Embed Size (px)
Citation preview

UNIVERSIDAD DE CÓRDOBA
Facultad de Ciencias
Grado de Bioquímica
Trabajo Fin de Grado
Análisis metabolómico de frutos de encina (Quercus ilex)
Có digó del TFG: BQ15-24-BBM
Autor: Cristina López Hidalgo
05/09/2016


pág. 1
Agradecimientos
Durante este año he tenido la oportunidad de aprender y evolucionar, tanto a nivel
personal como profesional. Hace dos años tuve la suerte de ser acogida en este grupo
personas que me han ayudado y acompañado en este proceso. He de decir que no exagero
cuando digo que para mí es como mi segundo hogar (en cual paso mucho más tiempo que en
el primero). A todas las personas que han pasado por este lugar les agradezco su compañía y
sus consejos.
A Jesús, le estoy enormemente agradecida. Me siento muy afortunada ya que me ha
permitido pertenecer a su grupo, me ha motivado a trabajar, y su invalorable paciencia han
hecho posible la realización de esta memoria. Gracias por confiar en mí durante este año.
A Maricarmen, por preocuparte siempre de que las condiciones en el laboratorio
fueran siempre las mejores. Muchas gracias.
No me puedo olvidar de Rosa. De ella puedo afirmar que es la persona más generosa
que he conocido y sin ella, el laboratorio no es el mismo lugar. Te agradezco tu ayuda y sabes
que siempre puedes contar conmigo para cualquier cosa. A su hermana, Silvia y José Antonio
también les agradezco su compañía, que hicieron amenas esas dos semanas larguísimas en el
invernadero.
A Ana Maldonado y Manolo Rodríguez que aunque no he pasado tanto tiempo con
ellos, siempre han estado disponibles para ayudar y que junto a Jesús han hecho los
momentos de descanso muy agradables y en los que he aprendido muchas cosas.
También debo agradecer buenos momentos a Andrés, Jon, Cris, Bárbara, Sabina,
Juanjo, Rocío, Leila, Isa, Daniel, Irene, Omar y a muchas personas que han pasado por el
laboratorio. Espero no olvidarme de nadie. A todas las personas del SCAI debo agradecer su
atención y ayuda.
Por último, a mis padres y hermano por apoyarme en todas y cada una de mis
decisiones, por su inmenso trabajo que valoro mucho, que ha permitido y permite que
continúe formándome.

pág. 2
Índice general
Índice de Ilustraciones ............................................................................................................ 4
Índice de tablas. ...................................................................................................................... 4
Resumen ................................................................................................................................. 5
Palabras clave ......................................................................................................................... 5
Abstract and conclusions ........................................................................................................ 6
Key words ................................................................................................................................ 6
1. Introducción………………………………………………………………………………………………………………….7
2. Materiales y Métodos….……………………………………………………………………………...……………..13
Material vegetal.………..……………………………………………………………………………………..…….14
Morfometría y contenido hídrico.….……..…………………………...………………………….….…….15
Viabilidad.……………………………………………………………………………………………………….………17
Obtención de la harina de bellota.…………………………………………………..…………..………….17
Análisis por Espectroscopía de Infrarrojo Cercano.…………………….……………………….……17
Análisis por Resonancia Magnética Nuclear.…………………………………………………………….18
Extracción de metabolitos……………………………………………………………………………………….19
Cuantificación de metabolitos por técnicas colorimétricas………………………………………19
Análisis por Cromatografía de Gases acoplada a Espectrometría de Masas.……….….…20
3. Presentación de los resultados y discusión….………………………………………….…………..…....21
Análisis por métodos colorimétricos……………………………………….……………………………….22
Análisis por Espectroscopía de Infrarrojo Cercano………………………….……………….……….23
Análisis por Resonancia Magnética Nuclear.…………………………………………………………….26
Análisis por Cromatografía de Gases acoplada a Espectrometría de Masas.………..……29
Relación entre la composición química y las características morfométricas……………..32

