Embed Size (px)
Citation preview

A new cause of progressive intrahepatic cholestasis: 3/3-Hydroxy'C27-steroid dehydrogenase/isomerase deficiency
E. Jacquemin , MD, K. D. R. Setchell, PhD, N. C. O'Connel l , MS, A. Estrada, MD, G. Magg iore , MD, J. Schmitz, MD, M. Hadchouel , MD, and O. Bernard, MD
From the Hepatology Service, Department of Pediatrics and iNSERM U.347, H6pital de BicOtre, Le Kremlin BicOtre, and the Department of Pediatrics, H6pital Necker-Enfants Malades, Paris, France; the Clinical Mass Spectrometry Laboratory, Children's Hospital, Cincinnati, Ohio; Pe- diatria Medica, Hospital de D. Estefania, Lisbon, Portugal; and Clinica Pediatrica, Istituto di Ri- covero e Cura a Carattere Scientifico Policlinico San Matteo, Pavia, Italy
There have been a few reports of infants with severe neonatal cholestasis related to a defect in primary bi le acid synthesis. To assess the importance of such def ic iency among children with progressive intrahepatic cholestasis (Byler disease), screening for inborn errors in bi le acid synthesis was performed by fast atom bombardment ionization-mass spectrometry of urine samples from 30 af- fected children. Bile acid analysis revealed a specif ic fast atom bombardment ionizat ion-mass spect rometry profi le for 3~-hydroxy-C27 steroid dehydroge- nase/isomerase def ic iency in f ive children who had jaundice, hepatospleno- megaly, and fatty stools beginning at ages ranging from 4 to 46 months. None of nthem had pruritus. Liver function tests showed persistently normal serum 7-glutamyltransferase activity, low serum cholesterol and vitamin E levels, nor- mal serum bile acid concentrations despite raised serum bil irubin levels, and decreased prothrombin time and clott ing factor V. In four of the cases a similar disease was observed in siblings. Liver function returned to normal after oral ur- sodeoxychol ic acid therapy. We conclude that 3~-hydroxy-C27-steroid dehy- drogenase/ isomerase def ic iency should be considered when idiopathic chole- static l iver disease with c l in ical features akin to Byler disease iS characterized by the association of normal serum y-glutamyltransferase activi ty, normal serum bile acid concentration, absence of pruritus, and a return to normal l iver func- tion during ursodeoxychol ic acid therapy. Early ident i f icat ion of these children is essential because they benefit from bi le acid therapy and might thus avoid the need for l iver transplantation. (J PEDIATR 1994;125:379-84)
Supported in part by the Assistance Publique-H6pitaux de Paris (CRC no. 920608), Paris, France; the March of Dimes Birth De- fects (grant no. 6-532), and the Falk Foundation.
Submitted for publication Jan. 6, 1994; accepted March 29, 1994.
Reprint requests: Emmanuel Jacquemin, MD, Service d'H6pa- tologie, D6partement de P~diatrie, Centre Hospitalier Universi- taire de Bic&re, 78 rue du G6n~ral Lecler6, 94275 Le Kremlin Bic%tre Cedex, France.
Copyright | 1994 by Mosby-Year Book, Inc. 0022-3476/94/$3.00 + 0 9/20/56324
Progressive intrahepatic cholestasis of childhood (Byler
disease) is a clinical entity of unknown origin that results in
death as a result of iiver failure at ages ranging from infancy
to adolescence and often requires liver transplantation. 1-5 It
is likely to be familial in origin in most instances and is
transmitted as an autosomal recessive trait. ~,2,6 Several
studies provide support for the heterogeneity of this clinical
entity.3, 6, 7 Progressive intrahepatic cholestasis has been
attributed to a defect in bile acid metabolism 2, 6, 8 and, in
some patients, to a defect in primary bile acid secretion: 9, !0
3 7 9

3 8 0 Jacquemin et al. The Journal of Pediatrics September 1994
ALT Alanine aminotransferase FAB-MS Fast atom bombardment ionization-mass
spectrometry GGT ~-Glutamyltransferase 3/3-HSD 3/3-Hydroxy-C27-steroid dehydrogenase/
isomerase UDCA Ursodeoxycholic acid
Recently two inborn errors in bile acid synthesis, namely 3/3-hydroxy-Cz7-steroid dehydrogenase/isomerase and A 4- 3-oxosteroid 5/3-reductase deficiencies, have been identified as causes of progressive cholestasis.ll, 12 These hepatic en- zymes are involved in the early steps in the conversion of cholesterol to the prima~y bile acids cholic and chenodeox- ycholic acids, which provide the major driving force for the promotion and secretion of bile. TM 14 It has been proposed that these defects in primary bile acid synthesis account for 2% to 5% of the cases of idiopathic cholestatic diseases of childhood and that, once recognized, patients with these defects may benefit from bile acid therapy. TM 12, 15-17 We report the clinical and biochemical characterization of five patients with progressive intrahepatic cholestasis caused by 3/3-HSD deficiency.
