Embed Size (px)
Citation preview

REVISTA ROMÂNÅ DE PEDIATRIE – VOL. LVIII, NR. 2, AN 2009 111
CE SUNT BOLILE GENETICE DE METABOLISM(BGM)?
Bolile genetice de metabolism (BGM) sunt ungrup de afec¡iuni ereditare ce interfereazå cu o seriede cåi metabolice, determinând o func¡ionareinadecvatå a acestor procese speciale. Interferen¡aîn cåile enzimatice sau metabolice normale areconsecin¡e variate, ce includ deficien¡a unuiprodus terminal particular/special sau acumulareaexcesivå a unei substan¡e care poate fi toxicå.Oricare din aceste douå situa¡ii determinå omorbiditate ¿i mortalitate semnificativå prin „afec-
tarea“ func¡iei normale a unei cåi metabolicespeciale.
BGM sunt cunoscute de aproximativ 100 deani, denumirea acestora fiind prima datå folositåde Sir Archibad Garrod în 1902 (Lancet). Entitå¡ileini¡ial descrise au fost: alcaptonuria, pentozuriabenignå, albinismul ¿i cistinuria, urmate apoi dedescrierea unor entitå¡i majore, respectivfenilcetonuria (PKU) de Folling în 1934. De atunciprogresele în medicinå au dus la descoperirea amai mult de 500 de afec¡iuni (Saudubray JM,Chappentier C, 2001).
3REFERATE GENERALE
ABORDAREA CLINICÅ A DIAGNOSTICULUIBOLILOR GENETICE DE METABOLISM
Clinical approach to the diagnosis of inborn errors ofmetabolism (IEM)
Dr. Andrei ZamfirescuClinica de Pediatrie, Spitalul Clinic de Copii „Dr. Victor Gomoiu“, Bucure¿ti
Adreså de coresponden¡å:Dr. Andrei Zamfirescu, Spitalul Clinic de Copii „Dr. Victor Gomoiu“, Bulevardul Basarabia, Nr. 21, Sector 2, Bucure¿ti
REZUMATArticolul trece în revistå: diferitele tipuri de boli genetice de metabolism, frecven¡a acestor entitå¡i, modul detransmisiune ereditarå a acestora, cum se realizeazå diagnosticul BGM (bolilor genetice de metabolism), careeste rolul NS (newborn screening – screening-ul neonatal) program în diagnosticul precoce al BGM ¿ipreven¡ia morbiditå¡ii ¿i mortalitå¡ii acestora, principiile de tratament ¿i måsurile terapeutice specifice în BGM,prognosticul pe termen lung la BGM.
Cuvinte cheie: boli genetice de metabolism, abordare clinicå a diagnosticului; copil
ABSTRACTIn this article the authors present:– what are IEMs?– what are the different types of IEMs?– what is the relative frequency of these IEMs?– what is the inheritance pattern of these IEMs?– how can these IEMs be diagnosed in a timely manner?– what is the role of newborn screening (NS) programe in early diagnosis and prevention of morbidity and
mortality?– what are the treatment and management options for these IEMs?– what is the long-term prognosis for patients with IEMs?
Key words: inborn errors of metabolism; clinical approach to the diagnosis; child

112 REVISTA ROMÂNÅ DE PEDIATRIE – VOL. LVIII, NR. 2, AN 2009
CARE SUNT DIFERITELE TIPURI DE BGM?
Unele dintre BGM comune sunt prezentate întabelul 1.
