Embed Size (px)
Citation preview

Pergamon Neuromusc. Disord.. Vol. 4, No. 2, pp. 139-142. 1994
Copyright (' 1994 Elsevier Science Ltd Pnnted in Great Britain. All rights reserved
0960,-8966,'94 $6.00 + .0~
A D A N I S H F A M I L Y W I T H L I M B - G I R D L E M U S C U L A R D Y S T R O P H Y
W I T H A U T O S O M A L D O M I N A N T I N H E R I T A N C E
J. F. NIELSEN* and J. JAKOBSEN Department of Neurology, Aarhus University Hospital. Aarhus. Denmark
( Received 22 April 1993: revi.wd 28 June 1993.' accepted 16 Ju(v 1993)
Abstract--We describe a Danish family of five generations showing a slowly progressive autosomal dc~minant limb-girdle myopathy with onset in early childhood. Three patients from two generations were examined and showed weakness predominantly of the proximal muscles of upper and lower extremities. Early contractures of wrist, tight heel cords, metacarpophalangeal and interphalangeal joints were present. Walking ability was partly preserved. Serum creatine kinase activity was normal or slightly elevated. EMG and biopsy showed mildly myopathic changes. Magnetic resonance images of the thigh showed severe atrophy of all compartments but with musculus semimembranosus, sartorius, and gracilis relatively preserved. This myopathy has a clinical picture similar to the Bethlem myopathy.
Key words: Limb-girdle muscular dystrophy, autosomal dominant inheritance, contractures, Beihlem myopathy.
I N T R O D U C T I O N
Limb-girdle muscular dystrophy (LGMD) has normally autosomal recessive inheritance: how- ever, some autosomal dominant (AD) cases have been reported in recent years in the literature. In 1976, Bethlem and van Wijngaarden [1] described three unrelated Dutch families, where the onset of limb-girdle weakness occurred in childhood with slow progression and widespread contractures. Other authors have reported families with limb-girdle muscular dystrophy with autosomal inheritance with manifest symptoms in adulthood [2-6] or in childhood [7 9]. We describe a Danish family with a slowly progressing myopathy appearing in early childhood that is clinically predominantly restricted to proximal limb muscles characterized by early contractures. This myopathy has a clinical picture similar to the myopathy described by Bethlem and van Wijngaarden.
P A T I E N T S A N D M E T i l O D S
Five generations of 34 members have been identified. Eleven members are known to have
* A u t h o r to w h o m c o r r e s p o n d e n c e shou ld be a d d r e s s e d at: Neurologisk afd . F, ~ r h u s K o m m u n e h o s p i t a l , N o r r e b r o n a d e 44. 8 0 0 0 / ~ r h u s C, D e n m a r k .
been affected, of whom three are alive (Fig. 1). The three members of the family still alive were interviewed, clinically examined, and evaluated with magnetic resonance images, serum creatine kinase (CK) determinations, and with EMG and muscle biopsy. Information concerning affected members of generation I, II and Ill was almost exclusively obtained from subject IV2.
Magnetic resonance imaging (MR[)
With the Philips Gyroscan SI 5 cross-sectional images of the thigh musculature were obtained from three patients.
Musch, biopsy
A biopsy was obtained in two of the three individuals in 1941 and 1961, respectively, Tis- sues from the patient VI were resectioned in 1992 and stained with haematoxylin-eosin, PAS, Gomori, and Well.
Electrono'ograph.v
Three patients underwent routine needle elec- tromyographic studies using standard techniques.
R E S U L T S
Pedi,gree anah'sis
II12 died at the age of 67. She suffered from
139

140 J, F. NIELSEN and J. JAK~ aSEN r ,
Iv
v t¢ 1¢
v! ?
• ta.I~t.LAR SYI~TOMB/OI~At~
O NO Mi~O.II.~dcl 8YMPI'OM8
Fig. 1. Pedigree data of a family with limb-girdle muscular dystrophy in five generations.
mild muscle weakness but retained the capability to walk with the help of a stick. 1116 could not climb stairs from early childhood and became a wheelchair user at the age of 50. Her daughter IV5 also suffered from mild muscle weakness. In the first generation 12 was affected but worked as a postman and died in old age. One of his sons, 114, was affected but practised as a tailor until he died at the age of 64. He had severe difficulties climbing stairs to his workshop.
Clinical evaluation
Subjects IV2, VI, and V2 were born after an uneventful pregnancy. They walked at the age of 1-2 years. At the age of 3--4 years they gradually developed a tendency to fall and difficulties in getting up and climbing stairs. In childhood they gradually developed pes equinovarus, tight heel cords leading to toe walking, as well as contractures of the metacarpophalangeal, interphalanageal, and metatarsophalangeal joints and/or the wrist. At the age of 2, IV2 developed a left-sided torticollis. In adulthood they all gradually developed mild contractures of the knee, hip, ankle, and elbow.
