Embed Size (px)
Citation preview

American Journal of Medical Genetics 135A:186–189 (2005)
Clinical ReportAdams–Oliver Syndrome and Hepatoportal Sclerosis:Occasional Association or Common Mechanism?Muriel Girard,1 Jeanne Amiel,4 Monique Fabre,2 Daniele Pariente,3 Stanislas Lyonnet,4*and Emmanuel Jacquemin1**1Pediatric Hepatology Bicetre University Hospital, AP-HP, Paris France2Pathology, Bicetre University Hospital, AP-HP, Paris France3Pediatric Radiology, Bicetre University Hospital, AP-HP, Paris France4Department of Genetics, Medical Genetics, Necker-Enfants Malades University Hospital, AP-HP, Paris France
Adams–Oliver syndrome (AOS) is characterizedby the association of scalp and skull defects andabnormalities of terminal limbs. Congenital heartmalformations have also been reported. Hepato-portal sclerosis (HPS) is a rare cause of portalhypertension in children characterized by ab-normalities of intra-hepatic portal veins, portalfibrosis, and nodular regeneration. Etiopathogen-esis of these rare disorders remains unclear, butthe hypothesis of vascular thrombotic mechanismhas been suggested. Association of both syn-dromes has been reported in only one child. Wenow report on two unrelated children with AOSand HPS, one child harboring a factor V Leidenmutation. We hypothesize that the association ofboth disorders may not be fortuitous and rein-forces the idea that AOS and HPS may share avascular thrombotic mechanism.� 2005 Wiley-Liss, Inc.
KEY WORDS: Adams–Oliver syndrome; hepa-toportal sclerosis; portal hy-pertension; pulmonary arterialhypertension; thrombosis; factorV Leiden; children
INTRODUCTION
Adams–Oliver syndrome (AOS, OMIM 100300) was firstdescribed in 1945 [Adams and Oliver, 1945]. It is a rarecondition characterized by the combined occurrence of scalpand skull defects: aplasia cutis congenita and terminal limbsabnormalities. The most frequent limb malformations aresyndactyly (bony/cutaneous), brachydactyly, poly and oligo-dactyly, and hypoplasic finger and toe nails [Kuster et al.,1988]. Congenital heart defects (ventricular and atrial septaldefects, tetralogy of Fallot, coarctation of the aorta, bicuspidaortic valve, pulmonary venous stenosis and unspecified
congenital heart disease) are associated and have beenreported in 15 out of 112 cases of AOS [Zapata et al., 1995].AOS is mostly inherited as an autosomal dominant trait[Bonafede and Beighton, 1979], but an autosomal recessivemode of inheritance has also been suggested [Koiffmann et al.,1988]. Whenever AOS is dominantly inherited, expression andpenetrance are highly variable [Bamsforth et al., 1994]. Thepathogenesis of AOS is still unclear. It is suggested that vas-cular abnormalities and thrombotic mechanism are involved inAOS [Hoyme et al., 1982; Toriello et al., 1988; Keymolen et al.,1999]. ALX4 and MSX2 genes, both involved in vasculardevelopment, have been considered candidate genes in AOSand further excluded [Verdyck et al., 2003].
