Embed Size (px)
Citation preview

Journal of Molecular Structure (Theochem), 167 (1988) 321-329 Elsevier Science Publishers B.V., Amsterdam - Printed in The Netherlands
321
A PRELIMINARY AB INITIO INVESTIGATION OF RETINAL ANALOGS
RAYMOND A. POIRIER*, ARPITA YADAV and PETER R. SURJAN**
Memorial University of Newfoundland, St. John’s, Newfoundland, AlB 3X7 (Canada)
(Received 18 August 1987 )
ABSTRACT
Optimized ab initio STO-3G geometries and various properties are presented for the largest protonated retinal Schiff base analog reported to date for ab initio calculations. The results indi- cate that completion of the cyclohexene ring has little effect on bonding and other properties of the molecule, but simply causes local torsional distortions. Starting with the four double bond analog, the bonding and net charges of the protonated Schiff base end do not alter upon further addition to the chain. The results also indicate that a conformational change around the C6-C7 single bond is accompanied by a small red shift which is important in understanding the opsin shift in bacteriorhodopsin.
INTRODUCTION
The photoisomerization process, which is known as the primary event, is one step in a series of complex steps leading to vision. The process of vision is far from being understood. An excellent review by Sandorfy and Vocelle of both the experimental and theoretical efforts made in this area has been published recently [ 11. The photoisomerization process is a challenging problem, both experimentally, due namely to the existence of a number of short-lived species, and theoretically, due to the size of the molecules and the methodological dif- ficulties of dealing with excited states of conjugated olefins [ 21. A first step, from a theoretical point of view, is to achieve a better understanding of the properties of retinal and retinal analogs. A knowledge, for example, of the con- formational hypersurface for the ground and excited states of the retinal Schiff base (RSB ) and the protonated retinal Schiff base (RPSB ) can be of value in predicting the feasibility of the different mechanisms that have been proposed for the cis-tram isomerization [ 31. Although the molecules involved are large, the authors have shown recently [4] that studying the analogs given below, using standard ab initio methods is feasible and useful.
*Author for correspondence. **Permanent address: Chinoin Research Centre, H-1325 Budapest, POB 110, Hungary.
0166-1280/88/$03.50 0 1988 Elsevier Science Publishers B.V.

322
The results [4] indicated that for retinal and retinal Schiff base analogs, the bond lengths, bond orders and atomic charges were essentially independent of chain length. On the other hand, the same properties for the protonated Schiff base analogs were dependent on chain length, due to a conjugational effect in the vicinity of the NH; group. Post Hartree-Fock, calculations with a Strictly Localized Geminals (SLG) wavefunction [ 51 indicated that the increase in atomic charge alternation at the carbon atoms for RPSB was the result of a considerable increase in the induced local dipoles. The properly correlated SLG results also showed that the previously reported sudden charge polarization observed when rotating around a double bond was simply an artifact of closed shell HF based methods. These small RSB analogs, even if based on simple orbital energies, show a red shift in n-n* excitation [ 41 energy upon protona- tion, in agreement with experimental observations. Recent experimental re- sults [ 61 suggest that part of the opsin shift is bacteriorhodopsin (1200 cm-’ out of 5100 cm-‘) arises because the chromophore changes upon binding to the protein from a 40” twisted 6-7-s& conformation to a planar 6-7-s-truns conformation.
In the present paper, the results of ab initio calculations are presented for a RPSB analog in which the only simplification made is the replacement of the methyl groups by hydrogen atoms. Both the 6-7-s-& (I) and 6-7-s-t~~n~ con- formations were studied.
METHOD
The calculations were performed with the basis set and program described in ref. 4. The difficulty of optimizing the geometry of such large molecules was reduced by combining the optimized geometry for the largest analog (II) pre- viously reported [ 41, with
7 \ \ \ \ \ p..fH2
the optimized geometry for the cyclohexene ring including an ethylenic (C7-C8) branch at C6 (III). Two conformations of the cyclohexene ring (IIIa and IIIb ) were optimized and the lowest energy conformation (IIIb, see Table 2 ) was used to construct the full RPSB (I).
323
ma IItb
The geometrical parameters common to both units were averaged. All geo- metrical parameters were relaxed, except the C9 to NH: part of the molecule which was kept planar. Since the C9 to NH,+ part of the molecule is conjugated, no significant torsional distortions were expected. (The gradients for the fixed torsional angles were all well within the convergence criteria.)
