Embed Size (px)
Citation preview

Chapter 1Nanostructured Macromolecules
Maria Vittoria Russo, Ilaria Fratoddi, and Iole Venditti
Abstract Macromolecules with nanoscale size are actually object of dramaticinterest due to the expectations in several technological applications ranging fromoptoelectronics to biomedicine. In this chapter the most investigated methods suit-able for the achievement of nanostructured macromolecules are reported togetherwith a variety of examples of chemical structures and properties. Self-assembly,template assisted, grafting, electrochemical and emulsion polymerizations, as wellas electrospinning technique are described highlighting the variety of materials,mainly polymers, that are prepared in a range of shapes and dimensions whichare most appropriate for a desired property. The same macromolecule can beobtained, for example, with the structure of a nanosphere or of a nanorod, whichin turn can be hollow or solid, thus being promising for different applications. Thestructure-property correlation will be outlined for many of the cited macromoleculesthroughout the chapter. Moreover, the mix of methods based on different approachesto generate nanostructures is also reported since often there is not a defined line ofseparation between them. Finally, a sub-chapter is dedicated to the advances in sev-eral fields of emerging technology and to the perspectives of future applications fornanostructured macromolecules.
1.1 Introduction
In the last decade, nanoscience and nanotechnology have been object of anoutstanding burst owing to expectations of benefits for health and quality of lifein a variety of fields: nanoelectronics, nanodevices, nanocomposite materials, alter-native energy resources, biotechnology and nanomedicine, besides breakthroughs inbasic science. The interface between science and technology is a peculiar feature forthis field of the research, involving the expertise of scientists of different education.
M.V. Russo (B)Department of Chemistry, University of Rome “Sapienza”, P.le A. Moro 5, Rome 00185, Italye-mail: [email protected]
1M.V. Russo (ed.), Advances in Macromolecules, DOI 10.1007/978-90-481-3192-1_1,C© Springer Science+Business Media B.V. 2010

2 M.V. Russo et al.
The focus of this chapter is mainly devoted to the most suitable proceduresfor the preparation of nanostructured macromolecules with particular emphasison polymeric materials of synthetic origin. Another chapter of the book will dealwith the methods for the attainment of nanostructured polymers of natural andbiological origin. Several challenges in the field of technological applications arementioned in appropriate contexts, highlighting the role of the nanostructure onthe properties and performances of polymers and macromolecules with a glanceto structural features. In general, two main approaches can be envisaged for theattainment of nanostructures: bottom-up (i.e. growth induced from the monomerto the macromolecule) and top-down (i.e. nano size induced from bulk material).To the bottom-up methods belong the self-assembly procedure, electrochemical,template assisted, grafting, emulsion, gamma-radiation induced, and chemical oxi-dation, among the preferential ones reported in a wide number of papers dealingwith nano polymers. The top-down methods involve electrospinning technology,Langmuir-Blodgett deposition, osmosis and laser micro/nanopatterning. However,this classification is sometimes ambiguous, because the two methods are oftencomplementary and the techniques to achieve nanoparticles are usually borderlineor overlapping.
One of the main goals of the recent research relies on the preparation andapplication of materials with the desired nanoscale morphology [1]. In fact, thedirect control of morphology is a fundamental request for the fine tuning of thesize, shape and extension of the nano-feature and this has to be in turn combinedto the achievement of desired optical and electronic properties. Furthermore, theobtained morphology should be stable in time and thermally. The nanoparticlesof conjugated and non-conjugated polymers exhibit a variety of morphologies, i.e.spheres, rods, fibers, ribbons, flakes and other ones which resemble the architec-tures in nature, such as cauliflowers, raspberries, fractals, that have inspired thefantasy of scientists. The nanoparticles are in turn able to build 1D, 2D and 3Dstructures.
Nanotechnologies are essential to fabricate highly integrated, tiny, andlightweight electronic devices with high performance and nanostructured materialsalso endow with intrinsically exceptional properties such as the energy conver-sion and storage [2]. The precise control over the nanostructure formation is oftenobtained through indirect methods, as for instance, thermal or solvent annealing[3, 4]. A good control of the morphology at nanometric scale is also accessiblewith methods based on the ability of certain classes of materials to self-assembly orcrystallize with the desired shape, i.e. spheres or rods [5] or organize themselves inemulsions with a solvent such as water, where the composition rules the predomi-nant phase in a predictable arrangement [6]. A different approach based on templateshas been widely explored, where the nanostructure is generated by using an organicor inorganic sacrificial material that is removed at a later stage of the process [7].Tethered polymer phases can be formed either by polymer grafting (“grafting to”)or graft polymerization (“grafting from”) and dense surface coverage are generallyobtained. Emulsion polymerization has proved to be effective for the formation ofspherical nanoparticles and experimental parameters drive regularity of shapes and
1 Nanostructured Macromolecules 3
sizes. Electrochemical methods were also applied for the preparation of nanotubesand nanowires by using template and template free methods. Electrospinning hasbeen extensively used for the preparation of nanofibres and nanotubes, by using anhigh voltage source to induce fibres formation from natural and synthetic polymers.
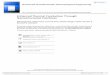
The chapter has not the ambition of being exhaustive because the current researchon this topic is rapidly evolving by the publication of hundreds of papers. However,the aim is to offer a glance on the efforts and challenges in the constantly growingfield of macromolecular nanostructures with some related applicative perspectives.As an example, a variety of nanostructured synthetic and natural polymers, withdifferent chemical and physical properties is reported in Fig. 1.1.
Hybrid systems, carbon nanotubes and composites are not reviewed in thischapter, our attention was mainly devoted to the preparation of different macro-molecules by means of the most investigated methods.
Fig. 1.1 SEM images of different morphologies obtained for synthetic polymers (a: polymethyl-methacrylate; b: polystyrene; c: polyphenylacetylene; d, e: poly(N,N-dimethylpropargylaminederivatives; f: Pt-polymetallayne) and biopolymers (g: chitosan; h, i, l, m: hyaluronic acid deriva-tives; n: dextran) (Reprinted with permission from Chronopoulou et al. [8]. Copyright 2009American Chemical Society)

4 M.V. Russo et al.
Self-assembly, template, grafting, electrochemical, emulsion polymerization andelectrospinning are reported with particular attention to new applications andperspectives for nanoscale materials with unforeseen properties due to the nanosize.
1.2 Self-Assembly
1.2.1 General Features
Self-organization of macromolecules is one of the most popular way to achievenanostructured features because it can be in principle applied to every kind of poly-mer, natural or synthetic [8]. The recent advances in design criteria for the attain-ment of well-defined polymers and nanostructures allow to produce macromoleculeswith specific functionalities which are tailored for potentials in development of cap-sules, drug delivery systems and nanoscale electro-optical devices [9]. Upon thispremise, the methods that are able to induce the self-assembly of macromoleculesare related to the chemico-physical properties of the selected polymer, of the sub-strate on which the nanostructure grows and on their combination. Obviously, thispremise envisages the variety of different morphologies, nanostructures and relatedapplications that can be obtained by the versatility of self-assembly.
The concept of self-assembly was introduced in a pioneering paper, wherethe idea was applied to biomolecules [10]; the authors report “Molecular self-assembly is the spontaneous association of molecules under equilibrium conditionsinto stable, structurally well-defined aggregates joined by noncovalent bonds.Molecular self-assembly is ubiquitous in biological systems and underlies theformation of a wide variety of complex biological structures”. This statementcan be straightforwardly transferred to non-biological macromolecules. A self-assembling system consists of a group of molecules or segments of a macromoleculethat interact each other. These molecules or molecular segments may be simi-lar or different. Their interaction flows from some less ordered state (a solution,disordered aggregate, or random coil) to a final more ordered state (a crystal orfolded macromolecule).
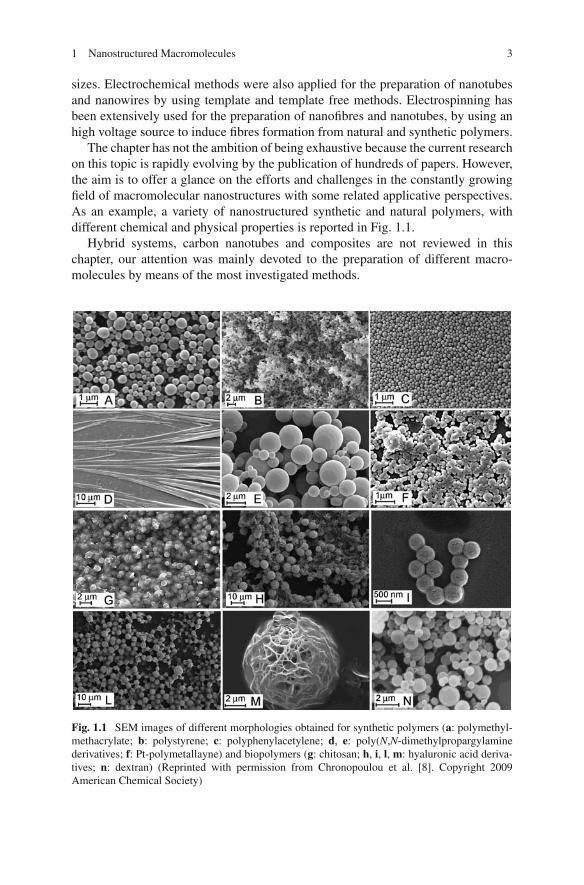
Aggregation occurs when there is a net attraction and an equilibrium sepa-ration between the components. The equilibrium separation normally representsa balance between attraction and repulsion. The following Fig. 1.2 simply illus-trates this concept. In a wide context, two-dimensional (2D) and three-dimensional(3D) structures are built with the self-assembly of macroscopic components ofdifferent nature via capillary interactions. The main topics proving the versatilityof the method are reported in a comprehensive paper [11] with many examples;open hexagonal arrays and hexagonal lattices are formed around circular templatesself-assembled from poly(dimethylsiloxane) plates; spherical structures grow byself-assembly of hexagonal metal plates on the surface of a drop of perfluodecalinin water; compact 3D structures are obtained by self-folding of a string of teth-ered, polymeric polyhedra and large crystals self-assemble from micrometer-sizedhexagonal metal plates; aggregates with electrical connectivity can be produced and
1 Nanostructured Macromolecules 5
E
d
r
a
eq
A
random
B C
aggregationequilibria
D
Fig. 1.2 (a) Schematic picture of aggregation equilibria: (a) The equilibrium curve (eq) repre-sents a balance between attraction (a) and repulsion (r); (b), (c) and (d) represent the aggregationequilibria from random to ordered assembling
assembled from polyhedral, polymer components bearing solder patterns of wiresand dots.
According to the above cited general principles, self-assembly of macro-molecules relies on some universal features and is mainly concerned with chemistryprinciples, design and selection of molecules and looks at the world of biologi-cal processes. It is interesting the comparison with the top-down “size-shrinking”(e.g. nanolithography), based on physical approach and particularly suitable for thedevelopment of the microelectronics technology, that lowers the limits of the sizeof components and devices, and with the nanofabrication and nano-manipulationbottom-up approach to molecular nanotechnology that also relies on physicalmethods (e.g., near-field scanning microscopes).
An attempt to provide a rationale to the features related to the self-assemblyhas been reported in a recent paper that has faced the investigation of thermody-namic parameters, in particular entropic terms, which are drivers for the polymernanoparticle self-assembly, with a theoretical approach based on fluids densityfunctional theory (DFT) calculations [12].
It is note worthy, however, that the concepts and methods and principles whichare on the basis of the self-assembly process often overlap each other and arecomplementary, so as it will be described in the text.

6 M.V. Russo et al.
1.2.2 1D, 2D and 3D Self-Assembled Macromolecular Structures:General Methods and Examples
1.2.2.1 Supramolecular Chemistry and Hierarchical Self-Organization;Polymers and Block Copolymers
Quite often, nanoparticles assembling is based on evaporation procedures, i.e. thedrying on a substrate of a drop of suspension or solution containing the nanoparticlesthus inducing the particles organization [13]. This is a dynamic process governed bya variety of factors, such as interactions between nanoparticles, substrates and sol-vents [14, 15], drying kinetics [16–18], hydrodynamic effects [19, 20] and diffusionprocesses [21], which lead to unusual transitory structures [22]. The morpholo-gies as well as the size of the domains of these self-assembled particles dependon the solvents used for the procedure, the evaporation time, temperature and par-ticle diffusion, and can be qualitatively evaluated with theoretical simulations. Thedrying-mediated self-assembly of nanoparticles on diblock copolymer substrateswas deeply investigated with the aid of a coarse-grained lattice gas model and MonteCarlo simulation techniques [23].
The “bottom-up” approach, that in principle is based on the ability of func-tional building blocks to assemble into defined superstructure arrays, is one ofthe most widely used method to achieve materials at the scales between 1 and100 nm. Supramolecular chemistry and self-organization is a fundamental topic ofthis approach “where the goal is not smaller size or individual addressing but com-plexity through self-processing, which strives for self-fabrication by the controlledassembly of ordered, fully integrated, and connected operational systems by hier-archical growth” as reported by J. M. Lehn [24] and is a convenient alternative tonanofabrication and nanomanipulation.
In the framework of hierarchical self-organization it is note worthy the synthesisand self-assembly of polymer coated ferromagnetic nanoparticles, where the use ofdipolar nanoparticles as building blocks with inherent dipole moment enables thepreparation of organized hierarchical materials in one- and two-dimensional assem-blies, which represent a promising area of application in materials chemistry. Asan example, a review on this topic reports on polymer-coated ferromagnetic cobaltnanoparticles (core shell nanoparticles self-assembled in aligned chains, reported inFig. 1.3) that were synthesized by using end-functionalized polystyrene (PS) sur-factants with amine, carboxylic acid or phosphine oxide end-groups as stabilizingagents of the ferromagnetic Co nanoparticles [25].
A different approach to the synthesis of nanosized macromolecules through hier-archical self-assembly is based on Layer-by-Layer (LbL) chemistry. LbL allowsthe deposition of ultra thin films whose thickness can be controlled by the chem-ical structure of the molecules and number of deposited layers. The interactionsbetween layers can be ionic, covalent, hydrogen-bonding, and charge-transfer,depending upon the nature of the polymer used in the preparation.The layer-by-layer assembly of an electroactive polymer nanocomposite thin film of cationiclinear poly(ethyleneimine) and Prussian Blue nanoparticles, has been exploided

1 Nanostructured Macromolecules 7
Fig. 1.3 TEM images ofself-assembled ferromagneticPSCoNPs (DPS-CoNPs21–31 nm) at low (a) andhigh magnification (b),prepared from a mixture ofPS-NH2 (3) and PSCOOH (6)in the thermolysis ofCo2(CO)8. The PS-CoNPswere cast onto supportingsurfaces from a particledispersion in toluene(Reprinted with permissionfrom Keng et al. [25].Copyright 2009 AmericanChemical Society)
showing mechanical and swelling properties [26]. Also the biomedical purposestake advantage of the LbL technique for the assembling of nanomaterials; for exam-ple a hydrophobic drug can be deposited by the sequential adsorption of oppositelycharged polyelectrolytes onto a charged substrate [27]. This technique is a suitabletool for the self assembly of other materials e.g. polydiacetylene (PDA) films andnanotubes organized on flat surfaces and inside of nonporous alumina templates[28] and porphyrin arrays [29].
The supramolecular self-assembly approach in the solid state from solution, lead-ing to well defined nanostructures has been discussed in a comprehensive paperthat describes the main features related to this method; interactions of macro-molecules with the substrate surface, design of well defined molecular structure, anduse of block copolymers have been considered in a joint experimental-theoreticalapproach, in view of understanding the structure-property relationship of conjugatednanostructures [30].
The hierarchical self-assembly approach has been proposed as a valuable methodin many examples of macromolecules nanodesign. Nanoparticles assembly at

8 M.V. Russo et al.
liquid-liquid interface can be controlled by tuning the size, the volume fractionand the chemical characteristics of the ligands; this method is suited to gener-ate nanoparticle-polymer composites, whose spatial distribution can be controlledby enthalpy or entropy, thereby producing auto responsive materials. An interest-ing paper illustrates how the self-assembly of polymeric supramolecules inducesthe synthesis of functional materials with peculiar properties and shows niceexamples of the way macromolecules are induced to self-assembly [31]: flexiblepolymers, such as comb-shaped supramolecules, are assembled through hydrogenbonds, while rod-like polymers require a combination of bonds (recognition); theconnection of amphiphiles to one of the blocks of a diblock copolymer induces self-organization in hierarchical structures; a lamellae-within-cylinders structure can becleaved to produce nanoporous materials, can lead to disk-like morphology by crosslinking the slices within the cylinders or can deliver nanorods by cleaving the sidechains.
The self-assembly of polymer nanoparticles (spheres and wire-like threads)can occur in solution by using dendrimer macroinitiators in atom transfer radicalpolymerization (ATRP) [32] that will be extensively discussed in Section 1.4.4.Ring opening polymerization (ROP) is successfully used for the self-assemblyof amphiphilic graft polyphosphazenes with different mole ratios of hydrophobicgroups to hydrophilic segments to yield supramolecular aggregates (nanospheres,high-genus particles, macrophage-like) [33]. These examples show the role ofthe chemical structure on designing nanoscale objects through supramolecularself-assembly.
Among macromolecules, porphyrins are particularly attractive building blocksbecause the intimate packing of these aromatic macrocycles can lead to new photo-physical and photochemical properties. Self-assembling of porphyrin molecules intohollow hexagonal nanoprisms with uniform size and shape and controllable aspectratio was recently achieved by the self-assembly technique assisted with surfactant.Nanoprisms can readily self-organize into an ordered, smectic three-dimensional(3D) architecture through simple evaporation of the solvent [34]. Free-standingporphyrin nanosheets with high aspect ratios were recently obtained by reprecip-itation method [35]. These results should be significant in porphyrin crystallizationand porphyrin application in optoelectronic devices, catalysis, drug delivery, andmolecular filtration.
About a decade ago, the main features (experimental and applicative) governingthe self-organization of nanostructured macromolecules were highlighted, with par-ticular emphasis on block copolymers, envisaging the future perspectives for thesematerials [36].
Since then, the research has dramatically grown and many goals have beenachieved. A review reports an organized and detailed overview on theoretical aspectsand basic principles of self-assembly and micellization of block copolymers in solu-tion, together with a wide number of examples concerning the methods for thestabilization of macromolecular aggregates and their applications, mainly focusedon biomedical field, in the perspective of “smart” nano-objects production [37]. Theself-organization of block copolymers in different shapes is depicted in Fig. 1.4.

1 Nanostructured Macromolecules 9
Fig. 1.4 Examples of structures obtained from block copolymers: (i) direct micelle, (ii) vesicles,and (iii) other morphologies: (iiia) inverse micelle, (iiib) lamellar structures, and (iiic) cylindricalor tubular micelle (Reprinted from Rodríguez-Hernández JR et al. [37], with permission fromElsevier)
Examples of structures obtained from block copolymers range from micelle,vescicles to lamelle or cylindrical and tubular structures, suitable for drug deliverysystems and, in general, as host-guest systems.
In fact, an emerging field of nanoscale science is envisaged in molecular capsuleswhich can host guest molecules through noncovalent interactions. These syntheticmolecular receptors exert their peculiar activity upon the conjugation of parameterssuch as size, shape, and chemical complementarity and are proposed for applica-tions in catalysis of chemical reactions and for the stabilization of reactive species[38]. For example, hollow hydrophilic metal functionalized nanostructures can beproduced from an amphiphilic metallic diblock copolymer which supramolecularlyself-assemble into monodisperse noncovalently connected micelle and can be usedas nanocages [39].
Interestingly, functionalized block copolymers in solution can provide the order-ing of nanoparticles in a variety of distinct phases, i.e. cubic, layered hexagonal,hexagonal columnar, gyroid and square columnar, as developed by moleculardynamics studies and by experimental investigations based on the solvent composi-tion and valence of the organic counter ion, respectively [40, 41]. A peculiar exam-ple of colloidal stable micelle formation is represented by core-shell organometallic1D nanocylinders obtained from the self-assembly of polyferrocenylsilane coresand polyisoprene coronas crosslinked block copolymers; these micelle are suited

10 M.V. Russo et al.
to be used in microfluidic alignment, nanoceramic fabrication and other advancedtechnologies [42].
Recently, self-assembled block copolymers have attracted the interest of sci-entists as masks for nanolithography, templates for the synthesis of nanoparti-cles [43] and membranes for ultrafiltration [44]. Block copolymers films madefrom polystyrene-block-poly (4-vinylpiridine) and 2-(4′-hydroxybenzeneato) ben-zoic acid form cylinders with hexagonal order aligned along the normal directionto the substrate and embedded into the PS matrix. The nanoporous films areobtained by removing the benzoic acid from the cylinders, and periodic hexag-onal moiré superstructures are obtained when the films with long range orderare superimposed to small misorientation angles, producing labyrinth-like patterns[45]. Similar moirè-type superstructures are reported for partly tert-butoxycarbonyl(BOC) and tert-butyl (TBU) protected block copolymers based on 4-hydroxystyrenewith varying block ratios; these materials give rise to the transformation of a partlyBOC-protected block copolymer into the homopolymer poly(4-hydroxystyrene) byannealing at moderate high temperature [46].
In a different context, complex coacervate core micelle can be obtained by thereaction of a polyion-neutral diblock copolymer with an oppositely charged poly-electrolyte. These micelle are formed upon hierarchical self-assembly in water ofthe two polymeric components and, more interestingly, upon self-assembly of metalion coordination polymers [47].
Self-organization of block copolymers into regular patterns has been inves-tigated with the aim of finding high performance applications in microelec-tronics [48]. In this review most of the reported studies deal with polystyrene(PS) and polymethylmethacrylate (PMMA) diblock copolymers (PS-b-PMMA)that are materials compatible with the semiconductor fabrication infrastructureand also suitable for the understanding of materials properties. PS-b-PMMA,alike diblock copolymers, spontaneously form patterns at molecular scale dimen-sions through microphase separation. For lithography applications, it is impor-tant the control of the orientation of the self-assembled pattern, e.g. cylindricaland lamellar phases, which can be obtained by coupling the self-assembly pro-cess with an external bias. The methods for the control of pattern orientationand pattern transfer processes, together with examples of device fabricationsuch as shallow-trench-array capacitors, of controlled optical index materials,of nanoporous membranes and nanocrystal Flash memories are reported. A listof features that must be considered as a guide for the development of poly-mer self-assembly-based high–resolution patterning methods for high-performancesemiconductor electronics at the nanoscale is provided and can be observed thatthe polymer self-assembly procedure is a challenging substitute for high-resolutionlithography.
It is noteworthy that a theoretical approach, i.e. the dissipative particle dynamics(DPD) method provides the understanding of the self-assembling behavior of blockcopolymers with two molecular architectures made from an A-homopolymer blockcombined with a BC-comb block or a BC-alternating block; hierarchical structures,such as spheres-within-lamellae, cylinders-within-lamellae, gyroid-within-lamellae,

1 Nanostructured Macromolecules 11
lamellae-within-lamellae, lamellae-within-cylinders, and lamellae within-spheres,can be foreseen for the development of photoelectron based devices [49].
1.2.2.2 Self-Assembly of Dendrimers
Dendritic molecules, which are three-dimensional branched compounds, have theproperty of self-assembling into complex arrays by non covalent (supramolecular)interactions, giving rise to controllable nanomaterials. Recent reviews emphasizethe use of these peculiar macromolecules as building-blocks to generate highlybranched complex nanoscale assemblies, represented in Fig. 1.5, and highlight thepotentials of these assemblies in chemistry and biology [50, 51].
Dendrons may exhibit the self-assembly ability trough hydrogen bonds insolution to produce controlled geometries (i.e. well defined assemblies of building-blocks) [52]. An alternative way to induce non covalent interactions of individualdendritic branches is the assembly mediated by templates, which can be organicmolecules interacting with the dendrons trough hydrogen bonds or acid-base
Fig. 1.5 Schematic illustration of the self-assembly of dendritic building blocks. (a) Untemplatedassembly of dendrons. (b) Templated assembly of dendrons. (c) Nanoparticles with assembled den-dritic surface groups. (d) One-dimensional, fibrous, gel-phase assemblies of dendritic molecules.(e) Liquid crystalline assemblies of dendritic molecules.(Reprinted from Smith et al. [50], withpermission from Elsevier)

12 M.V. Russo et al.
reactions [53, 54]. Dendrimers functionalized with rotaxane or dibenzo-24-crown-8 macrocycle are also favored for a spontaneous assembly into interlockedarchitectures [55, 56].
The assembly of dendritic superstructures can be carried out also by means ofmetal coordination chemistry, by a number of different key strategies. Since thepioneering work of Balzani group [57], the research has been developed by intro-ducing the idea that metal centers act as “building block connectors” and someselected, but not exhaustive, examples of literature reports are given [58–62]. Theproperties of these assembled dendritic superstructures range from electrochemical,light-harvesting, phosphorescent and electroluminescent to biochemical ones.
Clusters of metals are also cores for the assembly of dendrimers which showelectrochemical and biomimetic properties [63, 64]. A peculiar case is representedby the stabilization of gold nanoparticles with sulfur containing dendritic ligandswhich provide the control of nano-architecture dimensions for stable assemblies[65].
In the field of bio-nanotechnology, dendritic disulfides made from biocompat-ible L-lysine building-blocks were also found as useful ligands for the controlledassembly of gold nanoparticles [66] with anion sensing properties [67]. The synthe-sis of CdSe dendron stabilized nanoclusters with high stability and biocompatibility(box-nanocrystals) is also noteworthy [68].
Gene vectors which can deliver DNA to target cells are object of wide scien-tific interest for the development of gene therapy. In particular, polyamidoamine(PAMAM) dendrimers belong to a class of nano polymers with highly branchedspherical structure and a unique surface of primarily positively charged aminogroups. PAMAM can transport DNA into a large variety of cell types and hasemerged as a promising non-viral gene vector [69]. The increasing number of paperson this topic highlights the importance of this field of research and only some repre-sentative ones will be hereafter reported. Since the pioneering work of Tomalia andco-workers [70] who demonstrated that PAMAM-DNA complex dendrimers exhibitthe highest in vivo gene transfer efficiency, the research developed the formation ofnanoscale complexes which provide DNA protection and enhanced activity of bio-conjugates [71, 72]. Globular nanostructures were achieved from plasmid DNA-copolymers (dendritic poly L-lysine and linear PEG blocks) self-assembly [73], anda poly(azobenzene) dendrimer based on a calyx-4-arene core functionalized withperipheral L-lysine units provides a UV-switch able framework, thus showing thatthe affinity of the system for DNA can be controlled by using UV irradiation [74].Amphiphilic dendrimers are reported to be vectors for gene delivery with an inherentself-assembling potential with DNA [75].
Other morphologies can be obtained, i.e. dendritic nanoclusters and nanotubes,with different chemical approaches which generate a wide variety of differentnanoscale architectures and have a promising potential in host-guest chemistry andnanotechnology [76, 77]. Asymmetrically functionalized dendritic blocks, e.g. den-drons with polar and apolar groups, self-assemble to produce macromolecules withsurfactant properties [78–81].

