Embed Size (px)
Citation preview

Ab initio calculations for exploring hydration patterns of adenine
Ho-Tae Kim*
Department of Applied Chemistry, Kumoh National Institute of Technology, 188 Shinpyung-Dong, Kumi city Kyungbuk 730-701, South Korea
Received 5 September 2003; revised 5 September 2003; accepted 3 December 2003
Abstract
Density functional theory (DFT) has been used to optimize the hydrated structures of adenine. The systematic investigation in the
hydration process of adenine is performed. The optimized geometries and hydration energies in the adenine–(H2O)1 – 4 have been obtained
by ab initio calculations at B3LYP/6-311þþG(d, p) level. Adenine–(H2O)1 – 2 complexes are mostly optimized to the structure of planar
geometry. The optimized structures of adenine–(H2O)3 – 4 complexes are mixed with planar and non-planar geometries. The hydration
energies of adenine show that the hydration process of adenine is competed by two ways within a 8 kcal/mol energy difference.
q 2004 Elsevier B.V. All rights reserved.
Keywords: Density functional theory; DNA bases; Adenine–water complex; Hydration
1. Introduction
The hydration process of molecules has been studied in
many research groups. Several experimental techniques
have been applied to study the role of hydration in the
hydrogen bonding complexes [1 – 6]. Theoretical
approaches about the hydration process of molecules have
been also performed by semi-empirical and ab initio
calculations with reasonable accuracy [2,4,5,7–10].
The hydrogen-bonded complexes of DNA bases have
been studied both theoretically and experimentally because
DNA is an essential molecule in the organism. The four
bases of DNA are known to play important roles in the
mechanism of the molecular recognition system of living
things [11–13]. In the system of protein–nucleic acid base
recognition, water molecules are indeed nearly always
present and can make effects on that system [14].
In this paper, an adenine–water system is chosen as a
sample system to study water effects in the hydration
process of nucleic acid bases. The adenine–water system
has been studied in the gas-phase experiment at nano-
second and femto-second time scale under molecular beam
conditions [15–17]. The optimized structures about ade-
nine–(H2O)1 – 3 on the neutral and anion clusters were
obtained by means of semi-empirical model calculations
[18]. Ab initio calculations on the neutral and anion
complexes were also performed about adenine–(H2O)1 – 3
at Hartree–Fock (HF) and second-order Moller–Plesset
(MP2) levels of theory [19]. The prototype HF level
calculations for the hydrated clusters of adenine cation
[15] and the optimized structure of adenine–(water)6 with
the small basis sets [20] were also reported.
In the current study, a systematic investigation about the
stepwise hydration of adenine has been carried out by ab
initio calculations. The investigation on structures and
stabilization energies in the adenine–(H2O)2 – 4 are per-
formed to give some clue to the question, which hydration
process is favorable between single bind site hydration and
two or three bind sites hydration in energy viewpoint.
2. Results and discussion
Geometry optimizations are performed for adenine–
(H2O)1 – 4 using DFT theory at the B3LYP/6-311þþG(d, p)
level. The GAUSSIAN 98 package [21] is employed. The
structural features of adenine are shown in Fig. 1.
The adenine is seen to possess three primary binding
sites A, B, and C because a water molecule can form
two hydrogen bonds between a nitrogen atom and the
hydrogen atom connected to another nitrogen atom. For
example, the two hydrogen bonds of site A are formed as
a N1· · ·H–O(H)–H11–N10 structure.
0166-1280/$ - see front matter q 2004 Elsevier B.V. All rights reserved.
doi:10.1016/j.theochem.2003.12.006
Journal of Molecular Structure (Theochem) 673 (2004) 121–126
www.elsevier.com/locate/theochem
* Tel.: þ82-54-467-4372; fax: þ82-54-467-4477.
E-mail address: [email protected] (H.-T. Kim).

