Embed Size (px)
Citation preview

PHYSICAL REVIEW B 15 MARCH 1998-IIVOLUME 57, NUMBER 12
Ab initio study of structural, dielectric, and dynamical properties of GaN
K. Karch,* J.-M. Wagner, and F. BechstedtInstitut fur Festkorpertheorie und Theoretische Optik, Friedrich-Schiller-Universita¨t, Max-Wien-Platz 1, 07743 Jena, Germany
~Received 1 August 1997!
We report first-principles calculations of the structural, dielectric, and lattice-dynamical properties for wurtz-ite and zinc-blende GaN. The structural properties are calculated using a plane-wave-pseudopotential methodof the density-functional theory. A linear-response approach to the density-functional theory is used to deriveBorn effective charges, dielectric constants, phonon frequencies, and eigenvectors. The results are discussed interms of ionic and covalent bonding. The computed values are in reasonable agreement with experimental data.@S0163-1829~98!04112-5#
lyn-anonthninp
na
ip
heu
c-betsn
it
-icad
c
ikeanti
eded
ak-er-
.as
hecal-oft
al-ar
aen-
nc-l-ion
iesofII,
hefor
ve-ity-oftion
e of
-the
reveic-
I. INTRODUCTION
The group-III nitrides AlN, GaN, and InN are currentbeing actively investigated in view of their promising potetial for short-wavelength electroluminescence deviceshigh-temperature, high-power, and high-frequency electrics. This holds especially for GaN as a basic material forrealization of light-emitting diodes and lasers in the blue aUV region.1 Under ambient conditions GaN crystallizesthe hexagonal wurtzite~2H! structure with the space grouC6v
4 . However, recent epitaxy2 of thin GaN films has beendemonstrated also to result in the cubic zinc-blende~3C!structure with the space groupTd
2 . Remarkable progress ithe synthesis of pure 3C-GaN films is related to plasmassisted molecular-beam epitaxy on GaAs~001! substrates.3,4
In the 3C and 2H structures, each atom of one kindtetrahedrally surrounded by atoms of the other chemical scies. Both phases have the same nearest-neighbor sThey differ in the stacking of the Ga-N bilayers perpendiclar to the cubic@111# and hexagonal@0001# directions, re-spectively. The stacking in thisc-axis direction isABC inthe cubic case orAB in the hexagonal structure. In the zinblende material the atomic coordinates are fixed by the culattice constanta05&a. In the wurtzite case, however, thatomic positions are characterized by two lattice constanaand c. Moreover, because of the reduced symmetry oneeds an additional parameteru, which is usually defined asthe length of the Ga-N bond parallel to thec axis in units ofthe lattice constantc. In the case of ideal tetrahedrons,
holds thatc/a5A8/3 andu5 38 . Because of the similar sur
roundings of an atom, one expects quite similar physproperties of the two structures. However, there are alsoviations. For instance, the zinc-blende material with@001#orientation is more convenient in device production, sinthe cleavage faces of the type~110! are parallel.
Despite of the interest in GaN, several properties—lthe lattice-dynamical and dielectric ones—are at prespoorly known. On the other hand, phonons are importelementary excitations for standard sample characterizaby means of Raman spectroscopy5–15 or infrared reflectiv-ity.5,12,16 First-principles calculations have been performonly for nonpolar zone-center phonons using either a mixbasis method17 or the linear muffin-tin orbital method18–20
570163-1829/98/57~12!/7043~7!/$15.00
d-
ed
-
se-lls.-
ic
e
le-
e
entt
on
-
within the frozen-phonon approach. Reasons for the bredown of the frozen-core approximation in GaN are the engetical resonance of Ga 3d and N 2s levels21 and the signifi-cant overlap of 3d states with the valence electron states22
Therefore, such shallow core states are best dealt withvalence electrons. However, it is difficult to converge twave functions in a plane-wave basis. Because of the loization of the 3d functions in real-space energy cutoffsabout 240 Ry are needed.23 Nevertheless, one importansymptom of the shallowness of the Ga 3d electrons, the sig-nificant overlap with the valence states, can be treatedready within the frozen-core approximation, if the nonlinecore correction~NLCC! of Louie, Froyen, and Cohen24 istaken into account. The NLCC involves the addition ofmodel of the core charge density to the valence charge dsity before the evaluation of the exchange-correlation futional. In a recent Brief Report,25 we demonstrated the quaity of the NLCC approach also in the case of the calculatof zone-center phonons of 3C-GaN.
In this paper we present extensive first-principles studof structural, dielectric, and lattice-dynamical propertiesGaN in both zinc-blende and wurtzite structures. In Sec.we briefly review the underlying theoretical methods. In tfollowing sections, results are presented and discussedgeometry parameters and elastic moduli~Sec. III!, Born ef-fective charges and dielectric constants~Sec. IV!, and pho-non frequencies and eigenvectors~Sec. V!, respectively. Fi-nally, conclusions are given in Sec. VI.
II. METHOD
The calculations are performed using the plane-wapseudopotential approach within the framework of densfunctional theory~DFT!. The exchange-correlation energythe electrons is described in the local-density approxima~LDA ! by the Perdew-Zunger interpolation.26 We generatethe Ga and N pseudopotentials according to the schemTroullier and Martins27 including the NLCC24 to include theeffect of the semicore Ga 3d electrons on the bonding properties. Particular attention is also paid to the choice ofreference configuration for the excited higher-lying atomicdstates. The Troullier-Martins procedure yields soft-copseudopotentials, which allow a restriction of the plane-waexpansion of the electronic eigenfunctions by a kinet
7043 © 1998 The American Physical Society
s
tio
re
ur
-esu
reusio
ti
ente
y-
so
eath
l
m.o
ioletha
ew
pehtor
thro
hefic
enthiss.1
ula-m-u-is
l
ndez-
eal
in-nly
di-s ofata
l-ro-theca,ld
vol--th
7044 57K. KARCH, J.-M. WAGNER, AND F. BECHSTEDT
energy cutoff of 50~wurtzite! or 60 ~zinc-blende! Ry. Therelatively high cutoff energy is due to the lack ofp corestates in the nitrogen core@cf. calculations for BN and AlN~Ref. 28!#. The summations over the Brillouin zone~BZ! isperformed using sets of Chadi-Cohen special points.29 Be-cause of the semiconducting character of the crystals conered here convergence is reached for 12~10! special points inthe irreducible wedge of the BZ of the wurtzite~zinc-blende!structure.