1 Nanostructured Macromolecules 13
Many other features and properties are typical of dendrimers; assembly of largeaggregates is achieved when the dendrimer is linked to a different multi-functionalsystem, for example thiolated phosphorous dendrimers are suitable stabilizersof Au55 clusters leading to the formation of gold cluster superstructures [82].Supramolecular fibrillar architectures, of prominent interest for applications in neu-rodegenerative diseases, are achieved through hydrophobic and hydrophilic contactsin solvents which promote the aggregation in gel-phase material [50].
Percec and coworkers have developed a dramatic amount of research on dendriticself-ordering that gives rise to supramolecular dendromesogens packed in hexagonalor cubic structures of nanoscale dimensions with liquid crystal properties [83, 84].The self-assembly of dendritic molecules into liquid crystalline materials is favoredalso by the presence of mesogenic groups; nice examples of this approach werereported by Ponomarenko [85], Serrano [86] and Hult [87] research groups.
In this framework, an interesting paper reports the role of dendritic self-complementary hydrogen-bonding units that are used as noncovalent cross-linkingagents who promote the chain entanglement of linear polymers (PMMA derivatives)into polymeric nanoparticles [88].
1.2.3 Self-Assembly Induced in π-Conjugated Polymers
Among one dimensional nanomaterials, synthetic procedures, properties and appli-cations of polymers have been extensively reported in a dedicated chapter of a recentreview [89].
A class apart of macromolecules is represented by π-conjugated polymers, whichare the basis of the development of organic electronics whose performance isgoverned by their degree of order. The main properties related to conductive poly-mers in their nanozize morphology and the more reliable methods to induce nanosized structures based on intermolecular and intramolecular effects are extensivelyreviewed by Wessling [90] and by Kim [91]. The self-assembly of these materi-als is then an important topic in the field of nano macromolecules. In the followingsub-chapters, the main features concerning the most investigated π-conjugated poly-mers will be described, with examples on different synthetic methodologies for theattainment of nanostructures for several applications.
1.2.3.1 Polyaniline
Polyaniline (PANI) represents one of the most cited examples of nanostructuredpolymers, due to its outstanding electronic properties and technological applica-tions that have promoted a wide number of studies and publications. Although avariety of different and peculiar morphologies have been reported, such as brainlike [92], cauliflowers [93], nanoflakes, nanospheres and nanorods [94], chrysan-themum flower-like [95], plate-like structures and flower-like superstructures [96],

14 M.V. Russo et al.
the nanorods morphology seems to be the favorite one and has been obtained witha multiplicity of methods, some of which will be hereafter reported as examples.A simple dispersion polymerization in a PVA matrix allows the formation of PANIassembled nanorods with a tubular orientation. The electrical conductivity of thenanorods is interpreted by the two-dimensional variable-range hopping model, dueto the fact that the rods in the film are not strictly aligned in one particular direc-tion [97]. Polyaniline nanostructures with controlled morphology of different shapes(sheets-, fiber- and spherical-like) are synthesized by using p-toluene sulphuric acid(p-TSA) aqueous solutions and a rational mechanism based on the self-assemblyof micelle is proposed for the formation of PANI nanostructures [98]. Bundles of aPANI copolymer, i.e. poly(aniline-co-anthranilic acid) (PANANA), can be assem-bled by using proper amounts of anthranilic acid that plays the roles of monomer,acid-media provider, and dopant in the reaction system [99].
Emeraldine base (EB) and emeraldine salt (ES) forms of poly(o-methoxyaniline)(POMA), are able to construct biomolecular hybrids with DNA showing a fibrillarnetwork structure of invariant fibrillar diameter for different hybrid compositions.An approximate model of the Na-DNA/POMA-ES system indicates nanostructuredself-organized assembly of the components in the hybrid [100]. However, the mostdesired property of fibers for the electronic devices technology is their orientationin a definite direction. Self-assembly of oriented PANI arrays can be achieved inthe presence of inorganic acids and by changing the PANI/acid concentration ratio,(Fig. 1.6) [101, 102].
The choice of acids has also other effects on the nanostructure; for exam-ple, the use of tetrachloroaurate as an efficient oxidant of aniline in the presenceof a chiral inducing agent, i.e. (1S)-(+)-10-camphorsulfonic acid ((S)-(+)-CSA)or its enantiomer (R)-(–)-CSA, allows the formation of optically active PANInanorods, together with the further self-assemblies into monodispersed hierarchicalAu (0) microspheres [103]. Recently, aniline oligomers have also received atten-tion, because they can be envisaged as the building blocks of block architectures
Fig. 1.6 A large number ofPANI arrays with averagediameter of 1.2 μm andhighly ordered structure areproduced by change of theaniline/acid concentrationratio (Reprinted from Wuet al. [101], with permissionfrom Elsevier)

1 Nanostructured Macromolecules 15
with promising structure-function properties based on supramolecular constructionprinciples [104].
1.2.3.2 Polypyrrole
Among conjugated polymers, polypyrrole (PPy) and its derivatives represent a classof technologically important macromolecules mainly due to their conducting prop-erties and applications in molecular electronics. A remarkable review accounts forthe advanced research on the mono and multilayer deposition on different sur-faces of these polymers in their nanometer-size with one-dimensional resolutionand hybrids formation with gold nanoparticles [105].
In the framework of this research topic, PPy chains self-assembled in nanowireswith a coral-like shape can be obtained by FeCl3 induced oxidative polymerizationand dodecil-benzenic sulphonic acid (DBSA) dopant [106]; oxidative polymer-ization is a widely used method for the attainment of polymeric nanostructures.For example, bundles of self-assembled PPy nanotubes have been fabricated bypolymerization reaction with bis(2-ethylhexyl) sulfosuccinate reverse (water-in-oil)emulsions [107] and rods with enhanced electrical conductivity and thermal stabil-ity are reported to be formed via a self-assembly process of micelle obtained from aoxidative polymerization in the presence of p-toluensulfonic acid used as surfactantand doping agent [108, 109]. A further example of PPy nanotubes synthesized byoxidative polymerization in octane is reported in Fig. 1.7 [107].
A variety of synthetic procedures for the achievement of PPy nanostructures(spheres, rods, tubules, core-shells) are reported in the literature where the conceptof self-assembly is mixed with that of template synthesis and composites fabrica-tion, because the methods often show overlapping features. Most of the examplesdeal with the template assisted procedure and are reported in the subchapter“Templates”.
Fig. 1.7 TEM images of PPy nanotubes prepared in octane (a) and enlargement (b) (Adapted withpermission from Jang and Yoon [107]. Copyright 2009 American Chemical Society)

16 M.V. Russo et al.
1.2.3.3 Polythiophene
The surface morphology of polythiophene (PTh) films is important for themechanical and electrical properties of this widely investigated material. Differentsynthetic strategies were carried out for the preparation of nanostructured PTh,for example, nanotubules of PTh were obtained with electrochemical polymer-ization giving rise to self-ordered nanostructures with fractal dimensionality andnanowires with diameters in the range 50–100 nm have been produced with gammaradiation-induced oxidative polymerization [110]. Hollow spheres of PEDOT,poly(3,4-ethylenedioxythiophene), can be self-assembled through a “grow frommembrane” process; the self-assembly is promoted by the hydrogen bond between3,4-ethylenedioxythiophene monomers and acetic acid used as dopant agent dur-ing the oxidative polymerization [111]. Spherical PEDOT particles can be achievedby functionalization of the polymeric structure with specifically designed PEO-based reactive stabilizers in aqueous media. These self-assembled PEDOT particlesexhibit high conductivity for applications ranging from PLEDs to flexible organicsolar cells [112].
PEDOT aggregates of hollow microspheres were also obtained and SEM andTEM images, representatives of these morphologies, are reported in Fig 1.8 [113].
Different morphologies, i.e. vesicles and lamelle are formed by an amphiphilicconjugated diblock copolymer made from polyfluorene-b-polythiophene units; thismaterial shows the property of forming aggregates at the air-water interface inducedby the Langmuir-Blodgett (LB) technique and of tuning the optical properties uponmodification of the aggregation state [114].
When 3,4-ethylenedioxythiophene (EDOT) is chemically polymerized in thepresence of polyacrylic acid (PAA) as a template, conducting nanowires can beassembled from smaller nanowires in a side-by-side manner and exhibit excellentconductivity [115]. The electronic properties of PTh have promoted a wide interestin the development of organic/polymer light-emitting diodes (OLEDs/PLEDs) and itis note worthy that the performance of these devices is dramatically enhanced by the
Fig. 1.8 SEM (a) and TEM (b) micrographs of PEDOT hollow aggregates (the exterior size dis-tribution of hollow microsphere is shown in inset of (b) (Reprinted from Xia et al. [113], withpermission from Elsevier)

1 Nanostructured Macromolecules 17
use of integrated self-assembled nanowires of a PEDOT-organic molecule (highlysubstituted condensed benzothiophene) [116]. A further important application is thefabrication of polymer based sensors and indeed the chemical sensing responsesof organic field-effect transistors (FET) based on nanostructured regioregular poly-thiophene have been recently investigated reporting an in depth study of the sensingmechanism [117].
1.2.3.4 Polyacetylenes and Polyynes
Functional polyacetylenes show a variety of properties such as liquid crystallinity,photoconductivity, light emission, ionic susceptibility, photoresistance, chromism,helical chirality, optical nonlinearity, self-assembly, cytocompatibility, and bioactiv-ity [118] and have been the object of thousands of scientific reports. Quite recently,the research has been extended to the study of these materials in nanostructuredfashion with the aim of improving their performance and the few examples hereafterreported will give a glance on this emerging topic.
Polyphenylacetylene (PPA) is a π-conjugated semiconducting polymer, it ishighly stable and can be synthesized in cis or trans configurations, depending onthe polymerization procedure [119]; it shows NLO properties [120] and is a suit-able matrix for the immobilization of lipolytic enzymes [121]. Upon emulsionpolymerization it self-assembles into nanospheres with photonic crystal properties[122]. Mono-substituted helical poly(phenyl)acetylene structures have been pre-pared through living polymerization; the molecular structure looks like a narrowspiral with a conjugated electron system and with the attached side groups spiral-ing in the opposite sense. This polymer feature is able to self-assemble and may bedeposited in an oriented fashion, showing electrical conductivity [123].
An amphiphilic PPA carrying L-leucine pendants was self-assembled intonanospheres and nanorods and it was assessed that the morphology depends onthe polarity or solvating power of the solvent mixture, i.e. on the affinity orlikeness of the solvent molecules with the hydrophobic PPA backbone or thehydrophilic Leu pendant group; morphological transition processes from micellarnanopearls, via rings, globules, loops and cages to extended nanofibers are detectedon the course of the self-assembly process [124]. Amphiphilic polyacetylenes, suchas poly(N-octadecyl-2-ethynylpyridinium bromide), self-assemble through layer-by layer deposition within aluminosilicate (saponite) nanosheets, leading to adouble layer of polymer where the alkyl chains are arranged in interdigitated fea-tures, thus producing a hydrophobic barrier that hinders the transport of watermolecules [125].
Other conjugated materials with optoelectronic properties give rise to nanos-tructures. Uniaxially ordered films of a rigid rod conjugated polymer, namelypoly(para-phenylene ethynylene) with thioacetyl end groups (TA-PPE), are alignedonto friction-transferred poly(tetrafluoroethylene) substrates; the achievement ofhighly ordered structures self-assembled by simply drop casting of the polymersolution, dramatically enhances the performance of the photoswitcher devices dueto the efficient charge transfer along the aligned polymer structure [126].

18 M.V. Russo et al.
Metal containing polyynes are multifunctional materials which combine theproperties of organic polymers with those of metal centers coordinated to the organicmoiety and are able to form nanotemplates, colloidal photonic crystals, multilayercapsules and hollow vesicles [127, 128]. An example of a rod-like polymetallayneself-assembly in hollow nanorods has been recently reported [129]; the computersimulations of the nanostructure show that the polymer chains are ordered in par-allel lines that give rise to a tubular morphology rather unusual for these materials,but promising for sensor devices applications.
1.3 Templates
1.3.1 General Features
The word “template” in the contest of polymer science means that a structuredirected agent is able to replicate a shape into another under structural inversion.
A quite widely used method for the achievement of nanostructured polymersdeals with the assistance of templates. Direct templating is particularly suited forgetting mesostructures of organic and soft materials such as polymers that, ingeneral, can be easily replicated by adopting hard templates which allow a greatsynthetic flexibility.
If direct templating is carried out, the templated material is an inverse copy ofthe original template structure and this technique is then useful for the achievementof nanostructured or porous materials. Moreover, the dimensions and structures canbe tuned or modified by a proper choice of the template. A simple scheme of thetemplate technique is reported in Fig. 1.9.
Due to the feasibility of the technique, dramatic efforts have been recentlyexplored by researchers to exploit templating methods which can give rise tostructure controlled materials with functional advanced properties. The templat-ing strategies that induce the nano morphology build up include a variety ofpolymerization procedures such as photopolymerization, linear polymer chain tem-plating, particle dispersion templating, molecularly imprinted polymers, templatingin vesicles, and templating within liquid crystal surfactant assemblies [130].
1.3.2 Template Techniques
Most of the template techniques for the achievement of nanostructured macro-molecules are described more extensively in the sub-chapters 3 “Grafting polymer-ization” and 4 “Electrochemical methods”. Hereafter, some examples of polymericmaterials obtained in nano-size dimension through the use of different template-assisted polymerization methods will be shown.
Among the variety of procedures, photopolymerization [131] is widely adoptedto perform templating reactions due to its characteristic of control on the structuralevolution of the templated polymer structure, through the kinetic parameters.

1 Nanostructured Macromolecules 19
Fig. 1.9 Schematic presentation of templating approaches toward nanostructured soft materialsby using endo and exotemplates (Reprinted with permission from Thomas et al. [130]. Copyright2009 American Chemical Society)
When the polymerizations are carried out in highly organized mesoscale templates,well defined and controlled network architectures can be achieved. The combina-tion of templating and photopolymerization is of peculiar interest in the field ofbiomedicine, where nanostructured materials play a relevant role in tissue engi-neering and drug delivery applications. In general, due to a greater control overthe template process, this approach is most suitable for linear chain templatingor catalytic polymerization, organized particle templating, molecular imprinting,templating of assembled vesicles and polymer templating in liquid crystals.
Mesostructured inorganic solids, originated from self-assembling of supramolec-ular structures, represent a class of suitable templates for the achievement of nanos-tructured materials. Ordered mesoporous polymers and carbonaceous frameworks

20 M.V. Russo et al.
supramolecular aggregates act as templates for block copolymers introduction[132]. Polymeric templates are also widely described. For example, a porous poly-meric membrane can be obtained by the bombardment (irradiation) of a polymericfilm with high energy heavy ions, followed by chemical etching. The pore density(number of pores per square unit) depends on the intensity and duration of the irra-diation and the diameter of the pores is related to the intensity of the etching process[133].
A further way to accomplish nanostructured conducting polymers through tem-plate technique is the use of “soap-bubble” template; for example pyrrole iselectrochemically polymerized along the walls of soap bubbles giving patternedconducting microcontainers for biomolecules encapsulation [134]. In a similarmethod, resorcinol-formaldehyde nanopolymers, precursors of carbon nanoparti-cles, can be obtained by using surfactant-templated vesicular assemblies [135]. Theoxidative chemical polymerizations of polypyrrole (PPy), poly(N-methylpyrrole)(PNMPy), polythiophene (PTh) and poly(3,4-ethylenedioxythiophene) (PEDOT),performed with polycarbonate and alumina membranes as templates, lead to highlyoriented nanofibers and nanotubes whose diameter can be tailored with the pore sizeof the membrane [136] and linear aggregates of nano PPy blobs were achieved byalumina-membrane templated polymerization [137].
PTh nanostructures have been also produced by using metal nanoparticle tem-plate, i.e. copper nanoparticles are mixed with soluble PTh to yield thin films that arefurther subjected to thermal treatment so that insoluble PTh films with Cu nanoparti-cles included are obtained. The Cu nanoparticles are removed with a proper solvent,leaving voids that can be filled with spherical molecules such as fullerene derivatives[138]. PANI and PPy with controlled nano-morphologies are achieved by manipu-lating the length of the hydrophobic surfactant or by changing the chemical structureof the template adsorbing substrate [139]. The formation of PPy wires and ribbons isinduced by lamellar inorganic/organic mesostructures as templates that are shapedin situ during the polymerization between surfactant cations, such as cetyltrimethy-lammonium bromide (CTAB), and oxidizing anions, while by using short chainor nonionic surfactants sphere-like nanostructures are produced [140, 141]. Thesame procedure can be applied for the controlled growth of poly(N-methylaniline)nanowires and microspheres [142]. CTAB can also be used for the modificationof a fibrillar complex made by FeCl3 and methyl orange, acting as reactive self-degraded template that induces the formation of nanotubular structures of PPy[143].
Quite recently, biomolecules have became promising templates for the synthesisof 1D nanostructures; for example DNA promotes the assembling of Au and Ag par-ticles in nanotubes, nanowires and nanorods and proteins and polypeptides are alsocandidates for analogous purposes. In this contest, heparin and sodium alginate aremorphology-directing agents for the achievement of PPy and PANI nanowires andfibers; likewise starch is a convenient template for the electrochemical polymeriza-tion leading to PPy nanowires [144]. The synthesis of PPy and hybrid (Au-PPy-Au)nanowire arrays of controlled dimension can be performed by an all electrochemicaltemplate method, within the pores of homemade polycarbonate membranes [145].
1 Nanostructured Macromolecules 21
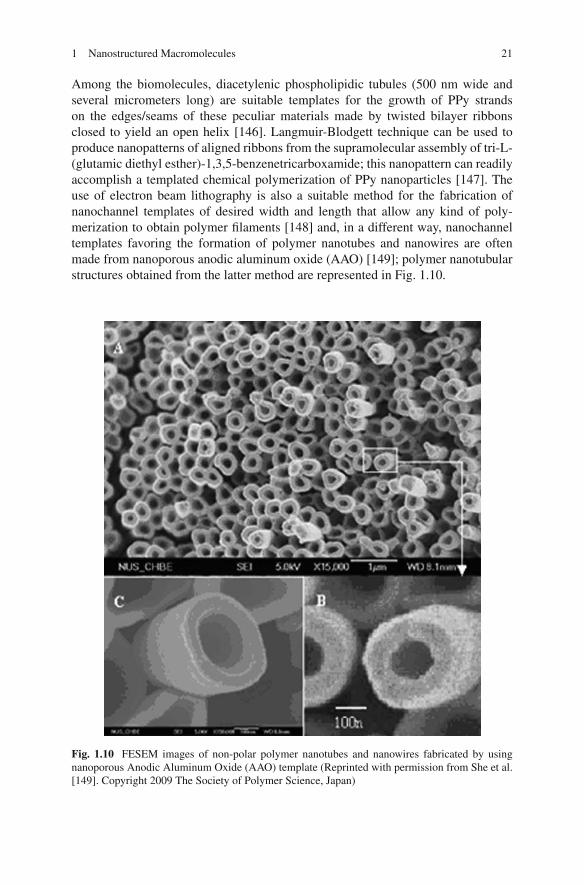
Among the biomolecules, diacetylenic phospholipidic tubules (500 nm wide andseveral micrometers long) are suitable templates for the growth of PPy strandson the edges/seams of these peculiar materials made by twisted bilayer ribbonsclosed to yield an open helix [146]. Langmuir-Blodgett technique can be used toproduce nanopatterns of aligned ribbons from the supramolecular assembly of tri-L-(glutamic diethyl esther)-1,3,5-benzenetricarboxamide; this nanopattern can readilyaccomplish a templated chemical polymerization of PPy nanoparticles [147]. Theuse of electron beam lithography is also a suitable method for the fabrication ofnanochannel templates of desired width and length that allow any kind of poly-merization to obtain polymer filaments [148] and, in a different way, nanochanneltemplates favoring the formation of polymer nanotubes and nanowires are oftenmade from nanoporous anodic aluminum oxide (AAO) [149]; polymer nanotubularstructures obtained from the latter method are represented in Fig. 1.10.
Fig. 1.10 FESEM images of non-polar polymer nanotubes and nanowires fabricated by usingnanoporous Anodic Aluminum Oxide (AAO) template (Reprinted with permission from She et al.[149]. Copyright 2009 The Society of Polymer Science, Japan)

22 M.V. Russo et al.
1.3.3 Materials in Template Techniques
The template assisted synthesis is based, among the others, on three main categoriesof materials, i.e. diblock copolymers [150–152], anodized alumina layers [153] andorganic or inorganic colloidal particles [154–156] which act as scaffolds or support-ing structures with desired size for the polymer that has to be templated and can beafterwards removed by dissolution in common solvents.
Polystyrene templating particles are particularly suited for the preparation ofnanostructured materials and, among these, for the synthesis of PANI nanos-tructures. The PS template assisted electrochemical preparation of PANI isbased on a general procedure that proceeds firstly with the formation of a PStemplate on a conducting substrate, followed then by electropolymerization ofaniline and finally with the removal of the PS template [157]. By using oppo-sitely charged PS nanoparticles (i.e. charged by addition of negatively chargedor positively charged polyelectrolytes, i.e. poly(sodium-4-styrenesulfonate, PSS,or poly(diallyldimethylammonium chloride), PDDA, respectively) as templates,PS/PANI core/shell particles, PANI hollow spheres, PANI/PS nanocomposite andnanoporous PANI can be obtained due to the different growth mechanism [158].A modification of this procedure, i.e. by using templating PS nanoparticles self-assembled onto a PANI modified screen-printed electrode, leads to the formation ofPANI nanostructures with the shape of cauliflowers. These peculiar PANI nanoparti-cles have found an interesting application in an amperometric enzyme biosensor forhydrogen peroxide [159]. As a curiosity, an unusual brain-like morphology of PANIis obtained by using aniline/citric acid salts as the template in a gas/solid reactionusing chlorine gas as the oxidant [160].
Amphiphilic micelle of azobenzenesulfonic acid are often used as templatesfor tuning PANI morphology, obtaining nanofibers, rods, spheres, and tubes,depending on the polymerization conditions; the solid state properties of PANI arehighly dependent on the size and shape of the polymerization templates employedfor the synthesis [161]. Analogous procedure has been proposed for the preparationof PANI micro/nanostructures through the supramolecular self-assembly attained byprotonated PANI intercalated nanoclays; inter-chain hydrogen bonding, inter-planephenyl stacking and electrostatic layer by layer self-assembling between polar-ized alkyl chains aided by dopant anions (3-pentadecyl phenol-4-sulphonic acid,PDPSA) lead to PANI nanostructures [162, 163].
In a wide context, the synthesis of inherently conducting polymers, PTh,PANI and PPy, and their properties and applications (capacitance, sensors,artificial muscles, biomolecular interactions, cell growth) related to the attainment ofnanodimension, have been reviewed and a section is dedicated to physical templates(pore sized membranes, synthetic opals) that induce the doped polymer fibrillarmorphology [164].
A peculiar type of templates is represented by phospholipids. Lipid tubules wereintroduced by Schnur and coworkers [165] a couple of decades ago and morerecently have been investigated as promising templating materials for the selectivegrowth of PPy nanostructures which surprisingly self-assemble at the edges (not atthe surface) of the phospholipidic tubules [146].

