Embed Size (px)
Citation preview

1828 G. Himbert 1979
Liebigs Ann. Chem. 1979, 1828 - 1846
(Aminoethinyl)metallierungen, 6 ’)
Umsetzungen von (Silylethiny1)- und (Germylethiny1)aminen mit Ketoketenen Gerhard Himbert
Fachbereich Chemie der Universitat Kaiserslautern, Postfach 3049, D-6750 Kaiserslautern
Eingegangen am 20. April 1979
Die P-silylierten und !3-germylierten Inamine 1 addieren die Ketoketene 2 bevorzugt zu den (3- Siloxy- bzw. (3-Germyloxy-3-alken-1-inyl)aminen 4. Nebenprodukte sind die Cycloaddukte 5i und 5 j sowie das C-silylierte Acylinamin 6k. Bei der sauren Hydrolyse von 4a- f erhalt man die 3-Siloxy-3-butenamide 9, die Acetoacetamide 10 und in einigen Fallen die Cyclisierungsprodukte 12; die basische Hydrolyse liefert Acylinamine wie 13. Das Eninamin 4d reagiert mit Sulfonylazi- den 7 iiber die Diazoverbindungen 15 unter Stickstoffabspaltung und intramolekularer Sauer- stoffverschiebung zu den 2-0xo-3-siloxy-N2-sulfinyl-3-butenamidinen 16. Mit Ketenen liefern die Eninamine 4 Cyclobutenone vom Typ 17.
(Aminoethynyl)metaUations, 6’). - Reactions of (Silylethyny1)- and (Germylethyny1)amines with Ketoketenes The P-silylated and P-germylated ynamines 1 add the ketoketenes 2 to give preferentially the (3- siloxy- and (3-germyloxy-3-alken-1-ynyl)amines 4, respectively. By-products are the cycloadducts 5i and j and the C-silylated acylynamine 6k. Acid hydrolysis of 4a- f affords the 3-siloxy-3- butenamides 9, the acetoamides 10, and - in few cases - the cyclization products 12; base hy- drolysis furnishes acylynamines, e. g. 13. Reaction of 4d with the sulfonyl azides 7 via diazo com- pounds 15 gives on elimination of nitrogen and intramolecular migration of oxygen the 2-0x04- siloxy-N2-sulfinyl-3-butenamidines 16. Ketenes react with the enynamines 4 to yield cyclobuten- ones of type 17.
Inamine mit Alkyl-, Aryl- und Acylsubstituenten an der C = C-Bindung reagieren mit Ketenen generell zu 3-Amino-2-cyclobuten-1 -onen und/oder zu Allencarboxamiden2s3). Phosphorylgruppen-tragende Inamine cyclisieren bei Einwirken von Diphenylketen teilweise zu Naphthalinderivaten’). Welche Produkte entstehen, hangt von den 0- standigen Resten am Inamin und von den eingesetzten Ketenen ab. In diesem Zusam- menhang interessierte nun auch das bisher nicht bekannte Verhalten von (Silylethiny1)- und (Germylethiny1)aminen gegeniiber Diaryl- und Dialkylketenen.
Eninamine des Typs 4 Synthese und spektroskopische Daten
Inamine des Typs 1 addieren die Ketoketene 2 bevorzugt zu den neuartigen Eninami- nen 44). Deren Bildung 1Mt sich in plausibler Weise uber das Zwitterion 3” deuten, das durch nukleophile Einlagerung des Inamin-P-Kohlenstoffatoms in die Carbonylgruppe
0 Verlag Chemie, GmbH, D-6940 Weinheim, 1979
0170-2041/79/1111- 1828 $ 02.5010

1979 (Aminoethinyl)metallierungen, 6 1829
des Ketens entsteht3). Es schlierjt sich eine [1,3]-Wanderung des elementorganischen Restes MR: an, wobei das Inaminsystem zuriickerhalten wird.
3,4 a b c d e f g h i j k 1
m
1 2
R’ R2 M H3
E t E t S i Me M e P h Si M e P h P h Si M e Et E t Si P h M e P h S i P h
~ P h P h S i P h M e P h G e P h P h P h G e P h ,
R1 R2 M R3 CR;
Et Et S i Me CPhz M e P h S i Me C P h z Ph P h S i Me CPhz E t Et Si P h CPhz Me P h S i P h CPhz P h P h S i P h CPhz M e P h G e P h CPhz P h P h G e P h CPhz M e P h Si P h 9-Fluorenyl iden P h P h Si P h 9-Fluorenyl iden Et Et Si Me CMez M e P h S i Me CMez Et E t Si Me Cyclopentyl iden
3 4
Die [1,3]-Wanderung verlauft so rasch, da13 andere Reaktionen, wie z. B. Cyclisie- rungen oder Cycloadditionen, nicht oder nur in eingeschranktem M a e (s. die Bildung von 5) konkurrieren kbnnen. Diese Reaktionsweise der Inamine 1 gegeniiber elektro- philen x-Systemen wurde als (Aminoethiny1)metallierung bezeichnet (zur Definition s. Lit.@). Es lassen sich nicht nur Ketene (oder deren Carbonylgruppen), sondern auch die entsprechenden x-Systeme von Isocyanaten, Isothiocyanaten und Carbodiimiden’) und die C = C-Bindung von AcetylendicarbonsPureestern1,6*8) und von Arinen’) einsetzen.
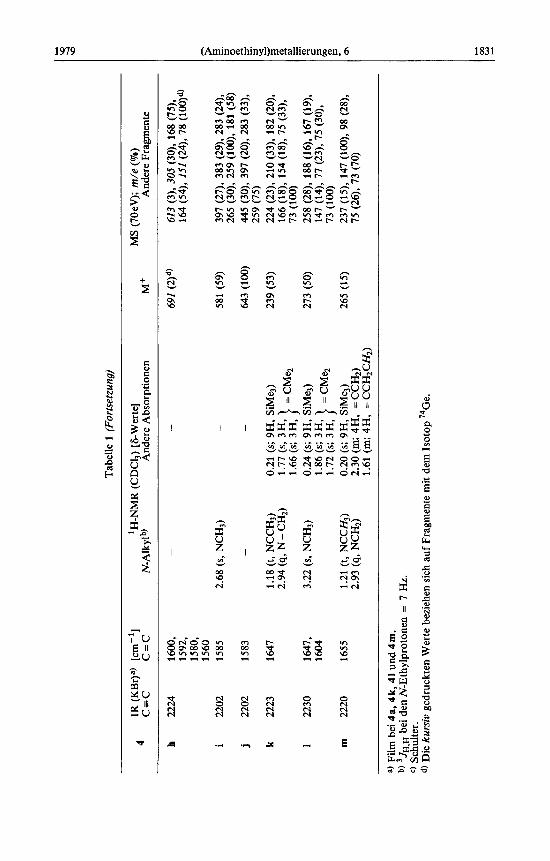
Die Konstitution der erhaltenen Eninamine 4 ist durch spektroskopische Daten be- legt, siehe Tabelle 1.
Nebenprodukte der Synthese
Bei den Umsetzungen mit Diphenylketen 2a werden keine Nebenreaktionen beob- achtet, wenn man die Keten-Lbsung zum Inamin tropft. 1st dagegen bei umgekehrter Zugabe das Keten im Uberschun vorhanden, treten neben 4 auch [I : 21-Addukte auf, da 4 als Inaminderivat mit Keten weiterreagieren kann.
Bei den beiden in-situ-Umsetzungen mit 9-Carbonylfluoren 2 b (erzeugt in Gegen- wart der Inamine l e und 1 f auf photochemischem Weg aus Diazophenanthrenchinon) werden neben den Eninaminen 4i und 4j die durch [2 + 21-Cycloaddition entstandenen Cyclobutenone 5i und 5j isoliert.

Tabe
lle 1
. Spe
ktro
skop
isch
e D
aten
der
Eni
nam
ine
4a- m
IR (
KBr
)=)
[cm
-’1
‘H-N
MR
(CD
C13
) [g-
Wer
te]
MS
(70e
V);
m/e
(Yo)
c=
c c=
c N
-Alk
yl b,
And
ere
Abs
orpt
ione
n M
+ A
nder
e Fr
agm
ente
a 22
16
1587
, 0.
95 (
t, N
CC
H,)
0.21
(s;
9H, S
iMq)
36
3 (6
) 34
8 (6
), 33
4 (6
), 14
7 (l
oo),
1569
2.
74 (
q, N
CH
2)
105
(26)
, 75
(27)
, 73 (44)
b 22
18
1599
, 3.
02 (s, N
CH
,) 0.
25 (
s; 9
H,
SiM
q)
1582
, 39
7 (3
0)
382
(II)
, 291
(49
), 18
8 (1
4),
75 (
21),
73 (1
00)
1566
c
2218
15
85,
-
0.13
(s;
9H
, SiM
q)
459
(40)
444
(6),
291
(loo
), 21
7 (1
9),
1 56O
C)
180
(19)
, 77
(19)
, 73
(60)
d
2210
15
92
0.71
(t,
NC
CH
3)
-
e 22
16
1600
2.
50 (s
, NC
H3)
-
f 22
35
1593
, -
-
2.44
(q,
“W
)
159O
C)
1565
c)
549
(85)
583
(25)
645
(51)
534
(20)
, 520
(60)
, 387
(21)
. 26
7 (2
2), 2
59 (
loo)
, 18
1 (4
0)
568
(13)
, 506
(19)
, 477
(44),
399 (60), 2
59 (
100)
56
8 (1
6), 4
77 (
62),
399 (64),
283
(39)
, 259
(68
), 18
0 (1
00)
g 22
08
1602
, 2.
