Embed Size (px)
Citation preview

8/12/2019 Analysis of a Kinetic Model for Melanin Biosynthesis Pathway
http://slidepdf.com/reader/full/analysis-of-a-kinetic-model-for-melanin-biosynthesis-pathway 1/10
THE O U R N A L OF BIOLOGICALHE MIST R Y
1992 by The American Society
for
Biochemistry and Molecular Biology, Inc
Vol. 261, No.
6 ,
Issue
of February 25, p. 3801-3810,1932
Printed
in
U.
S.
A .
Analysis
of
a Kinetic Model for Melanin Biosynthesis Pathway*
(Received for publication, October
11,
1991)
Jose
Neptuno Rodriguez-Lopez ,
Jose
Tudelap, Ramon VaronS, Francisco Garcia-Carmonap,nd
Francisco Garcia-Canovaspll
Fr o m the $Departamento de Quimica-Fisica, Escuela Universitaria Politecnica de Albacete, Universidad de Castilla-La Mancha,
Albace te and the §D epartam ento de Bi oquim ica Biologiu Molecular, Facultad de Biologiu, Universidad de Murcia,E-30100
Espinardo, Murc ia , Spain
Thekineticbehavior of the melaninbiosynthesis
pathway from L-tyrosine up to dopachrome has been
studied from experimental and simulation assays. The
reaction mechanism proposed
is
based on
a
single ac-
tive site f tyrosinase . The diphenolase andonophen-
olase activities of tyrosinase involve one single (oxi-
dase) and two overlapped (hydroxylase and oxidase)
catalytic cycles, respectively. The stoichiometryf the
pathway implies that one molecule of tyrosinase must
accomplish two turnovers in the hydroxylaseycle for
each one in the oxidaseycle. Furtherm ore, the steady-
sta te rat es f dopachrome production and
2
onsump-
tion from tyrosine and L-dopa, also fulfill the stoichi-
ometry of the pathway: V /V = 1.5 and V g J V k
=
1.0,where T represen ts L-tyrosine, DC represents do-
pachrome, and
L)
epr esents L-dopa. It has been ascer-
tained by high performance iquid chromatography
that in the steady-state, quan tity of dopa is accumu-
lated
([DlBB)
hich fulfills the constant ra tio
[DlBB
R[mlo.
aking this ratio into account, an analytical
expression has been deduced for the monophenolase
activity of tyrosinase.In hisexpression kTat = (21
3)k3(K1/K2)R,evealing thatkTatis not a true catalytic
constant, since itlso depends on equilibrium constants
and on the experimental
R
=
0.057.
This low value
explains the lower catalyticfficiency of tyrosinase on
tyrosine than on dopa,
(VZ,JKI)/(VE.,/KE)= (2 /3 )R,
since
a
significant portion of tyrosinase
is
scavenged
from the catalytic turnover
s
dead-end complex
EmetT
in the steady-state of th e monophenolase activity of
tyrosinase.
M elan ins are eterogeneous polymers of polyphenolic char-
acterand ittle defined struc ture with color varying rom
yellow t o black 1).Melanins o r ig inate the enzymat icrown-
ing in frui ts and vegetables a s well as the pigm entat io n of
animals . Hum an deficiency in melanins causes albinism and
vit il igo, and grea t in terest has beenhown in the involvem ent
of melan ins in malign ant elanomes, th e carcinogenic tum ors
of the skin. The re have also been s tudie s on he possible
relat ionship between neuromelanins and damage of neurons
and their selective vulnera bility in Parkin son’s disease (2).
Melanogenesis s tarts with the oxid at iony
O 2
of monophenols
and/or o-diphenols th at yield the corresponding o-quinones,
which evolve through coupling nonen zym atic reactions o-
* This paper has been partially supported by a grant from the
Comisi6n Interministerial de Ciencia y Tecnologia (Spa in), project
CICYT ALI89-674. Th e costs of publication of thi s article were
defrayed in part by the payment of page charges. This article must
therefore he hereby marked “advertisement” in accordance with 18
U.S.C. Section 1734 solely o indicate thi s fact.
Y
T o
whom the correspondence should be addressed.
ward the formationof melanins 1).
Tyro sinase mon ophen ol, L-dopa:oxygen oxidoreductase,
EC 1.14.18.1) is a copper enzyme pr ese nt in microorganisms,
plants , and animals . Different tyrosinases obtainedrom sev-
eral biological sources have simila r stru ctura l and func tiona l
characteris t ics (3). The act ive s i te of tyrosinase consis ts of
two copper a toms and th ree s t a tes , “met ,”deoxy,” an d “oxy”
(4-11). S tructu ral models for the activ e site of these three
forms of tyrosinase have beenroposed (12-14) and con firm ed
by t ransformations into other derivat ives (15-17). T he mon-
ophenolase activity of tyrosinase iscoupled to i ts dipheno lase
act iv i ty and the nonenzymat ic reac t ionsrom the correspon d-
ing o-quinones. T hes e processes ca n be studie d by using a
“bottom-up’’ approa ch, in order o ob tai na successful insight
into the increasingomplexity of th e pathway.
T he o-quinones suffer nonenzymatic breakdown through
polymerizat ion a nd react ion with a number of reagents such
as inorganic ions, reductant agents , hiol and amino com-
pounds, and biological macromolecules (1, 3, 4). The amino
group of the side chain of o-dopaquinone is involved in an
intramolecular 1,4-addi t ion f Micha el into the enzene ring,
causing its cyclization into leukodopachrome’ (4). This inter-
mediate is quickly oxidized to dop ach rom e by an othe r mole-
cule of o-d opaq uinone -H+, which is reduced t o L-dopa
(Scheme I . T hi s process has been kinetically charac terized
from spectrophotometric data
(18)
and has also been verified
from studieswithelectronspin esonance (19) and pulse
radiolysis (20) techniq ues. On the other hand, a similar se-
quence of react ions has been reported for noncycl izable
o -
quinones, s tart ingwith the intermolecu lar addi t ionf nucleo-
philic reagents (21, 22). In act , he hydroxylation of
u-
dopaquinone-H’ can also be significant
at
acid pH (Scheme
I),
as has been detected(23)and kinetically charac terized
(24). Thus, the elanogenic o-quinones evolve through cycli-
zation and hydroxylation branch es involving regeneration of
the respectiveo-diphenol, bu t he hydroxylation bran ch is
only significant at acid pH, as has been detected in melano-
somes an d melanome cells (25).
A
similar kinetic behavior
has been d etected and a nalyzed from a-methy ldopa (26, 27)
an d dopamine
28,
29).
Once the nonen zym atic conversion of o-dopa quinon e-H+
up todopachrome has beenclarified (Scheme
I),
it is possible
to s tudy the diphenolase act ivi tyf tyrosinase. The s t ructural
mec hanism of this reac tion has bee n widely studied (4, 13,
30-32), and the three form sf the enzyme considered Schem e
11). Early kinet ic s tudies into the s teady-statef the pathway
’
The abbreviations and trivial names used are: leukodopachrome,
2,3-dihydro-5,6-dihydroxyindole-2-carboxylate;
yrosine, L-tyrosine;
dopa, ~-3,4-dihydroxyphenyIalanine;-dopaquinone, 4-(2-carboxy-2-
aminoethyl)-1,2-benzoquinone;opachrome, 2-carboxy-2,3-dihydro-
indole-5,6-quinone;HPLC, high performance liquid chromatography.
3801
8/12/2019 Analysis of a Kinetic Model for Melanin Biosynthesis Pathway
http://slidepdf.com/reader/full/analysis-of-a-kinetic-model-for-melanin-biosynthesis-pathway 2/10
3802
Kinetic Mechanism of
Tyrosinase
O\
O m C O O -c
HO -7 -2 0
~~~
T OH
E
Pa HD
SCHEME.
Sequence of rea ct ions
of
the melanin biosynthesis
pathway f rom tyros ine up
o
dopachrome.
k k k
k,
k - 2 k.8 k.. T m et
met
+
D EmstD
Ersc~ 0 2 orv
+
DE
orvD
OH QH
SCHEME1. Reaction mechanism for the diphen olase ctivity
of tyro sinase, coupled
to
nonenzyma tic react ions from o-do-
paquinone-H+ up
to
dopachrome
in
the melanin b iosynthesis
pathway.
2 ~ n
+ D
+n+
SCHEME
11. General eact ion mechanisms for yrosinase
activities
on
tyrosine and dopa.
repor ted an apparen t p ing-pong mechanism
33,34),
a part ic-
ular case of the t rue Ter Bi m echanism Uni Unii Uni Ping
Pong, with two subs trates and two produc ts equal between
them
35).
This rea ct ion me chanism has be en useful for t h e
kineticchara cterization of the nhib ition of tyrosinase by
halides
36)
and by s low binding inhibi tors such as m-cou-
maric acid and mimosine
37, 38).
Fur thermore , th i s reac t ion
E
red
+
0 2
ka
EoxvT
QH
ir:
La,,
D
2 QH +
D
+
H*
DC
SCHEME
V. Reaction mechanism for themonophenolase
ac-
t ivity of tyrosinas e, coupled
to
nonenzymatic react ions from
o-dopaquinone-H+ up to dopachrome in the melanin biosyn-
thesis pathway.
mechanism has served as the ba seor the u nders tand ing and
kinet ic characterizat ion
of
th e suicide inactivation of tyrosin-
ase by o-diphenols such
as
catechol , L-dopa, and dopam ine
39-43).
Th is process is not s ignificant in the t ime rangef a
few minutes , as is usual in s teady-state kinet ic s tudies , but
must be tak en into account to preven t biased recordings of
enzymatic activity.