RESULTS AND DISCUSSION
Geometry
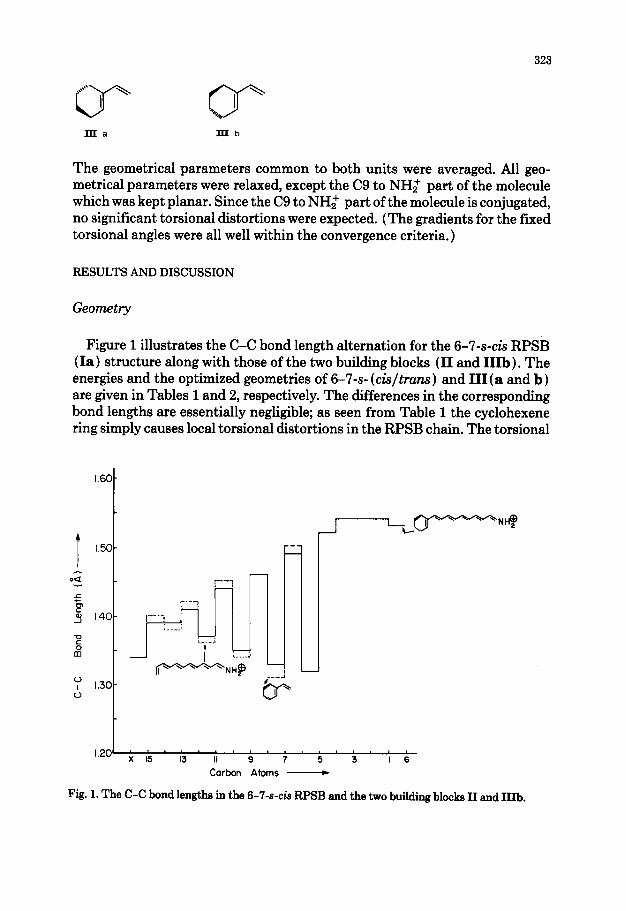
Figure 1 illustrates the C-C bond length alternation for the 6-7-s4.s RPSB (Ia) structure along with those of the two building blocks (II and IIIb). The energies and the optimized geometries of 6-7-s- (ck/truns) and III(a and b) are given in Tables 1 and 2, respectively. The differences in the corresponding bond lengths are essentially negligible; as seen from Table 1 the cyclohexene ring simply causes local torsional distortions in the RPSB chain. The torsional
1.60.
f
1.50-
I-“‘-‘! 8
P “-7 : -1 I-- I
: : _ . .
i,._ ,..A
f c j
I---
1 1.2oL m * ’ ’ ’ ’ ’ ’ ’ ’ ’ ’ 1 m ’ ’ *
x 15 13 Ii 9 7 5 3 I 6
Carbon Atoms -
Fig. 1. The C-C bond lengths in the 6-7-s-& RPSB and the two building block II and IIIb.
324
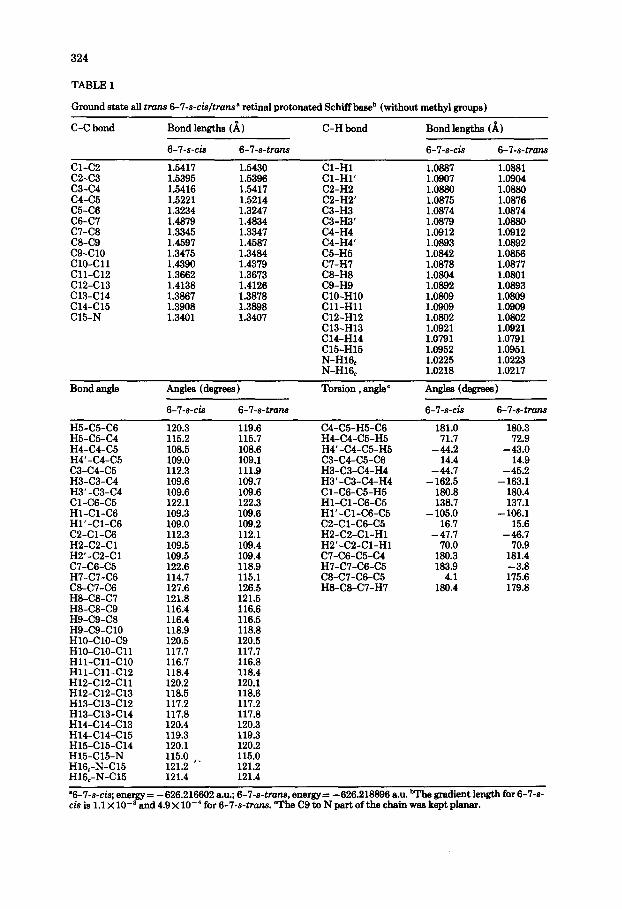
TABLE 1
Ground state all trans 6-7-s-&/tram9 retinal protonated Schiffba& (without methyl IVOUDS)
C-C bond Bond lengths (A)
6-7-s-& 6-7-s-trans
C-H bond Bond lengths (b)
6-7-s-& 6-7-s-trans
Cl-C2 1.5417 1.5430 Cl-H1 1.0887 1.0881 C2-C3 1.5395 1.5396 Cl-Hl’ 1.0907 1.0904 c3-c4 1.5416 1.5417 C2-H2 1.0880 1.0880 c4-c5 1.5221 C5-C6 1.3234 C6-C7 1.4879 c7-C8 1.3345 C&C9 1.4597 c9-Cl0 1.3475 ClO-Cl1 1.4390 Cll-Cl2 1.3662 C12-Cl3 1.4138 c13-Cl4 1.3867 c14-Cl5 1.3908 C15-N 1.3401
