Embed Size (px)
Citation preview

Clinical Genetics 1989: 36: 4 5 1 4 5 5
An apparently new autosomal recessive syndrome with facial dysmorphism,
macrocephaly, myopia and Dandy-Walker malformation
M. BUT TI ENS'.^, J. P. FRYNS' AND H. VAN DEN BERGHE' 'Centre for Human Genetics, University of Leuven and 'Stichting Marguerite-Marie Delacroix,
Tienen. Belgium
In this report we describe an apparently new MCA-MR syndrome with Dandy-Walker malfor- mation in three severely mentally retarded siblings born to normal, non-consanguineous par- ents. In addition, they presented macrocephaly, facial dysmorphism, extreme myopia and brachytelephalangy with short and broad finger-nails.
Received 22 May. accepted for publication 27 July 1989
Key wordr: autosomal recessive inheritance; Dandy-Walker malformation; macrocephaly; men- tal retardation
The Dandy-Walker malformation combines partial or complete absence of the cerebellar vennis with hydrocephalus and a posterior fossa cyst communicating with the fourth ventricle, and has mostly been reported as an isolated malformation of the Central Nervous System with low recurrence risk (Murray et al. 1985). It has also been de- scribed in a number of chromosomal abnor- malities and Mendelian-inherited, mono- genic syndromes, e.g. Warburg syndrome, Coffin-Siris syndrome, Fryns syndrome and oro-facio-digital syndrome type 2 (for re- View, see Murray et al. 1985).
Report of Patlents
Patient I B.J.L., a 26-year-old male, is the index pa- tient of this report. He was examined during a systematic clinical genetic survey of a p o p
ulation of institionalized, severely mentally handicapped. No information is available on the pen- or postnatal period. He was admitted to the institute at the age of 19 months because of severe psychomotor re- tardation and spastic quadriplegia.
Now, at the age of 26 years he is a deeply mentally retarded and bedridden adult with spastic quadriplegia, most pronounced in the lower legs. In addition to extreme ma- crocephaly (head circumference 66.7 cm, height 137 cm and weight 46 kg), there is transversal flattening of the skull, high and broad forehead and bushy hair with frontal upsweep. The facial features are somewhat coarsened (Fig. la) with bushy eyebrows, synophrys, bilateral epicanthal folds, broad nose, high-arched palate, maldentition and low-set, poorly lobulated ears. The terminal phalanges of all fingers are short and broad- ened with short fingernails. Secondary sex-

452 B U T T I E N S E T A L .
ual development is normal but the scrotum is hypoplastic with bilateral cryptorchidism. There is bilateral searching nystagmus and ophthalmological examination showed se-
vere myopia (-10 D), optic atrophy and a left side cataract which became eviderit after the age of 20 years. Neurological exami- nation revealed generalized hypertonicity,
Fig. l a and b. The facial features In Patients 1 (a) and 2 (b).

NEW M C A - M R S Y N D R O M E 453
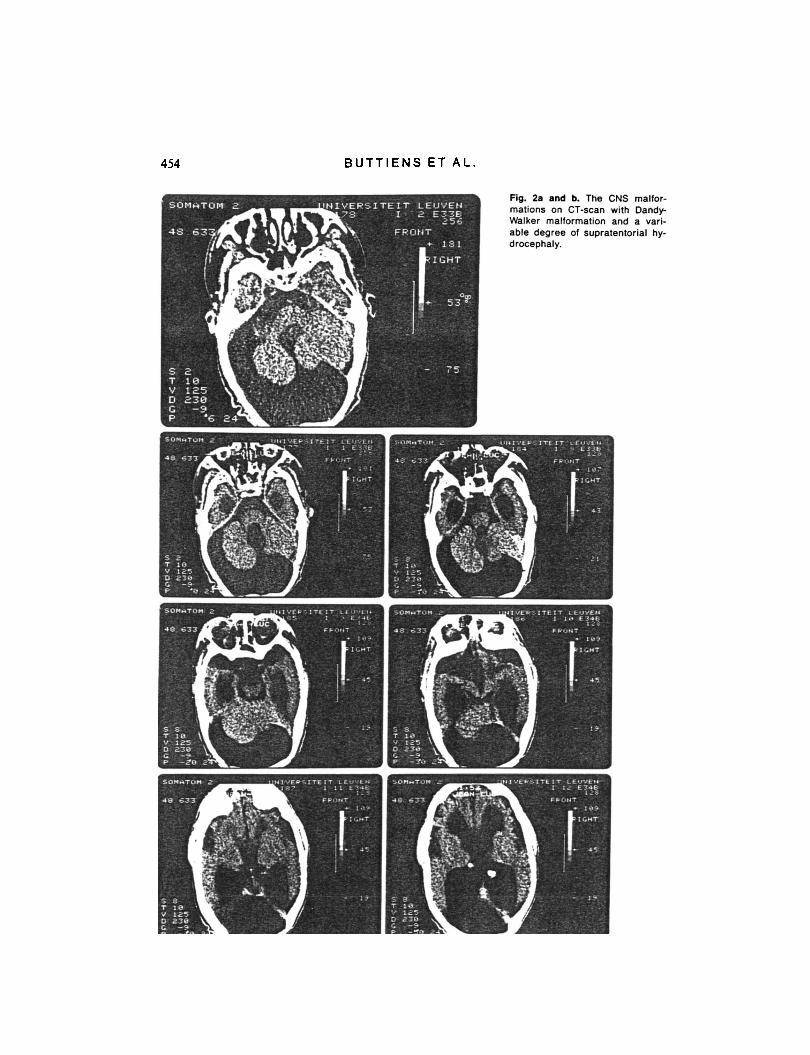
