Embed Size (px)
Citation preview

A Rigid-Rotor Potential Surface for the Hydrogen Molecule Dimer
D. L. MERRIFIELD AND N. S. OSTLUND Department of Chemistry, University of Arkansas, Fayetteville, Arkansas 72701,
U.S.A.
Abstract
A potential surface is presented for two interacting rigid-rotor hydrogen molecules. The calcu- lations include correlation effects by perturbation theory. A ghost orbital treatment is used to minimize limitations in the basis set. Ten intermolecular distances are included and the angular dependence of the interaction is presented in analytical form.
1. Introduction
Rotational energy transfer is a subject of considerable current interest [ I ] and scattering calculations of rotational inelastic cross sections have advanced to the point that, in conjunction with experiment, they can be used to test the accuracy of potential energy hypersurfaces [2]. Because of its simplicity, the gaseous H2-Hz system is of fundamental interest as a prototype system for studying molecule-molecule interactions. In the process of calculating the dipole moment and polarizability of the hydrogen molecule dimer, for a theoretical investigation of the dielectric properties and pressure-induced absorption of gaseous hydrogen [3], we have generated, by relatively simple means, an ab initio potential surface for two rigid-rotor hydrogen molecules. This potential surface has been used by Ramaswamy, Rabitz, and Green [2] to calculate rotational relaxation times and it appears to give reasonable agreement with experiment, better than might be expected considering the simplicity of the calculation. The purpose of this article is to present this rigid-rotor potential surface, for possible use by other investigators. A detailed discussion of the energy, dipole moment, and polarizability surfaces for H2-H2 will be the subject of a future publication
I n Section 2 we briefly describe the calculation method and then in Section [31.
3 give the results and an analytical fit to the angular part of the potential.
2. Methodology
I f space-fixed coordinates are used, as in scattering calculations, the inter- action between two diatomic molecules can be written [4] as
where rI and r2 are internuclear vectors for each molecu!e and R is the inter-
Intcrn;itional Journal of Quantum Chemistry: Quantum Chemistry Symposium I I . 245-250 (1977) 0 1977 by John Wiley &Sons. Inc. 245

246 MERRlFlELD A N D OSTLUND
TABLE I. Standard orientations for homonuclear diatomin.
Weight cos 01 cos e2 41 - 4 2
I 4 I I 0 2 40 I -5- IJ2 0 3 20 5-112 -5-112 0 4 40 5-112 -5-112 0 . 4 ~ 5 40 5-112 -5-112 0.8a
8 2 e- Figure l . Coordinate system for interacting diatomic molecules.
molecular vector (cf. Fig. 1). The angular dependence of the potential is con- tained in the factor
. 7 / l / 2 / (R . i1 , i2 ) = c (11rn l12rnzI lm) Y / I m t ( i I ) ~ / 2 m 2 ( i 2 ) Gm<R> (2)
where ( l lm I / 2 m 2 1 1 m ) is a Clebsch-Gordon vector coupling coefficient and the Yl,,, are spherical harmonics.
I f one is only interested in describing the intermolecular potential, which depends on six coordinates, a body-fixed coordinate system may be more con- venient [ 51.
m i m m
A E ( K . ~ I ~ 2 . ' 3 I s '32 .4) = C B/l/2m ( R J I J ~ ) Y/ lm( '3 I 9 4 1 ) Yr2-m ('32.42) (3) 1 I 12m
Here ' 3 1 and '32 are the angles that i l and i2 make with R, and 4 = 41 - 4 2 is the dihedral angle about the intermolecular axis (cf. Fig. I ) . The relation between these A and B coefficients has been discussed by Green [6].
In the rigid-rotor calculations reported here rl and r2 are fixed at 0.7416 A and hence, for a given intermolecular distance R , the potential depends only on the angular variables e l , e2, and 4. A convenient way of describing this depen- dence is to perform calculations which scan the angular space and then least- squares fit these to either Eq. ( I ) or (3). For a pair of homonuclear diatomics, only even values of 11 and 12 appear in Eqs. ( I ) and (3) and there are thus five unique coefficients u p to and including I I = 12 = 2. For reasons of economy we have not considered higher terms in these expansions; however, the hydrogen molecule is not highly anisotropic. To scan the angular space we have used an icosahedral averaging procedure [7] in which each molecule is directed towards the 12 corners of an icosahedron. This leads to 144 relative orientations of which only 5 are unique. These 5 orientations and their statistical weights are shown
RIGID-ROTOR SURFACE OF Hz DIMER 241
Figure 2. Monomer basis set
r--- -
I I 2.6 3.4 4.2 5 .o
Figure 3. SCF calculations of the interaction energy AE (Hartrees X I 03) as a function of the intermolecular distance R (A) for the linear orientation I : (- - - -) supermolecule cal- culation; (-) ghost orbital calculation.
in Table 1. Calculations were performed for ten intermolecular distances at these 5 orientations and an exact fit obtained to either harmonic expansion.
Calculations were performed in the Self-Consistent Field (sCF) approximation as well as in the approximation of second-order Rayleigh-Schroedinger (RS) perturbation theory. If the Moller-Plesset (MP) unperturbed Hamiltonian for N electrons is