Tabelul 1. Tipuri comune de BGM (dupå Kamboj M,2008)
Anomalii ale metabolismului glucidelor:– Boala de tezaurizare glicogenicå– GalactozemiaAnomalii ale metabolismului aminoacizilor (amino-
acidopatii):– Fenilcetonuria (PKU)– Leucinoza (Maple syrup urine disease-MSUD)– Homocistinuria– Tirozinemia– Hiperglicinemia noncetoticåAnomalii ale metabolismului acizilor organici
(acidemii organice):– Acidemia metilmalonicå– Acidemia propionicåAnomalii ale oxidårii acizilor gra¿i:– deficit al acyl CoA dehidrogenazei acizilor gra¿i
cu lan¡ scurt (SCAD)– deficit al acyl CoA dehidrogenazei acizilor gra¿i
cu lan¡ mediu (MCAD)– deficit al acyl CoA dehidrogenazei acizilor gra¿i
cu lan¡ lung (LCAD)– deficit al acyl CoA dehidrogenazei acizilor gra¿i
cu lan¡ foarte lung (VLCAD)Anomalii ale metabolismului mitocondrial:– MELAS (miopatie mitocondrialå, encefalopatie,
acidozå lacticå, stroke-like)– Aciduria glutatricå– Deficien¡a de piruvat dehidrogenazåAnomalii ale ciclului ureei:– Deficien¡a de carbamyl fosfat sintetazå– Deficien¡a de ornitin transcarbamilazå– Deficien¡a de argininosuccinatAnomalii peroxizomale:– Adrenoleucodistrofia– Sindromul Zellweger– Condrodistrofia punctatå– Boala Refsum de tip adultAnomalii ale cåii steroidiene:– Hiperplazia adrenalå congenitalå– Sindromul Smith-Lemli-OpitzAnomalii de tezaurizare a lipidelor:– Boala Tay-Sachs– Boala Gaucher– Leucodistrofia metacromaticåAnomalii ale metabolismului purinelor– Sindromul Lesch-NyhanAnomalii de transport:– Cistinoza– HipercolesterolemiaAnomalii lizozomale:– Mucopolizaharidoze (MPS)– MPS I (bolile Hurler ¿i Scheie)– MPS II (boala Hunter)– MPS III (boala Sanfilippo)– MPS IV (boala Morquio)– MPS VI (boala Maraoteux-Lamy)
– MPS VII– Glicoproteinoze– Sfingolipidoze– Anomalii combinate– Boala celularå IAnomalii ale metabolismului metalelor:– Boala Wilson– Hemocromatoza– Boala MenkesAlte BGM:– Hipotiroidismul– Hemoglobinopatiile– Sindromul MELAS
CARE ESTE FRECVENºA RELATIVÅ AINCIDENºEI BGM?
Inciden¡a BGM este foarte variabilå în cadruldiverselor entitå¡i clinice specifice, variind de la1/400 la afro-americanii din SUA pentru hemo-globinopatii, la 1/45.000 pentru cele mai multedintre anomaliile acizilor gra¿i (cu excep¡iaMCAD) ¿i acidemiile organice. Inciden¡a unorBMG comune este prezentatå în tabelul 2.
CARE ESTE „PATTERN“-UL DE TRASMITERE ABGM?
Sunt posibile diverse modalitå¡i de transmitereîn BGM. Este important de a detalia arborele ge-nealogic (pedigree) la a treia – a patra genera¡ie,pentru a evalua cu acurate¡e modul de transmitere.
Transmiterea autozomal recesivå (AR) este mo-dalitatea cea mai comunå de transmitere în BGM.Consanguinitatea are o ¿anså crescutå de expresiea unei transmiteri AR. Rar, aceste muta¡ii pot apåreade novo (Clarke JTR, 2006).
Transmiterea recesiv X-linkatå poate, de ase-menea, så existe în unele BGM, în care o „copie“a genei mutante pe cromozomul X este suficientåpentru a „cauza“/determina anomalia. De aceea,
Tabelul 2. Inciden¡a unor BGM

113REVISTA ROMÂNÅ DE PEDIATRIE – VOL. LVIII, NR. 2, AN 2009
în acest mod de transmisiune, anomalia geneticåprovine de la o mamå „purtåtoare“ ¿i afecteazåcopiii de sex masculin. De asemenea, muta¡iile denovo sunt observate cu o mai mare inciden¡å înacest „model“ de transmisiune (Clarke JTR, 2006).
Transmisiunea autozomal dominantå (AD) esteo modalitate de transmisiune mai pu¡in comunåîn BGM. Inciden¡a muta¡iilor de novo, ce determinåanomalii AD, este mult mai ridicatå decât în altemodele de transmisiune.