1.5214 1.3247 1.4834 1.3347 1.4587 1.3484 1.4379 1.3673 1.4126 1.3878 1.3898 1.3407
C2-H2’ 1.0875 1.0876 C3-H3 1.0874 1.0874 C3-H3’ 1.0879 1.0880 C4-H4 1.0912 1.0912 C4-H4’ 1.0893 1.0892 C5-H5 1.0842 1.0856 C7-H7 1.0878 1.0877 C8-H8 1.0804 1.0801 C9-H9 1.0892 1.0893 ClO-HlO 1.0809 1.0809 Cll-HI1 1.0909 1.0909 C12-H12 1.0802 1.0802 C13-H13 1.0921 1.0921 C14-H14 1.0791 1.0791 C15-H15 1.0952 1.0951 N-H16, 1.0225 1.0223 N-H16, 1.0218 1.0217
Bond angle Angles (degrees )
6-7-s-& 6-7-s-tram
Torsion , angle ’ Angles (degrees )
6-7-s-& 6-7-s-tram
H5-C5-C6 H5-C5-C4 H4-C4-C5 H4’-C4-C5 c3-c4-c5 H3-C3-C4 H3’-C3-C4 Cl-C6-C5 Hl-Cl-C6 Hl’-Cl-C6 C2-Cl-C6 H2-C2-Cl H2’-C2-Cl C7-C6-C5 H7-C7-C6 C8-C7-C6 H8-C8-C7 HS-C8-C9 H9-C9-C8 H9-C9-Cl0 HlO-ClO-C9 HlO-ClO-Cl1 Hll-Cll-Cl0 Hll-Cll-Cl2 H12-C12-Cl1 H12-C12-Cl3 H13-C13-Cl2 H13-C13-C14 H14-Cl4-Cl3 H14-C14-Cl5 H15-C15-Cl4 H15-C15-N H16,-N-Cl5 H16.-N-Cl5
120.3 119.6 115.2 115.7 108.5 108.6 109.0 109.1 112.3 111.9 109.6 109.7 109.6 109.6 122.1 122.3 109.3 109.6 109.0 109.2 112.3 112.1 109.5 109.4 109.5 109.4 122.6 118.9 114.7 115.1 127.6 126.5 121.8 121.5 116.4 116.6 116.4 116.5 118.9 118.8 120.5 120.5 117.7 117.7 116.7 116.8 118.4 118.4 120.2 120.1 118.5 118.6 117.2 117.2 117.8 117.8 120.4 120.3 119.3 119.3 120.1 120.2 115.0 121.2 t.