more pronounced in the lower legs, with pyramidal signs and secondary deforma- tions, i.e. thoracolumbar scoliosis, luxation of the left hip, equinocalcaneovalgus posi- tion of both feet and 30" extension deficit of the knees. Axial computerized tomo- graphy of the brain showed a large fossa posterior cyst communicating with an en- larged fourth ventricle (Fig. 2a) and extreme supratentorial hydrocephaly with severe cortical atrophy (hydranencephaly). Chro- mosomal investigation was normal after G- and R-banding in 25 cells of a peripheral lymphocyte culture.
Patient 2 B.C. is the 30-year-old sister of Patient 1. She is severely mentally handicapped and was admitted to an institute for the severely mentally retarded at the age of 2 years. We know that she had a normal head circumfer- ence (36 cm) at birth and that she was ad- mitted to the institute because of severe de- velopmental delay, progressive hydrocepha- ly and generalized convulsions.
Now, she is macrocephalic (head circum- ference 59 cm, weight 60 kg, height 158 cm) with coarse face (Fig. lb) and heavy eyebrows, epicanthal folds, broad nose, short philtrum, full, thickened lips, high- arched palate and maldentition. The ter- minal phalanges of all fingers are short and broad with short nails. Despite spastic di- plegia, she can walk without support and, except for thoracic hyperkyphosis, there are no other notable skeletal derfoxnities. Oph- thalmological examination revealed severe myopia (right eye - 13 D, left eye - 6 D), rotatory nystagmus, strabismus divergens, optic atrophy and macular degeneration. Axial computerized tomography of the brain showed discrete widening of the lat- eral ventricles and a cystic structure, 4 cm in diameter, in the posterior fossa communi- cating with an enlarged fourth ventricle (Fig. 2b).
Patient 3 B.M., a younger sister of Patients 1 and 2, died at the age of 10 years. She was born at the expected term without complications. Birthweight was 3900 g. We do not know a t what age hydrocephaly was clinically sus- pected, but at the age of 16 months she was admitted to University Hospital because of developmental retardation and convulsions. Marked hydrocephaly was evident (head circumfepce 53 cm), length and weight were 77 cm (25th percentile for age) and 7650 g (third percentile is 8500 g), respec- tively. The fontanelles were large without widening of the sutures, the ears low set and the palate high arched. She was severely hypotonic, without head control and with- out eye contact. Encephalography showed enlarged ventricles and ophthalmological examination signs of intracranial hyperten- sion, which disappeared a few months later, and continuous, horizontal nystagmus.
Family HLFtory The three siblings (Patie'nts 1 ,2 and 3) were born in a family with five children. The two others, a boy and a girl, were physically and mentally normal. Both parents were also physically normal and nonconsanguineous. They were alcoholic and mentally border- line, as is their only, l0-year-old grand- daughter. Computerized axial tomography of the brain was normal in this child.