248 MERRlFlELD A N D O S T L U N D
TABLE II. SCFenergy surface (mH).
N A ) AMXI A 202 A 220 A222 A 224
2.2 343.99757 2.6 92.55219 3 .O 23.58364 3.2 11.65010 3.4 5.64943 3.6 2.67 I94 3.8 1.2 I990 4.2 0.20571 4.6 0.00906 5.0 -0.0 I I 56
1 1.04482 3.24622 0.75921 0.33427 0. I2906 0.03601
-0.00192 -0.01 536 -0.00938 -0.00414
1.39441 0.67558 0.2 I I28 0.1 I706 0.06452 0.03745 0.023 10 0.01 I34 0.00663 0.00424
-0.00232 -0. I6196 -0.05523 -0.03 144 -0.01739 -0.0 1006 -0.00632 -0.003 I2 -0.001 91 -0.001 3 I
3.81671 1.77762 0.84921 0.61689
0.34735 0.267 I2 0.16354 0. I044 I 0.069 I7
0.45855
N
p= I H ( 0 ) = c f(p) (4)
where the molecular spin orbitals are eigenfunctions of a Fock operator
f( 1 )+i ( 1 ) = t i+ ; ( 1 ( 5 )
E(SCF) = -t E ( ’ ) (6)
(7) The basis set was a simple “double-zeta’’ Gaussian set, the 4-31G basis of
Ditchfield et al. [9]. In addition, six primitive s-type Gaussians were arranged octahedrally about each nucleus with the subsequent elimination of two orbitals from each monomer, the two which are in the “bond,” as shown in Figure 2. Off-center s-typc Gaussians like this have been used by us previously [ 101 to obtain reasonable polarizabilities with small basis sets. The distance r from the nucleus and the exponent of the off-center Gaussians were varied to maximize the monomer polarizability, with some attention paid to obtaining an accurate anisotropy. The final exponent was 0.15 with r = 0.10 A. This basis has linear dependence which was eliminated, using canonical orthogonalization [ 1 I], by throwing away those orthogonalized basis functions corresponding to eigenvalucs of the overlap matrix less than The final monomer basis set consists of 14 nonorthogonal s-type Gaussian basis functions ( 1 1 orthogonalized basis func- tions).
Basis set extension effects are relatively large with this small basis. If a qualitatively correct description of the interaction is to be obtained, a ghost or- bital treatment [ 101 is necessary. I f the monomer basis sets are incomplete, then as the two molecules approach, the basis orbitals of one can be used to lower the effective monomer energy of the other and a false minimum in the intermolecular potential occurs. The ghost orbital