51 (s,
NC
H3)
62
9 (3
2)d)
55
1 (3
2), 4
35 (
24),
358
(28)
, 30
5 (8
9), 2
07 (80), 1
54 (1
00)
-
1587
15
1 (6
5)d)
Tabe
lle 1
(For
tset
zung
)
IR (
KBr
)a)
[cm
-'1
'H-N
MR
(C
DC
l,) [
ti-W
erte
] M
S (7
0eV
); m
/e (T
o)
c=c
c=
c
N-A
lkyl
b)
And
ere
Abs
orpt
ione
n M
+
And
ere
Frag
men
te
-
h 22
24
1600
, 15
92,
1580
, 15
60
i 22
02
1585
2.
68 (
s, N
CH
3)
691
(2)d
)
581
(59)
j 22
02
1583
-
-
643
(loo
)
k 22
23
1647
1.
18 (t
, NC
CH
3)
0.21
(s;
9H, S
iMq)
2.
94 (9. N
- C
Hz)
1.
77 (s
, 3 H
, } =
CM
e2
1.66
(s;
3H,
239
(53)
1 22
30
1647
, 3.
22 (s
, N
CH
3)
0.24
(s;
9H
, SiM
q)
273
(50)
16
04
1.86
(s;
3H,
1.72
(s;
3 H, }
= C
Mez
2.30
(m
; 4H
, =
CC
Hz)
1.
61 (m
; 4H
, =
CC
HzC
Hz)
m
2220
16
55
1.21
(t, N
CC
H3)
0.
20 (s
; 9H
, Si
Me3
) 26
5 (1
5)
2.93
(9.
NC
H,)
a) F
ilm b
ei 4
a, 4k, 41
und
4m
.
C) Sc
hulte
r. d,
Die
kur
siu g
edru
ckte
n W
erte
bez
iehe
n si
ch a
uf F
ragm
ente
mit
dem
Iso
top
74G
e.
b, 3
&3 be
i den
N-E
thyl
prot
onen
= 7
Hz.
613
(3),
305
(30)
, 168
(73,
16
4 (5
4), 1
51 (2
4), 7
8
397
(27)
, 383
(29)
, 283
(24
), 26
5 (3
0), 2
59 (l
oo),
181
(58)
44
5 (3
0), 3
97 (
20),
283
(33)
, 25
9 (7
5)
224
(23)
, 210
(33)
, 182
(20)
, 16
6 (1
8),
154
(18)
, 75
(33)
, 73
(lo
o)
258
(28)
, 188
(16)
, 167
(19)
, 14
7 (1
4), 7
7 (2
3), 7
5 (3
0),
73 (
loo)
23
7 (1
5), 1
47 (l
oo),
98 (
28),
75 (
26),
73 (7
0)
Y
W 4
W
QI

1832 G. Himbert 1979
Die Dialkylketene 2c und 2d reagieren sehr vie1 trager als Diarylketene. Da zudem - zumindest im Fall von 2c - eine genaue Dosierung schwer zu erreichen ist, kann man das Auftreten der [l : 21-Addukte 17d und e nicht unterdriicken (s. Experimenteller Teil) .
Bei einigen Ansatzen zur Darstellung von 4k, 1 und m treten in den IR- und 'H- NMR-Spektren schwache Absorptionen auf, die den isomeren Acylinaminen 6 zuzu- ordnen sind. So kann bei der Umsetzung von l a mit 2c manchmal neben 4k auch 6k nachgewiesen werden und durch mehrmalige Kugelrohrdestillation angereichert werden (ca. 15%; 6k scheint einen etwas hoheren Siedepunkt zu haben als 4k). Die spektrosko- pischen Daten [IR (Film): 2178 cm-' (C=C). - 'H-NMR (CDCl,): 6 = 1.26 (t, J = 7 Hz; 6H, NCH,CH,), 1.39 (s; 6H, CMq), 0.16 (s; 9H, SiMe,)] harmonieren mit den bei Acylinaminen beobachteten Werten', lo).
5i: R' = Me
6-SiMe, ,. / I $: M e 1: 111 + TosN:, - N
E< 'Et 7 a f: /ci\
Et7N N\ Et Tos
5j: R' = P h 6k 8
Fur die Bildung von 6k neben 4k ist es naheliegend anzunehmen, da8 auf der Stufe des Zwitterions 3k als Endprodukt der Silylwanderung nicht nur der Enolatsauerstoff, sondern in untergeordnetem Ma13 auch das die negative Ladung mittragende Keten- Kohlenstoffatom fungieren kann. Nicht ganz auszuschliefien ist eine nachtragliche Iso- merisierung der Siloxyverbindung 4 zum Acylinamin 6, auch wenn bisher gezielte Iso- merisierungsversuche 4 -+ 6 fehlschlugen.
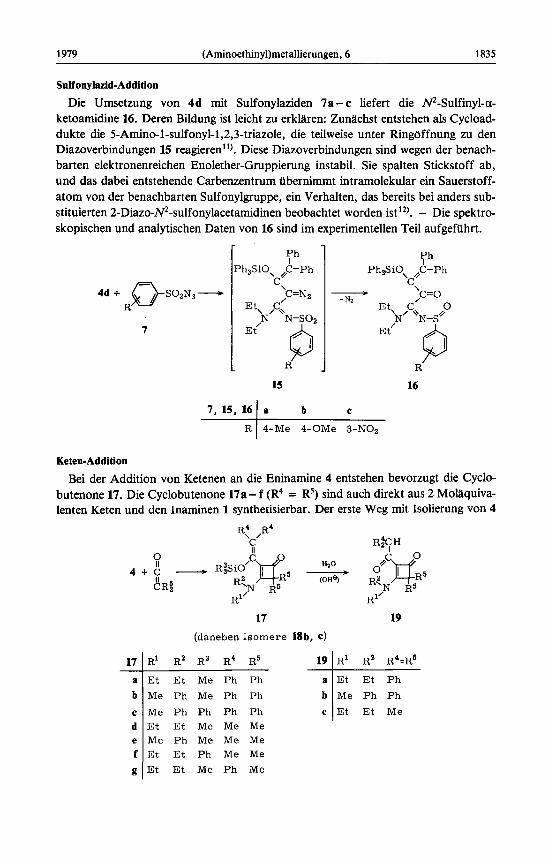
Eine Derivatisierung - und damit ein etwas uberzeugenderer Nachweis fur die Exi- stenz von 6k - gelingt durch Umsetzung des Gemisches von 4k und 6k mit Tosylazid (7a). Wahrend 4k zum a-Ketoamidin oder m Folgeprodukten reagiert (vgl. Bildung von 16), addiert 6k das Sulfonsaureazid zum 2-Diazo-3-ketoamidin 8, dessen spektro- skopische Daten (s. Experimenteller Teil) die Konstitution bestatigen").
Hydrolyse und Isomerisierung
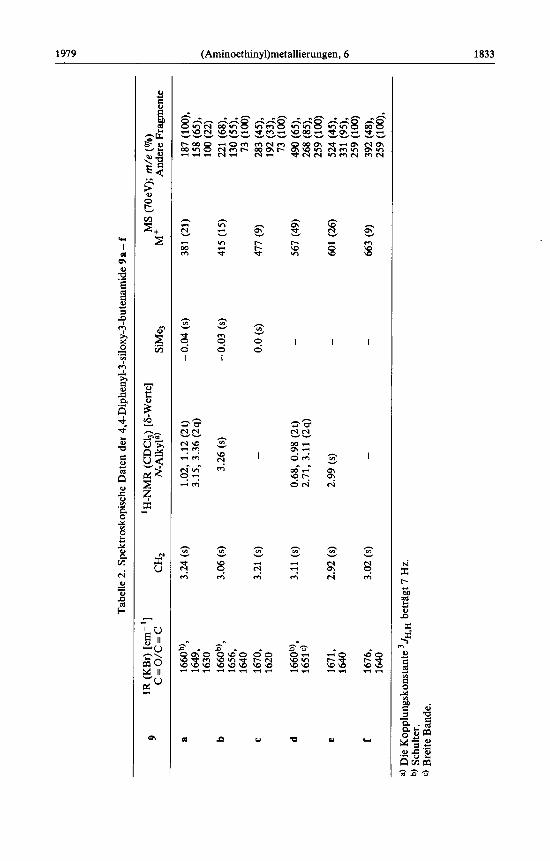
Bei vorsichtiger Hydrolyse der Eninamine 4a - f (Chromatographie an Kieselgel) ge- lingt die Isolierung der 3-Siloxy-3-butenamide 9a- f . Das Auftreten von IR- Absorptionen der Carboxamidgruppierung und der C = C-Bindung, eines 2-H- Singuletts im 'H-NMR-Spektrum und der Molekiilionen im Massenspektrum belegen deren Struktur, siehe Tabelle 2.
Die Trimethylsilylverbindungen 4a - c liefern selbst unter den voranstehend be- schriebenen, vorsichtigen Hydrolysebedingungen neben 9 auch die silylfreien Acetoacet- amide 10a - c. Wahrend die beiden ersten als Enole B kristallisieren, hat 1Oc im kristal- linen Zustand die Ketonform A. In CDCl,-Lbsung sind bei allen drei Verbindungen 'H- NMR-spektroskopisch Keto/Enol-Gleichgewichte (60 - 66% A) nachzuweisen. Quali-
1979 (Aminoethinv1)metallieruneen. 6 1833
h e % I-
h 0) W
8
I
h v) W - p! rn
m a V
1834 G. Himbert 1979
tativ h o t sich die Existenz der Enole B durch blauviolette Eisen(II1)-chlorid-Reakti- onen belegen.