IV2 is 64 yr old and retired from her position as high-school teacher because of muscle weakness. She still has the capability to walk with the help of sticks. Her son VI retired as a joiner at the age of 30 because of troublesome contractures. He can still walk miles without any support. V2 works as a counsellor in the Danish organization for patients with muscular dystrophy. He can still walk, but prefers to use a wheelchair.
At clinical examination they were all slender with atrophy especially of thigh and gluteal muscles, and with slight hyperlordosis of the back. Weakness was predominantly of the prox- imal muscles of upper and lower extremities, especially the hip flexors and knee extensors. Neck flexors revealed a moderate weakness. Facial musculature and sensation for touch, pain and vibration were preserved.
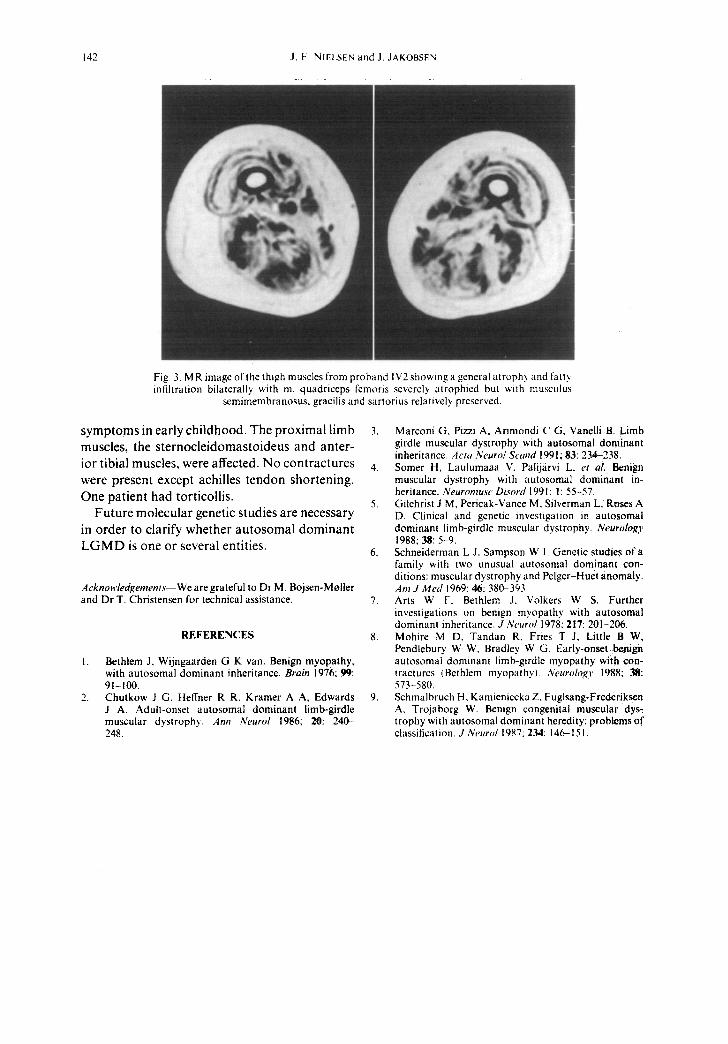
MRI. The images of the thighs obtained in 1992 show a general atrophy of all compartments but with a characteristic distribution of the musculature seen in all three probands with m. quadriceps femoris severely atrophied and the knee flexors, adductors, m. gracilis, and m. sartorius relatively preserved (Fig. 2).
EMG. Muscles examined by EMG in proband V2 at the age of 18 included m. deltoideus sin. and m. vastus medialis dxt. No spontaneous activity was seen. On voluntary activity short duration potentials 7.5 ms (normal values: 11.6 4- 0.5 ms), and 6.0 ms (normal values: 11.5 4- 0.8 ms), respectively, with small amplitudes ap- peared. No fasciculations or fibrillations were observed. Proband VI at the age of 4 yr and proband IV at the age of ! 3 yr showed an almost normal EMG.
Creatine kinase analysis. Serum CK con- centration was normal or mildly devated 120--228 U 1 - I (range), 124--448 U 1- i, and 235--555 U I- 1 in IV2, V2, and VI, respectively (normal values: female < 150 U 1-~, male < 270 U 1-1.
Biopsy. Biopsy undertaken from proband V1 at the age of 4 yr showed mild myopathic changes

Limb-girdle Muscular Dystrophy 141
"It
41 8' •11
q~
f, , d i p
9
w
- ~ " Q qD t •
Fig. 2. Muscle biopsy from musculus vastus lateralis (proband V1) stained with haematoxylin-eosin showing a mild myopathic picture with small variation in muscle fibre diameter, slender fibres, fatty tissue infiltration and several central nuclei. No cellular infiltrates were present. (HE, x
240.)
(Fig. 3). Small variations in muscle fibre diameter with slender fibres were present together with fatty tissue infiltration and scattered necrotic fibres. No cellular infiltrates were pre- sen t.
Cardiological examinat ions . Clinical exam- ination of the three probands: cardiac aus- cultation, thoracic X-rays, and standard ECG recordings showed no signs of cardiomyopathy or cardiac arrhythmias.
