Embed Size (px)
Citation preview

Journal of Immunological Methods 344 (2009) 58–63
Contents lists available at ScienceDirect
Journal of Immunological Methods
j ourna l homepage: www.e lsev ie r.com/ locate / j im
Research paper
A streamlined method for rapid and sensitivechromatin immunoprecipitation☆
Michael L. Sikes⁎, Justin M. Bradshaw, Wendell T. Ivory, Jessica L. Lunsford,Ruth E. McMillan, Clayton R. MorrisonDepartment of Microbiology, North Carolina State University, Raleigh, NC 27695, United States
a r t i c l e i n f o
Abbreviations: ChIP, chomatin immunoprecipitatioRNA polymerase 2; Ab, antibody; cad, carbamoylpaspartate carbamyltransferase/dihydroorotase genepromoter; Rag, Recombination activating gene; TCRdouble negative.☆ This work was supported by a grant from the Natioand Infectious Diseases (R56AI070848-01A1).⁎ Corresponding author. Department of Microbiolog
University, P.O. Box 7615, Raleigh, NC 27695, United S0528; fax: +1 919 515 7867.
E-mail address: [email protected] (M.L. Sikes).
0022-1759/$ – see front matter © 2009 Elsevier B.V.doi:10.1016/j.jim.2009.03.007
a b s t r a c t
Article history:Received 10 February 2009Received in revised form 16 March 2009Accepted 18 March 2009Available online 26 March 2009
We report a streamlined procedure to efficiently carry samples from chromatin to qPCR-compatible DNA in as little as 4 h. We use this streamlined ChIP to quantify histone H3modifications at active (cad) and repressed (T early alpha) promoters in a Rag1-deficient pro-Tcell line after 1–2 h IP. We further show that the protocol readily quantified histonemodifications in chromatin from 104 Rag-deficient DN thymocytes. Taken together, these dataoutline a simple, cost-effective procedure for efficient ChIP analysis.
© 2009 Elsevier B.V. All rights reserved.
Keywords:Chromatin immunoprecipitationHistoneEpigeneticscadTEAThymocyteRag1. Introduction
The fast-growing fields of epigenetics and epigenomicshave necessitated development of assays that probe interac-tions between DNA, structural histone proteins, and regula-tory transcription factors. One of the primary tools used tounravel the so-called “histone code” has been chromatinimmunoprecipitation, or ChIP (Dedon et al., 1991; O'Neill andTurner, 1996; Kuo and Allis, 1999). Indeed, ChIP, coupled withrealtime PCR (qPCR) has become the gold standard assay forchromatin organization (Jenuwein and Allis, 2001), and is
n; H3, histone 3; Pol IIhosphate synthetase/; TEA, T early alpha, T cell receptor; DN
nal Institute of Allergy
y, North Carolina Statetates. Tel.: +1 919 513
All rights reserved.
,
,
increasingly used to demonstrate differential transcriptionfactor recruitment to various promoters (Morshead et al.,2003; O'Neill et al., 2006). Despite such widespread use, thecomplexity, lengthiness, and scale of the standard ChIPprotocol, which can take up to 3 days and require ≥106 cellsper reaction, make it extremely sensitive to experimenter-induced variability and to contamination, and limit its utilityfor scarce cell populations, such as those found in selectcompartments of the immune system.
A number of groups have now proposed modifications tothe standard ChIP protocol (Nelson et al., 2006; O'Neill et al.,2006; Attema et al., 2007; Dahl and Collas, 2007, 2008). Thenewer protocols have demonstrate that ChIP is amenable tovariations in virtually every aspect of the assay from the sizeof chromatin input, the time dedicated to immunoprecipita-tion, washing, elution, and crosslink reversal, to Proteinase Ktreatment regiments. By replacing agarose or sepharose beadswith Protein A- or Protein G-coupled paramagnetic beads,newer approaches minimize the need to preclear inputchromatin of antibody-independent bead binding activities.At the same time, the ability to easily and quantitativelycapture magnetic bead complexes eliminates the need for

59M.L. Sikes et al. / Journal of Immunological Methods 344 (2009) 58–63
centrifugation, which both reduces the time required for eachof the many washes in the ChIP protocol, and reduces thepotential for sample loss or contamination during washaspiration.
We have designed a streamlined protocol that incorpo-rates improvements offered by the Q2-ChIP (Dahl and Collas,2007), FastChIP (Nelson et al., 2006), ChIP-IT Express(ActiveMotif), and miniChIP (Attema et al., 2007) into asimplified format. We find this streamlined protocol is easilymastered, rapid, and highly reproducible. Demonstrating itsadaptability to reduced sample size, our streamlined ChIPreadily quantitated histone modifications in as few as 104
CD4/CD8 double negative thymocytes harvested from a Rag-2deficient mouse. Though we find each of the existing ChIPprotocols can be highly effective, we propose our streamlinedprotocol as a simplified approach for those new to ChIP or forhigher throughput ChIP screens.