1 Nanostructured Macromolecules 23
PPy inverse opal patterns (ordered two-dimensional rings, hexagonal or honey-comb monolayers) over wide areas are accomplished by using a colloidal templatemethod. The templates are made of poly(styrene/sodium p-styrene sulphonate) latexparticles that drive the opal structure upon modulation of their packing density, thusinducing a modulation of the polymer properties [166, 167].
Unlike hard templates (alumina, zeolites, etc.) which require many syntheticsteps, surfactant templates may be a convenient alternative. The morphology ofPANI and PPy (spheres, wires, flat films) can be modulated though the useof adsorbed surfactants aided by co-adsorbing molecules; aligned nanowires ofPANI produced by this template assisted method can be self-assembled over largeareas for the improvement of microelectronic and sensor devices, as depicted inFig. 1.11 [139].
In this framework, reverse micelle also show easy feasibility for tem-plate self-assembly. For example, reverse cylindrical micelle systems were pre-pared from aggregates of sodium bis(2-ethylhexyl) sulfosuccinate, containing ananometer-sized water pool in the oil phase, and have been successfully exploitedfor the template oxidative polymerization of PPy nanotubes [107].
Fig. 1.11 Illustration of the process to fabricate morphologically controlled nanostructures of elec-trically conducting polymers on surfaces by using surfactant templates. This particular schematicdraw represents the proposed scheme of wire formation on (a) chemically treated HOPG and (b)HOPG (Reprinted with permission from Carswell et al. [139]. Copyright 2009 American ChemicalSociety)
1.3.4 Nanopatterning of Polymers (Top Down Methods)
From the point of view of the applicability of nanopatterning to a wide range ofmaterials, patterns of selected shape can be fabricated by assembly of nanopar-ticles without covalent interactions, as stated in a paper that provides the mainfeatures related to this topic; the assembly is performed: (i) in the absence of specific

24 M.V. Russo et al.
interactions, depending on the relation between the particle size and the patternfeature size, shape of the confining features and type of confinement; (ii) in the pres-ence of electrostatic interactions which act trough chemical functionalization on thenon-covered areas of the substrate to link nanoparticles with the appropriate com-plementary functionalization; (iii) in the presence of supramolecular interactionsthat involve host-guest chemistry [168].
In the case of polymeric materials, the patterning is in general based on twonanolithografic methods: reactive ones and non reactive ones; in the first approachthe polymer is synthesized during the patterning, in the latter one the polymer isdeposited or modified by a local perturbation.
The nanopatterning of conjugated polymers is based on general requirementssuch as the control of the dimension and position of the structures that aredeposited. The hierarchical organization of the macromolecules across multiplelength scales allows supramolecular charge transport and integration in electronicdevices [89]. The patterning reactive techniques are extensively mentioned in theliterature as Area Selected Polymerization, Chemical Amplified Soft-Lithography,Photochemical Patterning (where the patterning feature is a defined stamp) andnon reactive patterning as well, Microcontact Printing (by using the polymer as“ink”), Microtransfer Molding, Lithographically Induced Self-Construction, GridAssisted Self-Organization, Inkjet Printing, Lithographically Controlled Wettingand Nanorubbing, among the most popular.
Diblock copolymers self-organize to form patterns through minimization of freeenergy, i.e. trough a procedure called microphase separation. The up to date fea-tures and methods leading to high resolution patterning performed with polymerself-assembly at IBM are reviewed [48]; the processes for pattern orientation andtransfer, the engineering of polymer based patterns for the development of opticalwaveguides, fabrication of nanoporous membranes, the improvement of patterns forhigh resolution lithography and flash memory transistors are extensively presentedwith a particular emphasis to the expectations in future technology advances.
Colloidal nanolithography, deep silicon etching and nanomolding are the tech-niques used to achieve fibrillar polymer structures which mimic the gecko foothairs; these nanofibrils are densely packed, perpendicular and strongly adhesiveto a synthetic surface, and due to these characteristics are promising materials forintegration in flexible membranes and exploitation of new adhesives [169].
Direct Laser Interference Micro-Nanopatterning (DLIP) has been used to buildnanometer sized PANI arrays (as thin as 600 nm) self-assembled on dielectricpolymers; the width of the polymer lines can be modulated by changing the laserbeam intensity, without loss of the chemical and electronic properties of PANI. It isinteresting that the dielectric substrate can be ablated, exploiting its optical proper-ties at the working wavelength. The authors believe this technique relevant for thedevelopment of polymer based sensors [170].
The Electrochemical Dip-Pen Nanolithography (E-DPN) leads to direct writingof PTh nanowires (diameter less than 100 nm) on the surface of semiconductingor insulating materials, thus allowing the fabrication of complex structures whichare proposed for the design of devices with multipurpose applications (electronics,defense, pharmaceutics, and biotechnology) [171].
1 Nanostructured Macromolecules 25
1.4 Grafting Polymerization
1.4.1 General Features
The nanotechnology applied to chemistry provides new tools for the research in thefield of macromolecules and emerged from the desire of control on the physical andbiological functions of materials at the molecular level and of radically improvingthe physical properties of traditional materials.
Chemical surface modification with grafted polymers is a well known methodfor the tailoring of the surface properties of polymeric and ceramic membranes,thus improving their performance. In recent years, porous inorganic oxide substrates(e.g. silica, zirconia, or alumina) have been proposed as chemically and thermallystable materials for the graft polymerization processes. The covalent bonding andthe nanostructure of the polymeric phase deposited onto inorganic substrates hasbeen used to create membranes that can resist to swelling effects and operate at hightemperatures.
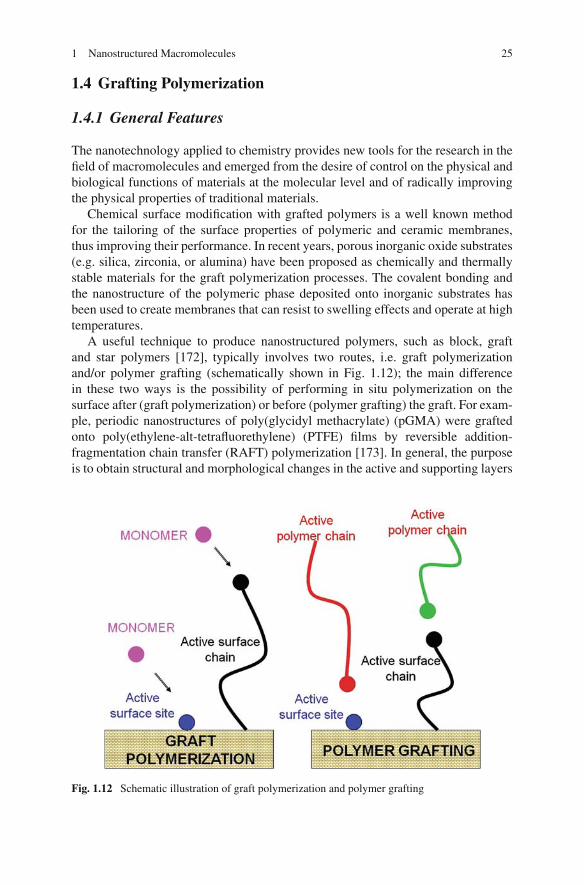
A useful technique to produce nanostructured polymers, such as block, graftand star polymers [172], typically involves two routes, i.e. graft polymerizationand/or polymer grafting (schematically shown in Fig. 1.12); the main differencein these two ways is the possibility of performing in situ polymerization on thesurface after (graft polymerization) or before (polymer grafting) the graft. For exam-ple, periodic nanostructures of poly(glycidyl methacrylate) (pGMA) were graftedonto poly(ethylene-alt-tetrafluorethylene) (PTFE) films by reversible addition-fragmentation chain transfer (RAFT) polymerization [173]. In general, the purposeis to obtain structural and morphological changes in the active and supporting layers
Fig. 1.12 Schematic illustration of graft polymerization and polymer grafting
26 M.V. Russo et al.
of the material upon chemical modification. Apart from the chemical modifica-tions, the research was devoted to assess the effect of modifications on the surfaceroughness by using nanomorphology.
Fundamental for the characterization of polymer grafting surface are microscopytechniques, i.e. Atomic Force Microscopy (AFM), Scanning Electron Microscopy(SEM), Transmission Electron Microscopy (TEM), Attenuated Total Reflection–Fourier transform infrared spectroscopy (ATR-FTIR spectroscopy).
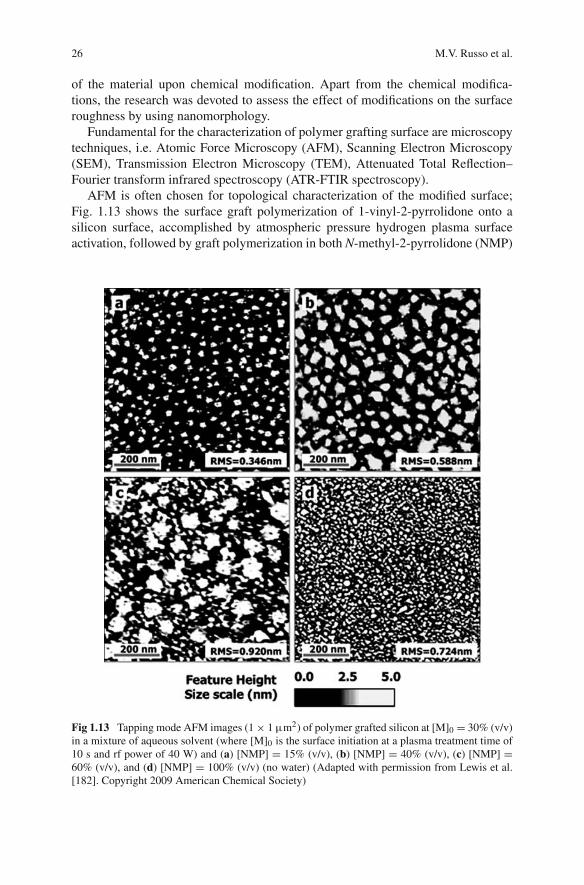
AFM is often chosen for topological characterization of the modified surface;Fig. 1.13 shows the surface graft polymerization of 1-vinyl-2-pyrrolidone onto asilicon surface, accomplished by atmospheric pressure hydrogen plasma surfaceactivation, followed by graft polymerization in both N-methyl-2-pyrrolidone (NMP)
Fig 1.13 Tapping mode AFM images (1 × 1 μm2) of polymer grafted silicon at [M]0 = 30% (v/v)in a mixture of aqueous solvent (where [M]0 is the surface initiation at a plasma treatment time of10 s and rf power of 40 W) and (a) [NMP] = 15% (v/v), (b) [NMP] = 40% (v/v), (c) [NMP] =60% (v/v), and (d) [NMP] = 100% (v/v) (no water) (Adapted with permission from Lewis et al.[182]. Copyright 2009 American Chemical Society)

1 Nanostructured Macromolecules 27
and in an NMP/water solvent mixture. SEM is often a complement of AFM andpoints out the attention on the mesoscopic morphology of the modified surface.TEM seems to be the most suitable technique to visualize the internal structure ofthe non-modified and modified active layer of a surface, due to its high resolutionand chance to achieve contrast between the areas having different chemical struc-ture: the latter property is most easily achieved through selective incorporation ofheavy elements. ATR–FT-IR spectroscopy provides an easy and convenient wayfor the determination of the relative amounts of different polymeric species presentin the outmost part of a polymer grafting surface; the depth of penetration of thereflected IR beam in the ATR technique is typically somewhat below 1 μm andtherefore the observed spectrum represents the average composition of this layer.Since the thickness of the active layer in the polymer grafting surface is far below1 μm, the method proves to be highly suitable for analyzing the active layer ofmodified and non-modified polymer grafting surfaces. The relative amount of thepolymers is determined by analyzing the IR adsorption bands specific for each poly-mer. This specificity renders the method sensitive even to relatively small amountsof the polymers attached to the surface.
Other typical features of grafted materials are investigated by studying thehydrophilic modification of the surface of the samples assessed, for example, bymeasuring the water droplet contact angle and the water droplet adsorption time.
The graft copolymerization plays a key role in the field of nanotechnologymainly because of the synthetic flexibility. In graft copolymerization the controlover the polymerization reaction is driven by important features: when the num-ber of growing chains is constant and chain transfer or termination reactions areavoided, the functional groups at the polymer terminus will be maintained, allow-ing for additional chemistry to take place. Such transformation reactions resultin the production of a macroinitiator which can initiate the polymerization ofa different monomer, thereby producing block copolymers; if a difunctional ini-tiator is used, the same technique can be applied toward triblock copolymerssynthesis. Furthermore, the presence of a functional group on a monomer inconjunction with another monomer in a statistical copolymerization results inpendant species which can be transformed to initiate the polymerization of graftcopolymers.
Of particular interest are copolymers which contain an inorganic block thatallows to exploit the best properties of the individual materials and to generate newclasses of compounds. For example, polysiloxanes show high oxygen permeabilityand favorable water and weather resistance, and polyphosphazenes exhibit a broadrange of physical properties leading to applications in biomedicine as well as flameretard, based on the substituents bonded to phosphorus.
For carbon-based vinyl monomers, controlled polymerization has been tradition-ally achieved by ionic mechanisms [174]. The living anionic polymerizations ofstyrene and methyl methacrylate are quite common, resulting in preservation of thepolymer functionality. However, alike the inorganic analogues the ionic polymeriza-tion mechanism is limited to a rather narrow class of monomers, under conditionsof the most stringent purity. Therefore, the aim to develop a controlled free radical

28 M.V. Russo et al.
polymerization system has driven the research in this area during the last decadeand atom transfer radical polymerization (ATRP) has been proposed as a favourableroute for graft polymerizations. If an activated alkyl halide is stirred with a vinylmonomer in the presence of a copper catalyst, well-defined polymers are obtainedwith predetermined functionality and molecular weight, making them ideal for thesynthesis of a variety of copolymers. The key of the control is the achievement ofa rapid equilibrium between active and dormant propagating species. The main-tenance of a low steady-state concentration of radicals ensures that terminationreactions are limited to nearly insignificant values until very high monomer conver-sions are attained. ATRP has demonstrated to provide controlled polymerizations ofmonomers such as styrenes, acrylates and methacrylates [175].
A large number of different methods for the achievement of surface graft poly-merization were developed including several patented techniques (often based onsurface activation with organosilanes), and controlled free-surface graft polymer-ization or plasma-assisted graft polymerization. The yield of polymer graft, as wellas chain length and density, are sensitive functions of the reaction conditions.
The engineering of the surfaces, consisting of a terminally anchored polymerlayer, requires a careful control of the surface density and molecular weight of thepolymeric chains. The resulting physicochemical and transport properties of thegrafted polymer layer depend on the conformation and topology of the tetheredpolymer phase. The polymer chain configuration will vary from the extreme brushlike configuration (for high density coverage) to separated chains in the so-calledmushroom regime.
In order to control the structure of the grafted polymer phase (both chain densityand chain length) in nanosize dimension, various graft polymerization methods havebeen proposed and hereafter reported:
• Free radical graft polymerization• Plasma surface treatment and Plasma-Induced Graft Polymerization (PIGP)• Atom Transfer Radical Polymerization (ATRP)
1.4.2 Free Radical Graft Polymerization
Grafted polymers offer unique opportunities to tailor and manipulate interfacialproperties and produce nanostructured devices while retaining the basic mechanicalstrength and geometry of the supporting solid substrate. For example, a substrate canbe modified with a polymer, which is completely miscible with the surrounding fluidmedium, mean while the polymer detachment is prevented by the covalent attach-ment of the polymer chains to the substrate. Surface engineering can be achieved byeither physically adsorbing or chemically bonding functional polymer chains [176].A tethered polymer phase can be formed either by polymer grafting (“grafting to”)or graft polymerization (“grafting from”) [177, 178]. Surface chain coverage andspatial uniformity achieved by polymer grafting may be limited by steric hindrance.

1 Nanostructured Macromolecules 29
In contrast, graft polymerization proceeds by sequential monomer addition, therebyallowing for the formation of a denser surface coverage. Among several methodsfor covalent bonding of polymer chains onto a substrate, free-radical surface graftpolymerization is a simple strategy to obtain a high surface coverage.
Free-radical graft polymerization typically involves the formation of both freepolymer chains (in the solution) and grafted polymer chains (on the substrate). Inthis approach, the sequential monomer addition to the surface occurs through thepropagation growth of terminally anchored surface chains (surface propagation)and coupling termination reactions between free polymer chains and growing sur-face chains (polymer grafting). In polymer grafting, the homopolymer radicals mustdiffuse toward the solid surface to react with the grafted polymer radicals. As aresult, the diffusion limitations of macromolecules may reduce the contribution ofpolymer grafting to the overall polymer graft yield. In contrast, in surface propaga-tion, diffusion and steric limitations are diminished because of the smaller size ofthe monomer molecules.
Free-radical method usually requires a surface activation by a direct attachmentof initiator molecules or by the introduction of surface active sites (i.e., vinyl groupsin surface graft polymerization of vinyl monomers). In particular, organosilane cou-pling agents (i.e., chloro- and alkoxysilanes) are commonly employed to introduceactive sites onto inorganic oxide surfaces.
For example, modification of amorphous silica surfaces with organosilanes hasbeen well studied in both gas and liquid phases for applications such as adsorption,adhesion and chromatography [179, 180]. Gas-phase silylation typically results in alower conversion and is cumbersome when a large scale silylation is desired. Liquid-phase silylation, can be performed in water or in anhydrous environment, and thechoice of solvent greatly affects the resulting silylation coverage.
Specifically, the chloro and alkoxy groups of multifunctional organosilanesundergo bulk hydrolysis and condensation, forming polysilane networks in an aque-ous environment prior to depositing onto the substrate. As a result, the fraction ofinitial surface silanols that reacts with the functional organosilane is quite small, andthe silylation process is usually non-uniform and difficult to control [181].
In contrast, in an anhydrous silylation reaction (i.e., in xylene) of a hydrated sil-ica substrate, condensation and hydrolysis between one or more functional groupsof neighboring silane molecules occur mainly on the surface with a minimal inter-condensation between silane molecules in the bulk phase. As a consequence, thislatter technique leads to a more dense and uniform silylation coverage.
After the whole activation, the vinylsilane-modified substrate can be free-radicalgraft polymerized with a desired functional monomer, producing polymer chainsthat are chemically bonded to the substrate along with homopolymer chains in solu-tion. In this step, the formation of grafted polymer chains is typically attributedto both propagation of growing surface chains (surface propagation) and couplingtermination between growing homopolymer chains and growing surface chains(polymer grafting).
Grafting with poly(vinyl acetate) is of particular interest since the grafted poly-mer layer can render the modified substrate hydrophobic or hydrophilic (by a

30 M.V. Russo et al.
post-grafting hydrolysis converting poly(vinyl acetate) into poly(vinyl alcohol) ata desired degree of hydrolysis).
There has been a growing interest in the surface modification of inorganicoxide substrates with covalently bonded polymer phases for a variety of practi-cal applications, such as filler–polymer control in polymer composites, supportpacking for liquid and gas chromatography, biocompatible surfaces, colloid stabil-ity and modified inorganic membranes, and for fundamental studies of interfacialphenomena.
1.4.3 Plasma Surface Treatment and Plasma-Induced GraftPolymerization (PIGP)
Surface nanostructuring by grafting functional polymers to a substrate surface isa surface modification approach that provides the enhancement of the chemicalfunctionality and alters the surface topology of native inorganic and organicmaterials [182].
Plasma surface treatment, which is used for metal oxide surface etching/cleaningin microelectronics, [183] has been proposed in several studies as a suitableapproach to both alter the surface chemistry and potentially supplant previoussolution-phase initiator strategies with high-density surface activation. Early stud-ies have focused on the use of plasma treatment to modify the surfaces in orderto reduce the adsorption of organics and biofoulants in separation membranes, toimprove the surface wettability in microcontact printing for poly(dimethylsiloxane)(PDMS) stamps, and to enhance the adhesive bonding strength in advanced mate-rials [184]. It was demonstrated that this versatile and environmentally benigntechnique has the propensity of modifying the surface chemistry with high effi-ciency for both organic and inorganic materials. Plasma treatment alone, however,proved to be an insufficient surface modification tool, and it has been noted thatpolymeric plasma-treated surfaces do not retain their modified chemical proper-ties over time and to air exposure. Vapor-phase plasma polymerization, in whichthe monomer feed through plasma is initiated in the gas phase and then themonomer polymerizes on a substrate surface, has also been investigated as asurface modification method [185]. Furtherly, surface-adsorbed radical monomerspecies, which are designed to polymerize with condensing monomer radicalsfrom the vapor phase, may be modified by continuous plasma bombardment,leading to highly cross-linked, chemically and physically heterogeneous polymerfilms, noncovalently adsorbed to the surface. It must be considered that the localconcentration of monomer species in the plasma afterglow is highly dependenton the radial dimensions of the plasma source, and the resulting spatial varia-tions in monomer deposition rate may lead to non uniform film structure andmorphology.
Plasma-induced graft polymerization (PIGP) is an alternative surface modifi-cation approach in which plasma is used to activate the surface; the monomer

1 Nanostructured Macromolecules 31
in the liquid phase is sequentially grafted to the initiation sites via free radicalgraft polymerization. This approach allows to engineer a grafted polymer phasecharacterized by a high surface density of polymer chains that are initiated andpolymerized directly from the substrate surface, thus minimizing polydisperse chaingrowth and improving the stability under chemical, thermal, and shear stresses[186]. To date, PIGP has focused primarily on low-pressure plasma initiation andsurface grafting onto polymeric materials, with limited studies on inorganic oxides[187].
Various studies have inferred and quantified, through surface binding assaysusing radical scavengers such as 1,1-diphenyl-2-picrylhydrazyl (DPPH), the pres-ence of surface radicals that serve as initiators for graft polymerization. Thesestudies have also reported that the surface radical number density that results fromplasma treatment can be controlled and optimized by tuning the plasma treatmenttime and the radio frequency (rf) power of the plasma generator.
Moreover, an excessive treatment time and/or rf power results in poor surfaceactivation, plausibly, as argued by Choi, because of the formation of stable inactivespecies [188].
A notable limitation for the achievement of PIGP on inorganic substrates, unlikepolymeric materials, has been the requirement of a sufficiently dense layer of surfaceactivation sites, created through silylation and macroinitiator grafting, that may formsurface radicals for the polymer initiation upon plasma treatment [189]. Given thecomplex surface chemistry and limited lifetime of reactive plasma-initiated surfacespecies, the exact chemical nature of these plasma-generated organic moieties needsto be established yet.
The surface preparation required for such technique combined with the diffi-culties related to surface hydroxyl chemistry limits the large-scale adaptation ofthis method and the level of chain density that can be achieved. Direct plasma ini-tiation and grafting without the use of surrogate surfaces has been demonstratedqualitatively on titanium oxide particles and silicone rubber materials [190] withcharacteristic surface radical formation noted as a function of treatment time andrf power, similar to that assessed for organic materials. Kai and coworkers [191]demonstrated that, under low-pressure plasma surface treatment of Shirasu porousglass, a direct correlation between silanol density and grafted polymer density isobserved. This suggests that the number density of surface radicals that may be pro-duced in the low pressure plasma surface activation of inorganic oxide substrates,may be limited by the native oxide surface chemistry. These findings, combined withthe added requirement of ultrahigh vacuum chambers necessary for low-pressureplasma processing, indicate that the current approach is insufficient for achiev-ing high-density surface activation and graft polymerization for large surface areamodification of inorganic substrates.
In a recent study [182], an atmospheric plasma (AP) composed of a mix-ture of hydrogen (1 vol%) and helium was used to activate silicon substratesdirectly, creating surface-bound radicals that can then initiate the liquid-phase graftpolymerization from these anchoring sites (see Fig. 1.14).

32 M.V. Russo et al.
Fig. 1.14 Illustration of multistep process plasma-induced graft polymerization (Reprinted withpermission from Lewis et al. [182]. Copyright 2009 American Chemical Society)
The AP plasma source selected in this study operated at a low breakdown volt-age, produced a highly uniform glow discharge, and maintained low processing gastemperatures (<80◦C), which is advantageous for the graft polymerization onto ther-mally sensitive materials. The monomer chosen for this study, 1-vinyl-2-pyrrolidone(VP), is of interest because poly(vinyl pyrrolidone) has excellent biocompatibleproperties, has been proposed as a surface modifier to reduce membrane fouling,and is miscible in both aqueous and organic media. Experimental results are reportedfor both plasma surface initiation and VP graft polymerization, focusing on the con-trol and optimization of the surface initiator density and on the impact of surfacegraft polymerization conditions on the resulting surface topology of the terminallyanchored polymer surface layer.