115.0 121.2
121.4 121.4
C4-C5-H5-C6 H4-C4-C5-H5 H4’-C4-C5-H5 C3-C4-C5-C6 H3-C3-C4-H4 H3’-C3-C4-H4 Cl-C6-C5-H5 Hl-Cl-C6-C5 Hl’-Cl-C6-C5 C2-Cl-C6-C5 H2-C2-Cl-H1 H2’-C2-Cl-H1 C7-C6-C5-C4 H7-C7-C6-C5 C8-C7-C6-C5 H8-C8-C7-H7
181.0 180.3 71.7 72.9
-44.2 -43.0 14.4
-44.7 - 162.5
180.8 138.7
- 105.0 16.7
-47.7 70.0
180.3 183.9
4.1 180.4
14.9 -45.2
- 163.1 180.4 137.1
- 106.1 15.6
-46.7 70.9
181.4 -3.8 175.6 179.8
“6-7-s-cis; energy= -626.216602 au.; 6-‘l-s-tram, energy= - 626.218896 B.U. bathe gradient length for 6-7-s-
cis is 1.1 X 10e3 and 4.9 X lo-’ for 6-7-s-tMn.s. “rhe C9 to N part of the chain was kept planar.
325
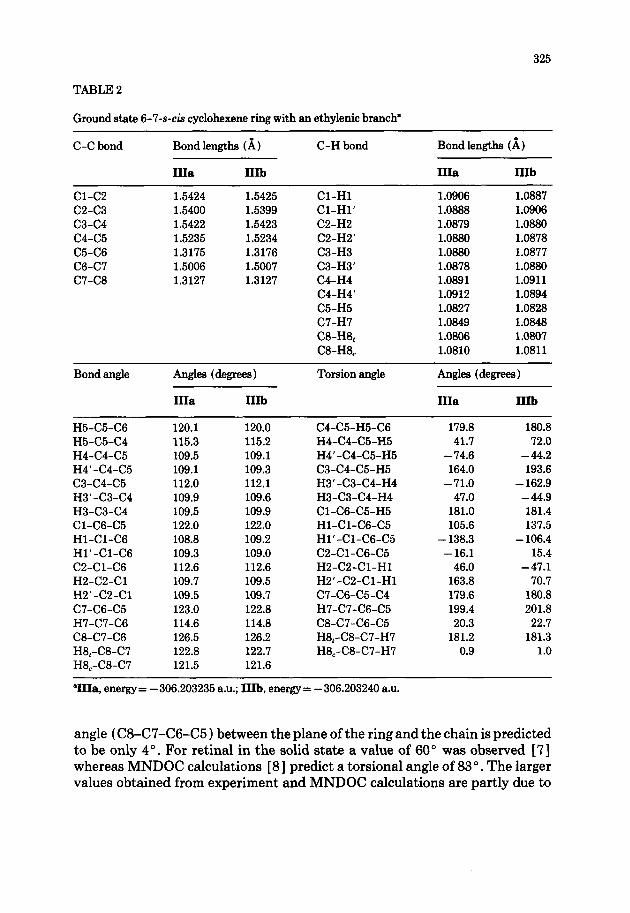
TABLE 2
Ground state 6-7-s-& cyclohexene ring with an ethylenic branch”
C-C bond
Cl-C2 C2-C3 c3-c4 c4-c5 C5-C6 C6-C7 C7-C8
Bond lengths (A)
IIIa IIIb
1.5424 1.5425 1.5400 1.5399 1.5422 1.5423 1.5235 1.5234 1.3175 1.3176 1.5006 1.5007 1.3127 1.3127
C-H bond
Cl-H1 Cl-Hl’ C2-H2 C2-H2’ C3-H3 C3-H3’ C4-H4 C4-H4’ C5-H5 C7-H7 CB-H8, C8-H8,
Bond lengths (A)
IIIa IIIb
1.0906 1.0887 1.0888 1.0906 1.0879 1.0880 1.0880 1.0878 1.0880 1.0877 1.0878 1.0880 1.0891 1.0911 1.0912 1.0894 1.0827 1.0828 1.0849 1.0848 1.0806 1.0807 1.0810 1.0811
Bond angle Angles (degrees)
IIIa IIIb
Torsion angle Angles (degrees)
IIIa IIIb
H5-C5-C6 120.1 120.0 H5-C5-C4 115.3 115.2 H4-C4-C5 109.5 109.1 H4’-C4-C5 109.1 109.3 c3-c4-c5 112.0 112.1 H3’-C3-C4 109.9 109.6 H3-C3-C4 109.5 109.9 Cl-C6-C5 122.0 122.0 Hl-Cl-C6 108.8 109.2 Hl’-Cl-C6 109.3 109.0 C2-Cl-C6 112.6 112.6 H2-C2-Cl 109.7 109.5 H2’-C2-Cl 109.5 109.7 C7-C6-C5 123.0 122.8 H7-C7-C6 114.6 114.8 C8-C7-C6 126.5 126.2 H8,-CS-C7 122.8 122.7 H8,-C8-C7 121.5 121.6
C4-C5-H5-C6 179.8 180.8 H4-C4-C5-H5 41.7 72.0 H4’-C4-C5-H5 - 74.6 - 44.2 C3-C4-C5-H5 164.0 193.6 H3’-C3-C4-H4 - 71.0 - 162.9 H3-C3-C4-H4 47.0 -44.9 Cl-C6-C5-H5 181.0 181.4 Hl-Cl-C6-C5 105.6 137.5 Hl’-Cl-C6-C5 - 138.3 - 106.4 C2-Cl-C6-C5 - 16.1 15.4 H2-C2-Cl-H1 46.0 -47.1 H2’-C2-Cl-H1 163.8 70.7 C7-C6-C5-C4 179.6 180.8 H7-C7-C6-C5 199.4 201.8 C8-C7-C6-C5 20.3 22.7 H8,-C8-C7-H7 181.2 181.3 H8,-CS-C7-H7 0.9 1.0
“IIIa, energy= -306.203235 a.u.; IIIb, energy= -306.203240 a.u.