The ground-state properties are obtained by minimizaof the total energy with respect to the unit-cell volumeV.Whereas in the zinc-blende case the volume is directlylated to the lattice constanta0 , for the wurtzite structure thisminimization has to be performed by a two-step proceddue to three independent parametersc and a ~lattice con-stants! andu ~internal cell parameter!. For a given unit-cellvolume V5)a2c/2 the total energy is minimized with respect toc/a andu. This step is repeated for other volumnear the experimental equilibrium one. The theoretical eqlibrium volumeV0 , the static bulk modulus at zero pressuB0 , and first-order pressure derivative of the bulk modulB08 , are determined by fitting the total energy as a functof volume to the Vinet equation of state.30 In the wurtzitecase of 2H-GaN we have also used the Murnaghan equaof state31 for test reasons.
The lattice-dynamical properties, i.e., the eigenfrequcies and eigenvectors of the lattice vibrations, are calculawithin the framework of the self-consistent densitfunctional perturbation theory~DFPT!.32–34 The DFPT alsoallows the calculation of the high-frequency dielectric tene~`!, and the Born effective charge tensorZB, for each in-equivalent atom in the unit cell. These quantities are necsary for the nonanalytic part of the dynamical matrix ofpolar semiconductor as GaN. The DFPT is generalized toinclusion of NLCC~Ref. 25! according to the work of DaCorsoet al.35 for II-VI compounds with shallowd electronsof the group-II atoms. In the case of the DFPT the BZ sumations are improved using sets of 28 special pointskinetic-energy cutoff and a special point set ensure the cvergence of the phonon frequencies to within 3 (2) cm21 forwurtzite ~zinc-blende! GaN.
III. STRUCTURAL PROPERTIES
The calculated ground-state energies plotted as functof volume for 3C- and 2H-GaN are shown in Fig. 1. In TabI, the calculated structural properties as obtained fromVinet equation of state are compared with experimental vues. In the 2H case the Murnaghan equation of statchecked to give practically the same values. Table I shothat the calculated structural parameters approach the exmental ones. The calculated lattice constants are sligsmaller than the experimental ones. The DFT-LDA errwidely cancel each other in the ratioc/a of the lattice con-stants. The theoretical data agree not only with respect toquantity but also in the case of the internal cell parameteu.The corresponding data are close to the ‘‘ideal’’ valueu50.375. For AlN we previously28 found a value ofu50.3816, deviating significantly from the ideal values. Texperimental values39–41 available for the bulk modulus owurtzite GaN scatter to a larger extent than the theoret
id-
n
-
e
i-,,
n
on
-d
r
s-
e
-An-
ns
el-issri-lys
is
f
al
ones using different approaches. Thus a distinct statemabout the accuracy of the calculations is not possible. Tholds also for the pressure derivative of the bulk modulu
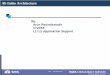
The energetical ordering of the two polytypes in Fig.depends sensitively on the details of the numerical calction. For that reason the graph for 3C in Fig. 1 is also coputed with a 50-Ry cutoff. The energy gain per pair calclated for 2H-GaN with respect to the zinc-blende polytype20 meV, close to the values calculated by other authors~cf.the collection in Ref. 42!. The energetical ordering followsthe usual rule forANB82N compounds related to the criticaparameterD(c/a)5c/a2A8/3.43 In the case of AlN~BN!this parameterD(c/a)520.029~0.024! ~Ref. 28! correlateswith the computed energy difference between wurtzite azinc blende. We find a slightly negative valuD(c/a)520.007, which suggests the stability of the wurtite structure.
The bonding tetrahedrons in GaN are close to the idones. This is also indicated by the relative bond lengthuclose to the ideal value. In GaN the lengths of the twoequivalent bonds are practically the same. They differ oby 0.2%.
IV. DIELECTRIC PROPERTIES
In Table II we compare the values calculated for theelectric tensor and the tensor of the Born effective charge3C- and 2H-GaN with theoretical and experimental d
available. In addition, the average valuese(`)5 13 Tr e(`)
and ZB5 13 Tr ZB are also given. In our calculations loca
field effects are included, so that we compute the macscopic dielectric constants. Therefore, it is surprising thatvalues are larger than those of Christensen and Gorczy21
who do not take local-field effects into account. Local-fieeffects reduce the dielectric constant by about 20%.45 The
FIG. 1. Calculated total energies per Ga-N pair vs reducedume. The solid~dotted! line indicates the total energy of the wurtzite ~zinc-blende! structure. For this figure 3C is also calculated wia cutoff of 50 Ry.
57 7045AB INITIO STUDY OF STRUCTURAL, DIELECTRIC, . . .