The adenine molecule has a planar geometry [22]. The
planar geometry is not changed by the single water
molecule which is hydrated in the adenine primary
binding site with the two hydrogen bonds. The plane
geometry is maintained until two water molecules are
hydrated to the A or B site of adenine. However, the
distortion is started when two water molecules are
hydrated cooperatively to the C primary binding site.
In the case of primary binding sites A and B, the
distortion is started when three water molecules are
hydrated cooperatively to a single binding site. The
optimized bond distances, bond angles, and dihedral
angles of the adenine molecule at the DFT level of
theory were reported elsewhere [22].
The optimized geometries of adenine–(H2O)2 – 4 are
reported here with the combination of a number of water
molecules on the primary binding sites. Few selected
structures among the optimized geometries of adenine–
(H2O)1 – 4 are listed in the table and figures. Triangle, square,
and rhombus patterns occur in the structure of hydrated
adenine when three or four water molecules are hydrated
cooperatively at the single binding site of adenine. This
pattern is in conformity with the findings on the hydration of
benzene, phenol, and water [23–26].
2.1. Adenine· · ·H2O complex
In the first series of calculations the adenine complexes
with a single water molecule are performed. Three
structures where a water molecule is connected to adenine
through two hydrogen bonds were reported in the previous
ab initio calculations [19]. One of three structures, adenine–
(H2O)1–B which possesses a water molecule in B primary
binding site, is shown in Fig. 2 as (I). The hydrated
configuration of site A or C is similar to (I) and hence is not
displayed separately.
The hydrogen-bond distances and angles of adenine–
(H2O)1 complexes are listed in Table 1. The hydrogen-bond
distances of the three complexes are at the 2.814–2.835 A
range, and the hydrogen-bond angles are in the 156.4–
163.48 range at the first hydration step. From the
consideration of typical hydrogen bond distance (2.92–
2.94 A) and angle (1808) in water dimmer [27–29], it seems
that there is some geometric constraint in the adenine–
(H2O)1 complex. Table 1 also shows hydrogen bonds and
angles in H3N· · ·HOH, H2C(H3C)N· · ·HOH complexes. The
hydrogen bond configuration of H2C(H3C)N· · ·HOH com-
plex is similar to that of adenine–(H2O)1. The hydrogen
bond distances (2.932 and 2.922 A) and angles (174.4 and
173.48) in the two complexes also support that there is some
geometric constraint in the adenine–(H2O)1 complex.
The stabilization energies in each of the hydration
complexes are listed in Table 2. The energies of the
adenine–(H2O)1 complexes show that B site is the most
energetically favorable binding site in the single water
hydration step of adenine. B site was also the most stable
binding site in the MP2 level calculations [19]. The
hydrogen bond moiety of adenine–(H2O)1–A complex
has a six-membered ring configuration like that of (I).
However, adenine–(H2O)1–B complex is more stable than
adenine – (H2O)1 – A complex by 1.2 kcal/mol at the
B3LYP/6-311þþG(d, p) level. It is regarded that the
electrical environment of B site for hydrogen bonds is better
than that of A site. The stabilization energy of adenine–
(H2O)1–C(210.7 kcal/mol) is almost the same as the
stabilization energy of B site.
2.2. Adenine–2H2O complex
Three structures where two water molecules are
connected cooperatively to the primary binding sites are
also possible in the adenine–2H2O hydration step. The
adenine–(H2O)2–B2 which possesses two water molecules
in B site is shown as (II) in Fig. 2. Two water molecules are
located at the adenine molecular plane when they are
connected cooperatively in the A or B primary binding site.
However, two oxygen atoms are a little bit off from the
adenine molecular plane when the two water molecules are
connected cooperatively in the C primary binding site. The
side view of adenine–(H2O)2–C2 structure is denoted as
(III) in Fig. 2. H12 atom is 9.98 off from the adenine
molecular plane in the adenine–(H2O)2–C2 structure. It
seems that C site does not have enough space for three
hydrogen bonds with two water molecules.