Transmisiunea AD înseamnå, în general, cåunul dintre pårin¡i are boala; 50% dintre descenden¡iau aceea¿i anomalie ¿i existå o distribu¡ie egalå laambele sexe (Clarke JTR, 2006).
Un mod mitocondrial de transmisiune este, deasemenea, våzut în unele BGM. Este interesantde notat cå ADN-ul mitocondrial este totdeaunanatural ca origine; prin urmare, o muta¡ie în ADN-ulmitocondrial este transmiså numai de la mamå.ADN-ul mitocondrial este „înclinat“ (prone) sprenoi muta¡ii; ca urmare, bolile transmise în acestmod pot fi sporadice ca inciden¡å (Clarke JTR,2006).
Tabelul 3 prezintå modelele de transmisiune aBGM comune.
Tabelul 3. Modalitå¡i de transmisiune a unor BGMcomune (dupå Kamboj M, 2008)
Transmisiune AR:– Fenilcetonurie (PKU)– Leucinoza (MSUD – Maple syrup urine disease)– Boala de tezaurizare glicogenicå– Galactozemie– Aciduriile organice– Deficit al AcylCoA dehidrogenazei acizilor gra¿i
cu lan¡ mediu (MCAD)– Sindromul ZellwegerTransmisiune X-linkatå:– Deficien¡a de ornitin carbamilazå– Maladia Fabry– Deficien¡a de piruvat dehidrogenazåTransmisiune AD:– Sindromul Marfan– Porfiria intermitentå– Hipercolesterolemia familialåTransmisiune mitocondrialå:– Sindromul Kearns-Sayre– Sindromul Leigh
CUM SE POATE DIAGNOSTICA O BGM ÎN TIMPUTIL
Semne ¿i simptome clinice
BGM pot fi prezente precoce în perioada denou-nåscut, mai tardiv în copilåria precoce sau mai
târziu în perioada de adult. Un index crescut desuspiciune a unei BGM este necesar de a fi pus îndiscu¡ie, deoarece simptomatologia unora dintreBGM este adesea nespecificå ¿i, ca urmare, poateconduce la cercetarea altor entitå¡i medicale. Ta-bloul clinic al unei BGM poate fi foarte variat ¿ipoate fi subclasificat într-o serie de categorii largi(tabelul 4) (Burton BK, 1998; SuttonVR, 2008).
Tabelul 4. Semne ¿i simptome importante în BGM(dupå Kamboj M, 2008)
• Dismorfism:– Boli peroxizomale– Sindromul Zellweger– Boli de stocaj lizozomal– Mucopolizaharidoze– Mucolipidoze– Gangliozidoze– Homocistinuria– Sindromul Smith-Lemli-Opitz• Manifeståri neurologice:– Convulsii – în orice BGM datoritå metaboli¡ilor
toxiciPiridoxino-dependen¡aAcid-folinic-dependen¡aSecundare hipoglicemiei:– Neuropatie perifericåAnomaliile mitocondrialeAnomaliile de tezaurizare lizozomalå:– HipotonieAnomalii ale oxidårii acizilor gra¿iAnomaliile peroxizomaleAnomaliile ciclului ureeiAnomalii mitocondrialeBoala PompeAnomalii ale tezaurizårii glicogenice:– MiopatieBolile de stocaj glicogenicAnomaliile oxidårii acizilor gra¿iAnomalii mitocondriale:– AtaxieAnomaliile peroxizomaleAnomaliile mitocondrialeAnomaliile de stocaj lizozomal:– Letargie sau comåAminoaciduriileAcidemiile organiceAnomaliile ciclului ureeiAnomaliile de oxidare a acizilor gra¿iAnomalii mitocondriale• Manifeståri gastrointestinale– Hepatomegalie ¿i/sau splenomegalieBolile de tezaurizare lizozomaleBolile de tezaurizare glicogenicå:– Icter sau disfunc¡ie hepaticåGalactozemieAnomaliile de oxidare a acizilor gra¿iTirozinemiaAnomaliile peroxizomaleDeficitul de alfa-1-antitripsinåBoala Niemann-Pick• Manifeståri cardiace

