Embed Size (px)
Citation preview

An Approach to Renal Massesin PediatricsAlpin D. Malkan, MDa, Amos Loh, MDa, Armita Bahrami, MDb, Fariba Navid, MDc,d, Jamie Coleman, MDe, Daniel M. Green, MDf,Andrew M. Davidoff, MDa, John A. Sandoval, MDa
abstractRenal masses in children may be discovered during routine clinical examinationor incidentally during the course of diagnostic or therapeutic procedures forother causes. Renal cancers are rare in the pediatric population and includea spectrum of pathologies that may challenge the clinician in choosing theoptimal treatment. Correct identification of the lesion may be difficult, and theappropriate surgical procedure is paramount for lesions suspected to bemalignant. The purpose of this article is to provide a comprehensive overviewregarding the spectrum of renal tumors in the pediatric population, both benignand malignant, and their surgical management.
Renal cancers are rare in children,accounting for 6% to 7% of allchildhood tumors.1 They can bedetected by a parent bathing orholding the child, during routinephysical examination or screening ofchildren with known clinicalsyndromes with predispositions torenal disease,2 or incidentally duringinvestigations for otherintraabdominal processes.3–5 The keychallenge is distinguishing malignantneoplasms from benign masses. Athorough understanding of the profileof common renal masses in children,as well as their associated clinical andimaging features, can facilitateaccurate preoperative diagnosis andoptimize patient care.
This article comprehensively reviewsthe approach to identifying renalcancers in children, individuallysummarizes the history, diagnosis,histopathology, and management ofmalignant and benign pediatric renalmasses (Table 1), and provides therationale for choice of the correctsurgical procedure.
APPROACH
The priority of management is thedifferentiation of nonneoplastic
processes from benign and malignantneoplasms (Fig 1). A thorough reviewof the patient’s clinical history andphysical examination may revealadditional signs or symptoms to aid inthe diagnosis. This may be useful incharacterizing nonneoplastic renalpseudotumors, which are masslikeimaging findings that mimicneoplasms.6 The presence of flank pain,fever, or pyuria, for example, issuggestive of an infective process, suchas pyelonephritis, instead of a tumor.However, the absence of symptomsneither suggests nor refutesa diagnosis of malignancy. This will beparticularly true of congenitalanomalies such as prominent columnsof Bertin or dromedary humps.
Although masslike renal lesions inchildren can sometimes be suspectedon plain radiographs and evaluatedwith ultrasound, subsequent computedtomography (CT) is usually necessaryfor further characterization andconfirmation of the diagnosis.7 Severalimage-based criteria can help to guidethe differentiation of malignant frombenign lesions.8 For solid lesions, oncepseudotumors are excluded, thepresence of contrast enhancement maybe indicative of a neoplasm although
Departments of aSurgery, bPathology, cOncology, eRadiology,and fEpidemiology and Cancer Control, St. Jude Children’sResearch Hospital, Memphis, Tennessee; and dDepartmentof Pediatrics, College of Medicine, University of TennesseeHealth Science Center, Memphis, Tennessee
Drs Malkan, Loh, Green, Davidoff, and Sandovalconceptualized, designed, wrote, and revised themanuscript; Drs Bahrami, Navid, and Colemancoordinated the collection of histopathologic andradiologic imaging and critically reviewed themanuscript for important intellectual content; andall authors approved the final manuscript assubmitted.
www.pediatrics.org/cgi/doi/10.1542/peds.2014-1011
DOI: 10.1542/peds.2014-1011
Accepted for publication Jul 23, 2014
Address correspondence to John A. Sandoval, MD,Department of Surgery, St. Jude Children’s ResearchHospital, MS 133, Room D3045, 262 Danny ThomasPlace, Memphis, TN 38105-3678. E-mail: [email protected]
PEDIATRICS (ISSN Numbers: Print, 0031-4005; Online,1098-4275).
Copyright © 2015 by the American Academy ofPediatrics
FINANCIAL DISCLOSURE: The authors have indicatedthey have no financial relationships relevant to thisarticle to disclose.
FUNDING: No external funding.
POTENTIAL CONFLICT OF INTEREST: The authors haveindicated they have no potential conflicts of interestto disclose.
PEDIATRICS Volume 135, number 1, January 2015 STATE-OF-THE-ART REVIEW ARTICLE by guest on June 10, 2018www.aappublications.org/newsDownloaded from

several other abnormalities includinginfection (pyelonephritis) andabscess require consideration. Theclaw sign is classically seen as thetumor grows due to the normalkidney being splayed around thetumor and indicative of renal origin.9
CT and especially MRI can
characterize the tissue composition ofthese solid lesions and candifferentiate soft tissue from fat, fluid,or hemorrhage. On serial imaging, anenlarging mass can spur worriesabout a neoplasm, although absolutetumor size may not correlate wellwith malignancy risk.
For cystic masses, the Bosniak renalcyst classification is well establishedin the literature for malignancy riskprediction in image-detected renalcysts in adults.10 Based on thisclassification system, the lesion’simaging (originally based on CTfindings, but ultrasound and MRI arecommonly applied) morphology andenhancements characteristics areused to categorize each lesion into1 of 5 groups (I and II are benign anddo not require further evaluation; IIFrequires follow-up to prove benignity;III are indeterminate and consideredsurgical lesions although some mayprove to be benign; IV are clearlymalignant cystic masses that requiresurgical removal).8,11–13 A modifiedBosniak classification has been shownto correlate fairly well withpathologic outcomes of complex renalcysts in children but has not beenformally validated.14
IMPLICATIONS FOR BIOPSY
Despite thorough clinical andradiographic evaluation, some renalmasses will remain indeterminate,and their management is subject toindividual clinical opinions. Carefulcorrelation of clinical and imagingfindings may facilitate thepreoperative diagnosis of most renallesions. Although histologicevaluation is the gold standard forpathologic diagnosis, obtaining tissuebiopsy via percutaneous (using Tru-cut biopsy, open biopsy, or fine-needle aspiration) or open surgicalapproach can have seriousimplications that requireconsideration. If a renal mass issuspicious for malignancy, themultidisciplinary team in addition tothe surgeon typically determinesthe potential for completeresectability of the primary mass, andwhether there is any evidence ofmetastatic disease on preoperativeimaging. If the mass is determined tobe unresectable then the issue ofbiopsy becomes more apparent toguide therapy.
In children, general anesthesia istypically needed to perform either anopen or percutaneous biopsy. Anopen biopsy requires an incision,allows for large specimens to beobtained as compared witha percutaneous needle approach;however, this method inevitablycauses the patient more discomfort,increased recovery time, and scarring.Even more problematic is obtainingan inadequate sample with potentialfor sampling error. In many cases,a percutaneous needle biopsy cannotdifferentiate renal lesions such asWilms tumor (WT) from hyperplasticnephrogenic rests.15 More disturbingare the consequences of upstaging thetumor, which is used to describea patient’s cancer from a lower stage(less extensive) to a higher stage(more extensive),15 necessitatingadministration of additionalchemotherapeutic agents and/orradiation therapy. For instance, in
FIGURE 1Renal mass algorithm.
TABLE 1 Renal Lesions in Children
Benign Malignant
Angiomyolipomas WTRenal pseudotumor Clear cell sarcoma
Renal cell carcinomaMAClear cell
Multicystic nephroma Medullary carcinomaMetanephric stromal
tumorMulticystic
ORTI ChromophilReninoma Chromophobe
Collecting ductPapillaryTranslocationXp11.2t(6;11)
NephroblastomatosisRhabdoid tumorAnaplastic sarcomaCongenital mesoblasticnephroma
DSRCTEwing sarcoma/PNETSynovial sarcoma
DSRCT, desmoplastic small round cell tumor; MA, metanephricadenoma; ORTI, ossifying renal tumor of infancy; PNET,primitive neuroectodermal tumor; WT, Wilms tumor.
2 MALKAN et al by guest on June 10, 2018www.aappublications.org/newsDownloaded from

lower stage WT without anaplasia,complete resection of the mass, alongwith adjuvant chemotherapy, resultsin cure rates of over 90% at 5 years,whereas resection alone results in20% long-term survival rate.16,17
A preoperative needle biopsy in thisparticular scenario would upstage themalignancy, and the possible need formore aggressive treatment, includingchemotherapy and radiation.
An emphasis on the multidisciplinaryapproach is essential to theevaluation and treatment of thesepatients including the pediatrician,pediatric radiologist, pediatricsurgeon, pediatric urologist, pediatriconcologist, pediatric radiationoncologist, and pathologist. The keycharacteristics of a comprehensivespectrum of pediatric renal massesare outlined below.
MALIGNANT NEOPLASMS
Wilms tumor
WT, also called nephroblastoma, isthe most common pediatric renalmalignancy,18–21 accounting for ∼6%to 7% of all childhood renalcancers,18 and is observed ata frequency of 1:10 000 newborninfants,18,22 occurring mostlybetween 2 and 5 years of age.19 Mostcases are sporadic, although it canoccur in hereditary form withautosomal dominant inheritance,variable penetrance, and severalaffected generations.18 Symptomsmay include abdominal pain,distension, and hypertension;however, the patient can also beasymptomatic. Bilaterality occurs in4% to 13% of cases. WT may beassociated with genitourinaryanomalies and some congenitalsyndromes such as WAGR, Denys-Drash, and Beckwith-Wiedemannsyndromes (BWS).15
WAGR syndrome includes WT,aniridia (absence of the colored partof the eye), genitourinaryabnormalities (gonadal tumors), andmental retardation.15 Interstitial
deletion on chromosome 11p13(prevalence of 0.4% of children withWT),15,23 and bilateral WT (15%)may be seen in association withWAGR syndrome.15,24 Denys-Drashsyndrome is characterized by thetriad of WT, pseudohermaphroditism,and chronic renal failure. Individualswith Denys-Drash syndrome manifestearly nephrotic syndrome, whichprogresses to diffuse mesangialsclerosis and subsequent renal failureusually within the first 3 years of life.Germline missense mutations in theWT1 gene are responsible for mostWTs that occur as part of the Denys-Drash syndrome25 including the 90%risk of developing WT.15,26 BWSincludes macroglossia, macrosomia(birth weight and length greater thanthe 90th percentile), midlineabdominal wall defects (omphalocele,umbilical hernia, diastasis recti), earcreases or ear pits, and neonatalhypoglycemia. It is also characterizedby the development of WT(bilateral),27 rhabdomyosarcoma, andhepatoblastoma.15 BWS results fromconstitutional loss of imprinting orheterozygosity of WT2.15
Approximately 1 of 5 patients withBWS and WT may present withbilateral disease; however,metachronous bilateral disease is alsoobserved.15,28–30
WT typically appears as a large,smooth, well-defined intrarenal masswith uniform echogenicity that oftendisplaces neighboring structures. WTis usually seen first on ultrasound, andsubsequently with less enhancementon CT than adjacent normalparenchyma (Fig 2).7 Calcifications, fatdensity, patches of renal parenchymalenhancement, invasion of the renalcollecting system, vascular invasionwith tumor thrombus extension intothe renal vein and inferior vena cava(most accurately evaluated on MRI),or the presence of distant lungmetastases may be seen.7 Rarely, itmay be largely cystic, an entityreferred to as cystic partiallydifferentiated nephroblastoma, thatcannot be differentiated from cystic
nephroma on imaging, but outcomesof both are favorable regardless oftreatment.31 Current imagingmodalities may fail to detect occultsynchronous contralateral tumors atthe time of initial diagnosis, but theseoccur in only a very small percentageof patients with WT, and outcomes forthese patients have also beenexcellent.32
Preoperative staging studies inpatients suspected of having WTinclude CT (or MRI) scans of thechest, abdomen, and pelvis.15 Thestage is determined by the results ofimaging studies, as well as surgicaland pathologic findings at time ofnephrectomy, and is the same fortumors with favorable or anaplastichistology. In stage I (43% of patients)and II (20%) WTs, the tumor iscompletely resected without evidenceof tumor at or beyond the margins ofresection.15 Stage III (21%) and IV(11%) WTs have either residualnonhematogenous tumor presentafter surgery that is confined to theabdomen or hematogenousmetastases or lymph node metastasesoutside the abdominopelvic regionare present, respectively.15 Lymphnode and major blood vesselinvolvement (encasement and/orthrombus) are reflective of advanced,potentially unresectable disease andhigher stage (III or IV).15
Histologically, WT can be separatedinto prognostic groups includingfavorable histology, anaplastichistology, and nephrogenic rests.Anaplastic histology is characterizedby multipolar polypoid mitotic figureswith marked nuclear enlargementand hyperchromasia and is the singlemost important predictor of responseand survival.15 Anaplasia correlatesbest with responsiveness to therapyrather than to aggressiveness, and ismost consistently associated withpoor prognosis when it is diffuselydistributed or identified at advancedstages.15 Nephrogenic rests areabnormally retained embryonickidney precursor cells arranged in
PEDIATRICS Volume 135, number 1, January 2015 3 by guest on June 10, 2018www.aappublications.org/newsDownloaded from