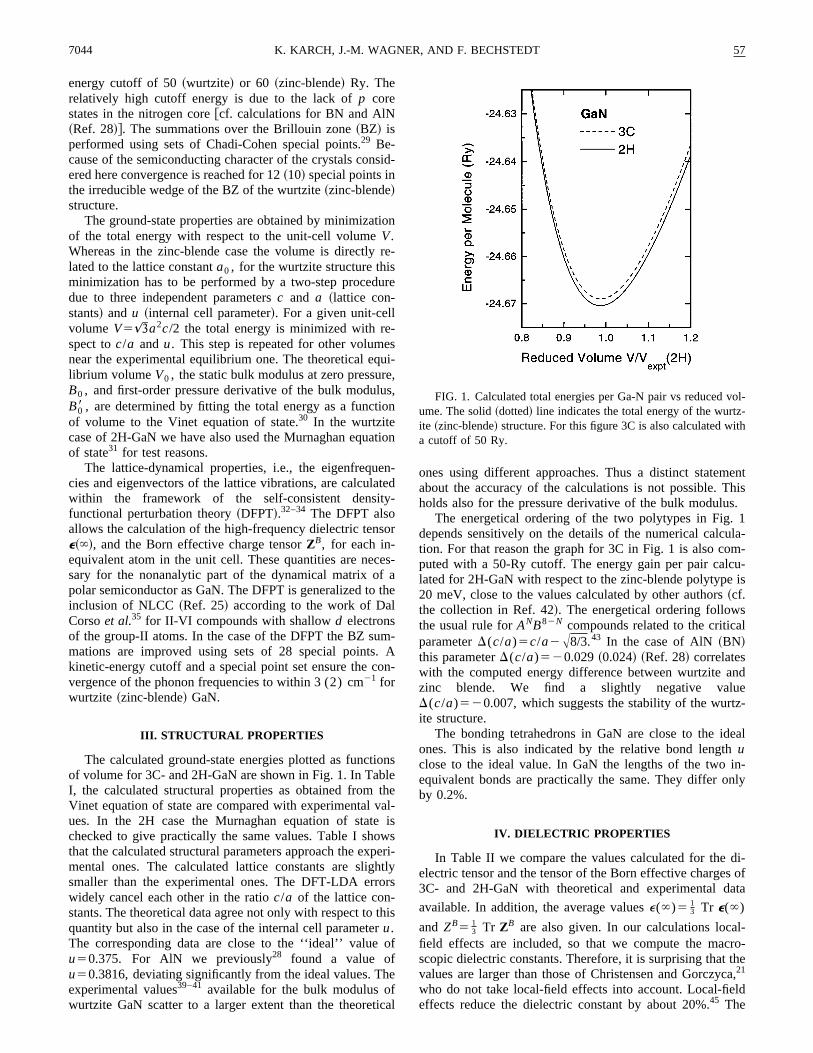
TABLE I. Comparison of calculated and measured structural properties of GaN in zinc-blende~3C! andwurtzite ~2H! phases: lattice constantsa0 and a ~Å!, ratio c/a, internal parameteru, bulk modulusB0
~Mbar!, and its pressure derivativeB08 .
Polytype Method Ref. a0 a c/a u B0 B08
3C present 4.447 2.07 3.94other calc. 17 4.446 1.95
20 4.46 2.01 3.921 4.4623 4.46 1.84 4.636 1.87
expt. 37 4.49 1.73 3.7
2H present 3.143 1.626 0.377 2.15 5.9other calc. 17 3.146 1.629 0.377 1.95
20 3.170 1.620 0.379 2.07 4.521 2.00 3.823 3.162 1.626 0.377 2.0236 3.126 1.638 0.377 1.90 2.9
expt. 9 2.45 4.038 3.189 1.62639 3.160 1.62140 1.88 3.241 2.37 4.3
nthtnnre-e
om
t.lle
s
rgebyallelhhe
o-iderri-a
c-
th
anisotropy for 2H follows the findings in the BN and AlNcases.28 Apart from the local-field effects no explanatiomay be given with respect to the reversed ordering ofcomponentse'(`) and e i(`) in Ref. 21. The agreemenwith the experiment is reasonable. A detailed comparisoimpossible, since no anisotropies have been considered idiscussion of the infrared-reflectivity measurements. Moover, the values ofe~`! in the cubic case arise from measurements in which the structure of the material was not wdefined. By means of the optical phonon frequencies frSec. V and the Lyddane-Sachs-Teller~LST! relation, we ob-tain the valuee(0)59.70 for the static dielectric constanFor the wurtzite structure, the perpendicular and paracomponents result from a generalized LST relation46 and arefound to bee'(0)59.25 ande i(0)510.34. These valueagree reasonably with experimental data,e(0)51262 ~Ref.
e
isthe-
ll
l
5! and, respectively,e'(0)59.5 ande i(0)510.4,16 takenfrom a fit of 2H-GaN IR spectra.
In the zinc-blende structure the tensor of the~transverse!Born effective charge is isotropic. Because of the chaneutrality the charges of the cation and anion only differthe sign. In the wurtzite case the tensor components parand perpendicular to thec axis are independent of eacother. Although there are four atoms in the unit cell, tnonsymmorphic space groupC6v
4 with a screw axis along thec axis enforces that only two of them are independent. Tgether with the charge neutrality one has again to consonly one tensor. Our results agree well with the few expemental data available. The Born effective charge followsclear chemical trend within the nitrides series. In the zinblende case it holdsZB51.93 ~BN!, 2.56 ~AlN !, and 2.65~GaN!. However, the screened effective chargeZ*5ZB/Ae(`) exhibits a more pronounced linear relation wi
TABLE II. Macroscopic dielectric tensore~`! and Born effective charge tensorZB. The average valuesare also given.