114 REVISTA ROMÂNÅ DE PEDIATRIE – VOL. LVIII, NR. 2, AN 2009
– Cardiomiopatia hipertroficå– Boala de tezaurizare glicogenicå tipul 2– Boala Pompe– Mucopolizaharidoze– Cardiomiopatie dilatativå– Anomaliile de oxidare a acizilor gra¿i– Aciduriile organice– Anomaliile mitocondriale• Manifeståri oftalmologice:– Cataractå lenticularåGalactozemiaAnomaliile mitocondriale:– Opacitå¡i corneeneBoala FabryMucopolizaharidozeCistinoza:– Pata ro¿ie-cire¿ie macularåBoala Tay-SchsGalactosialidozaBoala Niemann-PickGangliozidoza GM1:– Dislocarea cristalinuluiHomocistinuriaSindromul MarfanDeficien¡a cofactorului MolibdenuluiDeficien¡a de sulfit oxidazå:– Retinita pigmentaråAbetalipoproteinemiaAnomaliile peroxizomaleAnomaliile mitocondriale• Hidrops fetalis nonimun:– Anomaliile de tezaurizare lizozomalå– Anomaliile mitocondriale– Hemocromatoza neonatalå– Boala de tezaurizare glicogenicå tip IV• Manifeståri dermatologice:– Rash cutanatAcrodermatita enteropaticå:– IchtiozåIchtiosis X-linkatåSindromul Sjögren-Larson:– AngiokeratoameBoala Fabry:Anomaliile de stocaj lizozomal
ANOMALII CU DEBUT PRECOCE – ÎN PERIOADANEONATALÅ
Acestea sunt prezente în perioada neonatalå ¿ipot – în continuare – fi subgrupate în urmåtoarelecategorii: tulburåri u¿oare, tulburåri traduse printr-untablou de encefalopatie metabolicå acutå, tulburåricaracterizate prin acidozå metabolicå; tulburåricaracterizate prin hiperamoniemie ¿i tulburåriprezente mai tardiv sau în copilårie.
Tulburårile cu debut precoce, ce apar în pe-rioada neonatalå, sunt subgrupate în urmåtoarelecategorii:
– Tulburåri u¿oare (silent disorders): BGM dinaceastå categorie nu determinå manifeståri
amenin¡åtoare de via¡å ¿i simptome în perioadade sugar, dar se prezintå mai târziu în perioadacopilåriei precoce cu retard mental ¿i întârziereîn procesul de dezvoltare. În acest grup suntincluse: fenilcetonuria ¿i hipotiroidismulcongenital (Burton BK, 1998).
– Tulburåri cu tablou de encefalopatie meta-bolicå. În acest grup sunt incluse tulburårileîn ciclul ureei, acidemiile organice ¿i amino-aciduriile. Aceste entitå¡i prezintå tulburårimetabolice determinate de acumularea pre-cursorilor sau metaboli¡ilor, care sunt expri-ma¡i precoce în perioada neonatalå prin:inapeten¡å, letargie, vårsåturi persistente,convulsii, hipotonie, apnee, insuficien¡å res-piratorie, tahipnee ¿i tahicardie. Acestemanifeståri sunt atribuite efectului toxic lametaboli¡ilor asupra SNC, realizând untablou de encefalopatie metabolicå. Manifes-tårile biochimice sunt semnificative pentruacidoza metabolicå, hiperamoniemie saualte anomalii metabolice (Popescu V, 2008).
– Tulburåri/manifeståri ce se prezintå cu aci-dozå metabolicå. Acest grup include, în ge-neral, acidemiile organice. Ace¿ti nou-nåscu¡i prezintå acidozå metabolicå severåcu o cre¿tere a „gåurii anionice”; acidozalacticå este prezentå în deficien¡a de piruvatdehidrogenazå, anomaliile gluconeogenezei,deficien¡a de piruvat carboxilazå ¿i înanomaliile mitocondriale (Sutton VR, 2008).