2. Materials and methods
2.1. Cells
The RAG1−/−, p53−/− pro-T cell line, P5424, has beenpreviously described (Mombaerts et al., 1995). P5424 cellswere cultured at 37 °C/5% CO2 in RPMI 1640 medium sup-plemented with 10% fetal calf serum, 2 mM L-glutamine,0.01% penicillin/streptomycin, and 50 µM β-mercaptoetha-nol. Thymii were isolated from 4–8 weeks old Rag2−/− mice,crushed and filtered to yield a single cell suspension, and redblood cells were removed by hypotonic lysis. The mousestudies described here were reviewed and approved by theinstitutional animal care and use committee at North CarolinaState University.
2.2. Antibodies
Rabbit polyclonal antisera to acetylated H3K9 (06-599)and dimethylated H3K4 (07-030), along with rabbit controlIgG (12-370) were purchased from Upstate. Rabbit polyclonalantisera to dimethylated H3K9 (ab1772) and trimethylatedH3K4 (ab8580), and mouse monoclonal antibody to RNApolymerase II (ab5408) were purchased from Abcam.
2.3. Chromatin preparation
Protein:DNA complexes in 4×106 P5424 or freshly isolatedthymocyte suspension cells were cross-linked using formalde-hyde (1% final) in either tissue culture dishes or conicalcentrifuge tubes with gentle shaking for 10 min at roomtemperature. Crosslinking was stopped by drop-wise additionof glycine (125mM final concentration) and gentle shaking for5 min at room temperature. Cells were pelleted, washed in 1XPBS(5ml). Pelletedcellswere resuspended in500µl lysis buffer(10 mM Tris–HCl, pH7.5, 10 mM NaCl, 3 mM MgCl2, and 0.5%NP-40) supplemented with 1 mM PMSF and 1X ProteaseInhibitor Cocktail (PIC, Roche), and incubated for 30min on ice.Nuclei fromthe lysedcellswerepelletedbymicrocentrifugation(5000 rpm for 10 min at 4 °C). Nuclei were resuspended in100 µl MNase reaction buffer (10 mM Tris–HCl, pH 7.5, 10 mMNaCl, 3 mMMgCl2, 1 mM CaCl2, 4% NP-40) supplemented with1 mM PMSF and 1X PIC, and chromatin was sheared with the
addition of 1–2 UMNase (Sigma) for 10min at 37 °C. Digestionwas stopped with the addition of EDTA (10 mM final), and theresultant chromatin was stored at−80 °C.
2.4. Chromatin immunoprecipitation
For ChIP, paramagnetic Dynabeads (Dynal) separatelycoupled to Protein A (10 µl/IP) and Protein G (10 µl/IP)were combined in a 1.5 ml microcentrifuge tube, captured byplacing the tube against a strong magnet, and the suspensionmedium was immediately aspirated while the beads wereheld in place by the magnet. Beads were washed as described(Dahl and Collas, 2007) with the addition of 100 µl/IP 1X RIPAbuffer (10 mM Tris–HCl, pH 7.5, 150 mM NaCl, 1 mM EDTA,0.5 mM EGTA, 1% Triton X-100, 0.1% SDS, 0.1% NaDeoxycho-late) containing BSA (50 mg/ml) and sheared salmon spermDNA (0.5 mg/ml), denoted as RBD hereafter. Beads werevortexed (3 s), and recaptured. Wash solution was aspirated,and the wash was repeated. Beads were resuspended in RBD(50 µl/IP) containing PMSF and PIC, aliquoted into sterile200 µl PCR strip tubes containing 1–5 µg antibody, androtated 1 h at 4 °C. Conjugated antibody:bead complexes werewashed 2X in RBD as described above, and resuspended in75 µl/IP volumes of RBD supplemented with PMSF and PIC.
For each IP, antibody:bead suspensions were mixed with25 µl undiluted chromatin (106 cell equivalents) or chromatindiluted in RBD containing PMSF and PIC as indicated (Fig. 3).Protein–DNA complexes were immunoprecipitated for 2 h(except Fig. 1) at 4 °C with rotation. Bead complexes werecaptured by placing the PCR strip tubes on a horizontal barmagnet (attached to an empty 200 µlmicropipettor tip rack), IPsolution was aspirated, and beads were resuspended in 180 µlRBD. To wash the bead complexes, the strip tubes were rapidlymoved back and forth across the magnet 5 times (sequentiallyforcing the beads from one side of the tube to the other). Beadswere recaptured and thewashwas repeated 3×. Following RBDwash, bead complexes were washed 2× in 180 µl TE, pH 8.0.