Polytype Method Reference e'(`) e i(`) e~`! Z'B Zi
B ZB
3C present 5.41 2.65calc. 21 4.78expt. 10 5.29
12 5.344 5.29 5.7
2H present 5.21 5.41 5.28 2.60 2.74 2.64calc. 21 4.71 4.62 4.68expt. 5 5.860.4 3.260.5
16 5.35 2.65 2.82
n.
e-
th
inttefrin
-d
Ln
ec-le
N.
en-
alttothecity
ndeeiteing
nd-
psd
s oflines,
of
7046 57K. KARCH, J.-M. WAGNER, AND F. BECHSTEDT
ionicity ~or, more strictly, charge asymmetry coefficient! gcalculated according to the method of Garcia and Cohe22
One has to compareZ* 50.91 ~BN!, 1.21 ~AlN !, and 1.14~GaN! with g50.49~BN!, 0.80~AlN !, and 0.78~GaN!. Boththe dynamic (Z* ) and static (g) ionicities indicate that GaNis a slightly less ionic material than AlN. This fact is somwhat in contrast to an estimation of the bond polarities.47
V. PHONONS
A. Eigenmodes
Whereas in the cubic case one has to deal only withtwo frequenciesvLO andvTO at the zone centerG, the situ-ation is more complicated for 2H-GaN because of thecreased unit-cell size and the uniaxial symmetry. The laone induces an angular dispersion of the polar-modequencies due to the nonanalyticity of the accompanylong-range electric field. In the wurtzite case with theC6v
4
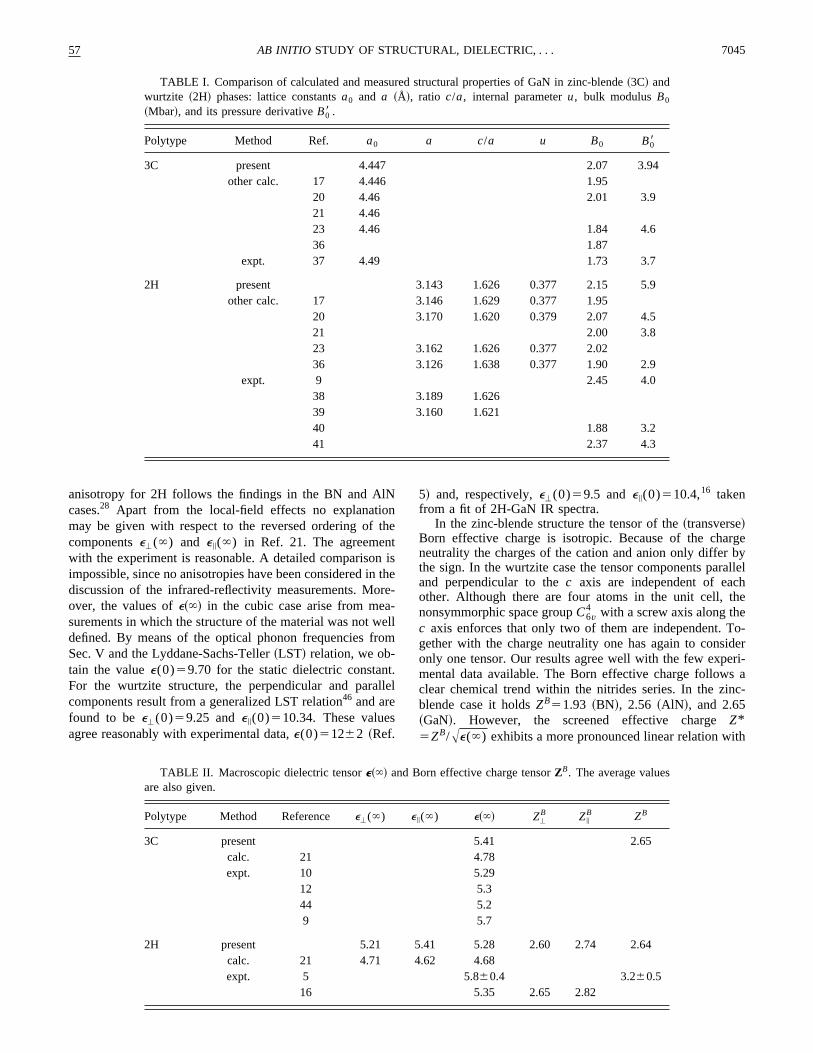
space group, the group theory predicts twoA1 , two E1 , twoE2 , and twoB1 modes with normal propagation. OneA1 andE1 are acoustic vibrations. TheE modes are twofold degenerate. Taking into account the degeneracy the optical moof wurtzite GaN~more strictly the eigenfrequenciesv andthe polarization vectorse of the nine optical modes! are pre-sented in Table III for two propagation directions, parallel~i!and perpendicular~'! to thec axis. Table III makes it obvi-ous that the strong modes, which correspond to TO andin the cubic case, exhibit the mentioned angular dependeAccording to Loudon,48 for the strong modes it holds that
vLO2 ~A1l1E1l !5cos2 Q3v2~A1l !1sin2 Q3v2~E1l !,
vTO2 ~E1t!5v2~E1t!, ~1!
vTO2 ~A1t1E1t!5sin2 Q3v2~A1t!1cos2 Q3v2~E1t!,
TABLE III. Eigenfrequencies and possible eigenvectors of otical G phonons in 2H-GaN for two different propagation directionThe polarization vector of the first Ga atom in the unit cell is listeValues in parentheses are calculated for a crystal with only15Nisotopes.
Mode
i '
v/cm21 e v/cm21 e
E2 143 ~20.704,20.710, 0! 143 ~0, 1, 0!~142! ~20.710, 0.704, 0! ~142! ~21, 0, 0!
B1 337 ~0, 0, 21! 337 ~0, 0, 1!~337! ~337!
TO 568 (E1t) ~20.703,20.711, 0! 541 (A1t) ~0, 0, 21!
~552! ~0.711,20.703, 0! ~526!568 (E1t) ~0, 21, 0!
~552!E2 579 ~0.707, 0.707, 0! 579 ~1, 0, 0!
~562! ~0.707,20.707, 0! ~562! ~0, 21, 0!B1 720 ~0, 0, 21! 720 ~0, 0, 1!