M E T H O D S
Patients, Thirty children with progressive intrahepatic cholestasis who were followed for at least 6 months in the pediatric hepatology unit of Bic&re Hospital were included in the study. All 30 children had a history of chronic chole- static liver disease with hepatomegaly or hepatospleno- megaly for which aU known causes of childhood cholestasis were excluded by appropriate investigations. 18, 19 Tables I and II list the clinical characteristics and the results of the biochemical liver function tests retrospectively collected at the time of diagnostic investigation at Bic~tre Hospital. Be- cause serum 3,-glutamyltransferase activity may be a use- ful tool in separating two forms of progressive intrahepatic cholestasis with or without bile duct involvement, 3, 7 the 30 children were classified into two groups according to their serum GGT levels; group 1 consisted of 20 children (including the 5 children with a 3~-HSD deficiency) who persistently had normal serum GGT activity, and group 2 consisted of 10 children with increased serum GGT activ- ity.
A cholangiogram obtained after percutaneous transhe- patic cholecystography was done in five children in group 1 (including one child with 3/3-HSD deficiency) and nine children in group 2, and showed in all cases normal intra- hepatic and extrahepatic bile ducts. Nineteen children in group 1 (including the five children with 3/3-HSD defi- ciency) and all children in group 2 underwent a liver biopsy. From June 1992 to March 1993, urine samples from the 30 children with progressive intrahepatic cholestasis were col-
lected in our institution, stored frozen, and then analyzed in a single batch by fast atom bombardment ionization-mass spectrometry for the presence of bile acid conjugates.
Before the results of the FAB-MS analysis were obtained, the patients were given ursodeoxycholic acid orally at the dose of 600 mg/m 2 of body area per day. Twenty-six patients received UDCA for at least 2 months.
Analysis of bile acids from serum. Total bile acid concen- tration in serum was measured by an enzymic technique using 3a-hydroxysteroid dehydrogenase (Enzabile, Ny- corned AS, Oslo, Norway.) This method determines only unconjugated and conjugated 3a-hydroxylated bile acids and does not quantify atypical bile acids or intermediates that lack a 3c~-hydroxy group in the steroid structure. It is used to provide a measure of primary bile acid concentra- tions.
Analysis of bile acids from urine. Bile acids were extracted and concentrated from urine samples by liquid-solid ex- traction using cartridges of octadecyl silane-bonded silica, as described in detail previously. 2~ The methanolic extract of urine was analyzed by FAB-MS as described previous- ly.21, 22 This technique is a highly specific method for the detection of bile acids in urine samples and for the identi- fication of disorders of bile acid synthesis. The presence of an inborn error in bile acid synthesis is revealed by a unique FAB-MS profile that shows accumulated intermediates, their metabolites, or both in the biosynthetic pathway proximal to the enzyme block.ll, 12 Definitive characteriza- tion of the intermediates, their metabolites, or both was confirmed by gas chromatography-mass spectrometry: 23
