Embed Size (px)
Citation preview

A novel xenograft model of cutaneous T-cell lymphoma
Thorbjørn Krejsgaard1,2, Katharina Kopp1,2, Elisabeth Ralfkiaer3, Ayelah E. Willumsgaard1,2, Karsten
W. Eriksen2, Tord Labuda2, Susanne Rasmussen2, Anne-Merete Mathiesen1,2, Carsten Geisler2,
Britt Lauenborg1,2, Jurgen C. Becker4, Qian Zhang5, Mariusz A. Wasik5, Niels Odum1,2 and
Anders Woetmann1,2
1Department of Biology, University of Copenhagen, Copenhagen, Denmark;2Institute of International Health, Immunology and Microbiology, University of Copenhagen, Copenhagen, Denmark;3Department of Pathology, University Hospital of Copenhagen, Copenhagen, Denmark;4Department of Dermatology, Julius-Maximilians-University, Wurzburg, Germany;5Department of Pathology and Laboratory Medicine, University of Pennsylvania, Philadelphia, PA, USA
Correspondence: Anders Woetmann, MSc, PhD, Department of Biology, Division of Immunology, IHIM, Panum institute 22.5.28, University of
Copenhagen, Blegdamsvej 3c, DK2200 Copenhagen N, Denmark, Tel.: +4535327868, Fax: +4535327876, e-mail: [email protected]
Accepted for publication 6 May 2010
Abstract: Cutaneous T-cell lymphomas (CTCLs) are characterized
by accumulation of malignant T cells in the skin. Early disease
resembles benign skin disorders but during disease progression
cutaneous tumors develop, and eventually the malignant T cells
can spread to lymph nodes and internal organs. However, because
of the lack of suitable animal models, little is known about the
mechanisms driving CTCL development and progression in vivo.
Here, we describe a novel xenograft model of tumor stage CTCL,
where malignant T cells (MyLa2059) are transplanted to
NOD ⁄ SCID-B2m) ⁄ ) (NOD.Cg-Prkdcscid B2mtm1Unc ⁄ J) mice.
Subcutaneous transplantation of the malignant T cells led to rapid
tumor formation in 43 of 48 transplantations, whereas
transplantation of non-malignant T cells isolated from the same
donor did not result in tumor development. Importantly, the
tumor growth was significantly suppressed in mice treated with
vorinostat when compared to mice treated with vehicle.
Furthermore, in most mice the tumors displayed subcutaneous
and ⁄ or lymphatic dissemination. Histological,
immunohistochemical and flow cytometric analyses confirmed
that both tumors at the inoculation site, as well as distant
subcutaneous and lymphatic tumors, originated from the
transplanted malignant T cells. In conclusion, we describe a novel
mouse model of tumor stage CTCL for future studies of disease
dissemination and preclinical evaluations of new therapeutic
strategies.
Key words: cutaneous T-cell lymphoma – metastasis – mouse
model – mycosis fungoides – vorinostat
Please cite this paper as: A novel xenograft model of cutaneous T-cell lymphoma. Experimental Dermatology 2010; 19: 1096–1102.
Introduction
Cutaneous T-cell lymphomas (CTCLs) are a heterogeneous
group of neoplasms that primarily affect the skin. Mycosis
fungoides (MF) is the most common form of CTCL
accounting for more than 50% of all primary non-Hodgkin
lymphomas of the skin (1). The early stages of MF are
characterized by an infiltrate of reactive inflammatory cells
in the skin with rare malignant T cells present in the
affected areas. As the disease progresses, the number of
malignant T cells increases and they tend to form intrader-
mal and often ulcerating tumors as well as spread to lymph
nodes and internal organs (2–4). Chromosomal aberrations
and increased prevalence of certain viral and bacterial infec-
tions in patients with MF have been observed, but the role
in the disease aetiology is essentially unknown (5,6). The
pathogenesis of MF is also poorly understood, however, its
typical features include an increase in the production of
certain cytokines (e.g. IL-5, IL-6, IL-7, IL-15) (7–11) and
angiogenic factors (12), a selective expression of chemokine
receptors (13), an up-regulation of matrix metalloprotein-
ase expression (14) as well as an impaired function of
apoptotic pathways (15–17). Furthermore, B-lymphoid
kinase (Blk) is ectopically expressed in the CTCL cells and
fosters their aberrant proliferation (18). The molecular
mechanisms also involve an abnormal activation and func-
tion of other tyrosine kinases (Jak3, Brk, Hck) (19–23),
transcription factors (c-Jun, NF-jB, Stat3, FOXP3) (24–
29), and suppressors of cytokine signalling (30,31). Finally,
a deficient expression and function of protein tyrosine
phosphatases and tumor suppressors is also believed to play
a key role in the pathogenesis (32–35). MF is not curable
but early stages can be controlled to a certain degree by
skin-directed therapies such as corticosteroids, UV therapy
or nitrogen mustard. Advanced disease is normally treated
systemically (e.g. IFN-alpha, bexarotene, denileukin diftitox
or chemotherapy), but these therapies generally do not
result in full remission or cure (36–38).
DOI:10.1111/j.1600-0625.2010.01138.x
www.blackwellpublishing.com/EXDOriginal Article
1096 ª 2010 John Wiley & Sons A/S, Experimental Dermatology, 19, 1096–1102

Despite recent progress in the understanding of the path-
ogenesis of CTCL, research into the mechanisms driving
extra-dermal dissemination, as well as the development of
new therapeutic agents, has been hampered by the lack of
suitable animal models. Although a few preclinical in vivo
models of CTCL have already been described, they suffer
from several shortcomings. Charley et al. (39) succeeded in
transplanting human skin grafts from patients with CTCL
to mice with severe combined immune deficiency (SCID).