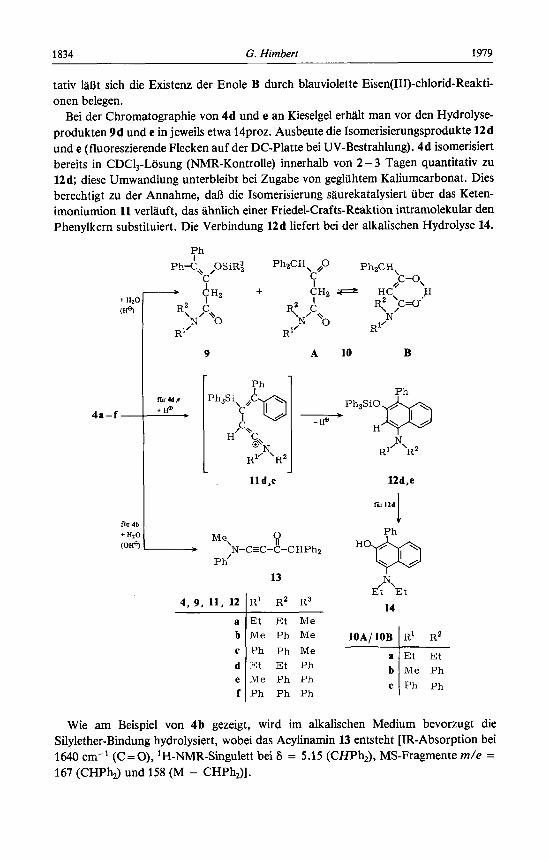
Bei der Chromatographie von 4d und e an Kieselgel erhalt man vor den Hydrolyse- produkten 9d und e in jeweils etwa 14proz. Ausbeute die Isomerisierungsprodukte 12d und e (fluoreszierende Flecken auf der DC-Platte bei UV-Bestrahlung). 4d isomerisiert bereits in CDCl,-Losung (NMR-Kontrolle) innerhalb von 2 - 3 Tagen quantitativ zu 12 d; diese Umwandlung unterbleibt bei Zugabe von gegliihtem Kaliumcarbonat. Dies berechtigt zu der Annahme, daR die Isomerisierung saurekatalysiert iiber das Keten- imoniumion 11 verlauft, das ahnlich einer Friedel-Crafts-Reaktion intramolekular den Phenylkern substituiert. Die Verbindung 12d liefert bei der alkalischen Hydrolyse 14.
P h
4a-f
9 A 10 B
13
R' R2 R3
Et Et M e Me P h M e P h P h M e Et Et P h M e P h P h P h P h P h
12d,e
I f i r 12d
N E't \Et
14
10A/ 1OB I R' R2
Wie am Beispiel von 4b gezeigt, wird im alkalischen Medium bevorzugt die Silylether-Bindung hydrolysiert, wobei das Acylinamin 13 entsteht [IR-Absorption bei 1640 cm-I (C=O), lH-NMR-Singulett bei 6 = 5.15 (CHPh,), MS-Fragmente m/e = 167 (CHPh,) und 158 (M - CHPh,)].
1979 (Aminoethinyl)metallierungen, 6 1835
4d + D S O Z N 3 - R
7
-
Yh c\ c/ 0
P h I
Ph3Si0, "C-Ph Ph3SiO\ "C-Ph
c=o C=N2 - c\ - N2
Et\ /c: Et\ / \\ N N-SOz N N-S"
R Et' R P - E ( 0-
Keten-Addition
Bei der Addition von Ketenen an die Eninamine 4 entstehen bevorzugt die Cyclo- butenone 17. Die Cyclobutenone 17a - f (R4 = R') sind auch direkt aus 2 Mokiquiva- lenten Keten und den Inaminen 1 synthetisierbar. Der erste Weg mit Isolierung von 4
R: /R4 R:CH $ I
4 + c 'i: - R $ S i O / c v R 5 Hzo ~ o"C)$5 !RZ R'N ~5 ( O H 9 R t N R5
R'/ R'/
17 a b
-
C
d e f
g
17 19
(daneben Isomere 18b, C)
R1 R2 R3 R4 R5
Et Et M e P h Ph M e P h M e P h Ph b M e P h P h M e P h P h P h P h c Et Et M e Et Et M e M e M e M e P h M e M e M e Et Et P h M e M e Et Et M e P h M e
1: 1:: :: ztR5

1836 G. Himbert 1979
gestattet jedoch den Einsatz verschiedener Ketene (R4 # R5) und damit die Synthese unsymmetrischer Cyclobutenone des Typs 17g. Bei der Bildung von l7b und c tritt auf beiden Reaktionswegen noch eine zu 17 konstitutionsisomere Substanz 18b bzw. 18c auf, deren Struktur bisher nicht aufgeklart werden konnte. Sie besitzen eine OH-Funk- tion, die auf eine Cyclisierung uber eine Phenylgruppe hinweist (spektroskopische und analytische Daten s. Experimenteller Teil)',).
Die Konstitution der Cyclobutenone 17a - g ist durch die Spektren belegt (vgl. Ta- belle 3). Im 'H-NMR-Spektrum spricht vor allem die Verdopplung der Signale der N- Ethyl-protonen bei 17a, d, fund g fur die Cyclobutenonkonstitution, da das vinyloge Carboxamidsystem der N - Ring-Bindung einen grol3en Doppelbindungscharakter ver- leiht und somit fur die Nichtlquivalenz beider Ethylgruppen am Stickstoff verantwort- lich ist.
Tabelle 3. IR- und 'H-NMR-Daten der Cyclobutenonderivate 17a- g und 19a - c
17 IR (KBK) [cm-'] 'H-NMR (CDCI,) [&Werte] 19 c=o4 C=cb) N-Alk ylC) Andere Signalec)
17 a
17b
17 c 17 d
17 e
17 f
17 g
19 a
19b
19 c
1739
1746
1747 1746
1742
1735
1748
1737 (1650) 1741
(1657) 1748
(1 649)
1575 (1 622) 1565
(1638) 1567 1588
(1673) (1 649) 1575
(1 666)
1580 (1671)
1585 (1630) 1592
1573
1580 1570
3.65, 3.10 (2q, je 2H), 1.30, 0.39 (2t, je 3H) 3.00 (s, 3H)
2.98 (s, 3H)d) 3.37 (q, 4H)d), 1.26, 1.15 (2t, je 3H)
3.49 (s, 3H)
3.18 (4, 4H)d), 1.16, 1.10 (2t, je 3H)
3.49, 3.17 (2q, je 2H), 1.17, 1.06 (2t, je 3H) 4.11, 3.31 (2q, je 2H), 1.22, 0.43 (2t, je 3H) 3.99 (s, 40V0)~), 3.20 (s, 60%) 4.00 (s, 3H)
-0.03 (s; 9H, SiMe,)
-0.10 (s; 9H, SiMe,)
- 0.13 (s; 9H, SiMq),
1.61 (s; 6H, Vinyl MQ) 0.19 (s; 9H, SiMq), 1.06 (s; 6H, ~ - M Q ) ~ ) , 1.61, 1.69 (2s; 6H, Vinyl Mq) 0.90 (s; 6H, ~ - M Q ) ~ ) , 1.59, 1.45 (2s; je 3H, Vinyl
0.11 (s; 9H, SiMe,),
1.30 (s; 6H, 4-M%),
Mez)
1.10 (s; 6H, 4-Mq) 6.13 (s; 1 H, CHPh2)
6.18 (s; 1 H(4O%), CNPhz)e), 5.92 (s; 1 H(6O%), CHPh2) 0.96 (s; 6H, ~ -MQ) , 1.12 (d, 6H, CHMe2), 3.43 (hept, 1 H, CHMq)
a) Absorptionen der Ringcarbonylgruppe; die in Klammern angegebenen Werte beziehen sich auf die 2-standige Acylgruppe.
b, Sehr intensive Absorptionen der Ringdoppelbindung; die in Klammern gesetzten Werte (schwache bis mittlere Intensitat) werden der exo-standigen Vinylgruppe zugeordnet. Nicht an- gegeben sind die aromatischen Ringschwingungsabsorptionen um 1600 und 1500 cm-'.
c, ,JH d, Brehe Absorptionen. e, Rotamerengleichgewicht.
bei den Ethyl- und Isopropylgruppen = 7 Hz.
Die basische Hydrolyse von 17a, b und d liefert die Acylcyclobutenone 19a- c. Auf- fallend ist, da8 im 'H-NMR-Spektrum des N-Methylanilino-Derivates 19b zwei rota-

1979 (Aminoethinyl)metallierungen, 6 1837
mere Formen beobachtet werden konnen, wahrend 17 b keine Signalverdopplung zeigt. Die Konstitutionszuordnung wird durch eine unabhangige Synthese (13 + 2a -+ 19b) gestutzt. Die IR-Daten von 19 unterscheiden sich von denen der Edukte 17 vor allem durch die grorjere Intensitat der im Bereich um 1650 cm-' auftretenden Absorption, da diese nun nicht mehr durch eine C = C-, sondern durch eine C = 0-Bindung verursacht wird.
Der Deutschen Forschungsgemeinschuft schulde ich Dank fur finanzielle Unterstutzung. Frau M. Alester danke ich fiir die Ausfiihrung der Elementaranalysen, Herrn G. Huuge fur die Auf- nahme der Massenspektren, Fraulein M. Jung fur die Anfertigung der IR-Spektren und fur die praparative Mitarbeit.
Experimenteller Teil Die Schmelzpunkte wurden im Heizblock bestimmt und sind unkorrigiert. - Saulenchromato-
graphische Trennungen wurden an Kieselgel der Fa. Woelm (0.05 - 0.2 mm) vorgenommen und durch DC rnit den fur die Saulentrennung angegebenen FlieDmitteln kontrolliert. - IR-Spektren: Gerat Beckman IR-20 A. 'H-NMR-Spektren: Gerate Varian NV 14 und Perkin Elmer R 24 (Te- tramethylsilan als interner Standard). Massenspektren (70eV): Gerat Varian MAT 31 1. - Ele- mentaranalysen: Perkin-Elmer Elemental Analyzer 240. - Der verwendete Petrolether siedete im Bereich 40-70°C.
Ausgangsoerbindungen: Die Inamine 1 a- h werden nach Literaturvorschrift hergestellt14). Di- phenylketen (2a), 9-Carbonylfluoren (2 b) und Carbonylcyclopentan (2 d) werden aus den ent- sprechenden Diazoverbindungen durch Wolff-Umlagerung erzeugt15). 2a wird redestilliert bei den Umsetzungen eingesetzt16), wahrend 2b und 2d in Gegenwart der Inamine photolytisch er- zeugt und in situ umgesetzt werden. Dimethylketen (2c) (in Ether) wird durch Dehalogenierung rnit Zink aus 2-Brom-2-methylpropionylbromid gewonnen17).