To elute protein–DNA complexes, beads were resuspendedin 100 mM NaHCO3 (100 µl), strip tubes were resealed withfresh strip caps, and were gently vortexed 15 min at roomtemperature. Tubes were returned to the magnet, and protein–DNA eluates were transferred to fresh PCR strip tubes contain-ing 4 µl 5 M NaCl. To prepare matched input samples, 10 µl ofinput chromatin was diluted in 90 µl 100 mM NaHCO3,transferred to a strip tube containing 4 µl 5 M NaCl, and carriedalong with the IP samples for all subsequent steps. Crosslinkswere reversed at 95 °C for 15 min, tubes were cooled to roomtemperature, and then incubated with Proteinase K (10 µg/mlfinal) 1 h at 45 °C. Digestionwas stoppedwith addition of PMSF(2 mM final), DNA was purified using Qiaquick nucleotideremoval columns (Qiagen) according to the manufacturer'sinstructions, and eluted in 100 µl Qiagen elution buffer.
2.5. Q-PCR and data analysis
For realtime PCR, bound (3 µl) and input (3 µl of 1:100dilution) samples were amplified in a MyIQ thermal cycler(Bio-RAD) using 1X SensiMix Plus (Quantace) and primersspecific for the cad promoter (forward: 5'-GTCTGCGTGCT-TGCCCTGTCTCAGC-3'; reverse: 5'-CGGGCTTGCTTACCCACTTC-CCCAGC-3'), or TEA (McMurry and Krangel, 2000; Sikes et al.,
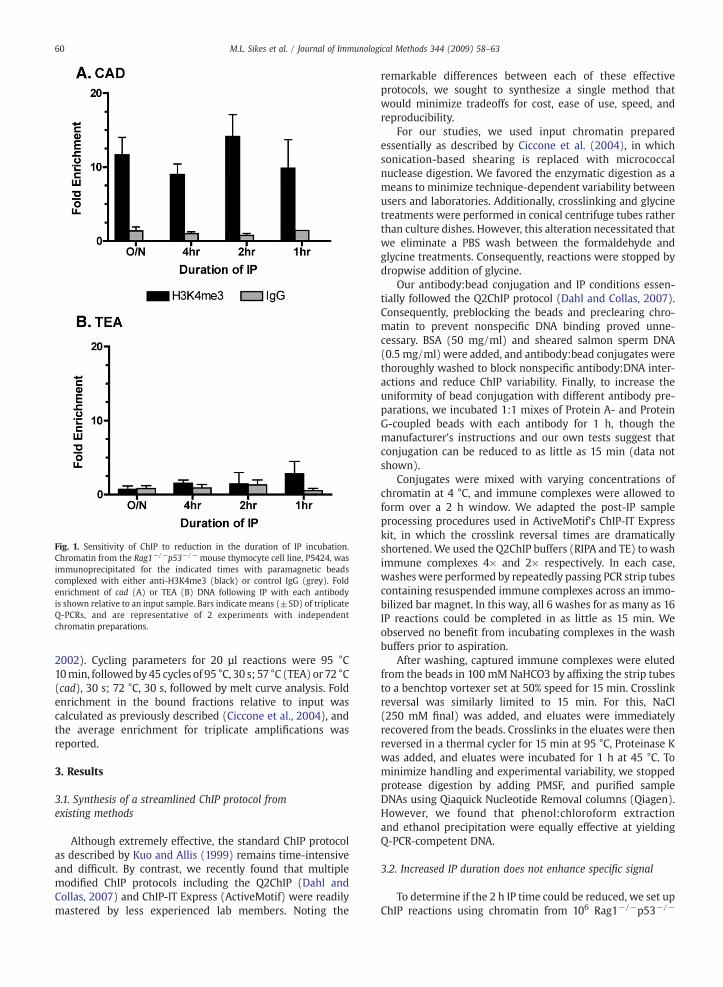
Fig. 1. Sensitivity of ChIP to reduction in the duration of IP incubation.Chromatin from the Rag1−/−p53−/− mouse thymocyte cell line, P5424, wasimmunoprecipitated for the indicated times with paramagnetic beadscomplexed with either anti-H3K4me3 (black) or control IgG (grey). Foldenrichment of cad (A) or TEA (B) DNA following IP with each antibodyis shown relative to an input sample. Bars indicate means (±SD) of triplicateQ-PCRs, and are representative of 2 experiments with independentchromatin preparations.
60 M.L. Sikes et al. / Journal of Immunological Methods 344 (2009) 58–63
2002). Cycling parameters for 20 µl reactions were 95 °C10min, followedby45 cycles of 95 °C, 30 s; 57 °C (TEA) or 72 °C(cad), 30 s; 72 °C, 30 s, followed by melt curve analysis. Foldenrichment in the bound fractions relative to input wascalculated as previously described (Ciccone et al., 2004), andthe average enrichment for triplicate amplifications wasreported.