~696! ~696!LO 748 (A1l) ~0, 0, 21! 757 (E1l) ~1, 0, 0!
~727! ~735!
e
-r
e-g
es
Oce.
whereQ is the angle between the phonon propagation dirtion and thec axis. We have checked the analytical angdependence in Eq.~1! by ab initio calculations~Fig. 2! andfound it to be a very accurate approximation for 2H-GaFor normal propagation the two transverseE1t branches aredegenerate. This degeneracy is lifted for propagation perpdicular to thec axis. The second modeA1t is lower in en-ergy. The behavior of the LO mode is reversed. For normpropagation the frequency of theA1l mode is lower than thaof the E1l mode occurring for propagation perpendicularthe c axis. The strength of the angular dispersion andmode splitting represents the strength of the nonanalytiof the long-range electric field.
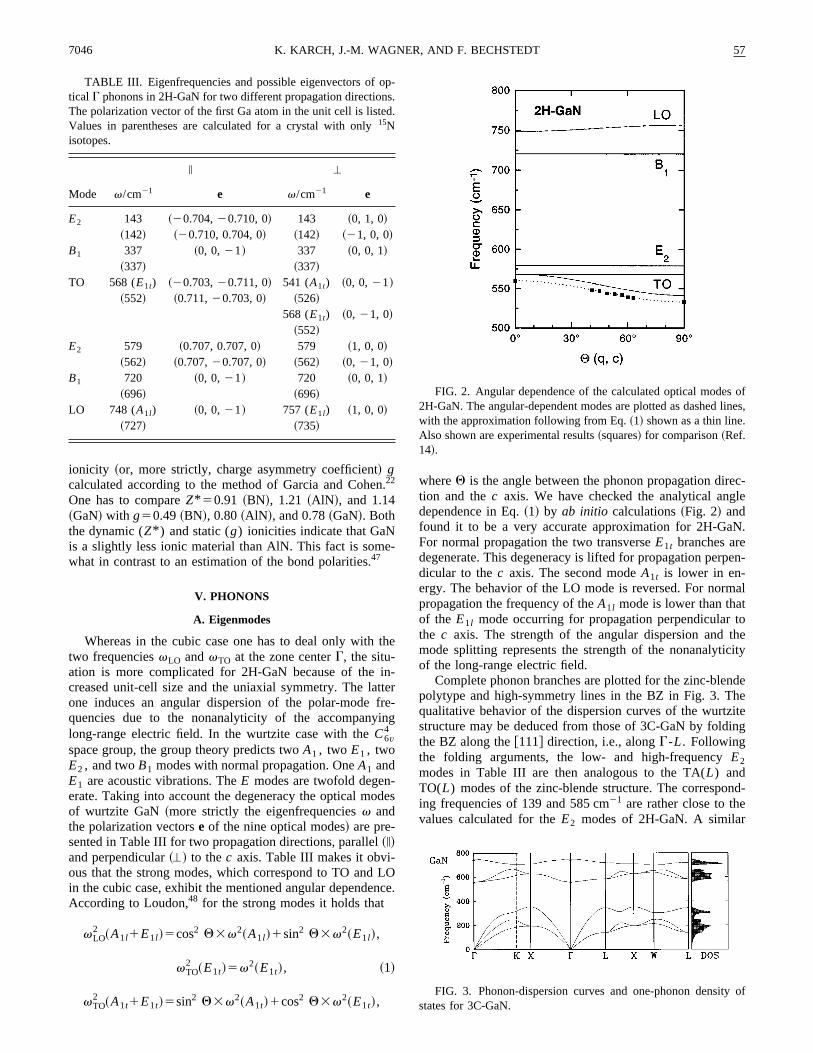
Complete phonon branches are plotted for the zinc-blepolytype and high-symmetry lines in the BZ in Fig. 3. Thqualitative behavior of the dispersion curves of the wurtzstructure may be deduced from those of 3C-GaN by foldthe BZ along the@111# direction, i.e., alongG-L. Followingthe folding arguments, the low- and high-frequencyE2modes in Table III are then analogous to the TA(L) andTO(L) modes of the zinc-blende structure. The correspoing frequencies of 139 and 585 cm21 are rather close to thevalues calculated for theE2 modes of 2H-GaN. A similar
-..
FIG. 2. Angular dependence of the calculated optical mode2H-GaN. The angular-dependent modes are plotted as dashedwith the approximation following from Eq.~1! shown as a thin line.Also shown are experimental results~squares! for comparison~Ref.14!.
FIG. 3. Phonon-dispersion curves and one-phonon densitystates for 3C-GaN.