clusters that are considered toincrease the risk for tumor formationin the remaining kidney.15
Unilateral WT is generally treatedwith nephrectomy followed byadjuvant chemotherapy. Neoadjuvantchemotherapy may be used topromote tumor shrinkage and is usedin bilateral (Fig 3) or inoperable WTsand in selected treatment protocols.Postoperative radiation therapy maybe administered to the tumor bed forlocal control, or to the wholeabdomen in cases of gross spillage ordissemination. Metastatic disease andunfavorable histology are poorprognostic factors in WT.15
Children with WAGR syndrome orother germline WT mutations requiremonitoring throughout their lifetimebecause of the increased risk ofdeveloping hypertension, nephropathy,and renal failure.15,33 Currentscreening recommendation’s forchildren with WAGR, Denys-Drash, andBWS or any genetic predisposition
(overgrowth syndrome, sporadicaniridia, or isolated hemihypertrophy)that increases the chance of developingWT include abdominal ultrasoundsevery 3 months until 8 years ofage.15,28,34–36 Approximately 5% to10% of individuals with WT havebilateral or multicentric tumors, andthose with genetic predisposition havean increased prevalence.15 Eighty-fivepercent of patients with either WAGRor BWS have unilateral tumors.15,37
Because ∼10% of patients with BWSwill develop a malignancy, mostcommonly WT or hepatoblastoma,screening with abdominal ultrasoundin addition to serum a-fetoprotein isrecommended until age 4 (becausemost hepatoblastoma occur before thisage), with renal ultrasoundsthereafter.38 Currently, there is nostandard approach for the treatment ofthose predisposed to developingbilateral WT.
Patients with Denys-Drash syndromeare faced with a significant challengeregarding the controversial need for
early prophylactic bilateralnephrectomies as a means todiminish the potential developmentof WT,39–44 the adverse effects ofchemotherapy, including theprolongation of time to considerationfor transplantation, and to reducemetabolic and nutritional sequelae ofchronic renal failure.40 Some authorspropose careful monitoring of patientsevery 4 to 6 months by abdominalultrasonography and performingnephrectomy after the onset of end-stage renal failure45,46; however,others feel it is preferable to proceedwith bilateral nephrectomies “early”rather than “late” in the course of theunderlying renal disease.40
Clear Cell Sarcoma of the Kidney
Clear cell sarcoma of the kidney(CCSK) is the second most commonrenal tumor in children, accountingfor 3% to 5% of all childhoodcancers.47–50 It commonly appears inchildren younger than 4 years ofage.18,22,51 CCSK is an aggressive
FIGURE 2Anaplastic WT. A, Axial contrast-enhanced CT of the abdomen demonstrates a large, heterogeneous left renal mass (white arrow) with adjacent metastaticadenopathy (black arrow). Note solid and cystic/necrotic areas within the renal mass. B, Longitudinal panoramic view of the left kidney reveals the largesize of the solid mass (white arrows) relative to the spared lower pole (black arrows). C, Axial fused 18F-fluorodeoxyglucose (FDG) positron emissiontomography (PET)/CT demonstrates marked increased FDG avidity of both the primary mass (white arrows) as well as the metastatic adenopathy (blackarrows). D, Whole body maximum intensity projection image from FDG-PET/CT reveals the extent of the abdominal disease, as well as activity in thoracicadenopathy and numerous pulmonary metastases. E, A complex cystic and solid mass with large areas of necrosis and hemorrhage in a 2-year-old girl.F, Hematoxylin and eosin section reveals poorly differential epithelial and blastemal cells with pronounced nuclear pleomorphism and large atypicalmitotic figures characteristic of anaplastic or unfavorable histology WT.
4 MALKAN et al by guest on June 10, 2018www.aappublications.org/newsDownloaded from

tumor with a unique predilection forbone and brain metastasis,20,22,49,51
but can also spread to the lung andabdomen. Symptoms may includeabdominal pain, hypertension, andhematuria. Typical presentationincludes a large, unilateral, well-circumscribed, sharply demarcatedmass49 that compresses thesurrounding normal renalparenchyma and displaces thecollecting system7 (Fig 4). CCSK isdescribed as an enigmatic tumor typebecause its morphologic appearancedoes not resemble that of thekidney.52 It can mimic and bemimicked by WT, congenitalmesoblastic nephroma, and rhabdoidtumor of the kidney.18
The classic microscopic patternincludes cords of round or spindle-shaped cells with clear cytoplasm andovoid to rounded vesicular nuclei withinconspicuous nucleoli.47,52 The cellsare surrounded by fibrovascular septaranging from a thin “chicken-wire”arrangement to broad sheetscontaining an arborizing capillaryvasculature.47,52 Cytogeneticabnormalities involving recurringtranslocations in t(10;17)(q22;p13/p12)53 and deletion of 14q24q31 havebeen described in CCSK.18 Stagingaccording to the National Wilms TumorStudy Group (NWTS) 5 definitiondemonstrated that stages I to III arenearly equally represented (25%, 37%,34%, respectively), whereas 4%present with stage IV and evidence ofmetastatic disease.47 Approximately
29% of CCSK cases may have evidenceof lymph node metastases.47 Ipsilateralrenal hilar lymph nodes are the mostcommon site of metastases andunderscore the importance of lymph
node sampling in staging. Currentmultimodal treatment consists ofradical nephrectomy for resectabletumors followed by intensivechemotherapy and radiotherapy.49,54
FIGURE 3Bilateral WT. A, Axial short TI inversion recovery (STIR) MRI sequence through the upper abdomen demonstrates large heterogeneous masses (blackarrows) arising from both kidneys. Note the compressed renal parenchyma (white arrows). B, Axial and (C) coronal STIR MRI sequences of the abdomendemonstrate the size of the large heterogeneous bilateral renal masses, which essentially fill the abdomen, and half-Fourier acquisition single-shot turbospin-echo MRI sequence clearly reveals the small cystic areas within the masses.
FIGURE 4Clear cell sarcoma. A, Axial postcontrast CT through the upper abdomen demonstrates an extremelylarge, heterogeneous mass arising from the right kidney (white arrows). Note the claw sign (blackarrows) indicating its renal origin. B, Coronal postcontrast image through the abdomen demonstratesthe size of the large right renal mass. C, A large solid mass with a homogenous tan cut surface anda yellow focus of necrosis with a sharply defined tumor-kidney junction in a 14-month-old girl.D, Hematoxylin and eosin section reveals tumor cells with uniform plump nuclei with delicate chro-matin arranged in cords and nests separated by an arborizing vascular network.
PEDIATRICS Volume 135, number 1, January 2015 5 by guest on June 10, 2018www.aappublications.org/newsDownloaded from