The hydrogen-bond angles in three adenine–(H2O)2
complexes are at the 173.9–178.78 range in Table 1. These
hydrogen-bond angles in the three adenine–(H2O)2 struc-
tures are similar to the typical hydrogen-bond angle in the
water dimmer or in the H3N· · ·HOH, H2C(H3C)N· · ·HOH
complexes. The decrease of geometric constraint (increase
of hydrogen-bond angle) in the second hydration step is
resulted to the bigger stabilization energy at the site A or B
in Table 2. The stabilization energies per water in the
adenine–(H2O)2 turn out to be 210.3, 212.0, and
29.9 kcal/mol, respectively, in the A, B, and C sites. The
stabilization energy per water in adenine–(H2O)2–B2 is
bigger than that of adenine–(H2O)1–B by 1.1 kcal/mol.
However, The stabilization energy per water in adenine–
(H2O)2–C2 is smaller than that of adenine–(H2O)1–C by
Fig. 1. Structure of adenine. A, B, and C indicate the primary water binding
sites in adenine molecule.
H.-T. Kim / Journal of Molecular Structure (Theochem) 673 (2004) 121–126122
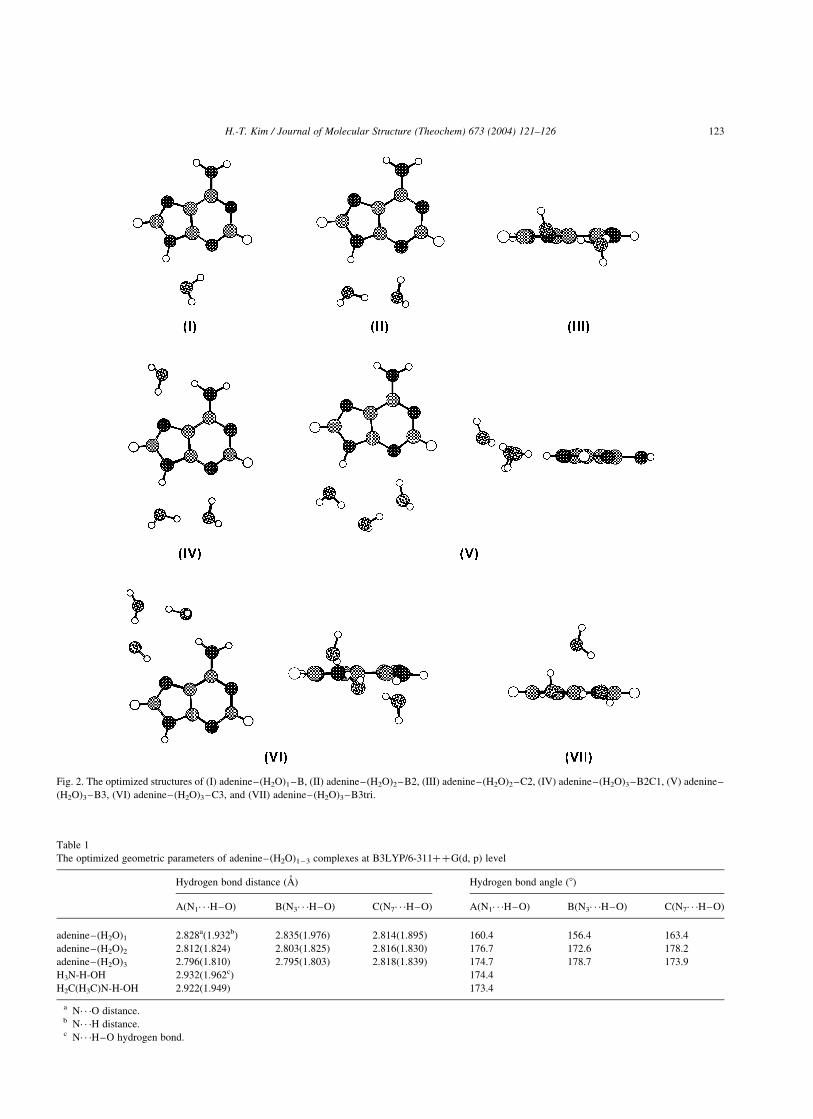
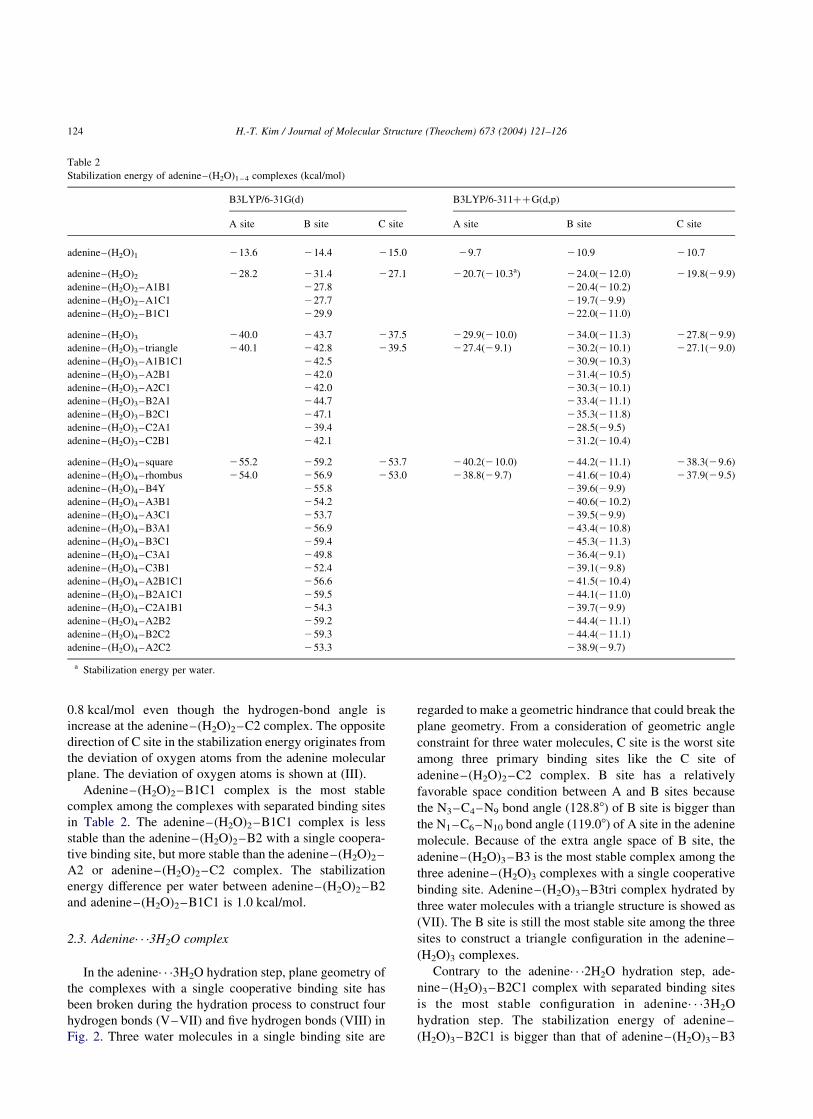
Table 1
The optimized geometric parameters of adenine–(H2O)1 – 3 complexes at B3LYP/6-311þþG(d, p) level
Hydrogen bond distance (A) Hydrogen bond angle (8)
A(N1· · ·H–O) B(N3· · ·H–O) C(N7· · ·H–O) A(N1· · ·H–O) B(N3· · ·H–O) C(N7· · ·H–O)
adenine–(H2O)1 2.828a(1.932b) 2.835(1.976) 2.814(1.895) 160.4 156.4 163.4
adenine–(H2O)2 2.812(1.824) 2.803(1.825) 2.816(1.830) 176.7 172.6 178.2
adenine–(H2O)3 2.796(1.810) 2.795(1.803) 2.818(1.839) 174.7 178.7 173.9
H3N-H-OH 2.932(1.962c) 174.4
H2C(H3C)N-H-OH 2.922(1.949) 173.4
a N· · ·O distance.b N· · ·H distance.c N· · ·H–O hydrogen bond.