treatment of Ref. [ 101 uses an identical basis set in both monomer and dimer calculations. With respect to the usual super- molecule calculation, the ghost orbital treatment lowers the monomer energy to compcnsate for basis set extension in the dimer, to give a correct description of the interaction. This is illustrated in Figure 3 for the linear orientation 1 which
then calculations are reported in the following two approximations (81.
E(RS,MP) = E(0) + E(1) + E(* )
RIGID-ROTOR SURFACE OF H 2 DlMER 249
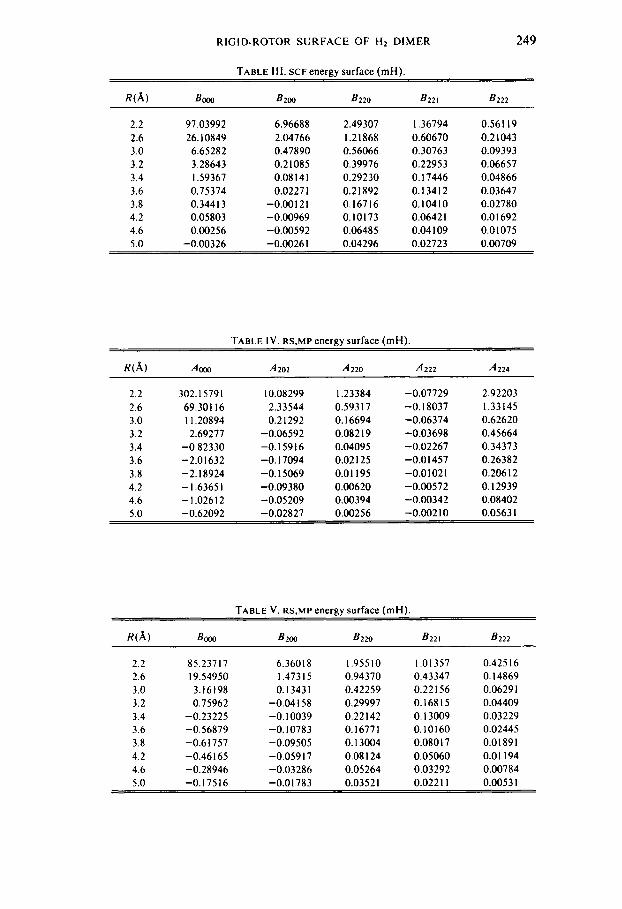
TABLE 111. SCF energy surface (mH).
W A ) BOO0 B 2 w 8 2 2 0 B 2 2 I B222
2.2 2.6 3.0 3.2 3.4 3.6 3.8 4.2 4.6 5.0
97.03992 26.10849 6.65282 3.28643 1.59367 0.75374 0.344 I 3 0.05803 0.00256
-0.00326
6.96688 2.04766 0.47890 0.21085
0.02271 -0.00121 -0.00969 -0.00592 -0.00261
0.08 I 4 I
2.49307 1.21868 0.56066 0.39976 0.29230 0.21892 0.16716 0.10173 0.06485 0.04296
1.36794 0.60670 0.30763 0.22953 0.17446 0. I3412 0.10410 0.0642 I 0.04 I09 0.02723
0.56 I I9 0.21043 0.09393 0.06657 0.04866 0.03647 0.02780 0.01692 0.0 I075 0.00709
TABLE Iv. RS.MP energy surface (mH).
K ( ' 4 AOLM A 202 A 220 A 2 2 2 A 224
2.2 2.6 3.0 3.2 3.4 3.6
4.2 4.6 5.0
3.8
302. I5791 10.08299 1.23384 -0.07729 2.92203 69.301 16 2.33544 0.59317 -0. I8037 1.33145 1 1.20894 0.2 1292 0. I6694 -0.06374 0.62620 2.69277 -0.06592 0.082 I9 -0.03698 0.45664
-0.82330 -0. I591 6 0.04095 - 0.0 2 2 6 7 0.34373 -2.01632 -0. I7094 0.02 I25 -0.01457 0.26382 -2.18924 -0.15069 0.01 195 -0.0 102 1 0.206 I2 - 1.6365 1 -0.09380 0.00620 -0.00572 0.12939 - I .02612 -0.05209 0.00394 - 0.00 3 4 2 0.08402
0.0563 I -0.62092 -0.02827 0.00256 -0.00210
TABLE V. RS,MP energy surface (mH).
M A ) BOO0 B 2 0 0 B 2 2 o 6 2 2 1 B222
2.2 2.6 3.0 3.2 3.4 3.6 3.8 4.2 4.6 5.0
85.237 I7 19.54950 3.16198 0.75962
-0.23225 -0.56879 -0.6 1757 -0.461 65 -0.28946 -0.1 7516
6.36018 1.473 I5 0. I343 I
-0.04 I58 -0.10039 -0.10783 -0.09505 -0.059 I7 -0.03286 -0.01 783
1.95510 0.94370 0.42259 0.29997 0.22 I42 0. I677 I 0.13004 0.08 124 0.05264 0.03521
1.01357 0.43347 0.22 I56 0. I681 5 0. I3009 0.101 60 0.080 I7 0.05060 0.03292 0.0221 I
0.425 I6 0. I4869 0.0629 1 0.04409 0.03229 0.02445 0.01891 0.01 194 0.00784 0.0053 I