Umsetzungen der Inamine l a - h mit Ketenen
4,4-Diphenyl-3-siloxy-3-buten-l-inylamine 4a - f. - Allgemeine Vorschr$t: Zu einer Ldsung von 20 mmol (Si1ylethinyl)amin 1 a - f in 30 ml wasserfreiem Ether tropft man unter Riihren bei - 20°C innerhalb 10 min 3.88 g (20.0 mmol) Diphenylketenl@ in 30 mlEther und ruhrt anschlie- Dend 4 h bei Raumtemp. Im Falle der Umsetzungen von l e und I f kristallisieren die Addukte 4e und 4f aus; bei den iibrigen entfernt man das Ldsungsmittel i.Vak., destilliert den Ruckstand im Kugelrohr (4a) bzw. nimmt ihn in 50 ml Petrolether auf, kuhlt und reibt an (4b-d). Ausbeuten und analytische Daten von 4a- f s. Tabelle 4.
3-Germyloxy-4,4-diphenyl-3-buten-I-inylamine (4g und h): Zu einer Ldsung von 2 mmol der (Germylethiny1)amine l g und h in 20 ml wasserfreiem Ether gibt man bei -20°C 0.39 g (2 mmol) Diphenylketen'@ und b e l a t anschlieflend 4 h bei Raumtemp. Nach Kiihlen auf - 70°C und Anreiben isoliert man durch Abnutschen die Eninamine 4g und h; Ausbeuten und analyti- sche Daten s. Tabelle 4.
Umetzung oon 1 e und 1 f rnit 9-Carbonylfuoren. - Allgemeine Vorschriyt: Die Mischung von 10 mmol l e bzw. I f und 2.20 g (10.0 mmol) DiazophenanthrenchinonlQ in 100 ml Benzol wird in einem Pyrex-Bestrahlungsgefa rnit Wasserkuhlung 7 h rnit einer Hg-Hochdrucklampe vom Typ Philips HPK 125 W bestrahlt. Es entweichen in dieser Zeit etwa 230 ml Stickstoff. Es wird i. Vak. eingedampft und in 50 ml Ether aufgenommen.

1838 G. Himbert 1979
A) Im Fall 1 e + 2b erhalt man ein Gemisch zweier Substanzen (4.15 g), das an 50 g Kieselgel rnit zunachst 200 ml Chloroform und anschlieoend ni t 500 ml ChlorfordEther (9: 1) aufgetrennt wird. Man isoliert nacheinander: a) 2 g (34%) Eninamin 4i (gelbe Fraktion); analytische Daten s. Tabelle 419). b) 1.1 g (19%) Fluoren-9-spiro-l'-[4'-N-methylanilino)-3 '-triphenylsilyl-3'-cyclobuten-2 'on] (Si); aus ChlorofordEther farblose Kristalle rnit Schmp. 208 - 209 "C (schwarzbraune Schmelze, Verhrbung der Kristalle ab 180°C). - IR (KBr): 1735 (C=O), 1545 breit (C=C), 1435 cm-' (Si-Phenyl). - 'H-NMR(CDCl3): 6 = 2.70 (s; 3H, NMe). - MS (70eV): m/e = 581 (63%, M'), 323 (13'70, M + H - SiPh3), 259 (100%, SiPh3), 199 (26V0), 181 (27%), 165 (13Vo), 134 (17%), 106 (16%); das Entstehen des Peaks bei m/e = 840 (<1%, Mf + SiPh,) wird durch Molekiilion-Reaktion gedeutet.
C41H31NOSi (581.8) Ber. C 84.64 H 5.37 N 2.41 Gef. C 83.8 H 5.44 N 2.4
B) Im Fall I f + 2b kristallisieren aus Ether 3.26 g (51%) reines Eninamin 4j; aus ChlorofordEther gelbe Kristalle rnit Schmp. 193 - 196OC. Analytische Daten s. Tabelle 4. - Das Filtrat von 4j wird i. Vak. eingedampft und der Riickstand an 50 g Kieselgel rnit 200 ml Chlo- roform und 500 ml ChlorofordEther (9: 1) chromatographiert. Man isoliert nacheinander: a) 0.54 g (8Vo) Eninamin 4j (Gesamtausbeute 3.80 g P 59%). b) 0.8 g (1 2%) Fluoren-9-spiro-l'-(4'-diphenylamino-3 '-triphenylsilyM '-cyclobuten-2'-on) (5 j); aus ChlorofordEther farblose Kristalle rnit Schmp. 227 - 228 "C (braune Schmelze, Sintern ab 220°C). - IR(KBr): 1739(C=O), 1531 (C=C), 1435 cm-' (Si-Phenyl). - 'H-NMR(CDC13): nur Signale aromatischer Protonen. - MS (70eV): m/e = 643 (31%, M'), 566 (13%, M - Ph), 402 (43@/'0), 259 (100V0, SiPh3), 181 (23%), 168.(18%), 105 (17%).
C46H33NOSi (643.9) Ber. C 85.80 H 5.17 N 2.17 Gef. C 85.3 H 5.22 N 2.1
Llmsetzung oon l a und 1 b rnit Dimethylketen 2c. - Allgemeine Vorschrvt: 10 mmol Inamin l a bzw. l b werden rnit 30 ml etherischer Dimethylketen-LOs~ng~~) versetzt und 12 h bei Raum- temp. geriihrt. Man entfernt das Lbsungsmittel i. Vak. und destilliert im Kugelrohr bei olpum- penvakuum (IR- und 'H-NMR-Spektren zeigen, d d die Rohgemische jeweils 1, 4 und 17 erhal- ten).
A) Im Fall l a + 2c erhalt man durch fraktionierende Kugelrohrdestillation: a) bei 20-80°C/0.4 Torr 0.3 g vorwiegend l a ; b) bei 80- 12O0C/O.4 Torr 0.93 g (39%) vorwiegend 4k, die bei der Redestillation 0.7 g (29%) reines 4k als fast farbloses 0 1 mit Sdp. 80-85 "U0.3 Torr liefern. Analytische Daten s. Tabel- le 4. c) Der Destillationsriickstand liefert in einer gesonderten Destillation 0.32 g (10%) 17d als farb- loses 0 1 mit Sdp. 110-120°C/0.3 Torr. - MS (70eV): m/e = 309 (13'70, M'), 294 @yo, M -Me), 242 (140/0), 147 (25%), 100 (24%, CONEtZ), 75 (100'7'0, HOSiMq), 73 (53%, SiMq), 72 (25%, NEt2). Weitere spektroskopische und die analytischen Daten s. Tabellen 3 und 6. B) Im Fall 1 b + 2c erhiilt man durch fraktionierende Kugelrohrdestillation:
a) bei 20- 100"C/0.3 Torr 0.4 g vorwiegend 1 b; b) bei 100- 14O0C/O.3 Torr 1.25 g (46070) vorwiegend 41, die bei der Redestillation 1.15 (42'70) reines 41 als gelbliches 61 mit Sdp. 110- 12OoC/0.3 Torr liefern. Analytische Daten s. Tabelle 4. c) Der Destillationsriickstand liefert bei 140- 180 "C/0.3 Torr (gesonderte Destillation) 0.45 g (9010) 17e, die beim Versetzen mit Petrolether kristallisieren. - MS (70eV): m/e = 343 (32%, M'), 328 (22%, M - Me), 300 (23Vo), 224 (44%), 160 (20%), 119 (26%), 77 (19'-70), 75 (190/0), 73 (100%, SiMq). Weitere spektroskopische und die analytischen Daten s. Tabellen 3 und 6.
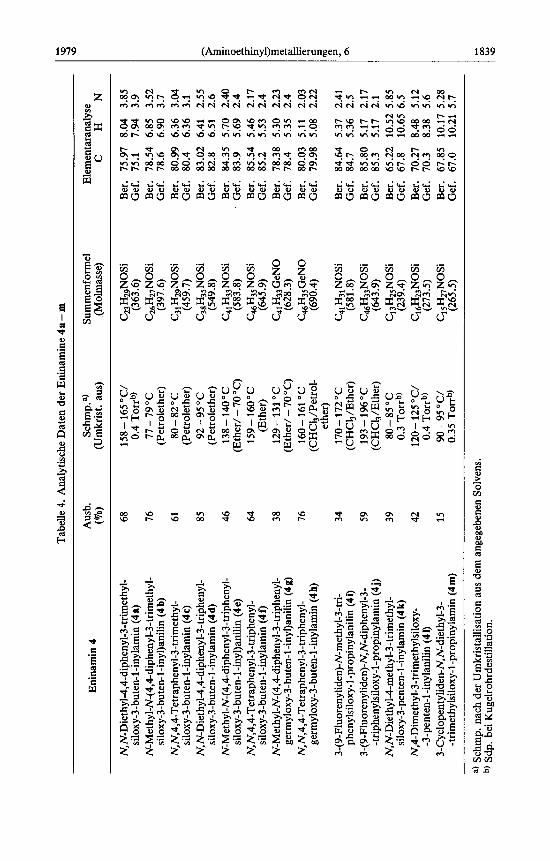
Tabe
lle 4.