angle (C8-C7-C6-C5) between the plane of the ring and the chain is predicted to be only 4”. For retinal in the solid state a value of 60” was observed [7] whereas MNDOC calculations [ 81 predict a torsional angle of 83 ‘. The larger values obtained from experiment and MNDOC calculations are partly due to
326
0” I 2.0. : ----I
- 1 .._““._. r--v -_(
I-_._._.) I---”
1.6 I.-‘-” 1 fj -
1.2-
1 -
.._.^. 4+&i ., _._
m vNt@
0.6 -
x 15 13 II 9 7 5 3 I 6
Carbon atoms -
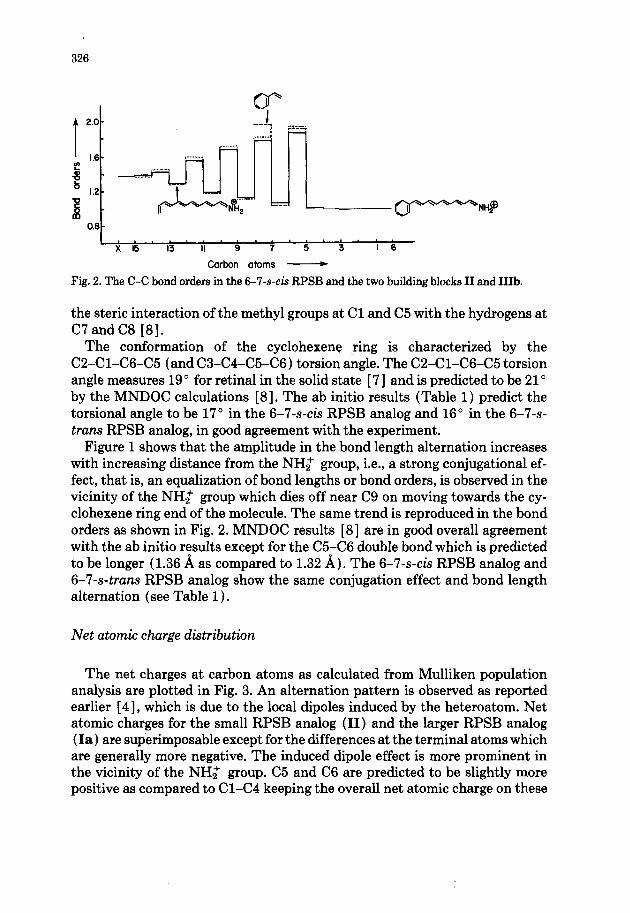
Fig. 2. The C-C bond orders in the 6-7-s-& RPSB and the two building blocks II and 11%
the steric interaction of the methyl groups at Cl and C5 with the hydrogens at C7 and C8 [8].
The conformation of the cyclohexene ring is characterized by the C2-Cl-C6-C5 (and C3-C4-C5-C6) torsion angle. The C2-Cl-C6-C5 torsion angle measures 19’ for retinal in the solid state [ 71 and is predicted to be 21” by the MNDOC calculations [8]. The ab initio results (Table 1) predict the torsional angle to be 17” in the 6-7-s-& RPSB analog and 16” in the 6-7-s- truns RPSB analog, in good agreement with the experiment.