Hepatoportal sclerosis (HPS), also referred as idiopathicportal hypertension or non-cirrhotic portal fibrosis, is a rarecause of portal hypertension in childhood. It was first andmainly described in adults [Mikkelsen et al., 1965], and fewreports exist in children [Carson et al., 1981; Maksoud et al.,1986; Hessel et al., 1997; Dhiman et al., 2002]. HPS is ofintrahepatic origin in its typical form, although it may beassociated with extrahepatic portal vein obstruction. Bycontrast, in isolated (primary) extrahepatic portal vein throm-bosis, as a cause of portal hypertension, there is no underlyingliver disease, and portal hypertension is entirely prehepatic[Alonso et al., 1997]. Histology represents the gold standardmethod for HPS diagnosis. It is likely the consequence ofobliteration of small-sized portal vein radicles, leading to thecollapse of the underlying parenchyma with zonal scarring andnodular regeneration with a pseudocirrhotic pattern. Typi-cally, liver histology shows portal/septal fibrosis and injuries ofsmall distal portal veins: subintimal sclerosis, thickening of thevessel walls, loss of intrahepatic portal veins in the portaltracts with presence of dilated neocapillaries and neoveinulesin place of the host veins. Liver function tests are usually onlyslightly disturbed. In most cases, evolution is characterizedby portal hypertension leading to gastrointestinal bleeding[Dhiman et al., 2002]. If hepatopulmonary syndrome developsa liver transplantation is needed [de Marsillac et al., 2000].Familial cases have been reported [Barnett et al., 1992] as theassociation with Turner syndrome [Ledinghen et al., 1994;Roulot et al., 2004] and chemotherapy [de Marsillac et al., 2000].
Association of AOS and HPS has been reported in only onechild [Swartz et al., 1999]. We now report on two unrelatedchildren, with AOS and HPS. These data reinforce the ideathat AOS and HPS can result from a common vascular and/orthrombotic pathological mechanism.
CLINICAL REPORT
Patient 1
A female of French origin is the second of two sisters born tonon-consanguinous healthy parents. Family history is unre-markable. She was born at 41 weeks of gestation with a birth
*Correspondence to: Stanislas Lyonnet, Departement deGenetique Medicale, Centre Hospitalier Universitaire Necker-Enfants Malades, 149, rue de Sevres, 75015 Paris, France.E-mail: [email protected]