The above advancesconcern ing heact ivesite of me t,
deoxy, and oxy forms of tyrosinase, the nonenz yma tic reac-
t ions from o-dopaquinone(Scheme I) and he d iphenolase
activity of tyrosinase(Scheme 11) have notbeen properly
considered in any paper on i ts mono phenolase a ct ivi ty
44-
46).
The s t ruc tura l mechanism
for
the m onophenolase ac t iv i ty
of tyrosinase hasbeen widely studied
4,13,30-32)
by consid-
ering the three forms of the enzyme (Scheme 111). Several
kinet ic s tudies
of
the s t eady-s ta te o f the pa thway repor t the
appare nt inhibition by a n excess of tyrosine
31, 44-46)
a s
well as the lower catalytic efficiency of tyrosinase on mono-
phenols than on-diphenols
(8).
Fur thermore , the appearance
of a agperiod has been reported
4, 14, 47-51).
This ag
period depend s on the enzyme and tyrosine concentrat ions,
as has been described from a simplified model
of
Scheme
IV
for frog epidermis tyrosinase
52).
In th i s paper
52)
nei ther
was the oxygen consum ption aken ntoaccount ,nor he
turnover of the enzyme in the pathwa y established. For this
reason no analytica l expression was derived for the mo no -
phenolaseactivity involving the hre esubstrates yrosine,
oxygen, and dopa.
The aim of this paper is the quant i tat ive characterizat ion
of thekin etic behavior of th e monophenolase activity of
tyrosinase. The react ion mechanism (Scheme
V)
involves all
the essen t ial s teps of the c atalyt ic cycle (Schem e 111) and is
coupled to the nonenzymatic react ions fromo-dopaquinone-
H' that yield dopachrome and regenerated L-dopa. The reli-
abi li ty of this react ion m echanism and the establ ishmen t of
the enzymat ic tu rnover in the pa thway has beenverified by
experimentalandsimulation assays. In hi s way, a valid
analytica l expression
for
the s teady-state rate
of
the mono-
phenolase activity
of
tyrosinase
is
derived for the fi rs t t ime
in he i terature.T h ecorrespondingkinet icconstants are

8/12/2019 Analysis of a Kinetic Model for Melanin Biosynthesis Pathway
http://slidepdf.com/reader/full/analysis-of-a-kinetic-model-for-melanin-biosynthesis-pathway 3/10
Kinetic
M e c h a n i s m
of
T y r o s i n a s e
dete rm ined and their physical significance discussed. In ad-
di t ion,as egards he elat ionship between theanalyt ical
expressions obtained or the m onoph enolase and d iphenolase
activities, the lower catalytic efficiency of the enzyme on
monophenols than on d iphenols i sxplained.
EXPERIMENTALPROCEDURES
Materials-Mushroom tyrosinase (3300 units/mg), tyrosine, and
dopa were purchased fromSigma. All other chemicalswere of analyt-
ical grade and suppliedby Merck. Mushroom tyrosinase was purified
by the procedure of Duckworth and Coleman (33). Protein concen-
trat ion was determined by a modified owry method (53). Th e enzyme
concentration was calculated takinga value of
M ,
120,000.
Kinetic Assays-The dopachrome accumulation was spectropho-
tometrically followed at 475 nm c = 3600 M-' cm ) using a Perkin-
Elmer Lambda-2 spectrophotometer interfaced on-line with an AM-
STRAD PC2086 computer. Th e reaction medium was 10 mM sodium
phosphate buffer, pH 7.0. Toestimate hekineticparameterson
tyrosine, care was taken hat recordings reached steady-s tate cor-
rectly, with no significant consumption of substrate, suicide inacti-
vation, or dopachrome breakdown. This aspect was solved by the
addition to the reaction medium of a [Dl0
<
[D],,/[qo, as well as
shortassays imes. neachassay he act that he pathway had
reached the steady-sta te was checked by quantifying the [Dl accu-
mulated in the reaction medium. Th is was done by measur ing the
absorbance increase a t 475 nm produced at each reaction time after
addition of 2 mM NaIO,.
HPLC Assays-Oxidation of tyrosine with tyrosinase in the pres-
ence of different amounts of dopa was carried out n a Beckman
System Gold liquid chromatography system equipped with a pump
model llOB for socratic elution, a programmable 168-diode array
detector (monitoring 250 nm; scanning 200-600 nm) , and a system
gold IBM PS/2 model 5.1. Samples were introducedvia a fixed-
volume injector (20 pl) Rheodyne. The compounds were sepa rated in
an Ultrasphere-ODS 4.6 X 250 mm; i.d. 5 pm) reversed-phasecolumn
and eluted at flow rate of 1 ml/min with ammonium acetate 50 mM
and Na2-EDTA, mM, adjusted
at
pH 3.0,25 C. The different peaks
were characterized by their absorption spectra. The purity of the
peaks was determined by the Real Time Purity Algorithm of the 168-
diode array detector.Th e values of [ T I Dl, and DC] were quantified
by interpolation of the peak areas on the linear calibration curves
obtained from their standards, that for dopachrome being obtained
by the oxidation of dopa with Na I0 4 in a 1:2 stoichiometry (dopa/
NaI04).
Oxygen Determination-Oxygen consumption was followed by a
Hansatech DWoxymeter, based on the Clarklectrode. Temperature
was controlled at 25 C using a Haake D1G circulating bath with a
heater/cooler and checked using a Cole-Parmer digital thermomet er
with a precision of kO.1 C.
Simulated Assays-The kinetic behavior of the reaction mecha-
nism is described by a system of differential equations, whose nu-
merical integration was carried out by using the predictor-corrector
algorithm of Adams-Moulton, starting with a fourth order Runge-
Kutta method (54). Th e algorithm was implemented andcompiled in
TurboBASIC 1.0 on a n I NVES PC-640A computer (IBM AT-com-
patible) with an Int el 80287 arithmetic coprocessor.
The reaction mechanism of the monophenolase activityf tyrosin-
ase (Scheme IV) involves the di fferentia l equations as ollows.
[&net1 =
k-JEmetTI + k-,[EmeDI + k,[EoxyD]
-
(k l [q + kz[Dl)[Emet]
[Eredl =
M E m e D I
+ k-~[Eoxy]
-
k,[Oz][Ered]
[ E m d l= kn[E,,t][D]
+
kt,[Eoryq
- (k--8 +
ks)[EmetD]
[Zrnetq =
kl[q[Emetl
-
k-JEmetq
[EOXY] k-4[EoxyTj + k-e[&x$I ks[Ered][Oz]
-
(k4[rrl+ k[Dl + k-d[EoxyI
[EoxyD]k~[Eoxy][D]- k-6
+
k~)[EoxyDl
[ B o w r r l = ~4[Eoxy1[q (k-4 + k ~ ) [ E o x y T I
4QHl = kdEmetD1+ k7[Eox,Dl
-
kapp[QHl
P C 1
= kaPp[QM/2
[bl
=
k--8[EmetDl+ k-e[EoxyD]
-
(kz[E,.t]
+
k6[Eox~1)[Dl
+
(kapp[QW/2)
The initial conditions are [E], = [Ern& [E,,,],, [ T I
=
[ T I o [Dl =
[Dl,, [OP]O 0.26 mM and [E,,t]~/[E,,y]o = 9O:lO. The [ T I was
considered constant throughout the simulations, in accordancewith
experimental data.
Th e mechanism of the diphenolase activity of tyrosinase (Scheme
11) is described by the following system of differential equations.
The initial conditions are E], = [Ern&+ [E,,,]o, [Dl = [Dlo, 02]0=
0.26 mM and [E,,t]o/[E,,y]o = 9O : lO . Th e [Dl was considered constant
throughout the si mulations, in ccordance with experimental data.
Assignment
of
Constants-The values of the equilibrium and rate
constants of the model were assigned by taking nto account he
kinetic analysis of the mechanism s as well as he experimen tally
determined kinetic parameters. By nonlinear regression of the Vo
values versus [Dl, and
[ g o ,
the parameters
K g ,
VgaX,
Z ,
and
V z e x
were determined. The fitt ing f the integratedMichaelis equation for
oxygen consumption gave
K?.
From VE., the catalytic constant k3
was determined. For theMichaelis constants of
Eoxy
n tyrosine and
dopa he magnitude order was taken from he iterature (8). kapp
corresponds to the processes of protonation-deprotonation of o-do-
paquinone-H+, cyclization, andheurther xidation-reduction,
which yield the formation of dopachrome and the regenera tion of
dopa in themedium (18). Thus, the setf values for he rate constants
of the reaction mechanisms f tyrosinase was obtained (Table I).
RESULTSANDDISCUSSION
Stoichiometry
of
the Pathway-The mela nin biosynth esis
pathway from tyrosine to dopach rome consis t of enzymatic
react ions of tyrosina se on tyrosine and on dopa yielding
o-
dopaquinone-H', which evolves nonenzy matically tow ard do-
pachro me (Schem e I) . T he verall s toichiometry of the pa th-
way involves, therefore, the catalyt ic turnover of the enzy-
mat ic reac t ions aswell as further nonenzymatic s teps. Thus,
the conversion of tyrosine up to dopach rome isefined by the
following mass balance (Scheme IV);
7'+ E,,,
+
2H+ D + E,,, + H20
D
+
Erne,
QH +
Edeory
+ 2H'
0 2 + Edeoxy E,,,
T + E,,, + 2H' D E,,, H20
D
+ E,,,
QH + Edeory
+
2H'
0 2 +
Ed eo x y
E,,,
2 Q H * D + D C + H +
D
+
Eo,,
+
4H' QH
E,.t
+
2H' 2H20
D
+
E m e t -+QH + Ed eo ry + 2H'
0 2
+
&eoxy E,,,
2 Q H + D + D C + H +
27'
30, +
2DC
+
2H+ 4H20
whereas the mass balance for the conversion of d opa up to
dopachrome (Sche me 11) is as follows.