The addition of doxorubicin to thetreatment protocol has led to actuarial6-year survival approaching 98% forlocalized disease.47
Renal Cell Carcinoma
Renal cell carcinoma (RCC) accountsfor ,0.3% of all childhood tumors,55
is the most common renal tumor inadolescents,56 and the average age ofpresentation is approximately 10–11years old.57,58 RCC is a malignancythought to arise from epithelial cellsof the renal tubule.57 Severalhistologic subtypes of RCC exist,including clear-cell and medullarycarcinoma (Fig 5), multicystic,59
chromophil, chromophobe,60 andcollecting duct.2,61,62 Papillary andtranslocation, Xp11.263 (Fig 6) or thet(6;11), types are more common inchildren.18,64,65 RCCs are seen withgreater frequency in childhoodcancer survivors55,66,67 and ingenetic syndromes such as vonHippel-Lindau disease, tuberoussclerosis,20,55,68 familial clear-cellrenal cancer, hereditary papillaryrenal carcinoma, hereditaryleiomyomatosis, and renal cell cancersyndrome, in individuals with cysticor end-stage renal diseases,69,70
sickle cell hemoglobinopathies, andin pediatric kidney transplantrecipients.61
Unlike WT, RCC is rarelyasymptomatic (only 12% of cases).56
Diagnostic clues may include theclassic triad of gross, painlesshematuria, flank pain, and thepresence of a palpable mass.20,56,71,72
Metastases most commonly occur inthe lungs (40% to 65%) and bones(10% to 42%)57; however, the liver(35% to 57%), bladder, brain, orpleura (7% to 15%) may also beinvolved.58 Abdominal ultrasoundwith a subsequent CT scan is used tobetter define the neoplasm.73
Distinguishing between RCC andother renal tumors requires histologicexamination, and usually few if anyhints come from imaging.56 Althoughon CT, RCC typically reveals a large,heterogeneous, solid mass with either
well-circumscribed or poorly definedborders.7 Intravenous enhancementof the tumor is usually less than theadjacent normal parenchyma.7,51
RCC tends to be smaller than WT,invades tissues locally withdistortion of normal renalarchitecture, and formation ofa pseudocapsule that contain foci of
calcification. Regionallymphadenopathy and vascularinvasion are commonly seen.7
RCC histology demonstrates epithelialcells of renal tubule origin withmoderate to large amounts of clear andvariably eosinophilic cytoplasm withnested, solid, acinar, and/ortubulopapillary architectural growth
FIGURE 5Renal medullary carcinoma. A, Axial contrast-enhanced CT of the abdomen reveals a hypodensemass in the upper pole of the right kidney (white arrow) and an adjacent pathologically enlargedretrocaval lymph node in keeping with metastatic disease (black arrow). B, Coronal reconstructionfrom the same CT reveals the mass involves the upper pole of the right kidney. The remainder of thekidney appears normal. Note the claw sign (black arrow), indicating a renal origin of the mass.C, Longitudinal color Doppler ultrasound image demonstrates a relatively hyperechoic mass in theupper pole of the right kidney (white arrows). The mass does demonstrate internal vascularity.D, Axial chest CT lung algorithm demonstrates an irregular nodule in the right lung, consistent withmetastatic disease. E, An ill-defined white-tan solid mass in the upper pole of the kidney in a 12-year-old boy with sickle cell trait. F, Hematoxylin and eosin section reveals epithelial cells with abundanteosinophilic cytoplasm and prominent nucleoli admixed with numerous neutrophils.
6 MALKAN et al by guest on June 10, 2018www.aappublications.org/newsDownloaded from
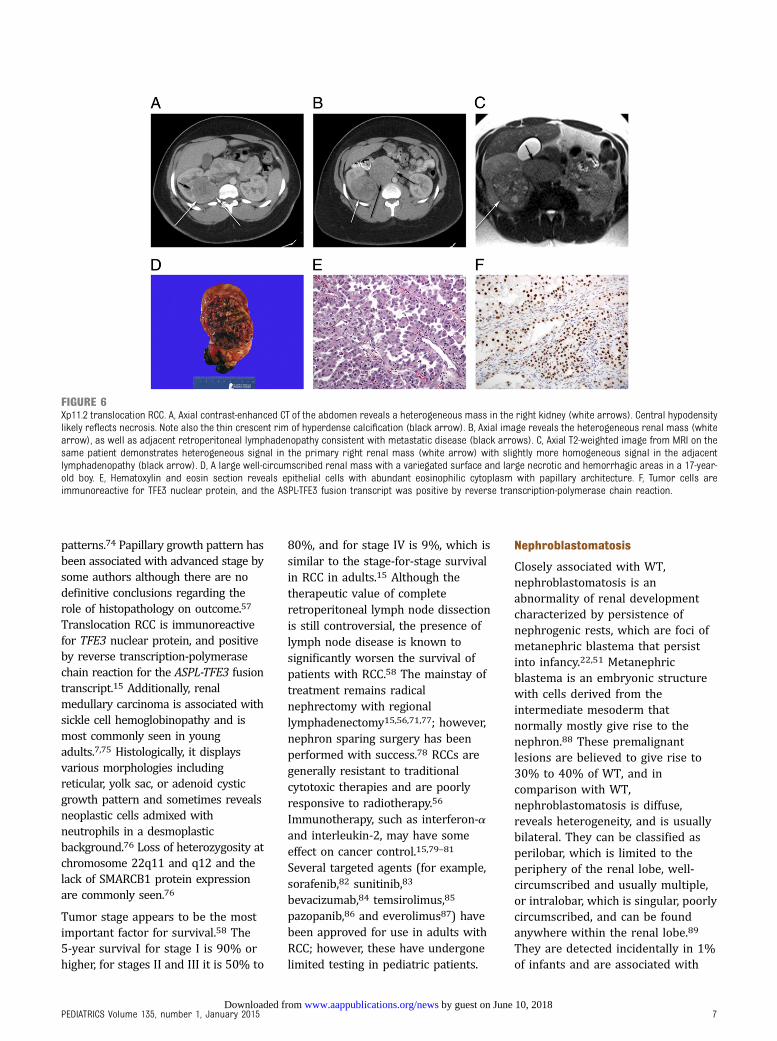
patterns.74 Papillary growth pattern hasbeen associated with advanced stage bysome authors although there are nodefinitive conclusions regarding therole of histopathology on outcome.57
Translocation RCC is immunoreactivefor TFE3 nuclear protein, and positiveby reverse transcription-polymerasechain reaction for the ASPL-TFE3 fusiontranscript.15 Additionally, renalmedullary carcinoma is associated withsickle cell hemoglobinopathy and ismost commonly seen in youngadults.7,75 Histologically, it displaysvarious morphologies includingreticular, yolk sac, or adenoid cysticgrowth pattern and sometimes revealsneoplastic cells admixed withneutrophils in a desmoplasticbackground.76 Loss of heterozygosity atchromosome 22q11 and q12 and thelack of SMARCB1 protein expressionare commonly seen.76
Tumor stage appears to be the mostimportant factor for survival.58 The5-year survival for stage I is 90% orhigher, for stages II and III it is 50% to
80%, and for stage IV is 9%, which issimilar to the stage-for-stage survivalin RCC in adults.15 Although thetherapeutic value of completeretroperitoneal lymph node dissectionis still controversial, the presence oflymph node disease is known tosignificantly worsen the survival ofpatients with RCC.58 The mainstay oftreatment remains radicalnephrectomy with regionallymphadenectomy15,56,71,77; however,nephron sparing surgery has beenperformed with success.78 RCCs aregenerally resistant to traditionalcytotoxic therapies and are poorlyresponsive to radiotherapy.56
Immunotherapy, such as interferon-aand interleukin-2, may have someeffect on cancer control.15,79–81
Several targeted agents (for example,sorafenib,82 sunitinib,83
bevacizumab,84 temsirolimus,85
pazopanib,86 and everolimus87) havebeen approved for use in adults withRCC; however, these have undergonelimited testing in pediatric patients.
Nephroblastomatosis
Closely associated with WT,nephroblastomatosis is anabnormality of renal developmentcharacterized by persistence ofnephrogenic rests, which are foci ofmetanephric blastema that persistinto infancy.22,51 Metanephricblastema is an embryonic structurewith cells derived from theintermediate mesoderm thatnormally mostly give rise to thenephron.88 These premalignantlesions are believed to give rise to30% to 40% of WT, and incomparison with WT,nephroblastomatosis is diffuse,reveals heterogeneity, and is usuallybilateral. They can be classified asperilobar, which is limited to theperiphery of the renal lobe, well-circumscribed and usually multiple,or intralobar, which is singular, poorlycircumscribed, and can be foundanywhere within the renal lobe.89
They are detected incidentally in 1%of infants and are associated with
FIGURE 6Xp11.2 translocation RCC. A, Axial contrast-enhanced CT of the abdomen reveals a heterogeneous mass in the right kidney (white arrows). Central hypodensitylikely reflects necrosis. Note also the thin crescent rim of hyperdense calcification (black arrow). B, Axial image reveals the heterogeneous renal mass (whitearrow), as well as adjacent retroperitoneal lymphadenopathy consistent with metastatic disease (black arrows). C, Axial T2-weighted image from MRI on thesame patient demonstrates heterogeneous signal in the primary right renal mass (white arrow) with slightly more homogeneous signal in the adjacentlymphadenopathy (black arrow). D, A large well-circumscribed renal mass with a variegated surface and large necrotic and hemorrhagic areas in a 17-year-old boy. E, Hematoxylin and eosin section reveals epithelial cells with abundant eosinophilic cytoplasm with papillary architecture. F, Tumor cells areimmunoreactive for TFE3 nuclear protein, and the ASPL-TFE3 fusion transcript was positive by reverse transcription-polymerase chain reaction.
PEDIATRICS Volume 135, number 1, January 2015 7 by guest on June 10, 2018www.aappublications.org/newsDownloaded from

BWS, hemihypertrophy and Perlmansyndrome in their perilobar form, andwith Denys-Drash syndrome, WAGRsyndrome, and sporadic aniridia intheir intralobar form.7
The diagnosis can be maderadiographically and may appear onCT as low-attenuation, multifocalnodules located at the renal peripherywith mild contrast enhancement or asdiffuse nephromegaly with retentionof the kidney’s reniform shape.22 MRIis particularly useful for diagnosis ofnephroblastomatosis when itdemonstrates homogeneity of thehypointense rindlike lesion oncontrast enhancement, whichdifferentiates it from WT.15
Microscopic findings range frombenign small nests of blastemal cells tolarge areas with frank malignanttransformation (WT), and is extremelydifficult to differentiate between the 2based on needle or even incisionalbiopsies alone.90,91 Currentrecommendations are for treatmentwith chemotherapy (vincristine anddactinomycin) until nearly completeresolution as determined byimaging.15 However, given the highincidence of bilaterality and thesubsequent development of WT, renalsparing surgery is indicated (partialnephrectomy).15,89
Rhabdoid Tumor
Rhabdoid tumor of the kidney isa highly aggressive neoplasmaccounting for ,2% of all pediatric
renal malignancies.18 This typicallyoccurs in children younger than 2years of age and is one of the mostlethal, malignant solid tumors inchildren, with an overall survival rate,25%.92 A distinct clinicalpresentation with fever, hematuria,young age (mean age 11 months), andhigh tumor stage at presentationsuggests a diagnosis of rhabdoidtumor of the kidney15,93; however,hypercalcemia due to elevatedparathormone levels may also befound.7,22 Characteristic CT featuresinclude hemorrhage, necrosis, andtumor lobules lined bycalcification.22,51 A peripheralsubcapsular crescent withattenuation of fluid may be seen in71% of patients (Fig 7).94
Synchronous or metachronousprimary intracranial masses or brainmetastases have been established asdistinctive features.22 Germlinemutations in SMARCB1 (also knownas SNF5 or INI1) gene95,96 are seen in∼35%97 of patients with rhabdoidtumor consistent with a geneticpredisposition.15,98 The most distincthistologic findings are rather largecells with large vesicular nuclei,a prominent single nucleolus, and insome cells, the presence of globulareosinophilic cytoplasmicinclusions.15,99 Treatment modalitiesinclude chemotherapy andnephrectomy; however, currenttherapy has limited efficacy for thispatient population.100,101 Four-year
overall survival rates for stages I and IIare 42% and stage III and IV are 16%.92
Congenital Mesoblastic Nephroma
Mesoblastic nephroma, the mostcommon solid renal tumor ininfancy,20,21,102,103 is a low gradefibroblastic sarcoma most commonlyaffecting infants younger than 3months, and .90% of cases appearwithin the first year of life.15,18,21 Theassociated manifestations includehypercalcemia with polyuria,congestive heart failure, andhypertension.7 Prenatal ultrasoundmay be helpful because in most casesthe diagnosis can be made in theneonatal period as it can lead topolyhydramnios (71% of gestationsassociated with this tumor), andpremature birth.21,104,105 A concentrichyperechoic and hypoechoic ringpattern known as the vascular “ring”sign may also be seen surrounding thetumor.106 Postnatal CT typicallyreveals a large, heterogeneous, solidintrarenal mass with smooth marginsthat enhances to a lesser degree thanthe adjacent normal renal parenchymaafter intravenous contrastadministration (Fig 8).7,104
Based on histopathologicappearances, congenital mesoblasticnephroma is subtyped into classic(24%), cellular (66%), and mixed(10%) types.103,105,107–109 Theclassic form is morphologicallyidentical to infantile fibromatosis ofthe renal sinus109 with fusiform
FIGURE 7Rhabdoid tumor. A, Axial contrast-enhanced CT of the abdomen demonstrates a hypodense solid mass in the right kidney (white arrow). B, Another imagefrom the same CT reveals a second solid mass in the contralateral left kidney (white arrows). C, Axial gadolinium-enhanced T1 weighted image from thispatient’s brain MRI confirms an enhancing mass in the left frontal lobe, in keeping with a synchronous atypical teratoid rhabdoid tumor.
8 MALKAN et al by guest on June 10, 2018www.aappublications.org/newsDownloaded from