– Tulburåri/manifeståri ce se prezintå cu ta-blou de hiperamoniemie. Mu¡i nou-nåscu¡icu anomalii în ciclul ureei, acidemii orga-nice ¿i hiperamoniemie tranzitorie a nou-nåscutului se prezintå cu tablou de encefa-lopatie metabolicå ¿i hiperamoniemie.
ANOMALII CE SE PREZINTÅ MAI TÂRZIU ÎNCOPILÅRIE
Acest grup de BGM include: anomaliile de te-zaurizare lizozomalå, boala Tay-Sachs, boalaGaucher ¿i leucodistrofia metacromaticå. Acesteentitå¡i se prezintå, în general, cu deteriorare neuro-logicå progresivå (Batshaw ML et al, 2002).
Alte semne ¿i simptome clinice, incluzând mani-feståri generalizate nespecifice sunt: semne ¿isimptome neurologice; tulburåri de dezvoltare;fenotipuri dismorfice; tulburåri gastrointestinale,hematologice ¿i dermatologice (a se vedea tabelul 4).
Una dintre cele mai speciale ¿i discutabile mani-feståri ale BGM este prezen¡a unor mirosuri spe-cifice întâlnite într-o serie de BGM (tabelul 5).

115REVISTA ROMÂNÅ DE PEDIATRIE – VOL. LVIII, NR. 2, AN 2009
CUM SE FACE UN DIAGNOSTIC CORECT
Tabloul clinic trebuie så ridice suspiciunea uneiBGM. Detaliile anamnestice, incluzând istoriculfamilial de consanguinitate, anomalii similare înfamilie, istoricul unor decese neonatale trebuie såfie cercetate.
Semnele clinice eviden¡iate la examinarea pesisteme ¿i aparate, ¿i anume hepato-splenomegalie,leziuni cutanate, deficitele neurologice, trebuie såghideze cåtre investiga¡ii paraclinice.
Investiga¡iile generale de laborator sunt pre-zentate în tabelul 6 (Burton BK, 1998; Sutton VR,2008).
Tabelul 6. Investiga¡ii de laborator in¡iale în BGM
Sânge:– Hemograma– Explorarea func¡iei hepatice ¿i renale– Electroli¡ii– Acidul uric– Studiul gazelor în sângele arterial– AmoniemiaUrinå:– Examenul de urinå– pH– culoare– miros– densitate– corpi cetonici– substan¡e reducåtoare în urinå
Analize biochimice specifice vor fi efectuatepe baza datelor din istoric, tabloul clinic, a inves-tiga¡iilor de laborator preliminare ¿i a suspiciuniiunei BGM specifice. O a doua rundå de investiga¡iide laborator poate fi efectuatå, în cazul cå estenecesarå (tabelul 7).
Tabelul 7. Investiga¡ii de laborator adi¡ionale în BGM
Sânge sau plasmå:– Examenul cantitativ al aminoacizilor– Lactat– Aldolaza, creatinkinaza– Acylcarnitina (profil)– Lipide (profil)Urinå:– Examenul cantitativ al aminoacizilor– Acizii organici– MioglobinaImagistica:– MRI – sistemul nervos– EcocardiogramaBiopsii:– muscularå– cutanatåStudii genetice:– cu indica¡ii specifice
Precau¡ii speciale pot fi necesare în recoltarea¿i procesarea unora dintre aceste specimene/mostre.Mostrele/specimenele pentru amoniacul, lactatul¿i piruvatul seric trebuie så fie recoltate fårå utili-zarea garoului ¿i este necesar så fie transportate„în ghea¡å“ pentru o analizå imediatå în laborator[specimenele/mostrele/e¿antioanele de piruvattrebuie colectate pe „perclorat“, pentru a prevenidegradarea sa (Sutton VR, 2008).
Tabelul 8 prezintå corela¡ia dintre dateleparaclinice cu diverse BGM.
CARE ESTE ROLUL PROGRAMULUI DESCREENING NEONATAL ÎN DIAGNOSTICUL
PRECOCE ªI ÎN PREVENIREA MORBIDITźII ªIMORTALITźII?