Ana
lytis
che
Dat
en d
er E
nina
min
e 4a
-m
Enin
amin
4
N, N-Diethyl-4,4-diphenyl-3-trimethyl-
doxy
-3-b
uten
-1-in
ylam
in (
4 a)
N-Methyl-N-(4,4-diphenyl-3-trimethyl-
siloxy-3-buten-I-iny1)anilin (4
b)
N, N,4,4-Tetraphenyl-3-trimethyl-
siloxy-3-buten-I-inylamin (4
c)
N, N-Diethyl-4,4-dipheny1-3-triphenyl-
silo
xy-3
-but
en-I
-inyl
amin
(4d)
N-Methyl-N-(4,4-diphenyl-3- triph
enyl
- siloxy-3-buten-1-inyl)anilin
(4e)
N,N,4,4-Tetraphenyl-3-triphenyl-
siloxy-3-buten-1-inylamin (4
0 N-Methyl-N-(4,4-diphenyl-3-triphenyl-
germyloxy-3-buten-1-iny1)anilin (
4 g)
N, N,4,4-Tetraphenyl-3-triphenyl-
germyloxy-3-buten-I-inylamin (4
h)
3-(9-Fluorenyliden)-N-methyl-3-tri-
phen
ylsi
loxy
-1 -p
ropi
nyla
nilin
(4 i
) 3-
(9-F
luor
enyl
iden
)-N
, N-d
iphe
nyl-3
- -tr
iphe
nyls
iloxy
-1 -p
ropi
nyla
min
(4 j
) N.N-Diethyl-4-methyl-3-trimethyl-
siloxy-3-penten-I-inylamin (4 k)
N,4-Dimethyl-3-trimethylsiloxy-
-3-p
ente
n-1-
inyl
anili
n (4
I) 3-
Cyc
lope
ntyl
iden
-N, N
-die
thyl
-3-
-trim
ethy
lsilo
xy-I
-pro
piny
lam
in (
4 m)
Aus
b.
VO)
68
76
61
85
46
64
38
76
34
59
39
42
15
Schm
p. a)
(Um
kris
t. au
s)
158-
165"
C/
0.4
Tom
b)
77 - 7
9°C
(P
etro
leth
er)
80-8
2°C
(P
etro
leth
er)
(Pet
role
ther
) 13
8- 1
40°C
(E
ther
/ - 70
"C)
159-
160°
C
(Eth
er)
129-
131
"C
(Eth
er/ -
70°
C)
(CH
CI3
/Pet
rol-
ethe
r)
(CH
CI3
/Eth
er)
(CH
C13
/Eth
er)
0.3
Tom
b)
120-
125
"C/
0.4
Ton
b)
90 - 95
"C/
0.35
Tom
b)
92 - 95
"C
160-
161
"C
170-
172°
C
193 -
196
°C
80-8
5°C
Sum
men
form
el
(Mol
mas
se)
q3H
z9N
OSi
(3
63.6
) C
&H
z7N
OSi
(3
97.6
) C
3, H
z9N
OSi
(4
59.7
) C
38H
35N
OSi
(5
49.8
) C4
1 H33
NO
Si
(583
.8)
C6H
3, N
OSi
(6
45.9
) C
dl H
33G
eN0
(628
.3)
C, H
35 G
eNO
(6
90.4
)
C41 H
31N
OSi
(5
81.8
) C
46H
33N
OSi
(6
43.9
) C1
Hz5
NO
Si
(239
.4)
Cl6
HZ3
NO
Si
(273
.5)
C15
Hz7
NO
Si
(265
.5)
Elem
enta
rana
lyse
C
HN
Ber
. 75
.97
8.04
3.
85
Gef
. 75
.1
7.94
3.
9 B
er.
78.5
4 6.
85
3.52
G
ef.
78.6
6.
90
3.7
Ber
. 80
.99
6.36
3.
04
Gef
. 80
.4
6.36
3.
1 B
er.
83.0
2 6.
41
2.55
G
ef.
82.8
6.
51
2.6
Ber
. 84
.35
5.70
2.
40
Gef
. 83
.9
5.69
2.4
B
er.
85.5
4 5.
46
2.17
G
ef.
85.2
5.
53
2.4
Ber
. 78
.38
5.30
2.
23
Gef
. 78
.4
5.35
2.4
B
er.
80.0
3 5.
11
2.03
G
ef.
79.9
8 5.
08
2.22
Ber
. 84
.64
Gef
. 84
.7
Ber
. 85
.80
Gef
. 85
.3
Ber
. 65
.22
Gef
. 67
.8
Ber
. 70
.27
Gef
. 70
.3
Ber
. 67
.85
Gef
. 67
.0
5.37
2.
41
5.36
2.5
5.
17
2.17
5.
17
2.1
10.5
2 5.
85
10.6
5 6.
5 8.
48
5.12
8.
38
5.6
10.1
7 5.
28
10.2
1 5.
7
a) S
chm
p. n
ach
der
Um
kris
talli
satio
n au
s de
m a
ngeg
eben
en S
olve
ns.
b, S
dp. b
ei K
ugel
rohr
dest
illat
ion.
C
L
00
w
W

1840 G. Himbert 1979
3-Cyclopentyliden-N,N-diethyl-3-trimethyIsiloxy-l-propinylamin (4m): Die Mischung von 1.24 g (10 mmol) 2-Diazocyclohexanon20) und 1.7 g (10 mmol) l a in 80 ml Ether wird in einem Pyrex-Bestrahlungsgefu rnit einer Hg-Hochdrucklampe vom Typ Philips HPK 125 W unter Wasserkiihlung 1.5 h bestrahlt, wobei etwa 260 ml Stickstoff entwickelt werden. Man ruhrt noch 30 min bei Raumtemp., dampft das Losungsmittel ab und destilliert den Riickstand i. Vak. Nach dreimaliger Kugelrohrdestillation erhalt man 0.4 g (15%) 4m als gelbes 01 mit Sdp. 88 - 95 "U0.35 Torrlg). Das bei der Umsetzung mitentstandene [l : 21-Addukt vom Typ 17 wird nicht isoliert. Analytische Daten von 4m s. Tabelle 4.
2-Diazo-N',N'-diethyl-4-methyl-3-oxo-N2-tosyl-4-(trimethylsi~l)pentanamidin (8): Ein Ge- misch von 0.7 g 4 k/6k und 0.5 g Tosylazid 7a2') in 3 ml Ether wird 12 h bei Raumtemp. geriihrt. Nach Entfernen des LiSsungsmittels i. Vak. wird der Riickstand an 50 g Kieselgel zunlchst rnit 150 ml Chloroform und anschlieRend rnit 500 ml Chloroform/Ether (9: 1) chromatographiert. Nach nicht verbrauchtem Azid 7 a isoliert man in der 20. bis 30. Fraktion (je 14 ml) 130 mg 01, aus dem nach Behandlung rnit Petrolether etwa 70 mg 8 auskristallisieren; aus Petrolether hellgel- be Kristalle rnit Schmp. 64-66°C. - IR (KBr): 2118 (CNz), 1661 (C=O), 1554 (C=N), 1315, 1302, 1279, 1146 cm-' (SQ-Bereich). - 'H-NMR (CDCI,): 6 = 3.43 (unstrukturiert; 4H, NCH2), 2.38 (s; 3H, Aryl-Me), 1.48 (s; 6H, CMQ), 1.15 (t, J = 7 Hz; 6H, NCH2CH3), 0.22 (s; 9H, SiMq). - MS (70eV): m/e = 408 (1010, M - N2), 221 (14%), 155 (16%), 139 (330/0), 131 (74%), 115 (26070, MQCSiMq), 91 (51%), 73 (100%, SiMq).
CMH32N403SSi (436.7) Ber. C 55.01 H 7.39 N 12.86 Gef. C 53.9 H 7.04 N 12.7
Hydrolyse und Isomerisierung
3-Trimethylsiloxy-3-butenamide 9 a - c und Acetoacetamide 10a - c. - Allgemeine Vorschrift: Jeweils 1 g 4a- c werden auf eine Saule mit 50 g Kieselgel gegeben und rnit 200 ml Chloroform und 500 ml ChlorofordEther (9: 1) chromatographiert. Man isoliert nacheinander die Buten- amide 9a-c und die Acetamide 10a-c. Analytische Daten s. Tabelle 5 .
10Aa/lOBa: IR (KBr): 1630 cm-' breit (C=O) [B]. - 'H-NMR (CDCl3): 6 = 15.2 (s; OH, B), 5.02, 4.88 (2s; Vinyl-H und CHPh2, B), 1.02 (t, J = 7 Hz; NCHzCH3, B), 5.53 (s; CHPh2, A), 3.52(s;CHzC0,A),1.10(t,J= 7Hz;NCH2CH,,A),3.34,3.12(2q,J= 7Hz;je2H,NCHz,A und B) [63% A/37% B]. - MS (70 eV): m/e = 309 (209'0, M'), 167 (loo%, CHPh2), 142 (73%,
10Ab/lOBb: IR(KBr): 1628cm-'breit(C=O)[B]. - 'H-NMR(CDC13): 6 = 14.42(s;OH,B), 4.76,4.66 (2s; Vinyl-H und CHPh,, B), 5.23 (s; CHPh2, A), 3.34 (s; CH2C0, A), 3.23 (s; NCH3, A und B) [66% A/34% B]. - MS (70eV): m/e = 343 (12%. M'), 176 (53%, M - CHPhz), 167
10Ac/lOBc: IR (KBr): 1720 (Keto-C=0), 1662 (Amid-C=0) [A]. - 'H-NMR (CDC13): 6 = 14.22(s; OH,B),4.88,4.74(2~; Vinyl-HundCHPh2,B), 5.22(s; CHPh2, A),3.56(s;CH2C0, A) [60% A/40% B]. - MS(70eV): m/e = 405 (2010, M'), 196 (9010, M - CONPhz), 169(77%), 167
3-Triphenylsiloxy-3-butenamide 9d - f und Naphthylamine 12d und e. - AIIgemeine Vor- schrift: Man tragt 1 g 4d - f auf eine Saule rnit 50 g Kieselgel auf und eluiert rnit 200 ml Chloro- form und 500 ml Chloroform/Ether (9: 1). In den beiden ersten Fallen isoliert man zuntichst die Isomerisierungsprodukte 12d und e (jeweils 14%) und anschlieoend die 3-Butenamide 9d und e. Bei der letzten Umsetzung erhPlt man zunPchst 0.68 g 4f zuruck und anschlieRend 0.26g 9f. Analytische Daten von 9d-f s. Tabelle 5 .