spindle cells and rare mitoses.105,107
The cellular form is identical toinfantile fibrosarcoma,109 ischaracterized by a highly cellularspindle cell proliferation withminimal intervening collagen/supporting stroma, and high nuclear-to-cytoplasm ratios.108 The cellularvariant carries a specifictranslocation, t(12;15)(p13;q25),involving the ETV6 and NTRK3 genes,similar to the translocation seen ininfantile fibrosarcoma.110 The mixedtype is composed of areas of low(formed by elongated spindle cellsresembling fibroblastic cells seen inbenign myofibromas) and highcellularity.108 Complete surgicalresection via radical nephrectomy isadequate therapy for most patientsand reduces the risk of localrecurrence.102,105,107 In stage IIIpatients (incomplete resection and/or histologically positive resectionmargin), or those with cellularsubtype, and aged 3 months or olderat diagnosis are at increased risk forlocal and eventually metastaticrecurrence. This select group ofpatients are recommended to haveadjuvant chemotherapy.15,107 Five-year event-free survival rate is 94%,and the overall survival rate is 96%when diagnosed within the first 7months of life.111
Anaplastic Sarcoma
Anaplastic sarcoma of the kidney isa recently recognized pediatrictumor112 seen in children oradolescents younger than 15 years ofage113 with a female predominance.114
Patients typically present with a largerenal mass, and the most common sitesof metastases are the lung, liver, andbones.15,114 The main histologicfeatures are the presence ofundifferentiated spindle cells arrangedin short fascicles as well asundifferentiated small round primitivemesenchymal cells, very markedcellular anaplasia with giantpleomorphic cells, and usuallyprominent areas of benign ormalignant cartilage or chondroiddifferentiation.114,115 Optimal therapyfor this tumor is currently unclear.15,114
Other Renal Neoplasms
Several other renal malignant tumorsare even less common in children.Desmoplastic small round cell tumor(DSRCT) and Ewing sarcoma/primitive neuroectodermal tumor(PNET) of the kidney are extremelyrare in children.7 DSRCT is aggressive,malignant, typically affects children6 to 8 years of age but may also beseen in young adolescents,18 and isdiagnosed by its characteristicEWS-WT1 translocation.116 CT may
reveal a hypovascular, heterogeneous,and well-circumscribed mass withinternal punctate calcifications.117 Itreveals similar histologic features as inthe extrarenal sites consisting of nests,cords, or sheets of smallundifferentiated cells with foci ofnecrosis, calcification, anddesmoplasia.115 However, thedefinitive diagnosis of DSRCT is basedon the presence of the typicaltranslocation t(11;22)(p13;q12).18
Patients usually present at anadvanced stage with a poor prognosis.
Ewing sarcoma/PNETs of the kidneyare small round blue cell tumors. Thetissue of origin is unknown; however,molecular profiling of these tumorssuggests a mesenchymal cellorigin.118–121 They are very aggressive,usually large and invasive,19 andtypically have poorly defined marginswith areas of hemorrhage andnecrosis.122,123 The distinct vascularnature is manifested by an arborizingvascular pattern, perithelialarrangement of tumor cells, andperivascular pseudorosettes.124 Morethan 90% of renal Ewing sarcoma/PNETs have characteristictranslocations involving rearrangementof the ESWR1 gene on chromosome 22,t[11;22][q24;q12].18,124 The treatmentof these tumors is similar to theircounterparts at other sites andincludes a combination ofchemotherapy, surgery, and/orradiation therapy. Synovial sarcoma ofthe kidney may present with anabdominal mass and flank pain.115
Histologically they demonstratea monophasic, spindle cell appearance,with these stromal areas composed ofrelatively monomorphic, plump, ovoidspindle cells with an intersectingfascicular or solid sheetlike pattern.47
This rare renal tumor is geneticallycharacterized by translocation t[X;18][p11;q11].18
RENAL TUMOR AS A SECONDMALIGNANCY
In pediatric patients with cancer,renal tumors may be detected on
FIGURE 8Congenital mesoblastic nephroma. A, Axial contrast-enhanced CT of the abdomen demonstratesa complex mass in the left kidney (white arrows). Note hypodense fluid component and hyperdensecalcification. B, Coronal image from the same study.
PEDIATRICS Volume 135, number 1, January 2015 9 by guest on June 10, 2018www.aappublications.org/newsDownloaded from

imaging studies conducted during thecourse of their treatment or duringcancer survivorship follow-upsurveillance. RCC has been detectedas a second malignancy duringroutine surveillance in survivors ofleukemia,66,125 supratentorialPNET,66 WT,67 andneuroblastoma.55,126 In theChildhood Cancer Survivor Study,51 kidney cancers were detected assecond malignancies among 14 3595-year survivors and another 28 asthird or subsequent malignancies.127
SECONDARY INVOLVEMENT OF THEKIDNEY
Primary lymphoma of the kidney isextremely rare.128 However, diffuseinfiltration of the kidney may beobserved in children with leukemia.Involvement of the kidney as seen innon-Hodgkin lymphoma and B-celllymphomas is usually associated withhematogenous spread or directextension of retroperitoneal disease.7
On imaging, lymphoma may appear ina variety of forms: as a solitary mass,diffuse nephromegaly, or bilateralmultifocal nodules.7 The lattermostform may mimic nephroblastomatosisbut is unusual in infants and youngchildren. In contrast, leukemicinvolvement of the kidneys is seen onCT imaging as bilateral diffusesymmetric nephromegaly associatedwith decreased corticomedullarydifferentiation and decreased corticalenhancement.51 Adjacent or systemiclymphadenopathy is often alsovisualized on imaging when suchrenal lesions are detected.
BENIGN NEOPLASMS
Angiomyolipoma
Renal angiomyolipoma is considereda benign mesenchymal tumor,accounts for 3% of solid renalmasses,129,130 and contains varyingamounts of a disordered arrangementof blood vessels, smooth muscle, andadipose tissue.4,22,129 Angiomyolipomamay occur sporadically,22,131 although
it is seen in up to 80% of patientswith tuberous sclerosis (usuallybilateral and/or multiple renalangiomyolipomas).130–132 It can be alsoseen in von Hippel-Lindau syndrome,neurofibromatosis,22 Sturge-Webersyndrome, autosomal dominantpolycystic kidney disease, andpulmonary lymphangiomyomatosis.133
Presentation at extrarenal sites such asin the liver, spleen, abdominal wall,retroperitoneum, lung, and genitalregion may also be seen.134 Althoughlargely asymptomatic, patients maypresent with flank pain, fever, nausea,vomiting, hematuria, hypertension, oreven retroperitoneal hemorrhage dueto aneurysm formation because of theabundant abnormal, elastin-poorvascularity of the tumor.22,130,135
Imaging appearance variesconsiderably based on the amount andtype of histologic elements present.Diagnosis can usually be made by CT orMRI that reveals macroscopic fat131;however, fat is not pathognomonic orunique to angiomyolipoma and isoccasionally seen in WT and RCC.22 Infact, radiologic techniques usingenhancement patterns and histogramanalysis at CT and chemical shift MRIremain controversial to rule out RCC inhistologically proven fat poorangiomyolipomas.136–138 A solid renaltumor without fat content on imagingstudies is regarded as RCC and treatedas such.138
Microscopically, angiomyolipomas arevery cellular consisting mainly ofmyoid cells with vacuolatedcytoplasm spinning off of largevessels in a background of maturefat.130 In asymptomatic lesions,4 cm, evaluation with abdominalultrasound or CT scan should beperformed biannually or yearly. Inlesions that are .4 cm, symptomatic,or bilateral, arterial embolization andnephron sparing surgery are thetreatments of choice130 because ofthe risk of hemorrhage.132
Angiomyolipoma of the kidney tumorremains a benign tumor with rarepossibility of transformation toa malignancy139; therefore, follow-up
of these patients remains animportant concern.
Follow-up evaluation requirement isproposed for asymptomatic patientswith abdominal ultrasound or CTscan of the abdomen (risk of radiationexposure) or MRI at every 6- or12-month intervals130 to monitor thenumber and size of lesions, and foraneurysm development. The modalityused to image children remainsa challenge. Ultrasonography istechnician dependent, as comparedwith CT, which requires exposure toradiation, intravenous contrast, andthe potential need for generalanesthesia. Alternatively, MRI alsorequires intravenous contrast, withthe potential need for generalanesthesia and longer time requiredto complete imaging study. However,the significant advantage of MRI is itsdetailed imaging, and even moreimportantly is the elimination ofradiation exposure, which makes thismore optimal in children. Cost andthe availability of these imagingmodalities also impact the type offollow-up that may be provided.
Pseudotumors
Renal pseudotumors are masslikeimaging findings (normal renaltissue) that mimic neoplasms.96
These are caused by a variety ofconditions including renal congenitalanomalies (prominent renal columnsof Bertin, dromedary humps),inflammatory masses (focalpyelonephritis, renal abscess,autoimmune disease),7 vascularstructures (renal artery aneurysmor arteriovenous fistula), orabnormalities secondary to trauma orhemorrhage.8 A combination ofpatient demographics, clinicalpresentation, careful evaluation ofspecific imaging appearances on CTand/or MRI may provide usefulinformation for evaluating thisdiverse group of etiologies. Forexample, a history of blunt abdominaltrauma with microscopic hematuriamay support a diagnosis of renalhematoma. CT features to support an
10 MALKAN et al by guest on June 10, 2018www.aappublications.org/newsDownloaded from