D
+
Erne, QH + Edeory 2H+
0 2
+
Edeoxy E,,,
D
+
Eoxy 4H+ QH + E,,, + 2H' + 2H,O (2)
2QH D + DC + H'
D
+
0-8
4 U C
+
H'
+
2H20
8/12/2019 Analysis of a Kinetic Model for Melanin Biosynthesis Pathway
http://slidepdf.com/reader/full/analysis-of-a-kinetic-model-for-melanin-biosynthesis-pathway 4/10
3804 Kineticechan ism of Tyrosinase
TABLE
Values
of
the rate constants used or simulation
of
the melanin
biosynthesis pathway, taking into consideration the reaction
mechanism
of
tyrosinase
Michaelis
constants
Transformation
constantsonstants
K I = 1.9
X
lo-' M k l = 5.5
x
l o7
M-'
s-' kZi= lo's-'
k-l = 1.0 X 104
s-1
k s = 10
s-'
KE
=
1.4
X
M
k,
=
7.3
X
l o7
M-I
sC1
ki
=
l o 3
-'
k-, =
1.0 X
lo s-' k,, =
0.41 s-'
KZxy= 1.0 X M
k4
= 2.0 X 10 M-I s-'
k-' = 1.0 X lo3 s-'
~t~~1.2 x 10-6 M k6 = 1.6
x
109 M-1
s-1
K?
= 1.9
x
M k s =
5 .5 x
10'
M - I s-1
k-6
=
1.0
X 103
8-1
k-s = 1.0 X 103
s-l
Th e melanogenesis pathway from tyrosine s tarts with the
monophenolase activity of tyrosinase Sche me V), which
consist of two catalytic cycles overlapping through three com -
mon interm ediates . The s toichiome try of the pathw ay (bal-
ance
l )
implies tha t one molecule of tyrosinase m ust accom-
plish two turnovers in the hydroxylase cycle for each one in
the oxidase cycle (Schem e IV). Thus, yrosinaseopera tes
three t imes, twice a nd once thro ugh the s teps control led by
k3, k g ,
an d
k7,
respectively. Th erefore , in the steady -state of
the pathwa y, the ol lowing rate rat ios mustbe fulfilled.
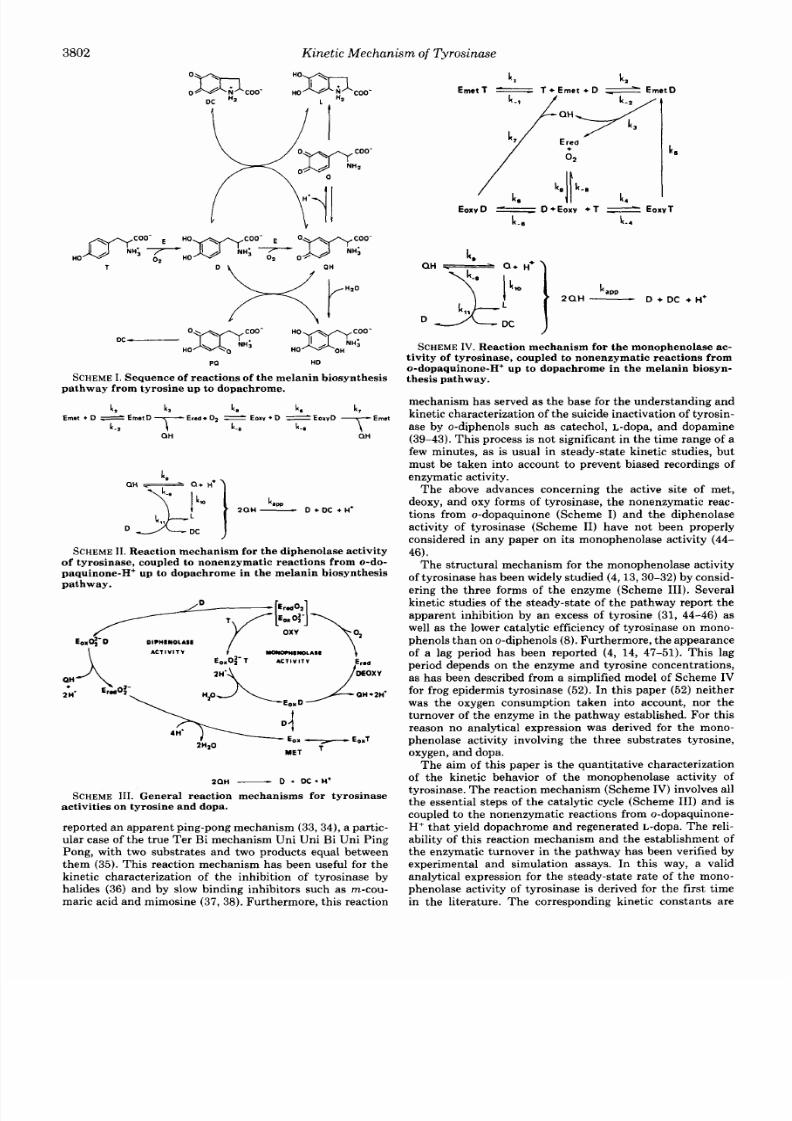
k,[Eme,Dl = (3/2)kdEoxsr l = [&&I ( 3 )
T he reliability of these ratios has bee n erified from simu -
lation assays (Fig.
1, A-C)
in the final s teady-state of the
pathway (from
600 s).
At
a
shorter t ime range
(2.0
s) there is
an early s teady-state restricted to theydroxylase cycle (Fig.
1A)
with
k3[E,,tD]/k5[E,,,~1.0,
which evolves toward the
overal l s t eady-s ta te of the pa thway with k3 [E , , t~ ] /k 5[ &, , ,~
=
1.5.
On the other hand, the melanogenesis pathway from
dopa tarts with the iphenolase ct ivi ty of tyrosinase
(Scheme 11),defined only by the oxidase cycle and on esingle
turnover balance 2) . In hestead y-state of thepathway,
therefore, the rate rat ios verified as follows,
k,[EmetDl =
k 7 [ E o x y D ] ,
(4)
in accordancewith simulationassays (Fig.
1 D ) .
T h e a t e
rat ios (Equat ions
3
a n d
4)
are useful in the derivat ion of th e
steady-state rate equat ions for the catalyt ic act ivi t ies of ty-
rosinase (see "Appendix").
The s toichiome try of the melan in biosynthesis pathw ay
from tyrosine (balance
1 )
and from dopa (balance
2)
implies
thatV$/Vgc = 1.5 and V&/V& =
1.0,
respectively, values
also obtained in experimental (Fig.
2 A
and simulat ion (Fig.
2B)
assays. Thes e results Figs.
1
an d
2)
sup po rt the eliability
of the react ion mech anisms roposed for the monophenolase
(Scheme IV) and d iphenolase (Scheme
11)
activities of tyro -
sinase, involved in the pathw ay u nder s tudy (Schem e I) .
Accumulation of [Dl,-The ope ratio n of th e melanog enesis
pathway from tyrosine (Scheme IV) can be monitored
at 475
nm, and showsa t ransie nt pha se hat evolves toward he
overal l s teady-state of the pathw ay,with linear produ ction of
dopachrome (Fig.
3A).
Th is process can also be followed in a
discont inuous way by t i t rat ion of the [Dl accu mulated in the
assay medium w ith Na I04. Thu s, a s igmoid pat tern of
[Dl
uersus t ime, whose final plateau corresponds to the level
of
[Dl,, is obtaine d (Fig.
3A) ,
s imul taneous ly wi th he inear
formation of dopachrome. Th e [Dl ,, i s not depen dent on E],
(Fig.
3 B ) ,
whereas i t is proport ional to
[ T I o
(Fig.
3 B ) ,
[Dlss
=
R [ r l s S R[ , ( 5 )
since the consum ption of tyrosine is negligible during he
assay time (Fig.
3A).
The same behavior and dependencies
were obtained from simulat ion assays (resul ts not shown).
The t ransient phase of the pathw ay (Fig.
3A)
involves the
regenerat ion of dopa in the nonenzy matic react ions from
o-
dopaqu inone-H +, as well
as
the reported competi t ion of ty-
rosine and dopa on the
Emet
n d
E,,,
forms
14)
(Scheme IV),
which is consis ten t w ith the nondep enden ce of
[Dlss
on
[El0
(Fig.
3B) .
Thus , the increas ingevels of [Dl remove
Emet
rom
~
0 600 1200
t
(SI
0
600
1200
t s )
0 50 100
t
(SI
FI G . 1. Evolution with time
of
severa l rate ratios of the
catalyt ic
cycle. A-C, monophenolase activity (Scheme IV), with 0.2
mM tyrosine, 0.26 mM
O,,
and 0.1 P M tyrosinase. D, iphenolase
activity (Scheme
II),
with
0.2
mM dopa, 0.26 mM
0 2 ,
and
0.1
y M
tyrosinase.