N, N-Diethyl-4-phenyl-3-triphenylsiloxy-I-naphthylamin (12 d): Aus Petrolether farblose Kristalle mit Schmp. 141 - 143°C. - IR(KBr): 1582cm-' (C=C). - 'H-NMR(CDC13): 6 = 0.85 (t, J =
M - 167), 100 (78'70, CONEt2).
(100V0, CHPhz), 134 (74%), 106 (53%).
(100%).
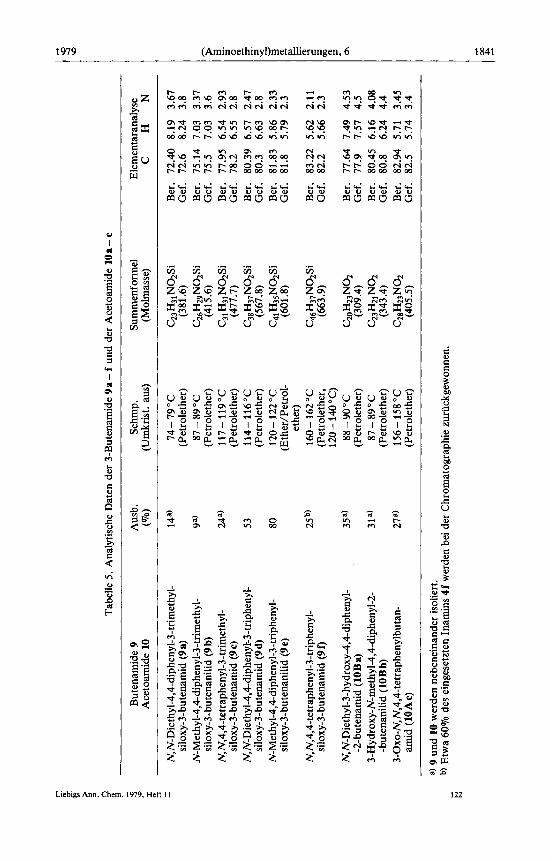
Tabe
lle 5
. Ana
lytis
che D
aten
der
3-B
uten
amid
e 9a
- f
und
der
Ace
toam
ide
10a-
c
But
enam
ide
9 A
ceto
amid
e 10
A
usb.
Sc
hmp.
Su
mm
enfo
rmel
E
lem
enta
rana
lyse
(VO)
(Um
kris
t. au
s)
(Mol
mas
se)
CH
N
N,N-Diethyl-4,4-diphenyl-3-trimethyl-
N-Methyl-4,4-diphenyl-3-trimethyl-
N,N,4,4-tetraphenyl-3-trimethyl-
N,N-Diethyl-4,4-diphenyk-3- triph
enyl
-
N-Methyl-4,4-diphenyl-3-triphenyl-
silo
xy-3
-but
enam
id (9
a)
silo
xy-3
-but
enan
ilid
(9 b)
silo
xy-3
-but
enam
id (9
c)
silo
xy-3
-but
enam
id (9
d)
silo
xy-3
-but
enan
ilid
(9e)
N, N,4,4-tetraphenyl-3-triphenyl-
silo
xy-3
-but
enam
id (9
f)
N, N-D
ieth
yl-3
- hyd
roxy
-4,4
-dip
heny
l-
3-Hydroxy-N-methyl-4,4-diphenyl-2-
3-0xo-N,N,4,4-tetraphenylbutan-
-2-b
uten
amid
(10
Ba)
-but
enad
id (
10B
b)
amid
(10
Ac)
14a)
9 a)
24a)
53
80
25 b,
35a)
31 a)
27 a)
74-7
9°C
(P
etro
leth
er)
87-8
9°C
(P
etro
leth
er)
(Pet
role
ther
) 11
4- 1
16°C
(P
etro
leth
er)
120-
122°
C
(Eth
edPe
trol
- et
her)
(Pet
role
ther
, 12
0- 1
40°C
)
117-
119°
C
160-
162°
C
88-9
0°C
(P
etro
leth
er)
(Pet
role
ther
)
(Pet
role
ther
)
87 - 8
9°C
156-
158
°C
c3H
31N
@S
i
c6H
29N
@si
c3 1
H3
1 Nqsi
C38
H37
N@
Si
C41
H35
N@
Si
(381
.6)
(4 1 5
.6)
(477
.7)
(567
.8)
(601
.8)
C6H
3,N
@Si
(6
63.9
)
GO
H23
N@
c23
H21
N@
%H
23N
@
(405
.5)
(309
.4)
(343
.4)
Ber
. G
ef.
Ber
. G
ef.
Ber
. G
ef.
Ber
. G
ef.
Ber
. G
ef.
72.4
0 8.
19
3.67
72
.6
8.24
3.
8 75
.14
7.03
3.
37
75.5
7.
03
3.6
77.9
5 6.
54
2.93
78
.2
6.55
2.
8 80
.39
6.57
2.
47
80.3
6.
63
2.8
81.8
3 5.
86
2.33
81
.8
5.79
2.
3
Ber
. 83
.22
5.62
2.
11
Gef
. 82
.2
5.66
2.
3
Ber
. 77
.64
7.49
4.
53
Gef
. 77
.9
7.57
4.
5 B
er.
80.4
5 6.
16
4.08
G
ef.
80.8
6.
24
4.4
Ber
. 82
.94
5.71
3.
45
Gef
. 82
.5
5.74
3.
4
a) 9
und
10
wer
den
nebe
nein
ande
r is
olie
rt.
b) E
twa
60%
des
ein
gese
tzte
n In
amin
s 4f
wer
den
bei d
er C
hrom
atog
raph
ie z
uriic
kgew
onne
n.

1842 G. Himbert 1979
7 Hz; 6H, NCH2CH,), 2.90 (9, J = 7 Hz; 4H, NCH2), 6.77 (s; l H , Naphthalin-2-H). - MS (70eV): m/e = 549 (100V0, M'), 534 (%Yo), 378 (39%), 259 (78070), 199 (19%), 181 (23%).
C,,H,,NOSi (549.8) Ber. C 83.02 H 6.41 N 2.55 Gef. C 83.3 H 6.42 N 2.6
N-Methyl-4-phenyl-3-triphenylsiloxy-l-naphthylanilin (12e): Aus Petrolether farblose Kristalle mit Schmp. 129-131°C. - IR (KBr): 1616, 1604, 1590, 1580 cm-l (C=C). - 'H-NMR (CDCl,): 6 = 3.17 (s; 3H, NCH,). - MS (70eV): m/e = 583 (100V0, M'), 259 (44S), 181 (17%), 105 (10%).
C41H33NOSi (583.8) Ber. C 84.35 H 5.70 N 2.40 Gef. C 83.9 H 5.74 N 2.7
Gezielte Isomerisierung uon 4d zu 12d: Man be ld t 0.5 g 4d in 3 ml CDCl, 5 Tage bei Raum- temp. Man entfernt das Losungsmittel i.Vak., nimmt in 15 ml Petrolether auf, kiihlt und reibt an. 12d kristallisiert in 84proz. Ausbeute (0.42 g) aus. Die Isomerisierung unterbleibt unter den angegebenen Bedingungen, wenn man 0.2 g gegliihtes Kaliumcarbonat zusetzt.
4-Diethylamino-I-phenyl-2-naphthol(l4): 1 g 12d wird in 20 ml Ethanol und 4 ml Wasser 1 h mit 1 g Kaliumhydroxid geriihrt. Man neutralisiert mit verd. Salzsaure, schiittelt mit vie1 Ether aus, trocknet die organische Phase mit Calciumchlorid und entfernt das Losungsmittel i. Vak. Bei Zugabe von Ether/Petrolether kristallisieren 0.2 g (400/0) Triphenylsilanol. Das Filtrat wird einge- dampft und an 60 g Kieselgel mit Chloroform chromatographiert. In der 8. bis 35. Fraktion (je 11 ml) isoliert man 0.4 g (75%) 14 als gelbliches 01 mit Sdp. 170- 180 "C/0.4 Torr (Kugelrohrde- stillation). - IR (Film): 3500, 3420 cm-' (OH). - 'H-NMR (CDC1,): 6 = 6.92 (s; 1 H, 3-H),
(70eV): m/e = 291 (52%, M'), 276 (100%, M - Me). 5.08 (s; l H , OH), 3.23 (q, J = 7 Hz; 4H, NCH,), 1.10 (t, J = 7 Hz; 6H, NCH~CHY,). - MS
CmHZINO (291.4) Ber. C 82.44 H 7.26 N4.81 Gef. C 82.2 H7.27 N5.0
4-(N-Methylanilino)-I, I-diphenyC3-butin-2-on (13): 2 g (5 mmol) 4b werden in einem Gemisch von 20 ml Methanol und 10 ml Wasser suspendiert, mit 2 Kaliumhydroxidpastillen versetzt und 15 min bei Raumtemp. geriihrt. Man gibt 50 ml Eis/Wasser hinzu und erhalt durch Absaugen 1.5 g (92%) 13; aus Ether farblose Kristalle mit Schmp. 105-106°C. - IR (KBr): 2200, 2180 (C=C), 1640cm-' (C=O). - 'H-NMR(CDC1,): 6 = 5.15 (s; 1 H, CHPhZ), 3.20(s; 3H, NMe). - MS (70eV): m/e = 325 ( 5 % , M'), 167 (10070, CHPhZ), 158 (100V0, M - CHPh2).
C2,H1,NO (325.4) Ber. C 84.90 H 5.88 N4.31 Gef. C 85.5 H 5.99 N4.4
Umsetzungen mit Sulfonylaziden
N',N'-Diethyl-3-oxo-4,4-diphenyl-N2-(p-tolylsulfinyl)-3-triphenylsiloxy-3-butenamidin (16a): 2.2 g (4 mmol) 4d in 5 ml Ether werden mit 2 g (10 mmol) Tosy1azidz1) (7a) versetzt und 4 h bei Raumtemp. geriihrt. Man isoliert durch Absaugen 2.42 g (84%) 16a als gelbe Kristalle mit Schmp. 211-212°C. - IR (KBr): 1643 (C=O), 1546 (C=N), 1117, 1100, 1074 cm-' (SO- Bereich). - 'H-NMR (CDCI3): 6 = 2.68 (m; 2H, NCH2), 2.32 (q, J = 7 Hz; 2H, NCH2), 2.34 (s; 3H, Aryl-Me), 0.62, 0.40 (2t, J = 7Hz; je 3H, NCH2CH,). - MS (70eV): m/e = 718 (< 170, M'), 580 (24%, M - SOC,H,), 503 (330/0), 259 (100%, SiPh,), 91 (29%).