inflammatory process may include ill-defined margins and perinephricstranding, and abnormalities in therenal vasculature such as vesselhypertrophy may support a diagnosisof vascular malformation. In mostcases, a familiarity with this group ofmasses in addition to a thoroughclinical and radiologic evaluationshould reveal the correct diagnosis.
Metanephric Adenoma
Metanephric adenoma (MA) is a rare,benign tumor of the renal cortexcomposed exclusively of epithelial andstromal elements.140–142 Patients canbe asymptomatic143,144 or can havesigns and symptoms related topolycythemia, hypertension,hematuria, dysuria, flank or abdominalpain, or a palpable abdominal mass.3
The lesion has radiologic similaritiesto RCC and WT, making preoperativediagnosis difficult.141 MAs have beendiagnosed by using ultrasound141,145
or CT.146 Histologic features of MAreveal small uniform epithelial cells ofan acinar, tubular, glomeruloid, orpapillary growth pattern with a highnuclear-to-cytoplasmic ratio.146
Surgical resection is recommended forMA due to the rarity of the tumor,potential for malignant degeneration,and inability to establish a diagnosiswithout histologic evaluation141;therefore, nephron-sparing surgery orpartial nephrectomy are consideredsufficient in most cases.141,147
Although MA is considered benignwith a good prognosis,141 reports ofatypical histologic features, bonemetastasis,148 and lymphaticdissemination149,150 may portenda poor prognosis.151
Cystic Nephroma
Multicystic nephroma is anuncommon, noninheritable,unilateral, benign tumor thatrepresents 2% to 3% of all primaryrenal tumors in children and ispredominantly seen in boys.152,153
Children typically present witha painless, progressively enlarging,palpable abdominal or flank mass that
has a variable growth rate and may bediscovered incidentally.152,153 Flankpain, urinary tract infection orobstruction, hematuria, andhypertension may also be seen.154 CTmay reveal a well-defined,nonenhancing cystic mass without solidcomponents involving the kidney withmultiple enhancing thin internal septacausing compression and stretchingof the remaining enhancing renalparenchyma.152 The tumor is bulky,well-encapsulated, noninfiltrating,and composed of multiplenoncommunicating, fluid-filled cysts.152
The presence of blastemal cells andpoorly differentiated cells must beruled out to diagnose this lesion.Complete resection either withnephrectomy152 or nephron-sparingsurgery152,154 remains the treatment ofchoice because carcinomatousdegeneration may occur within the wallof such tumors.152
Metanephric Stromal Tumor
Metanephric stromal tumor isa benign stromal tumor of the kidneyseen mostly within the first decade oflife155 with a mean age atpresentation of 2 years.155,156 Themost common presentation is anabdominal mass.155,156 This tumor,MA, and metanephric adenofibromarepresent a spectrum of well-differentiated nephroblastic lesionsthat appear to be related toWT.142,155,157 The most characteristichistologic features include an onionskin ring formation around entrappedtubules and blood vessels withvascular changes, includingangiodysplasia/epithelioidtransformation of medial smoothmuscle and myxoid changes.102 It iscomposed primarily of spindle cellswith a low proliferation index, anddiffusely infiltrates the perirenalfat.158 Nephrectomy is usuallycurative,102 and patients have anoverall excellent prognosis.158
Ossifying Renal Tumor of Infancy
Ossifying renal tumor of infancy(ORTI) is a rare, benign renal tumor
that may present as an abdominalmass typically in infants. Boys aremore commonly affected, and grosshematuria is almost alwaysobserved7,159 due to bulging of thetumor into the pelvicalycealsystem.160 A soft tissue mass withcalcifications near the renal pelvisand no significant contrastenhancement may be seen onCT.7,21,160 Histologically, ORTI ischaracterized by 3 majorcomponents: an osteoid core,osteoblastlike cells, and spindlecells.160 The best treatment approachfor this tumor involves resection6
with kidney preservationtechniques.161
Reninoma
Reninoma, also known asjuxtaglomerular cell tumor, is a rare,benign cause of curable hypertensionin children and adolescents.162,163
Patients may be found to havehypertension incidentally, whichprompts further investigation leadingto the discovery of clinical findingsincluding hypokalemia,hyperaldosteronism, and high reninactivity.162,163 Reninoma can bedivided into typical, atypical, andnonfunctioning types depending onthe blood pressure and serumpotassium levels.164 Althoughultrasound may in somecircumstances be helpful, CT andeven MRI remain the most usefulimaging modalities to document thepresence of a reninoma. It mayappear isodense or hypodense ascompared with the renalmedulla162,163; however, there is nodefinitive radiologic examination forthe diagnosis of reninoma. Therefore,histopathologic evaluation withimmunohistochemical staining aids inthe correct diagnosis.
Reninoma is composed of polygonaltumor cells within vascular stromaand thick-walled vessels.Immunohistochemical staining isparticularly useful in differentiatingreninoma from RCC andangiomyolipoma.162,165 Features
PEDIATRICS Volume 135, number 1, January 2015 11 by guest on June 10, 2018www.aappublications.org/newsDownloaded from

unique to reninoma include thetumor staining positively for CD34,and negatively for CD117, HMB-45,and S100 protein. Because these areusually small and rarely associatedwith metastasis,166 nephron sparingsurgery is the standard treatmentoption allowing for preservation ofrenal function.162,163
SUMMARY
Upon satisfactory exclusion of renalpseudotumors, renal neoplasmsinevitably require some form ofintervention to obtain a histologicdiagnosis through biopsy orresection. Intervention for thesemasses is easily justified in view oftheir significant mass effect and theparental concern that such dramaticlesions would understandablygenerate. Management decisions needto be tempered with anunderstanding of the difference inprofile of renal masses in children.Renal masses that are seen within thepediatric population, as discussedhere, remain a delicate and sensitiveissue. A combination of pathologicrarity, complexity, and uncertainmalignant potential may lead totreatment that is either tooaggressive, and, in retrospectunnecessary; or that is entirely tooconservative when in fact aggressivemeasures are warranted. Weencourage continued advancementsin research to better define evidence-based approaches and treatmentstrategies for these challengingmasses.
REFERENCES
1. Howlader N, Noone AM, Krapcho M,et al, eds. SEER Cancer StatisticsReview, 1975–2010. Bethesda, MD:National Cancer Institute. Available at:http://seer.cancer.gov/csr/1975_2010/.Accessed October 7, 2014
2. Cameron C, Greenbaum L, Sato T, TrostB, Lundeen B, Kelly ME. Renal cellcarcinoma in a patient with cystinosisand inflammatory bowel disease:a case report. Pediatr Nephrol. 2008;23(7):1167–1170
3. Amodio JB, Shapiro E, Pinkney L, et al.Metanephric adenoma in an 8-year-oldchild: case report and review of theliterature. J Pediatr Surg. 2005;40(5):e25–e28 [Review]
4. Joniau S, Goeman L, Kreuzbauer S, et al.Benign renal mesenchymoma in thepediatric age group: a novel pathologicand karyotype entity. Urology. 2004;63(5):981–984
5. Akil I, Ozguven A, Canda E, et al. Co-existence of chronic renal failure, renalclear cell carcinoma, and Blausyndrome. Pediatr Nephrol. 2010;25(5):977–981
6. Sotelo-Avila C, Beckwith JB, Johnson JE.Ossifying renal tumor of infancy:a clinicopathologic study of nine cases.Pediatr Pathol Lab Med. 1995;15(5):745–762 [Review]
7. Lee EY. CT imaging of mass-like renallesions in children. Pediatr Radiol. 2007;37(9):896–907
8. Israel GM, Silverman SG. The incidentalrenal mass. Radiol Clin North Am. 2011;49(2):369–383
9. Kim J, Cha S, Daldrup-Link HE, GoldsbyR. Pediatric tumors. In: Daldrup-Link HE,Gooding CA, eds. Essentials of PediatricRadiology: A Multimodality Approach.New York, NY: Cambridge UniversityPress; 2010:204
10. Bosniak MA. The use of the Bosniakclassification system for renal cystsand cystic tumors. J Urol. 1997;157(5):1852–1853
11. Israel GM, Bosniak MA. An update of theBosniak renal cyst classificationsystem. Urology. 2005;66(3):484–488
12. Bosniak MA. The Bosniak renal cystclassification: 25 years later. Radiology.2012;262(3):781–785
13. Bosniak MA. The current radiologicalapproach to renal cysts. Radiology.1986;158(1):1–10
14. Wallis MC, Lorenzo AJ, Farhat WA, BägliDJ, Khoury AE, Pippi Salle JL. Riskassessment of incidentally detectedcomplex renal cysts in children:potential role for a modification of theBosniak classification. J Urol. 2008;180(1):317–321
15. National Cancer Institute at the NationalInstitutes of Health. Wilms tumor andother childhood kidney tumorstreatment (PDQ). Available at: www.
cancer.gov/cancertopics/pdq/treatment/wilms/HealthProfessional/page2. Accessed March 19, 2013
16. Green DM, Jaffe N. The role ofchemotherapy in the treatment ofWilms’ tumor. Cancer. 1979;44(1):52–57
17. Gratias EJ, Dome JS. Current andemerging chemotherapy treatmentstrategies for Wilms tumor in NorthAmerica. Paediatr Drugs. 2008;10(2):115–124 [Review]
18. Royer-Pokora B. Genetics of pediatricrenal tumors. Pediatr Nephrol. 2013;28(1):13–23
19. Tagarelli A, Spreafico F, Ferrari A, et al.Primary renal soft tissue sarcoma inchildren. Urology. 2012;80(3):698–702
20. Castellino SM, Martinez-Borges AR,McLean TW. Pediatric genitourinarytumors. Curr Opin Oncol. 2009;21(3):278–283
21. Glick RD, Hicks MJ, Nuchtern JG,Wesson DE, Olutoye OO, Cass DL. Renaltumors in infants less than 6 months ofage. J Pediatr Surg. 2004;39(4):522–525[Review]
22. Lowe LH, Isuani BH, Heller RM, et al.Pediatric renal masses: Wilms tumorand beyond. Radiographics. 2000;20(6):1585–1603 [Review]
23. Fischbach BV, Trout KL, Lewis J, Luis CA,Sika M. WAGR syndrome: a clinicalreview of 54 cases. Pediatrics. 2005;116(4):984–988
24. Breslow NE, Norris R, Norkool PA, et al;National Wilms Tumor Study Group.Characteristics and outcomes ofchildren with the Wilms tumor-Aniridiasyndrome: a report from the NationalWilms Tumor Study Group. J Clin Oncol.2003;21(24):4579–4585
25. Royer-Pokora B, Beier M, Henzler M,et al. Twenty-four new cases of WT1germline mutations and review of theliterature: genotype/phenotypecorrelations for Wilms tumordevelopment. Am J Med Genet A. 2004;127A(3):249–257
26. Pelletier J, Bruening W, Kashtan CE,et al. Germline mutations in the Wilms’tumor suppressor gene are associatedwith abnormal urogenital developmentin Denys-Drash syndrome. Cell. 1991;67(2):437–447
27. Scott RH, Douglas J, Baskcomb L, et al;Factors Associated with Childhood
12 MALKAN et al by guest on June 10, 2018www.aappublications.org/newsDownloaded from