250 MERRIFIELD A N D OSTLUND
has a repulsive quadruple interaction. In the SCF approximation (no dispersion) the interaction should be strictly repulsive. The usual supermolecule calculation has, however, the spurious minimum arising from an extension of the monomer basis set. All calculations reported here thus use the ghost orbital treatment.
3. Results
The A and B coefficients for both the SCF and RS,MP potentials are shown in Tables 11-V. I t is only necessary to discuss the B coefficients. The spherically averaged interaction is Booo/(4~). In the S C F approximation this is repulsive; the very small attraction at long range represents induction. After adding the correlation energy, the spherically averaged interaction is attractive, because of dispersion; the well depth, however, is only about one-half the value deduced from experiment [ 121. The reason for this is probably an underestimation of the dispersive R-6 and R-8 interactions [3]. B2m = Bozo, the leading term in the anisotropy, represents long range induction in the SCF calculation and is small relative to the quadruple interactions. At small intermolecular distances it gives the repulsive nonbonded interactions which favor a rectangular configuration of the 4 atoms. With the correlation energy included, the larger negative value at long range represents the favorable linear arrangement of the molecules for largest dispersion. The Bz~, , , = B22-, terms represent quadruple-quadrupole interactions in the SCF calculations at long and medium range. The ratio [ 5 ] B22o:B221:B222 of 6:4:1 is evident. These electrostatic interactions are larger than dispersive interactions and the favored conformation of the van der Waals molecule is a T-shape.
Acknowledgment
The authors would like to thank Mr. Harrell Sellers for help with some of the calculations and Professor H. Rabitz for supplying them with a copy of his fitting program. They are grateful to the University of Arkansas Computer Center for a generous allocation of machine time. One of the authors (N.S.O.) would like to thank Professor A. D. Buckingham and NATO for their encouragement and partial financial support.
Bibliography
[I ] J. P. Toennies, Ann. Rev. Phys. Chem. 27,225 (1976). [2] R . Ramaswamy, H. Rabitz, and S. Green, J . Chem. Phys. 66,3021 (1977). [3] N . S. Ostlund and A. D. Buckingham, to be published. (41 G . Zarur and H . Rabitz, J . Chem. Phys. 60,2057 (1974). [5] J . A. Pople. Proc. Roy. Soc. A221,498 (1954). 161 S. Green, J . Chem. Phys. 62,2271 (1975). 171 J. Del Bene and J. A. Pople, J. Chem. Phys. 55,2296 (1971). 181 N. S. Ostlund and M. F. Bowen. Theor. Chim. Acta 40, 175 (1975). 191 K. Ditchfield. W. J . Hehre,and J . A . Pople, J. Chem. Phys. 54,724 (1971).
[ 101 N. S. Ostlund and D. 1.. Merrifield, Chem. Phys. Lett. 39,612 (1976). [ 1 I] P. 0. LBwdin, Advan. Quant. Chem. 5, 185 (1970). [ I21 W. Bauer, B. Lantzsh, J . P. Toennies. and K. Walaschewski. Chem. Phys. 17, 19 (1976)
Rcccived February 8, 1977




![Review Article Understanding the Functional Plasticity in ...downloads.hindawi.com/journals/np/2016/4827268.pdf · analysis and the -HTC receptor di used as a dimer [ ] . Single uorescent-molecule](https://img.pdfslide.net/doc/110x75/603a8db43e5cc21c9827b9a1/review-article-understanding-the-functional-plasticity-in-analysis-and-the-htc.jpg)



![STANDARD™ F D-dimer FIA CAUTIOND-dimer levels rise during pregnancy and high levels are associated with complications. [Intended use] STANDARD F D-dimer FIA is an in vitro diagnostic](https://img.pdfslide.net/doc/110x75/5e245a9d8893db4d306c0295/standarda-f-d-dimer-fia-caution-d-dimer-levels-rise-during-pregnancy-and-high.jpg)








![[홍보자료Œ] Vcheck D-dimer PPT EN](https://img.pdfslide.net/doc/110x75/61e11e2b440fb31d77387e5a/-vcheck-d-dimer-ppt.jpg)