Fig. 2. The optimized structures of (I) adenine–(H2O)1–B, (II) adenine–(H2O)2–B2, (III) adenine–(H2O)2–C2, (IV) adenine–(H2O)3–B2C1, (V) adenine–
(H2O)3–B3, (VI) adenine–(H2O)3–C3, and (VII) adenine–(H2O)3–B3tri.
H.-T. Kim / Journal of Molecular Structure (Theochem) 673 (2004) 121–126 123
0.8 kcal/mol even though the hydrogen-bond angle is
increase at the adenine–(H2O)2–C2 complex. The opposite
direction of C site in the stabilization energy originates from
the deviation of oxygen atoms from the adenine molecular
plane. The deviation of oxygen atoms is shown at (III).
Adenine–(H2O)2–B1C1 complex is the most stable
complex among the complexes with separated binding sites
in Table 2. The adenine–(H2O)2–B1C1 complex is less
stable than the adenine–(H2O)2–B2 with a single coopera-
tive binding site, but more stable than the adenine–(H2O)2–
A2 or adenine–(H2O)2–C2 complex. The stabilization
energy difference per water between adenine–(H2O)2–B2
and adenine–(H2O)2–B1C1 is 1.0 kcal/mol.
2.3. Adenine· · ·3H2O complex
In the adenine· · ·3H2O hydration step, plane geometry of
the complexes with a single cooperative binding site has
been broken during the hydration process to construct four
hydrogen bonds (V–VII) and five hydrogen bonds (VIII) in
Fig. 2. Three water molecules in a single binding site are
regarded to make a geometric hindrance that could break the
plane geometry. From a consideration of geometric angle
constraint for three water molecules, C site is the worst site
among three primary binding sites like the C site of
adenine–(H2O)2–C2 complex. B site has a relatively
favorable space condition between A and B sites because
the N3–C4–N9 bond angle (128.88) of B site is bigger than
the N1–C6–N10 bond angle (119.08) of A site in the adenine
molecule. Because of the extra angle space of B site, the
adenine–(H2O)3–B3 is the most stable complex among the
three adenine–(H2O)3 complexes with a single cooperative
binding site. Adenine–(H2O)3–B3tri complex hydrated by
three water molecules with a triangle structure is showed as
(VII). The B site is still the most stable site among the three
sites to construct a triangle configuration in the adenine–
(H2O)3 complexes.
Contrary to the adenine· · ·2H2O hydration step, ade-
nine–(H2O)3–B2C1 complex with separated binding sites
is the most stable configuration in adenine· · ·3H2O
hydration step. The stabilization energy of adenine–
(H2O)3–B2C1 is bigger than that of adenine–(H2O)3–B3
Table 2
Stabilization energy of adenine–(H2O)1 – 4 complexes (kcal/mol)
B3LYP/6-31G(d) B3LYP/6-311þþG(d,p)
A site B site C site A site B site C site
adenine–(H2O)1 213.6 214.4 215.0 29.7 210.9 210.7
adenine–(H2O)2 228.2 231.4 227.1 220.7(210.3a) 224.0(212.0) 219.8(29.9)
adenine–(H2O)2–A1B1 227.8 220.4(210.2)
adenine–(H2O)2–A1C1 227.7 219.7(29.9)
adenine–(H2O)2–B1C1 229.9 222.0(211.0)
adenine–(H2O)3 240.0 243.7 237.5 229.9(210.0) 234.0(211.3) 227.8(29.9)
adenine–(H2O)3–triangle 240.1 242.8 239.5 227.4(29.1) 230.2(210.1) 227.1(29.0)
adenine–(H2O)3–A1B1C1 242.5 230.9(210.3)
adenine–(H2O)3–A2B1 242.0 231.4(210.5)
adenine–(H2O)3–A2C1 242.0 230.3(210.1)
adenine–(H2O)3–B2A1 244.7 233.4(211.1)