Malignant T cells were maintained for a month in the
human skin graft but never spread outside the graft
suggesting that this model is not applicable to study disease
dissemination (39). More recently, Thaler et al. (40) devel-
oped an alternative murine xenograft model using subcuta-
neous transplantation of the malignant T-cell line, MyLa,
to the flanks of athymic nude mice lacking T cells and
T-cell dependent antibody responses. In this model, serial
passages and selections of cells displaying the fastest growth
in vivo were performed to obtain growth rates that allowed
development of palpable tumors within 40–60 days (tumor
take 80%). Malignant T cells were observed in the lymph
and blood circulation. Single T cells displayed perivascular
localization in the liver and lung but macroscopically visi-
ble metastases were not detected. Likewise, subcutaneous
tumors did not develop outside of the inoculation site
indicating that the CTCL cells had limited capacity to dis-
seminate in nude mice (40).
Here, we describe a novel xenograft transplantation
model of CTCL utilizing a NOD ⁄ SCID-B2m) ⁄ ) mouse
strain that is deficient in T and B cells, lacks expression of
major histocompatibility complex (MHC) class I and has
impaired natural killer (NK) cell activity as the recipient of
the malignant T-cell line MyLa2059 (41). In this model,
malignant CTCL cells rapidly formed tumors that grew to
substantial sizes and subsequently ulcerated. Notably, most
of these tumors developed subcutaneous and ⁄ or lymphatic
dissemination. Vorinostat (Zolinza, suberoylanilide hy-
droxamic acid), an HDAC inhibitor recently approved by
the FDA for treatment of patients with MF, suppressed the
tumor growth when compared to vehicle (42). Thus, this
model displays some features similar to those observed in
patients with tumor stage CTCL and could provide a valu-
able tool for preclinical testing of new therapeutic options.
Materials and methods
Cell linesThe malignant T-cell line MyLa2059 and the non-malignant
T-cell line MyLa1850 were obtained from a plaque biopsy of
a patient diagnosed with MF (43). MyLa2059 cells were
cultured in conditional media (CM) (RPMI 1640, 2 mm
l-glutamine, 100 lg ⁄ ml penicillin ⁄ streptomycin all from
Sigma-Aldrich, St Louis, MO, USA) supplemented with
10% foetal bovine serum (Life technologies, Roskilde,
Denmark). MyLa1850 was cultured in CM supplemented
with 10% pooled human serum (Blood Bank, State Univer-
sity Hospital, Copenhagen, Denmark), 103 units ⁄ ml IL-2
(Proleukin) (Chiron, Emeryville, CA, USA) and 2.5 ng ⁄ ml
IL-4 (Leinco, St. Louis, MO, USA).
CTCL xenograft modelThe murine xenograft model of CTCL is based on the
immunodeficient NOD.Cg-Prkdcscid B2mtm1Unc ⁄ J strain
(NOD ⁄ SCID-B2m) ⁄ )) (The Jackson Laboratory, Bar Har-
bor, ME, USA). Eight to twelve weeks old mice were
injected subcutaneously (s.c.) with varying numbers (as
indicated) of malignant (MyLa2059) or non-malignant
(MyLa1850) T cells in a volume of 200 ll saline in each
flank. In one experiment, malignant T cells were trans-
planted to the flanks of outbred athymic aplastic nu ⁄ numice
of Naval Medical Research Institute background (M&B A ⁄ S,
Ry, Denmark) as used in the model of Thaler et al. (40).
Tumor onset and growth were monitored continuously
by slide calliper measurements. At experiment termination,
the mice were killed and the tumors excised, measured and
weighed. In the study investigating the effect of vorinostat
on tumor growth, eight NOD ⁄ SCID-B2m) ⁄ ) mice were
inoculated s.c. on each flank with 1 · 106 MyLa2059 cells.
When a mouse had established palpable tumors it was allo-
cated alternately to the group receiving vehicle or the group
receiving vorinostat and the treatment was initiated (day 1).
Unfortunately, one mouse died before treatment initiation.
The mice received 60 mg ⁄ kg vorinostat (Selleck Chemicals,
Houston, TX, USA) or vehicle intraperitoneally (i.p.) five
consecutive days a week, and the tumor growth was moni-
tored continuously by slide calliper measurements. All ani-
mal experiments were performed according to the rules
and regulations of the Danish Animal Licence regulations
(Forsøgsdyrstilsynet).
Histology and immunohistochemistrySamples of primary and secondary lesions were fixed in
formalin and processed for paraffin embedding. The biop-
sies were then examined by histology and immunohisto-
chemistry for CD2 (G11; Leinco) and Blk (C-20; Santa
Cruz Biotechnology, Santa Cruz, CA, USA). Prior to stain-
ing, the sections were heated in a microwave oven in a
TEG buffer (pH: 9) for 15 min. The staining was per-
formed in the Techmate 500 Immunostainer, using the
DAKO Envision K5007 as a secondary antibody.
StatisticsFor analysis of the data presented in Fig. 1b, a two-tailed
two-sample t test with Welch’s correction and a signifi-
cance level (a) of 0.05 was performed to compare the aver-
age tumor volume in mice inoculated with 1 · 106 cells
A xenograft model of CTCL
ª 2010 John Wiley & Sons A/S, Experimental Dermatology, 19, 1096–1102 1097

and 2.5 · 106 cells at each measured time-point. Owing to
previous studies showing that vorinostat is effective in
treatment of patients with MF, our a priori hypothesis in
the experiment shown in Fig. 3 was that a reduced tumor
volume would be observed in mice treated with vorinostat
when compared with mice treated with vehicle (42).
Accordingly, a one-tailed two-sample t test with Welch’s
correction was employed to compare the average tumor
volume in mice treated with vorinostat and vehicle at the
measured time-points. These data were also subjected to a
one-tailed Mann–Whitney U test which gave the same sig-
nificance results as the two-sample t test. In both tests, a
significance level (a) of 0.05 was used. Data are shown as
the mean ± the standard error of the mean (SEM) and *
denotes a significant difference (P < 0.05).