Tumours (FACT) Collaboration.Constitutional 11p15 abnormalities,including heritable imprinting centermutations, cause nonsyndromic Wilmstumor. Nat Genet. 2008;40(11):1329–1334
28. Green DM, Breslow NE, Beckwith JB,Norkool P. Screening of children withhemihypertrophy, aniridia, andBeckwith-Wiedemann syndrome inpatients with Wilms tumor: a reportfrom the National Wilms Tumor Study.Med Pediatr Oncol. 1993;21(3):188–192
29. DeBaun MR, Siegel MJ, Choyke PL.Nephromegaly in infancy and earlychildhood: a risk factor for Wilmstumor in Beckwith-Wiedemannsyndrome. J Pediatr. 1998;132(3 pt 1):401–404
30. DeBaun MR, Tucker MA. Risk of cancerduring the first four years of life inchildren from The Beckwith-WiedemannSyndrome Registry. J Pediatr. 1998;132(3 pt 1):398–400
31. Luithle T, Szavay P, Furtwängler R, GrafN, Fuchs J; SIOP/GPOH Study Group.Treatment of cystic nephroma andcystic partially differentiatednephroblastoma—a report from theSIOP/GPOH study group. J Urol. 2007;177(1):294–296
32. Ritchey ML, Shamberger RC, Hamilton T,Haase G, Argani P, Peterson S. Fate ofbilateral renal lesions missed onpreoperative imaging: a report fromthe National Wilms Tumor Study Group.J Urol. 2005;174(4 pt 2):1519–1521,discussion 1521
33. Lange J, Peterson SM, Takashima JR,et al. Risk factors for end stage renaldisease in non-WT1-syndromic Wilmstumor. J Urol. 2011;186(2):378–386
34. Gracia Bouthelier R, Lapunzina P.Follow-up and risk of tumors inovergrowth syndromes. J PediatrEndocrinol Metab. 2005;18(suppl 1):1227–1235
35. Lapunzina P. Risk of tumorigenesis inovergrowth syndromes:a comprehensive review. Am J MedGenet C Semin Med Genet. 2005;137C(1):53–71
36. Scott RH, Walker L, Olsen ØE, et alSurveillance for Wilms tumour in at-risk children: pragmaticrecommendations for best practice.Arch Dis Child. 2006;91(12):995–999
37. Porteus MH, Narkool P, Neuberg D, et al.Characteristics and outcome ofchildren with Beckwith-Wiedemannsyndrome and Wilms’ tumor: a reportfrom the National Wilms Tumor StudyGroup. J Clin Oncol. 2000;18(10):2026–2031
38. Tan TY, Amor DJ. Tumour surveillance inBeckwith-Wiedemann syndrome andhemihyperplasia: a critical review ofthe evidence and suggested guidelinesfor local practice. J Paediatr ChildHealth. 2006;42(9):486–490
39. Hu M, Zhang GY, Arbuckle S, et al.Prophylactic bilateral nephrectomies intwo paediatric patients with missensemutations in the WT1 gene. Nephrol DialTransplant. 2004;19(1):223–226
40. Rudin C, Pritchard J, Fernando ON, DuffyPG, Trompeter RS. Renaltransplantation in the management ofbilateral Wilms’ tumour (BWT) and ofDenys-Drash syndrome (DDS). NephrolDial Transplant. 1998;13(6):1506–1510
41. Aronson DC, Slaar A, Heinen RC, et alLong-term outcome of bilateral Wilmstumors (BWT). Pediatr Blood Cancer.2011;56(7):1110–1113
42. Hassinger AB, Garimella S. Refractoryhypotension after bilateralnephrectomies in a Denys-Drash patientwith phenylketonuria. Pediatr Nephrol.2013;28(2):345–348
43. Auber F, Jeanpierre C, Denamur E, et al.Management of Wilms tumors in Drashand Frasier syndromes. Pediatr BloodCancer. 2009;52(1):55–59
44. Schumacher V, Schärer K, Wühl E, et al.Spectrum of early onset nephroticsyndrome associated with WT1missense mutations. Kidney Int. 1998;53(6):1594–1600
45. Nso Roca AP, Peña Carrión A, BenitoGutiérrez M, García Meseguer C, GarcíaPose A, Navarro M. Evolutive study ofchildren with diffuse mesangialsclerosis. Pediatr Nephrol. 2009;24(5):1013–1019
46. Eddy AA, Mauer SM.Pseudohermaphroditism,glomerulopathy, and Wilms tumor(Drash syndrome): frequency in end-stage renal failure. J Pediatr. 1985;106(4):584–587
47. Argani P, Perlman EJ, Breslow NE, et al.Clear cell sarcoma of the kidney:a review of 351 cases from the National
Wilms Tumor Study Group PathologyCenter. Am J Surg Pathol. 2000;24(1):4–18
48. Hiradfar M, Zabolinejad N, Shojaeian R,et al. Pediatric clear cell sarcoma of thekidney with atriocaval thrombus.Pediatr Surg Int. 2012;28(11):1141–1145
49. Gooskens SL, Furtwängler R, VujanicGM, Dome JS, Graf N, van den Heuvel-Eibrink MM. Clear cell sarcoma of thekidney: a review. Eur J Cancer. 2012;48(14):2219–2226 [Review]
50. Furtwängler R, Gooskens SL, vanTinteren H, et al. Clear cell sarcomas ofthe kidney registered on InternationalSociety of Pediatric Oncology (SIOP) 93-01 and SIOP 2001 protocols: a report ofthe SIOP Renal Tumour Study Group. EurJ Cancer. 2013;49(16):3497–3506
51. Siegel MJ. Pediatric Body CT.Philadelphia, PA: Lippincott Williams &Wilkins; 1999:226–252
52. Karlsson J, Holmquist Mengelbier L,Ciornei CD, Naranjo A, O’Sullivan MJ,Gisselsson D. Clear cell sarcoma of thekidney demonstrates an embryonicsignature indicative of a primitivenephrogenic origin. GenesChromosomes Cancer. 2014;53(5):381–391
53. O’Meara E, Stack D, Lee CH, et al.Characterization of the chromosomaltranslocation t(10;17)(q22;p13) in clearcell sarcoma of kidney. J Pathol. 2012;227(1):72–80
54. Kalapurakal JA, Perlman EJ, Seibel NL,Ritchey M, Dome JS, Grundy PE.Outcomes of patients with revised stageI clear cell sarcoma of kidney treated inNational Wilms Tumor Studies 1-5. Int JRadiat Oncol Biol Phys. 2013;85(2):428–431
55. Fleitz JM, Wootton-Gorges SL, Wyatt-Ashmead J, et al. Renal cell carcinomain long-term survivors of advancedstage neuroblastoma in earlychildhood. Pediatr Radiol. 2003;33(8):540–545
56. Spreafico F, Collini P, Terenziani M,Marchianò A, Piva L. Renal cellcarcinoma in children and adolescents.Expert Rev Anticancer Ther. 2010;10(12):1967–1978
57. Indolfi P, Terenziani M, Casale F, et al.Renal cell carcinoma in children:a clinicopathologic study. J Clin Oncol.2003;21(3):530–535
PEDIATRICS Volume 135, number 1, January 2015 13 by guest on June 10, 2018www.aappublications.org/newsDownloaded from

58. Kumar S, Sharma P, Pratap J, Tiwari P,Bera MK, Kundu AK. Renal cellcarcinoma in children and adolescence:our experience. Afr J Paediatr Surg.2014;11(2):101–104
59. Atilgan D, Uluocak N, Erdemir F,Parlaktas BS, Koseoglu RD, Boztepe O.Multicystic renal cell carcinoma: a rarekidney tumor in children. Urol J. 2013;10(1):811–814
60. Kesik V, Yalçin B, Akçören Z, SenocakME, Talim B, Büyükpamukçu M. A raretype of renal cell carcinoma in a girl:hybrid renal cell carcinoma. PediatrHematol Oncol. 2010;27(3):228–232
61. Greco AJ, Baluarte JH, Meyers KE, et al.Chromophobe renal cell carcinoma ina pediatric living-related kidneytransplant recipient. Am J Kidney Dis.2005;45(6):e105–e108
62. Said JW, Thomas G, Zisman A. Kidneypathology: current classification ofrenal cell carcinoma. Curr Urol Rep.2002;3(1):25–30
63. Jing H, Tai Y, Xu D, Yang F, Geng M. Renalcell carcinoma associated with Xp11.2translocations, report of a case.Urology. 2010;76(1):156–158
64. Wu A, Kunju LP, Cheng L, Shah RB. Renalcell carcinoma in children and youngadults: analysis of clinicopathological,immunohistochemical and molecularcharacteristics with an emphasis onthe spectrum of Xp11.2 translocation-associated and unusual clear cellsubtypes. Histopathology. 2008;53(5):533–544
65. Geller JI, Argani P, Adeniran A, et al.Translocation renal cell carcinoma: lackof negative impact due to lymph nodespread. Cancer. 2008;112(7):1607–1616
66. Schafernak KT, Yang XJ, Hsueh W,Leestma JL, Stagl J, Goldman S. Pediatricrenal cell carcinoma as secondmalignancy: reports of two cases anda review of the literature. Can J Urol.2007;14(6):3739–3744 [Review]
67. Rich BS, McEvoy MP, La Quaglia MP.A case of renal cell carcinoma aftersuccessful treatment of Wilms tumor.J Pediatr Surg. 2010;45(9):1883–1886
68. Kubo M, Iwashita K, Oyachi N, Oyama T,Yamamoto T. Two different types ofinfantile renal cell carcinomasassociated with tuberous sclerosis. JPediatr Surg. 2011;46(10):E37–E41
69. Cohen HT, McGovern FJ. Renal-cellcarcinoma. N Engl J Med. 2005;353(23):2477–2490 [Review]
70. Tsui KH, Shvarts O, Smith RB, Figlin R,de Kernion JB, Belldegrun A. Renal cellcarcinoma: prognostic significance ofincidentally detected tumors. J Urol.2000;163(2):426–430
71. Labanaris AP, Schott GE, Zugor V. Renalcell carcinoma in children under 10years old: a presentation of four cases.Pediatr Surg Int. 2007;23(4):327–330
72. Baek M, Jung JY, Kim JJ, Park KH, RyuDS. Characteristics and clinicaloutcomes of renal cell carcinoma inchildren: a single center experience. IntJ Urol. 2010;17(8):737–740
73. Leveridge MJ, Bostrom PJ, Koulouris G,Finelli A, Lawrentschuk N. Imaging renalcell carcinoma with ultrasonography,CT and MRI. Nat Rev Urol. 2010;7(6):311–325 [Review]
74. Perlman EJ. Pediatric Renal CellCarcinoma. Surg Pathol Clin. 2010;3(3):641–651
75. Simpson L, He X, Pins M, et al. Renalmedullary carcinoma and ABL geneamplification. J Urol. 2005;173(6):1883–1888
76. Liu Q, Galli S, Srinivasan R, Linehan WM,Tsokos M, Merino MJ. Renal medullarycarcinoma: molecular,immunohistochemistry, andmorphologic correlation. Am J SurgPathol. 2013;37(3):368–374
77. Pratt CB, Pappo AS. Management ofinfrequent cancers of childhood. In:Pizzo PA, Poplack DG, eds. Principlesand Practice of Pediatric Oncology, 4thed. Philadelphia, PA: Lippincott Williams& Wilkins; 2004:1158–1160
78. Cook A, Lorenzo AJ, Salle JL, et al.Pediatric renal cell carcinoma: singleinstitution 25-year case series andinitial experience with partialnephrectomy. J Urol. 2006;175(4):1456–1460, discussion 1460
79. Fyfe G, Fisher RI, Rosenberg SA, et alResults of treatment of 255 patientswith metastatic renal cell carcinomawho received high-dose recombinantinterleukin-2 therapy. J Clin Oncol. 1995;13(3):688–696
80. Motzer RJ, Mazumdar M, Bacik J, RussoP, Berg WJ, Metz EM. Effect of cytokinetherapy on survival for patients with
advanced renal cell carcinoma. J ClinOncol. 2000;18(9):1928–1935
81. Pizzocaro G, Piva L, Colavita M, et al.Interferon adjuvant to radicalnephrectomy in Robson stages II and IIIrenal cell carcinoma: a multicentricrandomized study. J Clin Oncol. 2001;19(2):425–431
82. Escudier B, Michaelson MD, Motzer RJ,et al. Axitinib versus sorafenib inadvanced renal cell carcinoma:subanalyses by prior therapy froma randomised phase III trial. Br JCancer. 2014;110(12):2821–2828
83. Motzer RJ, Escudier B, Bukowski R,et al. Prognostic factors for survival in1059 patients treated with sunitinib formetastatic renal cell carcinoma. Br JCancer. 2013;108(12):2470–2477
84. Feldman DR, Baum MS, Ginsberg MS,et al. Phase I trial of bevacizumab plusescalated doses of sunitinib inpatients with metastatic renal cellcarcinoma. J Clin Oncol. 2009;27(9):1432–1439
85. Patel PH, Senico PL, Curiel RE, MotzerRJ. Phase I study combining treatmentwith temsirolimus and sunitinib malatein patients with advanced renal cellcarcinoma. Clin Genitourin Cancer.2009;7(1):24–27
86. Escudier B, Porta C, Bono P, et al.Randomized, controlled, double-blind,cross-over trial assessing treatmentpreference for pazopanib versussunitinib in patients with metastaticrenal cell carcinoma: PISCES Study.J Clin Oncol. 2014;32(14):1412–1418
87. Molina AM, Hutson TE, Larkin J, et al.A phase 1b clinical trial of themulti-targeted tyrosine kinase inhibitorlenvatinib (E7080) in combination witheverolimus for treatment of metastaticrenal cell carcinoma (RCC). CancerChemother Pharmacol. 2014;73(1):181–189
88. Glassberg KI. Normal and abnormaldevelopment of the kidney: a clinician’sinterpretation of current knowledge.J Urol. 2002;167(6):2339–2350, discussion2350–2351 [Review]
89. Perlman EJ, Faria P, Soares A, et al;National Wilms Tumor Study Group.Hyperplastic perilobarnephroblastomatosis: long-termsurvival of 52 patients. Pediatr BloodCancer. 2006;46(2):203–221
14 MALKAN et al by guest on June 10, 2018www.aappublications.org/newsDownloaded from

