Embed Size (px)
Citation preview

Thrombosis Research 124 (2009) 508–511
Contents lists available at ScienceDirect
Thrombosis Research
j ourna l homepage: www.e lsev ie r.com/ locate / th romres
Letter to the Editors-in-Chief
ArarecaseofMYH9disorderspresentingwithmacrothrombocytopeniaand deafness caused byMYH9-R702C mutation
Introduction
Giant platelets often appear in acquired thrombocytopenia, such asidiopathic thrombocytopenic purpura (ITP) and myelodysplastic syn-drome1. Congenital macrothrombocytopenia is rare, and consists of avariety of diseases [1], including 4 autosomal dominant disorders, May-Hegglin anomaly, and Sebastian, Fechtner, and Epstein syndromes,which have neutrophil inclusion bodies (Döhle-like bodies), or Alport-like symptoms (nephritis, deafness, or cataract) [1]. These diseases havebeendifferentiated by expressionpattern andmorphology of neutrophilinclusionbodies, or the clinical features ofAlport-likemanifestations [1].Mutations of the non-muscle myosin heavy chain 9 (MYH9) gene whichencodes non-muscle myosin heavy chain IIA (NMMHC-IIA), however,were identified in all 4 disorders [1–4]. From this viewpoint, these 4diseases were proposed as MYH9 disorders [4–7]. Recently, genotype-phenotype correlation in MYH9 disorders has also emerged [8,9] andeffective therapy for nephropathy has been suggested [10].
AmongMYH9 disorders, it is difficult to diagnose Epstein syndromeearly because this disease lacks neutrophil inclusion bodies andhas late-onset of Alport-like symptoms [11]. Immunofluorescence analysis ofneutrophil NMMHC-IIA localization, however, demonstrated thatneutrophil inclusion bodies existed in Epstein syndrome [1,12].
Wedescribe a sporadic case ofmacrothrombocytopeniawith deafnesshavingMHY9-R702C mutation and faint neutrophil inclusion bodies.
Case report
A 31-year-old woman registered at our hospital. There was nofamily history of thrombocytopenia, nephritis, deafness, or cataract.She had suffered from endometriosis since 24 years old.
She had done well with no bleeding tendency from birth. At theage of 2, thrombocytopenia was noted when she had pneumonia. Shewas diagnosed with ITP from examination results. After diagnosis, shewas treated with steroids and immunoglobulin, but the platelet countremained from10 to 50×109/μL. At the age of 7, her platelet countwasless than 10×109/μL, and she began to bleed easily; therefore, shewasadministered with interferon, 6-mercaptopurine, cepharanthin, andvitamin C in addition to steroids and immunoglobulin. These drugsdid not show reasonable efficacy. Splenectomywas performed becausemedical treatment showed no response; however, her platelet counthas remained low.
Sensorineural hearing loss was noted at the age of 7. Her bilateralhearingdisturbance gradually progressed, and completely lost at the ageof 25, resulted inundergoing cochlear implantation.Neither cataract nornephritis (negative urinary protein, no casts and RBCs by urinarymicroscopic examination, BUN 11.9 mg/dL, Cr 0.55 mg/dL, Cystatin-C0.81 mg/L, urinary-NAG 1.7 IU/L, and urinary-β2MG 19 μg/L) hasoccurred.
0049-3848/$ – see front matter © 2009 Elsevier Ltd. All rights reserved.doi:10.1016/j.thromres.2009.02.001
At the age of 25, she was diagnosed with Epstein-like syndromedue to macrothrombocytopenia, deafness, and absence of neutrophilinclusion bodies. She is now suffering fromanemia duringmenstruationand could not stop bleedingwith hemostatic drugs. Platelet transfusionsshowed no response because she had many allo-antibodies to plateletsfrom frequent platelet transfusions. Administration of activated recom-binant factor VIIa (rFVIIa) for her bleeding was not effective.