Figure 1 shows that the amplitude in the bond length alternation increases with increasing distance from the NH2 group, i.e., a strong conjugational ef- fect, that is, an equalization of bond lengths or bond orders, is observed in the vicinity of the NH; group which dies off near C9 on moving towards the cy- clohexene ring end of the molecule. The same trend is reproduced in the bond orders as shown in Fig. 2. MNDOC results [8] are in good overall agreement with the ab initio results except for the C5-C6 double bond which is predicted to be longer (1.36 A as compared to 1.32 A). The 6-7-s-& RPSB analog and 6-7-s-trans RPSB analog show the same conjugation effect and bond length alternation (see Table 1).
Net atomic charge distribution
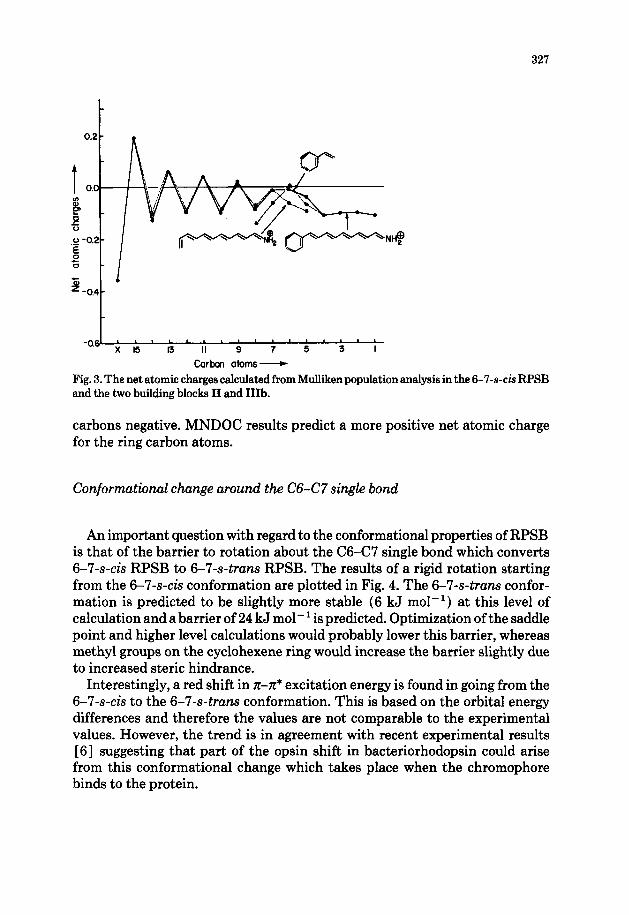
The net charges at carbon atoms as calculated from Mulliken population analysis are plotted in Fig. 3. An alternation pattern is observed as reported earlier [4], which is due to the local dipoles induced by the heteroatom. Net atomic charges for the small RPSB analog (II) and the larger RPSB analog (Ia) are superimposable except for the differences at the terminal atoms which are generally more negative. The induced dipole effect is more prominent in the vicinity of the NH,+ group. C5 and C6 are predicted to be slightly more positive as compared to Cl-C4 keeping the overall net atomic charge on these
327
t -o.$" * ” ” “I ’ ” * "
x 15 13 II 9 7 5 3
Carbon atoms-
Fig. 3. The net atomic charges calculated from Mulliken population analysis in the 6-7-s-& RPSB and the two building blocks II and IIIb.
carbons negative. MNDOC results predict a more positive net atomic charge for the ring carbon atoms.