1 Nanostructured Macromolecules 33
Moreover, active polymer radicals generated by electric discharges initiate thegraft polymerization of a vinyl monomer, and a composite structure can be formedon the polymer surface.
Glycidyl methacrylate (GMA) [192] has a pendant epoxy group, through whichthe grafted polymer can be chemically coupled with a selected compound, havinga functional group such as –OH; cyclodextrin (CyD) is composed of a cyclic ringstructure with six to eight glucopyranose units, and has a unique chemical prop-erty holding a molecule in the ring cavity. CyD contains many –OH groups, andthe molecule can be chemically bound to the GMA grafted polymers through theopening of the epoxy group. More interests have been paid to the applications ofthe CyD-immobilized materials such as the expected ability of selective adsorptionby formation of inclusion compounds. The reported examples highlight how thematerial’s function is determined by the chemistry of the grafted polymers.
1.4.4 ATRP: Atom Transfer Radical Polymerization
ATRP or Atom Transfer Radical Polymerization is a powerful synthetic techniquein polymer science and involves free radicals; it was introduced as an extension toATRA or Atom Transfer Radical Addition by Jin-Shan Wang and Mitsuo Sawamotoin the nineties.
As a living radical polymerization which is a form of living polymerization, itallows the reaction to be carried out in a controlled way, and can be used to obtainpolymers with high molecular weight and low polydispersity index, besides longrange order of the nanophases.
This control is accomplished by the use of a transition metal based catalyst. Thiscatalyst provides an equilibrium between active, and therefore propagating, polymerand an inactive form of the polymer known as the dormant form. Since the dormantstate of the polymer is vastly preferred in this equilibrium, side reactions are sup-pressed. By lowering the concentration of radicals, the termination is suppressedand the control is achieved.
Similarly to the other controlled-living radical polymerization (CRP) methods,ATRP allows the synthesis of polymers with desired composition and moleculararchitecture. The polymers prepared by ATRP are highly chain end-functionalizedand can therefore participate in various post-polymerization modifications and serveas macroinitiators in the synthesis of block copolymers. Several nanostructuredpolymers and organic/inorganic nanocomposites have also been synthesized bythis technique [193, 194]. For example Fig. 1.15 shows a series of well-definedliquid crystalline (LC) homopolymers and amphiphilic LC-coil diblock copoly-mers with functional azobenzene units and narrow polydispersities, synthesizedby using the ATRP method. Among block copolymers, cylinder and/or spheri-cal morphologies of PEG block dispersed into LC block were clearly observedafter annealing at 105◦C (smectic phase) for 24 h. The sizes of the separatedstructures are in the range of 10–20 nm, increasing with the increase of LCfraction [195].

34 M.V. Russo et al.
Fig. 1.15 TEM micrographs of the amphiphilic LC-coil diblock copolymers with a defined lengthof a flexible poly(ethylene glycol) segment as the hydrophilic coil, prepared by the ATRP method.PEG blocks exhibit as cylinders and/or spheres with a width/diameter about 2–3 nm dispersed intothe LC matrix: PEG block appeared as dark and the solid lines in a, b, and c represent the domainboundaries (Reprinted with permission from Tian et al. [195]. Copyright 2009 American ChemicalSociety)
A general mechanism for ATRP is shown in Fig. 1.16.The homolytic cleavage of the alkyl (pseudo)halogen bond of (Pn X) by a tran-
sition metal complex in the lower oxidation state (Mtm Lz) generates an alkyl
radical (Pn•) and a transition metal complex in the higher oxidation state (XMt
m+1 Lz). The formed radical can initiate the polymerization through additionacross the double bond of a vinyl monomer, promote the propagation and induce thetermination by either coupling or disproportionation reactions, or can be reversiblydeactivated by the transition metal complex in the higher oxidation state. The for-mation of radicals during the ATRP process is reversible. An example is the ATRPfor the polymerization of styrene, initiated with alkyl halide 1-phenylethyl chloride,giving PnX, in the presence of the activator CuBr complexed by 2,8′-bipyridine(Mt
m Lz). Furthermore, their stationary concentration is low because the equilib-rium between the activation (ka) and deactivation (kd) processes is shifted to the
Fig. 1.16 Mechanism for the metal catalyzed ATRP

1 Nanostructured Macromolecules 35
left-hand side, which reduces the termination reactions. As a result of persistent rad-ical effect, polymers with predictable molecular weights, narrow molecular weightdistributions and high functionalities have been synthesized [175].
ATRP technique allows the controlled polymerization of a wide range of vinylmonomers including styrenics, acrylates, acrylonitrile, vinyl acetate.
To obtain consistent results, special handling procedures are required, and thepreformed catalysts must be stored under an inert atmosphere; oxygen or other oxi-dants should be removed from the system prior to the addition of the catalyst inthe lower oxidation state because the process of catalyst complex handling can bechallenging.
Although the ATRP is a convenient method for the synthesis of block copoly-mers, however it undergoes some limitations, such as the easy oxidation of thetransition metal complex (Fig. 1.17a). To rise above this drawback, the reverseATRP was developed (Fig. 1.17b); it uses the more stable Cu(II) complexes in theinitiating step. However, a drawback occurs also in reverse ATRP: since the transfer-able atom or group (X) is added to the reaction as part of the copper salt, thereforehighly active catalysts should still be used in the amount comparable to the con-centration of the radical initiator; for this reason, complex concentration cannot beindependently reduced and block copolymers cannot be formed.
Fig. 1.17 Methods for conducting ATRP (Reprinted with permission from Jakubowski andMatyjaszewski [175]. Copyright 2009 American Chemical Society)

36 M.V. Russo et al.
Simultaneous normal and reverse initiation (SR&NI) ATRP (Fig. 1.17c) wasdeveloped to allow the precursors of highly active catalytic complexes to be added tothe reaction in the higher oxidation state and at lower concentration. SR&NI ATRPcomprises a dual initiation system i.e. standard free radical initiators and initiatorscomprising a transferable atom or group in conjunction with the stable precursor ofan active catalyst complex. This initiation system can be used to prepare any typeof polymer that can be obtained by normal ATRP, and can be conducted in bulk,solution, emulsion, miniemulsion, and by heterogeneous polymerization.
Problems arise for SR&NI ATRP as well, since a standard free radical initia-tor is still added to the polymerization mixture to form radicals that reduce Cu(II).A new method for the formation of an active catalyst was recently introduced byJakubowski and Matyjaszewski to induce chain initiation [175]. The procedureconsists of the preparation of an “activator generated by electron transfer” for ATRP(AGET ATRP, Fig. 1.17d), which overcomes the previously cited problems by usingan electron transfer rather than organic radicals to reduce the higher oxidation statetransition metal. This procedure has all the benefits of normal ATRP plus the ben-efits of adding a more stable catalyst complex to the reaction mixture. The use ofoxidatively stable catalyst precursors can allow the more facile preparation, storage,and shipment of ATRP catalytic systems. The universal character of this approachwas proved by applying the new activation/initiation process to a wide range ofpolymers.
1.5 Electrochemical Methods
1.5.1 General Features
The electron transfer process is one of the most important synthetic methodsin organic and polymer chemistry [196, 197] and among several pathways forelectron transfer reactions, the electrochemical method is a straightforward andpowerful one requiring mild polymerization conditions. The reversible interchangebetween the redox states in the conductive polymer gives rise to the changes inits chemico-physical properties including polymer conformation, doping level, con-ductivity and color. New compounds have been synthesized in order to enhancethe stability and optimize and tune the electroluminescent properties of macro-molecules [198, 199]. Among the materials prepared by electrochemical synthesis,conducting polymers, such as poly(para-phenylenevinylene) (PPV), polythiophene(PTh), polypyrrole (PPy), polyaniline (PANI), polyacetylene (PA) and poly(3,4-ethylenedioxythiophene) (PEDOT) are nothe worthy. After the pioneering studieson the electroluminescence properties of PPV [200], conducting polymers havebeen deeply investigated in view of their potential applications in optoelectron-ics, for example as light-emitting diodes [201] or displays, energy storage devices,actuators, sensors, etc [202]. However, the interchange rate is usually slow, dueto the rate-determining process of counter-ion transport into the polymer layer

1 Nanostructured Macromolecules 37
for charge balance [203]. It usually takes a few 100 ms or more to completethe charge in a regular conductive polymer film [204]. This slow interconver-sion rate is the main obstacle for applications of these polymer films to devicesrequiring fast charge/discharge capability such as electrochromic devices andsupercapacitors.
Electrochemistry has recently emerged as a powerful tool in a different fieldof research, i.e. the preparation of nanostructured conducting polymers [205]; infact electrochemical synthesis offers several advantages with respect to chemicalpolymerization, first of all the easy control of the thickness of a deposited poly-mer layer on any conducting surface. In modern synthesis, mild conditions such aslow temperature and anhydrous atmosphere are often employed in order to achieveselectivity and high yields and electrochemical reactions generally fit this request.
Electrosynthesis is based on the use of an electrochemical cell and a requisitefor this technique is the use of electroactive molecules, for example thiophene, pyr-role and aniline among organic monomers. The experimental setup is based on agalvanic cell, a potentiostat and two electrodes [206], as schematized in Fig. 1.18.The monomer is solubilized typically into organic solvents or in aqueous media andusually an electrolyte (e.g. lithium perchlorate or tetrabutylammonium acetate) isadded. Platinum, carbon rods, magnesium, mercury, stainless steel can be used aselectrodes and the electrosynthesis can be carried out with constant potential or con-stant current. The choice of the electrodic material, its shape and size play a crucialrole in many electrochemical reactions.
The initial reaction takes place at the surface of the electrode and then the inter-mediates diffuse into the solution where they participate to secondary reactions. Theoxidations take place at the anode with initial formation of radical cations as reac-tive intermediates and the reductions occur at the cathode, with formation of radicalanions. At a sufficiently high positive (i.e. anodic) electrode potential, monomersundergo electrochemical oxidation, the polymerization process starts and cationradicals or other reactive species are formed.
Fig. 1.18 Initial steps in the electropolymerization of thiophenes

38 M.V. Russo et al.
The tendency of the substrates or monomers to undergo electron transfer is rep-resented by their oxidation and reduction potentials. The electron-transfer process ismore favorable when the oxidation potential is less positive or the reduction poten-tial is less negative. In order to achieve electron-transfer-driven reactions selectively,this process should occur choosy at the position that is needed for the subsequentchemical process, i.e. to cleave the specific bond or make a bond in the specific posi-tion in the starting molecule. From the viewpoint of synthetic efficiency and atomeconomy, the control of the electron transfer reactions can be achieved by the use offunctional groups and templates to drive and facilitate the selectivity in predictablemanner.
For example, in the case of polymerization of thiophene, scheme reported inFig. 1.18, the oxidation of the monomer produces a radical cation which can couplewith a second radical cation or with another monomer to produce a dication dimeror a radical cation dimer, respectively. By successive steps, polymeric chains growup and can be recovered.
The electron density in the π-system of the thiophene ring drives the potentialrequired to oxidize the monomer itself; electron-donating groups lower the oxida-tion potential, while electron-withdrawing groups increase the oxidation potential.For example, unsubstituted thiophene polymerizes at about 1.7 V vs. SCE (satu-rated calomel electrode), while 3-methylthiophene polymerizes at lower potentialdue to electron-donor effects and steric hindrance. This trend can be observed alsofor polythiophene with respect to thiophene monomer: the oxidation potential of themonomer is higher than the oxidation potential of the resulting polymer. This is oneof the critical points of the electrochemical polymerization, that makes the processdifficult to be performed in terms of regioregularity, stability and solubility of theproduct.
Functional groups that make the electron-transfer process more favorable areusually called electroauxiliary (EA) [207] and drive the oxidation potential tobecome less positive or the reduction potential less negative. From a molecularorbital point of view, the oxidation process can be explained by electron trans-fer from the highest occupied molecular orbital (HOMO) of a monomer to theelectrode. The increase of the HOMO enegy level by the introduction of an EAis the most straightforward method for activating the substrate toward oxidation.Alternatively it is possible to stabilize the radical cation generated by one-electronoxidation of the substrate. The choice of the best EA depends on the nature ofthe substrate and the reagent. Silyl derivatives, sulphides, stannyl or arylthio (ArS)groups serve as electroauxiliaries for the oxidation of heteroatom compounds [208].Electroauxiliaries based on intramolecular coordination can also be advantageouslyused: if a substrate molecule has a specific coordinating site that can be stabilized bythe developing of a charge, the electron transfer could be assisted by intramolecularcoordination [209]. As an example, the pyridyl group is effective as a coordinatinggroup for the oxidation of compounds containing heteroatoms such as O, S, andSe [210].
An advantage of the electrochemical polymerization is that the polymer doesnot need to be isolated and purified, but it produces structures onto the electrode

1 Nanostructured Macromolecules 39
surface with various degrees of structural irregularities. The nucleation and growthmechanism leading to the deposition of polymeric chains onto the anode has beenwidely studied; depending on the monomer concentration, electrolyte used, cur-rent density, temperature, solvent, electrode material, electrode potential and otherexperimental parameters, different morphologies for conducting polymers can beisolated. Nanostructured morphologies were obtained for example with the aid oftemplates during the polymerization process or, in general, with a proper control ofthe experimental parameters. In fact, the control of the growth process, starting fromthe nucleation of colloidal particles can be achieved by a fine tuning of the experi-mental conditions through which complex structures can be obtained. For example,disc-shaped nanoparticles of polyaniline have been prepared with the use of a pulsepotentiostatic method at a highly ordered pyrolytic graphite (HOPG) electrode, fromlow concentration of aniline solution. The diameters of the obtained discs were inthe range 20–60 nm depending on the variation of the electropolymerization chargeapplied, from 5.7 to 19.3 μC cm−2 [211].
The use of ultrasounds in chemical synthesis has also attracted significantresearch interest and has been applied in electrochemical polymerization. It is sug-gested that, in homogeneous solutions, the mass-transport process is acceleratedby ultrasounds because of the macroscopic streaming [212] and microscopic cav-itations phenomena [213]. The effect of ultrasounds on homogeneous electrolysisprocesses has been studied and a characteristic change in the distribution from one-electron to two-electron oxidation products was observed together with a fasterconversion under ultrasound conditions, although the detailed mechanism is notclear at present. For example, Kolbe electrolysis was found to be enhanced byultrasound under biphasic conditions, in emulsion systems [214].
Recent progress was achieved by using electrochemical reactors enabled toperform electrochemical reactions under high-temperature and/or high-pressureconditions [215], opening also perspectives to the use of supercritical fluids asreaction media. Supercritical fluids have attracted significant research interest asefficient reaction media and in particular, supercritical carbon dioxide (scCO2) hassignificant potential as an environmentally benign solvent. Supercritical conditionsfor CO2 can be readily attained (Tc = 31◦C, Pc = 7.3 MPa) [216] and scCO2 mightreplace hazardous organic solvents because it is non-toxic, inexpensive, misciblewith organic compounds, and non-flammable. In addition, scCO2 can be recov-ered and reused after the reaction. Early experimental studies on electrolysis inscCO2 by Silvestre and coworkers showed that carbon dioxide is poorly conductingunder supercritical conditions [217]. It was found, however, that the use of a smallamount of water as co-solvent led to sufficient conductivity and the voltammetry offerrocene in scCO2 containing tetrahexylammonium hexafluorophosphate could beachieved by the addition of water [218]. It has been reported that the scCO2/wateremulsion system is also effective for the electrochemical polymerization of pyrroleto form PPy and PTh films [219, 220]. Moreover, the use of scCO2 in electro-chemical polymerizations has other actractive features because it is well-known thatcarbon dioxide penetrates into the polymeric materials and often reduces interchaininteractions. Recently, homogeneous scCO2/cosolvent systems have been applied

40 M.V. Russo et al.
to electrochemical polymerization. For example, electrochemical polymerizationof pyrrole to form PPy films was successfully achieved in the scCO2/acetonitrilesystem [221].
1.5.2 Role of Reaction Media
Reaction media such as solvents and supporting electrolytes play a fundamental rolein the control of electrochemical reactions; in fact they serve as an environment formolecular events in the electron transfer processes. Among others, trifluoroethanolor trifluoroacetic acid and in general unconventional fluorine-containing organicsolvents, have recently received significant research interest and exhibited inter-esting features in the anodic oxidation of organic compounds. Ionic liquids andsupercritical fluids can also be used for organic electrochemical reactions.
Ionic liquids are salts that form a stable fluid at or near room temperature andare expected to replace hazardous and volatile organic solvents because they havelow vapor pressure, non flammability, high polarity and relative inertness. Mostionic liquids consist of bulky organic cations, for example N,N-dialkylimidazolium,N-alkylpyridinium, quaternary ammonium, quaternary phosphonium, and com-mon weakly coordinating anions such as AlCl4−, BF4
−, PF6−, CF3SO3
−, TfO−,(CF3SO3)2N− and some of them, such as N,N-dialkylimidazolium salts, showexcellent conductivity [222]. Typical chemical structures of cations and anions arereported in Fig. 1.19.
Ionic liquids can be recovered and reused after the completion of reactions,although there are some practical problems associated with these processes [223].
Ionic liquids themselves play the role of supporting electrolyte, and therefore,electrolysis can be conducted without any intentionally added electrolyte. The useof ionic liquids as electrolytes, doping agents, and practical recycling media forelectrochemical polymerization has been developed. It is evident that the electro-chemical synthesis of π-conjugated polymers takes also advantage of the fact thatthe anion component of the electrolyte can be introduced into the polymer as adoping agent to improve the conductivity.
An early investigation reports about reactions carried out in chloroaluminateionic liquids and π-conjugated polyarenes, polythiophene and polyaniline filmswere prepared in ionic liquids [224]. However, the use of chloroaluminate ionic
NN R'R + N R+
1-alkyl-3-alkylmidazolinum cations pyridinum cations
AlCl4 BF4 PF6 Cl (CF3SO3)2N typical anions–– – – –
Fig. 1.19 Typical chemical structures of cations and anions for ionic liquids

1 Nanostructured Macromolecules 41
liquids is problematic because their handling requires a special apparatus due totheir inherent moisture sensitivity. An important improvement has been achievedby using N,N-dialkylimidazolium compounds having stable counter anions suchas BF4
−, PF6−, and CF3SO3
−, which exhibit air and moisture stability [225].For example, electrochemical polymerization of pyrrole was carried out by using1-ethyl-3-methylimidazolium bis(trifluoromethanesulfonyl)amide, with a good con-trol of the morphological structure of the film formed on the anode, and markeddifferences between the morphology of the upper and lower surface of the poly-mer film were observed. SEM analysis of the polymer films grown by using voltagepulses (Fig. 1.20) indicate that the polymer appears first as a series of fibrils, whichcan extend over a significant portion of the film before filling to form a completefilm. This fine structure imparts a larger surface area to the polymer that can beprepared as a solid, homogeneous film [226].
The electrochemical synthesis of conductive polymers is accompanied by thedoping of counterions into the polymer network. Considering that the conductivepolymer properties are mainly affected by their doping states, it is of particularimportance the choice of an appropriate reaction medium for the electropolymeriza-tion in order to attain conductive polymers with the desirable properties for differentapplications.
Fig. 1.20 SEM images ofdifferent fine-structuredpoly(pyrrole) films formed onthe ionic liquid surface byusing voltage pulses (100 mspulses, top; 10 ms, lowerfour) (Reprinted withpermission from Pringle et al.[226]. Copyright 2009American Chemical Society)

42 M.V. Russo et al.
For example, aniline monomer can be easily oxidized in the presence of var-ious electrolytes but good conductivities can be achieved only at low pH values,suggesting the use of acidic media for the electrochemical synthesis of polyaniline.Different acids have been used for this purpose and among others, perchloric orsulphoric acids are common examples [227]. Less attention has been paid to the useof phosphoric acid; however, a mixed electrolyte based on H3PO4 and H2SO4 mix-tures has been used for the electropolymerization of aniline, giving rise to films withgood stabilities and grain like morphologies. It has been assessed that H2SO4 actsas a required agent for successful polymer growth, whereas H3PO4 has an action asdoping agent [228].
Environmentally friend and electrochemically stable waterproof ionic liquidslike imidazolium cations with stable anions were also used. It is known that,unlike highly polar media, a hydrophobic medium facilitates the deposition of uni-form well-adherent thick films. For example, the ionic liquid used to synthesizepoly(3,4-ethylenedioxythiophene) (PEDOT) films leads to the formation of ran-domly oriented nanofibers and particles confined to submicrometer-sized domainsin the film microstructure [229].
The role of the electrosynthesis parameters (monomer concentration and elec-trolyte nature) on the kinetics and on the morphology of the polymeric nanoma-terials has been deeply investigated with particular attention to the incorporationof polyelectrolytes into conducting polymers, in view of potential application inthe biomedical field. Composite films of polypyrrole and polysaccharides (hep-arin, hyaluronic acid and derivatives) were electrosynthesized as nanotubes and itwas demonstrated that the conductivity and the morphology of the resulting poly-mers strongly depend on the negative charge distributions in the polysaccharidemacromolecules backbone [230].
1.5.3 Nanowires and Nanotubes by Template and TemplateFree Methods
Conductive polymeric nanostructures can be prepared by using hard or soft tem-plates or with template-free methods. The template method has been extensivelyused because of its simplicity, versatility and controllability. Some further featureson this topic are reported in Section 1.3. A typical hard template material can bea thin porous film of aluminum oxide or polycarbonate and polymeric materialscan be deposited into the pores to form nanotubes or nanowires. The electrochemi-cal template method enables a better control of the dimensions compared with thechemical methods. In addition, the nanostructures produced by the electrochemi-cal method are in solid contact with a base electrode that is beneficial for furtherprocessing steps when building an electrochemical device.
Since the first report of nanowire synthesis by Possin in 1970, many nanowireshave been made, but the synthesis of nanotubes requires a delicate control ofexperimental parameters, such as concentration and reaction times. Only in the

1 Nanostructured Macromolecules 43
90thies Martin and co-workers prepared nanotubes in template pores [231]. Theyhave shown that PPy nanofibers (diameters ca. 200 nm) have higher charge trans-port rates with respect to a conventional film under the same conditions. Recently,Demoustier-Champagne and co-workers chemically and electrochemically syn-thesized nanotubes and nanowires of polyaniline, polypyrrole, and poly(3,4-ethylenedioxythiophene) [232]. Joo and co-workers have also electrochemicallysynthesized partially filled, long (up to 40 μm) nanotubes of conductive polymerslike PANI, PPy and PEDOT, by controlling the polymerization time and the current,for potential applications of these materials as nanotip emitters in field emissiondisplays and polymer-based transistors [233].
Usually, when a template is used, the electrodic surface is covered with an elec-trically insulating template having cavities with different shapes and sizes throughwhich the solute species get through and catch up with the electrode surface. Theelectropolymerization starts at the electrodic surface, and propagates through thepores towards the outside solution. Nanowires or nanofibrils of different length,thickness and shape can be obtained although the nature of the nanosized tem-plates and the control of the electrodic potential and solution concentration arefundamental for the attainment of nanofeatured films.
It has been observed that an increase of electropolymerization rate occurs at acontrolled electrode potential with increase of the feed solution concentration [234]and the thickness of PPy nanotubules strongly depends on the pore size of the tem-plate and on the kind of used electrolyte [235]. Moreover, the increasing of theelectric conductivity of polypyrrole nanotubules with respect to the bulk materialhas been observed, probably due to an increased conjugation length in polypyrrolemolecules packed into nanotubules. The increase in conductivity was observed alsofor other conducting polymer based nanotubes, probably because the polymeriza-tion inside the confined space of the pores, combined with electrostatic interaction,ensures the alignment of the resulting polymers on the walls of the pores [236]. Indepth SEM studies of electrochemically prepared polyaniline suggested that the for-mation of hollow nanotubes can be driven by the deposition of polyaniline on thesurface of the pore walls during the electropolymerization process [237].
Among oxides, alumina template has been widely used because of its easy prepa-ration by anodic treatment of aluminum metal. Nanofibrils with uniform and wellaligned structure made of copolymers based on PANI and PPy have been preparedon this template [238] and aligned nanotubular heterojunctions of poly(p-phenylene)and PTh have been isolated; it has been demonstrated that the length and the wallthickness of the nanotubes can be controlled by varying experimental parameterssuch as the electropolymerization current, the duration, and the nature of the dopinganion present in the electropolymerization solution [239, 240].
Martin’s group proposed a mechanism based on electrostatic and solvophobicinteractions between the growing polymer and the pore walls to explain the nan-otube growth in template pores [241]. They observed that the interactions inducethe growing polymer to nucleate and the chains grow preferentially along the porewall to form tubular structures. As the polymerization proceeds further, the polymergrows inwardly and nanowires can be isolated.