3. Results
3.1. Synthesis of a streamlined ChIP protocol fromexisting methods
Although extremely effective, the standard ChIP protocolas described by Kuo and Allis (1999) remains time-intensiveand difficult. By contrast, we recently found that multiplemodified ChIP protocols including the Q2ChIP (Dahl andCollas, 2007) and ChIP-IT Express (ActiveMotif) were readilymastered by less experienced lab members. Noting the
remarkable differences between each of these effectiveprotocols, we sought to synthesize a single method thatwould minimize tradeoffs for cost, ease of use, speed, andreproducibility.
For our studies, we used input chromatin preparedessentially as described by Ciccone et al. (2004), in whichsonication-based shearing is replaced with micrococcalnuclease digestion. We favored the enzymatic digestion as ameans to minimize technique-dependent variability betweenusers and laboratories. Additionally, crosslinking and glycinetreatments were performed in conical centrifuge tubes ratherthan culture dishes. However, this alteration necessitated thatwe eliminate a PBS wash between the formaldehyde andglycine treatments. Consequently, reactions were stopped bydropwise addition of glycine.
Our antibody:bead conjugation and IP conditions essen-tially followed the Q2ChIP protocol (Dahl and Collas, 2007).Consequently, preblocking the beads and preclearing chro-matin to prevent nonspecific DNA binding proved unne-cessary. BSA (50 mg/ml) and sheared salmon sperm DNA(0.5 mg/ml) were added, and antibody:bead conjugates werethoroughly washed to block nonspecific antibody:DNA inter-actions and reduce ChIP variability. Finally, to increase theuniformity of bead conjugation with different antibody pre-parations, we incubated 1:1 mixes of Protein A- and ProteinG-coupled beads with each antibody for 1 h, though themanufacturer's instructions and our own tests suggest thatconjugation can be reduced to as little as 15 min (data notshown).
Conjugates were mixed with varying concentrations ofchromatin at 4 °C, and immune complexes were allowed toform over a 2 h window. We adapted the post-IP sampleprocessing procedures used in ActiveMotif's ChIP-IT Expresskit, in which the crosslink reversal times are dramaticallyshortened. We used the Q2ChIP buffers (RIPA and TE) to washimmune complexes 4× and 2× respectively. In each case,washes were performed by repeatedly passing PCR strip tubescontaining resuspended immune complexes across an immo-bilized bar magnet. In this way, all 6 washes for as many as 16IP reactions could be completed in as little as 15 min. Weobserved no benefit from incubating complexes in the washbuffers prior to aspiration.
After washing, captured immune complexes were elutedfrom the beads in 100 mM NaHCO3 by affixing the strip tubesto a benchtop vortexer set at 50% speed for 15 min. Crosslinkreversal was similarly limited to 15 min. For this, NaCl(250 mM final) was added, and eluates were immediatelyrecovered from the beads. Crosslinks in the eluates were thenreversed in a thermal cycler for 15 min at 95 °C, Proteinase Kwas added, and eluates were incubated for 1 h at 45 °C. Tominimize handling and experimental variability, we stoppedprotease digestion by adding PMSF, and purified sampleDNAs using Qiaquick Nucleotide Removal columns (Qiagen).However, we found that phenol:chloroform extractionand ethanol precipitation were equally effective at yieldingQ-PCR-competent DNA.