Results
Dose-dependent tumor formation followingsubcutaneous transplantation of MyLa2059 cellsinto NOD ⁄ SCID-B2m) ⁄ ) miceTo investigate if malignant CTCL cells formed tumors when
transplanted to NOD.Cg-Prkdcscid B2mtm1Unc ⁄ J (NOD ⁄ S-
CID-B2m) ⁄ )) mice, we initially inoculated 1 · 106, 5 · 106,
10 · 106 or 20 · 106 MyLa2059 cells s.c. into both flanks of
NOD ⁄ SCID-B2m) ⁄ ) mice and measured the tumor load
and volume after 3 weeks. As shown in Fig. 1a, all mice
inoculated with MyLa2059 cells developed tumors in a
dose-dependent manner and, expectedly, no tumors were
observed on the flanks of mice injected with saline. To
examine the kinetics of the tumor formation, we next inoc-
ulated NOD ⁄ SCID-B2m) ⁄ ) mice with 1 · 106 or 2.5 · 106
MyLa2059 cells and measured the tumor size two or three
times a week for up to 6 weeks. Tumor formation could
clearly be detected as early as 7 to 10 days following inocu-
lation of 2.5 · 106 malignant T cells (Fig. 1b, upper panel).
By the end of the first week and onwards, the tumors grew
exponentially until the mice were killed on day 22. Inocula-
tion of 1 · 106 MyLa2059 cells resulted in detectable tumor
formation by the end of the second week and also gave rise
to an exponential (albeit slower) growth curve until the
mice had to be killed on day 37. The lower panel in Fig. 1b
shows two representative mice that received 1 · 106 (mouse
210) and 2.5 · 106 (mouse 190) malignant T cells and were
killed after 37 and 22 days, respectively. Subcutaneous
tumors were macroscopically visible in both mice and skin
ulceration was also observed in mouse 190. Owing to the
delayed tumor growth allowing a longer observation period
before the mice had to be killed, 1 · 106 cells per inocula-
tion was chosen for further studies.
Highly reproducible tumor formation followingtransplantation of 1 · 106 malignant T cellsTo confirm our initial results in a larger cohort, 11 mice
were inoculated s.c. into both flanks with 1 · 106 MyLa2059
cells and the tumor size and weight determined 15 days
later. Although we observed some variation in the tumor
formation and growth rates, an overall robust tumor devel-
opment was seen with an average tumor size of 64 mm3
and an average tumor load of 95 mg (Table S1). In total, 20
of the 22 (90%) transplantations resulted in macroscopically
visible tumors (Table S1). To investigate whether the tumor
formation was related to the neoplastic nature of the
T lymphocytes, mice were inoculated into both flanks with
either 1 · 106 malignant T cells (MyLa2059) or 5 · 106
non-malignant T cells (MyLa1850) derived from the same
(a) (b)
Figure 1. Tumor formation following s.c. transplantation of MyLa2059
cells to NOD ⁄ SCID-B2m) ⁄ ) mice. (a) Two mice per group were injected
s.c. into both flanks with saline or different numbers of MyLa2059 cells
suspended in saline. After three weeks, the mice were killed and the
weight, as well as the length, width and depth of the tumors
determined. The histograms show the average (upper panel) tumor
mass and (lower panel) volume per mouse and the error bars represent
SEM. The ellipsoid tumor volume was calculated using the formula
(a · b · c) · p ⁄ 6, where a, b and c designate tumor diameters (mm)
for length, width and depth, respectively. (b) Two groups of three mice
were injected s.c. with 1 · 106 or 2.5 · 106 MyLa2059 cells into both
flanks. The length and width of the tumors were measured at 2- to 3-
day intervals until killing on day 22 (2.5 · 106) or 37 (1 · 106) post
injection. (Upper panel) A histogram showing the accumulated average
tumor volume from both flanks for mice inoculated with 1 · 106 (grey
line) or 2.5 · 106 (black line) MyLa2059 cells. The tumor volume was
calculated using the formula V = (a · b2) ⁄ 2, where a defines the length
(mm) and b the width (mm) of the tumor. Error bars represent SEM.
* denotes a significant difference (P < 0.05) in the average tumor
volume between the two groups at the given time-point using a
two-tailed two-sample t test with Welch’s correction. (Lower panel)
Representative images of tumors in mouse 210 and mouse 190
inoculated with 1 · 106 or 2.5 · 106 MyLa2059 cells, respectively.
Krejsgaard et al.
1098 ª 2010 John Wiley & Sons A/S, Experimental Dermatology, 19, 1096–1102
patient with CTCL, and the tumor development was fol-
lowed for 36 days. In mice inoculated with malignant
T cells, tumor formation was detected in 16 of 18 transplan-
tations (88%), whereas tumor formation was not observed
in any of the ten animals inoculated with non-malignant
T cells (data not shown). Moreover, in a separate experi-
ment, tumor formation was not observed within 6 months
irrespective if 1 · 106 or 5 · 106 non-malignant T cells were
inoculated (data not shown).