adenine–(H2O)3–B2C1 247.1 235.3(211.8)
adenine–(H2O)3–C2A1 239.4 228.5(29.5)
adenine–(H2O)3–C2B1 242.1 231.2(210.4)
adenine–(H2O)4–square 255.2 259.2 253.7 240.2(210.0) 244.2(211.1) 238.3(29.6)
adenine–(H2O)4–rhombus 254.0 256.9 253.0 238.8(29.7) 241.6(210.4) 237.9(29.5)
adenine–(H2O)4–B4Y 255.8 239.6(29.9)
adenine–(H2O)4–A3B1 254.2 240.6(210.2)
adenine–(H2O)4–A3C1 253.7 239.5(29.9)
adenine–(H2O)4–B3A1 256.9 243.4(210.8)
adenine–(H2O)4–B3C1 259.4 245.3(211.3)
adenine–(H2O)4–C3A1 249.8 236.4(29.1)
adenine–(H2O)4–C3B1 252.4 239.1(29.8)
adenine–(H2O)4–A2B1C1 256.6 241.5(210.4)
adenine–(H2O)4–B2A1C1 259.5 244.1(211.0)
adenine–(H2O)4–C2A1B1 254.3 239.7(29.9)
adenine–(H2O)4–A2B2 259.2 244.4(211.1)
adenine–(H2O)4–B2C2 259.3 244.4(211.1)
adenine–(H2O)4–A2C2 253.3 238.9(29.7)
a Stabilization energy per water.
H.-T. Kim / Journal of Molecular Structure (Theochem) 673 (2004) 121–126124

by 1.3 kcal/mol in Table 2. The difference of configuration
corresponding to the most stable complex in adenine· · ·3H2-
O hydration step could be explained with the breakdown of
planar geometry in the complexes with a single cooperative
binding site. The broken plane geometry of adenine–
(H2O)3–B3 is showed in Fig. 2 as (VI). H12 atom is 22.58 off
from the adenine molecular plane in the adenine–(H2O)3–
B3 complex. However, the plane geometry is still
maintained in the case of adenine–(H2O)3–B2C1 even
though three water molecules are hydrated to the adenine
molecule.
From a consideration of stabilization energy in adenine–
(H2O)3 complexes, it seems most reasonable to conclude
that the relative stability of the adenine primary binding
sites depends on the number of water molecules on those
sites. Even adenine–(H2O)3–B2C1 is more stable than
adenine–(H2O)3 –B3, other complexes with separated
binding sites in three water hydration step are less stable
than adenine–(H2O)3–B3. The stabilization energy differ-
ence per water between adenine–(H2O)2 – B2C1 and
adenine–(H2O)2–B3 is 0.4 kcal/mol. Adenine–(H2O)3–
B3tri complex is less stable than adenine–(H2O)3–B3
complex. The stabilization energies in adenine–3H2O
hydration step are listed in the Table 2 within a 7 kcal/mol
energy difference.
2.4. Adenine–(H2O)4 complex
Many possible structures in the adenine· · ·4H2O
hydration step have been explored by the DFT method.
However, only a few selected complexes with separated
binding sites and three complexes with a single cooperative
binding site are listed in Table 2. Stabilization energies are
ranged from 236.4(29.1 per water) to 245.3(211.3 per
water) kcal/mol. The optimized structures with separated
binding sites and a single cooperative binding site are
displayed in Fig. 3. (II), (III), and (IV) have square,
rhombus, and Y shape structures, respectively, in each of the
primary binding sites. (II) is the most stable complex among
three complexes with a single cooperative binding site in
adenine· · ·4H2O hydration step. The square pattern was well
studied in the water tetramer [25], benzene–(H2O)4 [24],
and phenol–(H2O)4 hydration complex [26]. The square
plane of four water molecules in adenine–(H2O)4–B4sq has
a 1188 angle to the adenine molecular plane.