44 M.V. Russo et al.
Wire-shaped growth of nanostructured PPy with diameter ≤10 nm, has beenobtained by electropolymerization at naturally occurring step defects and artifi-cially formed pit defects of HOPG, in a template assisted electropolymerizationwhere the size of the nanostructures could be controlled by limiting the pyrrolepolymerization time at anodic potentials [242]. Electrochemical polymerization ofpyrrole within the confines of anodized alumina templates and subsequent metalnanoparticles immobilization on the surface of polymer pillars has been used tomake surfaces that show roughness on two independently controllable levels: sub-microscopic roughness from polymer pillar dimensions and nanoscopic roughnessfrom the appropriate size selection of metal NPs [243].
A combined two-step method of preparing PPy nanowires was reported, consist-ing of (i) electrochemical grafting of a thin polyacrylate film at a carbon electrodeand (ii) electropolymerization of pyrrole, resulting in polypyrrole wires with diam-eter 600 nm and length 300 μm, growing through the pores of a polyacrylatefilm [244].
Polystyrene nanospheres can be also used as a specific template for the prepara-tion of nanocomposites with organic materials. In fact, the electropolymerizationcan be carried out within the voids of polystyrene nanospheres giving rise toPANI honeycomb films with desirable pore size, pore wall widths, and filmthickness [245].
The use of template assisted electropolymerization is based on the preparationof nanosized cavities, channels or holes, on the electrodic surface, which drivesthe deposition of the resulting polymeric layer. Recent papers deal with aspectsof template-based and templateless electropolymerization [246, 247]. Althoughthe template based electropolymerization allows the preparation of well definedconducting polymers with different dimensions and morphologies (nanotubes ornanowires), the costs of the template synthesis are high and the handling andremoving processes are relatively complex. In the case of templateless electropoly-merization, the choice of experimental conditions such as monomer concentration,reaction time and working electrode material and porosity is crucial for the con-trolled growth of conducting polymer colloids. Electrochemical assembly methodhas been reported for the preparation of close packed ordered nanostructuresof polyaniline in the presence of p-aminobenzenethiol [248] and nanodot basedarrays of poly(o-phenylenediamine) were grown on Au(111) surfaces and on ap-aminobenzenethiol-modified Au surfaces [249].
PANI nanowires-gold nanoparticles hybrid networks were recently prepared andused as chemiresistive sensors. Initially, polyaniline nanowires with a diameter of250–320 nm, bridging the gap between a pair of microfabricated gold electrodes,were synthesized by using templateless electrochemical polymerization with a twostep galvanostatic technique. Polyaniline nanowires were then electrochemicallyfunctionalized with gold nanoparticles by using the cyclic voltammetry technique[250] and tested as hydrogen sulphide sensors with excellent limit of detection(0.1 ppb).
Conductive polymer nanotubes with different inner morphologies ranging fromhollow nanotubes to solid nanowires were prepared, to develop fast color-switching

1 Nanostructured Macromolecules 45
electrochromic devices. Since the switching rate depends on the ion-transportrate occurring in the polymer layer, an easy pathway to slow interchange rateis to reduce the ion-transport resistance in the polymer. A suitable approach isto introduce molecular-scale porosity, by a proper tailor of the polymer struc-ture [251]. Up to now, the Reynolds group obtained the fastest electrochromicresponse (ca. 90 ms) by using a bulky conductive polymer such as poly(dimethyl-3,4-propylenedioxythiophene) [252].
These nanostructured materials show intrinsically high surface area and can leadto high charge/discharge capacities and short diffusion distances for ion transport,which in turns leads to fast charge/discharge rates. Among the others, the nan-otubular structure is particularly attractive because it is possible to control thecharge/discharge rate and charge/discharge capacity, by a proper choice of the wallthickness and length of the nanotubes. Furthermore, the inside surface of hollowtubes can be chemically modified to further enhance their functions [253].
A further controllable approach for preparing conducting polymer nanostruc-tures is the molecular template-assisted electrosynthesis, where some of the dopinganions or other active species used in a “templateless” synthesis can themselvesact like templates with molecular dimensions. In addiction, the electrode surfacecan be modified with an adsorbate and the electropolymerization is driven toproceed in a template-like manner. For example, a gold electrode modified withtwo component self-assembled monolayers consisting of p-aminobenzenethiol andn-octadecanethiol has been used for the growth of polyaniline nanostructures on p-aminobenzenethiol islands embedded into an octadecanethiol layer [254]. Similarlya gold electrode modified with thiolate β-cyclodextrin self-assembled monolay-ers have been exploited for the preparation of PPy films in acetonitril, where thepolymer growth sites are restricted within the β-cyclodextrin cavities [255]. PPyand PANI based nanodots and nanowires have also been prepared by using agold electrode modified with self-assembled monolayers of either β-cyclodextrinor p-aminobenzenethiol [256].
1.5.4 Aligned Nanostructures
One of the key factors to obtain high performance functional nanomaterials is theirordering at a large scale, providing the alignment and orientation of the nanostruc-tures. Oriented PANI nanowires, deposited in large arrays have been prepared by aproper control of the nucleation and growth process, by using a high initial currentdensity in a first step. A large number of polyaniline nuclei were firstly depositedonto the electrode and in a successive step, with a reduction of the polymeriza-tion current density, the growth of oriented nanowires was achieved [257]. PPynanowires have been obtained by one-dimensional growth at a graphite-paraffincomposite electrode from bidimensional nucleation sites onto electrodic surface[258]. Oriented PPy nanofibers with fairly uniform diameters (in the range of120–200 nm) and average length of up to microns were produced on a pre-roughedplatinum plate electrode. Tubules of PPy with diameters ranging from 0.8 to 2.0 μm

46 M.V. Russo et al.
and length 15–30 μm were prepared in the presence of either β-naphthalenesulfonicacid or p-toluenesulfonic acid as a doping agent, on a stainless steel electrode. Thegrowth mechanism has been studied and it was hypothized that an assembling ofmicelle of doping molecules or pyrrole–dopant clusters occurs, acting like templateson the electrode surface and giving rise to the tubules [259, 260].
By using a biphasic aqueous–organic system, one-step electrochemical strategyhas been fixed up to synthesize polypyrrole nanofiber arrays. The experimental pro-tocol ensures a very low concentration of pyrrole monomer in the polymerizationprocess and provides good control on the deposition of nanofibers with diametersranging from hundreds of nanometers to micrometers. In this procedure, the pyrrolemonomer was dissolved in the organic phase and slowly allowed to diffuse into anaqueous phase through the interface, and then PPy was deposited on the electrodeby electropolymerization. Owing to the very low concentration of the monomer nearthe electrode controlled by the interface, the lengths of PPy nanofibers could be eas-ily tailored [261]. Aligned polypyrrole nanofibers were produced with successiveCV electropolymerizations as schematized in Fig. 1.21.
Fig. 1.21 Schematic illustration of biphasic electropolymerization. Pyrrole diffused from organicphase into aqueous phase slowly (indirect diffusion). Then polypyrrole was after deposited onthe electrode by electropolymerization by the diffusion of pyrrole to the electrode (direct diffu-sion). Aligned polypyrrole nanofibers were produced with successive CV electropolymerizations(Reproduced with permission of The Royal Society of Chemistry, from Li et al. [261]. Copyright2009)
1.5.5 Coaxial Nanowires and Nanotubes
Coaxial nanowires and nanotubes have attracted great attention due to the poten-tial synergic properties or functionalities arising from the combination of differentmaterials in core/shell systems [262]. Among others, coaxial nanowires based on

1 Nanostructured Macromolecules 47
conductive polymers in the presence of transition metal oxides could be promisingelectroactive materials for electrochemical energy storage [263]. As an example,due to its high energy density and low cost, MnO2 is one of the most used aselectrochemical energy storage material, and to prepare coaxial nanowires with con-ducting polymers. MnO2/poly(3,4-ethylenedioxythiophene) coaxial nanowires wereprepared by co-electrodeposition in a porous alumina template. While MnO2 pro-vides high energy storage capacity, the highly conductive and flexible PEDOT shellfacilitates the electron transport and ion diffusion into the core MnO2 [264].
Controlled electrochemical synthesis of conductive polymer nanotubes in aporous alumina template has been studied as a function of monomer concentra-tion and potential; in the case of PEDOT the electropolymerization leads either tosolid nanowires or to hollow nanotubes depending on the template pore diameter,the applied oxidation potential and the monomer concentration [265]. Nanowiresare formed at slow reaction rate and high concentration monomer supply; in factmonomeric molecules should have enough time to diffuse into and fill the pores,from the bulk solution. On the other hand, nanotubes are predominantly formedwith fast reaction rate and low monomer concentration, because the monomers thatdiffuse from the bulk solution can be deposited along the pore wall thanks to theinteraction of the polymer with the wall surface.
Deep investigation on electrodeposition of 3,4-ethylenedioxythiophene (EDOT)as a model monomer and inner morphologies of PEDOT nanostructures synthe-sized at various monomer concentrations (10−100 mM) and applied potentials(1.0−1.8 V) have been reported and the growth mechanism is depicted in Fig. 1.22[251]. At potentials higher than 1.4 V the reaction is limited by the diffusion ofthe monomers and tubular portions increase along with the applied potentials andagainst monomer concentrations. If low potential and high monomer concentra-tion are used (e.g., 1.4 V, 100 mM EDOT), the polymerization reaction occurs onthe whole electrode surface without notable preference, because the monomer candiffuse into the pore bottom under the slow reaction rate, leading to rigid, dense
Fig. 1.22 Growthmechanism of PEDOTnanostructures based ondiffusion and reactionkinetics for high oxidationpotential region (≥1.4 V).Two extreme cases areconsidered: (a) slow reactionrate under sufficient monomersupply and (b) fast reactionrate under insufficientmonomer supply (Reprintedwith permission from Choet al. [251]. Copyright 2009American Chemical Society)

48 M.V. Russo et al.
nanowires. On the other hand, if high potential and low monomer concentration(e.g., 1.8 V, 10 mM EDOT) is used, the polymerization reaction becomes very fast,and monomer supply is not sufficient to fill out the pores. Long, porous, thin-wallednanotubes are obtained because the monomer is immediately consumed to elongatethe polymer chain. Since the reaction initiates along the electrode surface at thepore bottom, the conductive polymers are continuously deposited along the porewall. Other parameters, such as electrolyte concentration, pore diameter, templatethickness, stirring, temperature, also have influence on the inner morphology of thenanotubes. The growth mechanism of nanotubes was also studied at very low oxida-tion potentials (<1.4 V) where the reaction occurs very slowly. It was observed thatnanotubular structures are favored and are nearly independent of monomer con-centrations at potentials lower than 1.4 V. In this case, the morphology of baseelectrodes at the pore bottom becomes critical for the polymerization and the syn-thesis of PEDOT on the flat-top electrodes at the very low oxidation potential of1.2 V leads to solid nanowires.
1.6 Emulsion Polymerization
1.6.1 General Features
Emulsion polymerisation is a chemical technique widely used to produce particleswith various colloidal and physicochemical properties and with nanodimensions.
This is in general a heterogeneous free radical polymerization that involvesthe emulsification of the relatively hydrophobic monomer in water and some-times an organic phase-in-water emulsifier, followed by the initiation reaction witheither a water soluble initiator (e.g. sodium persulfate (NaPS)) or an oil-solubleinitiator (e.g. 2-20-azobisisobutyronitrile (AIBN)) [266]. Typical monomers usedin the emulsion polymerization include butadiene, styrene, acrylonitrile, acrylateester and methacrylate ester, vinyl acetate, and vinyl chloride, but also biopoly-mers are now obtained by this versatile technique in several mesodimensionatemorphologies [267].
In the common procedure extremely large oil–water interfacial area is gener-ated and the particle nuclei grow in size with the progress of the polymerization.Thus, effective stabilizers such as ionic and non-ionic surfactants and protective col-loids (e.g. hydroxyethyl cellulose and polyvinyl alcohol), which can be physicallyadsorbed or chemically incorporated onto the particle surface, are often required toprevent the interactive latex particles from coagulation. Under the circumstances,satisfactory colloidal stability can be achieved via the electrostatic stabilizationmechanism [268], the steric stabilization mechanism [269] or both.
The emulsion polymerization process is rather complex because nucleation,growth and stabilization of the polymer particles are controlled by the free radicalpolymerization mechanisms in combination with various colloidal phenomena.

1 Nanostructured Macromolecules 49
Perhaps, the most striking feature of emulsion polymerization is the segrega-tion of free radicals among the discrete monomer-swollen polymer particles. Thiseffect will greatly reduce the probability of bimolecular termination of free radi-cals and, thereby, results in a faster polymerization rate and yields polymers withhigher molecular weight. This advantageous characteristic of the emulsion poly-merization cannot be achieved simultaneously in bulk or solution. Although thenucleation period is quite short, generation of particle nuclei during the early stageof the polymerization plays a crucial role in determining the final latex particle sizeand particle size distribution and it has also a significant influence on the qualityof the latex products. The way to effectively control the particle nucleation pro-cess represents a very challenging task to those who are involved in this fascinatingresearch area. The transport of monomer, free radicals and surfactant to the growingparticles and partition of these reagents among the continuous aqueous phase, theemulsified monomer droplets (monomer reservoir), the monomer swollen polymerparticles (primary reaction loci) and the oil–water interface are the key factors thatgovern the particle growth stage.
The batch emulsion polymerization is commonly used in the laboratory to studythe reaction mechanisms, to develop new latex products and to obtain kineticdata for the process development and the reactor scale-up. Most of the commer-cial latex products are manufactured by semibatch or continuous reaction systemsdue to the very exothermic nature of the free radical polymerization and therather limited heat transfer capacity in large-scale reactors. One major differenceamong the above reported polymerization processes is the residence time distri-bution of the growing particles within the reactor. The broadness of the residencetime distribution in decreasing order is continuous>semibatch>batch. As a conse-quence, the broadness of the resultant particle size distribution in decreasing orderis continuous>semibatch>batch, and the rate of polymerization generally followsthe trend: batch>semibatch>continuous. Furthermore, the versatile semibatch andcontinuous emulsion polymerization processes offer the operational flexibility toproduce latex products with controlled polymer composition and particle morphol-ogy. This may have an important influence on the application properties of latexproducts [270].
Moreover the miniemulsion, microemulsion and conventional emulsion polymer-izations techniques show quite different particle nucleation and growth mechanismsand kinetics [271]
Despite several advantages (water as the dispersion medium is environmentallyfriendly) [272], i.e. higher polymerization rates [273] and the relative simplicity ofthe process, the emulsion polymerization involves many mechanistic events, andthe understanding of the events that dictate the rate of formation and the growth ofpolymer particles is difficult [274]. The mechanism of emulsion polymerization isshown in Fig. 1.23.
This topic is still studied and in recent papers Thickett and Gilbert focused theirinvestigations on the aspects concerning the mechanism of the emulsion polymer-ization, for both electrostatically and electrosterically stabilized particles [275, 276];various kinetic limits has been described to explain an emulsion polymerization

50 M.V. Russo et al.
Fig. 1.23 Full scheme ofkinetics process in a typicalemulsion polymerizationreaction (Reprinted fromThickett and Gilbert [274].Copyright 2009, withpermission from Elsevier)
system and their applicability under various conditions, and this allows accuratedetermination of the rate parameters.
The colloidal properties of latex products are of great importance from both aca-demic and industrial points of view. Some representative characteristics include theparticle size and particle size distribution, the particle surface charge density (or zetapotential), the particle surface area covered by one stabilizer molecule, the confor-mation of the hydrophilic polymer physically adsorbed or chemically couplet ontothe particle surface, the type and concentration of functional groups on the parti-cle surface, the particle morphology, the optical and rheological properties and thecolloidal stability.
1.6.2 Theoretical Overview
A typical batch emulsion polymerization reaction contains three distinct intervals,labeled Interval I, II and III, such as reported in a pioneering paper by Harkins [277].
Interval I is that where the particle formation takes place and monomer droplets,surfactant micelle (if above the critical micelle concentration, CMC) and precur-sor particles (a small, colloidally unstable particle that upon further propagationalgrowth, coagulation and adsorption of surfactant will eventually grow to a col-loidally stable “mature” particle) are present.

1 Nanostructured Macromolecules 51
Interval II occurs after the conclusion of the particle formation period wherebyonly mature latex particles now exist; the particle number density (Np, the numberof particles per unit volume of the continuous phase) remains constant and the par-ticles grow by propagation in the presence of monomer droplets. As the diffusionof monomer from a droplet to a particle is rapid on the timescale of polymerization,the droplets act as monomer stores that ensure the monomer concentration within aparticle to be essentially constant.
Upon the exhaustion of these monomer droplets, Interval III starts, where theremaining monomer contained within the particles is polymerized. This often, butnot always, corresponds to an increase in the polymerization rate and above a cer-tain weight fraction of polymer (wp) within the particle a “gel” effect exists wherethe effective termination rate is reduced [278]. These three intervals are showngraphically in Fig. 1.24.
The rate varies as a function of Np, of the particle size and of the initiator con-centration [I]; in the ideal experiment for the understanding of the mechanism, eachof these can be changed independently while all other quantities are kept constant.However, the early studies used systems where both Np and size changed togetheras [I] was changed. The complicated nature of this process means that the rate coef-ficients for entry and exit could not be determined unambiguously. Then the methodchosen for the kinetic experiments devoted to the understanding of mechanismsconsists in seeded experiments that begin in Interval II (by-passing particle forma-tion), wherein Np and particle size can be controlled independently. It is essentialin such experiments that the particle formation be avoided during Interval II, sinceotherwise Np will change during the experiment, which sometimes creates difficultexperimental constraints.
The value of Np is obtained from the size measurements of the latex by using:
Np = mp/(4/3π r3 dp) (1.1)
Fig. 1.24 The three intervals of a typical emulsion polymerization reaction, showing surfactantmolecules ( ), large monomer droplets, micelle (indicated by clusters of surfactant moleculeswithin Interval I), radicals (R′), initiator (I) and surfactant-stabilized latex particles (Reprinted fromThickett and Gilbert [274]. Copyright 2009, with permission from Elsevier)

52 M.V. Russo et al.
where mp is the mass of the polymer per unit volume of the continuous phase, 4/3π r3 the volume-average, t the unswollen radius of the seed latex and dp the densityof the polymer.
A detailed understanding of the particle formation mechanism is complex butcrucial for a complete evaluation of the emulsion polymerization, and the knowl-edge of the conditions that avoid the formation of new particles in seeded systems(secondary nucleation) is vital for a number of industrial procedures.
The rate of a polymerization is normally defined as the rate of consumption ofthe monomer:
d[M]/dt = −Kp[M][R] (1.2)
where [M] is the concentration of the monomer and [R] the total radicalconcentration.
In an emulsion where the polymerization only takes place within the particleinterior, [M] is replaced by Cp (concentration of monomer in the particle phase); thetotal radical concentration is ñNp = NA, where ñ is the average number of radicalsper particle, Np is the number of particles per unit volume of the continuous phase,and NA is Avogadro’s constant. Since it is experimentally convenient to measure thefractional conversion of monomer into polymer (denoted by x, where 0≤ x ≤1), achange in variable is made and the rate of fractional conversion is now considered,giving:
dx/dt = n[(KpCpNp)/(n◦MNA)] (1.3)
where n◦M is the initial number of moles of monomer per unit volume of the con-
tinuous phase (all other parameters as defined previously). Equation (1.3) showsthat ñ, including its time dependence, can be obtained experimentally via accuratemonitoring of the polymerization rate.
The synthesis of well-characterized electrosterically stabilized latexes presentsa more difficult challenge. Electrosteric stabilization (by using ionizable water-soluble polymers grafted to the particle surface to impart colloidal stability) is acommon technique in the synthesis of industrial polymers to be used in surfacecoatings and adhesives. While easy to synthesize, the characterization of the “hairylayer” on the particle surface is extremely complicated. A new route to the synthesisof well-defined electrosteric latexes has recently been developed through the adventof the successful controlled-radical polymerization in emulsion [279], in particularof the reversible addition-fragmentation chain transfer (RAFT) technique. Fergusonet al. [280, 281] developed the first electrosterically stabilized emulsion under com-plete RAFT control through the use of an amphipathic RAFT agent that allows thesynthesis of relatively monodisperse hydrophilic block polymers in water as the firststep. Subsequent starved-feed addition of a hydrophobic monomer into the aque-ous phase eventually results in the self-assembly of diblock copolymer chains (thebeginning of particle formation), after which the particles continue to grow to anysize. The reaction scheme of this procedure is shown in Fig. 1.25.

1 Nanostructured Macromolecules 53
Fig. 1.25 Synthesis of an electrosterically stabilized seed latex by using RAFT (Reprinted fromThickett and Gilbert [274]. Copyright 2009, with permission from Elsevier)
1.6.3 Critical Parameters for Emulsion Polymerization
Some typical features of the nanobeads and an experimental parameter wich allowto control and modulate the chemical physical properties are shown in Fig.1.26a,b respectively. In particular the shape and dimensions of the spheres are influ-enced by the reaction time, by cosolvent and initiator concentration. In the emulsiontechnique for copolymerization the monomer/comonomer ratio plays the main roleto determinate the chemical features and dimensions [282, 283].
Fig. 1.26 (a) SEM image of PPA nanobeads with d = 280 nm and larger ordered domain ofabout 12 × 12 μ, obtained using toluene/PA= 2/1, KPS = 0.008 g, and reaction time = 90 min.;(b) average diameter of PPA (�) (toluene/PA= 2/1; reaction time 90 min) and P(PA/HEMA) (•)(PA/HEMA = 10/1; toluene/PA= 2/1; reaction time 150 min) nanobeads, as a function of KPSamount. Adapted from Venditti et al. [283]. Copyright 2009, with permission from Elsevier

54 M.V. Russo et al.
The particle size (often nano) and polydispersion are usually confirmed viascanning electron microscopy (SEM), transmission electron microscopy (TEM),dynamic light scattering (DLS) or hydrodynamic chromatography, a separationtechnique that keeps apart the particles on the basis of the size; sometimes DLSand SEM data are not directly comparable (SEM images are collected from driedsamples while DLS measurements are in solution and a swelling effect occurs) butthe trends of growth of the nanoparticles are similar for the two methods.
The cosolvent effect on the dimensions and shape of the particles is due to theformation of a layer around the monomer droplets and this layer controls the trans-fer of the monomer molecules from inside the droplets to the surrounding aqueousphase and can absorb the monomer molecules that are dispersed in the solvent; e.g.in the presence of toluene, the size of the drop limits the dimension of the par-ticle and also reduces the probability of collision, responsible of the coalescence.Moreover, the cosolvent acts as a hydrophobic layer and reduces the potential energyof the interface, leading to an overall stabilization of the emulsion and hence toregularly shaped particles; a high polydispersity and irregular particle shapes werefound for those samples prepared in the absence of toluene, while very low poly-dispersity and a regular round shape were found for the others; this behavior isconsistent with the study of Tanrisever et al. [284] on the polymerization kinetics ofPMMA.
The role of the cosolvent is analogous to the role of nonionic and ionic/nonionicemulsifiers in the radical polymerization of unsaturated monomers (styrene,alkyl(methacrylates), etc.) in aqueous medium, as reported by Capek [285] wherethe increased stability of the emulsions containing a nonionic emulsifier wasattributed to the effects produced by the presence of a layer of the emulsifier aroundthe monomer droplets.
The effect of KPS concentration in the emulsion polymerization is shown inFig. 1.26b for a hydrofobic monomer, phenylacetylene; the increase of the initiatoramount produces larger polymeric particles, because in this situation the polymer-ization rate is high and the polymer chain growth is completed in short times, so thatthe growth rate of the particles is controlled by the coalescence.
In the emulsion copolymerization, when a hydrophilic monomer is involved,the initiator concentration has a different influence on the particles size, such asin P(PA/HEMA) emulsion copolymerization (see Fig. 1.26b); when the initiatorconcentration increases, the particles dimension decreases, because the density ofpolymerization sites of nucleation increases.
In general, it is assessed that the initiator is important not only for the tuning ofthe rate of polymerization and of the molecular weight but also the particle size andlatex viscosity can be tuned by changing the concentration and the kind of initiator[286].
Finally an important experimental parameter to be considered in the emulsioncopolymerization is the concentration of hydrophilic monomer; it has several effectson the particles features, in particular on the particle size, particle size distributionand superficial charges, liable for swelling behavior and adhesion properties.