We reevaluated her hematological features. Platelet counts rangedfrom 5 to 20×109/μL. Plateletswere as large as or larger than red bloodcells (Fig. 1-B, C). Bleeding time by the Ivy method was elongated(15 min.), and platelet aggregation response to ADP and collagen wasbelow the normal range. Leukocyte inclusion bodies were mostlyundetectable on peripheral blood smears stained by conventionalMay-Grünwald-Giemsa (MG) staining (Fig. 1-D). We could, however,identify faint Döhle-like bodies in some neutrophils by smearingimmediately after taking a fresh blood sample (Fig. 1-E). Moreover,immunofluorescence analysis of NMMHC-IIA localization revealedDöhle-like bodies to be vivid speckles in the neutrophil cytoplasm(Fig. 2). We also performed mutational analysis of the MYH9 gene.Heterozygous missense mutation, R702C, within exon 16 of theMYH9gene was found (Fig. 3).
Discussion
MYH9 disorders are autosomal-dominant diseases caused byMYH9mutations [5–7]. Until now, the correlation between the genotypeand phenotype was not clear [4–7]. Recently, MYH9 mutations in theC-terminal coiled coil region or truncation of the tailpiece has beenassociated with a hematological-only phenotype, while mutations ofthe head ATPase domain have frequently been related to nephritisand/or hearing loss8. Mutations of other regions have an intermediateexpression of non-hematological characteristics [8]. Moreover, thereport clarified that the position of MYH9 mutations predicts thenatural history of MYH9 disorder [9]. All subjects with mutations inthemotor domain present with severe thrombocytopenia and developnephritis and deafness before the age of 40 years, while thosewith mutations in the tail domain have a much low risk of non-hematological complications and significantly higher platelet counts[9]. The R702 mutations detected in our patient frequently result insevere thrombocytopenia and develop nephritis and deafness at ajuvenile age [9]. Our patient had severe thrombocytopenia and bilateralsensorineural deafness, but has not developed nephritis, possiblybecause it shows late onset or the involvement of other factors,including genetic or environmental backgrounds. We therefore needto monitor this patient carefully because she is likely to progress tonephritis. These results demonstrated that genotype-phenotype corre-lation was useful for the patient's clinical management.
MYH9 disorders are characterized by macrothrombocytopeniawith neutrophil inclusion bodies and Alport-like symptoms [1].Among them, Epstein syndrome shows a lack of neutrophil inclusionbodies and the late onset of Alport-like manifestation [11]; therefore,diagnosis of Epstein syndrome is often late and is frequently mistaken
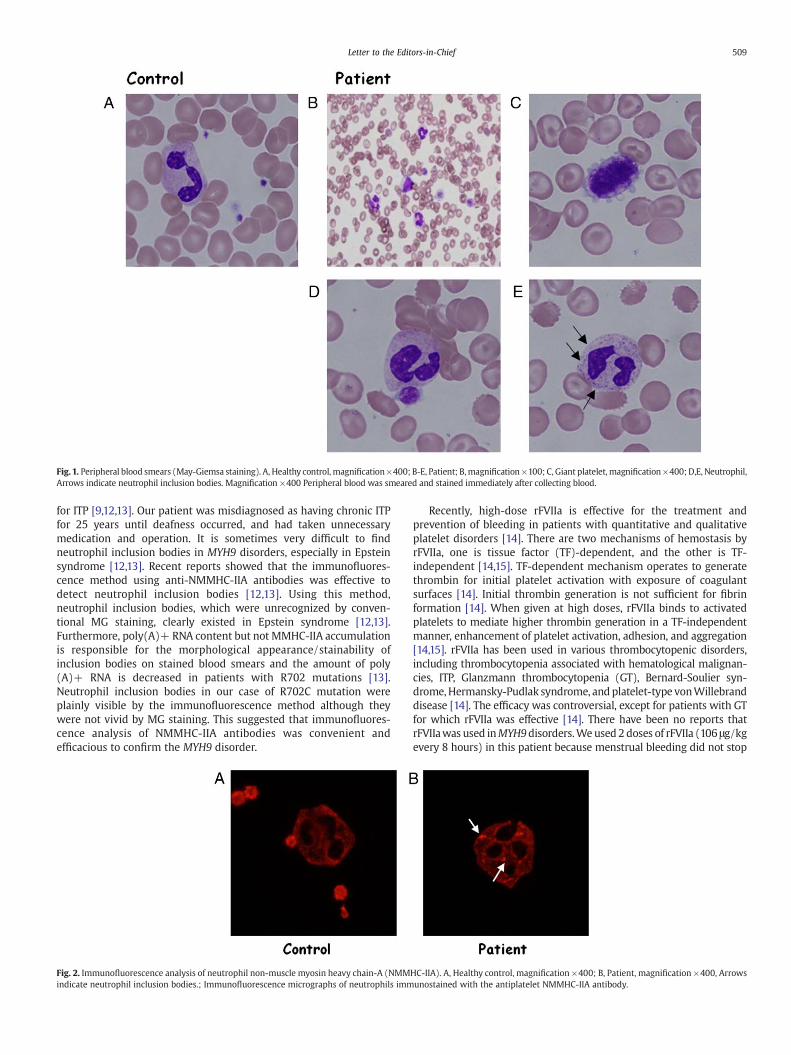