**Correspondence to: Emmanuel Jacquemin, HepatologiePediatrique, Centre Hospitalier Universitaire de Bicetre, 78, ruedu General Leclerc, 94275 Le Kremlin Bicetre Cedex, France.E-mail: [email protected]
Received 20 July 2004; Accepted 16 December 2004
DOI 10.1002/ajmg.a.30724
� 2005 Wiley-Liss, Inc.
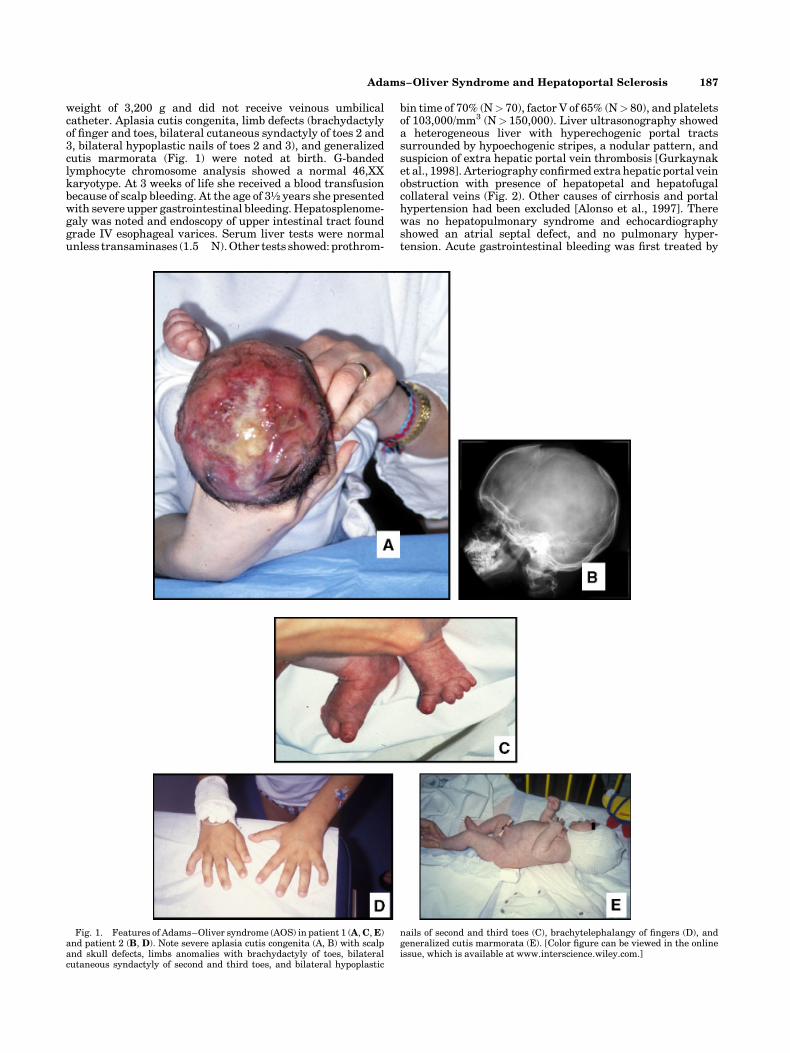
weight of 3,200 g and did not receive veinous umbilicalcatheter. Aplasia cutis congenita, limb defects (brachydactylyof finger and toes, bilateral cutaneous syndactyly of toes 2 and3, bilateral hypoplastic nails of toes 2 and 3), and generalizedcutis marmorata (Fig. 1) were noted at birth. G-bandedlymphocyte chromosome analysis showed a normal 46,XXkaryotype. At 3 weeks of life she received a blood transfusionbecause of scalp bleeding. At the age of 3½ years she presentedwith severe upper gastrointestinal bleeding. Hepatosplenome-galy was noted and endoscopy of upper intestinal tract foundgrade IV esophageal varices. Serum liver tests were normalunless transaminases (1.5�N). Other tests showed: prothrom-
bin time of 70% (N> 70), factor V of 65% (N> 80), and plateletsof 103,000/mm3 (N> 150,000). Liver ultrasonography showeda heterogeneous liver with hyperechogenic portal tractssurrounded by hypoechogenic stripes, a nodular pattern, andsuspicion of extra hepatic portal vein thrombosis [Gurkaynaket al., 1998]. Arteriography confirmed extra hepatic portal veinobstruction with presence of hepatopetal and hepatofugalcollateral veins (Fig. 2). Other causes of cirrhosis and portalhypertension had been excluded [Alonso et al., 1997]. Therewas no hepatopulmonary syndrome and echocardiographyshowed an atrial septal defect, and no pulmonary hyper-tension. Acute gastrointestinal bleeding was first treated by
Fig. 1. Features of Adams–Oliver syndrome (AOS) in patient 1 (A, C, E)and patient 2 (B, D). Note severe aplasia cutis congenita (A, B) with scalpand skull defects, limbs anomalies with brachydactyly of toes, bilateralcutaneous syndactyly of second and third toes, and bilateral hypoplastic
nails of second and third toes (C), brachytelephalangy of fingers (D), andgeneralized cutis marmorata (E). [Color figure can be viewed in the onlineissue, which is available at www.interscience.wiley.com.]