8/12/2019 Analysis of a Kinetic Model for Melanin Biosynthesis Pathway
http://slidepdf.com/reader/full/analysis-of-a-kinetic-model-for-melanin-biosynthesis-pathway 5/10
Kineticechanism
of
Tyrosinase 3805
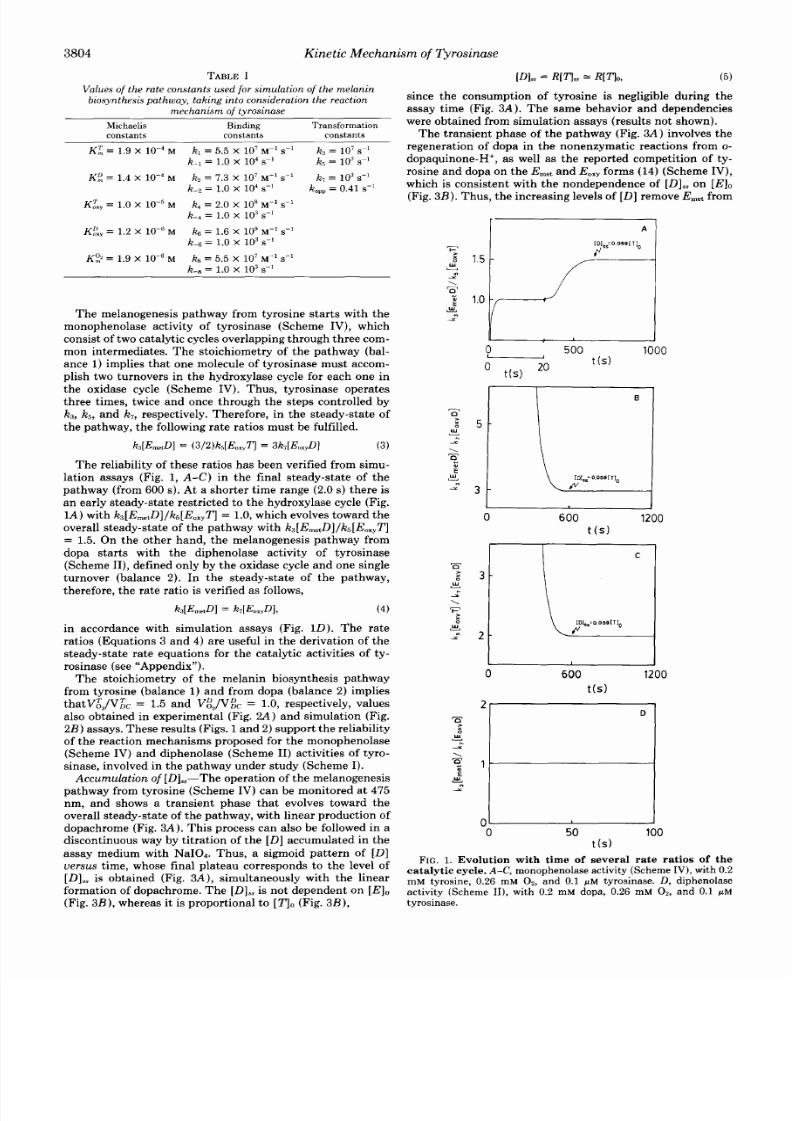
2
V
>
\
P
2
>
\
9
1
6
O) CD l , mM)
FIG. 2.
A ,
experimental results of relation between steady-state of
2 consumption and dopachrome formation:
0 ,
dependence on [Dlo ,
with [E10 = 1.6 nM; A, dependence on [qo ith [ E ] ,=
5.0
nM; B ,
relation between steady-state rates
of
0,
onsumption and dopa-
chrome formation obtained by simulation: 0 , dependence on [Dl0 ,
with [ E ] = 0.1
PM;
A, dependence on [qo,ith
[E l o
= 0.1 FM.
the dead-en d omplex
EmetT,
ielding
E,,,&,
which is involved
in the xidase cycle (Scheme
IV).
This increases the operat ive
[E] s wel l as the form ation of o-dopaquinone-H+ and do-
pach rom e up to the at tainm ent f the overal l s teady-state of
the pathw ay Fig. 3 A ) .Therefore , themon'ophenolase activity
of tyrosinase (Scheme IV) s tarts with the ingle operat ion of
the hydroxylase cycle an d evolves toward the s teady-state,
with two tur nov ers in the hydroxylase cycle for each one in
the oxidase cycle, as wel l as a ne t decrease in the early EmetT]
due to the regenera t ionf dopa in the nonenzymat ic reac t ions
from o-dopaquinone-H+. For this reason, t higher values of
[7 l0
there i s an n crease n he ear ly
[EmetTJ,
nd grea ter
values of [Dlssare required for the at ta inm ent of the s tea dy-
sta te of the pathw ay Fig.
3 3 ) .
Th e removal of
Emet
rom the dead-endomplex EmetTould
be c arried o ut by any dipheno l presen t in the assay medium
such as d opa or eukodopachrome, according to the ir respec-
t ive levels in the s teady -state of the pathw ay (Schem e IV).
The quan t i ty o f [Dl,,as been experimental ly determined
(Fig. 3, A a n d B ) , whereas leukodopachrome has not been
detected in the react ion medium 18, 4). T he value of [L],,
can be calcu lated from the ol lowing.
[LI = k1o[Q1
-
~ I I [ Q W [ L I0
6 )
Therefore
(24),
I I
0.11
I
0
1.0 2 .o
0 )
TI, mM)
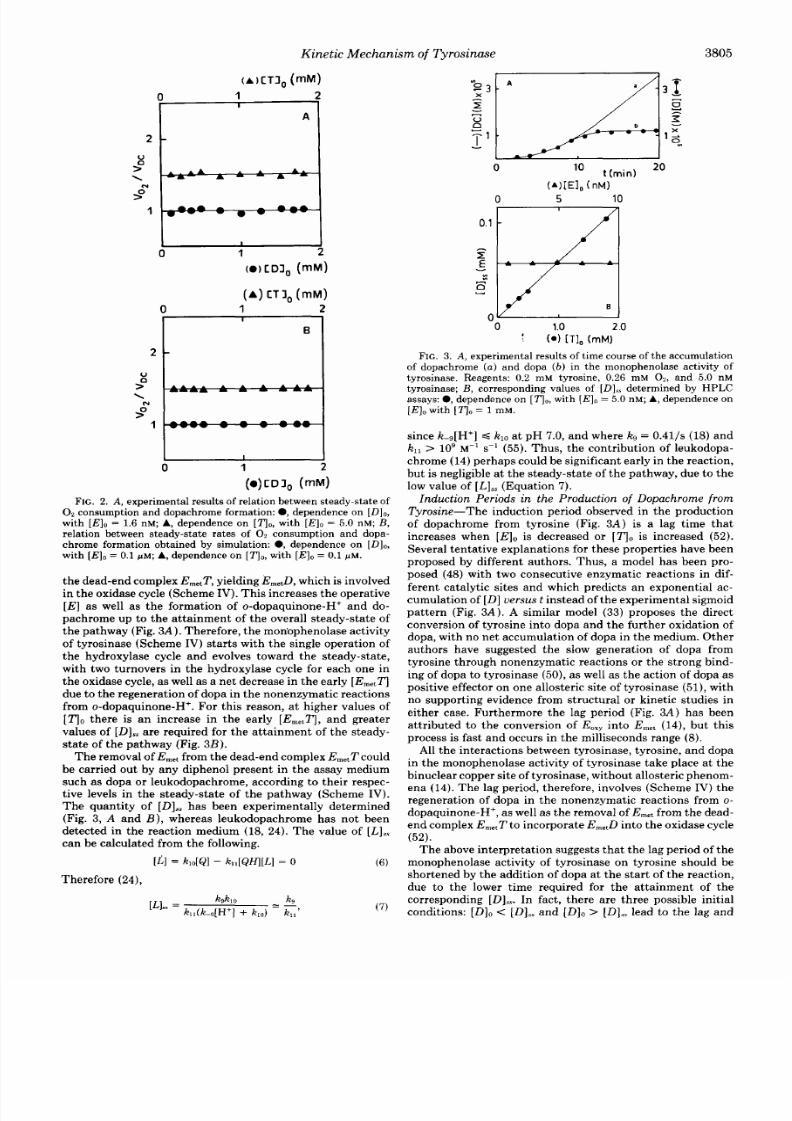
FIG.
3.
A ,
experimental results of time course of the accumulation
ofdopachrome
a )
and dopa ( b ) in the monophenolase activity of
tyrosinase. Reagents: 0.2 mM tyrosine, 0.26 mM 0 2 , and 5. 0 nM
tyrosinase;
B ,
corresponding values of
[Dl,,
determined by HPLC
assays:
0 ,
dependence on [TI,, ith ( E ] ,= 5.0 nM; A , dependence on
[E l o
with [ T I o = 1 mM.
since k-9[H']
s ,,
a t p H
7.0,
and where
k,= 0.41/s (18)
an d
k > lo9 M
s-'
(55).
Thu s, the con tributio n of leukodopa-
chrome
(14)
perhaps could be s ignificant early n the react ion,
but isnegligible
at
the s t eady-s ta te f the pa thway , d ue to the
low value of
[L],,
Equat ion
7).
Induct ion Periods in the Product ion
of
Dopachrome
f r o m
Tyrosine-The induction period observed in the pro duc tion
of dopachrome rom tyrosine (Fig. 3 A ) i s a ag t ime hat
increaseswhen
[El0
is decreased or
[ T I 0
is increased
(52).
Several tentative explan ations for these prope rties have been
proposed by different authors . Thus, a m odel h as been pro-
posed
(48)
with two consecutive enzym atic reaction s in dif-
ferent catalyt ic s i tes and which predicts an exponent ial ac-
cum ulation of [Dluersus t instead of the exp erim enta l igmoid
pat tern (Fig. 3 A ) . A similar model
(33)
proposes th e direc t
conversion of tyrosine into dopa and the further oxidat ionf
dopa, with no net accumulat ion f dopa in the medium. Other
authors have suggested the slow generation of dopa from
tyrosine through no nenzymatic react ions or the s t rong bind-
ing of dopa to tyrosinase 50), s well as the act ion f dop a as
positive effector on one allosteric site of tyrosinase
(51),
with
no support ing evidence from structural or kinet ic s tudies in
ei ther case. Fur therm ore the lag period (Fig.