Conformational change around the C6-C7 single bond
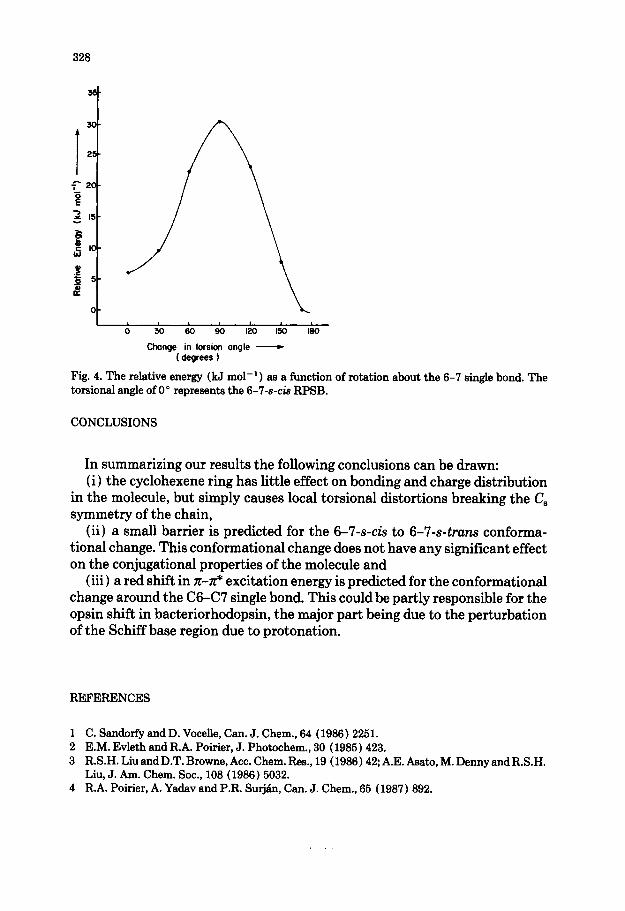
An important question with regard to the conformational properties of RPSB is that of the barrier to rotation about the C6-C7 single bond which converts 6-7-s-cis RPSB to 6-7-s-truns RPSB. The results of a rigid rotation starting from the 6-7-s-& conformation are plotted in Fig. 4. The 6-7-s-truns confor- mation is predicted to be slightly more stable (6 kJ mol-l ) at this level of calculation and a barrier of 24 kJ mol- ’ is predicted. Optimization of the saddle point and higher level calculations would probably lower this barrier, whereas methyl groups on the cyclohexene ring would increase the barrier slightly due to increased steric hindrance.
Interestingly, a red shift in rr-ti excitation energy is found in going from the 6-7-s-& to the 6-7-s-trans conformation. This is based on the orbital energy differences and therefore the values are not comparable to the experimental values. However, the trend is in agreement with recent experimental results [6] suggesting that part of the opsin shift in bacteriorhodopsin could arise from this conformational change which takes place when the chromophore binds to the protein.
328
o-
0 30 60 90 120 150 I80
Change in torsion angle - ( degrees )
Fig. 4. The relative energy (kJ mol-‘) as a function torsional angle of 0” represents the 6-7-s-k RPSB.
of rotation about the 6-7 single bond. The
CONCLUSIONS
In summarizing our results the following conclusions can be drawn: (i ) the cyclohexene ring has little effect on bonding and charge distribution
in the molecule, but simply causes local torsional distortions breaking the C, symmetry of the chain,
(ii) a small barrier is predicted for the 6-7-s-& to 6-7-s-truns conforma- tional change. This conformational change does not have any significant effect on the conjugational properties of the molecule and
(iii) a red shift in n-ti excitation energy is predicted for the conformational change around the C6-C7 single bond. This could be partly responsible for the opsin shift in bacteriorhodopsin, the major part being due to the perturbation of the Schiff base region due to protonation.
REFERENCES
1 C. Sandorfy and D. Vocelle, Can. J. Chem., 64 (1986) 2251. 2 E.M. Evleth and R.A. Pokier, J. Photochem., 30 (1985) 423. 3 R.S.H.LiuandD.T.Browne,Acc.Chem.Res.,19 (1986)42;A.E.Asato,M.DennyandR.S.H.
Liu, J. Am. Chem. Sot., 108 (1986) 5032. 4 R.A. Pokier, A. Yadav and P.R. SU~&I, Can. J. Chem., 65 (1987) 892.

329
5 P.R. Surj&, Phys. Rev. A, 30 (1984) 43; P.R. Surje;l, Croat. Chem. Acta, 57 (1984) 833; P.R. Surj& I. Mayer end I. Lukovits, Phys. Rev. A, 32 (1985) 748; R.A. Pokier and P.R. SurjBin, J. Comput. Chem., 8 (1987) 436.
6 R. van der Steen, P.L.Biesheuvel, R.A. Mathies and J. Lugtenburg, J. Am. Chem. kc., 108 (1986) 6410.
7 T. Hamanaka, T. Mitaui, T. Ashida and M. Kakudo, Acta Crystallogr., Sect. B., 28 (1972) 214. 8 P. Tavan, K. Schulten and D. Oesterhelt, Biophys. J., 47 (1985) 415.