Adams–Oliver Syndrome and Hepatoportal Sclerosis 187
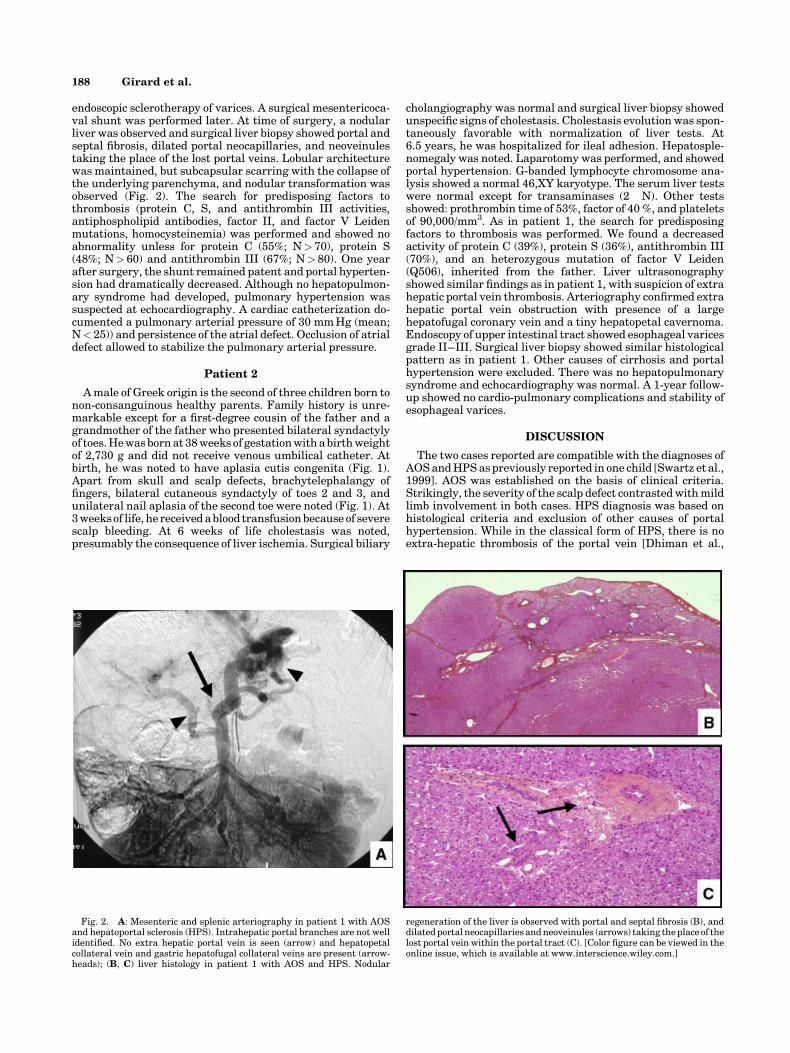
endoscopic sclerotherapy of varices. A surgical mesentericoca-val shunt was performed later. At time of surgery, a nodularliver was observed and surgical liver biopsy showed portal andseptal fibrosis, dilated portal neocapillaries, and neoveinulestaking the place of the lost portal veins. Lobular architecturewas maintained, but subcapsular scarring with the collapse ofthe underlying parenchyma, and nodular transformation wasobserved (Fig. 2). The search for predisposing factors tothrombosis (protein C, S, and antithrombin III activities,antiphospholipid antibodies, factor II, and factor V Leidenmutations, homocysteinemia) was performed and showed noabnormality unless for protein C (55%; N> 70), protein S(48%; N> 60) and antithrombin III (67%; N> 80). One yearafter surgery, the shunt remained patent and portal hyperten-sion had dramatically decreased. Although no hepatopulmon-ary syndrome had developed, pulmonary hypertension wassuspected at echocardiography. A cardiac catheterization do-cumented a pulmonary arterial pressure of 30 mm Hg (mean;N< 25)) and persistence of the atrial defect. Occlusion of atrialdefect allowed to stabilize the pulmonary arterial pressure.