1 Nanostructured Macromolecules 55
1.7 Electrospinning
1.7.1 General Overview
Self-assembly has been defined in a stretchable fashion and it is possible to simplydescribe it as the process by which spontaneous arrangements of ordered structuresare generated, based on several different interactions at various length scales. Forexample, if we consider a length scale ranging from the angstrom to the millime-ter we would experience interactions ranging from covalent and hydrogen bonds,to Van der Waals and electrostatic force and finally surface tensions. The control ofthese forces and interactions is fundamental for the achievement of the desired depo-sition [287]. There are several jet-based approaches, which have been demonstratedto have a significant impact on the preparation of materials at nanometric level,ranging from ink-jet printing to electrospraying. Ink-jet printing has contributed agreat deal to this endeavor, although it shows limitations in the processing of highlyconcentrated suspensions from which nanoscaled deposits are derived.
Electrospray explores high intensity electric fields to generate droplets and formself-assemblies from a wide range of conducting nanoparticles. Polymeric fibershaving diameters ranging from a few nanometers up to microns can be success-fully prepared by means of the electrospinning based method [288]. The use ofpolymeric fibers has several applications, varying from the tissue engineering [289]to reinforcements in nanocomposites [290], nanowires and nanotubes [291]. Thepioneering work of Martin reported the extraordinary increase in the electronic con-ductivity of polymeric nanofibers, due to the confinement of dimension and size inthe nanometric range [292].
Electrospinning is a suitable technique for the production of fibers with smalldiameter, and was introduced for the first time by Formhals in 1934 by developinga method for the production of artificial filaments, applied to spun cellulose acetatefibers from an acetone/alcohol solution [293]. Electrospinning easily tunes the pos-sibility of assembling polymeric fibers with diameters less than 100 nm and lengthup to microns for a wide choice of polymeric materials with different molecularweights and functionalities. This technique was in depth reinvestigated in the recentyears [294, 295] and applied to a variety of functional polymeric materials [296]and to biopolymers, suited for tissue engineering applications [297, 298].
This method is based on the feed of a polymeric solution or melt, maintained ata high positive potential, through a thin metallic needle. A typical electrospinningsetup consists of a capillary through which the liquid to be electrospun is forced, ahigh voltage source with positive or negative polarity which injects charge into theliquid and a grounded collector; the set up is shown in Fig. 1.27.
In a prototypical electrospinning experiment, a characteristic jet path is createdwhen a fluid polymer solution, supplied to a drop attached to an orifice by surfacetension and viscoelastic stresses, is electrified by a sufficiently high electrical poten-tial applied between the drop and a collector placed some distance away. The shapeof the drop approaches a cone and an electrically charged jet of fluid emanates from

56 M.V. Russo et al.
collector
V needle
Taylor cone
syringe
polymer solutionor melt
Highvoltage supply
Liquid jet
Fig. 1.27 Schematic representation of the electrospinning technique
the tip of the cone. The electrical forces from the charge carried within the jet, inducethe jet to continue to elongate as it cools and finally the thin fluid jet solidifies intoa nanofibre onto the collector.
A syringe pump, gravitational forces, or pressurized gas are typically used toforce the liquid through a small-diameter capillary, forming a pendant drop at thetip. An high voltage electrode is then immersed in the liquid or can be directlyattached to the capillary if a metal needle is used. The voltage source (typicallydirect currents, in the range 1–30 kV) is then turned on and the charge is injectedinto the polymer solution. The increase of the electric field strength gives ori-gin to the repulsive interactions between homologue charges in the liquid and theattractive forces between the oppositely charged liquid and the collector beginsto exert tensile forces on the fluid, elongating the pendant drop at the tip of thecapillary.
As the electrostatic attraction between the oppositely charged liquid and the col-lector and the electrostatic repulsions between homologue charges in the liquidbecome stronger, the leading edge of the solution changes from a rounded meniscusto a cone (the Taylor cone) [299]. By applying a high voltage difference betweenthe polymeric droplet coming out of the metallic needle and a collecting cathode, itis possible to force the formation of a controlled stream, and to prepare polymericfibers.
Examining how the polymer droplet at the end of a capillary behaves when anelectric field is applied, it was found in further literature studies that the pendantdroplet develops into a cone and the fiber jet is emitted from the apex of the cone[300]. This is one reason why electrospinning can be used to generate fibers withdiameters significantly smaller than the diameter of the capillary from which theyare ejected. At critical values of the applied voltage, the drop on the tip of theTaylor cone becomes a jet and this effect is strictly connected to the overcomingof the surface charge on the surface tension of the droplets. The fibers are formedduring the flight of the stream towards the collecting plate, as a consequence of

1 Nanostructured Macromolecules 57
solvent evaporation. The just formed fibers can be collected on the cathode and usu-ally highly disordered depositions are observed, depending on several parameterssuch as splitting of the stream, applied voltage, rate of the stream and chemico-physical characteristics of the polymers such as viscosity, solubility, surface tension.Fibers produced by using this process typically have diameters in the order of a fewmicrometers down to the tens of nanometers. Despite a relatively easy access to thisoperation, a precise control of the parameters affecting the morphology of the fibersis necessary to obtain specificity in the product shape, size or properties. It is noteworty that nanofibres with small diameters have a large surface area per unit massand different molecules, particles, and biomolecules can be sequestered and pro-tected inside or on the surface of the nanofibers, remaining accessible for use whenneeded. Moreover, nanofibers can be used as convenient packages and supports forreagents and catalysts.
1.7.2 Process and Mechanism
Typically, molecular weights of polymers suitable for electrospinning are in therange from 100,000 to several millions u.m.a. and the concentrations in the range5–15 wt%. The electrical charge that is important in electrospinning is an excess oruncompensated charge, usually in the form of positive or negative ions. Although allionic solutions contain charged molecules or ions, the solution is electrically neutralbecause the number of positive and negative ions is exactly equal.
The essential excess ions are usually created near the interface between a metallicconductor and the molecules in the solution. The electrons moving into the solutionfrom the metal electrode create excess negative ions in the polymeric solution andthe electrons moving from the solution into the metal leave excess positive ions.Once created, the ions move by diffusive and convective processes to reduce therepulsive interactions between the similarly charged excess ions and to maintain thesame electrical potential everywhere on the surface of the fluid body. The addition ofa salt to the uncharged solution preserves the electrical neutrality, although the saltmolecules may dissociate into positive and negative ions which move independentlyand thereby increase the electrical conductivity.
The process of collecting fluid jets involves coils formed by electrical bending,branching, conglutinated networks of fibers and garlands and the solidification ofthe thin jets produces nanofibers with the occurrence of nucleation and crystal-lization inside the nanofibers. Phase separation of both polymer blends and blockcopolymers can be observed.
The formulation proposed by Taylor to explain the conical shape of the dropletbefore the jet issues a mathematical singularity at the tip of the cone where the jetbegins. The singularity is avoided assuming a parabolic shape for the tip and theshape and the size of the drop may vary with time so that the flow into the drop andthe flow out of the drop are not necessarily equal at every instant. For a constant feedrate, if the voltage is too high, the drop becomes smaller and the beginning of thejet often moves to the edge of the orifice before the jet finally stops. If the voltage

58 M.V. Russo et al.
is too small, the size and the shape of the drop and the diameter of the jet tend tooscillate in time. If the voltage is far too low, the drop grows large and changes theshape as the gravitational forces became important. Ultimately, a drop flows overits support and a pendant drop drips. The length of a short, essentially straight seg-ment, measured along its own axis, increases in response to the Coulomb repulsionbetween the excess charge distributed along and carried with the segment. The repul-sive Coulomb forces between the charges of homologue sign on adjacent segmentsof the jet work against the axial component of the viscoelastic stress and elongatethe jet in the direction of its axis. Polymer solutions which support the motion offree electrons as in a metallic conductor are not commonly encountered. PANI pro-vides an interesting exception in which the electrons move through dry polyanilinenanofibers [301]. The electrical conductivity of the PANI fibers becomes evidentduring the collection of the dry nanofibers, because of the tendency of the networkof nanofibers to extend in the direction of the applied electric field rather than form-ing a flat non-woven mat typical of insulating polymers. This extension happensbecause the characteristic redistribution time for the electrons along the length ofthe electronically conducting polyaniline nanofibres is short. The electronic chargequickly accumulates at favorably oriented ends or bends of the fibers and stretchesthe dry fiber network in the direction of the applied field.
1.7.3 Parameters Affecting the Electrospinning Process
1.7.3.1 General Features
The choice of the flow rate of the polymer, of the distance between needle and col-lecting cathode, of the applied potential, of the temperature and humidity should gowith the account of the concentration and viscosity of the solution, of the molecularweight and molecular weight distribution of the polymer and of the functionalitieson the polymeric chains, which in turn affect the dielectric constant and the solubil-ity of the polymers. For example, it was observed that the polymer solution shouldhave a concentration high enough to cause the entanglement of the chains but not sohigh to reach very high viscosity values that can inhibit the polymer motion in theelectric field.
Systematic researches on the influence of the parameters were carried out [302]showing that, for instance, the size of the polymeric fibers is strongly affected by theconcentration i.e. the diameters increase by increasing the concentration. In recentyears, a greater understanding of the processing parameters has developed the for-mation of fibers with diameters in the range of 100–500 nm, typically referred to asnanofibers. The achievement of nanofibers has led to reneved interest on the electro-spinning process due to potential applications in filtration, protective clothing, andbiological purposes in tissue engineering scaffolds and drug delivery devices [303].
Polyaniline-based nanofibers with diameter below 30 nm were obtained with theelectrospinning process [304] which is suitable not only for conjugated polymers,but also for non conjugated, water soluble polymers such as poly-ethylen oxide(PEO), polyvinyl alcohol (PVA) and poly-lactic acid (PLA). In fact, one of the

1 Nanostructured Macromolecules 59
Fig. 1.28 Optical and FE-SEM images of electrospun fibers (c) Optical micrograph of alignedPEO nanofibers 20×; (d) FE-SEM image of aligned PEO nanofibers (5,000×); (e) FE-SEMimage of isotropic PEO nanofibers (5,000×) (Adapted with permission from Kakade et al. [306].Copyright 2009 American Chemical Society)
advantages of this technique is that it can be applied to a wide variety of polymers,bearing polar or non-polar pending groups [305]. In Fig. 1.28 patterns of alignedPEO electrospun fibers are shown [306].
Polymeric porous and wrinkled fibers can also be obtained, as in the case ofpolystyrene electrospun from DMF solutions [307]; moreover, polystyrene micro-and nanospheres with low dimensional polydispersion have been produced byelectrospray and the effect of PS molecular weight and concentration on thebeads morphology has been investigated [308].The interior of these fibers, whenelectrospun in a high-humidity environment, was found to be highly porous ratherthan polished, despite the smooth and nonporous appearance of the fibers surfaces.The formation of interior porosity is attributed to the miscibility of water, a non-solvent for the polymers in solution, with DMF. The resulting morphology is aconsequence of the relatively rapid diffusion of water into the jet, leading to aliquid−liquid phase separation that precedes the solidification due to the evapo-ration of DMF from the jet. The fibers exhibit a wrinkled morphology that can beexplained by a buckling instability, when they are electrospun in a low-humidityenvironment. Understanding which morphology is formed under a given set of con-ditions is achieved through the comparison of three characteristic times: the dryingtime, the buckling time, and the phase separation time. The morphology has impor-tant consequences for the main properties of the fibers, among the others theirmechanical strength and stiffness.
1.7.3.2 Applied Voltage, Distance Between Capillary Tip and Collectorand Flow Rate
An increase of the applied voltage alters the shape and the morphology of the dropat which the Taylor cone and fiber jet are formed, as in the case of polyethyleneoxide (PEO)/water system studied by Deitzel et al. [309]. At low applied voltagesthe Taylor cone is formed at the tip of the pendent drop and as the applied volt-age is increased, the volume of the drop decreases until the Taylor cone is formedat the tip of the capillary. Meechaisue et al. [310] examined the effects of theprocessing parameters, including the applied electric field strength, on electrospunpoly(desaminotyrosyl- tyrosine ethyl ester carbonate) (poly(DTE carbonate). The

60 M.V. Russo et al.
authors investigated the behavior of a solution of poly(DTE carbonate) at concen-tration 15% w/v, varying the applied electric field strength from 10 to 25 kV/10 cm.Beaded fibers were primarily observed when the applied electric field strengthwas below 20 kV/10 cm, while mostly smooth fibers were obtained above thisfield strength. The increase of the electric field strength from 10 to 15 kV/10 cmdecreased the bead density, while by increasing the field strength from 20 to25 kV/10 cm the average fiber diameter increased from 1.9 to 2.2 mm. The authorsattribute these latter results to the increase of the mass flow rate, related to theenhancement of the electrostatic force. Based on literature reports, it is evident thatthere is an optimal range of electric field strengths for a certain polymer/solvent sys-tem, since either a too weak or a too strong field will lead to the formation of beadedfibers.
While playing a much smaller role, the distance between capillary tip and collec-tor can also influence the fiber size by 1–2 orders of magnitude. The fiber diametergenerally decreases with increasing the distance from the Taylor cone, as in the caseof electrospun fibers from a PEO/water solution; examining the fiber diameter asa function of the distance from the Taylor cone, it was observed that the diameterof the fiber jet decreased approximately 2-fold, from 19 to 9 mm after travelingdistances of 1 and 3.5 cm, respectively [311]. Beaded morphology for electrospunpolystyrene fibers upon shortening the distance between the capillary tip and thecollector was observed, attributed to inadequate drying of the polymer fiber prior toreaching the collector [312].
The polymeric fibers are deposited onto a grounded collector and, depending onthe application, a number of collector configurations can be used, including a sta-tionary plate, rotating mandrel, solvent (e.g. water), etc, and different morphologiescan be obtained. Typically the use of a stationary collector will lead to the formationof a randomly oriented fiber mat. A rotating collector can be used to generate matswith aligned fibers, with the rotation speed playing an important role in determiningthe degree of anisotropy. The use of rotating collector plates allows the deposition offibers with a higher degree of alignment [309]. Additionally, both the conductivityand the porosity of the collector play also a role in determining the packing den-sity of the collected fibers [313]. The polymer flow rate has an impact on the fibersize, and additionally can influence the fiber porosity as well as the fiber shape andmorphology. The effect of the flow rate on the structure of the electrospun fibers ofpolystyrene in tetrahydrofuran solution was studied, demonstrating that both fiberdiameter and pore size increase with increasing the flow rate. Moreover, at high flowrates significant amounts of bead defects become noticeable, due to the inability ofthe fibers to dry completely before reaching the collector. Incomplete fiber dryingalso leads to the formation of ribbon like (or flattened) fibers as compared to fiberswith a circular cross section [314].
1.7.3.3 Choice of Solvent, Polymer Concentration and Solution Conductivity
The choice of solvent is also a critical parameter, influencing whether the fibers arecapable of forming as well as the fiber porosity. In order to have solvent evaporation

1 Nanostructured Macromolecules 61
during the jet flight between capillary tip and collector, a volatile solvent is required.For example, polystyrene fibers electrospun from solutions containing various ratiosof DMF and THF were examined. The produced electrospun PS fibers containedirregular beads and the electrospinning certainly was enhanced with increasing theDMF content. The bead concentration was also controlled by DMF content. Theaspect ratio of the formed beads and the diameter of the fibers were increased withincreasing the solution concentration and when PS was dissolved in THF only, anunexpected half hollow spheres (HHS) structure appeared. Further different shapeforms of PS non-woven mats have been prepared by controlling the electrospinningparameters [315].
Between these two extremes it was observed that as the solvent volatilitydecreases, the pore size increases with decreased pore depth (thus decreasing poredensity). A phase separation occurs as the polymer jet is traveling through theatmosphere. This phase separation can be vapor-induced, which occurs when thenon-solvent from the vapor phase penetrates the polymer solution. However, trans-port of the non-solvent into the polymer solution is limited by the slow diffusion ofthe non solvent adjacent to the fiber surface. For very volatile solvents, the regionadjacent to the fiber surface can be saturated with solvent in the vapor phase, whichfurther limits the penetration of non-solvent. This can hinder the skin formationleading to the development of a porous surface morphology.
The polymer concentration is a fundamental parameter and determines thespinnability of a solution, namely whether a fiber forms or not. The polymer con-centration influences either the viscosity and the surface tension of the solution,both of which are very important parameters in the electrospinning process. In fact,if the solution is too dilute, the polymer fiber will break up into droplets beforereaching the collector due to the effects of the surface tension. However, if the solu-tion is too concentrated, fibers cannot be formed due to the high viscosity, whichmakes it difficult to control the solution flow rate through the capillary. The solu-tion must have a high enough polymer concentration for the chain entanglements tooccur, although it cannot be either too dilute or too concentrated. Thus, an optimumrange of polymer concentrations exists in which fibers can be electrospun when allother parameters are held constant. In many experiments it has been shown thatwithin the optimal range of polymer concentrations, the fiber diameter increaseswith increasing the polymer concentration; for example, increasing the concentra-tion of polystyrene in THF, the fiber diameter increased and the distribution of poresizes became narrower [312].
The solution conductivity plays a less important role although it can influence thefibers size within 1–2 orders of magnitude. If a solution possesses high conductivity,it will have a greater charge carrying capacity with respect to solutions with low con-ductivity and as a consequence, the fiber jet of highly conductive solutions will besubjected to a greater tensile force in the presence of an electric field than a fiber jetwill from a solution with a low conductivity. In general, the radius of the fiber jet isinversely related to the cube root of the solution conductivity [316]. Highly conduc-tive solutions are extremely unstable in the presence of strong electric fields, whichlead to a dramatic bending instability as well as a broad diameter distribution [317].

62 M.V. Russo et al.
The effect of adding ions to PVA/water solution on the diameters of the electrospunfibers was studied and it was observed that by adding increasing concentrationsof NaCl (ranging from 0.05 to 0.2 wt%) to a PVA/water solution, a decrease ofthe mean fiber diameter from about 200 to 160 nm occurs [318]. The authorsattribute this decrease in the mean fiber diameter to the increased net charge densityimparted by NaCl, which increases the electric force exerted on the jet. Some effortswere made also to incorporate biomolecules like bovine serum albumin (BSA) intodextran fibers for potential drug delivery or tissue engineering applications [319].It was observed that adding 5% BSA, a decrease of the mean fiber diameter occursfrom approximately 2.5 mm–500 nm and that the viscosity of the dextran solutionis unchanged by the addition of up to 10% BSA, indicating that the decrease in fiberdiameter is due to the increased net charge found in the polymer jets.
Another possible technique is the direct electrospinning of a polymer melt. Ingeneral, the solution spinning results in a greater range of fiber sizes, while meltspun fibers are typically limited to micron size or larger [320]; however, there arespecific advantages and disadvantages for each method. Melt electrospinning elimi-nates the need for harsh organic solvents, which is ideal for scaled-up processes, butmelts must be kept at elevated temperatures to be electrospun, whereas stable solu-tions can typically be electrospun at room temperature. The melt electrospinningof block copolymers of PEG and PCL (polycaprolactone) with various molecu-lar weights was studied and it was observed that the optimum melt temperaturesranged between 60 and 90◦C [321]. These high temperatures may preclude theiruse for tissue engineering or drug delivery applications. The use of polymer meltsalso eliminates the problem of inadequate solvent evaporation between the capillarytip and the collector; however, the polymer must be able to cool suitably over thisdistance in order to generate fibers with a cylindrical morphology.
1.7.3.4 Nozzle Configuration
In addition to adjusting the solution or processing parameters, the type of elec-trospinning process can greatly influence the resulting product. This can includechoices in nozzle configuration such as single, side-by-side, or coaxial nozzles. Anumber of nozzle configurations have been employed, and perhaps the simplest andmost common is the single nozzle technique. In this configuration a charged poly-mer solution or melt flows through a single capillary. This configuration is veryversatile and has been used to electrospin single polymer solutions [322] as well aspolymer blends out of polymers soluble in a common solvent [323] and was usedalso for the electrospinning of composite fibers containing bovine serum albumin(BSA) loaded Ca-alginate micro spheres microencapsulated in poly(L-lactic acid)(PLLA) fibers [324].
While the electrospinning of polymer blends is often desirable in order to achievethe proper combination of properties, it may not be possible by using a single needleconfiguration if the polymers of interest are not soluble in a common solvent. Thus,it may be necessary to use a side-by-side mode. In this configuration two separatepolymer solutions flow through two different capillaries, which are set side-by-side.

1 Nanostructured Macromolecules 63
This configuration was used to electrospin bicomponent systems out of poly(vinylchloride)/segmented polyurethane and poly(vinylchloride)/poly(vinylidene fluo-ride) [325] and it was observed that the solution conductivity plays a more importantrole in the ability to form a single fiber jet under a strong electric field in the side-by-side configuration. The conductivity of the PVC solution was significantly higherthan either of the other two solutions, and thus two distinct Taylor cones, one fromeach solution, were formed when subjected to a strong electric field and the ratioof the two components varied along the length of the fiber, which was attributed tofluctuations of the jet on the surface of the Taylor cone.
A relatively new nozzle configuration is the coaxial configuration, which allowsthe simultaneous coaxial electrospinning of two different polymer solutions. Inthis set up two separate polymer solutions flow through two different capillaries,which are coaxial with a smaller capillary inside a larger capillary. This technique,called coaxial-electrospinning, has received great interest because by using this noz-zle configuration smaller fibers can essentially be encapsulated into larger fibers,leading to what is known as core-shell morphology. For example, PEO–PEO andPEO–PDT type core–shell nanofibers were produced by this method [326]. In gen-eral, two coaxial capillary tips are used to simultaneously feed the two liquids. At ashort distance from this two-capillary nozzle, typically several centimeters, a metal-lic plate is placed as counter-electrode or collector [327]: in Fig. 1.29 the process isschematically represented.
Fig. 1.29 Two immiscible liquids (red and blue in this Figure) are injected through two concen-tric electrified needles which are placed a few centimeters away from a grounded electrode (thecollector). A compound Taylor cone is developed from whose tip a coaxial nanojet is emitted.Upon solidification of the outer liquid, a sheathed fiber, or a liquid-filled hollow fiber, is formed(Reprinted with permission from Loscertales et al. [327]. Copyright 2009 American ChemicalSociety)

64 M.V. Russo et al.
1.7.4 Core-Shell Nanofibers and Nanotubes by CoaxialElectrospinning
Modifications of the electrospinning technique have been proposed in orderto obtain oriented fibers or core-shell nanofibers and nanotubes. The co-electrospinning allows the preparation of core-shell fibers or tubes: differentpolymeric solutions can be injected thought concentric annular needles and electro-spun through the formation of a double Taylor cone. The mixing of the two solutionscan be avoided by means of a very fast electrospinning process by which coax-ial nano and micro fibers can be obtained; with a proper choice of the polymericmaterials, the inner core can be removed and nanotubes isolated [328], as shown inFig. 1.30.
This technique was proved to be very versatile for the encapsulation of biorele-vant molecules and nanocomposites: for example, coaxial electrospinning of alignedPCL nanofibers encapsulated with bovine and platelet-derived growth factor-bb wascarried out for the demonstration of controlled release and bioactivity retention[329]. The encapsulation of a model protein, (fluorescein isothiocyanateconju-gated bovine serum albumin, fitcBSA), along with PEG within the biodegradablepoly(caprolactone) PCL nanofibers was achieved by using a coaxial electrospinningtechnique [330]. By varying the inner flow rate with a constant outer flow rate, theloading of fitcBSA could be controlled.
The coaxial electrospinning has also been used for the encapsulation of activebiological threads and scaffolds. In the cell/polymer electrospinning, a coaxial nee-dle arrangement is used with the flow of highly concentrated cellular suspensionin the inner needle and medical grade biocompatible polymer in the outer nee-dle. After electrospinning, the collected cells have been cultured, have been foundto be viable and showed no evidence of having incurred any cellular damageduring the bionanofabrication process. Electrospun poly(vinyl alcohol) nanofiberswere tested for the encapsulation of bacteria (Escherichia coli, Staphylococcus)and viruses that remained viable for months at low temperatures [331]. In general,complex biological objects can be prepared directly without total loss of biologicalfunctionality.
Fig. 1.30 (a) Simplenanofiber, (b) core-shellnanofiber, (c) nanotubes

1 Nanostructured Macromolecules 65
As a curiosity, spider and caterpillar silk were electrospun into nanofibersand characterized by electron microscopy [332, 333]. The diameter of these silknanofibers ranged from about 6.5 to 300 nm, making them several orders of mag-nitudes smaller than the silk fibers spun by silkworms or by many kinds of spiders.Electron diffraction patterns of annealed electrospun nanofibers exhibited diffrac-tion peaks with the molecules aligned along the axis of the fiber with a crystallineorder comparable to that of naturally spun silks.
1.7.5 Electrospun Fiber Properties
One of the most important advantages of the electrospinning technique is theproduction of very thin fibers with large surface areas together with the ease offunctionalization for various purposes, the superior mechanical properties and theeffortlessness of the process. These advantages provide a wide range of opportu-nities for their use in many different applications, ranging from tissue engineering,drug release, implants, to biotransformation, reinforced composites etc. [334]. Thepotentiality of the electrospinning is that the method can be varied in different waysto combine materials properties with different morphological structures suitable forthese applications.
A large number of biodegradable and biocompatible polymers have been elec-trospun by conventional electrospinning or modified electrospinning methods andloaded with different bioactive molecules. Moreover, the faculty to adjust the fibersize from micro to nano size is one of the strengths of electrospinning, since fiberswith diameters in the nanometer size range closely mimic the size scale of fibrousproteins of the natural extra cellular matrix (ECM), such as collagen. This abilityof electrospun nanofibers to mimic the ECM is vital, since previous studies haveshown that both the size scale of the structure and the topography play importantroles in cell proliferation and adhesion, respectively [335]. Additionally, the poros-ity of electrospun mats aids in nutrient transport. Although many researches haveencountered limitations concerning the cell infiltration into nanofiber mats, due tothe relatively small pores associated with such matrices, nanofibrous mats have thepotential to overcome mass transfer limitations seen in other polymer drug deliverysystems due to their high surface to volume ratio. Additionally, nanofiber systemscan afford greater drug loading as compared to other techniques. In addition tothese bio-properties, more usual chemical and mechanical properties should alsobe considered in relation with the electrospinning processes.
1.8 Applications and Perspectives
Nanostructured macromolecules, synthetic and natural, functional or structural,have been prepared by using several chemical, physical, and even biomimeticapproaches. New ways dedicated to investigate the growth of nanostructures

66 M.V. Russo et al.
continue to be developed in order to obtain the control over size, shape, and distri-bution [8]. The great challenge of organizing the nanostructures, assembling theminto larger complexes or connecting them into devices, becomes every day morerealistic.
Nanostructured polymers currently represent one of the most active areas ofresearch in the whole word of polymer science, represented not only by syntheticapproaches, but also by theory, structural analysis and applications developments.
At the interface of science and technology, electronic and optoelectronic devicesimpact many areas of business and society, i.e. communications, computing, andmedical devices and the demand for ever more compact and powerful systems isstrong, exciting a growing interest in the development of nanoscale devices thatcould combine new functions with greatly enhanced performances.
The extraordinarily large surface area of the nanoparticles and the opportunityof placing different functional groups on the surface, offers new and unespectedperspectives and applications. Nanostructured polymers can expand/contract withchanges of pH, or interact with biomolecules, enzymes, anti-bodies, cells, virusand bacteria in peculiar ways to provide rapid ex-vivo medical diagnostic tests; anoverview on the emerging biotechnological and medical applications is presented inChapter 5.
Synthetic macromolecules are inherently capable of organizing into a variety ofperiodic and nonperiodic morphologies, i.e spheres, wires, tubes, grains, spongeswith a characteristic length scale typically of the order of 10–1,000 nm. Significanttechnological applications for these materials can be envisaged in nanofiltra-tion membranes, in ionomer-membranes for fuel cells and electrodialysis, and inelastomeric multi-block-copolymer fibers.
For example, the lotus effect in macromolecules and in general superhydrophobicsurfaces can be achieved in the case of nanostructured polymers likewise super-hydrophobic surfaces, that occur naturally in some plant leaves and insect wings,eyes, and legs, and are characterized by a high contact angle (usually >150◦) andlow sliding angle less than 5◦ (low flow resistance). The water-repellening prop-erties of superhydrophobic surfaces strongly depend on the micro- and nanoscaledapproach and on the control of the wetting behavior of protecting layers and surfacestoward the detrimental effects of environmental water and moisture; it was recentlydemonstrated the formation of superhydrophobic poly(dimethylsiloxane) (PDMS),by exposing the surface to ultra fast laser pulses [336].
The development of nanostructured materials for ultra sensitive detection oforganic, inorganic and biological species has also received great attention con-cerning conjugated macromolecules [337]. For example, porphyrin colorimetricindicators were prepared in molecular and nano-architectures for the developmentof well-integrated systems for the sensing of particular chemical species [338].
Nanoscale π-conjugated organic and organometallic polymers can be used aswell for sensors [339, 340], biosensors [341], electrochemical devices, single elec-tron transistors [342], nanotips of field emission displays [343] and a recent reviewreports the technological advances in these important topics [344].