Screening-ul neonatal pentru depistarea unorboli genetice este efectuat în condi¡iile unor criterii:boala geneticå puså în discu¡ie trebuie så prezinte
Tabelul 5. Mirosuri caracteristice unor BGM
116 REVISTA ROMÂNÅ DE PEDIATRIE – VOL. LVIII, NR. 2, AN 2009
o morbiditate sau mortalitate seminifcativå, så prezinteun mecanism patogenic cunoscut, så constituie unmijloc poten¡ial de preven¡ie sau så beneficieze de untratament adecvat; så beneficieze de o testare u¿oarå¿i rapidå de screening, så fie cost-eficientå.
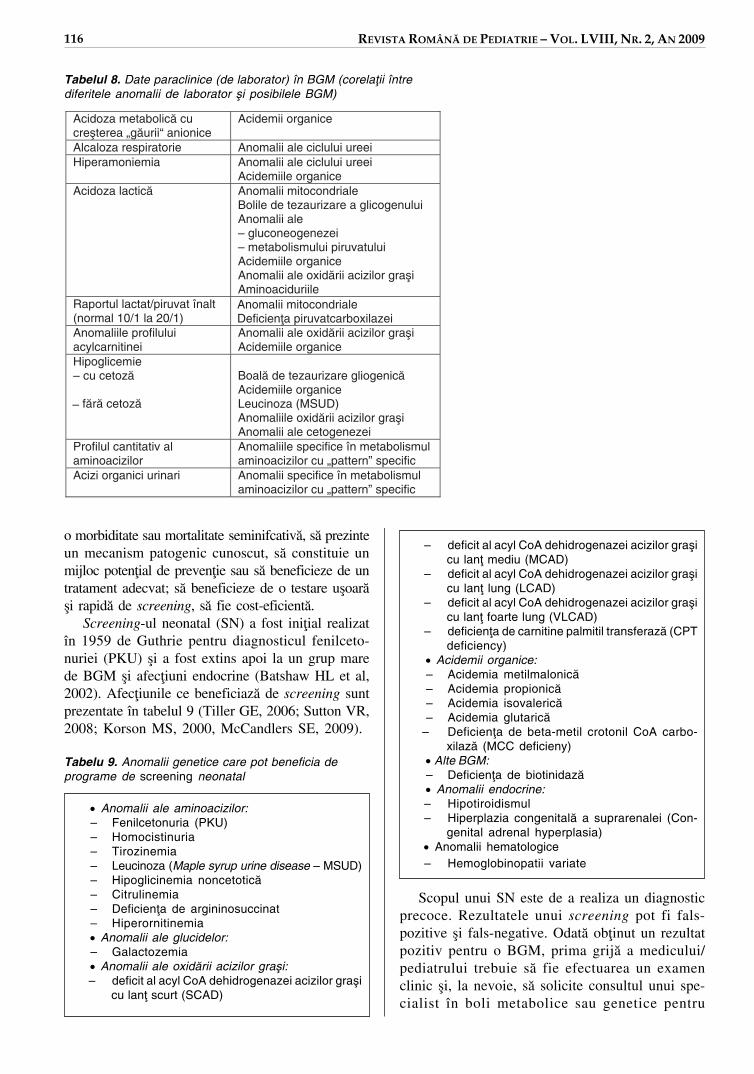
Screening-ul neonatal (SN) a fost ini¡ial realizatîn 1959 de Guthrie pentru diagnosticul fenilceto-nuriei (PKU) ¿i a fost extins apoi la un grup marede BGM ¿i afec¡iuni endocrine (Batshaw HL et al,2002). Afec¡iunile ce beneficiazå de screening suntprezentate în tabelul 9 (Tiller GE, 2006; Sutton VR,2008; Korson MS, 2000, McCandlers SE, 2009).