C4,h2N2O3SSi (719.0) Ber. C 75.17 H 5.89 N 3.90 Gef. C 74.7 H 5.89 N 3.8
N', N'-Diethyl-N2-(4-methoxyphenylsulfinyl)-3-oxo-4,4-diphenyl-3-triphenylsiloxy-3-buten- amidin (16b): 1.1 g (2 mmol) 4d in 10 ml Ether werden portionsweise mit 0.43 g (2 mmol) 4- Methoxybenzolsulfonylazid22) (7b) versetzt und 10 h bei Raumtemp. geriihrt. Man erhalt 1.23 g (87%) 16b; aus ChlorofordEther leuchtend gelbe Kristalle mit Schmp. 201 -202°C. - IR (KBr): 1642 (C=O), 1544 (C=N), 1115, 1101, 1079, 1071 cm-' Schulter (SO-Bereich). - 'H- NMR (CDCI,): 6 = 3.77 (s; 3H, OCH,), 2.68, 2.64 (2q, J = 7 Hz; je 1 H, NCH,), 2.37 (9. J = 7 Hz; 2H, NCH,), 0.76: 0.53 (t, J = 7 Hz; je 3 H, NCH2CH3). - MS (70eV): m/e = 734 (1070,

1979 (Aminoethinyl)metallierungen, 6 1843
M'), 657 (2%, M - Ph), 579 (22%), 551 (l6%), 481 (130/0), 414 (17Oro), 259 (100%), 199 (l8%), 181 (20%), 155 (220/0), 139 (28%).
C45&2N204SSi (735.0) Ber. C 73.54 H 5.76 N 3.81 Gef. C 73.0 H 5.75 N 3.8
N', N'-Diethyl-N2-(3-nitrophenylsulfinyl)-j-4-diphenyl-3-triphenylsiloxy-3-butenamidin (16c): Eine Mischung von 0.55 g (1 mmol) 4d und 0.46 g (2 mmol) 3-Nitrobenzols~lfonylazid~~) (7c) in 5 ml Ether wird 1 h bei Raumtemp. geriihrt. Man isoliert durch Abnutschen 0.44 g (57%) 16c; aus Chloroform/Ether gelbe Kristalle rnit Schmp. 184-186°C. - IR (KBr): 1661, 1645 (C=O), 1545 Schulter, 1533 (C=N, NOz), 1121, 1105, 1070 cm-' (SO-Bereich). - 'H-NMR (CDCI3):S = 2.72,2.70(2q,J= 7Hz;je1H,NCH2),2.46(q,J= 7Hz;2H,NCH2),0.81,0.54 (2t, J = 7 Hz; je 3H, NCH2CH3). - MS (70eV): m/e = 580 (109'0, M - SOC&N&), 457 (15%), 379 (26%), 276 (63%), 264 (400/0), 259 (880/0), 235 (88%), 199 (100%), 181 (400/0), 165 (72%), 132 (39%), 122 (470/0), 108 (33%), 104 (66%), 77 (94%).
C4H3,N305SSi (750.0) Ber. C 70.47 H 5.24 N 5.60 Gef. C 70.6 H 5.33 N 5.5
Umsetzungen rnit Ketenen
Diphenylketen-Derivate 17 a - c. - Allgemeine Vorschriften A) Zu einer Losung von 2 mmol Eninamin 4a, b oder e in 15 ml Ether (Benzol bei 4e) gibt man
0.39 g (2 mmol) Diphenylketen (2a) und riihrt 10 h bei Raumtemp. Nach Zugabe von 10 ml Pe- trolether, Kuhlen und Anreiben kristallisieren die Cyclobutenone 17a -c aus. Ausbeuten und analytische Daten s. Tabelle 6.
B) Alternative Darstellung von 17a- c: Zu einer Lbsung von 10 mmol (Silylethiny1)amin l a , b oder e in 50 ml Ether (Benzol bei l e ) gibt man 3.9 g (20 mmol) Diphenylketen und riihrt bei Raumtemp. 12-24 h. Nach Zugabe von 20 ml Petrolether, Kuhlen und Anreiben isoliert man durch Absaugen die Cyclobutenone 17a- c.
Isoliemng der zu 17 konstitutionskomeren Verbindungen 18b und c13): Das Filtrat von 17b bzw. c wird i. Vak. eingedampft. Bei der Chromatographie des Riickstandes an 50 g Kieselgel rnit Chloroform isoliert man in den ersten Fraktionen (groRer RF-Wert) 18b bzw. 18c. 18b: Ausbeute 10% bei Verfahren A), Ausbeute 11 - 17% bei Verfahren B); aus Ether (-70°C) farblose Kristalle rnit Schmp. 139- 143 "C. - IR (KBr): 3510 (OH), 860, 850 cm-' ( S M q ) . - 'H-NMR (CDC13): 6 = 5.49 (s; 1 H, OH; verschwindet beim Deuterieren), 3.57 und 2.89 (2s, 40 bzw. 60%; 3H, NMe), -0.10 und -0.25 (2s, 40 bzw. 60%; insgesamt 9H, SiMe3). - MS (70eV): m/e = 591 (30%, M+), 424 (100V0, M - CHPh2), 408 (87%), 393 (39010), 167 (300/0), 73 (74%, SiMe3).
C40H37N&Si (591.8) Ber. C 81.19 H 6.30 N 2.37 Gef. C 80.5 H 6.35 N 2.6
18c: Ausbeute 27% nach Verfahren A), Ausbeute 17% nach Verfahren B); aus Petrolether (120-140'C) farbloseKristalle rnit Schmp. 210-212°C. - IR (KBr): 3510 cm-' (OH). - 'H- NMR (CDC13): 6 = 4.91 (s; I H, OH; verschwindet beim Deuterieren), 3.64 und 2.89 (2s, je 50%; zusammen 3H, NMe). - MS (70eV): m/e = 778 (31%, M'), 611 (79%, M - CHPhZ), 532 (lOOVo), 259 (50%, SiPh3).
CS5&3N&Si (778.1) Ber. C 84.91 H 5.57 N 1.80 Gef. C 84.8 H 5.95 N 2.0
Dimethylketen-Derivate 17 d - g 17d und e werden als Nebenprodukte bei der Synthese von 4k bzw. 41 isoliert (s. dort). Durch
Einwirken eines Uberschusses von Dirneth~lketenl~) auf die Inamine 1 lassen sich die [l : 21- Addukte 17 gezielt herstellen, wie am Beispiel von 17e gezeigt wird.
122'
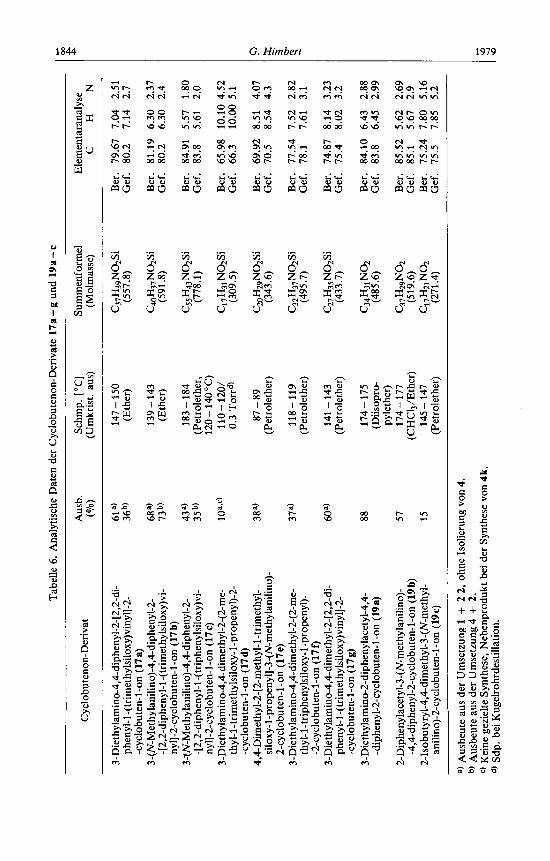
Tabe
lle 6
. Ana
lytis
che
Dat
en d
er C
yclo
bute
non-
Der
ivat
e 17
a - g
und
19a -
c
Cyc
lobu
teno
n-D
eriv
at
Aus
b.