3.2. Increased IP duration does not enhance specific signal
To determine if the 2 h IP time could be reduced, we set upChIP reactions using chromatin from 106 Rag1−/−p53−/−
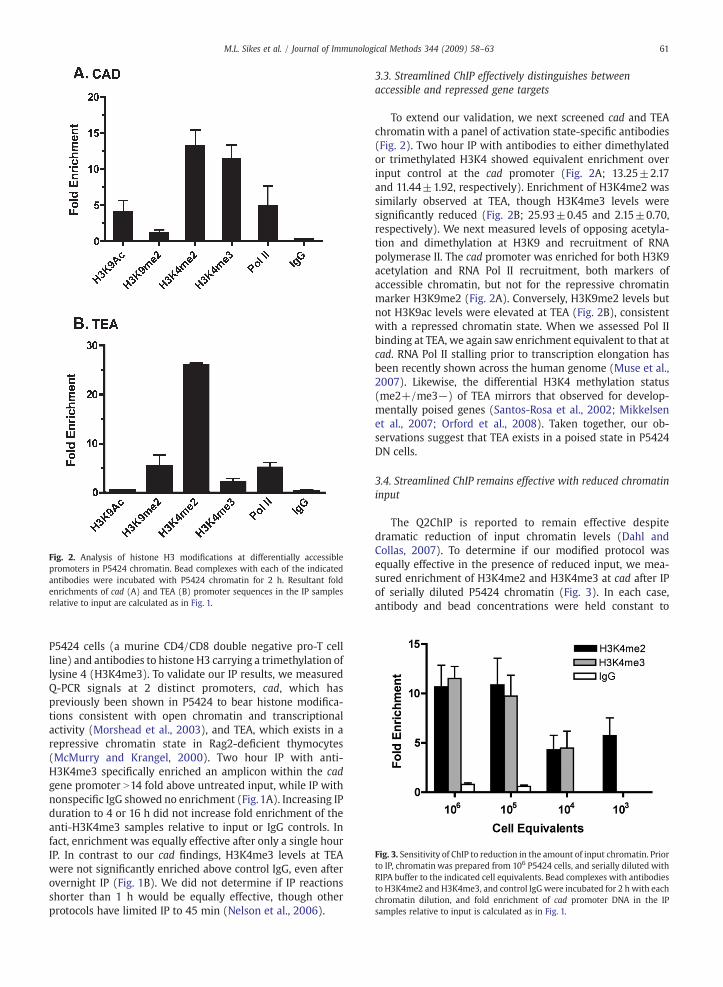
Fig. 3. Sensitivity of ChIP to reduction in the amount of input chromatin. Priorto IP, chromatin was prepared from 106 P5424 cells, and serially diluted withRIPA buffer to the indicated cell equivalents. Bead complexes with antibodiesto H3K4me2 and H3K4me3, and control IgGwere incubated for 2 h with eachchromatin dilution, and fold enrichment of cad promoter DNA in the IPsamples relative to input is calculated as in Fig. 1.
Fig. 2. Analysis of histone H3 modifications at differentially accessiblepromoters in P5424 chromatin. Bead complexes with each of the indicatedantibodies were incubated with P5424 chromatin for 2 h. Resultant foldenrichments of cad (A) and TEA (B) promoter sequences in the IP samplesrelative to input are calculated as in Fig. 1.
61M.L. Sikes et al. / Journal of Immunological Methods 344 (2009) 58–63
P5424 cells (a murine CD4/CD8 double negative pro-T cellline) and antibodies to histone H3 carrying a trimethylation oflysine 4 (H3K4me3). To validate our IP results, we measuredQ-PCR signals at 2 distinct promoters, cad, which haspreviously been shown in P5424 to bear histone modifica-tions consistent with open chromatin and transcriptionalactivity (Morshead et al., 2003), and TEA, which exists in arepressive chromatin state in Rag2-deficient thymocytes(McMurry and Krangel, 2000). Two hour IP with anti-H3K4me3 specifically enriched an amplicon within the cadgene promoter N14 fold above untreated input, while IP withnonspecific IgG showed no enrichment (Fig. 1A). Increasing IPduration to 4 or 16 h did not increase fold enrichment of theanti-H3K4me3 samples relative to input or IgG controls. Infact, enrichment was equally effective after only a single hourIP. In contrast to our cad findings, H3K4me3 levels at TEAwere not significantly enriched above control IgG, even afterovernight IP (Fig. 1B). We did not determine if IP reactionsshorter than 1 h would be equally effective, though otherprotocols have limited IP to 45 min (Nelson et al., 2006).
3.3. Streamlined ChIP effectively distinguishes betweenaccessible and repressed gene targets
To extend our validation, we next screened cad and TEAchromatin with a panel of activation state-specific antibodies(Fig. 2). Two hour IP with antibodies to either dimethylatedor trimethylated H3K4 showed equivalent enrichment overinput control at the cad promoter (Fig. 2A; 13.25±2.17and 11.44±1.92, respectively). Enrichment of H3K4me2 wassimilarly observed at TEA, though H3K4me3 levels weresignificantly reduced (Fig. 2B; 25.93±0.45 and 2.15±0.70,respectively). We next measured levels of opposing acetyla-tion and dimethylation at H3K9 and recruitment of RNApolymerase II. The cad promoter was enriched for both H3K9acetylation and RNA Pol II recruitment, both markers ofaccessible chromatin, but not for the repressive chromatinmarker H3K9me2 (Fig. 2A). Conversely, H3K9me2 levels butnot H3K9ac levels were elevated at TEA (Fig. 2B), consistentwith a repressed chromatin state. When we assessed Pol IIbinding at TEA, we again saw enrichment equivalent to that atcad. RNA Pol II stalling prior to transcription elongation hasbeen recently shown across the human genome (Muse et al.,2007). Likewise, the differential H3K4 methylation status(me2+/me3−) of TEA mirrors that observed for develop-mentally poised genes (Santos-Rosa et al., 2002; Mikkelsenet al., 2007; Orford et al., 2008). Taken together, our ob-servations suggest that TEA exists in a poised state in P5424DN cells.