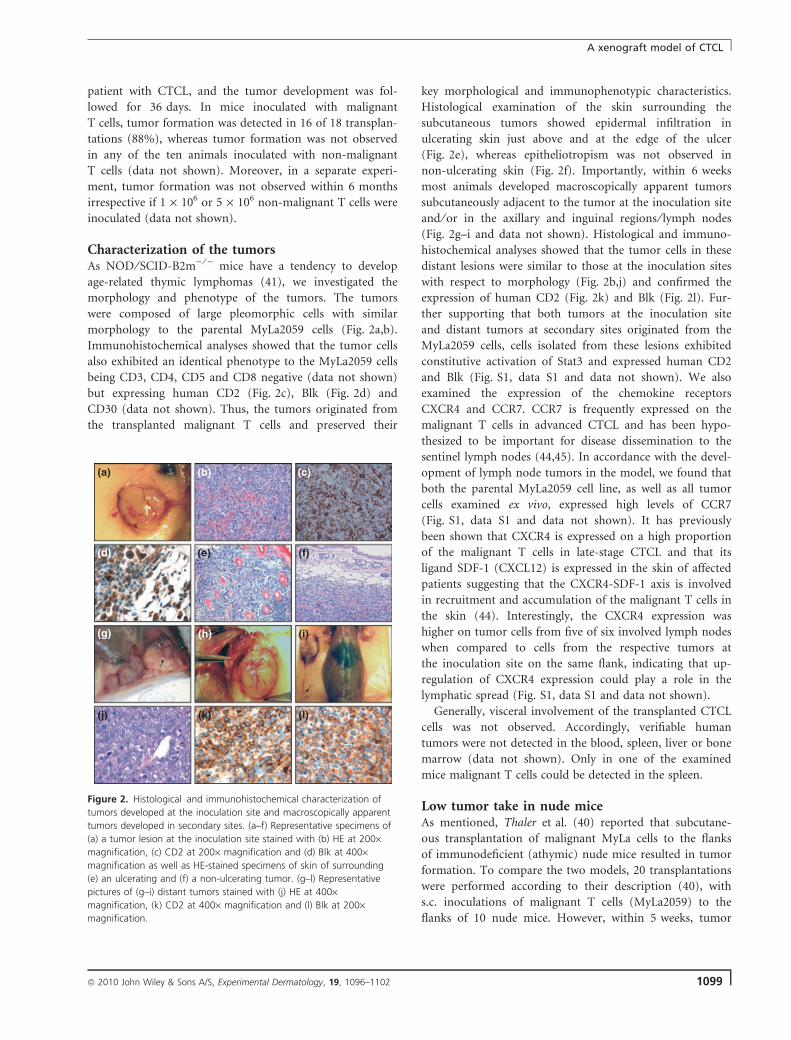
Characterization of the tumorsAs NOD ⁄ SCID-B2m) ⁄ ) mice have a tendency to develop
age-related thymic lymphomas (41), we investigated the
morphology and phenotype of the tumors. The tumors
were composed of large pleomorphic cells with similar
morphology to the parental MyLa2059 cells (Fig. 2a,b).
Immunohistochemical analyses showed that the tumor cells
also exhibited an identical phenotype to the MyLa2059 cells
being CD3, CD4, CD5 and CD8 negative (data not shown)
but expressing human CD2 (Fig. 2c), Blk (Fig. 2d) and
CD30 (data not shown). Thus, the tumors originated from
the transplanted malignant T cells and preserved their
key morphological and immunophenotypic characteristics.
Histological examination of the skin surrounding the
subcutaneous tumors showed epidermal infiltration in
ulcerating skin just above and at the edge of the ulcer
(Fig. 2e), whereas epitheliotropism was not observed in
non-ulcerating skin (Fig. 2f). Importantly, within 6 weeks
most animals developed macroscopically apparent tumors
subcutaneously adjacent to the tumor at the inoculation site
and ⁄ or in the axillary and inguinal regions ⁄ lymph nodes
(Fig. 2g–i and data not shown). Histological and immuno-
histochemical analyses showed that the tumor cells in these
distant lesions were similar to those at the inoculation sites
with respect to morphology (Fig. 2b,j) and confirmed the
expression of human CD2 (Fig. 2k) and Blk (Fig. 2l). Fur-
ther supporting that both tumors at the inoculation site
and distant tumors at secondary sites originated from the
MyLa2059 cells, cells isolated from these lesions exhibited
constitutive activation of Stat3 and expressed human CD2
and Blk (Fig. S1, data S1 and data not shown). We also
examined the expression of the chemokine receptors
CXCR4 and CCR7. CCR7 is frequently expressed on the
malignant T cells in advanced CTCL and has been hypo-
thesized to be important for disease dissemination to the
sentinel lymph nodes (44,45). In accordance with the devel-
opment of lymph node tumors in the model, we found that
both the parental MyLa2059 cell line, as well as all tumor
cells examined ex vivo, expressed high levels of CCR7
(Fig. S1, data S1 and data not shown). It has previously
been shown that CXCR4 is expressed on a high proportion
of the malignant T cells in late-stage CTCL and that its
ligand SDF-1 (CXCL12) is expressed in the skin of affected
patients suggesting that the CXCR4-SDF-1 axis is involved
in recruitment and accumulation of the malignant T cells in
the skin (44). Interestingly, the CXCR4 expression was
higher on tumor cells from five of six involved lymph nodes
when compared to cells from the respective tumors at
the inoculation site on the same flank, indicating that up-
regulation of CXCR4 expression could play a role in the
lymphatic spread (Fig. S1, data S1 and data not shown).
Generally, visceral involvement of the transplanted CTCL
cells was not observed. Accordingly, verifiable human
tumors were not detected in the blood, spleen, liver or bone
marrow (data not shown). Only in one of the examined
mice malignant T cells could be detected in the spleen.
Low tumor take in nude miceAs mentioned, Thaler et al. (40) reported that subcutane-
ous transplantation of malignant MyLa cells to the flanks
of immunodeficient (athymic) nude mice resulted in tumor
formation. To compare the two models, 20 transplantations
were performed according to their description (40), with
s.c. inoculations of malignant T cells (MyLa2059) to the
flanks of 10 nude mice. However, within 5 weeks, tumor
(a)
(d) (e) (f)
(i)
(l)(k)
(h)(g)
(j)
(b) (c)
Figure 2. Histological and immunohistochemical characterization of
tumors developed at the inoculation site and macroscopically apparent
tumors developed in secondary sites. (a–f) Representative specimens of
(a) a tumor lesion at the inoculation site stained with (b) HE at 200·magnification, (c) CD2 at 200· magnification and (d) Blk at 400·magnification as well as HE-stained specimens of skin of surrounding
(e) an ulcerating and (f) a non-ulcerating tumor. (g–l) Representative
pictures of (g–i) distant tumors stained with (j) HE at 400·magnification, (k) CD2 at 400· magnification and (l) Blk at 200·magnification.
A xenograft model of CTCL
ª 2010 John Wiley & Sons A/S, Experimental Dermatology, 19, 1096–1102 1099

formation was only detected in 1 of 20 transplantations in
nude mice, whereas tumor formation was observed in
seven of eight transplantations in NOD ⁄ SCID-B2m) ⁄ )
mice (Table S2). Thus, the difference in tumor formation
frequency between the two mouse strains was highly signi-
ficant (P < 0.0001, Fisher’s exact test).