3 A )
has been
at t r ibu ted to the conversion of
E oxy
nto
E,,,
(14),
but th i s
process is fas t and occurs in the milliseconds range
(8).
All the interac t ions etween tyrosinase, tyrosine, and dopa
in the monophenolase act ivi ty of tyrosinase take place a t th e
binuclear coppersite of tyrosinase , withou t llosteric phen om-
ena
(14).
The lag period, therefore, involves (Scheme
IV)
t h e
regeneration of dop a in the nonenz ymatic react ions from
o-
dopaquinone-H', as well as the emoval of
E,,,
f rom the dead-
end complex
E,,,T
to incorporate
E,,,D
into the xidase cycle
(52) .
T he above interpretat ion suggests th at th e ag periodof the
monophenolase activity of tyrosinase on tyrosine should be
shortened by the addi t ionof dopa a t the s t ar tf the react ion,
due to he lower t ime required or the at tainm ent of the
corresponding
[D],,.
n fact , there are three possible ini t ial
conditions:
[Dl0< [Dl,,
a n d
[Dl0> [ Dl s s
ead to the l ag and
8/12/2019 Analysis of a Kinetic Model for Melanin Biosynthesis Pathway
http://slidepdf.com/reader/full/analysis-of-a-kinetic-model-for-melanin-biosynthesis-pathway 6/10
3806
Kineticechanism of Tyrosinase
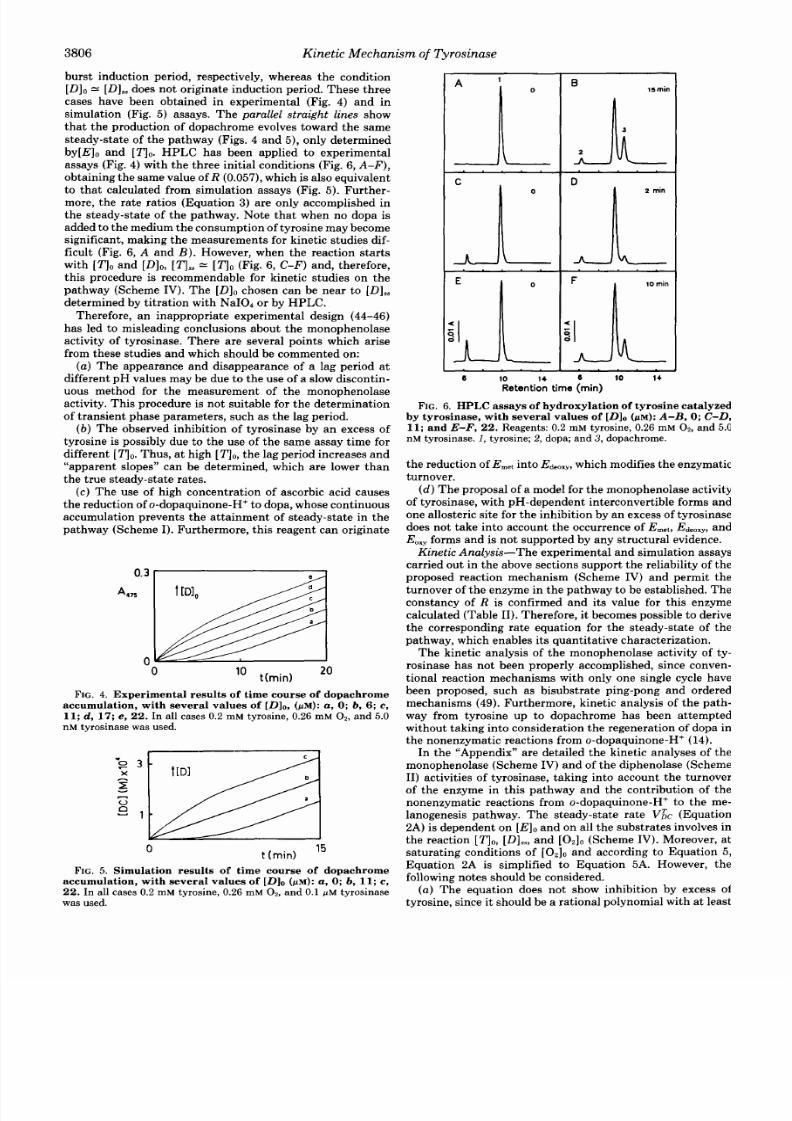
burst nduction period, respectively, wh erea s the con dition
[Dl0
=
[Dls3does not originate induct ion period. These three
caseshave beenobta ined nexperimental (Fig.
4)
a n d n
simulation (Fig.
5 )
assays. The
parallel
straig ht lines show
that the prod uct ion of dopachrome evolves towa rd the same
steady-state of the pathwa y (Figs.
4
an d 5), only d etermined
by[E]o and
[q0.
PLC has beenapplied toexper imenta l
assays (Fig. 4)with the three ini t ial condi t ionsFig.
6,
A-F),
ob ta in ing the samealue of
R
(0.057),
which is also equiva lent
to that calculated from simulat ion assays (Fig. 5 ) . Fur ther-
more, the rate rat ios (Equat ion
3)
are only accomplished in
the s t eady-s ta te o f the pa thw ay. Note tha t whe n no d opa i s
added to themedium the consum ption f tyrosine may become
significant, making the mea surem ents for kinetic studies dif-
ficult (Fig. 6,A a n d B ) . However, when the reac tion
starts
with
[q0
nd [Dl0,
[ l ‘ ls8 [q0
Fig.
6, C -F )
and , herefore ,
thi s procedure is recommendable for kinet ic s tudies on the
pathway (Scheme IV). The [D l0 chosen c an be n ear to [Dlss
determined by t i t rat ion with NaI04
or
by HPLC.
There fore, an nappropriate experim ental design (44-46)
has led to misleading conclusions about the monop henolase
act ivi ty of tyrosinase. There are several points which arise
from these s tudies andwhich shou ld be comm ented on:
a)
The appearance and disappea rance of a lag period
at
different p H values may be due to th e se of a slow discontin-
uous method for themeasu reme nt of themonophenolase
activity. Thi s procedure is not sui table for the determ inat ion
of t ransient pha se para meters , su chs the lag period.
( b )
The observed inhibition of tyrosinase by an excess of
tyrosine is possibly due to the se of the sam e assay t ime
or
different
[TI,.
hus,
at
high
[T I o ,
the lag eriod increases and
“appare nt s lopes” can be determined, which a re lower than
the t rue s t eady-s ta te ra tes .
(c) Th e use of high conc entrat ion of ascorbic acid causes
the reduct ion of o-dopa quinone-H + to dopa,hose continuous
accum ulat ion preven ts the at tainment of s teady-sta te in th e
pathway (Scheme
I).
Fur thermore , th i s reagen t can or ig inate
“0
lo
t (min)
20
FIG.4.
Experimental results of time course of dopachrome
accumulation, with several values of [Dl0, pM):
a, ;
b, 6; c,
11;
d ,
17;
,
22.
In all ca ses 0.2 mM tyro sine , 0.26 mM
02
n d
5.0
nM tyrosinase was used.
0
t (m i d
15
FIG.5.
Simulation results of time course
of
dopachrome
accumulation, with several values of [Dl0
(pM): a ; b, 11; ,
22.
In all cases
0.2
mM tyrosine, 0.26 mM
02
n d
0.1
p M
tyrosinase
was used.
6
10
14
D
6 10 14
Retention time min)
FIG. 6.
HPLC assays of hydroxylation of tyrosine catalyzed
by tyrosinase, with several values
of [Dl0
( p ~ ) :
-B,
0;
C-D,
11; and
E-F, 22. Re age nts: 0.2 mM tyro sine , 0.26 mM
02
nd 5.0
nM tyrosinase. 1, tyrosine; 2, dopa; an d 3, dopachrome.
th e reduction of Emetnto Edeoxy, hich m odifies the enzymatic
turnover.
d )
Th e proposal of a model for th e monophenolase activity
of tyrosinase, with pH-de pende nt in terconv ertible forms and
one allosteric site for the inhib ition y an excess of tyrosinase
does not take into acc oun t the ccurrence of Emet,Edeoxy,nd
Enryorms and s no t suppor tedby any struc tural evidence.
Kinetic Analysis-The exp erim enta l and sim ulatio n ssays
carried ou t in the bove sect ions support theeliability of the
proposed react ion mechanism (Scheme IV) an d perm it the
turnover of the enzyme in the pathw ay to be establ ished. The
constancy of
R
is confirm ed and i ts value for this enzyme
calculated (Table 11).Therefore, i tbecomes possible to derive
the corresponding rate equat ion for the s tead y-state of the
pathway, which enables i ts quant i tat ive charac terizat ion.
The k inet ic analysis of the mo nophen olase act ivi ty of ty-
rosinase has not been properly accomplished, since conv en-
tional reaction mechanism s with only one single cycle have
beenproposed, such as bisubstrate ping-po ng and ordered
mechanisms (49). urthermore, kinet ic analysisof the pa th-
way from tyrosineup odopachrome has been at tempted
without taking into considerat ion the regenerat ionf dopa in
the n onenzy matic react ions rom o-dopaquinone-HC(14).