pág. 3
Valoración de las diferentes técnicas empleadas………………………………………………….…..33
4. Conclusiones…………………………………………………………………………..…………………………………..35
5. Bibliografía…………………………………….………………………………….……………….……………………….36

pág. 4
Índice de Ilustraciones
ILUSTRACIÓN 1 - Flujo de trabajo de un análisis metabolómico. 10
ILUSTRACIÓN 2 - Etapas del flujo de trabajo. 14
ILUSTRACIÓN 3 - Área de muestreo. 15
ILUSTRACIÓN 4 - Selección según características morfométricas de los frutos. 16
ILUSTRACIÓN 5 - Ensayo de viabilidad en frutos germinados a las 0, 24 y 48 horas. 17
ILUSTRACIÓN 6 - Procedimiento experimental para la extracción de metabolitos. 20
ILUSTRACIÓN 7 - Rectas de calibrado. 21
ILUSTRACIÓN 8 - Espectro NIRS de muestras de harina de bellota para los tres individuos. 24
ILUSTRACIÓN 9 - Diagramas de caja de las características de los frutos. 26
ILUSTRACIÓN 10 - Espectro de 1D 13
C CP-MAS NMR sólida. 29
ILUSTRACIÓN 11 - Cromatogramas (TIC, total ion chromatogram) de GC-MS. 31
ILUSTRACIÓN 12 - Heatmap de los metabolitos identificados. 32
ILUSTRACIÓN 13 - Metabolitos identificados. 35
ILUSTRACIÓN 14 - Matriz de correlaciones. 35
Índice de tablas
TABLA 1 - Parámetros morfométricos. 17
TABLA 2. - Contenido en compuestos fenólicos, aminoácidos libres y azúcares. 23
TABLA 3 - Análisis de composición química de los frutos por NIRS. 25
TABLA 4 – Valoración de las técnicas empleadas. 34

pág. 5
Resumen
El presente trabajo fin de grado, titulado “Análisis metabolómico de frutos de encina
(Quercus ilex)” ha constituido una primera aproximación al análisis del perfil metabolómico
del fruto de la encina (Quercus ilex subsp. Ballota [Desf.] Samp), la bellota. Se han comparado
diferentes metodologías que pueden ser utilizadas en futuros estudios de variabilidad
poblacional y de respuesta a estreses ambientales, de tipo biótico y abiótico. Se analizaron
bellotas de individuos de una población de Aldea de Cuenca, Córdoba. Para ello, se utilizaron
las siguientes técnicas: técnicas no destructivas, como NIRS y NMR y técnicas de análisis in
vitro del extracto de harina, técnicas colorimétricas y GC-MS. Los componentes mayoritarios
determinados fueron polisacáridos como el almidón, azúcares, ácidos grasos y proteínas.
Mediante NIRS se cuantificaron el contenido en ceniza, ácido palmítico, ácido esteárico, ácido
linoleico, ácido oleico, la digestibilidad y capacidad calorífica. Mediante NMR se establecieron
las zonas del espectro que correspondieron a los carbonos constituyentes de carbohidratos,
grasa y proteína. Los análisis colorimétricos dieron valores en µg equivalentes de compuesto
de referencia (treonina, glucosa y ácido clorogénico, respectivamente) por g de harina de
aminoácidos libres (400.4 a valores inferiores al límite de detección), azúcares (2270.4 a 995.7)
y compuestos fenólicos (518.0 a 296.0). Mediante GC-MS se identificaron 23 compuestos
entre los cuales se encontraron diferentes aminoácidos, azúcares, ácidos orgánicos, alcoholes
y polioles. Finalmente, se discutieron las ventajas e inconvenientes de cada una de las
técnicas. A partir de los resultados obtenidos y el análisis de todos los métodos empleados, se
recomienda el uso de NIRS y técnicas de GC-MS para el estudio del perfil metabolómico y el
consecuente establecimiento de la variabilidad poblacional.
Palabras clave: Quercus ilex; Bellota; Metabolómica; NIRS; NMR; GC-MS