R E S U L T S
Fast atom bombardment ionization-mass spectrometry. Among the 30 children with progressive intrahepatic cholestasis, the analysis of bile acids from urine revealed a negative ion FAB-MS profile specific for a 3~-HSD deft- ciency 11 in five children (three girls) in group 1 (Tables I and II); this was confirmed by gas chromatography-mass spectrometry. None of the 30 children had a FAB-MS pro- file specific for a Lx4-3-oxosteroid 5/3-reductase deficiency. 12
Clinical, biochemical, and histologic findings. The pa- tients with a 3/3-HSD deficiency had jaundice (4/5) he- patomegaly (5/5), splenomegaly (1/5), and fatty stools (2/5) with onset at ages ranging from 4 to 46 months (Ta- bles I and II). None had pruritus. In these five patients liver function tests showed high serum alanine aminotransferase activity, normal serum GGT activity, normal or undetect- able concentrations of 3a-hydroxy bile acids in serum despite significantly elevated serum bilirubin concentra- tions, decreased prothrombin time (normal, >70%; [mean, 64%; range. 45% to 74%]) and decreased clotting factor V (normal, >80%; [mean, 70%; range. 30% to 100%]). Serum alkaline phosphatase activity (normal, <360 U/L) was
The Journal o f Pediatrics Jacquemin et al. 3 8 1 Volume 125, Number 3
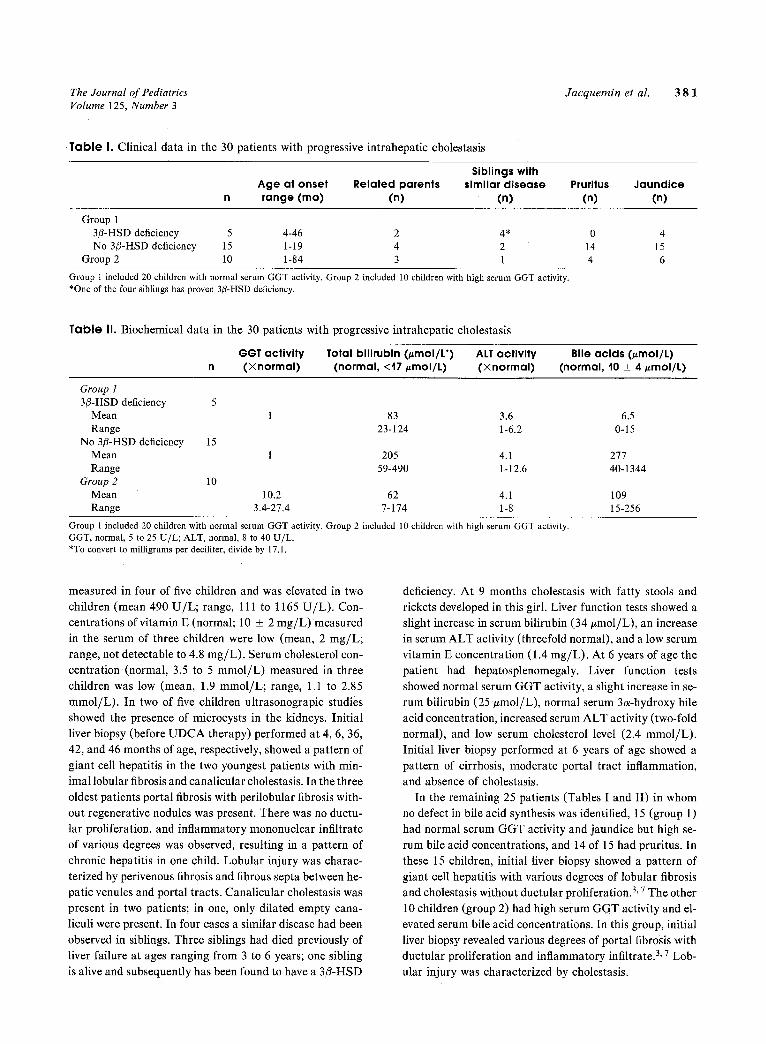
Table I. Clinical data in the 30 patients with progressive intrahepatic cholestasis
Siblings with Age at onset Related parents similar disease Pruritus Jaundice range (mo) (n) (n) (n) (n)
Group 1 3/3-HSD deficiency 5 4-46 2 4* 0 4 No 3/3-HSD deficiency 15 1-19 4 2 14 15
Group 2 10 1-84 3 1 4 6
Group 1 included 20 children with normal serum GGT activity. Group 2 included 10 children with high serum GGT activity. *One of the four siblings has proven 3/3-HSD deficiency.
Table II. Biochemical data in the 30 patients with progressive intrahepatic cholestasis
GGT activity Total bilirubin (/~mol/L*) ALT activity Bile acids (#mol/t) n (Xnormal) (normal, <17 ~mol/L) (Xnormal) (normal, 10 _+ 4/~mol/L)
Group ! 3/3-HSD deficiency 5
Mean 1 83 3.6 6.5 Range 23-124 1-6.2 0-15
No 3/3-HSD deficiency 15 Mean 1 205 4.1 277 Range 59-490 1-12.6 40-1344
Group 2 10 Mean 10.2 62 4.1 109 Range 3.4-27.4 7-174 1-8 15-256
Group 1 included 20 children with normal serum GGT activity. Group 2 included 10 children with high serum GGT activity. GGT, normal, 5 to 25 U/L; ALT, normal, 8 to 40 U/L. *To convert to milligrams per deciliter, divide by 17.1.