Patient 2
A male of Greek origin is the second of three children born tonon-consanguinous healthy parents. Family history is unre-markable except for a first-degree cousin of the father and agrandmother of the father who presented bilateral syndactylyof toes. He was born at 38 weeks of gestation with a birth weightof 2,730 g and did not receive venous umbilical catheter. Atbirth, he was noted to have aplasia cutis congenita (Fig. 1).Apart from skull and scalp defects, brachytelephalangy offingers, bilateral cutaneous syndactyly of toes 2 and 3, andunilateral nail aplasia of the second toe were noted (Fig. 1). At3 weeks of life, he received a blood transfusion because of severescalp bleeding. At 6 weeks of life cholestasis was noted,presumably the consequence of liver ischemia. Surgical biliary
cholangiography was normal and surgical liver biopsy showedunspecific signs of cholestasis. Cholestasis evolution was spon-taneously favorable with normalization of liver tests. At6.5 years, he was hospitalized for ileal adhesion. Hepatosple-nomegaly was noted. Laparotomy was performed, and showedportal hypertension. G-banded lymphocyte chromosome ana-lysis showed a normal 46,XY karyotype. The serum liver testswere normal except for transaminases (2�N). Other testsshowed: prothrombin time of 53%, factor of 40 %, and plateletsof 90,000/mm3. As in patient 1, the search for predisposingfactors to thrombosis was performed. We found a decreasedactivity of protein C (39%), protein S (36%), antithrombin III(70%), and an heterozygous mutation of factor V Leiden(Q506), inherited from the father. Liver ultrasonographyshowed similar findings as in patient 1, with suspicion of extrahepatic portal vein thrombosis. Arteriography confirmed extrahepatic portal vein obstruction with presence of a largehepatofugal coronary vein and a tiny hepatopetal cavernoma.Endoscopy of upper intestinal tract showed esophageal varicesgrade II–III. Surgical liver biopsy showed similar histologicalpattern as in patient 1. Other causes of cirrhosis and portalhypertension were excluded. There was no hepatopulmonarysyndrome and echocardiography was normal. A 1-year follow-up showed no cardio-pulmonary complications and stability ofesophageal varices.
DISCUSSION
The two cases reported are compatible with the diagnoses ofAOS and HPS as previously reported in one child [Swartz et al.,1999]. AOS was established on the basis of clinical criteria.Strikingly, the severity of the scalp defect contrasted with mildlimb involvement in both cases. HPS diagnosis was based onhistological criteria and exclusion of other causes of portalhypertension. While in the classical form of HPS, there is noextra-hepatic thrombosis of the portal vein [Dhiman et al.,
Fig. 2. A: Mesenteric and splenic arteriography in patient 1 with AOSand hepatoportal sclerosis (HPS). Intrahepatic portal branches are not wellidentified. No extra hepatic portal vein is seen (arrow) and hepatopetalcollateral vein and gastric hepatofugal collateral veins are present (arrow-heads); (B, C) liver histology in patient 1 with AOS and HPS. Nodular
regeneration of the liver is observed with portal and septal fibrosis (B), anddilated portal neocapillaries and neoveinules (arrows) taking the place of thelost portal vein within the portal tract (C). [Color figure can be viewed in theonline issue, which is available at www.interscience.wiley.com.]
188 Girard et al.

2002], such thrombosis as been reported in adults and childrenwith HPS [Mikkelsen et al., 1965; Maksoud et al., 1986; Hesselet al., 1997]. In HPS patients, with or without extrahepatic por-tal vein thrombosis, patients have clear intrahepatic compo-nent of portal hypertension and liver disease as reported here.
The pathological mechanism of HPS remains largely un-known. It has been suggested that the proliferation andthickening of subendothelial tissue of intrahepatic and distalportal veins were a possible cause [Mikkelsen et al., 1965].Other authors have proposed intra-hepatic thrombosis to bethe primary disorder [Maksoud et al., 1986]. AOS is believed tobe the consequence of an abnormality in small vessel structuremanifesting in embryogenesis and of a thrombotic mechanism.Therefore HPS and AOS may result from an endothelialabnormality that could favor thrombosis [Swartz et al., 1999].In this view, it is interesting to note that the two patientsreported here exhibit signs that may represent predispositionto thrombosis [Gurgey et al., 1996; Dubuisson et al., 1997].Indeed decreased activities of protein C, protein S, andantithrombin III were found in both patients and heterozygousfactor V Leiden mutation was present in patient 2. Althoughdecreased activity of these anticoagulant proteins may repre-sent a secondary event, being the consequence of portal veinobstruction and/or portal hypertension [Dubuisson et al., 1997;Mack et al., 2003] HPS has been recently reported in an adultpatient with heterozygous factor V Leiden mutation [Ishii andKatada, 2003]. When factor V Leiden mutation is associated toprotein S decreased activity, it represents an additional risk ofthrombosis [Gurgey et al., 1996]. Patient 1 developed pulmon-ary arterial hypertension that occurred very early in the courseof portal hypertension even if we consider the association toan atrial septal defect. In this contex, pulmonary arterialhypertension may be favored by vasculopathy, favoring initself micro thrombosis of pulmonary arteries.