pág. 6
Abstract and Conclusions
“Metabolomic analysis of the acorn in Holm oak (Quercus ilex)” is the tittle of the
present study. It has constituted a first attempt to characterize the Holm oak acorn
metabolomic profile (Quercus ilex subsp. Ballota [Desf.] Samp). Different approaches have
been analyzed. Considering different methodologies which could be highly alluring in future
researches on population variabilities and adaptive responses of plants against abiotic and
biotic stresses. Mature acorns of different trees from Aldea de Cuenca (Córdoba) have been
studied. The following techniques have been carried out: non-destructive techniques (NIRS
and NMR), and in vitro acorn flour extract analysis (colorimetric assays and GC-MS). The main
extracted components have been polysaccharides (starch), sugars, fatty acids and proteins. As
well as, ash, palmitic acid, stearic acid, oleic acid, linoleic acid, digestibility, and energy have
been NIRS-derived chemical data. By means of NMR, different spectral regions could be set
corresponding to carbons constituent of carbohydrates, fat and protein. Colorimetric assay
allows to obtain values of free amino acids (400.4 to detection limit), sugars (2270.4 to 995.7)
and phenolic compounds (518.0 to 296.0) by micrograms of reference compound (threonine,
glucose and chlorogenic acid, respectively) per flour gram. GC-MS enables to identify 23
metabolites among which different amino acids, sugars, organics acids, alcohols and polyols
could be found. Finally, advantages and disadvantages for each technique has been described.
Through the analysis of results, it could be stated that NIRS and GC-MS are the best option to
study the metabolomic profile and population variabilities.
Keywords: Quercus ilex, Acorn, Metabolomics, NIRS, NMR, GC-MS

pág. 7
1. Introducción
Tres son los elementos básicos que definen una investigación biológica, el sistema
experimental, la pregunta o hipótesis y la metodología. En la presente Tesis de Grado dichos
elementos quedan reflejados en el título de la misma ,“Análisis metabolómico de frutos de
encina (Quercus ilex)”: el fruto de la encina comúnmente conocido como bellota, como
sistema de estudio, cuál es su perfil metabolómico, como pregunta, y el uso de
aproximaciones clásicas y holísticas de análisis de biomoléculas, como aproximaciones
metodológicas.
El trabajo queda encuadrado dentro de la línea de investigación sobre aproximaciones
moleculares al estudio de especies forestales (Sghaier-Hammami et al., 2015, Romero-
Rodríguez et al., 2014) que se lleva a cabo en el Grupo de Investigación “Bioquímica y
Proteómica Vegetal y Agroforestal” (AGR-164). Es un trabajo básicamente metodológico, en
el que se ha incidido en estrategias holísticas de análisis de perfiles metabolómicos
(metabolómica). Se ha pretendido implementar otras técnicas utilizadas previamente
realizada (bioquímica clásica y proteómica, fundamentalmente), encaminando la
investigación en la dirección definida por la Biología de Sistemas.
La encina (Quercus ilex subsp. Ballota [Desf.] Samp) es la especie predominante del
bosque mediterráneo, en general, y del ecosistema agroforestal dehesa, en particular. Es de
enorme interés para nuestra zona y cuya biología, en particular a nivel molecular, es, en gran
medida, desconocida. Los problemas históricos de sobreexplotación, los actuales de pérdida
de masa forestal por enfermedades y estreses ambientales (la seca de la encina) y los
asociados a un escenario de futuro cambio climático, hacen que el estudio de su biología pase
a ser una prioridad de cara a diseñar programas de manejo y conservación que aseguren su
valor medioambiental y agrosilvopastoral (Jorrín-Novo et al., 2014).
De todas las actuaciones biotecnológicas que se pueden utilizar en el ámbito
agroforestal, es la caracterización de la biodiversidad y la selección de genotipos “élite” o
“plus” la más adecuada o única viable. Esto es debido a las características de las especies
forestales (ciclos de vida muy largos y no domesticadas), lo que dificulta los programas de
mejora clásica, o la posición europea frente a los transgénicos. En dicho marco, la selección
de individuos con determinadas características fenotípicas, como las de adaptación y
tolerancia a condiciones ambientales adversas (sequía y patógenos, fundamentalmente)