Fig. 1. Peripheral blood smears (May-Giemsa staining). A, Healthy control, magnification×400; B-E, Patient; B,magnification ×100; C, Giant platelet, magnification ×400; D,E, Neutrophil,Arrows indicate neutrophil inclusion bodies. Magnification ×400 Peripheral blood was smeared and stained immediately after collecting blood.
509Letter to the Editors-in-Chief
for ITP [9,12,13]. Our patient was misdiagnosed as having chronic ITPfor 25 years until deafness occurred, and had taken unnecessarymedication and operation. It is sometimes very difficult to findneutrophil inclusion bodies in MYH9 disorders, especially in Epsteinsyndrome [12,13]. Recent reports showed that the immunofluores-cence method using anti-NMMHC-IIA antibodies was effective todetect neutrophil inclusion bodies [12,13]. Using this method,neutrophil inclusion bodies, which were unrecognized by conven-tional MG staining, clearly existed in Epstein syndrome [12,13].Furthermore, poly(A)+ RNA content but not MMHC-IIA accumulationis responsible for the morphological appearance/stainability ofinclusion bodies on stained blood smears and the amount of poly(A)+ RNA is decreased in patients with R702 mutations [13].Neutrophil inclusion bodies in our case of R702C mutation wereplainly visible by the immunofluorescence method although theywere not vivid by MG staining. This suggested that immunofluores-cence analysis of NMMHC-IIA antibodies was convenient andefficacious to confirm the MYH9 disorder.
Fig. 2. Immunofluorescence analysis of neutrophil non-muscle myosin heavy chain-A (NMMindicate neutrophil inclusion bodies.; Immunofluorescence micrographs of neutrophils imm
Recently, high-dose rFVIIa is effective for the treatment andprevention of bleeding in patients with quantitative and qualitativeplatelet disorders [14]. There are two mechanisms of hemostasis byrFVIIa, one is tissue factor (TF)-dependent, and the other is TF-independent [14,15]. TF-dependent mechanism operates to generatethrombin for initial platelet activation with exposure of coagulantsurfaces [14]. Initial thrombin generation is not sufficient for fibrinformation [14]. When given at high doses, rFVIIa binds to activatedplatelets to mediate higher thrombin generation in a TF-independentmanner, enhancement of platelet activation, adhesion, and aggregation[14,15]. rFVIIa has been used in various thrombocytopenic disorders,including thrombocytopenia associated with hematological malignan-cies, ITP, Glanzmann thrombocytopenia (GT), Bernard-Soulier syn-drome,Hermansky-Pudlak syndrome, and platelet-type vonWillebranddisease [14]. The efficacy was controversial, except for patients with GTfor which rFVIIa was effective [14]. There have been no reports thatrFVIIawas used inMYH9disorders.Weused 2 doses of rFVIIa (106 μg/kgevery 8 hours) in this patient because menstrual bleeding did not stop
HC-IIA). A, Healthy control, magnification ×400; B, Patient, magnification ×400, Arrowsunostained with the antiplatelet NMMHC-IIA antibody.