Dlscurrlon The three siblings in this report present a medical history which was very similar in all three: postnatal onset of progressive hy- drocephaly, ocular abnormalities with nys- tagmus, a generalized neurological syn- drome with quadriplegia, severe to deep mental retardation and generalized con- vulsions. They were all admitted to an insti- tution between the ages of 1.5 and 2 years, and the youngest died at the age of 10 years.
454 B U T T I E N S E T A L
Fig. 2. and b. The CNS malfor- mations on CT-scan with Dandy- Walker malformation and a vari- able degree of supratentorial hy- drocephaly.
a

N E W M C A - M R S Y N D R O M E 455
The surviving, adult siblings present a pe- culiar combination of abnormal clinical findings: macrocephaly, high and broad forehead, coarse facial features with heavy eyebrows, a tendency to synophrys, broad nose, full lips, high-arched palate, ocular abnormalities with severe myopia, nystag- mus and optic atrophy, short and broad terminal phalanges with short nails, and a Dandy-Walker cyst on CT-scan.
Mental retardation was deep in Patient 1 with massive hydrocephaly (hydranence- phaly) and in this sibling secondary defor- mations, i.e. scoliosis, hip luxation and con- tractures, were also the most pronounced. It is also likely that the macrocephaly and part of the ocular abnormalities, i.e. the optic atrophy and the nystagmus, are sec- ondary to the variable degree of supratento- rial hydrocephaly.
In a retrospective study of 113 subjects with Dandy-Walker malformation, includ- ing 21 personal observations and 92 patients reported in the literature, Murray et al. ( 1 985) found seven recognisable syndromes, i.e. two with Meckel-Griiber syndrome, and one each with arthrogryposis 2B, Warburg syndrome, Ruvalcaba syndrome, Cornelia de Lange syndrome and congenital rubella infection. Dandy-Walker malformation has been reported as a more or less constant feature in a number of other Mendelian dis- orders with autosomal recessive inheritance, e.g. Coffin-Siris syndrome (Lucaya et al. 1981), Fraser cryptophthalmos syndrome (Behrens-Baumann et al. 1981), Joubert- Boltshauser syndrome (Boltshauser & Isler 1977), the oro-facialdigital syndrome type 2 (Mohr syndrome) (Gustavson et al. 1971) and Fryns syndrome (Fryns 1987).
As far as we know, the unusual combi- nation of dysmorphic facial stigmata, severe
myopia, brachytelephalangy with short, broad nails and Dandy-Walker malfor- mation of the CNS with secondary hydroce- phaly has not previously been reported. Its presence in two (three) of the five siblings born to physically normal parents suggests autosomal recessive inheritance of this MCA/MR syndrome in which Dandy-Wal- ker malformation is an important feature.
References Behrens-Baumann, W., G. Dust, K. Rittmeier, U.
Langenbeck & M. Vogel(l981). Okulozerebra- le Dysplasie: Aplasia Nervi Optici sowie famili- arer Mikr- und Kryophthalmus. Klin. Mbl. Au- genheilk. 179, 90-93.
Boltshauscr. E. & W. Isler (1977). Joubert syn- drome: episodic hyperpnea, abnormal eye movements, retardation and ataxia, associated with dysplasia of the cerebellar vermis. Neur- opaediatrie 8, 5 7-66.
Fryns, J. P. (1987). Fryns syndrome: a variable MCA syndrome with diaphragmatic defects, coarse face. and distal limb hypoplasia. J. Med. Genet. 24, 271-274.
Gustavson, K. H., A. Krueger & P. 0. Peterson (1971). Syndrome characterized by lingual mal- formation, polydactyly, tachypnea, and psy- chomotor retardation (Mohr syndrome). Cfin. Genet. 2, 261-266.
Lucaya, J., J. A. Garcia-Conesa, J. M. Bosch- Banyeras & G. Pons-Peradejordi (1981). The Comn-Siris syndrome. A report of four cases and review of the literature. Pediatr. Radiol. 11, 35-38.
Murray, J. C., J. A. Johnson & T. D. Bird (1985). Dandy-Walker malformation: etiologic hetero- geneity and empiric recurrence risks. Clin. Genet. 28, 272-283.
Address: J. i? Frym Centre for Human Genetics Herestraat 49 8-3000 Luuven Belgium





