Tabelu 9. Anomalii genetice care pot beneficia deprograme de screening neonatal
• Anomalii ale aminoacizilor:– Fenilcetonuria (PKU)– Homocistinuria– Tirozinemia– Leucinoza (Maple syrup urine disease – MSUD)– Hipoglicinemia noncetoticå– Citrulinemia– Deficien¡a de argininosuccinat– Hiperornitinemia• Anomalii ale glucidelor:– Galactozemia• Anomalii ale oxidårii acizilor gra¿i:– deficit al acyl CoA dehidrogenazei acizilor gra¿i
cu lan¡ scurt (SCAD)
– deficit al acyl CoA dehidrogenazei acizilor gra¿icu lan¡ mediu (MCAD)
– deficit al acyl CoA dehidrogenazei acizilor gra¿icu lan¡ lung (LCAD)
– deficit al acyl CoA dehidrogenazei acizilor gra¿icu lan¡ foarte lung (VLCAD)
– deficien¡a de carnitine palmitil transferazå (CPTdeficiency)
• Acidemii organice:– Acidemia metilmalonicå– Acidemia propionicå– Acidemia isovalericå– Acidemia glutaricå
– Deficien¡a de beta-metil crotonil CoA carbo-xilazå (MCC deficieny)
• Alte BGM:– Deficien¡a de biotinidazå• Anomalii endocrine:– Hipotiroidismul– Hiperplazia congenitalå a suprarenalei (Con-
genital adrenal hyperplasia)• Anomalii hematologice– Hemoglobinopatii variate
Scopul unui SN este de a realiza un diagnosticprecoce. Rezultatele unui screening pot fi fals-pozitive ¿i fals-negative. Odatå ob¡inut un rezultatpozitiv pentru o BGM, prima grijå a medicului/pediatrului trebuie så fie efectuarea un examenclinic ¿i, la nevoie, så solicite consultul unui spe-cialist în boli metabolice sau genetice pentru
Tabelul 8. Date paraclinice (de laborator) în BGM (corela¡ii întrediferitele anomalii de laborator ¿i posibilele BGM)

117REVISTA ROMÂNÅ DE PEDIATRIE – VOL. LVIII, NR. 2, AN 2009
confirmarea diagnosticului sau infirmarea sa ¿i,în caz pozitiv, pentru stabilirea måsurilor terapeuticeadecvate.
Pacien¡ii vor fi urmåri¡i în continuare, preferabilde speciali¿ti în boli metabolice. O terapie instituitåprecoce poate influen¡a pozitiv morbiditatea ¿imortalitatea prin BGM.
CARE SUNT PRINCIPIILE DE TRATAMENT ÎNBGM?
Principii generale de tratament
Måsurile generale de tratament sunt instituiteînainte de stabilirea unui diagnostic definitiv alunei boli specifice de metabolism. Unele din acesteinterven¡ii includ ab¡inerea de la orice aport oraldietetic pânå ce pot fi stabilite unele investiga¡iispecifice de ghidare a atitudinii, de preferat dupåun consult cu un specialist în boli metabolice. Înfoarte multe cazuri, poate fi ini¡iatå o perfuzie intra-venoaså cu solu¡ie glucozatå ¿i salinå dupå ce înprealabil s-a efectuat o hidratare cu solu¡ie salinånormalå. Acidoza poate necesita, de asemenea,corec¡ie cu solu¡ie de bicarbonat de sodiu (KwonKT et al, 2007).
Måsuri terapeutice specifice
Op¡iunile terapeutice specifice bolii suntinstituite dupå ce a fost diagnosticatå BGM. În
figura 1 este reprezentatå schema terapeuticåadaptatå tipului de BGM. În tabelul 10 esteprezentatå op¡iunea terapeuticå specificådisponibilå pentru variatele BGM (Batshaw MLet al, 2002).