Vorinostat inhibits the tumor growthTo examine the potential use of the xenograft model in pre-
clinical testing of novel drugs for treatment of advanced
CTCL, we investigated the effect of vorinostat on the tumor
growth. Vorinostat was recently approved by the FDA for
treatment of cutaneous manifestations in patients with MF
who have progressive, persistent or recurrent disease on or
after two systemic therapies (42). Briefly, NOD ⁄ SCID-
B2m) ⁄ ) mice were inoculated s.c. with 1 · 106 MyLa2059
cells into the flanks and after development of established
palpable tumors treatment was initiated. The mice received
either vehicle or 60 mg ⁄ kg vorinostat i.p. 5 days a week and
the tumor sizes were measured continuously. As seen in
Fig. 3, the tumor growth was suppressed in mice treated
with vorinostat when compared to mice treated with vehi-
cle. A significant reduction of the tumor volume was
already observed 4 days post treatment initiation and
onwards until the study was terminated at day 20 post treat-
ment initiation (P < 0.05 both using the two-sample t test
with Welch’s correction and the Mann–Whitney U test).
These data indicate that the NOD ⁄ SCID-B2m) ⁄ ) xenograft
model could be useful as part of preclinical evaluation of
novel therapeutic agents.
Discussion
In this study, we show that the malignant CTCL T-cell line
MyLa2059 forms tumors after s.c. inoculation into NOD ⁄ S-
CID-B2m) ⁄ ) mice. Specifically, inoculation of 1 · 106
malignant T cells resulted in a rapid tumor formation with
a tumor take of 90%. These subcutaneous tumors grew to
substantial sizes and eventually ulcerated. Importantly, the
transplanted malignant T cells disseminated outside the
inoculation site and formed subcutaneous tumors adjacent
to the transplantation site as well as in the axillary and
inguinal regions ⁄ lymph nodes. NOD ⁄ SCID-B2m) ⁄ ) mice
frequently develop spontaneous age-related thymic lympho-
mas (41). However, the human origin of tumors at both
primary and secondary sites, as well as the preservation of
the key features of the malignant CTCL cells, was demon-
strated by cell morphology and expression of human CD2.
As Blk is a novel diagnostic marker expressed in situ in
patients with CTCL and involved in the proliferation of the
malignant T cells, it was of particular interest that both
tumors at the inoculation site and distant tumors displayed
a clear expression of Blk in vivo (18). Furthermore, tumor
cells with similar phenotype and size as the parental
MyLa2059 cell line could be isolated from tumors at the
inoculation site and from involved lymph nodes. Both
MyLa2059 cells and all isolated cells expressed high levels
of CCR7 which could, at least partly, explain the propensity
of the malignant T cells to home to the lymphatic tissue.
Interestingly, CXCR4 was up-regulated on tumor cells from
five of six involved lymph nodes when compared to cells
isolated from the respective tumors at the inoculation site
on the same flank. A previous report has provided evidence
that the CXCR4-SDF-1 axis is involved in CTCL cell skin
recruitment and accumulation. SDF-1 is continuously pro-
duced by stromal cells in the lymph nodes and malignant
expression of CXCR4 correlates with lymph node metasta-
sis and poor survival in a number of malignancies (46,47).
Accordingly, the present data raise the possibility that the
CXCR4-SDF1 axis might not only play a role in the skin
homing of the malignant T cells but that it could also be
involved in lymph node metastasis in CTCL. Of notice,
SDF-1 is highly conserved between mouse and man, allow-
ing it to act across species barriers (47). In contrast to the
present NOD ⁄ SCID-B2m) ⁄ ) model, skin ulceration or
macroscopically apparent tumor dissemination was not
observed in either of the two previously described CTCL
models, i.e. the patient-skin-graft model in SCID mice and
the xenograft transplantation model in nude mice (39,40).
Because transplantation of immune competent T cells
into a severely immune-compromised host theoretically
Figure 3. Vorinostat inhibits the tumor growth. NOD ⁄ SCID-B2m) ⁄ )
mice were injected s.c. with 1 · 106 MyLa2059 cells into both flanks.
When the mice had developed established palpable tumors treatment
was initiated (day 1). The mice received either vehicle (black line) or
60 mg ⁄ kg vorinostat (grey line) i.p. five days a week. In total, there
were four mice in the group receiving vehicle and three mice in the
group receiving vorinostat. The length and width of the tumors were
measured continuously until the experiment was terminated on day 20
post treatment initiation. The tumor volume was calculated using the
formula V = (a · b2) ⁄ 2, where a defines the length (mm) and b the
width (mm) of the tumor. The histogram shows the average tumor
volume per flank ± SEM. * denotes a significant difference (P < 0.05) in
the average tumor volume per flank between mice treated with vehicle
and vorinostat at the given time-point both by using a one-tailed two-
sample t test with Welch’s correction and a Mann–Whitney U test.
Krejsgaard et al.
1100 ª 2010 John Wiley & Sons A/S, Experimental Dermatology, 19, 1096–1102

could lead to graft-versus-host reactions, we utilized a
malignant T-cell line (MyLa2059) with a deficient expres-
sion and function of the T-cell receptor (TCR) ⁄ CD3 com-
plex (48) which is often seen in malignant T cells in
advanced disease stages (17,48). Accordingly, MyLa2059
cells do not respond with an enhanced proliferation follow-
ing antibody-mediated CD3 cross-linking (48). Therefore,
the formation of tumors in vivo was most likely a result of
an ‘autonomous’ proliferation of the malignant T cells and
certainly not caused by TCR ⁄ CD3-mediated proliferation
of malignant T cells engaged in xenograft-versus-host reac-
tions. Because NOD ⁄ SCID-B2m) ⁄ ) mice have an impaired
function of antigen presenting cells together with a defi-
cient expression of MHC class I and complement factor
C5, it is reasonable to assume that these mice are less
potent inducers of xenograft-versus-host reactions when
compared to conventional SCID mice. This notion is sub-
stantiated by the observation that even transplantation of
non-malignant T cells did not lead to xenograft-versus-host
reactions. Despite the fact that the non-malignant T cells
express a functional TCR ⁄ CD3 complex and respond to
CD3 cross-linking by proliferation in vitro (48), they did
not induce an inflammatory response in vivo. Thus, these
observations suggest that the non-malignant T cells were
not properly activated to sustain their growth in vivo in
NOD ⁄ SCID-B2m) ⁄ ) mice. In contrast, the malignant T
cells readily formed tumors in vivo further indicating
that the malignant T-cell line isolated from the affected skin
of a patient with CTCL has retained its neoplastic nature.