Schm
p. [
"C]
Sum
men
form
el
Elem
enta
rana
lyse
(0
70)
(Um
kris
t. au
s)
(Mol
mas
se)
CH
N
~
3-Diethylamino-4,4-diphenyl-2-[2,2-di-
phenyl-l-(trimethylsiloxy)vinyl]-2-
-cyc
lobu
ten-
1-on
(17
a)
3-(N-Methylanilino)-4,4-diphenyl-2-
-[2,
2-di
phen
yl-l
-(tri
met
hyls
iloxy
)vi-
nyl]-
2-cy
clob
uten
-l-on
(17
b)
-[2,
2-di
phen
yl-1
-(tri
phen
ylsi
loxy
)vi-
nyl]-
2-cy
clob
uten
-l -o
n (1
7 c)
3-Diethylamino-4,4-dimethyl-2-(2-me-
thyl
-1 -tr
imet
hyls
iloxy
-1 -p
rope
nyl)-
2-
-cyc
lobu
ten-
1-on
(17
d)
silo
xy-1
-propenyl]-3-(N-methylanilino)-
2-cy
clob
uten
-1 -o
n (1
7 e)
3-Diethylamino-4,4-dimethyl-2-(2-me-
thyl
-1 -triphenylsiloxy-1-propeny1)-
-2-c
yclo
bute
n-1-
on (
17 f)
3-Diethylamino-4,4-dimethyl-2-[2,2-di-
phen
yl-1
-(tr
imet
hyls
iloxy
)vin
yl]-
2-
-cyc
lobu
ten-
1-on
(17
g)
3-D
ie th
ylam
ino-
2-di
p hen
ylac
etyl
-4,4
- -diphenyl-2-cyclobuten-l-on (1
9a)
3-@
V-M
ethy
lani
lin0)
-4,4
-dip
heny
l-t-
4,4-Dimethyl-2-[2-methyl-l-trimethyl-
2-Diphenylacetyl-3-(N-methylanilino)-
57
-4,4-diphenyl-2-cyclobuten-l-on (1
9 b)
anilino)-2-cyclobuten-l-on (1
9c)
2-Is
obu tyryl-4,4-dimethyl-3-(N-methyl-
15
147 -
150
(E
ther
)
139-
143
(E
ther
)
183 -
184
(P
etro
leth
er,
120-
140°
C)
110 -
120
/ 0.
3 T
ond)
87 - 89
(P
etro
leth
er)
118-
119
(Pet
role
ther
)
141 -
143
(P
etro
leth
er)
174 -
175
(D
iisop
ro-
pyle
ther
)
(CH
CI3
/Eth
er)
(Pet
role
ther
)
174-
177
145 -
147
C37
H39
NW
i (5
57.8
)
C,H3
, N
q Si
(591
.8)
CSSH
43N
%Si
(7
78.1
)
C17
H3,
NO
zSi
(309
.5)
qO
H2
9N
qS
i (3
43.6
)
C32
H37
NqS
i (4
95.7
)
q7H
35N
02Si
(4
33.7
)
C34
H31
N%
(4
85.6
)
c37
H2
9N
9
(519
.6)
c17H
21 N%
(271
.4)
Ber
. G
ef.
Ber
. G
ef.
Ber
. G
ef.
Ber
. G
ef.
Ber
. G
ef.
Ber
. G
ef.
Ber
. G
ef.
Ber
. G
ef.
Ber
. G
ef.
Ber
. G
ef.
79.6
7 80
.2
81.1
9 80
.2
84.9
1 83
.8
65.9
8 66
.3
69.9
2 70
.5
77.5
4 78
.1
74.8
7 75
.4
84.1
0 83
.8
85.5
2 85
.1
75.2
4 75
.5
7.04
2.
51
7.14
2.
7
6.30
2.
37
6.30
2.
4
5.57
1.
80
5.61
2.
0
10.1
0 4.
52
10.0
0 5.
1
8.51
4.
07
8.54
4.
3
7.52
2.
82
7.61
3.
1
8.14
3.
23
8.02
3.
2
6.43
2.
88
6.45
2.
99
5.62
2.
69
5.67
2.
9 7.
80
5.16
7.
85
5.2
a) A
usbe
ute
aus
der U
mse
tzun
g 1
+ 2
2, o
hne
Isol
ieru
ng v
on 4
. b) A
usbe
ute
aus
der U
mse
tzun
g 4
+ 2
. C
) K
eine
gez
ielte
Syn
thes
e, N
eben
prod
ukt
bei d
er S
ynth
ese
von
4k.
d, S
dp. b
ei K
ugel
rohr
dest
illat
ion.

1979 (Aminoethinyl)metallierungen, 6 1845
17e: 2.03 g (10.0 mmol) 1 b werden mit 125 ml etherischer Dimethylketen-Losung versetzt und 24 h bei Raumtemp. geriihrt. Man filtriert, entfernt das Losungsmittel i.Vak., nimmt in 30 ml Petrolether auf, kuhlt, reibt an und isoliert durch Absaugen 1.3 g (38%) 17e.
17f: 3.55 g (10.0 mmol) I d werden mit 100 ml etherischer Dimethylketen-Losung versetzt und 5 h bei Raumtemp. gertihrt. Nach Entfernen des Losungsmittels i. Vak. kristallisieren 1.83 g (37%) 17f aus 30 ml Petrolether.
17g: 0.73 g (2 mmol) 4d werden mit 40 ml etherischer Dimethylketen-Lbsung versetzt und 12 h bei Raumtemp. geriihrt. Nach Entfernen des Ldsungsmittels i. Vak. kristallisieren 0.52 g (60070) 17g aus 15 mi Petrolether.
Hydrolyse der Cyclobutenon-Derioate 17a, b und e zu 19a, b und c 19a: 0.56 g (1 mmol) 17a werden bei Raumtemp. mit einem Gemisch von 20 ml Ethanol, 3 mi
Wasser und 1 g Kaliumhydroxid geriihrt. Nach 30 min saugt man den Niederschlag ab, trocknet auf Ton und kristallisiert aus Diisopropylether um. Analytische Daten s. Tabelle 6.
19b: 0.59 g (1 mmol) 17b werden nach voranstehender Vorschrift behandelt. Nach Trocknen auf Ton wird 19b aus ChlorofordEther umgefallt. Analytische Daten s. Tabelle 6.
19c: 0.69 g (2 mmol) 17e werden bei Raumtemp. mit einem Gemisch von 20 ml Ethanol, 3 ml Wasser und 1 g Kaliumhydroxid geriihrt. Nach 45 min wird das Losungsmittel i. Vak. weitgehend entfernt und der Ruckstand mit je 100 ml Chloroform und Wasser versetzt. Die Chloroformpha- se wird uber Calciumchlorid getrocknet und i. Vak. eingedampft. Nach Zugabe von 10 ml Petrol- ether isoliert man 80 mg (15Vo) 19c (Im Filtrat lafit sich N-Methylanilin nachweisen, das die wei- tere Hydrolyse von 19c anzeigt.)
Unabhungige Synthese von 19b: Die Losung von 0.65 g (2 mmol) 13 wird in 3 ml Benzol mit 0.4 g (2 mmol) Diphenylketen 2a versetzt und 0.5 h bei Raumtemp. geruhrt. Nach Zugabe von 10 mlEther erhalt man0.18 g (17%) 19b; identisch mit der bei der Hydrolysevon 17b erhaltenen Substanz.
l) 5. Mitteilung: G. Himbert, J. Chem. Res. 1979, (S) 88, (M) 1201. 2, G. Himbert, Liebigs Ann. Chem. 1979, 829. 3, Zusammenfassung: J. Ficini, Tetrahedron, 32, 1448 (1976). 4, Teilweise veroffentlicht: G. Himbert, Angew. Chem. 88, 59 (1976); Angew. Chem., Int. Ed.
9 Analoge Zwitterionen werden bei nahezu allen Umsetzungen der Inamine mit Heterocumule- Engl. 15, 51 (1976).
nen formuliert; siehe dazu Lit.3). G. Himbert, J. Chem. Res. 1978, (S) 104, (M) 1445.
’) G. Himbert und W. Schwickerath, Tetrahedron Lett. 1978, 1951. 8, Y. Sato, Y. Kobayashi, M. Sugiura und H. Shirai, J. Org. Chem. 43, 199 (1978). 9, G. Himbert, Angew. Chem. 91, 432 (1979); Angew. Chem., Int. Ed. Engl. 18, 405 (1979).
lo) U. Lienhard, H.-P. Fahrni und M. Neuenschwander, Helv. Chim. Acta, 61, 1609 (1978). l) Zusammenfassende Darstellung der Sulfonylazid-Addition an Inamine siehe: M. Regitz, Di-
12) D. Frank, G. Himbert und M. Regitz, Chem. Ber. 111, 183 (1978) und dort zitierte Literatur. 13) Als Vorschlag kommt fur 18 die 4-Anilino-l-phenyl-3-(2,2-diphenyl-1-siloxyvinyl)-2-naph-
thol-Struktur in Frage; die Spektren von 18 und die der Hydrolyseprodukte gestatten bisher je- doch keine eindeutige Aussage.
14) G. Himbert, D. Frank und M. Regitz, Chem. Ber. 109, 370 (1976). Is) Zusammenfassung: H. Meier und K.-P. Zeller, Angew. Chem. 87, 52 (1975); Angew. Chem.,
azoalkane, 1. Aufl., S. 216ff, Thieme, Stuttgart 1977.
Int. Ed. Engl. 14, 32 (1975).

1846 G. Himbert 1979
16) L. J. Smith und H. H. Hoehn, Org. Synth., Coll. Vol. 111, 356 (1967). 17) D. Borrmann in Methoden der organischen Chemie (Houben- Weyl-Miiller), 4. Aufl., Bd.
VI1/4, S. 53ff, Thieme, Stuttgart 1968; der dort (S. 92) beschriebene Ansatz wird benutzt. 18) 0. Siis, H . Steppan und R. Dietrich, Liebigs. Ann. Chem. 617, 20 (1958). 19) K. -H. Busch, Teil der Fortgeschrittenenarbeit Univ. Kaiserslautern 1976. 20) M. Regitz und J. Riiter, Chem. Ber. 101, 1263 (1968). 21) M. Regitz, J . Hocker und A . Liedhegener, Org. Synth. 48, 36 (1968). 22) L. Horner und A . Christmann, Chem. Ber. 96, 388 (1963). 23) D. L. Rector und R. E. Harmon, J. Org. Chem. 31, 2837 (1966).
[91/79]








![Cancerogene Wirkung von Kunststoff-Folienzfn.mpdl.mpg.de/data/Reihe_B/7/ZNB-1952-7b-0353.pdf · foi's-[Chloräthyl]-alkyl-aminen u. Diese Substanzen geben in vitro wahrscheinlich](https://img.pdfslide.net/doc/110x75/5e0b56f81ad9a54dda0bd5ba/cancerogene-wirkung-von-kunststoff-fois-chlorthyl-alkyl-aminen-u-diese-substanzen.jpg)




