DISCUSSION
Characteristic features in this family were: (!) onset of symptoms during early childhood; (2) early joint contractures; (3) generalized muscle weakness: (4) normally or slightly elevated CK; (5) mild myopathic changes at biopsy and EMG: and (6) absence of cardiac involvement. Pedigree analysis shows affected probands of both sexes in successive generations suggesting autosomal dominant inheritance. Other dom- inantly inherited muscular dystrophies such as
facioscapulohumeral muscular dystrophy, myo- tonic dystrophy, scapulohumeral muscular dystrophy of Seitz can be excluded because of the clinical and electrophysiological findings. The lack of facial involvement with primary arm weakness excludes facioscapulohumeral muscular dystrophy. No clinical or electro- physiological signs of myotonia exclude myotonic dystrophy. The lack of cardiac involvement excludes scapulohumeral muscular dystrophy of Seitz. The existence of limb-girdle muscular dystrophy with autosomal inheritance is now well established. However, it is obvious that the clinical diagnosis of limb-girdle muscular dystrophy includes several disease entities. As suggested by Mohire et al. [8], there is a group of LGMD often called "'Bethlem myopathy", and, similar to the Danish patients described in the present report, characterized by early onset of symptoms, a relatively benign course, contractures especially of the ankle, elbow, interphalangeal joints, and a high frequency of torticollis. Bethlem and van Wijngaarden described three Dutch families with slowly progressing muscle weakness with proximal musculature more affected than distal, and extensors more affected than flexors. Early contractures of the elbow, interphalangeal joints, and achilles tendon shortening and a high in- cidence of congenital torticollis were prominent. The first symptoms were observed around the age of five. Two reports in the literature on dominant LGMD with early onset by Arts et al.
[7] and by Mohire et al. [8] describe a clinical picture similar to our cases, whereas the family described by Schmalbruch lacks contractures and has a different picture of muscular involvement. Arts et al. [7] described a Polish family with LGMD with onset in early infancy. In addition to a limb-girdle type of muscular weakness, the extensor digitorum communis muscle was invariably involved. Contractures of the ankle, elbow, and the interphalangeal joint of the last four fingers were frequently found. Congenital torticollis was present in one patient. Mohire et al, [8] described a family with early onset limb-girdle myopathy. Examination of affected members revealed mild to moderate weakness of the proximal limb musculature. All affected members showed contractures of the elbows and ankles. In addition, some showed contractures of the wrist, interphalangeal joints, metacarpophalangeal joints, knee, and hip. Schmalbruch et al. [9] described a slowly progressing dominant dystrophy with onset of
142 J.F. NIELSEN and J. JAKOBSEN
Fig. 3. MR image of the thigh muscles from proband IV2 showing a general atrophy and fatt~ infiltration bilaterally with m. quadriceps fcmoris severely atrophied but with musculus
semimembranosus, gracilis and sartorius relatively preserved.
symptoms in early childhood. The proximal limb muscles, the sternocleidomastoideus and anter- ior tibial muscles, were affected. No contractures were present except achilles tendon shortening. One patient had torticollis.
Future molecular genetic studies are necessary in order to clarify whether autosomal dominant L G M D is one or several entities.
Acknowledgements--We are grateful to Dr M. Bojsen-Moller and Dr T. Christensen for technical assistance.
REFERENCES
1. Bethlem J, Wijngaarden G K van. Benign myopathy, with autosomal dominant inheritance. Brain 1976; 99: 91-100.
2. Chutkow J G, Heffner R R. Kramer A A, Edwards J A. Adult-onset autosomal dominant limb-girdle muscular dystrophy. Ann Neurol 1986; 20: 240- 248.
3. Marconi G, Pizzi A, Arimondi C G. Vanelli B. Limb girdle muscular dystrophy with autosomal dominant inheritance. Acta Neurol Scand 1991 ; g3: 234-238.
4. Somer H, Laulumaaa V, Palij/irvi L, et al. Benign muscular dystrophy with autosomal dominant in- heritance. Neuromusc Disord 1991: 1: 55-57.
5. Gilchrist J M, Pericak-Vance M, Silverman L, Roses A D. Clinical and genetic investigation in autosomal dominant limb-girdle muscular dystrophy. Neurology 1988; 38: 5-9.
6. Schneiderman L J, Sampson W I. Genetic studies of a family with two unusual autosomal dominant con- ditions: muscular dystrophy and Pelger-Huet anomaly. Am J Med [969: 46: 380-393.
7. Arts W F. Bethlem J, Volkers W S. Further investigations on benign myopathy with autosomal dominant inheritance. J Neurol 1978: 217: 201-206.
8. Mohire M D, Tandan R, Fries T J, Little B W, Pendlebury W W, Bradley W G. Early-onset benign autosomal dominant limb-girdle myopathy with con- tractures (Bethlem myopathy). Neurolog)' 1988: 38: 573-580.
9. Schmalbruch H, Kamieniecka Z, Fuglsang-Frederiksen A, Trojaborg W. Benign congenital muscular dys- trophy with autosomal dominant heredity: problems of classification. J Neurol 1987: 234:146-151.