NOD ⁄ SCID-B2m) ⁄ ) mice display a profound immune
deficiency characterized by the absence of mature T and B
cells, a C5 deficiency, an absence of MHC class I molecules,
as well as severely diminished NK cell activity. These fea-
tures are believed to make this strain of mice particularly
suited for xenograft transplantations and the present study
supports this notion (41). Indeed, we observed a signifi-
cantly better tumor formation in NOD ⁄ SCID-B2m) ⁄ ) mice
(seven of eight transplantations) when compared to the a-
thymic nude mice (1 of 20 transplantations) within a per-
iod of 5 weeks. The observation of a substantial lower
tumor take in nude mice is in keeping with the previous
study by Thaler et al. (40) who reported on tumor forma-
tion of MyLa cells in only one of six transplantations into
nude mice within a 2-month observation period. As both
mouse strains lack mature T cells and have a deficient anti-
body response, the major immunological differences relate
to the compromised NK cell function together with the
MHC class I and complement factor C5 deficiency in
NOD ⁄ SCID-B2m) ⁄ ) mice. Accordingly, our observations
suggest that NK cells and ⁄ or complement factor C5 play a
key role in tumor rejection and growth control. In support
of this hypothesis, malignant CTCL cells express NK cell
ligands and are known targets of autologous NK cells in vi-
tro (49,50). Moreover, it is well established that the com-
plement system is a major barrier for successful xenograft
transplantation and C5 plays a key role in xenograft rejec-
tion in experimental models (51–53). This may also explain
why tumors in nude mice appeared much later
(>4 months) and required inoculation of 10 times more
MyLa cells per inoculation (40) when compared to the
present findings in NOD ⁄ SCID-B2m) ⁄ ) mice. To circum-
vent these problems, Thaler et al. (40) performed serial
passages and selections of cells displaying the fastest in vivo
growth risking an artificial cell selection. Importantly, in
the NOD ⁄ SCID-B2m) ⁄ ) model, such measures were not
necessary as tumors developed fast and with a robust
tumor take. Therefore, the present model is characterized
by several advantages over the previous models: (i) rapid
tumor growth following transplantation of an unselected
‘primary’ malignant T-cell line, (ii) a high, robust, and pre-
dictable tumor take and (iii) the formation of macroscopi-
cally apparent secondary tumors. These characteristics are
mandatory prerequisites for preclinical testing of any new
therapeutic strategy in vivo. Supporting the potential use of
the current xenograft model as part of preclinical evalua-
tion of new drugs for treatment of advanced CTCL, we
found that vorinostat suppressed the tumor growth when
compared to mice treated with vehicle. To our knowledge,
this is the first study demonstrating that vorinostat inhibits
the tumor growth in an animal model of CTCL.
In conclusion, the NOD ⁄ SCID-B2m) ⁄ ) mouse xenograft
model presented here displays some features similar to
those of tumor stage CTCL and could provide a valuable
model for future studies of disease dissemination and for
the evaluation of new therapies.
Acknowledgements
This work was supported by grants from The University of Copenhagen,
The Danish Research Councils, The Foundation of 17-12-1981, The Novo
Nordic Foundation, The Danish Cancer Society, The Neye Foundation,
The Lundbeck Foundation, and The National Cancer Institute (CA89194:
MA Wasik). We wish to thank Keld Kaltoft (Arhus University and Cell-
Cure Arhus, Denmark) for the generous gift of the MyLa cell lines. The
project part concerning establishment and study of CTCL cell lines by
Dr Keld Kaltoft has been approved by ‘Den videnskabsetiske Kommite i
Arhus Amt’ (The science-ethical committee in Arhus County).
Conflict of interest
The authors state no conflict of interest.
References
1 Olsen E, Vonderheid E, Pimpinelli N et al. Revisions to the staging and classifi-cation of mycosis fungoides and Sezary syndrome: a proposal of the Interna-tional Society for Cutaneous Lymphomas (ISCL) and the cutaneous lymphomatask force of the European Organization of Research and Treatment of Cancer(EORTC). Blood 2007: 110: 1713–1722.
2 Karenko L, Hahtola S, Ranki A. Molecular cytogenetics in the study of cutane-ous T-cell lymphomas (CTCL). Cytogenet Genome Res 2007: 118: 353–361.
A xenograft model of CTCL
ª 2010 John Wiley & Sons A/S, Experimental Dermatology, 19, 1096–1102 1101

3 Diamandidou E, Cohen P R, Kurzrock R. Mycosis fungoides and Sezary syn-drome. Blood 1996: 88: 2385–2409.
4 Robson A. The pathology of cutaneous T-cell lymphoma. Oncology (WillistonPark) 2007: 21: 9–12.
5 Ballanger F, Bressollette C, Volteau C, Planche L, Dreno B. Cytomegalovirus:its potential role in the development of cutaneous T-cell lymphoma. Exp Der-matol 2009: 18: 574–576.
6 Jackow C M, Cather J C, Hearne V et al. Association of erythrodermiccutaneous T-cell lymphoma, superantigen-positive Staphylococcus aureus,and oligoclonal T-cell receptor V beta gene expansion. Blood 1997: 89: 32–40.
7 Dobbeling U, Dummer R, Laine E et al. Interleukin-15 is an autocrine ⁄ para-crine viability factor for cutaneous T-cell lymphoma cells. Blood 1998: 92:252–258.