measured in four of five children and was elevated in two
children (mean 490 U / L ; range, 111 to 1165 U / L ) . Con-
centrations of vitamin E (normal; 10 _+ 2 m g / L ) measured
in the serum of three children were low (mean, 2 rag/L;
range, not detectable to 4.8 mg /L) . Serum cholesterol con-
centration (normal , 3.5 to 5 mmol /L ) measured in three
children was low (mean, 1.9 mmol /L ; range, 1.1 to 2.85
mmol /L) . In two of five children ultrasonograpic studies
showed the presence of microcysts in the kidneys. Initial
liver biopsy (before U D C A therapy) performed at 4, 6, 36,
42, and 46 months of age, respectively, showed a pattern of
giant cell hepatitis in the two youngest patients with min-
imal lobular fibrosis and canalicular cholestasis. In the three
oldest patients portal fibrosis with perilobular fibrosis with-
out regenerative nodules was present. There was no ductu-
lar proliferation, and inflammatory mononuclear infiltrate
of various degrees was observed, resulting in a pattern of
chronic hepatitis in one child. Lobular injury was charac-
terized by perivenous fibrosis and fibrous septa between he-
patic venules and portal tracts. Canalicular cholestasis was
present in two patients; in one, only dilated empty cana-
liculi were present. In four cases a similar disease had been
observed in siblings. Three siblings had died previously of
liver failure at ages ranging from 3 to 6 years; one sibling
is alive and subsequently has been found to have a 3/3-HSD
deficiency. At 9 months cholestasis with fatty stools and
rickets developed in this girl. Liver function tests showed a
slight increase in serum bilirubin (34 #mol /L) , an increase
in serum A L T activity (threefold normal), and a low serum
vitamin E concentration (1.4 mg/L) . At 6 years of age the
patient had hepatosplenomegaly. Liver function tests
showed normal serum GGT activity, a slight increase in se-
rum bilirubin (25 #tool /L) , normal serum 3a-hydroxy bile
acid concentration, increased serum A L T activity (two-fold
normal), and low serum cholesterol level (2.4 mmol /L) .
Initial liver biopsy performed at 6 years of age showed a
pattern of cirrhosis, moderate portal tract inflammation,
and absence of cholestasis.
In the remaining 25 patients (Tables I and II) in whom
no defect in bile acid synthesis was identified, 15 (group 1)
had normal serum GGT activity and jaundice but high se-
rum bile acid concentrations, and 14 of 15 had pruritus. In
these 15 children, initial liver biopsy showed a pattern of
giant cell hepatitis with various degrees of lobular fibrosis
and cholestasis without ductular proliferation. 3, 7 The other
10 children (group 2) had high serum GGT activity and el-
evated serum bile acid concentrations. In this group, initial
liver biopsy revealed various degrees of portal fibrosis with
ductular proliferation and inflammatory infiltrate. 3, 7 Lob-
ular injury was characterized by cholestasis.
3 8 2 Jacquemin et al. The Journal of Pediatrics September 1994
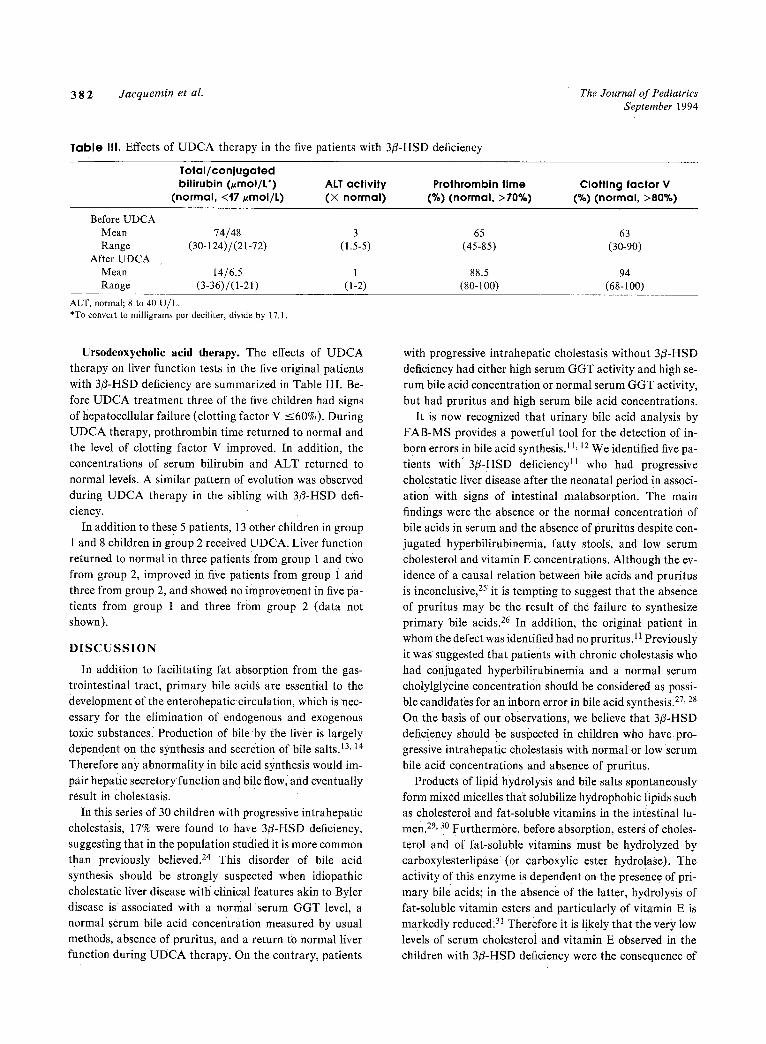
Table III, Effects of UDCA therapy in the five patients with 3/3-HSD deficiency
Total/conjugated bilirubin (#mol/L*)
(normal, <17/~mol/L) ALT activity (• normal)
Prothrombin time (%) (normal, >70%)
Clotting factor V (%) (normal, >80%)
Before UDCA Mean 74/48 3 65 Range (30-124)/(21-72) (1.5-5) (45-85)
After UDCA Mean 14/6.5 1 88.5 Range (3-36)/(1-21) (1-2) (80-100)
63 (30-90)
94 (68-100)
ALT, normal; 8 to 40 U/L. *To convert to milligrams per deciliter, divide by 17. I.