In the “Appendix” are detai led the kinet ic analysesof the
monophenolase (Scheme IV) and of the diphenolase Scheme
11)
activities of tyrosinase, taking into account the turnover
of the enzyme in this pathw ay and the contribut ion of the
nonenzymatic react ions from o-d opaqu inone-H + to the me-
lanogenes is pa thway . The s teady-s ta te ra te V& (Equa t ion
2A) is de pendent on
[Elo
nd on a l l the subs t ra tesnvolves in
the reaction
[ T I o , [Dlsb,
n d
[OZlO
Scheme IV). Moreover, a t
saturat ing condi t ions of [O2 lO and ccording to Equat ion
5 ,
Equation 2A is simplified toEquat ion 5A. However, the
following notes should be onsidered.
a )
The equa t ion does not show inhibi t ion by excess
of
tyrosine, since it shou ld be rational polynomial with at least
8/12/2019 Analysis of a Kinetic Model for Melanin Biosynthesis Pathway
http://slidepdf.com/reader/full/analysis-of-a-kinetic-model-for-melanin-biosynthesis-pathway 7/10
Kinet icechani sm
of
Tyrosinase
3807
1:2
degree in tyrosine.
( b )
The k inet i c cons tan t L,, (Equat ion
6A)
a n d K ; f , Equa-
t ion
7A)
have the same denominator, as corresponds to an
inhibi tor s toichiometric with the substrate.
c ) Th e analyt ical expression of
k z t
(Equat ion
6A)
is ap-
parent , s ince
kZt
depend s not only on rate constants but lso
on equi librium constants , whe reas kELdepends only on rate
cons tan t s (Equat ion 14A).
TABLE
1
Values of the k inet ic constants f the melanin biosynthesis
pathway determined from experimental data andalculated
f rom s im u la t i on da ta
constantsxperimentalataimulationata
Kinetic Value determined from Value calculated from
0.18 0.02
0.20 0.01
0.13 0.01
0.12 0.01
1.87? 0.20
1.97 0.11
(1.28 0.06) X lo-
(0.99
0.05)
X lo-*
(1.75 0.10) X
10
(1.50 0.04) X
10
8.03 0.10
6.24
0.05
107.40 1.70
90.20 1.60
(5.70
?
0.04)
X
lo-
(5.96
k 0.02)
X
lo-
(5.30 1.50)
X lo-
(3.96 0.83)
X lo-
"Values referred 'for active m onomer
(3).
0.4
0 1o 2.0
[Dl, mM)
[Dl, m M )
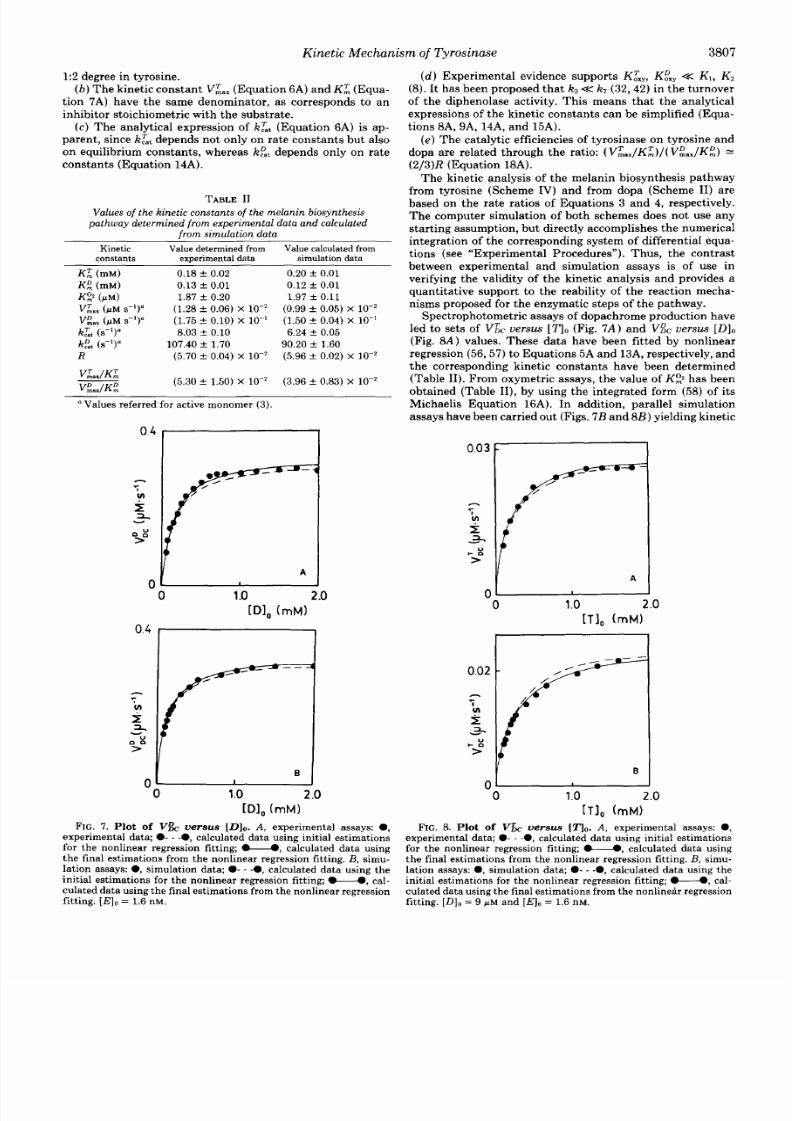
FI G . 7. Plot
of
V versus [Dl,,. A , experimental assays: 0 ,
experimental data; 0- -0, alculated data using init ial estimations
for the nonlinea r regression fitting;U alculated data using
the final estim ations from the nonlinea r regression fi t t ing. B, s imu-
lation assays: 0 , imulation data; 0 - -0, alculated data using the
initial estim ations for the nonlinea r regression fitting; . - cal-
culated datausing the final estimations from the nonlinear egression
fitting. [El 0= 1.6 nM.
d ) Experimental evidence supports KTxy,
K & <<
K1, K z
(8).
t has been roposed that k3
<< k7 ( 3 2 ,4 2 )
in the turnover
of the diphenolase activity. This mean s that the analy t ical
expressions of the k inet i c cons tan t s can beimplified (Equa-
t ions
SA, SA, 14A,
an d
15A).
(e ) T he catalyt ic efficiencies of tyrosinase on tyrosine and
dopa are related through the ratio:
(V L , J K ; f , ) / ( V k x / K g )
(2/3)R
(Equat ion
18A).
The kinet ic analysis
o f
the melan in b iosynthes i s pa thway
from tyrosine (Scheme IV) and from dopa (Scheme 11) are
based on the rate rat ios of Equat ions
3
a n d 4, espectively.
The com puter s imulat ion of both schemes does not use an y
start ing assumption, butdirectly accomplishes the num erica l
integrat ion of the correspo nding system f different ial equa-
t ions (see "Experime ntal Procedures"). Thu s, he contr ast
between experimentaland imulat ionassays is of use in
verifying th e validity of the kine tic analysis and provides
a
quant i tat ive support to the reabil ity of the react ion mecha-
nisms proposed for the enzym atic s teps
f
the pathway.
Spectrophotometric assays f dopachrome product ion have
led
to se t s of
V 6 , uersus [ T I o
(Fig.
7A)
a n d
V& uersus
[Dl0
(Fig. S A ) values. These data have been fitted by nonlinear
regression
(56 ,57)
to Equat ions
A
a n d
13A,
respectively, an d
th e corresponding kinet ic constants havebeen determined
(Table
11).
From oxymetric assays, the value of KII: has been
obtained (Table II) , by using the integrated form
(58)
of its
Michaelis Equation 16A). Inaddition,paral lel imulat ion
assays have been carried out Figs.
7 B
a n d 8 B )yielding kine tic
0.03 1
0
0
1.0
2.0
[ T I , mM)
v
B
0
0 1.o
2.0
[ T I , mM)
FIG.
8.
Plot
of
V versus [m .
,
experimental assays: 0 ,
experimental data; 0 - -0, alculated data using init ial estimations
for the nonlin ear regression fitting;
M
alculated data using
th e final estimations from the nonlinea r regression fi t t ing. B , simu-
lation assays:
0 ,
simulation data;
0- -0,
alculated data using the
initial estimations for the nonlinear regression fitting;
.
cal-
culated datausing the final estimations from the nonlinearegression
fitting.
[Dl0
=
9
p M an d [El0= 1.6 nM.

8/12/2019 Analysis of a Kinetic Model for Melanin Biosynthesis Pathway
http://slidepdf.com/reader/full/analysis-of-a-kinetic-model-for-melanin-biosynthesis-pathway 8/10
3808 Kineticechanism of Tyrosinase
cons tan t s s imi lar to tha t ca lcu la ted f rom exper imenta l assays
(Table 11).No te the verification (Ta ble
11)
on the simplified
expressions corresponding to
Vz,,, KE,
V”,.,,
K g
a n d
K
regarding Equat ions 8A, 9A, 14A, 15A, a n d 17A, respectively.
T he expression KE = K , is equivalent to a t rue inhibi t ion
cons tant , the dissociat ion con stant of the dead-en d complex
EmetT,
hereas
K f ; l = K 2
s thedissociation con stan t of a true
enzyme-substrate complex EmetD Scheme IV). In both cases,
th e overall affini ty of tyrosinase towa rd tyrosine and dopa is
determined by
E,,,,
with lower affini ty than
Eoxy
8) . Fur-
thermore, k g , = k3,whereas k:, = ( 2 / 3 ) k 3 ( K l / K 2 ) R ,evealing
t h a t k:, is not a t rue catalyt ic constant , s ince i t depends on
equilibrium constants
as
well as on the experimental rat io
R
=
0.057.