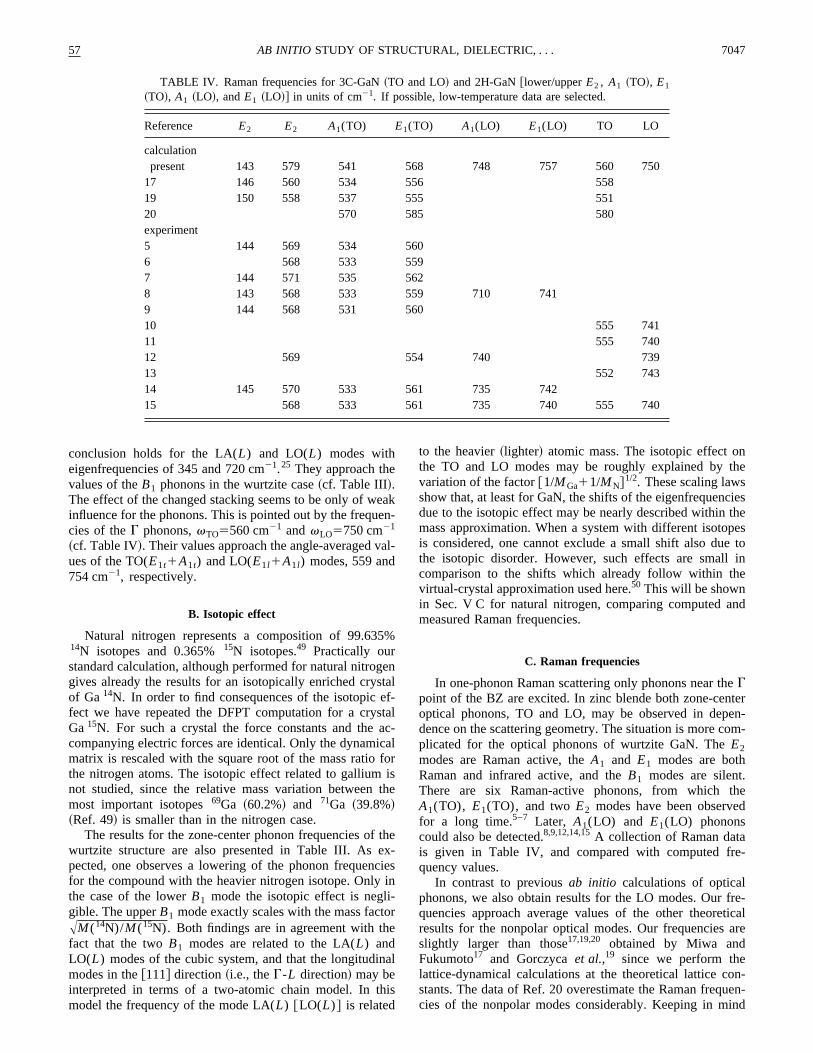
57 7047AB INITIO STUDY OF STRUCTURAL, DIELECTRIC, . . .
TABLE IV. Raman frequencies for 3C-GaN~TO and LO! and 2H-GaN@lower/upperE2 , A1 ~TO!, E1
~TO!, A1 ~LO!, andE1 ~LO!# in units of cm21. If possible, low-temperature data are selected.
Reference E2 E2 A1(TO) E1(TO) A1(LO) E1(LO) TO LO
calculationpresent 143 579 541 568 748 757 560 750
17 146 560 534 556 55819 150 558 537 555 55120 570 585 580experiment5 144 569 534 5606 568 533 5597 144 571 535 5628 143 568 533 559 710 7419 144 568 531 56010 555 74111 555 74012 569 554 740 73913 552 74314 145 570 533 561 735 74215 568 533 561 735 740 555 740
een
a
5%
estaefstacafoith
thxciin
-toe
na
is
nthe
ciesthepese to
ine
nd
eteren-om-
thed
e-
fre-ticalare
n-uen-ind
conclusion holds for the LA(L) and LO(L) modes witheigenfrequencies of 345 and 720 cm21.25 They approach thevalues of theB1 phonons in the wurtzite case~cf. Table III!.The effect of the changed stacking seems to be only of winfluence for the phonons. This is pointed out by the frequcies of theG phonons,vTO5560 cm21 andvLO5750 cm21
~cf. Table IV!. Their values approach the angle-averaged vues of the TO(E1t1A1t) and LO(E1l1A1l) modes, 559 and754 cm21, respectively.
B. Isotopic effect
Natural nitrogen represents a composition of 99.6314N isotopes and 0.365%15N isotopes.49 Practically ourstandard calculation, although performed for natural nitroggives already the results for an isotopically enriched cryof Ga14N. In order to find consequences of the isotopicfect we have repeated the DFPT computation for a cryGa15N. For such a crystal the force constants and thecompanying electric forces are identical. Only the dynamimatrix is rescaled with the square root of the mass ratiothe nitrogen atoms. The isotopic effect related to galliumnot studied, since the relative mass variation betweenmost important isotopes69Ga ~60.2%! and 71Ga ~39.8%!~Ref. 49! is smaller than in the nitrogen case.
The results for the zone-center phonon frequencies ofwurtzite structure are also presented in Table III. As epected, one observes a lowering of the phonon frequenfor the compound with the heavier nitrogen isotope. Onlythe case of the lowerB1 mode the isotopic effect is negligible. The upperB1 mode exactly scales with the mass facAM (14N)/M (15N). Both findings are in agreement with thfact that the twoB1 modes are related to the LA(L) andLO(L) modes of the cubic system, and that the longitudimodes in the@111# direction~i.e., theG-L direction! may beinterpreted in terms of a two-atomic chain model. In thmodel the frequency of the mode LA(L) @LO(L)# is related
ak-
l-
nl
-alc-lr
se
e-es
r
l
to the heavier~lighter! atomic mass. The isotopic effect othe TO and LO modes may be roughly explained byvariation of the factor@1/MGa11/MN#1/2. These scaling lawsshow that, at least for GaN, the shifts of the eigenfrequendue to the isotopic effect may be nearly described withinmass approximation. When a system with different isotois considered, one cannot exclude a small shift also duthe isotopic disorder. However, such effects are smallcomparison to the shifts which already follow within thvirtual-crystal approximation used here.50 This will be shownin Sec. V C for natural nitrogen, comparing computed ameasured Raman frequencies.
C. Raman frequencies
In one-phonon Raman scattering only phonons near thGpoint of the BZ are excited. In zinc blende both zone-cenoptical phonons, TO and LO, may be observed in depdence on the scattering geometry. The situation is more cplicated for the optical phonons of wurtzite GaN. TheE2modes are Raman active, theA1 and E1 modes are bothRaman and infrared active, and theB1 modes are silent.There are six Raman-active phonons, from whichA1(TO), E1(TO), and twoE2 modes have been observefor a long time.5–7 Later, A1(LO) and E1(LO) phononscould also be detected.8,9,12,14,15A collection of Raman datais given in Table IV, and compared with computed frquency values.