90. Machmouchi M, Bayoumi M, Mamoun I,Al-Ahmadi K, Kanaan H. Bilateraluniversal nephroblastomatosis in an8-month-old infant treated withchemotherapy. Pediatr Nephrol. 2005;20(7):1007–1010
91. Beckwith JB. Nephrogenic rests and thepathogenesis of Wilms tumor:developmental and clinicalconsiderations. Am J Med Genet. 1998;79(4):268–273
92. Tomlinson GE, Breslow NE, Dome J, et al.Rhabdoid tumor of the kidney in theNational Wilms’ Tumor Study: age atdiagnosis as a prognostic factor. J ClinOncol. 2005;23(30):7641–7645
93. Amar AM, Tomlinson G, Green DM,Breslow NE, de Alarcon PA. Clinicalpresentation of rhabdoid tumors of thekidney. J Pediatr Hematol Oncol. 2001;23(2):105–108 [Review]
94. Mehrain R, Nabahati M. A case ofrhabdomyosarcoma of kidneymimicking nephroblastoma. Caspian JIntern Med. 2013;4(1):621–623
95. Roberts CW, Biegel JA. The role ofSMARCB1/INI1 in development ofrhabdoid tumor. Cancer Biol Ther. 2009;8(5):412–416
96. Versteege I, Sévenet N, Lange J, et al.Truncating mutations of hSNF5/INI1 inaggressive paediatric cancer. Nature.1998;394(6689):203–206
97. Eaton KW, Tooke LS, Wainwright LM,Judkins AR, Biegel JA. Spectrum ofSMARCB1/INI1 mutations in familial andsporadic rhabdoid tumors. PediatrBlood Cancer. 2011;56(1):7–15
98. Biegel JA, Zhou JY, Rorke LB, StenstromC, Wainwright LM, Fogelgren B. Germ-line and acquired mutations of INI1 inatypical teratoid and rhabdoid tumors.Cancer Res. 1999;59(1):74–79
99. Radhika S, Bakshi A, Rajwanshi A, et al.Cytopathology of uncommon malignantrenal neoplasms in the pediatric agegroup. Diagn Cytopathol. 2005;32(5):281–286
100. Ahmed HU, Arya M, Levitt G, Duffy PG,Mushtaq I, Sebire NJ. Part I: Primarymalignant non-Wilms’ renal tumours inchildren. Lancet Oncol. 2007;8(8):730–737 [Review]
101. Ahmed HU, Arya M, Levitt G, Duffy PG,Sebire NJ, Mushtaq I. Part II: Treatmentof primary malignant non-Wilms’ renal
tumours in children. Lancet Oncol. 2007;8(9):842–848 [Review]
102. Toutain J, VuPhi Y, Doco-Fenzy M, et al.Identification of a complex 17qrearrangement in a metanephricstromal tumor. Cancer Genet. 2011;204(6):340–343
103. Chaudry G, Perez-Atayde AR, Ngan BY,Gundogan M, Daneman A. Imaging ofcongenital mesoblastic nephroma withpathological correlation. Pediatr Radiol.2009;39(10):1080–1086
104. Irsutti M, Puget C, Baunin C, Duga I,Sarramon MF, Guitard J. Mesoblasticnephroma: prenatal ultrasonographicand MRI features. Pediatr Radiol. 2000;30(3):147–150
105. Santos LG, Carvalho JS, Reis MA, SalesRL. Cellular congenital mesoblasticnephroma: case report. J Bras Neurol.2011;33(1):109–112
106. Kelner M, Droullé P, Didier F, Hoeffel JC.The vascular “ring” sign in mesoblasticnephroma: report of two cases. PediatrRadiol. 2003;33(2):123–128
107. Furtwaengler R, Reinhard H, LeuschnerI, et al; Gesellschaft fur PädiatrischeOnkologie und Hämatologie (GPOH)Nephroblastoma Study Group.Mesoblastic nephroma—a report fromthe Gesellschaft fur PädiatrischeOnkologie und Hämatologie (GPOH).Cancer. 2006;106(10):2275–2283
108. Bayindir P, Guillerman RP, Hicks MJ,Chintagumpala MM. Cellularmesoblastic nephroma (infantile renalfibrosarcoma): institutional review ofthe clinical, diagnostic imaging, andpathologic features of a distinctiveneoplasm of infancy. Pediatr Radiol.2009;39(10):1066–1074
109. Kulkarni MP, Gosavi AV, Anvikar AR,Ramteerthakar NA, Lanjewar DN.Cytodiagnosis of congenitalmesoblastic nephroma: a case report.Diagn Cytopathol. 2013;41(3):234–238
110. Vujani�c GM, Sandstedt B, Harms D,Kelsey A, Leuschner I, de Kraker J; SIOPNephroblastoma Scientific Committee.Revised International Society ofPaediatric Oncology (SIOP) workingclassification of renal tumors ofchildhood. Med Pediatr Oncol. 2002;38(2):79–82
111. van den Heuvel-Eibrink MM, Grundy P,Graf N, et al. Characteristics and
survival of 750 children diagnosed witha renal tumor in the first seven monthsof life: A collaborative study by theSIOP/GPOH/SFOP, NWTSG, and UKCCSGWilms tumor study groups. PediatrBlood Cancer. 2008;50(6):1130–1134
112. Watanabe N, Omagari D, Yamada T, et al.Anaplastic sarcoma of the kidney: casereport and literature review. PediatrInt. 2013;55(5):e129–e132
113. Gomi K, Hamanoue S, Tanaka M, et al.Anaplastic sarcoma of the kidney withchromosomal abnormality: first reporton cytogenetic findings. Hum Pathol.2010;41(10):1495–1499
114. Vujani�c GM, Kelsey A, Perlman EJ,Sandstedt B, Beckwith JB. Anaplasticsarcoma of the kidney:a clinicopathologic study of 20 cases ofa new entity with polyphenotypicfeatures. Am J Surg Pathol. 2007;31(10):1459–1468
115. Sebire NJ, Vujanic GM. Paediatric renaltumours: recent developments, newentities and pathological features.Histopathology. 2009;54(5):516–528
116. Wang LL, Perlman EJ, Vujanic GM, et al.Desmoplastic small round cell tumor ofthe kidney in childhood. Am J SurgPathol. 2007;31(4):576–584
117. Egloff AM, Lee EY, Dillon JE, Callahan MJ.Desmoplastic small round cell tumor ofthe kidney in a pediatric patient:sonographic and multiphase CTfindings. AJR Am J Roentgenol. 2005;185(5):1347–1349
118. Potikyan G, France KA, Carlson MR,Dong J, Nelson SF, Denny CT. Geneticallydefined EWS/FLI1 model systemsuggests mesenchymal origin ofEwing’s family tumors. Lab Invest. 2008;88(12):1291–1302
119. Riggi N, Suvà ML, Suvà D, et al. EWS-FLI-1expression triggers a Ewing’s sarcomainitiation program in primary humanmesenchymal stem cells. Cancer Res.2008;68(7):2176–2185
120. Tirode F, Laud-Duval K, Prieur A,Delorme B, Charbord P, Delattre O.Mesenchymal stem cell features ofEwing tumors. Cancer Cell. 2007;11(5):421–429
121. Dela Cruz FS. Cancer stem cells inpediatric sarcomas. Front Oncol. 2013;3:168
122. Antoneli CB, Costa CM, de Camargo B,Sredni ST, Alfer W Jr, Chojniak R.
PEDIATRICS Volume 135, number 1, January 2015 15 by guest on June 10, 2018www.aappublications.org/newsDownloaded from