1 Nanostructured Macromolecules 67
Biosensors based on nanomaterials exploit many new signal transduction tech-nologies in their manufacture [345]. In molecular electronics and sensors, conduct-ing polymers represent innovative systems for the immobilization of enzymes [346,347]. The entrapment of enzymes in polymeric films provides a controlled methodto fasten biologically active molecules in a defined area on the electrodes. Theseexamples show that conducting polymers in the area of bioanalytical sciences are ofgreat interest since their biocompatibility opens up the possibility of using them asin vivo biosensors.
Conducting polymers particularly in the form of thin films or blends or com-posites have also been widely investigated as membranes in sensors for air-bornevolatiles (alcohols, NH3, NO2, CO) detection. In particular, polyaniline and polypyr-role nanofibres have received a great deal of attention because of their vast surfacearea that allows fast diffusion of gas molecules into the structure [348] and poly-thiophene based sensors allowed the detection of ppb of hydrazine gases [349].Nanostructured organometallic polymers containing Pd(II) or Pt(II) sites and palla-dium dispersed nanoparticles were studied for the development of devices, SurfaceAcoustic Wave sensors, which show response towards relative humidity and hydro-gen [350]; the mode of interaction of these polymers with H2S gas was also deeplyinvestigated [351].
Among the variety of morphologies, nanowires are emerging as a powerfulclass of materials that, through controlled growth and organization, are opening upsubstantial opportunities for novel nanoscale photonic and electronic devices [352].
In this framework, nanostructured polyaniline is a most promising material forchemical sensors since significantly enhanced performance of nanofiber films overconventional materials can be observed in both sensitivity and response time [353].Nanostructured polyaniline is highly sensitive to many chemical vapors at the partsper million levels or less through several different mechanisms including dop-ing/dedoping, reduction/oxidation, swelling, and by the modulation of the polymerconformation. The modification of polyaniline nanofibers with selected additives isan effective strategy to improve the gas sensitivity; for example the mixing of CuCl2with polyaniline nanofiber films leads to an improved response (about 4 orders ofmagnitude) towards hydrogen sulfide gas [354].
Another interesting property that foresees advances in technology is flash weld-ing. When exposed to light, polyaniline converts most of the absorbed energy intoheat and if the polymer is in the form of nanofibers, the generated heat is trappedinside the individual nanofibers and, since the surrounding air is a very poor heatconductor, the heat dissipates slowly. If a camera flash intense light is used, a rapidtemperature rise occurs which can lead to instantaneous welding of the nanofibers,while under irradiation of moderate intensity, polyaniline nanofibers will rapidly“melt” to form a smooth and continuous film. This phenomenon, called flash weld-ing will open new applications for monolithic actuators, i.e. devices that make useof different forms of energy to induce motion [355].
The potentiality of nanofibers is not limited to electronic devices; in factnanoscale support materials for catalysis could be also developed. For example,

68 M.V. Russo et al.
palladium nanoparticles supported on polyaniline nanofibers behave as an activecatalyst for Suzuki coupling at relatively low temperatures and as water-dispersiblecatalyst supports to produce and stabilize Pd nanoparticles [356].
Nanostructured semiconducting block copolymers containing triphenylamine ashole transport moiety and perylene bisimide as dye and electron transport, have beeninvestigated in view of applications in photovoltaic devices. The polymers shownanowire like structure which formation is driven by the crystallization of perylenebisimides via π- π stacking and since this self-assembly gives rise to domains sizecomparable to the exciton diffusion length, these materials offer perspectives for theimplementation of organic solar cells [357].
Another fascinating property of conducting polymers is electrochromism thatenvisages applications to smart windows, rear-view mirrors, electronic paper, dis-plays, stealth technology, etc. A prerequisite, especially for display applications,is a fast color switching, property showed by the nanotubular structure of PEDOTwith an extremely fast electrochromic response without sacrificing the color contrast[358]. Electrochromic devices based on PEDOT nanotubes can be switched from theoxidized to the reduced states by applying alternating square potentials between 1.0and −1.0 V and show strong coloration and flexibility. The color is yellowish at theoxidized state due to the background of the sputtered gold electrode, while it is darkblue at the reduced state [359].
Conductive polymer nanotubes show further interesting applications for theconstructions of high-power energy storage devices such as supercapacitors andbatteries, by using their fast charge/discharge characteristic [360]. Devices basedon nanostructured materials can provide high power density by enhancing thecharge/discharge rates [2]. Redox-type supercapacitors provide high specific capac-itances, as in the case of PEDOT-nanotube-based supercapacitor, which maintainsat least 80% of its maximum energy density even when the power density is boostedto 25 kW/kg [361], getting high power density without significant loss of its energydensity.
Nanostructured macromolecules play also an important role in the developmentof materials for photonics [362]. In fact they can possess useful optical proper-ties such as electroluminescence, photoluminescence, nonlinear optical properties[363] or can be used as matrices for optically active species [364]. Moreover, poly-mers that show compositional patterns can coherently scatter light [365] or beexploited for producing photonic materials [366]. Photonic crystals are materialswhere the coherent scattering of light or modification of the modes of light propaga-tion occur [367, 368]. Photonic crystals obtained from polymers possess a relativelylow refractive index (n) and the intrinsically low value of n gives rise to materialswith full or incomplete bandgaps. On this basis, polymeric photonic crystals are ableto suppress the propagation of light in specific directions and wavelength range. Theself-assembly of polymeric nanospheres into colloid crystals with different packing,i.e. hexagonal close-packed (hcp) or face-centered cubic (fcc), is a typical approachfor the achievement of materials with periodic structures. Changes in the spectralposition of the stop band originate from a change in the average refractive indexand/or the lattice constant. For example, these changes occur in response to a change

1 Nanostructured Macromolecules 69
in pH, temperature and in the presence of analytes [369, 370]. Polymeric photoniccrystals can also show inverse opals structure and it is possible to obtain a twostate color switch in the case of poly(methacrylic-acid-co-N-isopropylacrylamide)hydrogel or functionalized polyacrylamide [371]. One of the potential applicationsof colloidal crystals based on polymeric nanostructures is the production of record-ing media for optical data storage [372] which open new perspectives, for example,in the fabrication of protecting secure documents.
The reported topics aim to give just several insights into the emerging fieldsof nanomaterials and related technology devoted to meet social needs such ascommunication and security, health and energy, here briefly outlined.
References
1. Van Gough D, Juhl AT, Braun PV (2009) Materials Today 12(6):28–352. Fallace GG, Chen J, Mozer AJ, Forsyth M, MacFarlane DR, Wang C (2009) Materials Today
12(6):20–273. Li G, Shrotriya V, Huang J, Yao Y, Moriarty T, Emery K, Yang Y (2005) Nat Mater
4:864–8684. Ma W, Yang C, Gong X, Lee K, Heeger AJ (2005) Adv Funct Mater 15:1617–16225. Beek WJE, Wienk MM, Kemerink M, Yang X, Janssen RAJ (2005) J Phys Chem B
109:9505–95166. Almgren M, Brown W, Hvidt S (1995) Colloid Polym Sci 273:2–157. Andreasen JW, Jorgensen M, Krebs FC (2007) Macromolecules 40:7758–77628. Chronopoulou L, Fratoddi I, Palocci C, Venditti I, Russo MV (2009) Langmuir
25:11940–119469. Hawker CJ, Wooley KL (2005) Science 309:1200–1205
10. Whitesides GM, Mathias JP, Seto CT (1991) Science 254:1312–131911. Whitesides GM, Boncheva M (2002) Proc Natl Acad Sci USA 99:4769–477412. McGarrity ES, Duxbury PM, Mackay ME, Frishknecht AL (2008) Macromolecules
41:5952–595413. Rabani E, Reichman DR, Geissler PL, Brus LE (2003) Nature 426:271–27414. Rabani E, Egorov SA (2002) J Phys Chem B 106:6771–677815. Saunders AE, Shah PS, Park EJ, Lim KT, Johnston KP, Korgel BA (2004) J Phys Chem B
108:15969–1597516. Redl FX, Cho KS, Murray CB, O’Brien S (2003) Nature 423:968–97117. Sztrum CG, Rabani E (2006) Adv Mater 18:565–57118. Martin CP, Blunt MO, Moriarty P (2004) Nano Lett 4:2389–239219. Maillard M, Motte L, Pileni MP (2001) Adv Mater 13:200–20420. Stowell C, Korgel BA (2001) Nano Lett 1:595–60021. Ge G, Brus LE, (2001) Nano Lett 1:219–22222. Tang J, Ge G, Brus LE, (2002) J Phys Chem B 106:5653–565823. Boeker A, He J, Emrick T, Russel TP (2007) Soft Matter 3:1231–124824. Lehn JM (2002) Proc Natl Acad Sci USA 99:4763–476825. Keng PY, Shim I, Korth BD, Douglas JF, Pyun J (2007) ACS Nano 1:279–29226. Schmidt DJ, Cebeci FC, Kalcioglu ZI, Wyman SG, Ortiz C, Van Vliet KJ, Hammond PT
(2009) ACS Nano 3:2207–221627. Uttam M, Satish P (2009) Langmuir 25:10515–1052228. Gatebe E, Herron H, Zakeri R, Rajasekaran PR, Aouadi S, Kohli P (2008) Langmuir
24:11947–1195429. Smith, ARG, Ruggles JL, Yu A, Gentle IR, (2009) Langmuir 25:9873–9878

70 M.V. Russo et al.
30. Leclère Ph, Surin M, Brocorens P, Cavallini M, Biscarini F, Lazzaroni R (2006) Mat SciEngin R 55:1–56
31. Ikkala O, ten Brinke G (2002) Science 295:2407–240932. Zhu Y, Xu Z, Yi C (2008) J Polym Sci A Polym Chem 46:2658–266633. Zhang JX, Qiu LY, Zhu KJ (2005) Macromol Rapid Commun 26:1716–172334. Hu JS, Guo YG, Liang HP, Wan LJ, Jiang L (2005) J Am Chem Soc 127:17090–1709535. Wang Z, Li Z, Medforth CJ, Shelnutt JA (2007) J Am Chem Soc 129:2440–244136. Spontack RJ, Alexandridis P (1999) Curr Opin Colloid Interface Sci 4:140–14637. Rodríguez-Hernández JR, Chécot F, Gnanou Y, Lecommandoux S (2005) Prog Polym Sci
30:691–72438. Hof F, Rebek J Jr (2002) Proc. Natl. Acad. Sci USA 99:4775–477739. Mougthon AO, O’Reilly RK (2008) J Am Chem Soc 130:8714–872540. Sknepnek R, Anderson JA, Lamm MH, Schmalian J, Travesset A (2008) ACS Nano
2:1259–126541. Hales K, Chen Z, Wooley KL, Pochan DJ (2008) Nano Lett 8:2023–202642. Wang X, Liu K, Arsenault AC, Rider DA, Ozin GA, Winnik MA, Manners IA (2007) J Am
Chem Soc 129:5630–563943. Hamley IW (2003) Nanotechnology 14:R39–R5444. Yang SY, Ryu I, Kim HY, Kim JK, Jang SK, Russel TR (2006) Adv Mater 18:709–71245. Luchnikov V, Kondyurin A, Formanek P, Lichte H, Stamm M (2008) Nano Lett 8:2315–232046. Messerschmidt M, Millaruelo M, Choinska R, Jehnichen D, Voit B (2009) Macromolecules
42:156–16347. Yan Y, Besseling NAM, de Kreizer A, Marcelis ATM, Drechsler M, Cohen Stuart MA (2007)
Angew Chem Int Ed 46:1807–180948. Black CT, Ruiz R, Breyta G, Cheng JY, Colburn ME, Guarini KW, Kim HC, Zhang Y (2007)
IBM J Res Dev 1:605–63349. Huang CI (2009) React Funct Polym 69:530–53850. Smith DK, Hirst AR, Love CS, Hardy JG, Brignell SV, Huang B (2005) Prog Polym Sci
30:220–29351. Tomalia AD (2005) Prog Polym Sci 30:294–32452. Gillies ER, Fréchet JMJ (2004) J Org Chem 69:46–5353. Wang Y, Zeng FW, Zimmermann SC (1997) Tetrahedron Lett 38:5459–546254. Dykes GM, Smith DK, Seeley GJ (2002) Angew Chem Int Ed 41:3254–325755. Jeong K-S, Park E-J (2004) J Org Chem 69:2618–262156. Gibson HW, Yamaguchi N, Hamilton L, Jones JW (2002) J Am Chem Soc 124:4653–466557. Denti G, Campagna S, Seroni S, Ciano M, Balzani V (1992) J Am Chem Soc 114:2944–295058. Newcome GR, Yoo KS, Kim HJ, Moorefield CN (2003) Chem Eur J 9:3367–337459. Armaroli N, Boudon C, Felder D, Gisselbrecht J-P, Gross M, Marconi G, Nicoud J-F,
Nierengarten J-F, Vicinelli V (1999) Angew Chem Int Ed 38:3730–373360. Krishna TR, Jayaraman N (2002) J Chem Soc Perkin Trans 1:746–75461. Stone DL, Smith DK, Whitwood AC (2004) Polyhedron 23:1709–171762. Anthopoulos TD, Frampton MJ, Namdas EB, Burn PL, Samuel IDW (2004) Adv Mater
16:557–56063. Enomoto M, Aida T (2002) J Am Chem Soc 124:6099–610864. Saalfrank RW, Deutscher C, Maid H, Ako AM, Sperner S, Nakajima T et al. (2004) Chem
Eur J 10:1899–190565. Wang RY, Yang J, Zheng ZP, Carducci MD, Jiao J, Seraphin S (2001) Angew Chem Int Ed
40:549–55266. Smith DK (2004) Speciality Chem Mag 2442–244367. Daniel MC, Ruiz J, Nlate S, Blais JC, Astruc D (2003) J Am Chem Soc 125:2617–262868. Guo W, Li J, Wang YA, Peng XG (2003) Chem Mater 15:3125–313369. Braun CS, Vetro JA, Tomalia DA, Koe GS, Koe JG, Middaugh CR (2005) J Pharm Sci
94:423–436

1 Nanostructured Macromolecules 71
70. Kukowska-Latallo JF, Bielinska AU, Johnson J, Spindler R, Tomalia DA, Baker JR (1996)Proc Natl Acad Sci USA 93:4897–4902
71. Abdelhady HG, Allen S, Davies MC, Roberts CJ, Tendler SJB, Williams PM (2003) NucleicAcids Res 31:4001–4005
72. Lee JH, Lim YB, Choi JS, Lee Y, Kim TI, Kim HJ, Yoon JS, Kim K, Park JS (2003)Bioconjugate Chem 14:1214–1221
73. Choi JS, Joo DK, Kim CH, Kim K, Park JS (2000) J Am Chem Soc 122:474–48074. Nagasaki T, Atarashi K, Makino K, Noguchi A, Tamagaki S (2000) Mol Cryst Liq Cryst
345:227–23275. Joester D, Losson M, Pugin R, Heinzelmann H, Walter E, Merkle HP, Diederich F (2003)
Angew Chem Int Ed 42:1486–149076. Choi Y, Mecke A, Orr BG, Banaszak Holl MM, Baker JR (2004) Nano Lett 4:391–39777. Kim Y, Mayer MF, Zimmerman SC (2003) Angew Chem Int Ed 42:1121–112678. Chapman TM, Hillyer GL, Mahan EJ, Shaffer KA (1994) J Am Chem Soc 116:11195–1119679. van Hest JCM, Delnoye DAP, Baars MWPL, Genderen MHP, Meijer EW (1995) Science
268:1592–159580. Uchiyama T, Ishii K, Nonomura T, Kobayashi N, Isoda S (2003) Chem Eur J 9:5757–576181. Kellermann M, Bauer W, Hirsch A, Shade B, Ludwig K, Böttcher C (2004) Angew Chem
Int Ed 43:2959–296282. Schmid G, Meyer-Zaika W, Pugin R, Sawitowski T, Majoral J-P, Caminal A-M, Turrin CO
(2000) Chem Eur J 6:1693–169783. Zeng XB, Ungar G, Liu YS, Percec V, Dulcey SE, Hobbs JK (2004) Nature 428:157–16084. Percec V, Mitchell CM, Cho WD, Uchida S, Glodde M, Ungar G (2004) J Am Chem Soc
126:6078–609485. Ponomarenko SA, Boiko NI, Shibaev VP, Magonov SN (2000) Langmuir 16:5487–549386. Serrano JL, Marcos M, Martin R, Gonzales M, Barberá J (2003) Chem Mater 15:3866–387287. Busson P, Ortegren J, Ihre H, Gedde UW, Hult A, Andersson G (2002) Macromolecules
35:1663–167188. Seo M, Beck BJ, Paulusse JMJ, Hawker CJ, Kim SY (2008) Macromolecules 41:6413–641889. Satyanarayana Kuchibhaltla VNT, Karakoti AS, Bera D, Seal S (2007) Prog Mat Sci
52:699–91390. Wessling B (2000) Conductive polymers as organic nanometals. In: Nalwa HS (ed)
Handbook of nanostructured materials and nanotechnology, Chapter 10. Wiley, New York,pp 501–575
91. Kim J (2002) Pure Appl Chem 74:2031–204492. Zhu Y, Li J, Wan M, Jiang L (2007) Macromol Rapid Commun 28:1339–134493. Luo X, Dominguez Vidal G, Killard AJ, Morrin A, Smyth MR (2007) Electroanalysis
19:876–88394. Amarnath CA, Kim J, Kim K, Choi J, Sohn D (2008) Polymer 49:432–43795. Zhang Z, Deng J, Yub L, Wana M (2008) Synth Met 158:712–71696. Zhou C, Han J, Guo R (2008) Macromolecules 41:6473–647997. Bhadra J, Sarkar D (2009) Mater Lett 63:69–7198. Wang J, Wang J, Yang Z, Wang Z, Zhang F, Wang S (2008) React Funct Polym
10:1435–144099. Han D, Song J, Ding X, Xu X, Niu L (2007) Mat Chem Phys 105:380–384
100. Dawn A, Nandi AK (2007) J Phys Chem C 111:6268–6274101. Wu J, Tang Q, Li Q, Lin J (2008) Polymer 49:5262–5267102. Wu J, Tang Zhang L, Zhang L, Wan M (2008) Eur Polym J 44(7):2040–2045103. Zhang X, Chechik V, Smith DK, Walton PH, Duhme-Klair A-K (2008) Macromolcules
41:3417–3421104. Wei Z, Faul CFJ, Tran HD, Wang Y, D’Arcy JM, Kaner RB (2008) ACS Nano 2:1841–1848105. Berlin A, Vercelli B, Zotti G (2008) Polymer Rev 48:493–530106. He C, Yang C, Li Y (2003) Synth Met 139:539–545

72 M.V. Russo et al.
107. Jang J, Yoon H (2005) Langmuir 21:11484–11489108. Kim DK, Oh KW, Ahn HJ, Kim SH (2008) J Appl Polym Sci 107:3925–3932109. Wu J, Tang Q, Li Q, Lin J, (2008) Polymer 49:5262–5267110. Zuo JP, Wang X-S (2005) Physica E 28:7–13111. Wu J, Li Y, Feng W (2007) Synth Met 157:1013–1018112. Mumtaz M, Lecommandoux S, Cloutet E, Cramail H (2008) Langmuir 24:11911–11920113. Xia Y, Wei M, Lu Y (2009) Synth Met 159:372–376114. Park JY, Koenen N, Forster M, Ponnapati R, Scherf U, Advincula R (2008) Macromolecules
41:6169–6175115. Sun X, Hagner M (2007) Macromolecules 40:8537–8539116. Niu Q, Zhou Y, Wang L, Peng J, Wang J, Pei J, CaoY (2008) Adv Mater 20:964–969117. Li B, Lambeth DN (2008) Nano Lett 8:3563–3567118. Lam JWY, Tang BZ (2005) Acc Chem Res 38:745–754119. Isopo A, Caminiti R, D’Amato R, Furlani A, Russo MV (2003) J Macromol Sci Physics
42:1061–1083120. Sone T, D’Amato R, Mawatari Y, Tabata M, Furlani A, Russo MV (2004) J Polym Sci A
Polym Chem 42:2365–2376121. Cernia E, D’Amato R, Palocci C, Panzavolta F, Russo MV, Soro S (2005) J Mol Catal
B-Enzym 32:67–76122. D’Amato R, Medei L, Venditti I, Russo MV, Falconieri M (2003) Mat Sci Engin C
23:861–865123. Albrecht O, Sone T, Kuriyama A, Educhi K, Yano K (2008) Nanotechnology
19:505201–505207124. Li BS, Cheuk KKL, Yang D, Lam JWY, Wan LJ, Bai C, Tang BZ (2003) Macromolecules
36:5447–5450125. Kim DW, Ku B-C, Steeves D, Nagarajan R, Blumstein A, Kumar J, Gibson PW, Ratto JA,
Samuelson LA (2006) J Memb Sci 275:12–16126. Dong H, Li H, Wang E, Wei Z, Xu W, Hu W, Yan S (2008) Langmuir 24:13241–13244127. Eloi J-C, Chabanne L, Whittel GR, Manners I (2008) Materials Today 11(4):28–36128. Whittel GR, Manners I (2007) Adv Mater 19:3439–3468129. Fratoddi I, Gohlke C, Cametti C, Diociaiuti M, Russo MV (2008) Polymer 45:3211–3216130. Thomas A, Goettmann F, Antonietti M (2008) Chem Mater 20:738–755131. Clapper JD, Sievens-Figuero L, Guymon CA (2008) Chem Mater 20:768–781132. Wan Y, ShiY, Zhao D (2008) Chem Mater 20:932–945133. Dauginet-De Pra L, Ferain E, Legras R, Demoustier-Champagne S (2002) Nucl Instrum
Methods Phys Res B 196:81–88134. Bajpai V, He P, Dai L (2004) Adv Func Mater 14:145–151135. Fujikawa D, Uota M, Sakai G, Kijima T (2007) Carbon 45:1289–1295136. Acik M, Sonmez G (2006) Polym Adv Technol 17:697–699137. Mativetsky JM, Datars WR (2002) Solid State Commun 122:151–154138. Andreasen JW, Jørgensen M, Krebs F (2007) Macromolecules 40:7758–7762139. Carswell ADW, O’Rear EA, Grady BP (2003) J Am Chem Soc 125:14793–14800140. Zhang X, Zhang J, Liu Z, Robinson C (2004) Chem Commun 16:1852–1853141. Zhang X, Zhang J, Song W, Liu Z (2006) J Phys Chem 110:1158–1165142. Mao H, Lu X, Chao D, Zhang W (2008) Mater Lett 62:998–1001143. Dai T, Yang X, Lu Y (2007) Nanotechnology 17:3028–3034144. Shi W, Liang P, Ge D, Wang J, Zhang Q (2007) Chem Commun 23:2414–2416145. Reynes O, Demoustier-Champagne S (2005) J Electrochem Soc 152:D130–D135146. Goren M, Qi Z, Lennox RB (2000) Chem Mater 12:1222–1228147. Chen P, Gao P, Liu M (2006) Polymer 47:7446–7454148. Peng C-Y, Nam WJ, Fonash SJ, Gu B, Sen A, Strawhecker K, Natarajan S, Foley HC, Kim
SH (2003) J Am Chem Soc 125:9298–9299149. She X, Song G, Li J, Han P, Yang S, Shulong W, Peng Z (2006) Polym J 38:639–642