3.4. Streamlined ChIP remains effective with reduced chromatininput
The Q2ChIP is reported to remain effective despitedramatic reduction of input chromatin levels (Dahl andCollas, 2007). To determine if our modified protocol wasequally effective in the presence of reduced input, we mea-sured enrichment of H3K4me2 and H3K4me3 at cad after IPof serially diluted P5424 chromatin (Fig. 3). In each case,antibody and bead concentrations were held constant to
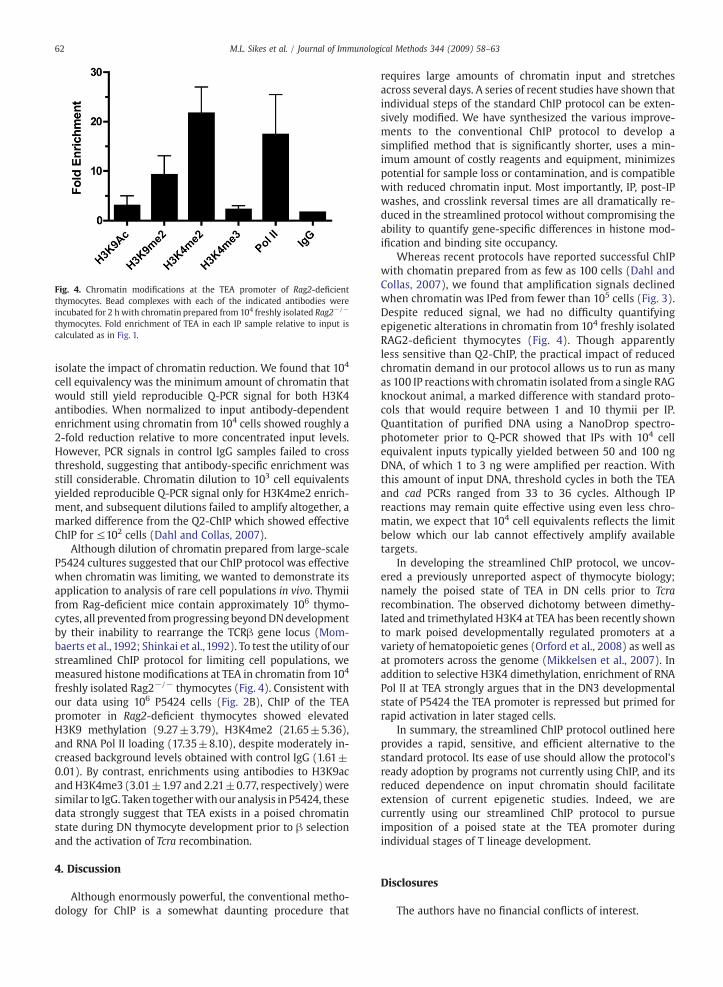
Fig. 4. Chromatin modifications at the TEA promoter of Rag2-deficientthymocytes. Bead complexes with each of the indicated antibodies wereincubated for 2 h with chromatin prepared from 104 freshly isolated Rag2−/−
thymocytes. Fold enrichment of TEA in each IP sample relative to input iscalculated as in Fig. 1.
62 M.L. Sikes et al. / Journal of Immunological Methods 344 (2009) 58–63
isolate the impact of chromatin reduction. We found that 104
cell equivalency was the minimum amount of chromatin thatwould still yield reproducible Q-PCR signal for both H3K4antibodies. When normalized to input antibody-dependentenrichment using chromatin from 104 cells showed roughly a2-fold reduction relative to more concentrated input levels.However, PCR signals in control IgG samples failed to crossthreshold, suggesting that antibody-specific enrichment wasstill considerable. Chromatin dilution to 103 cell equivalentsyielded reproducible Q-PCR signal only for H3K4me2 enrich-ment, and subsequent dilutions failed to amplify altogether, amarked difference from the Q2-ChIP which showed effectiveChIP for ≤102 cells (Dahl and Collas, 2007).
Although dilution of chromatin prepared from large-scaleP5424 cultures suggested that our ChIP protocol was effectivewhen chromatin was limiting, we wanted to demonstrate itsapplication to analysis of rare cell populations in vivo. Thymiifrom Rag-deficient mice contain approximately 106 thymo-cytes, all prevented fromprogressing beyondDNdevelopmentby their inability to rearrange the TCRβ gene locus (Mom-baerts et al., 1992; Shinkai et al., 1992). To test the utility of ourstreamlined ChIP protocol for limiting cell populations, wemeasured histone modifications at TEA in chromatin from 104
freshly isolated Rag2−/− thymocytes (Fig. 4). Consistent withour data using 106 P5424 cells (Fig. 2B), ChIP of the TEApromoter in Rag2-deficient thymocytes showed elevatedH3K9 methylation (9.27±3.79), H3K4me2 (21.65±5.36),and RNA Pol II loading (17.35±8.10), despite moderately in-creased background levels obtained with control IgG (1.61±0.01). By contrast, enrichments using antibodies to H3K9acandH3K4me3 (3.01±1.97 and 2.21±0.77, respectively)weresimilar to IgG. Taken togetherwith our analysis in P5424, thesedata strongly suggest that TEA exists in a poised chromatinstate during DN thymocyte development prior to β selectionand the activation of Tcra recombination.