Ursodeoxycholic acid therapy. The effects of UDCA therapy on liver function tests in the five original patients with 3/3-HSD deficiency are summarized in Table III. Be- fore UDCA treatment three of the five children had signs of llepatocellular failure (clotting factor V --<60%). During UDCA therapy, prothrombin time returned to normal and the level of clotting factor V improved. In addition, the Concentrations of serum bilirubin and ALT returned to normal levels. A similar pattern of evolution was observed during UDCA therapy in the sibling with 3fl-HSD defi- ciency.
In addition to these 5 patients, 13 other children in group 1 and 8 children in group 2 received UDCA. Liver function returned to normal in three patients from group 1 and two from group 2, improved in five patients from group i and three from group 2, and showed no improvement in five pa- tients from group 1 and three from group 2 (data not shown).
D I S C U S S I O N
In addition to facilitating fat absorption from the gas- trointestinal tract, primary bile acids are essential to the development of the enterohepatic circulation , which is nec- essary for the elimination of endogenous and exogenous toxic substances. Production of bile by the liver is largely dependent on the synthesis and secretion of bile salts. 13, a4 Therefore any abnormality in bile acid synthesis would im- pair hepatic secretory function an d bi!e flow, and eventually result in Cholestasis.
In this series of 30 children with progressive intrahepatic cholestasis, 17% were found to have 3fl-HSD deficiency, suggesting that in the population studied it is more common than previously believed.24 This disorder of bile acid synthesis should be strongly Suspected when idiopathic cholestatic liver disease with clinical features akin to Byler disease is associated with a normal serum GGT level, a normal serum bile acid concentration measured by usual methods, absence of pruritus, and a return to normal liver function during UDCA therapy. On the contrary; patients
with progressive intrahepatic cholestasis without 3/3-HSD deficiency had either high serum GGT activity and high se- rum bile acid concentration or normal serum GGT activity, but had pruritus and high serum bile acid concentrations.
It is now recognized that urinary bile acid analysis by FAB-MS provides a powerful tool for the detection of in- born errors in bile acid synthesis. TM 12 We identified five pa- tients with 3~-HSD deficiency 11 who had progressive cholestatic liver disease after the neonatal period in associ- ation with signs of intestinal malabsorption. The main findings were the absence or the normal concentration of bile acids in serum and the absence of pruritus despite con- jugated hyperbilirubinemia, fatty stools, and low serum cholesterol and vitamin E concentrations. Although the ev- idence of a causal relation between bile acids and pruritus is inconclusive. 25 it is tempting to suggest that the absence
of pruritus may be the result of the failure to synthesize primary bile acids. 26 In addition, the original patient in whom the defect was identified had no pruritus, l 1 Previously
it was suggested that patients with chronic cholestasis who had conjugated hyperbilirubinemia and a normal serum cholylglycine concentration should be considered as possi- ble candidates for an inborn error in bile acid synthesis. 27- 28
On the basis of our observations, we believe that 3/%HSD deficiency should be suspected in children who have pro- gressive intrahepatic cholestasis with normal or low serum bile acid concentrations and absence of pruritus.
