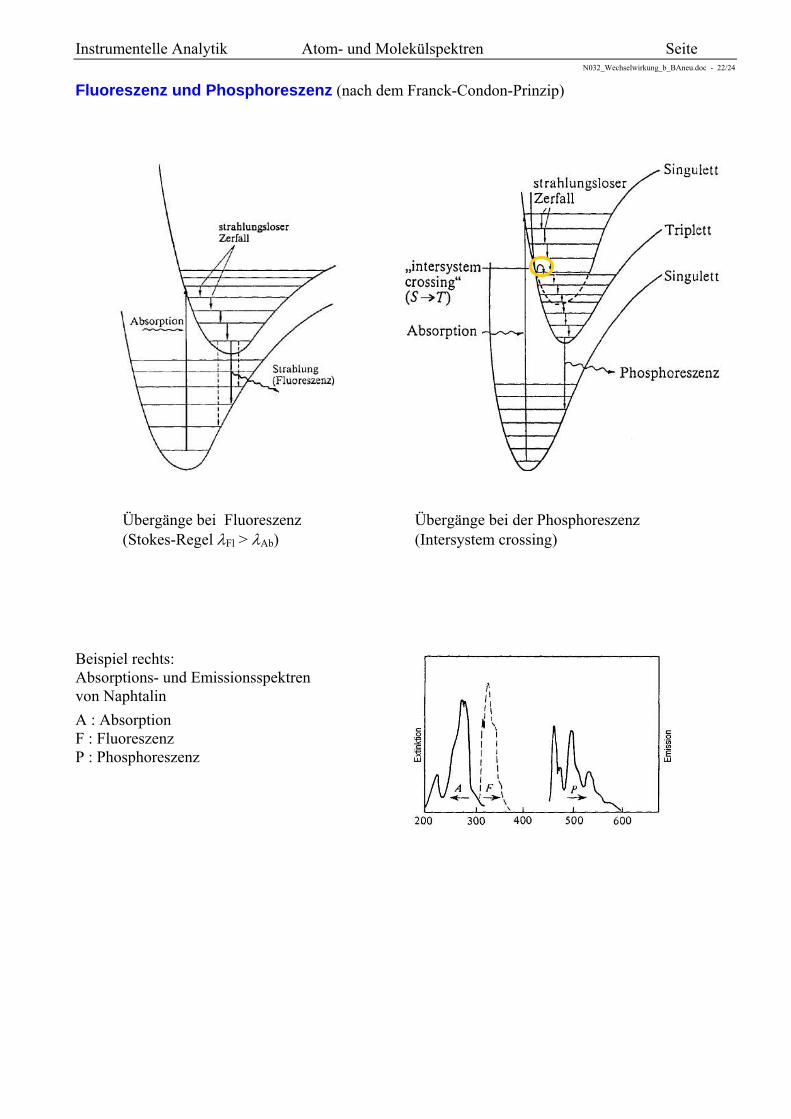
Fluoreszenz und Phosphoreszenz (nach dem Franck-Condon-Prinzip)
Übergänge bei Fluoreszenz Übergänge bei der Phosphoreszenz (Stokes-Regel Fl > Ab) (Intersystem crossing)
Beispiel rechts: Absorptions- und Emissionsspektren von Naphtalin
A : Absorption F : Fluoreszenz P : Phosphoreszenz

Instrumentelle Analytik Atom- und Molekülspektren Seite N032_Wechselwirkung_b_BAneu.doc - 23/24
Zusammenfassung: Prozesse Strahlungsabsorption und Emission
Instrumentelle Analytik Atom- und Molekülspektren Seite N032_Wechselwirkung_b_BAneu.doc - 24/24
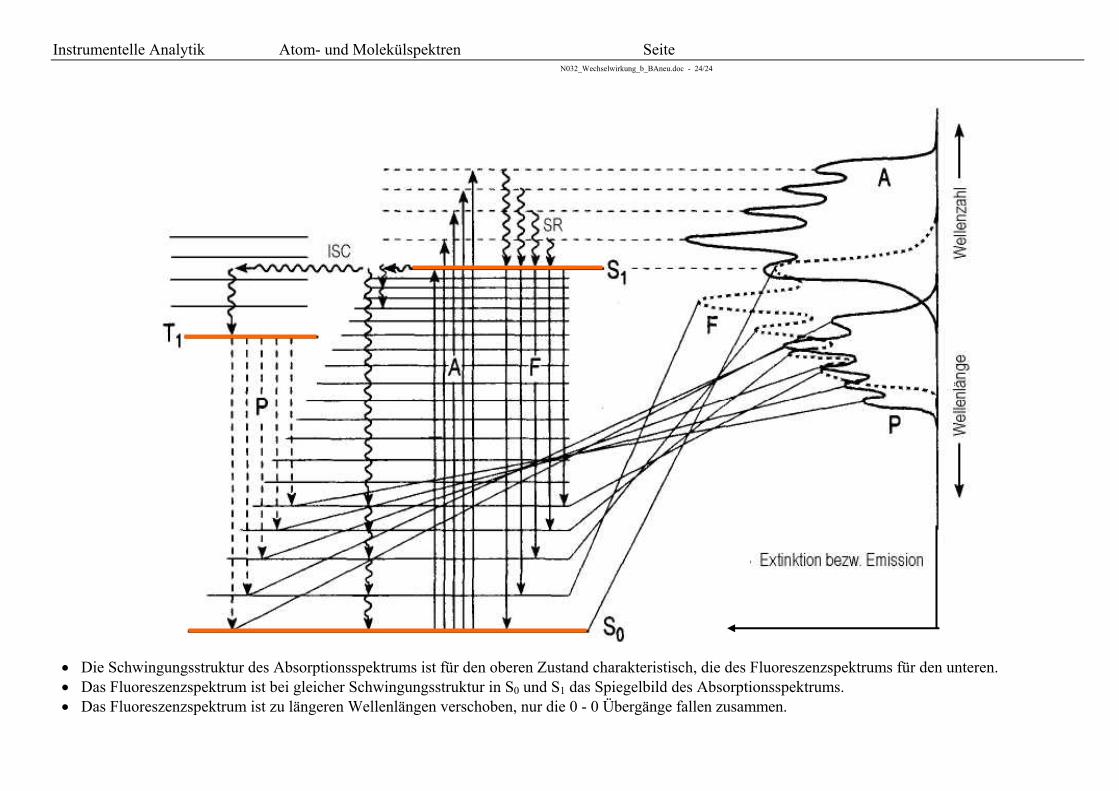
Die Schwingungsstruktur des Absorptionsspektrums ist für den oberen Zustand charakteristisch, die des Fluoreszenzspektrums für den unteren. Das Fluoreszenzspektrum ist bei gleicher Schwingungsstruktur in S0 und S1 das Spiegelbild des Absorptionsspektrums. Das Fluoreszenzspektrum ist zu längeren Wellenlängen verschoben, nur die 0 - 0 Übergänge fallen zusammen.




























![[Psilocybin de]Entheogene Blaetter Juli 07-2002 Drogen Thc Psilocybin Psilocin Xtc](https://img.pdfslide.net/doc/110x75/577dab601a28ab223f8c57f4/psilocybin-deentheogene-blaetter-juli-07-2002-drogen-thc-psilocybin-psilocin.jpg)