8 Qin J Z, Zhang C L, Kamarashev J et al. Interleukin-7 and interleukin-15 regu-late the expression of the bcl-2 and c-myb genes in cutaneous T-cell lym-phoma cells. Blood 2001: 98: 2778–2783.
9 Nielsen M, Nissen M H, Gerwien J et al. Spontaneous interleukin-5 productionin cutaneous T-cell lymphoma lines is mediated by constitutively activatedStat3. Blood 2002: 99: 973–977.
10 Yamanaka K, Clark R, Rich B et al. Skin-derived interleukin-7 contributes tothe proliferation of lymphocytes in cutaneous T-cell lymphoma. Blood 2006:107: 2440–2445.
11 Asadullah K, Haeussler-Quade A, Gellrich S et al. IL-15 and IL-16 overexpres-sion in cutaneous T-cell lymphomas: stage-dependent increase in mycosis fun-goides progression. Exp Dermatol 2000: 9: 248–251.
12 Krejsgaard T, Vetter-Kauczok C S, Woetmann A et al. Jak3- and JNK-depen-dent vascular endothelial growth factor expression in cutaneous T-cell lym-phoma. Leukemia 2006: 20: 1759–1766.
13 Kim E J, Hess S, Richardson S K et al. Immunopathogenesis and therapy ofcutaneous T cell lymphoma. J Clin Invest 2005: 115: 798–812.
14 Vacca A, Moretti S, Ribatti D et al. Progression of mycosis fungoides is associ-ated with changes in angiogenesis and expression of the matrix metallopro-teinases 2 and 9. Eur J Cancer 1997: 33: 1685–1692.
15 Wu J, Nihal M, Siddiqui J, Vonderheid E C, Wood G S. Low FAS ⁄ CD95 expres-sion by CTCL correlates with reduced sensitivity to apoptosis that can berestored by FAS upregulation. J Invest Dermatol 2009: 129: 1165–1173.
16 Contassot E, French L E. Targeting apoptosis defects in cutaneous T-cell lym-phoma. J Invest Dermatol 2009: 129: 1059–1061.
17 Klemke C D, Brenner D, Weiss E M et al. Lack of T-cell receptor-induced sig-naling is crucial for CD95 ligand up-regulation and protects cutaneous T-celllymphoma cells from activation-induced cell death. Cancer Res 2009: 69:4175–4183.
18 Krejsgaard T, Vetter-Kauczok C S, Woetmann A et al. Ectopic expression ofB-lymphoid kinase in cutaneous T-cell lymphoma. Blood 2009: 113: 5896–5904.
19 Nielsen M, Kaltoft K, Nordahl M et al. Constitutive activation of a slowlymigrating isoform of Stat3 in mycosis fungoides: tyrphostin AG490 inhibitsStat3 activation and growth of mycosis fungoides tumor cell lines. Proc NatlAcad Sci USA 1997: 94: 6764–6769.
20 Zhang Q, Nowak I, Vonderheid E C et al. Activation of Jak ⁄ STAT proteinsinvolved in signal transduction pathway mediated by receptor for interleukin 2in malignant T lymphocytes derived from cutaneous anaplastic large T-celllymphoma and Sezary syndrome. Proc Natl Acad Sci USA 1996: 93: 9148–9153.
21 Eriksen K W, Kaltoft K, Mikkelsen G et al. Constitutive STAT3-activation inSezary syndrome: tyrphostin AG490 inhibits STAT3-activation, interleukin-2receptor expression and growth of leukemic Sezary cells. Leukemia 2001: 15:787–793.
22 Kasprzycka M, Majewski M, Wang Z J et al. Expression and oncogenic role ofBrk (PTK6 ⁄ Sik) protein tyrosine kinase in lymphocytes. Am J Pathol 2006: 168:1631–1641.
23 Kennah E, Ringrose A, Zhou L L et al. Identification of tyrosine kinase, HCK,and tumor suppressor, BIN1, as potential mediators of AHI-1 oncogene inprimary and transformed CTCL cells. Blood 2009: 113: 4646–4655.
24 Mao X, Orchard G, Lillington D M et al. BCL2 and JUNB abnormalities in pri-mary cutaneous lymphomas. Br J Dermatol 2004: 151: 546–556.
25 Thakur S, Lin H C, Tseng W T et al. Rearrangement and altered expression ofthe NFKB-2 gene in human cutaneous T-lymphoma cells. Oncogene 1994: 9:2335–2344.
26 Sommer V H, Clemmensen O J, Nielsen O et al. In vivo activation of STAT3 incutaneous T-cell lymphoma. Evidence for an antiapoptotic function of STAT3.Leukemia 2004: 18: 1288–1295.
27 Dobbeling U. Transcription factor profiling shows new ways towards newtreatment options of cutaneous T cell lymphomas. Curr Drug Discov Technol2007: 4: 24–30.
28 Krejsgaard T, Gjerdrum L M, Ralfkiaer E et al. Malignant Tregs express lowmolecular splice forms of FOXP3 in Sezary syndrome. Leukemia 2008: 22:2230–2239.
29 Knol A C, Quereux G, Brocard A et al. Absence of modulation ofCD4+CD25(high) regulatory T cells in CTCL patients treated with bexarotene.Exp Dermatol 2009: DOI: 10.1111/j.1600-0625.2009.00993.x.
30 Brender C, Nielsen M, Kaltoft K et al. STAT3-mediated constitutive expressionof SOCS-3 in cutaneous T-cell lymphoma. Blood 2001: 97: 1056–1062.
31 Brender C, Lovato P, Sommer V H et al. Constitutive SOCS-3 expression pro-tects T-cell lymphoma against growth inhibition by IFNalpha. Leukemia 2005:19: 209–213.
32 Zhang Q, Raghunath P N, Vonderheid E, Odum N, Wasik M A. Lack of phosp-hotyrosine phosphatase SHP-1 expression in malignant T-cell lymphoma cellsresults from methylation of the SHP-1 promoter. Am J Pathol 2000: 157:1137–1146.