Fig. 3. Mutational analysis of the MYH9 gene DNA sequence analysis showed a C to Ttransition in exon 16 of MYH9. Arrow indicates the mutational location.
510 Letter to the Editors-in-Chief
and she had many anti-platelet antibodies. One reason why rFVIIa hadno effectwas that the dosage of rFVIIawas low; however, we speculatedthat rFVIIa should be carefully used forMYH9disorders since bleeding inother diseases with thrombocytopenia did not stop even after theadministration of substantial rFVIIa.
In conclusion, we describe a Japanese female who presented withmacrothrombocytopenia and deafness due toMYH9-R702C mutation.We need to monitor this patient carefully because most patients withan MYH9-R702C mutation develop nephritis. We also suggest thatimmunofluorescence analysis of neutrophil NMMHC-IIA is valuable toidentify a patient with MYH9 disorders who does not present withneutrophil inclusion bodies.
Conflict of interest
There is no any financial or other interest with regard to thismanuscript that might be construed as a conflict of interest.
Acknowledgements
This work was supported by grants to S.K. from the Japan Societyfor the Promotion of Science (18591094 and 20591161), the Ministryof Health, Labor andWelfare (Grant for Child Health and Development19C-2), Charitable Trust LaboratoryMedicine Foundation of Japan, andNational Hospital Organization (network research grant for congenitalthrombocytopenia).
We thank Ms. Yoshimi Ito for her skillful technical assistance.
References
[1] KunishimaS, SaitoH. Congenitalmacrothrombocytopenias. BloodRev2006;20:111–21.[2] Seri M, Cusano R, Gangarossa S, Caridi G, Bordo D, Lo Nigro C, et al. Mutations in
MYH9 result in the May-Hegglin anomaly, and Fechtner and Sebastian syndromes.The May-Heggllin/Fechtner Syndrome Consortium. Nat Genet 2000;26:103–5.
[3] Kelley MJ, JawienW, Ortel TL, Korczak JF. Mutation ofMYH9, encoding non-musclemyosin heavy chain A, in May-Hegglin anomaly. Nat Genet 2000;26:106–8.
[4] Kunishima S, Kojima T, Matsushita T, Tanaka T, Tsurusawa M, Furukawa Y, et al.Mutations in the NMMHCA- A gene cause autosomal dominant macro-thrombocytopenia with leukocyte inclusions (May-Hegglin anomaly/Sebastiansyndrome). Blood 2001;97:1147–9.
[5] Kunishima S, Matsushita T, Kojima T, Amemiya N, Choi YM, Hosaka N, et al.Identification of six novelMYH9mutations and genotype-phenotype relationshipsin autosomal dominant macrothrombocytopenia with leukocyte inclusions. J HumGenet 2001;46:722–9.
[6] Heath KE, Campos-Barros A, Toren A, Rozenfeld-Granot G, Carlsson LE, Savige J, et al.Nonmuscle myosin heavy chain IIA mutations define a spectrum of autosomal
dominantmacrothrombocytopenias:May-Hegglin anomaly and Fechtner, Sebastian,Epstein, and Alport-like syndromes. Am J Hum Genet 2001;69:1033–45.
[7] Seri M, Pecci A, Di Bari F, Cusano R, Savino M, Panza E, et al.MYH9-related disease:May-Hegglin anomaly, Sebastian syndrome, Fechtner syndrome, and Epsteinsyndrome are not distinct entities but represent a variable expression of a singleillness. Medicine (Baltimore) 2003;82:203–15.
[8] Dong F, Li S, Pujol-Moix N, Luban NL, Shin SW, Seo JH, et al. Genotype-phenotypecorrelation in MYH9-related thrombocytopenia. Br J Haematol 2005;130:620–7.
[9] Pecci A, Panza E, Pujol-Moix N, Klersy C, Di Bari F, Bozzi V, et al. Position ofnonmuscle myosin heavy chain IIA (NMMHC-IIA) mutations predicts the naturalhistory of MYH9-related disease. Hum Mutat 2008;29:409–17.