The rhombus shape of four water molecules in adenine–
(H2O)4–B4rhom is not a plane geometry contrary to the
plane geometry of a square shaped structure in the adenine–
(H2O)4–B4sq complex. Because two water molecules of
rhombus shape are distorted from the planar geometry of
rhombus, adenine–(H2O)4–B4rhom is less stable than
Fig. 3. The optimized structures of (I) adenine–(H2O)4–B3C1, (II) adenine–(H2O)4–B4sq, (III) adenine–(H2O)4–B4rhom, and (IV) adenine–(H2O)4–B4Y.
H.-T. Kim / Journal of Molecular Structure (Theochem) 673 (2004) 121–126 125

the adenine–(H2O)4–B4sq in Table 2. The geometry frame
of adenine–(H2O)4–B4Y which possesses four water
molecules with Y shaped configuration is the same with
that of adenine–(H2O)3–B3. The fourth water molecule is
added to the hydrogen atom of center water among the three
water molecules of B site. The stabilization energy of (IV)
complex is smaller than those of (II) and (III) because (IV)
has only five hydrogen bonds compared to the six hydrogen
bonds of (II) and (III) complexes.
In the case of adenine· · ·4H2O, the most stable complex
is adenine–(H2O)4–B3C1 with separated binding sites like
as adenine· · ·3H2O hydration step. However, the stabiliz-
ation energy difference per water between adenine–
(H2O)4 – B3C1 and adenine – (H2O)4 – B4sq is just
0.3 kcal/mol. The stabilization energy difference per
water between adenine–(H2O)4–B2A1C1 and adenine–
(H2O)4–B4sq is also negligible. On the basis of the energy
of hydration complexes which have a different number of
binding sites, it seems that there is no particularly
preferable hydration process in the energy viewpoint
between the process with a single binding site and two
or three binding sites at the adenine· · ·4H2O hydration step.
3. Conclusions
Hydration energies per water range from 29.0 to
212.0 kcal/mol in the adenine–(H2O)1 – 4 hydration pro-
cess. Adenine–(H2O)2–B2 with 212.0 kcal/mol hydration
energy per water seems to have the most favorable
configuration in the hydration process of adenine. The
hydration energies of adenine also show that the cooperative
hydration process with a single binding site and the
hydration process with separated binding sites are competing
within a 8 kcal/mol energy difference in each hydration step.
Acknowledgements
This paper was supported by Research Fund, Kumoh
National Institute of Technology.
References
[1] T. Ebata, A. Iwasaki, N. Mikami, J. Phys. Chem. A 104 (2000) 7974.
[2] Ch. Janzen, D. Spangenberg, W. Roth, K. Kleinermanns, J. Chem.
Phys. 110 (1999) 9898.
[3] T. Ebata, A. Fujii, N. Mikami, Intern. J. Mass. Spectrom. Ion
Processes 159 (1996) 111.
[4] C.J. Gruenloh, J.R. Carney, F.C. Hagemeister, C.A. Arrington, T.S.
Zwier, S.Y. Fredericks, J.T. Woods, K.D. Jordan, J. Chem. Phys. 109
(1998) 6601.
[5] P. Ilich, C.F. Hemann, R. Hille, J. Phys. Chem. B 101 (1997) 10923.
[6] S.K. Kim, W. Lee, D.R. Herschbach, J. Phys. Chem. 100 (1996)
7933.
[7] S.R. Gadre, K. Babu, A.P. Rendell, J. Phys. Chem. A 104 (2000) 8976.
[8] H.H.Y. Tsui, T. van Mourik, Chem. Phys. Lett. 350 (2001) 565.
[9] N.A. Besley, J.D. Hirst, J. Am. Chem. Soc. 121 (1999) 8559.
[10] D.M.A. Smith, J. Smets, L. Adamowicz, J. Phys. Chem. A 103 (1999)
4309.