We conclude and propose that vasculopathy and thrombosis,possibly favored by a genetic predisposition, could lead todifferent type of lesions and to AOS and HPS. The association ofAOS and HPS described by Swartz et al. [1999] and the twocases reported here could be the illustration of this phenomenaand reinforce the idea that HPS and AOS may share commonmechanisms, while we cannot exclude a fortuitous association.Despite the absence of macrocephaly in either child, we cannotexclude an overlap with the macrocephaly-cutis marmoratasyndrome, that may also result from intrauterine abnormalvascular development and/or postnatal vascular dysfunction[Clayton-Smith et al., 1997; Megarbane et al., 2003]. From apractical point of view, we propose to search for predisposingfactors to thrombosis in AOS or HPS. In addition we suggest toscreen for signs of portal hypertension and its cardiopulmon-ary complications, and for signs of liver disease in AOSpatients, in order to detect infra clinical liver involvement.Indeed, discovery of HPS may be acute and fatal due tobleeding from esophageal varices.
ACKNOWLEDGMENTS
We thank Dr. M. Dreyfus and Dr. E. Mselati (Laboratory ofHematology, CHU Bicetre, Paris, France) for the search forpredisposing factors to thrombosis.
REFERENCES
Adams FH, Oliver CP. 1945. Hereditary deformities in man due to arresteddevelopment. J Hered 36:3–7.
Alonso EM, Hackworth C, Whitington PF. 1997. Portal hypertension inchildren. Clin Liver Dis 1:201–222.
Bamsforth JS, Kaurah P, Byrne J, Ferreira P. 1994. Adams–Oliversyndrome: A family with extreme variability in clinical expression. AmJ Med Genet 49:393–396.
Barnett JL, Appelman HD, Moseley RH. 1992. A familial form of incompleteseptal cirrhosis. Gastroenterology 102:674–678.
Bonafede RP, Beighton P. 1979. Autosomal dominant inheritance of scalpdefects with ectrodactyly. Am J Med Genet 3:35–41.
Carson JA, Tunell WP, Barnes P, Altshuler G. 1981. Hepatoportal sclerosisin childhood: A mimic of extrahepatic portal vein obstruction. J PediatrSurg 16:291–296.
Clayton-Smith J, Kerr B, Brunner H, Tranebjaerg L, Magee A, HennekamRC, Mueller RF, Brueton L, Super M, Steen-Johnsen J, Donnai D. 1997.Macrocephaly with cutis marmorata, haemagioma and syndactyly—adistinctive overgrowth syndrome. Clin Dysmorphol 6:291–302.
De Marsillac M, Fabre M, Pariente D, Bayari M, Piloquet H, Bernard O.2000. Hepatoportal sclerosis (HPS) in children. A study of 15 patients.J Pediatr Gastroenterol Nutr 31:S2:s122–s123.
Dhiman RK, Chawla Y, Vasishta RK, Kakkar N, Dilawari JB, Trehan MS,Puri P, Mitra SK, Suri S. 2002. Non cirrhotic portal fibrosis (idiopathicportal hypertension): Experience with 151 patients and review of thelitterature. J Gastroenterol Hepatol 17:6–16.
Dubuisson C, Boyer-Neumann C, Wolf M, Meyer D, Bernard O. 1997.Protein C, protein S and antithrombin III in children with portal veinobstruction. J Hepatol 27:132–135.
Gurgey A, Mesci L, Renda Y, Olcay L, Kocak L, Erdem G. 1996. Factor V Q506 mutation in children with thrombosis. Am J Haematol 53:37–39.