In contrast to previousab initio calculations of opticalphonons, we also obtain results for the LO modes. Ourquencies approach average values of the other theoreresults for the nonpolar optical modes. Our frequenciesslightly larger than those17,19,20 obtained by Miwa andFukumoto17 and Gorczycaet al.,19 since we perform thelattice-dynamical calculations at the theoretical lattice costants. The data of Ref. 20 overestimate the Raman freqcies of the nonpolar modes considerably. Keeping in m

%t
n
f
u
tina
a
b
ng
hnar
py,
aNtz-
ns.ivere inDe-ntse
dsndtrichar-siesThisr-theseBN,
thein
ge-bleof
7048 57K. KARCH, J.-M. WAGNER, AND F. BECHSTEDT
the accuracy of our first-principles calculation of about 2our computed frequencies agree well with the average ofmeasured Raman frequencies. This holds especially forE2 and TO modes. The deviations in the LO modes aslightly larger. When we omit theA1(LO) frequency of Ref.8 because of the problem with the sample quality, one fia maximum deviation of 2%. Apart from the lowerE2 modethe theory slightly overestimates the zone-center phononquencies. This fact may be mainly traced back to the undestimation of the lattice constants.
D. Electrostatic forces
The agreement between our theory and the measuremis better for the LO-TO splittings. In the 3C case the calclated valuesv~LO!2v~TO!5190 cm21 ~cf. Table IV! givesan upper limit of the measured data 186,10 185,11,15 and191 cm21.13 For wurtzite 2H-GaN we havev@A1(LO)#2v@A1(TO)#5207 cm21 and v@E1(LO)#2v@E1(TO)#5189 cm21. The Raman data are 177 and 182 cm21,8 202and 181 cm21,14 and, 202 and 179 cm21,15 respectively.These differences characterize the strength of the elecstatic Coulomb forces between the atoms of the vibratlattice. The differences are related to the screened ion chZi /'* 5Zi /'
B /Ae i /'(`), where the parallel component applieto the A1 modes and the orthogonal one to theE1 modes.According to the values listed in Table II, it holds thZ* 51.14 ~3C!, as well asZi* 51.18 andZ'
* 51.14 ~2H!.According to Refs. 48 and 51, the uniaxial crystals can
divided into two classes considering the relative strengththe electrostatic forces over the anisotropy of the short-rainteratomic forces. The anisotropy is given by the splittinv@E1(LO)#2v@A1(LO)#59 cm21 and v@E1(TO)#2v@A1(TO)#527 cm21. These values are much smallethan the LO-TO splittings. Therefore, 2H-GaN belongs tsecond class, where the anisotropy due to the long-raelectrostatic forces is small. The same observation is mfor 2H-BN and -AlN,28 although their anisotropy is simila~BN! or larger ~AlN !. If „v@E1(TO)2v(A1(TO)#…/
,hethere
ds
re-er-
ent-
ro-g
rges
t
eofges
regede
v@E1(TO)# is used as a measure of the crystal anisotroone finds 4.831022. The corresponding values for BN~AlN ! are 4.531022 (8.631022).
VI. CONCLUSIONS
In conclusion, we have presentedab initio calculations ofstructural, dielectric, and lattice-dynamical properties of Gin both relevant crystal modifications, zinc blende and wurite. The effect of the shallow Ga 3d core electrons is in-cluded taking into account the nonlinear core correctioThe results for lattice constants, elastic coefficients, effectcharges, dielectric constants, and Raman frequencies areasonable agreement with experimental data available.spite the well-known underestimation of the lattice constawithin the local version of the density-functional theory, thbulk modulus and the other structural parametersc/a andufor wurtzite agree well with measured values. This holespecially for the tensor of the Born effective charge ahigh-frequency dielectric constant. Since the static dielecconstant is also derived, a complete set of parameters cacterizing the Fro¨hlich interaction of polar optical phononand electrons is given. The computed phonon frequencapproach the values measured by Raman spectroscopy.is especially true for the LO-TO splitting, which characteizes the long-range electrostatic forces on the atoms ofvibrating lattice. We find a nonmonotonous behavior of thesplittings and the screened ion charge along the seriesAlN, and GaN. The ionic bonding in GaN is smaller~larger!than in AlN ~BN!. A similar behavior is observed for theanisotropy of the crystals characterized by the strength ofangular dispersion of the zone-center LO and TO modesthe wurtzite case.
ACKNOWLEDGMENTS
This work was supported by the Deutsche Forschungsmeinschaft under Contract No. Be 1346/8-2. A considerapart of the computations was performed on the Cray-YMPthe Forschungszentrum KFA in Ju¨lich.
on-
.
g,
lz-
d
n,th
*Permanent address: Daimler-Benz Aerospace Dornier GmDept. F4T/T, 88039 Friedrichshafen, Germany.
1S. Nakamura,The Blue Laser Diode–GaN based Light Emittersand Lasers~Springer, Berlin, 1997!.
2M. J. Paisley, Z. Sitar, J. B. Posthill, and R. F. Davis, J. Vac. STechnol. A7, 701 ~1989!.
3O. Brandt, H. Yang, B. Jenichen, Y. Suzuki, L. Da¨weritz, and K.H. Ploog, Phys. Rev. B52, R2253~1995!.
4D. Schikora, M. Hankeln, D. J. As, K. Lischka, T. Litz, A. WaagT. Buhrow, and F. Henneberger, Phys. Rev. B54, R8381~1996!.
5D. D. Manchon, A. S. Barker, P. J. Dean, and R. B. ZetterstroSolid State Commun.8, 1227~1970!.
6V. Lemos, C. A. Argu¨ello, and R. C. C. Leite, Solid State Commun.11, 1351~1972!.