Note that the alue of k z t is lower tha n tha t of any
rate constants of t ransformation s teps of the react ion mech -
an i sm (Table
11).
T he value of R (Table 11)i s equ ivalen t o the xperimental ly
obtained value (Fig.
3 B )
according to Equat ion 5, and also
fulfills Equation 18A. This low value of
( 2 / 3 ) R
mplies the
lower efficiency of tyros inase on ty rosin e th an o n dopa, due
to the s ignificant port ionof tyrosinase as dead-endcomplex
E,,,T
in the mon opheno lase act ivi ty of tyrosinase (Scheme
IV)
.
In conclusion, the mono phenolase act ivi tyf tyrosinase can
be described by a reaction mechanism (Scheme V) involving
three e nzym atic form s w ith one s ingle act ive s i te, two over-
lappingcatalytic cycles, andonedead-en d complex. Th is
reaction is coupled to a series of none nzym atic reactions rom
o-dopaquinone-H+ which yield dopachrome and regenerated
dopa, unt i l the veral l s teady-state of the pathwa y iseached.
To maintain the s teady-state, thenzyme mu st realize aglobal
turnove r involving two turn ove rs in the ydroxylase cycle for
eachone in he oxidase cycle. Th isdetermines he a t io
between the ratesof the different s teps,which can beused to
deduce an analyt ical expression for the rate in the s teady-
state of the pathwa y, eading to i ts quant i tat ive characteriza -
tion.
Acknowledgments-JosB
Neptuno Rodriguez
Lbpez
has a
fellow-
ship
from
the
Comunidad
Aut6noma de Castilla-La
Mancha.
The
authors are grateful to Dr. Marino Baiibn for
the
technical assistance
in HPLC determinations.
APPENDI X’
Stea dy-s tate Rate for the Monophenolase
Ac tivi ty of Tyrosin ase
Themelan inbiosynthesispathwa y from tyrosinestarts
with monophenolase act ivi ty of tyrosinase (Scheme IV). In
the s t eady-s ta te (Equat ion
) ,
t
is
fulfilled th at
V & I kdEmetD1 + MEoxyDI = 4MEoxyD1,
1.4)
since
2QH
yields 1DC (Scheme IV)
V& = V&/2.
By applying
the s teady-state approach
(58)
to the d i f feren t in termedia te
species and solving the corres ponding system of linear eq ua-
~~ ~
The notation and definitions
are
as follows: T, -tyrosine;D , L-
dopa;
Q H , Q , o-dopaquinone-H’
and o-dopaquinone,
respectively;
L ,
leukodopachrome; DC, dopachrome; H D , topa
(~-2,4,5-trihydroxy-
phenylalanine); P Q , p-topaquinone [5-(2-carboxy-2-aminoethyl)-2-
hydroxy-1,4-benzoquinone]; A oncentration of the
species
X dur-
ing the course of the reaction; [XI ., concentration of the
species X
during the steady-stateof the
reaction; [Ao
nitial
concentration of
the species
X in the assay medium; E ,
tyrosinase;
E,,, oxidized form
of
tyrosinase with
Cui’
in the
active
site; Ered, reduced form
of
tyrosinase
with
Cui
in the
active
site; Emet,mettyrosinase
( E o x ) ;
de oxy ,
deoxytyrosinase (End);E,,,,
oxytyrosinase
(Eredo2
or E&): V ~ C ,
V&,
steady-state rate of the production of DC
from
T and D ,
respectively;
V&,
V&,
teady-state rate
of oxygen
consumption
from
T nd
D ,
respectively;
k ,
i = 1-8),
ate
constants
of the reaction
t ions, the analyt ical expressions for
V&
can be derived as
follows,
where
L Y ~= 2kakk7ks(k5 + k-4)KI
P o
=
ksk-a(k7
+
k-d(k.5
+
k-4lK1
PI =
3k3k~k7(k5
+
k-4)K1
0 2 = ks(k5 + k-,)KI[ks(ki
+
k-6) + 3kckXzI
Pa = kcks(k3 + 3k,)(kz
+
k-,)Kj
P
=
3ksk,ks(ks
+
k-4)KZ
+
k3k4ks(k7 + k-dK1.
(3.4)
Mushroom tyrosin ase is satura ted at 0.26 mM
O 2 ( 3 3 )
a n d
underhese ondi t ions (see “ExperimentalProcedures”),
Equat ion 2A becomes
4.4)
From e xperime ntal and s imulated data (Fig.
3 B ) ,
i t has
been obta ined the linea r ratio: [Dlss= R [ q o (Equat ion 5 ) ,
which can be introduced into Equat ion
A
yielding
Th e above expressions of these overal l kinet ic constants
can be simplified by taking into acc oun t everal exper imental
dat a as follows. a)R = [D] . . / [T10= 0.057 (Fig. 3 B ) ; b )K g , ,
K t x y<<
K , ,
K 2
8);
(c)
k3
<<
k7, k3
being the l imit ing s tep in
the reac t ion mechanismf the diphenolase act ivi tyf tyrosin-
ase (32 , 42) . Therefore, from Eq uat ions 6A an d 7A, VI,, a n d
KE
can be simplified to
V L ( 2/3 )k a( Kd K* )R [E Io
(8.4)
KK
= K ,
(9.4)
due to
(3k7K,K,T.,
+
k 3 K l K f , ) R<< ( 3 k j K z K L ,+ k 3 K l K fx , )
n d
k 3 K , K f x ,< 3k7K2Kgx, ince k3 << k7 an d K I K f x y K 2 K g x , .
mechanism of
tyrosinase;
K ,
=
k , / k - 9 ,
dissociation constant
of the
deprotonation/protonation equilibrium
between
QH and
Q;
klo, cycli-
zation constant
of
Q into L ; kn , rate
constant of the
production
of
DC D from
L +
QH; ,,,,
apparent
constant
for
the transformation
of QH into D DC + H’; K1, K 2 , dissociation constants of Emet
toward T
and
D ,
respectively
( K l = k- , /k l , K2 = k-z/kZ); KTxy,Ki iy ,
Michaelis constants of
E,,, toward T
nd
D ,
respectively
( K Z , = (k-4
+
k s ) / k 4 , K f x y ( k - 6
+
k , ) / 4 ; Vgax,V:ax,
maximal
steady-state rates
of
E
toward
T
nd
D ,
respectively
( V z , .
=
k z t [E]0,V k x= k g , [E],);
KK, K i , Michaelis
constants
of
E
toward T
and
D , respectively;
VL.,/K;I;, VD,.,/KZ,
catalytic efficiencies
of
E
toward
T
and
D ,
re-
spectively.

8/12/2019 Analysis of a Kinetic Model for Melanin Biosynthesis Pathway
http://slidepdf.com/reader/full/analysis-of-a-kinetic-model-for-melanin-biosynthesis-pathway 9/10
Kinet icechani sm of Tyrosinase 3809
Steady-stateateorheiphenolasectivity of Tyro sinase REF ERENCES
The melanin biosynthesis pathway from dopa starts with
diphenolase activity of tyrosinase (Scheme 11). In thesteady-
state, i t is fulfilled that Equation
4
V = k,[EmetD]
+
k,[Eox@l = 2h[EmetD] (IOAI
since
2QH
yields
1DC
(Scheme 11)
V& = V&H/2.
By applying
the steady-state approach
(58)
to the different intermediate
species and solving the corresponding system of linear equa-
tions, the analytical expressions for
V&
can be derived as
follows,
where
=
k3
+
4 k 7
8 -
‘
-
kdk,
+
k7)
kdk7
+
k-6) kiKz
kdk3 + k7) k3
+
k7
8 2 =
Mushroom tyrosinase is saturated at
0.26
mM
O 2 ( 3 3 ) .
Under
these conditions (see “Experimental Procedures”), Equation
1 1 A becomes the following.
Simplifications of
V”,,.
and KE-The above expressions
of
these overall kinetic constants can be simplified by taking
into account several experimental data such as K & <<
Kz
(8)
and k3 << k7 ( 3 2 ,42). Therefore, from Equations 1 2 A to 13A,
V”,,,
and
Kfr:
can be simplified to the ollowing.
V L (k [EIo)/(k3 + k7) kdE10 ( 1 4 4
and
Oxygen Consumption-The
affinity of tyrosinase toward O2
is inversely related with its corresponding Michaelis constant,
which expression can be derived from Equation
1 1 A
at satu-
rating [ D l ovalues as follows,
where
Catalytic Efficiencies of Tyr osin ase on Ty rosin e a nd opa
The catalytic efficiency of tyrosinase on tyrosine
(VLJ
KZ) and on dopa (VD,.JKD,), defined from Equations 8 A to
9 A and 1 4 A to 15A, respectively, are related through the
following expression:
Therefore, since
R
=
0.057
(Fig.
4B),
yrosinase shows a lower
efficiency on tyrosine than on dopa.
1. Prota, G. (1988) Med. Res. Rev. 8, 25-556
2. Langston , J . W. (1988) Trends Pharmacol. Sci .
9,
347-348
3. Robb, D.
A.
(1984) in Copper Proteinsand Copper Enzym es
4. Mason, H. S. (1956) Nature 1 7 7 , 7 9- 81
5.
Jolley, R. L., Jr., Evan s, L. H., and Mason , H.S.
(1972) Biochem.
6. Schoot-Uiterkamp, A. J. M., and Mason,H. S. (1973) Proc. Nat l .
7. Jolley, R. L., Jr. , Evans, L. H., Makino, N., an d Mas on, H. S.
(1974) J . Biol. Chem . 2 4 9 , 3 3 5- 34 5
8. Makino, N., and M ason, H. . (1973)
J.