4. Discussion
Although enormously powerful, the conventional metho-dology for ChIP is a somewhat daunting procedure that
requires large amounts of chromatin input and stretchesacross several days. A series of recent studies have shown thatindividual steps of the standard ChIP protocol can be exten-sively modified. We have synthesized the various improve-ments to the conventional ChIP protocol to develop asimplified method that is significantly shorter, uses a min-imum amount of costly reagents and equipment, minimizespotential for sample loss or contamination, and is compatiblewith reduced chromatin input. Most importantly, IP, post-IPwashes, and crosslink reversal times are all dramatically re-duced in the streamlined protocol without compromising theability to quantify gene-specific differences in histone mod-ification and binding site occupancy.
Whereas recent protocols have reported successful ChIPwith chomatin prepared from as few as 100 cells (Dahl andCollas, 2007), we found that amplification signals declinedwhen chromatin was IPed from fewer than 105 cells (Fig. 3).Despite reduced signal, we had no difficulty quantifyingepigenetic alterations in chromatin from 104 freshly isolatedRAG2-deficient thymocytes (Fig. 4). Though apparentlyless sensitive than Q2-ChIP, the practical impact of reducedchromatin demand in our protocol allows us to run as manyas 100 IP reactionswith chromatin isolated from a single RAGknockout animal, a marked difference with standard proto-cols that would require between 1 and 10 thymii per IP.Quantitation of purified DNA using a NanoDrop spectro-photometer prior to Q-PCR showed that IPs with 104 cellequivalent inputs typically yielded between 50 and 100 ngDNA, of which 1 to 3 ng were amplified per reaction. Withthis amount of input DNA, threshold cycles in both the TEAand cad PCRs ranged from 33 to 36 cycles. Although IPreactions may remain quite effective using even less chro-matin, we expect that 104 cell equivalents reflects the limitbelow which our lab cannot effectively amplify availabletargets.
In developing the streamlined ChIP protocol, we uncov-ered a previously unreported aspect of thymocyte biology;namely the poised state of TEA in DN cells prior to Tcrarecombination. The observed dichotomy between dimethy-lated and trimethylated H3K4 at TEA has been recently shownto mark poised developmentally regulated promoters at avariety of hematopoietic genes (Orford et al., 2008) as well asat promoters across the genome (Mikkelsen et al., 2007). Inaddition to selective H3K4 dimethylation, enrichment of RNAPol II at TEA strongly argues that in the DN3 developmentalstate of P5424 the TEA promoter is repressed but primed forrapid activation in later staged cells.
In summary, the streamlined ChIP protocol outlined hereprovides a rapid, sensitive, and efficient alternative to thestandard protocol. Its ease of use should allow the protocol'sready adoption by programs not currently using ChIP, and itsreduced dependence on input chromatin should facilitateextension of current epigenetic studies. Indeed, we arecurrently using our streamlined ChIP protocol to pursueimposition of a poised state at the TEA promoter duringindividual stages of T lineage development.
Disclosures
The authors have no financial conflicts of interest.

63M.L. Sikes et al. / Journal of Immunological Methods 344 (2009) 58–63
Acknowledgements
We gratefully thank Drs Hosni Hassan and Jose BrunoBarcena for use of their Q-PCR equipment.
References
Attema, J.L., Papathanasiou, P., Forsberg, E.C., Xu, J., Smale, S.T., Weissman, I.L.,2007. Epigenetic characterization of hematopoietic stem cell differentia-tion using miniChIP and bisulfite sequencing analysis. Proc. Natl. Acad.Sci. U.S.A. 104, 12371.
Ciccone, D.N., Morshead, K.B., Oettinger, M.A., 2004. Chromatin immunopre-cipitation in the analysis of large chromatin domains across murineantigen receptor loci. Methods Enzymol. 376, 334.
Dahl, J.A., Collas, P., 2007. Q2ChIP, a quick and quantitative chromatinimmunoprecipitation assay, unravels epigenetic dynamics of develop-mentally regulated genes in human carcinoma cells. Stem Cells 25, 1037.