7G. Burns, F. Dacol, J. C. Marinace, B. A. Scott, and E. BursteAppl. Phys. Lett.22, 356 ~1973!.
8A. Cingolani, M. Ferrara, M. Lugara´, and G. Scamarcio, SolidState Commun.58, 823 ~1986!.
9P. Perlin, C. Jauberthie-Carillon, J. P. Itie, A. San Miguel,
bH,
ci.
,
m,
-
in,
I.
Grzegory, and A. Polian, Phys. Rev. B45, 83 ~1992!.10T. Azuhata, T. Sota, K. Suzuki, and S. Nakamura, J. Phys.: C
dens. Matter7, L129 ~1995!.11H. Siegle, L. Eckey, A. Hoffmann, C. Thomson, B. K. Meyer, D
Schikora, M. Hankeln, and K. Lischka, Solid State Commun.96,943 ~1995!.
12M. Giehler, M. Ramsteiner, O. Brandt, H. Yang, and K. H. PlooAppl. Phys. Lett.67, 733 ~1995!.
13A. Tabata, R. Enderlein, J. R. Leite, S. W. da Silva, J. C. Gaerani, D. Schikora, M. Kloidt, and K. Lischka, J. Appl. Phys.79,4137 ~1996!.
14L. Filippidis, H. Siegle, A. Hoffmann, C. Thomsen, K. Karch, anF. Bechstedt, Phys. Status Solidi B198, 621 ~1996!.
15A. Cros, R. Dimitrov, H. Aagerer, O. Ambacher, M. StutzmanS. Christiansen, M. Albrecht, and H. P. Strunk, J. Cryst. Grow181, 197 ~1997!.
16A. S. Barker and M. Ilegems, Phys. Rev. B7, 743 ~1973!.17K. Miwa and A. Fukumoto, Phys. Rev. B48, 7897~1993!.18K. Kim, W. R. Lambrecht, and B. Segall, Phys. Rev. B50, 1502
~1994!.

hy
v.
ys
tate
a-
e-
s
L.
ys.
57 7049AB INITIO STUDY OF STRUCTURAL, DIELECTRIC, . . .
19I. Gorczyca, N. E. Christensen, E. L. Peltzer y Blanca´, and C. O.Rodriguez, Phys. Rev. B51, 11 936~1995!.
20K. Kim, W. R. Lambrecht, and B. Segall, Phys. Rev. B53,16 310~1996!.
21N. E. Christensen and I. Gorczyca, Phys. Rev. B50, 4397~1994!.22A. Garcia and M. L. Cohen, Phys. Rev. B47, 6751~1993!.23A. F. Wright and J. S. Nelson, Phys. Rev. B50, 2159~1994!.24S. G. Louie, S. Froyen, and M. L. Cohen, Phys. Rev. B26, 1738
~1982!.25K. Karch and F. Bechstedt, Phys. Rev. B56, 7404~1997!.26P. Perdew and A. Zunger, Phys. Rev. B23, 5048~1981!.27N. Troullier and J. L. Martins, Phys. Rev. B43, 1993~1991!.28K. Karch and F. Bechstedt, Phys. Rev. B56, 7404~1997!.29D. J. Chadi and M. L. Cohen, Phys. Rev. B8, 5747~1973!.30P. Vinet, J. Ferrante, J. R. Smith, and J. H. Rose, J. Phys. C19,
L467 ~1986!.31F. D. Murnaghan, Proc. Natl. Acad. Sci. USA30, 244 ~1944!.32S. Baroni, P. Giannozzi, and A. Testa, Phys. Rev. Lett.58, 1861
~1987!.33P. Giannozzi, S. de Gironcoli, P. Pavone, and S. Baroni, P
Rev. B43, 7231~1991!.34K. Karch, F. Bechstedt, P. Pavone, and D. Strauch, Phys. Re
53, 13 400~1996!.35A. Dal Corso, S. Baroni, R. Resta, and S. de Gironcoli, Ph
Rev. B47, 3588~1993!.
s.
B
.
36P. E. Van Camp, V. E. Van Doren, and J. T. Devreese, Solid SCommun.81, 23 ~1992!.
37T. Lei, M. Faneiulli, R. J. Molnar, T. D. Moustakas, R. J. Grham, and J. Scandon, Appl. Phys. Lett.59, 944 ~1991!.
38H. P. Maruska and J. J. Tietjen, Appl. Phys. Lett.15, 327~1969!.39O. Lagerstedt and B. Monemar, Phys. Rev. B19, 3064~1979!.40H. Xia, Q. Xia, and A. L. Ruoff, Phys. Rev. B47, 12 925~1993!.41M. Ueno, M. Yoshida, A. Onodera, O. Shimomura, and K. Tak
mura, Phys. Rev. B49, 14 ~1994!.42C.-Y. Yeh, Z. W. Lu, S. Froyen, and A. Zunger, Phys. Rev. B46,
10 086~1992!.43P. Lawaetz, Phys. Rev. B5, 4039~1972!.44E. Ejder, Phys. Status Solidi A5, 445 ~1971!.45V. I. Gavrilenko and F. Bechstedt, Phys. Rev. B55, 4343~1997!.46W. Cochran and R. A. Cowley, J. Phys. Chem. Solids23, 447
~1962!.47W. A. Harrison,Electronic Structure and the Properties of Solid
~Dover, New York, 1989!.48R. Loudon, Adv. Phys.13, 423 ~1963!.49American Institute of Physics Handbook, edited by D. E. Gray
~McGraw-Hill, New York, 1957!.50H. D. Fuchs, C. H. Grein, C. Thomsen, M. Cardona, W.
Hansen, E. E. Haller, and K. Itoh, Phys. Rev. B43, 4835~1991!.51D. W. Feldman, J. H. Parker, W. J. Choyke, and L. Patrick, Ph
Rev.173, 787 ~1968!.