Products of lipid hydrolysis and bile salts spontaneously form mixed micelles that solubilize hydrophobic lipids Such as cholesterol and fat-soluble vitamins in the intestinal lu- men.29, 30 Furthermore. before absorption, esters of choles-
terol and of fat-soluble vitamins must be hydrolyzed by carboxylesterlipase (or carboxylic ester hydrolase). The activity of this enzyme is dependent on the presence of pri- mary bile acids; in the absence of the latter, hydrolysis of fat-soluble vitamin esters and particularly of vitamin E is markedly reduced. 31 Therefore it is likely that the very low levels of serum cholesterol and vitamin E observed in the children with 3~-HSD deficiency were the consequence of

The Journal of Pediatrics Jacquemin et al. 3 8 3 Volume 125, Number 3
the absence of primary bile acids in the gut, because pan- creatic lipase activity was normal and could not account for the lipid malabsorption syndrome (data not shown).
Serum GGT activity may be a useful tool in separating children with progressive intrahepatic cholestasis into two groups.3, 7 It has been suggested that, in children with ele- vated serum GGT activity, cholestasis is caused by lesions of the bile ducts, whereas in the group with normal GGT activity cholestasis is caused by a hepatocellular disorder, either intracellular or canalicular. In the latter group, the concept of a canalicular disorder has been reinforced by data showing the presence of high levels of primary bile ac- ids in serum and absence or very low levels of primary bile acids in bile, suggesting a defect of primary bile acid secre- tion.9, lO Our results show that in children with progressive
intrahepatic cholestasis, 3/3-HSD deficiency is highly asso- ciated with normal serum GGT activityfl 7, 28 The remain-
ing patients with normal serum GGT activity had high lev- els of bile acids in serum and may be similar to the children previously described with a defect of primary bile acid se- cretion 9, 10; the reason that these children have normal se-
rum GGT activity is unclear. Primary bile acids excreted into the canalicular lumen are important factors for biliary excretion of GGT, by means of their detergent action. 32 During cholestasis the increase in serum GGT activity has been ascribed to a reflux of canalicular bile into sinusoidal blood through the leaky paracellular pathway. 33 In these patients the absence of primary bile acids in the canalicular lumen, as a result of a defect of synthesis or secretion, may explain the normal GGT activity in serum.
A similar disease occurred in four of five siblings of our five patients with 3/3-HSD deficiency. The absence of a pre- dominant gender, and the presence of consanguinity, sug- gest that the disease is transmitted in an autosomal reces- sive fashion. In addition, in fibroblasts from parents of chil- dren with 3/3-HSD deficiency, 3/3-HSD activity is decreased and thus compatible with a heterozygous phenotype. 34 Among the children previously reported 11, 28, 35 and those
described here, age at onset of the disease was quite vari- able, ranging from several months to 10 years. This suggests different degrees of the severity of the disease, which could be related to variable expression of the enzyme defect. Consequently 3/3-HSD deficiency should also be considered in children with unexplained late-onset cholestasis, chronic hepatitis, and a malabsorption syndrome. 35, 36
The mechanism of liver injury in 3/3-HSD deficiency is unknown, but may be related to the failure of the liver to synthesize primary bile acids or to the production of atyp- ical metabolites that are presumed to be hepatotoxic and cholestatic. The success of UDCA therapy in these patients is surprising because it has little or no effect on regulating endogenous bile acid synthesis 13, 37 and therefore does not
prevent further synthesis of atypical 3/3-hydroxy-A 5 bile acids. Previous experience has shown that primary bile acid administration provides the most appropriate therapy. 15-t7 It is probable that, as for other cholestatic liver diseases, 38 UDCA induces hypercholeresis, stimulates bile flow and thereby facilitates the canaticular secretion of the accumu- lated atypical bile acids. 39, 4o In the short term, UDCA
therapy appears to be of value, but primary bile acid ther- apy, when available, should be the first choice. However, in the absence of a defect of bile acid synthesis, primary bile acid therapy might worsen the evolution of progressive in- trahepatic cholestasis; thus definitive diagnosis should first be established.
The various clinical and biochemical patterns reported here in children with progressive intrahepatic cholestasis may be helpful in selecting patients who may have a 3/3-HSD deficiency and benefit from urine screening by FAB-MS. Early identification of these children is essential because they will benefit from primary bile acid therapy and might thus avoid liver transplantation. In the short term, UDCA, a safe and commercially available drug, could be considered in the initial therapeutic management of these patients because it appears effective in resolving the bio- chemical abnormalities in liver function. The precise cause of progressive intrahepatic cholestasis in children who have no 3/3-HSD deficiency remains to be defined.
R E F E R E N C E S
1. Clayton R J, Iber FL, Ruebner BH, McKusick VA. Byler dis- ease: fatal familial intrahepatic cholestasis in an Amish kindred. Am J Dis Child 1969;117:112-24.