Gurkaynak G, Yildirim B, Aksoy F, Temucin G. 1998. Sonographic findingsin non-cirrhotic portal fibrosis. J Clin Ultrasound 26:309–313.
Hessel G, Escanhoela CA, de Oliveira AD, De-Maria HK, Onishi E, YamadaRM, Ferreira AG. 1997. Hepatoportal sclerosis associated with portalvein thrombosis in children: Report of 3 cases. Arq Gastroenterol34:121–125.
Hoyme HE, Jones KL, Van Allen MI, Saunders BS, Benirschke K. 1982.Vascular pathogenesis of transverse limb reduction defects. J Pediatr101:839–843.
Ishii M, Katada Y. 2003. Idiopathic portal hypertension in a systemicsclerosis patient heterozygous for factor V Leiden mutation. RheumatolInt 23:44–46.
Keymolen K, De Smet L, Bracke P, Fryns JP. 1999. The concurrence of ringconstrictionsinAdams–Oliversyndrome:Additionalevidenceforvasculardisruption as common pathogenetic mechanism. Genet Counsel 10:295–300.
Koiffmann CP, Wajntal A, Huyke BJ, Castro RM. 1988. Congenital scalpskull defects with distal limb anomalies (Adams–Oliver syndrome):Further suggestion of autosomal recessive inheritance. Am J Med Genet29:263–268.
Kuster W, Lenz W, Kaariainen H, Majewski F. 1988. Congenitalscalp defects with distal limbs anomalies (Adams–Oliver syndrome):Report of ten cases and review of the literature. Am J Med Genet 31:99–115.
Ledinghen V, Levillain P, Besson I, Palazzo L, Fabre M, Silvain C, Morichau-Beauchamt M. 1994. Hyperplasie nodulaire du foie et syndrome deTurner. Gastroenterol Clin Biol 18:898–899.
Mack CL, Superina RA, Whitington PF. 2003. Surgical restoration of portaflow corrects procoagulant and anticoagulant deficiencies associatedwith extrahepatic portal vein thrombosis. J Pediatr 142:197–199.
Maksoud JG, Mies S, da Costa Gayotto LC. 1986. Hepatoportal sclerosis inchildhood. Am J Surg 151:484–488.
Megarbane A, Haddad J, Lyonnet S, Clayton-Smith J. 2003. Child withovergrowth, pigmentary streaks, polydactyly, and intestinal lymphan-giectasia: Macrocephaly-cutis marmorata telangiectasia congenitasyndrome or new disorder? Am J Med Genet 116A:184–187.
Mikkelsen WP, Edmondson HA, Peters RL, Redeker AG, Reynolds TB. 1965.Extra and intrahepatic portal hypertension without cirrhosis (hepato-portal sclerosis). Ann Surg Oct 162(4):602–620.
Roulot D, Degott C, Chazouilleres O, Oberti F, Cales P, Carbonell N,Benferhat S, Bresson-Hadni S, Valla D. 2004. Vascular involvment ofthe liver in Turner’s syndrome. Hepatology 39:239–247.
Swartz EN, Sanatani S, Sandor GG, Scheiber RA. 1999. Vascularabnormalities in Adams–Oliver syndrome: Cause or effect? Am J MedGenet 82:49–52.
Toriello HV, Graff RG, Florentine MF, Lacina S, More WD. 1988. Scalp andlimb defects with cutis marmorata telangiectatica congenita: Adams–Oliver syndrome ? Am J Med Genet 29:269–276.
Verdyck P, Holder-Espinasse M, Hul WV, Wuyts W. 2003. Clinical andmolecular analysis of nine families with Adams–Oliver syndrome.
Zapata HH, Sletten LJ, Pierpont MEM. 1995. Congenital cardiac malforma-tions in Adams Oliver syndrome. Clin Genet 47:80–84.
Adams–Oliver Syndrome and Hepatoportal Sclerosis 189


