Legenda figurilor: Tratamentul poate fi dirijat spre (1) limitareaaportului de compu¿i toxici poten¡iali; (2) suplimentarea produsuluideficient; (3) stimularea unei cåi metabolice alternative; (4) pro-curarea unui cofactor vitaminic pentru stimularea activitå¡ii enzimeireziduale; (5) suplimentare enzimaticå; (6) transplantarea unui ¡esut/organ con¡inând enzima deficitarå; (7) terapia genicå
Figura 1. Abordarea tratamentului în BGM (dupåBatshaw ML et al, 2002)
Tabelul 10. Exemple de abordåri terapeutice în BGM (dupå Batschaw ML, 2002)

118 REVISTA ROMÂNÅ DE PEDIATRIE – VOL. LVIII, NR. 2, AN 2009
CARE ESTE PROGNOSTICUL PE TERMENLUNG?
Evolu¡ia clinicå a copiilor cu BGM depinde defactori multipli. Ace¿tia includ: severitatea ano-maliei metabolice de bazå, capacitatea de a efectuadiagnosticul precoce, disponibilitatea unor op¡iuniterapeutice adecvate ¿i instituirea adecvatå a må-surilor terapeutice. În func¡ie de to¡i ace¿ti factori,
BIBLIOGRAFIE
1. Batshaw ML, Tuchman M – PKU and others inborn errors ofmetabolism. In: Batshaw ML (ed) Children with disabilities, 5th ed,p 333-345, Paul H Brookes Publishing Co, Baltimore (MD) 2002.
2. Burton BK – Inborn errors of metabolism in infancy: a guide todiagnosis. Pediatrics, 1998, 102(6), E69.
3. Clarke JTR – General principles. In: A clinical guide to inheritedmetabolis diseases, 3rd ed, p 1-27, Cambridge University Press,New York, 2006
4. Garrod AE – The incidence of alcaptonuria: a study in chemicalindividuality.Lancet, 1902, 2, 1616-1620.
5. Kamboj M – Clinical approach to the diagnosis of inborn errors ofmetabolism. Pediatr Clin N Am, 2008 55, 1119-1127.
6. Korson MS – Advances in newborn screening for metabolicdisorders: what the pediatrician needs to know. Pediatr Ann, 2000,29(5), 294-301.
7. Kwon KT, Tsai VW – Metabolic emergencies. Emerg Med ClinNorth Am, 2007, 25(4), 1041-1060.
8. McCandless SE – A primes on expanded newborn screening bytadem mass spectroscopy. Prim Care, 2004, 31(3), 583-604.
unele BGM au un prognostic relativ mai bun decâtaltele. Mul¡i dintre ace¿ti copii pot avea o via¡ålungå, dar mul¡i pot fi cu risc crescut pentru dez-voltarea progresivå a unor deficite neurologice,dizabilitå¡i în procesul de instruc¡ie ¿i retard mental.
Un studiu efectuat pentru evaluarea råspunsuluila tratament în BGM a eviden¡iat o ameliorare a para-metrilor clinici la aproximativ jumåtate dintre pa-cien¡ii care prezintå o BGM (Treacy E et al, 1995).
9. Popescu V – Abordarea clinico-paraclinicå în BGM. Când sesuspecteazå sau nu o BGM. Actualitå¡i în Pediatrie, vol I, cap2, p15-35, Ed Amaltea, Bucure¿ti, 2006.
10. Saudubray JM, Chappentier C – Clinical phenotypes diagnosis/algorithms. In: Servier CR, Beaudet AL, Sly WS et al (eds)Metabolic and molecular basis of inherited diseases, 8th ed, p 1327-1403, McGraw-Hill, New York, 2001.
11. Sielski LA – Newborn screening. Up-to-date. Available at: www:uptodate. com. Accessed March, 2008.
12. Sutton VR – Overview of the evaluation of inborn errors ofmetabolism. Up to date. Available at www. up to date.com.Accessed March 2008.
13. Tiller GE – Inborn errors of metabolism. In: Sabella C,Cunningham RJIII (eds) ntensive review of pediatrics, 2nd ed, p353-361, Lippincott Williams and Wilkins, Philadelphia, 2006.
14. Treacy E, Childs B, Servier CR – Response to treatment inhereditary metabolic diseases: 1993 survey and 10-year comparison(review). Am J Hum Genet, 1996, 56(2), 359-367.





