33 van D R, Zoutman W H, Dijkman R et al. Epigenetic profiling of cutaneousT-cell lymphoma: promoter hypermethylation of multiple tumor suppres-sor genes including BCL7a, PTPRG, and p73. J Clin Oncol 2005: 23: 3886–3896.
34 Gallardo F, Esteller M, Pujol R M et al. Methylation status of the p15, p16 andMGMT promoter genes in primary cutaneous T-cell lymphomas. Haematolog-ica 2004: 89: 1401–1403.
35 Nagasawa T, Zhang Q, Raghunath P N et al. Multi-gene epigenetic silencingof tumor suppressor genes in T-cell lymphoma cells; delayed expression of thep16 protein upon reversal of the silencing. Leuk Res 2006: 30: 303–312.
36 Bagot M. Introduction: cutaneous T-cell lymphoma (CTCL)–classification, stag-ing, and treatment options. Dermatol Clin 2008: 26 (Suppl. 1): 3–12.
37 Mestel D S, Assaf C, Steinhoff M et al. Emerging drugs in cutaneous T celllymphoma. Expert Opin Emerg Drugs 2008: 13: 345–361.
38 Klemke C D, Goerdt S, Schrama D, Becker J C. New insights into the molecu-lar biology and targeted therapy of cutaneous T-cell lymphomas. J DtschDermatol Ges 2006: 4: 395–406.
39 Charley M R, Tharp M, Locker J et al. Establishment of a human cutaneousT-cell lymphoma in C.B-17 SCID mice. J Invest Dermatol 1990: 94: 381–384.
40 Thaler S, Burger A M, Schulz T et al. Establishment of a mouse xenograftmodel for mycosis fungoides. Exp Dermatol 2004: 13: 406–412.
41 Christianson S W, Greiner D L, Hesselton R A et al. Enhanced human CD4 +T cell engraftment in beta2-microglobulin-deficient NOD-scid mice. J Immunol1997: 158: 3578–3586.
42 Olsen E A, Kim Y H, Kuzel T M et al. Phase IIb multicenter trial of vorinostat inpatients with persistent, progressive, or treatment refractory cutaneous T-celllymphoma. J Clin Oncol 2007: 25: 3109–3115.
43 Kaltoft K, Bisballe S, Dyrberg T et al. Establishment of two continuous T-cellstrains from a single plaque of a patient with mycosis fungoides. In Vitro CellDev Biol 1992: 28A: 161–167.
44 Narducci M G, Scala E, Bresin A et al. Skin homing of Sezary cells involvesSDF-1-CXCR4 signaling and down-regulation of CD26 ⁄ dipeptidylpeptidase IV.Blood 2006: 107: 1108–1115.
45 Picchio M C, Scala E, Pomponi D et al. CXCL13 is highly produced by Sezarycells and enhances their migratory ability via a synergistic mechanism involvingCCL19 and CCL21 chemokines. Cancer Res 2008: 68: 7137–7146.
46 Arai J, Yasukawa M, Yakushijin Y, Miyazaki T, Fujita S. Stromal cells in lymphnodes attract B-lymphoma cells via production of stromal cell-derived factor-1.Eur J Haematol 2000: 64: 323–332.
47 Burger J A, Kipps T J. CXCR4: a key receptor in the crosstalk between tumorcells and their microenvironment. Blood 2006: 107: 1761–1767.
48 Woetmann A, Lovato P, Eriksen K W et al. Nonmalignant T cells stimulategrowth of T-cell lymphoma cells in the presence of bacterial toxins. Blood2007: 109: 3325–3332.
49 Dulphy N, Berrou J, Campillo J A et al. NKG2D ligands expression and NKG2D-mediated NK activity in Sezary patients. J Invest Dermatol 2009: 129: 359–364.
50 Bouaziz J D, Ortonne N, Giustiniani J et al. Circulating natural killer lympho-cytes are potential cytotoxic effectors against autologous malignant cells insezary syndrome patients. J Invest Dermatol 2005: 125: 1273–1278.
51 Ryan U S. Complement inhibitory therapeutics and xenotransplantation. NatMed 1995: 1: 967–968.
52 Markiewski M M, DeAngelis R A, Benencia F et al. Modulation of the antitu-mor immune response by complement. Nat Immunol 2008: 9: 1225–1235.
53 Gaca J G, Appel J Z III, Lukes J G et al. Effect of an anti-C5a monoclonal anti-body indicates a prominent role for anaphylatoxin in pulmonary xenograft dys-function. Transplantation 2006: 81: 1686–1694.
Supporting information
Additional Supporting Information may be found in the online version of
this article:
Data S1. Method.
Figure S1. Characterization of isolated tumor cells by flow cytometry.
Representative flow cytometric analysis of parental MyLa2059 cells and cells
isolated from mouse 187 (M187) stained with antibodies against CD2,
CXCR4, CCR7 or respective isotype control antibodies.
Table S1. Tumor volume and weight after 15 days in mice inoculated
s.c. with 1 · 106 MyLa2059 cells into each flank.
Table S2. Tumor formation in NOD ⁄ SCID-B2m) ⁄ ) and nude mice.
Please note: Wiley-Blackwell are not responsible for the content or func-
tionality of any supporting materials supplied by the authors. Any queries
(other than missing material) should be directed to the corresponding
author for the article.
Krejsgaard et al.
1102 ª 2010 John Wiley & Sons A/S, Experimental Dermatology, 19, 1096–1102

















![Case Report Primary cutaneous γδ-T-cell lymphoma … cutaneous γδ-T-cell lymphoma (CGD-TCL) ... TCL [3]. Some other study reports that allogenic ... we reported a case of CGD-TCL](https://img.pdfslide.net/doc/110x75/5ae360cf7f8b9a495c8d272b/case-report-primary-cutaneous-t-cell-lymphoma-cutaneous-t-cell-lymphoma.jpg)