2. Linarelli LG, Williams CN, Phillips MJ. Byler's disease: fatal intrahepatic cholestasis. J PED~AT~ 1972;81:484-92.
3. Maggiore G, Bernard O, Riely CA, Hadchouel M, Lemonnier A, Alagille D. Normal "r-glutamyl-transpeptidase activity identifies groups of infants with idiopathic cholestasis with poor prognosis. J PEDIATR 1987;111:251-2.
4. Soubrane O, Gauthier F, Devictor D, et al. Orthotopic liver transplantation for Byler disease. Transplantation 1990;50: 804-6.
5. Whitington PF, Whitington GL. Partial external diversion of bile for the treatment of intractable pruritus associated with intrahepatic cholestasis. Gastroenterology 1988;95:130-6.
6. Riely CA. Familial intrahepatic cholestatic syndromes. Semin Liver Dis 1987;7:119-33.
7. Maggiore G, Bernard O, Hadchouel M, Lemonnier A, Alagille D. Diagnostic value of serum 7-glutamyl transpeptidase activ- ity in liver diseases in children. J Pediatr Gastroenterol Nutr 199!;12:21-6.
8. Williams CN, Kaye R, Baker L, Hurwitz R, Senior JR. Pro- gressive familial cholestatic cirrhosis and bile acid metabolism. J PED1ATR 1972;81:493-500.
9. Tazawa Y, Yamada M, Nakagawa M, Konno T, Tada K. Bile acid profile in siblings with progressive intrahepatic cholesta- sis: absence of biliary chenodeoxycholate. J Pediatr Gastroen- tern Nutr 1985;4:32-7.

3 8 4 Jacquemin et al. The Journal of Pediatrics September 1994
10. Jacquemin E, Dumont M, Bernard O, Erlinger S, Hadchouel M. Evidence for defective primary bile acid secretion in chil- dren with progressive familial intrahepatic cholestasis (Byler disease). Eur J Pediatr (in press).
11. Clayton PT, Leonard JV, Lawson AM, et al. Familial giant cell hepatitis associated with synthesis of 3/3, 7a-dihydroxy-and 313, 7c~, 12a-trihydroxy-5-cholenoic acids. J Clin Invest 1987;79: 1031-8.
12. Setchell KDR, Suchy FJ, Welsh MB, Zimmer-Nechemias L, Heubi J, Balistreri WF. ~4-3-Oxosteroid 513-reductase defi- ciency described in identical twins with neonatal hepatitis. A new inborn error in bile acid synthesis. J Clin Invest 1988; 82:2148-57.
13. Russell DW, Setchell KDR. Bile acid biosynthesis. Biochem- istry 1992;31:4737-49.
14. Nathanson MH, Boyer JL. Mechanisms and regulation of bile acid secretion. Hepatology 1991;14:551-66.
15. Setchell KDR, Balistreri WF, Piccoli DA, Clerici C. Oral bile acid therapy in the treatment of inborn errors in bile acid syn- thesis associated with liver disease. In: Paumgartner G, Stiehl A, Gerok W, eds. Bile acids as therapeutic agents. Dordrecht, The Netherlands: Kluwer Academic, 1991:367-73.
16. Ichimiya H, Nazer H, Gunasekaran T, Clayton P, Sj6vall J. Treatment of chronic liver disease caused by 3/3-hydroxy-AS- C27-steroid dehydrogenase deficiency with chenodeoxycholic acid. Arch Dis Child 1990;65:1121-4.
17. Daugherty CC, Setchell KDR, Heubi JE, Balistreri WF. Res- olution of liver biopsy alterations in three siblings with bile acid treatment of an inborn error of bile acid metabolism (A4-3-OX - osteroid 5B-reductase deficiency). Hepatology 1993;18:1096- 101.
18. Alagille D, Odi6vre M. Liver and biliary tract disease in chil- dren. New York: Wiley & Sons, 1979.
19. Mowat AP. Liver disorders in childhood. London: Butter- worth, 1987.
20. Setchell KDR, Worthington J. A rapid method for the quan- titative extraction of bile acids and their conjugates from se- rum using commercially available reverse octadecylsilane bonded silica cartridges. Clin Chim Acta 1982; 125:135-44.
21. Setchell KDR, Street JM. Inborn errors of bile acid synthesis. Semin Liver Dis 1987;7:83-99.
22. Sj6vall J, Lawson AM, Setchell KDR. Mass spectrometry of bile acids. In: Law JH, Rilling HC, eds. Methods and Enzy- mology; vol 1. London: Academic Press, 1985:63-113.
23. Setchell KDR, Matsui A. Serum bile acid analysis. The appli- cation of liquid-gel chromatographic techniques and capillary column gas chromatography and mass spectrometry. Clin Chim Acta 1983;127:l-17.