[11] I. Galetich, M. Kosevich, V. Shelkovsky, S.G. Stepanian, Y.P. Blagoi,
L. Adamowicz, J. Mol. Struct. 478 (1999) 155.
[12] Z. Tao, T. Fujiwara, I. Saito, H. Sugiyama, J. Am. Chem. Soc. 121
(1999) 4804.
[13] S. White, J.W. Szewczyk, J.M. Turner, E.E. Baird, P.B. Dervan,
Nature 391 (1998) 468.
[14] P.I. Nagy, G. Alagona, C. Ghio, J. Am. Chem. Soc. 121 (1999) 4804.
[15] N.J. Kim, Y.S. Kim, G. Jeong, T.K. Ahn, S.K. Kim, Int. J. Mass Spec.
219 (2002) 11.
[16] N.J. Kim, H. Kang, G. Jeong, Y.S. Kim, K.T. Lee, S.K. Kim, J. Phys.
Chem. A 104 (2000) 6552.
[17] H. Kang, K.T. Lee, S.K. Kim, Chem. Phys. Lett. 359 (2002) 213.
[18] V. Periquet, A. Moreau, S. Carles, J.P. Schermann, C. Desfrancois, J.
Electron, Spectrosc. Relat. Phenom. 106 (2000) 141.
[19] A.F. Jalbout, L. Adamowicz, J. Phys. Chem. A 105 (2001) 1033.
[20] N.U. Zhanpeisov, J. Leszczynski, Struct. Chem. 12 (2001) 121.
[21] M.J. Frisch, G.W. Tucks, H.B. Schlegel, G.E. Scuseria, M.A. Robb,
J.R. Cheeseman, V.G. Zakrzewski, J.A. Montgometry, R.E. Strat-
mann, J.C. Burant, S. Dapprich, J.M. Millam, A.D. Daniels, K.N.
Kudin, M.C. Strain, O. Farkas, J. Tomasi, V. Barone, M. Cossi,
R. Cammi, B. Mennucci, C. Pomelli, C. Adamo, S. Clifford, J.
Ochterski, G.A. Peterson, P.Y. Ayala, Q. Cui, K. Morokuma, D.K.
Malick, A.D. Rabuck, K. Raghavachari, J.B. Foresman, J. Cioslowski,
J.V. Ortiz, B.B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I.
Komaromi, R. Gomperts, R.L. Martin, D.J. Fox, T. Keith, M.A. Al-
Laham, C.Y. Peng, A. Nanayakkara, C. Gonzalez, M. Challacombe,
P.M.W. Gill, B.G. Johnson, W. Chen, M.W. Wong, J.L. Andres,
M. Head-Gordon, E.S. Replogle, J.A. Pople, Gaussian 98, Gaussian,
Inc., Pittsburgh, PA, 1998.
[22] X. Yan, P. Day, T. Hollis, A. Monzingo, E. Schelp, J.D. Robertus,
G.W.A. Milne, S. Wang, Prot. Struct. Funct. Genet. 31 (1998) 33.
[23] M.R. Viant, J.D. Cruzan, D.D. Lucas, M.G. Brown, K. Liu, R.J.
Saykally, J. Phys. Chem. A 101 (1997) 9032.
[24] R.N. Pribble, T.S. Zwier, Science 265 (1994) 75.
[25] J.D. Cruzan, M.R. Viant, M.G. Brown, R.J. Saykally, J. Phys. Chem.
A 101 (1997) 9022.
[26] Ch. Jacoby, W. Roth, M. Schmitt, Ch. Janzen, D. Spangenberg, K.
Kleinermanns, J. Phys. Chem. A 102 (1998) 4471.
[27] P.L. Silvestrelli, M. Parrinello, J. Chem. Phys. 111 (1999) 3572.
[28] S.J. Grabowski, Chem. Phys. Lett. 338 (2001) 361.
[29] F.H. Stillinger, Science 209 (1980) 451.
H.-T. Kim / Journal of Molecular Structure (Theochem) 673 (2004) 121–126126





