Primitive neuroectodermal tumor(PNET)/extraosseous Ewing sarcoma ofthe kidney. Med Pediatr Oncol. 1998;30(5):303–307
123. Perer E, Shanberg AM, Matsunaga G,Finklestein JZ. Laparoscopic removal ofextraosseous Ewing’s sarcoma of thekidney in a pediatric patient.J Laparoendosc Adv Surg Tech A. 2006;16(1):74–76
124. Karpate A, Menon S, Basak R, YuvarajaTB, Tongaonkar HB, Desai SB. Ewingsarcoma/primitive neuroectodermaltumor of the kidney: clinicopathologicanalysis of 34 cases. Ann Diagn Pathol.2012;16(4):267–274
125. Sausville JE, Hernandez DJ, Argani P,Gearhart JP. Pediatric renal cellcarcinoma. J Pediatr Urol. 2009;5(4):308–314
126. Medeiros LJ, Palmedo G, Krigman HR,Kovacs G, Beckwith JB. Oncocytoid renalcell carcinoma after neuroblastoma:a report of four cases of a distinctclinicopathologic entity. Am J SurgPathol. 1999;23(7):772–780
127. Wilson CL, Ness KK, Neglia JP, et al.Renal carcinoma after childhoodcancer: a report from the childhoodcancer survivor study. J Natl CancerInst. 2013;105(7):504–508
128. Dash SC, Purohit K, Mohanty SK, DindaAK. An unusual case of bilateral renalenlargement due to primary renallymphoma. Indian J Nephrol. 2011;21(1):56–58
129. Xi S, Chen H, Wu X, et al. Malignant renalangiomyolipoma with metastases ina child. Int J Surg Pathol. 2014;22(2):160–166
130. Astigueta JC, Abad MA, Pow-Sang MR,et al. Epithelioid angiomyolipoma:a rare variant of renal angiomyolipoma.Arch Esp Urol. 2009;62(6):493–497[Review]
131. Hilmes MA, Dillman JR, Mody RJ,Strouse PJ. Pediatric renal leukemia:spectrum of CT imaging findings.Pediatr Radiol. 2008;38(4):424–430
132. Choh NA, Choh SA, Yousuf R, Jehangir M.A giant aneurysm arising from renalangiomyolipoma in tuberous sclerosis.Arch Dis Child. 2009;94(1):47–48
133. Khaled A, Arif N, Zaman W, Nasir TA.Bilateral renal angiomyolipoma notassociated with tuberous sclerosis:
a case report. Available at: www.banglajol.info/index.php/PULSE/article/download/6965/529. Accessed May 30,2014
134. Ito M, Sugamura Y, Ikari H, Sekine I.Angiomyolipoma of the lung. ArchPathol Lab Med. 1998;122(11):1023–1025
135. Umeoka S, Koyama T, Miki Y, Akai M,Tsutsui K, Togashi K. Pictorial review oftuberous sclerosis in various organs.Radiographics. 2008;28(7):e32
136. Milner J, McNeil B, Alioto J, et al. Fatpoor renal angiomyolipoma: patient,computerized tomography andhistological findings. J Urol. 2006;176(3):905–909
137. Kim JK, Park SY, Shon JH, Cho KS.Angiomyolipoma with minimal fat:differentiation from renal cellcarcinoma at biphasic helical CT.Radiology. 2004;230(3):677–684
138. Seyam RM, Bissada NK, Kattan SA, et al.Changing trends in presentation,diagnosis and management of renalangiomyolipoma: comparison ofsporadic and tuberous sclerosiscomplex-associated forms. Urology.2008;72(5):1077–1082
139. Inci O, Kaplan M, Yalcin O, Atakan IH,Kubat H. Renal angiomyolipoma withmalignant transformation,simultaneous occurrence withmalignity and other complex clinicalsituations. Int Urol Nephrol. 2006;38(3-4):417–426
140. Kohashi K, Oda Y, Nakamori M, et al.Multifocal metanephric adenoma inchildhood. Pathol Int. 2009;59(1):49–52
141. Mei H, Zheng L, Zhou C, Tong Q.Metanephric adenoma in a 2-year-oldchild: case report andimmunohistochemical observations.J Pediatr Hematol Oncol. 2010;32(6):489–493
142. Küpeli S, Baydar DE, Canakl F, et al.Metanephric adenoma in a 6-year-oldchild with hemihypertrophy. J PediatrHematol Oncol. 2009;31(6):453–455
143. Davis CJ Jr, Barton JH, Sesterhenn IA,Mostofi FK. Metanephric adenoma.Clinicopathological study of fiftypatients. Am J Surg Pathol. 1995;19(10):1101–1114
144. Jones EC, Pins M, Dickersin GR, YoungRH. Metanephric adenoma of the
kidney. A clinicopathological,immunohistochemical, flow cytometric,cytogenetic, and electron microscopicstudy of seven cases. Am J Surg Pathol.1995;19(6):615–626
145. McNeil JC, Corbett ST, Kuruvilla S, JonesEA. Metanephric adenoma in a five-year-old boy presenting with chyluria: casereport and review of literature. Urology.2008;72(3):545–547
146. Liniger B, Wolf RW, Fleischmann A,Kluwe W. Local resection ofmetanephric adenoma with kidneypreservation. J Pediatr Surg. 2009;44(8):E21–E23
147. Navarro O, Conolly B, Taylor G, Bägli DJ.Metanephric adenoma of the kidney:a case report. Pediatr Radiol. 1999;29(2):100–103
148. Pins MR, Jones EC, Martul EV, Kamat BR,Umlas J, Renshaw AA. Metanephricadenoma-like tumors of the kidney:report of 3 malignancies with emphasison discriminating features. Arch PatholLab Med. 1999;123(5):415–420
149. Renshaw AA, Freyer DR, Hammers YA.Metastatic metanephric adenoma ina child. Am J Surg Pathol. 2000;24(4):570–574
150. Drut R, Drut RM, Ortolani C. Metastaticmetanephric adenoma with foci ofpapillary carcinoma in a child:a combined histologic,immunohistochemical, and FISH study.Int J Surg Pathol. 2001;9(3):241–247
151. Nakagawa T, Kanai Y, Fujimoto H, et al.Malignant mixed epithelial and stromaltumours of the kidney: a report of thefirst two cases with a fatal clinicaloutcome. Histopathology. 2004;44(3):302–304
152. Mehra BR, Thawait AP, Akther MJ,Narang RR. Multicystic nephromamasquerading as Wilms’ tumor:a clinical diagnostic challenge. Saudi JKidney Dis Transpl. 2011;22(4):774–778
153. Babu S, Agarwal R, Narayansamy K,Rajendranath R, Balasubramanian S.Cystic renal neoplasm causinghypertension in a 2-year-old child.Saudi J Kidney Dis Transpl. 2011;22(4):779–781
154. Bhardwaj AK, Sharma PD, Mittal A,Sharma A. Bilateral cystic nephromawith pleuropulmonary blastoma. BMJCase Rep. 2011
16 MALKAN et al by guest on June 10, 2018www.aappublications.org/newsDownloaded from

155. Kacar A, Azili MN, Cihan BS, Demir HA,Tiryaki HT, Argani P. Metanephricstromal tumor: a challengingdiagnostic entity in children. J PediatrSurg. 2011;46(12):e7–e10
156. Argani P, Beckwith JB. Metanephricstromal tumor: report of 31 cases ofa distinctive pediatric renalneoplasm. Am J Surg Pathol. 2000;24(7):917–926
157. Argani P. Metanephric neoplasms: thehyperdifferentiated, benign end of theWilms tumor spectrum? Clin Lab Med.2005;25(2):379–392 [Review]
158. De Pasquale MD, Diomedi-Camassei F,Serra A, et al. Recurrent metanephricstromal tumor in an infant. Urology.2011;78(6):1411–1413
159. Hu J, Wu Y, Qi J, Zhang C, Lv F. Ossifyingrenal tumor of infancy (ORTI): a casereport and review of the literature.J Pediatr Surg. 2013;48(2):e37–e40
160. Liu J, Guzman MA, Pawel BR, et al.Clonal trisomy 4 cells detected in theossifying renal tumor of infancy: studyof 3 cases. Mod Pathol. 2013;26(2):275–281
161. Vazquez JL, Barnewolt CE, ShambergerRC, Chung T, Perez-Atayde AR. Ossifyingrenal tumor of infancy presenting asa palpable abdominal mass. PediatrRadiol. 1998;28(6):454–457
162. Xu B, Zhang Q, Jin J. Hypertensionsecondary to reninoma treated withlaparoscopic nephron-sparing surgeryin a child. Urology. 2012;80(1):210–213
163. Gottardo F, Cesari M, Morra A,Gardiman M, Fassina A, Dal Bianco M.A kidney tumor in an adolescent withsevere hypertension and hypokalemia:an uncommon case—case report andreview of the literature on reninoma.Urol Int. 2010;85(1):121–124
164. Dong D, Li H, Yan W, Xu W.Juxtaglomerular cell tumor of thekidney—a new classification scheme.Urol Oncol. 2010;28(1):34–38
165. Markey RB, MacLennan GT.Juxtaglomerular cell tumor of thekidney. J Urol. 2006;175(2):730
166. Duan X, Bruneval P, Hammadeh R, et al.Metastatic juxtaglomerular cell tumorin a 52-year-old man. Am J Surg Pathol.2004;28(8):1098–1102
PEDIATRICS Volume 135, number 1, January 2015 17 by guest on June 10, 2018www.aappublications.org/newsDownloaded from

originally published online December 1, 2014; Pediatrics M. Green, Andrew M. Davidoff and John A. Sandoval
Alpin D. Malkan, Amos Loh, Armita Bahrami, Fariba Navid, Jamie Coleman, DanielAn Approach to Renal Masses in Pediatrics
ServicesUpdated Information &
014-1011http://pediatrics.aappublications.org/content/early/2014/11/25/peds.2including high resolution figures, can be found at:
Permissions & Licensing
http://www.aappublications.org/site/misc/Permissions.xhtmlin its entirety can be found online at: Information about reproducing this article in parts (figures, tables) or
Reprintshttp://www.aappublications.org/site/misc/reprints.xhtmlInformation about ordering reprints can be found online:
by guest on June 10, 2018www.aappublications.org/newsDownloaded from

originally published online December 1, 2014; Pediatrics M. Green, Andrew M. Davidoff and John A. Sandoval
Alpin D. Malkan, Amos Loh, Armita Bahrami, Fariba Navid, Jamie Coleman, DanielAn Approach to Renal Masses in Pediatrics
http://pediatrics.aappublications.org/content/early/2014/11/25/peds.2014-1011located on the World Wide Web at:
The online version of this article, along with updated information and services, is
ISSN: 1073-0397. 60007. Copyright © 2014 by the American Academy of Pediatrics. All rights reserved. Print the American Academy of Pediatrics, 141 Northwest Point Boulevard, Elk Grove Village, Illinois,has been published continuously since 1948. Pediatrics is owned, published, and trademarked by Pediatrics is the official journal of the American Academy of Pediatrics. A monthly publication, it
by guest on June 10, 2018www.aappublications.org/newsDownloaded from





