[10] Pecci A, Granata A, Fiore CE, Balduini CL. Renin-angiotensin system blockade iseffective in reducing proteinuria of patients with progressive nephropathy causedby MYH9 mutations (Fechtner-Epstein syndrome). Nephrol Dial Transplant2008;23:2690–2.
[11] Epstein CJ, SahudMA, Piel CF, Goodman JR, BernfieldMR, Kushner JH, et al. Hereditarymacrothrombocytopathia, nephritis and deafness. Am J Med 1972;52:299–310.
[12] KunishimaS,MatsushitaT,KojimaT, SakoM,KimuraF, JoEK, et al. Immunofluorescenceanalysis of neutrophil nonmusclemyosinheavychain-A inMYH9disorders: associationof subcellular localizationwithMYH9mutations. Lab Invest 2003;83:115–22.
[13] Kunishima S, Yoshinari M, Nishio H, Ida K, Miura T, Matsushita T, et al.Haematological characteristics of MYH9 disorders due to MYH9 R702 mutations.Eur J Haematol 2007;78:220–6.
[14] Poon MC. The evidence for the use of recombinant human activated factor VII inthe treatment of bleeding patients with quantitative and qualitative plateletdisorders. Transfus Med Rev 2007;21:223–36.
[15] LismanT, Adelmeijer J, Cauwenberghs S,Van Pampus EC,Heemskerk JW,DeGroot PG.Recombinant factor VIIa enhances platelet adhesion and activation under flowconditions at normal and reduced platelet count. J Thromb Haemost 2005;3:742–51.
Rumi KodamaCentral Clinical Laboratory, Shimane University Hospital, Shimane, Japan
Takeshi Taketani⁎Division of Blood Transfusion, Shimane University Hospital,
Shimane, JapanDepartment of Pediatrics, Shimane University School of Medicine,
Shimane, Japan⁎Corresponding author. Taketani is to be contacted at Division of Blood
Transfusion, Shimane University Hospital, 89-1, Enya, Izumo-city,Shimane 693-8501, Japan. Tel.:+81853 20 2424; fax:+81853 20 2423.
Kunishima, Department of Advanced Diagnosis,Clinical Research Center,
National Hospital Organization Nagoya Medical Center,4-1-1 Sannomaru, Naka-ku, Nagoya 460-0001, Japan.
Tel.: +81 52 9511111; fax: +81 52 951 0664.E-mail address: [email protected] (T. Taketani).
Shinji Kunishima⁎Department of Advanced Diagnosis, Clinical Research Center, National
Hospital Organization Nagoya Medical Center, Nagoya, Japan⁎Corresponding author. Taketani is to be contacted at Division of Blood
Transfusion, Shimane University Hospital, 89-1, Enya, Izumo-city,Shimane 693-8501, Japan. Tel.:+81853 20 2424; fax:+81853 20 2423.
Kunishima, Department of Advanced Diagnosis,Clinical Research Center,
National Hospital Organization Nagoya Medical Center,4-1-1 Sannomaru, Naka-ku, Nagoya 460-0001, Japan.
Tel.: +81 52 9511111; fax: +81 52 951 0664.E-mail address: [email protected] (S. Kunishima).
Seiji MishimaCentral Clinical Laboratory, Shimane University Hospital, Shimane, Japan
Yoko YoshikawaDepartment of Pediatrics, Shimane University School of Medicine,
Shimane, Japan
Rie KanaiDepartment of Pediatrics, Shimane University School of Medicine,
Shimane, Japan

511Letter to the Editors-in-Chief
Tamiko SuyamaIsao Yoshino
Hiroyuki KunishiHiroshi Shibata
Central Clinical Laboratory, Shimane University Hospital, Shimane, Japan
Atsushi NagaiDepartment of Laboratory Medicine, Shimane University School of
Medicine, Shimane, Japan
Seiji YamaguchiDepartment of Pediatrics, Shimane University School of Medicine,
Shimane, Japan
Junichi MasudaDivision of Blood Transfusion, Shimane University Hospital,
Shimane, JapanDepartment of Laboratory Medicine, Shimane UniversitySchool of Medicine, Shimane, Japan





