1 Nanostructured Macromolecules 73
150. Darling SB, Yufa NA, Cisse AL, Bader SD, Sibener SJ (2005) Adv Mater 17:2446–2450151. Minelli C, Geissbuehler I, Hinderling C, Heinzelmann H, Vogel H, Pugin R, Lylei M (2006)
J Nanosci Nanotechnol 6:1611–1619152. Weng CC, Hsu KF, Wei KH (2004) Chem Mater 16:4080–4086153. Maschmann MR, Franklin AD, Fisher TS (2006) Nanotechnology 17:3925–3929154. Yoon SB, Kim JY, Kim JH, Park SG, Kim JY, Lee CW, Yu JS (2006) Curr Appl Phys
6:1059–1063155. Hu XF, Kuai SL, Yu Y, Troung VV (2005) J Inorg Mater 20:1463–1468156. Wang CH, Yang C, Song YY, Gao W, Xia XH (2005) Adv Funct Mater 15:1267–1275157. Braun PV, Viltzius P (2002) Curr Opin Colloid Interface Sci 7:116–123158. Luo X, Killard AJ, Morrin A, Smyth MR (2007) Chem Commun 3207–3209159. Luo X, Vidal GD, Killard AJ, Morrin A, Smyth MR (2007) Electroanalysis 19:876–883160. Zhu Y, Li J, Wan M, Jiang L, Wei Y (2007) Macromol Rapid Commun 28:1339–1344161. Anilkumar P, Jayakannan M (2008) Macromolecules 41:7706–7715162. Sudha JD, Reena VL, Pavithran C (2007) J Polym Sci B Polymer Phys 45:2664–2673163. Sudha JD, Sasikala TS (2007) Polymer 48:338–347164. Wallace GG, Moulton SE, Misoska VJ, Kane-Maguire LAP, Innis PC (2002) Mater Forum
26:29–38165. Georger JH, Singh A, Price RR, Schnur JM, Yager P, Schem PE (1987) J Am Chem Soc
109:6169–6175166. Lee JM, Lee DG, Kim JH, Cheong IW (2007) Macromolecules 40:9529–9536167. Marquez M, Patel K, Carswell ADW, Schmidtke DW, Grady BP (2006) Langmuir 22:8010–
8016168. Maury PA, Reinhoudt DN, Huskens J (2008) Curr Opin Colloid Interface Sci 13:74–80169. Kustandi TR, Samper VD, Yi DK, Sing Ng W, Neuzil P, Sun W (2007) Adv Func Mater
17:2211–2218170. Acevedo DA, Lasagni AF, Barbero CA, Mücklich F (2007) Adv Mat 19:1272–1275171. Maynor BW, Filocamo SF, Grinstaff MW, Liu J (2002) J Am Chem Soc 124:522–523172. Matyjaszewski K, (1998) Appl Organometal Chem 12:667–673173. Farquet P, Padeste C, Solak HH, Grusel SA, Scherer GG, Wokaun A (2008) Macromolecules
41:6309–6313174. Webster O (1991) Science 251:887–893175. Jakubowski W, Matyjaszewski K (2005) Macromolecules 38:4139–4146176. Nguyen V, Yoshida W, Jou JD, Cohen Y (2002) J Polym Sci A Polym Chem 40:26–42177. Montse RB, Giralt F, Cohen Y (2001) J Colloid Interface Sci 235:70–79178. Papirer E, Nguyen VT (1973) Angew Makromol Chem 128:31–38179. Fireman-Shoresh S, Huesing N, Avnir D (2001) Langmuir 17:5958–5963180. Walker P (1992) In: Mittal KL (ed) Silanes and other coupling agents. VSP, Netherlands181. Yoshida W, Castro RP, Jou JD, Cohen Y (2001) Langmuir 17:5882–5888182. Lewis TG, Nowling GR, Hicks RF, Cohen Y (2007) Langmuir 23:10756–10764183. Pearton SJ, Norton DR (2005) Plasma Process Polym 2:16–37184. Langowski BA, Uhrich KE (2005) Langmuir 21:6366–6372185. Zhang Y, Kang ET, Neoh KG, Huang W, Huan ACH, Zhang H, Lamb RN (2002) Polymer
43:7279–7288186. Orefice RL, Clark E, Brenna AB (2006) Macromol Mater Eng 291:377–386187. Chen KS, Ku YA, Lin HR, Yan TR, Sheu DC, Chen TM (2006) J Appl Polym Sci
100:803–809188. Choi HS, Kim YS, Zhang Y, Tang S, Myung SW, Shin BC (2004) Surf Coat Technol
182:55–64189. Zhang Y, Tan KL, Liaw BY, Liaw DJ, Kang ET, Neoh KG (2001) Langmuir 17:2265–2274190. Zhong SF, Meng YD, Ou QR, Shu XS (2005) J Appl Polym Sci 97:2112–2117191. Kai T, Suma Y, Ono S, Yamaguchi T, Nakao SI (2006) J Polym Sci A Polym Chem
44:846–856

74 M.V. Russo et al.
192. Hirotsu T (2006) Thin Solid Films 506:173–175193. Babin J, Lepage M, Zhao Y (2008) Macromolecules 41:1246–1253194. Kowalewski T, McCullough RD, Matyjaszewski K (2003) Eur Phys J E Soft Matter 10:5–16195. Tian Y, Watanabe K, Kong X, Abe J, Iyoda T (2002) Macromolecules 35:3739–3747196. Yoshida J, Kataoka K, Horcajada R, Nagaki A (2008) Chem Rev 108:2265–2299197. Donohoe TJ (2000) Oxidation and reductions in organic synthesis. Oxford University Press,
Oxford198. Chao TC, Lin YT, Yang CY, Hung TS, Chou HC, Wu CC, Wong KT (2005) Adv Mater
17:992–996199. Lu W, Fadeev AG, Qi B, Smela E, Mattes BR, Ding J, Spinks GM, Mazurkiewicz J, Zhou
D, Wallace GG, MacFarlane DR, Forsyth SA, Forsyth M (2002) Science 297:983–987200. Burroughes JH, Bradley DDC, Brown AR, Marks RN, Mackay K, Friend RH, Burns PL,
Holmes AB (1990) Nature 347:539–541201. Evans NR, Devi LS, Mak CSK, Watkins SE, Pascu SI, Kohler A, Friend RH, Williams CK,
Holmes AB (2006) J Am Chem Soc 128:6647–6656202. Skotheim TA, Reynolds JR (2007) Handbook of conducting polymers, 3rd edn. CRC Press,
Boca Raton203. O’Neill M, Kelly SM (2000) J Phys D Appl Phys 33:R67–R84204. Jiang X, Patil R, Harima Y, Ohshita J, Kunai A (2005) J Phys Chem B 109:221–229205. Malinauskas A, Malinauskiene J, Ramanavicius A (2005) Nanotechnology 16:R51–R62206. Sperry JB, Wright DL (2006) Chem Soc Rev 35:605–621207. Yoshida J, Nishiwaki K (1998) J Chem Soc Dalton Trans 2589–2596208. Yoshida J, Sugawara M, Tatsumi M, Kise N (1998) J Org Chem 63:5950–5961209. Yoshida J, Izawa M (1997) J Am Chem Soc 119:9361–9365210. Watanabe M, Suga S, Yoshida J (2000) Bull Chem Soc Jpn 73:243–247211. Tang ZY, Liu SQ, Tang ZX, Dong SJ, Wang EK (2000) Electrochem Commun 2:32–35212. Kado Y, Atobe M, Nonaka T (2000) Electrochemistry 68:262–267213. Atobe M, Kaburagi T, Nonaka T (1999) Electrochemistry 67:1114–1117214. Wadhawan JD, Del Campo FJ, Compton RG, Foord JS, Marken F, Bull SD, Davies SG,
Walton DJ, Ryley S (2001) J Electroanal Chem 507:135–143215. Mazine V, Heinze J (2004) J Phys Chem 108:230–235216. Gouw T, Jentoft RJ (1986) J Chromatogr Sci 68:303–323217. Silvestri G, Gambino S, Filardo G, Cuccia C, Guarino E (1981) Angew Chem Int Ed Engl
20:101–102218. Niehaus D, Philips M, Michael A, Wightman RM (1989) J Phys Chem 93:6232–6236219. Jikei M, Yasuda H, Itoh H (2007) Polymer 48:2843–2852220. Atobe M, Ohsuka H, Fuchigami T (2004) Chem Lett 33:618–619221. Yan H, Sato T, Komago D, Yamaguchi A, Oyaizu K, Yuasa M, Otake K (2005) Langmuir
21:12303–12308222. Galinski M, Lewandowski A, Stepniak I (2006) Electrochim Acta 51:5567–5580223. Kubisa P (2005) J Polym Sci A Polym Chem 43:4675–4683224. Koura N, Ejiri H, Takeishi K (1993) J Electrochem Soc 140:602–605225. Pang Y, Li X, Ding H, Shi G, Jin L (2007) Electrochim Acta 52:6172–6177226. Pringle JM, Forsyth M, Wallace GG, MacFarlane DR (2006) Macromol 39:7193–7195227. Choi SJ, Park SM (2002) J Electrochem Soc 149:E26–E34228. Eftekhari A, Afshani R (2006) J Polym Sci A Polym Chem 44:3304–3311229. Ahmad S, Deepa M, Singh S (2007) Langmuir 23:11430–11433230. Serra Moreno J, Panero S, Scrosati B (2008) Electrochim Acta 53:2154–2160231. Martin CR (1998) Chem Mater 8:1739–1746232. Duvail JL, Dubois S, Demoustier-Champagne S, Long Y, Piraux L (2008) Intern J Nanotech
5:838–850233. Kang HS, Kang HS, Lee JK, Joo J, Lee MS, Kim MS, Lee JYJ (2004) Nonlin Opt Phys
Mater 13:633–636

1 Nanostructured Macromolecules 75
234. Demoustier-Champagne S, Ferain E, Jerome C, Jerome R, Legras R (1998) Eur Polym J34:1767–1774
235. Demoustier-Champagne S, Stavaux PY (1999) Chem Mater 11:829–834236. Kros A, Nolte RJM, Sommerdijk NAJM (2002) Adv Mater 14:1779–1782237. Delvaux M, Duchet J, Stavaux PY, Legras R, Demoustier-Champagne S (2000) Synth Met
113:275–280238. Li X, Zhang X, Li H (2001) J Appl Polym Sci 81:3002–3007239. Fu MX, Chen F, Zhang JX, Shi GQ (2002) J Mater Chem 12:2331–2333240. Joo JJ, Park KT, Kim BH, Kim MS, Lee SY, Jeong CK, Lee JK, Park DH, Yi WK, Lee SH,
Ryu KS (2003) Synth Met 135:7–9241. Wirtz M, Martin CR (2003) Adv Mater 15:455–458242. Noll JD, Nicholson MA, Van Patten PG, Chung C, Myrick ML (1998) J Electrochem Soc
145:3320–3328243. Bok HM, Shin TY, Park S (2008) Chem Mater 20:2247–2251244. Jerome C, Labaye D, Bodart I, Jerome R (1999) Synth Met 101:3–4245. Han SB, Briseno AL, Shi XY, Mah DA, Zhou FM (2002) J Phys Chem B 106:6465–6472246. Wallace GG, Innis PC (2002) J Nanosci Nanotechnol 2:441–451247. Jerome C, Demoustier-Champagne S, Legras R, Jerome R (2000) Chem Eur J 6:3089–3093248. Luo J, Zhang HP, Huang HG, Wu LL, Lin ZH (1999) Mol Cryst Liq Cryst 337:157–160249. Zhang HP, Luo J, Huang HG, Wu LL, Lin ZH (2000) Chem Phys Lett 326:169–174250. Shirsat MD, Bangar MA, Deshusses MA, Myung NV, Mulchandani A (2009) Appl Phys
Lett 94:083502–083505251. Cho SI, Lee SB (2008) Acc Chem Res 41:699–707252. Cirpan A, Argun AA, Grenier CRG, Reeves BD, Reynolds JR (2003) J Mater Chem
13:2422–2428253. Arico AS, Bruce P, Scrosati B, Tarascon JM, van Schalkwijk W (2005) Nat Mater 4:366–377254. Hayes WA, Shannon C (1998) Langmuir 14:1099–1102255. Lee JY, Park SM (2000) J Electrochem Soc 147:4189–4195256. Park SM, Lee JY, Choi SJ (2001) Synth Met 121:1297–1298257. Liu J, Lin YH, Liang L, Voigt JA, Huber DL, Tian ZR, Coker E, McKenzie B, McDermott
MJ (2003) Chem Eur J 9:605–611258. Ge D, Wang J, Wang Z, Wang S (2002) Synth Met 132:93–95259. Yang YS, Wan MX (2001) J Mater Chem 11:2022–2027260. Yang YS, Liu J, Wan MX (2002) Nanotechnology 13:771–773261. Li M, Wei Z, Jiang L (2008) J Mater Chem 18:2276–2280262. Kim DW, Hwang IS, Kwon SJ, Kang HY, Park KS, Choi YJ, Choi KJ, Park JG (2007) Nano
Lett 7:3041–3045263. Winter M, Brodd RJ (2004) Chem Rev 104:4245–4269264. Liu R, Lee SB (2008) J Am Chem Soc 130:2942–2943265. Xiao R, Cho SI, Liu R, Lee SB (2007) J Am Chem Soc 129:4483–4489266. Chern CS (2006) Prog Polym Sci 31:443–486267. Chena BH, Chenb C-I, Hoc M-L, Chend H-N, Chene W-C, Wang C-K (2009) Mater Chem
Phys 113:365–371268. Verwey EJW, Overbeek JThG (1943) Theory of the stability of lyophobic colloids. Elsevier,
New York269. Napper DH (1983) Polymeric stabilization of colloidal dispersions. Academic Press, London270. Nomura M, Tobita H, Suzuki K (2005) Adv Polym Sci 175:1–128271. Antonietti M, Landfester K (2002) Prog Polym Sci 27:689–757272. Urban D, Takamura K (2002) Polymer dispersions and their industrial applications. Wiley-
VCH, Weinheim273. Gilbert RG (1995) Emulsion polymerisation: A mechanistic approach. Academic Press, San
Diego274. Thickett SC, Gilbert RG (2007) Polymer 48:6965–6991

76 M.V. Russo et al.
275. Thickett SC, Gaborieau M, Gilbert RG (2007) Macromolecules 40:4710–4720276. Thickett SC, Morrison B, Gilbert RG (2008) Macromolecules 41:3521–3529277. Harkins WD (1947) J Am Chem Soc 69:1428–1444278. Norrish RGW, Smith RR (1942) Nature 150:336–337279. Save M, Guillaneuf Y, Gilbert RG (2006) Aust J Chem 59:693–711280. Ferguson CJ, Hughes RJ, Nguyen D, Pham BTT, Hawkett BS, Gilbert RG, Serelis AK, Such
CH, Hawkett BS (2005) Macromolecules 38:2191–2294281. Ferguson CJ, Hughes RJ, Pham BTT, Hawkett BS, Gilbert RG, Serelis AK, Such CH (2002)
Macromolecules 35:9243–9245282. D’Amato R, Venditti I, Russo MV, Falconieri M (2005) J Appl Polym Sci 102:4493–4499283. Venditti I, D’Amato R, Russo MV, Falconieri M (2007) Sens Actuators B 126:35–40284. Tanrisever T, Okay O, Sonmezoglu IC (1996) J Appl Polym Sci 61:485–493285. Capek I (2002) Adv Colloid Interfaces Sci 99:77–162286. Tauer K, Muller H (2003) Colloid Polym Sci 281:52–65287. Greiner A, Wendorff JH (2008) Adv Polym Sci 219:107–171288. Huang ZM, Zhang YZ, Kotaki M, Ramakrishna S (2003) Comp Sci Technol 63:2223–2253289. Xu CY, Inai R, Kotaki M, Ramakrishna S (2004) Biomaterials 25:877–886290. Deitzel JM, Kosik, W, McKnight SH, Beck Tan NC, De Simone JM, Crette S (2002) Polymer
43:1025–1029291. Hu J, Odom TW, Lieber CM (1999) Acc Chem Res 32:435–445292. Cai Z, Martin CR (1989) J Am Chem Soc 111:4138–4139293. Formals A (1934) Process and apparatus for preparing artificial threads. US patent n
1,975504294. Reneker DH, Yarin AL (2008) Polymer 49:2387–2425295. Dan L, Younan X (2004) Adv Mater 16:1151–1170296. Jayasinghe SN (2008) Physica E 40:2911–2915297. Matthews JA, Wnek GE, Simpson DG, Bowlin GL (2002) Biomacromol 3:232–238298. Li W, Laurencin CT, Caterson EJ, Tuan RS, Ko FK (2002) J Biomed Mater Res 60:613–621299. Taylor G (1969) Proc R Soc London Ser A Math Phys Eng Sci 313:453–475300. Baumgarten E (1997) J Colloid Interface Sci 36:1–9301. Cardenas J R, de Franca MGO, de Vasconcelos EA, de Azevedo WM, da Silva EFJ (2007) J
Phys D Appl Phys 40:1068–1071302. Deitzel JM, Kleinmeyer J, Harris D, Beck Tan NC (2001) Polymer 42:261–272303. Sill TJ, von Recum HA (2008) Biomaterials 29:1989–2006304. Zhou Y, Freitag M, Hone J, Stali C, Johnson ATJ, Pinto NJ, MacDiarmid AG (2003) Appl
Phys Lett 83:3800–3802305. Dersch R, Steinhart M, Boudriot U, Greiner A, Wendorff JH (2005) Polym Adv Technol
16:276–282306. Kakade MV, Givens S, Gardner K, Lee KH, Chase DB, Rabolt JF (2007) J Am Chem Soc
129:2777–2782307. Pai CL, Boyce MC, Rutledge GC (2009) Macromol 42:2102–2114308. Fantini D, Zanetti M, Costa L (2006) Macromol Rapid Comm 27:2038–2042309. Deitzel JM, Kleinmeyer J, Harris D, Tan NCB (2001) Polymer 42:261–272310. Meechaisue C, Dubin R, Supaphol P, Hoven VP, Kohn J (2006) J Biomater Sci Polym Ed
17:1039–1056311. Doshi J, Reneker DH (1995) J Electrostat 35:151–160312. Megelski S, Stephens JS, Chase DB, Rabolt JF (2002) Macromol 35:8456–8466313. Liu HQ, Hsieh YL (2002) J Polym Sci B Polym Phys 40:2119–2129314. Koombhongse S, Liu W, Reneker DH (2001) J Polym Sci B Polym Phys 39:2598–2606315. Lee KH, Kim HY, Bang HJ, Jung YH, Lee SG (2003) Polymer 44:4029–4034316. Baumgarten PK (1971) J Colloid Interf Sci 36:71–79317. Hayati I, Bailey AI, Tadros TF (1987) J. Colloid. Interface Sci 117:205–221318. Zhang CX, Yuan XY, Wu LL, Han Y, Sheng J (2005) Eur Polym J 41:423–432

1 Nanostructured Macromolecules 77
319. Jiang H, Fang D, Hsiao BS, Chu B, Chen W (2004) Biomacromol 5:326–333320. Larrondo L, Manley RSJ (1981) J Polym Sci B Polym Phys 19:909–920321. Dalton PD, Lleixa Calvet J, Mourran A, Klee D, Moller M (2006) Biotechnol J 1:998–1006322. Tan EP, Ng SY, Lim CT (2005) Biomaterials 26:1453–1456323. Stitzel J, Liu J, Lee SJ, Komura M, Berry J, Soker S (2006) Biomaterials 27:1088–1094324. Qi H, Hu P, Xu J, Wang A (2006) Biomacromol 7:2327–2330325. Gupta P, Wilkes GL (2003) Polymer 44:6353–6359326. Sun Z, Zussman E, Yarin AL, Wendorff JH, Greiner A (2003) Adv Mater 15:1929–1932327. Loscertales IG, Barrero A, Márquez M, Spretz R, Velarde-Ortiz R, Larsen G (2004) J Am
Chem Soc 126:5376–5377328. Kameoka J, Orth R, Yang Y, Czaplewski D, Mathers R, Coates GW, Craighead HG (2003)
Nanotechnology 14:1124–1129329. Ye Z, Mahato RI (2008) Nanomed 3:5–8330. Park KE, Kang HK, Lee SJ, Min BM, Park WH (2006) Biomacromol 7:635–643331. Salalha W, Kuhn J, Dror Y, Zussman E (2006) Nanotechnology 17:4675–4681332. Casper CL, Yamaguchi N, Kiick KL, Rabolt JF (2005) Biomacromol 6:1998–2007333. Zhang S, Chen D, Jiao X (2008) J Colloid Interface Sci 318:331–336334. Agarwal S, Wendorff JH, Greiner A (2008) Polymer 49:5603–5621335. Zhang YZ, Su B, Venugopal J, Ramakrishna S, CT Lim (2007) Int J Nanomed 2:623–638336. Yoon TO, Shin HJ, Jeoung SC, Park YI (2008) Opt Express 16:12715–12725337. Qiu H, Zhai J, Li S, Jiang J, Wan M (2003) Adv Funct Mater 13:925–928338. Xie Y, Hill JP, Charvet R, Ariga K (2007) J Nanosci Nanotech 7:2969–2993339. Hatchett DW, Josowicz M (2008) Chem Rev 108:746–769340. Bearzotti A, Macagnano A, Pantalei S, Zampetti E, Venditti I, Fratoddi I, Russo MV (2008)
J Phys Condens Matter 20:474207–474212341. Alsana S, Gabrielli C, Pierrot H (2003) J Electrochem Soc 150:E444–E449342. Kim BH, Kim MS, Park KT, Lee JK, Park DH, Joo J, Yu SG, Lee SH (2003) Appl Phys Lett
83:539–541343. Kim BH, Park DH, Joo J, Yu SG, Lee SH (2005) Synth Met 150:279–284344. Rajesh, Ahuja T, Kumar D (2009) Sens Actuat B Chem 136:275–286345. Jianrong C, Yuqing M, Nongyue H, Xiaohua W, Sijiao L (2004) Biotech Adv 22:505–518346. Venditti I, Fratoddi I, Russo MV, Bellucci S, Crescenzo R, Iozzino L, Staiano M, Aurilia V,
Varriale A, Rossi M, D’Auria S (2008) J Phys Cond Matter 20:474202–474205347. Ahuja T, Mir IA, Kumar D, Rajesh (2007) Biomaterials 28:791–805348. Huang J (2006) Pure Appl Chem 78:15–27349. McQuade DT, Pullen AE, Swager TM (2000) Chem Rev 100:2537–2574350. Caliendo C, Contini G, Fratoddi I, Irrera S, Pertici P, Russo MV, Scavia G (2007)
Nanotechnology 18:125504–125511351. Battocchio C, Fratoddi I, Russo MV, Polzonetti G (2008) J Phys Chem A 112:7365–7373352. Li D, Huang J, Kaner RB (2009) Acc Chem Res 42:135–145353. Virji S, Huang J, Kaner RB, Weiller BH (2004) Nano Lett 4:491–496354. Virji S, Fowler JD, Baker CO, Huang JX, Kaner RB, Weiller BH (2005) Small 1:624–627355. Huang JX, Kaner RB (2004) Nat Mater 3:783–786356. Gallon BJ, Kojima RW, Kaner RB, Diaconescu PL (2007) Angew Chem Int Ed
46:7251–7254357. Lindner SM, Kaufmann N, Thelakkat M (2007) Org Electron 8:69–75358. Cho SI, Kwon WJ, Choi SJ, Kim P, Park SA, Kim J, Son SJ, Xiao R, Kim SH, Lee SB
(2005) Adv Mater 17:171–175359. Cho SI, Choi DH, Kim SH, Lee SB (2005) Chem Mater 17:4564–4566360. Pushparaj VL, Shaijumon MM, Kumar A, Murugesan S, Ci L, Vajtai R, Linhardt RJ,
Nalamasu O, Ajayan PM (2007) Proc Natl Acad Sci USA 104:13574–13577361. Winter M, Brodd RJ (2004) Chem Rev 104:4245–4269362. Parquet C, Kumacheva E (2008) Materials Today 11(4):48–56

78 M.V. Russo et al.
363. Jiang HY, Kelch S, Lendlein A (2006) Adv Mater 18:1471–1476364. Lindsay GA, Ashley PR, Davis MC, Guenthner AJ, Sanghadasa M, Wright ME (2006) Mater
Sci Eng B 132:8–11365. Xia Y, Gates B, Li ZY (2001) Adv Mater 13:409–413366. Braun PV, Rinne SA, García-Santamaría F (2006) Adv Mater 18:2665–2678367. López C (2003) Adv Mater 15:1679–1704368. Joannopoulous JD (2001) Nature 414:257–258369. Ueno K, Matsubara K, Watanabe M, Takeoka Y (2007) Adv Mater 19:2807–2812370. Debord JD, Eustis S, Debord SB, Lofye MT, Lyon LA (2002) Adv Mat 14:658–662371. Matsubara K, Watanabe M, Takeoka Y (2007) Angew Chem Int Ed 46:1688–1692372. Siwick BJ, Kalinina O, Kumacheva E, Dwayne Miller RJD, Noolandi J (2001) J Appl Phys
90:5328–5334