24. Setchell KDR, O'Connell NC. Bile acid synthesis. In: Suchy F J, ed. Liver disease in children. St. Louis: Mosby-Year Book, 1994:835-51.
25. Bartholomew TC, Summerfield JA, Billing BH, Lawson AM, Setchell KDR. Bile acid profiles in human serum and skin in- terstitial fluid and their relationship to pruritus studied by gas chromatography-mass spectrometry. Clin Sei 1982;63:65-73.
26. Garden JM, Ostrow JD, Roenigk HH. Pruritus in hepatic cholestasis. Pathogenesis and treatment. Arch Dermatol 1985;121:1415-20.
27. Jacquemin E, Setchell KDR, Hadchouel M, Schmitz J, Ber- nard O. 3/3-Hydroxysteroid-dehydrogenase (3/3-HSD) defi- ciency in children: a new cause of progressive intrahepatic cholestasis (PIC) with normal serum gammaglutamyltrans- ferase (GGT) activity [Abstract]. J Hepatol 1993;18:$3.
28. Setchell KDR, Piccoli D, O'Connell NC, Jacquemin E, Ber- nard O. Progressive intrahepatic cholestasis with normal ~/-glutamyltransferase is highly associated with the 313-hydrox- ysteroid dehydrogenase/isomerase deficiency, an inborn error in bile acid synthesis--a new category of metabolic liver dis- ease [Abstract]. Hepatology 1993;18:178A.
29. Carey M, Small D. The characteristics of mixed micellar so- lutions with particular reference to bile. Am J Med 1970; 49:590-608.
30. Kayden H J, Traber MG. Absorption, lipoprotein transport, and regulation of plasma concentrations of vitamin E in humans. J Lipid Res 1993;34:343-58.
31. Lombardo D, Guy O. Studies on the substrate specificity of a carboxylester hydrolase from human pancreatic juice. II. Ac- tion on cholesterol esters and lipid-soluble vitamin esters. Bio- chim Biophys Acta 1980;611:147-55.
32. Hirata E, Inoue M, Morino Y. Mechanism of biliary secretion of membranous enzymes: bile acids are important factors for biliary occurrence of "/-glutamyltransferase and other hydro- lases. J Biochem 1984;96:289-97.
33. Boyer JL. Tight junctions in normal and cholestatic liver: does the paracetlular pathway have functional significance? Hepa- tology 1983;3:614-17.
34. Buchmann MS, Kvittingen EA, Nazer H, et al. Lack of 3/3-hydroxy-2xs-C27-steroid dehydrogenase/isomerase in fibro- blasts from a child with urinary excretion of 3/3-AS-hydroxy - bile acids. A new inborn error of metabolism. J Clin Invest 1990;86:2034-7.
35. Setchell KDR, Flick R, Watkins JB, Piccoli DA. Chronic hepatitis in a 10 yr old due to an inborn error in bile acid syn- thesis-diagnosis and treatment with oral bile acid [Abstract]. Gastroenterology 1990;98:631A.
36. Vanderpas JB, Koopman B J, Cadranel S, et al. Malabsorption of liposoluble vitamins in a child with bile acid deficiency. J Pediatr Gastroenterol Nutr 1987;6:33-41.
37. Heuman DM, Vlahcevic RZ, Bailey ML, Hylemon PB. Reg- ulation of bile acid synthesis. II. Effect of bile acid feeding on enzymes regulating hepatic cholesterol and bile acid synthesis in the rat. Hepatology 1988;8:892-7.
38. Balistreri WF, A-Kader HH, Heubi JE, Setchell KDR, Whi- tington P. Ursodeoxycholic acid (UDCA) decreases serum cholesterol levels, ameliorates symptoms, and improves bio- chemical parameters in pediatric patients with chronic intra- hepatic cholestasis. In: Paumgartner G, Stiehl A, Gerok W, eds. Bile acids as therapeutic agents. Dordrecht, The Nether- lands: Kluwer Academic, 1991:323-33.
39. Jacquemin E, Dumont M, Erlinger S. Ursodeoxycholic acid improves ethinyl estradiol induced-cholestasis in the rat. Eur J Clin Invest 1993;23:794-802.
40. Dumont M, Erlinger S, Uchman S, Hypercholeresis induced by ursodeoxycholic acid and 7-ketolithocholic acid in the rat. Possible role of bicarbonate transport. Gastroenterology 1980; 79:82-9.

















![[2015] post lt cholestasis](https://img.pdfslide.net/doc/110x75/58ee0ee21a28ab92198b4665/2015-post-lt-cholestasis.jpg)