Dahl, J.A., Collas, P., 2008. MicroChIP — a rapid micro chromatin immuno-precipitation assay for small cell samples and biopsies. Nucleic Acids Res.36, e15.
Dedon, P.C., Soults, J.A., Allis, C.D., Gorovsky, M.A., 1991. A simplifiedformaldehyde fixation and immunoprecipitation technique for studyingprotein–DNA interactions. Anal. Biochem. 197, 83.
Jenuwein, T., Allis, C.D., 2001. Translating the histone code. Science 293, 1074.Kuo, M.H., Allis, C.D., 1999. In vivo cross-linking and immunoprecipitation for
studying dynamic Protein:DNA associations in a chromatin environment.Methods 19, 425.
McMurry, M.T., Krangel, M.S., 2000. A role for histone acetylation in thedevelopmental regulation of VDJ recombination. Science 287, 495.
Mikkelsen, T.S., Ku, M., Jaffe, D.B., Issac, B., Lieberman, E., Giannoukos, G.,Alvarez, P., Brockman, W., Kim, T.K., Koche, R.P., Lee, W., Mendenhall, E.,O'Donovan, A., Presser, A., Russ, C., Xie, X., Meissner, A., Wernig, M.,Jaenisch, R., Nusbaum, C., Lander, E.S., Bernstein, B.E., 2007. Genome-wide maps of chromatin state in pluripotent and lineage-committedcells. Nature 448, 553.
Mombaerts, P., Iacomini, J., Johnson, R.S., Herrup, K., Tonegawa, S.,Papaioannou, V.E., 1992. RAG-1-deficient mice have no mature B and Tlymphocytes. Cell 68, 869.
Mombaerts, P., Terhorst, C., Jacks, T., Tonegawa, S., Sancho, J., 1995.Characterization of immature thymocyte lines derived from T-cellreceptor or recombination activating gene 1 and p53 double mutantmice. Proc. Natl. Acad. Sci. U. S. A. 92, 7420.
Morshead, K.B., Ciccone, D.N., Taverna, S.D., Allis, C.D., Oettinger, M.A., 2003.Antigen receptor loci poised for V(D)J rearrangement are broadlyassociated with BRG1 and flanked by peaks of histone H3 dimethylatedat lysine 4. Proc. Natl. Acad. Sci. U.S.A. 100, 11577.
Muse, G.W., Gilchrist, D.A., Nechaev, S., Shah, R., Parker, J.S., Grissom, S.F.,Zeitlinger, J., Adelman, K., 2007. RNA polymerase is poised for activationacross the genome. Nat. Genet. 39, 1507.
Nelson, J.D., Denisenko, O., Sova, P., Bomsztyk, K., 2006. Fast chromatinimmunoprecipitation assay. Nucleic Acids Res. 34, e2.
O'Neill, L.P., Turner, B.M., 1996. Immunoprecipitation of chromatin. MethodsEnzymol. 274, 189.
O'Neill, L.P., VerMilyea, M.D., Turner, B.M., 2006. Epigenetic characterizationof the early embryo with a chromatin immunoprecipitation protocolapplicable to small cell populations. Nat Genet 38, 835.
Orford, K., Kharchenko, P., Lai, W., Dao, M.C., Worhunsky, D.J., Ferro, A.,Janzen, V., Park, P.J., Scadden, D.T., 2008. Differential H3K4 methylationidentifies developmentally poised hematopoietic genes. Dev. Cell 14,798.
Santos-Rosa, H., Schneider, R., Bannister, A.J., Sherriff, J., Bernstein, B.E., Emre,N.C., Schreiber, S.L., Mellor, J., Kouzarides, T., 2002. Active genes are tri-methylated at K4 of histone H3. Nature 419, 407.
Shinkai, Y., Rathbun, G., Lam, K.P., Oltz, E.M., Stewart, V., Mendelsohn, M.,Charron, J., Datta, M., Young, F., Stall, A.M., 1992. RAG-2-deficient micelack mature lymphocytes owing to inability to initiate V(D)J rearrange-ment. Cell 68, 855.
Sikes, M.L., Meade, A., Tripathi, R., Krangel, M.S., Oltz, E.M., 2002. Regulationof V(D)J recombination: a dominant role for promoter positioning ingene segment accessibility. Proc. Natl. Acad. Sci. U.S.A. 99, 12309.






















![Techniques and strategies employing engineered …bleris/papers/2017-TALEs.pdfNCP [31,32]. Chromatin immunoprecipitation and sequencing (ChIP-seq) has revealed dCas9 binding from tens](https://img.pdfslide.net/doc/110x75/60accfbcf2c1682e39595fa9/techniques-and-strategies-employing-engineered-blerispapers2017-talespdf-ncp.jpg)