Biol. Chem.
248,
5731-
5735
9 . Makino, N., McMahill, P., Mason,
H. S.,
and Moss,
T. H.
(1974)
J . Biol. Chem. 2 4 9 , 6 0 6 2 - 6 0 6 6
10. Schoot Uiterkamp, A . J. M., Evans, L.
H.,
Jolley, R. L., and
Mason, H. S. (1976) Biochim. Biophys. Acta 4 5 3 , 2 0 0 - 2 0 4
11. Lerch, K. (1981) in Metal
I ons
in Biological Sys tems (Sigel, H.,
ed) pp. 143-186, Marcel Dekker, Inc., New York
12. Himmelwright, R. S., Eickm an, N. C., Lu Bien, C.
D.,
Lerch, K.
and Solomon, E. I. (1980) J . A m . Chem. SOC. 02, 7339-7344
13. Solomon, E. I. (1981) in Copper Proteins (Spiro, T . G., ed) Vol.
111,pp. 41-108, Wiley-Interscience, New York
14. Wilcox, D. E., Porras, A. G., Hwang, Y. T., Lerch, K., Winkler,
M. E., and Solomon, E. I. (1985) J. A m. C hem.
SOC.
0 7 , 4 0 1 5 -
4027
15. Beltramini, M., Salvato, B., Santa maria , M., and Lerch, K . (1990)
Biochim. Biophys. Acta 1 0 4 0 , 3 65 -3 72
16.
Menon, S.,Fleck, R. W., Yong, G., and Strothka mp,
K.
G.
(1990)
Arch. Biochem. Biophys. 2 8 0 , 2 7 - 3 2
17. Yong, G., Leone, C., and Strothkamp, K. G. (1990) Biochemistry
2 9 , 9 6 8 4 - 9 6 9 0
18. Garcia-Carmona,
F.,
Garcia-Canovas, F., Iborra, J. L., and Loz-
ano,
J.
A. (1982) Biochim. Biophys. Acta
717,
124-131
19. Kalyanaraman,
B.,
Felix, C. C., and Sealey,
R.
C. (1982) Photo-
chem . Photobiol. 3 6 , 5 - 12
20. Land, E. J. , Thompson,A., Truscott , T. G., Subbarao, K. V., and
Chedekel, M. R. (1986) Photochem . Photobiol. 4 4 , 6 9 7 - 7 0 2
21. Cabanes, J. , Garcia-Canovas, F., and G arcia-Carmo na, F. (1987)
Biochim. Biophys. Acta 9 1 4 , 1 9 0- 19 7
22. Garcia-Carm ona, F., Cabanes,
J.,
and Garcia-Canovas, F. (1987)
Biochim. Biophys. Acta
914,
198-204
23. Garcia-Cinova s, F., Garcia-C armona, F., Sanchez, J. , Iborra Pas-
tor, J. L., and Lozano Teruel, J . A. (1982)
J .
Biol. Chem. 257,
8738-8744
24. Rodriguez-Lopez, J. N., Tudela, J., Varon, R., and Garcia-Cano-
vas, F. (1991) Biochim. Biophys. Acta 1 0 7 6 , 3 79 -3 86
25. Wick, M. M. (1979) Cancer Treat.
Rep.
6 3 , 9 9 1 - 9 9 7
26. Jimenez, M., Garcia-Canovas, F., G arcia-Carm ona, F., Tudela,
J., and Iborra , J . L . (1986) Int . J . Biochem. 1 8 , 39-47
27. Serna,
P.,
Rodriguez-Lopez, J. N., Tudela, J.,Varon, R., and
Garcia-Canovas, F.
(1990)
Biochem. J .
2 7 2 , 4 5 9- 46 3
28. Jimenez, M., Garcia-Carm ona, F., Garcia-Canovas, F., Iborra,
J.
L., Lozano, J. A., and Mart inez-Ort iz ,F. (1984)Arch. Biochem.
Biophys. 2 3 5 , 4 3 8- 44 8
29. Garcia-Moreno, M., Rodriguez-Lbpez, J. N., Ma rtinez- Ortiz , F.,
Tudela,
J.,
Varbn, R., and Garcia-Canovas, F. (1991) Arch.
Biochem. Biophys. 2 8 8 , 4 2 7 - 4 3 4
(Lontie,
R.,
ed) , Vol.
11
pp. 207-240, CRC Press , Boca Raton
Biophys.
Res.
C ommun. 4 6 , 8 7 8 - 8 8 4
Acud. Sci.
U .
S.
A.
7 0 , 9 9 3 -9 9 6
30.
Ochiai,
E.
(1973)
Znorg. Nucl. Chem . Lett.
9 , 9 87 -9 91
31. Vanneste, W. H ., and Zuberbuhler, A. (1974) n Molecular Mech-
anisms of Oxygen Activation (Nayaishi, O., ed) pp. 371-404,
Academic Press, New York
32. Ochiai, E. (1985) in Quimica Bioinorgunica,pp. 245-247, Revertb,
Barcelona
33. Duckworth, H. W., and Coleman,
J.
E. (1970) J . Biol. Chem.
34. Gutteridge,
S.,
and Robb,
D.
A. (1975) Eur.
J.
Biochem. 5 4 , 1 0 7 -
116
35. Galindo, J., Pedreiio, E., Garcia-Carmona, F., Garcia-Canovas,
F., Solano, F., and Lozano, J. A. (1983) Znt.
J.
Biochem. 1 5 ,
1455-1461
36. Peiiafiel, R., Galindo, J.
D.,
Solano, F., Pedreiio, E., Iborra, J.
L.,
and Lozano, J . A. (1984)Biochim. Biophys. Acta7 8 8 , 3 2 7 - 3 3 2
37. Cabanes, J., Ga rcia-C arm ona, F., G arcia-Canovas, F., Iborra, J.
L., and Lozano, J. A. (1984) Biochim. Biophys. Acta 7 9 0 , 1 0 1-
107
2 4 5 , 1 61 3 -1 6 25

8/12/2019 Analysis of a Kinetic Model for Melanin Biosynthesis Pathway
http://slidepdf.com/reader/full/analysis-of-a-kinetic-model-for-melanin-biosynthesis-pathway 10/10
3810
Kineticechanism
of
Tyrosinase
38. Cabanes, J. , Garcia-Cinovas,
F.,
Tudela, J. , Lozano,
J.
A., and
Garcia-Carmona, F. (1987) Phytochemistry
26,
917-919
39. Tudela, J. , Garcia-Canovas,
F.,
Varbn, R., Garcia-Carmona, F.,
Galvez, J., and Lozano, J . A. (1987) Biochim. Biophys. Acta
9 1 2 ,4 0 8 -4 1 6
40. Tudela, J., Garcia-Canovas, F., Varon, R., Jimenez, M., Garcia-
Carmona, F., and Lozano, J. A. (1987) J . Enz. Inhibit. 2, 47-
56
41. Tudela, J. , Garcia-Canovas, F., Varon, R., Jimenez, M., Garcia-
Carmona,
F.,
and Lozano, J.
A .
(1988)
Biophys.
Chem.
30,
303-310
42. Garcia-Canovas,
F.,
Tudela, J., Martinez-Madrid, C., Varon,
R.,
Garcia-Carmona,
F.,
and Lozano, J. A. (1987) Biochim. Biophys.
Acta 9 1 2 ,4 1 7 -4 2 3
43. Escribano, J., Tudela, J., Garcia-Carmona, F., and Carcia-Cano-
vas, F. (1989) Biochem. J.
262,
597-603
44. Devi, C. C., Tripathi, R.
E.,
andRam aiah, A. (1987) Eur. J.
Biochem. 166,
705-711
45. Tripathi, R. E., Devi, C. C., and Ram aiah , A. (1988) Biochem. J.
252,481-487
46. Devi, C. C., Tripathi, R. E., and Ram aiah , A. (1989) Pigment.
Cell Res.
2,
1-6
47. Bright, H. J., Wood, B. J. B., and Ingraham , L. L. (1963) Ann.
48. Osaki, S 1963) Arch. Biochem. Biophys. 100, 378-384
49. Vaughan, P. F. T., and But t , V. S. (1972) Biochem. J . 1 2 7 , 6 4 1 -
50. Pomerantz,
S.
H., and Murphy ,V. (1974)Arch. Biochem. Biophys.
51. H earing, V. J., and Ekel, J. M. (1976) Biochem.
J.
157, 549-557
52. Cabanes, J., Garcia-Canovas,
F.,
Lozano, J. A,, andGarcia-
Carmona,
F.
(1987)
Biochim. Biophys. Acta 923,
187-195
53. Hartree, E. (1972) Anal. Biochem. 48, 422-427
54. Gerald, C. F. (1978) Applied Numerical Analysis Addisson-Wes-
ley, Reading, MA
55. Thompson, A., Land, E. J. , Chedekel,
M.
R., Subbarao, K . V.,
an d T ru sco t t , T .G. (1985) Biochim. Biophys. Acta 8 4 3 ,4 9 -5 7
56. Marqua rdt, D. W. (1963) J . Soc. Ind. Appl. Math. 11, 431-441
57. Jand el Scientific (1989) SigmaPlot 4 .0 , Jandel Scientific, Corte
58. Cornish-Bowden, A. (1979) in Fundamentals of Enzyme Kinetics,
N .
Y.
Acad. Sci. 180 ,96 5-966
647
160,
73-82
Madera, CA
pp. 34-37, Buttenvorth & Co., Ltd., London





