pág. 8
(Simova-Stoilova et al., 2015), sería un logro importante. Por otra parte, el estudio
metabolómico puede llevarnos a la identificación de compuestos que, por su actividad
biológica y potencial uso aplicado, pueden darle valor añadido a la especie de estudio.
Trabajos previos han demostrado la existencia de una elevada variabilidad poblacional
y polimorfismo en Quercus spp. Dicha conclusión se ha alcanzado gracias a estudios
anatómicos y morfométricos (Castro-Díez et al., 1997), químicos (Valero et al., 2010), de
marcadores de DNA (Jiménez et al., 2004, Lumaret et al., 2009) y proteínas (Jorge et al., 2005,
Valero et al., 2011). También se han llevado a cabo análisis del contenido en fenólicos en
Quercus ilex L., como componentes antioxidantes útiles para la preservación de alimentos en
la industria alimentaria (Cantos et al., 2003, Karioti et al., 2011, Yarnes et al., 2006).
La pregunta que justifica este trabajo ha sido la siguiente, ¿las diferencias fenotípicas
observadas estarían asociadas a una composición química diferencial a causa de un
metabolismo diferenciado? Para resolver dicha pregunta se necesita alcanzar un
conocimiento del perfil de metabolitos de bellotas de encina. Con ello, pretendemos abordar
los dos principales objetivos anteriormente señalados, la caracterización de la variabilidad
poblacional y la búsqueda de compuestos bioactivos.
Debido al importante papel del género Quercus en el sector agroalimentario, a causa de su
elevado potencial antioxidante y antibacteriano (Güllüce et al., 2004) de diversos extractos
alcohólicos, o su consideración como una fuente rica en fenólicos, como proantocianidinas,
taninos, etc., la identificación de estos compuestos empleando técnicas metabolómicas,
permiten alcanzar un mayor conocimiento de éstos, y por último, asociarlos a dicha
variabilidad fenotípica.
La aproximación experimental, la metabolómica, ha constituido el principal reto de
este trabajo. Las técnicas metabolómicas son una herramienta importante para el diagnóstico
y estudio de las respuestas fisiológicas de las especies vegetales a los cambios ambientales y
ofrecen la oportunidad de obtener conocimiento de los mecanismos implicados en la correcta
relación biodiversidad-ecosistema (Scherling et al., 2010).
La metabolómica, término empleado por primera vez por Oliver Fiehn (Fiehn, 2002),
entendida como la disciplina científica o aproximación metodológica del área de la bioquímica,
tiene por objeto el análisis cualitativo y cuantitativo de forma exhaustiva de todos los

pág. 9
metabolitos de un sistema biológico dado, bajo un conjunto de condiciones ambientales
determinadas (Sheth et al., 2014).
Analiza el metaboloma, término sugerido por Oliver et al. (1998) (Oliver et al., 1998). Este se
define como el conjunto completo de pequeñas moléculas, metabolitos, producidos por las
diversas unidades biológicas (desde organismos, órganos, tejidos, células y orgánulos o
compartimentos celulares), en un estado de desarrollo específico y bajo determinadas
condiciones ambientales.
Los diversos metabolitos son pequeñas moléculas resultantes de las múltiples reacciones
enzimáticas durante el metabolismo que constituyen un grupo de compuestos de bajo peso
molecular (normalmente menores de 1500 Da) que incluyen: aminoácidos, péptidos, ácidos
grasos, lípidos, purinas, pirimidinas, azúcares y otras moléculas.
La gran diversidad de moléculas se refleja en una amplia gama de polaridades, pesos
moleculares, grupos funcionales, estabilidad y reactividad química, entre otras propiedades.
Esto obliga irremediablemente la utilización de múltiples plataformas y configuraciones
analíticas que maximicen la cobertura del metaboloma analizado.
Considerada como la más reciente de las grandes ómicas, la metabolómica permite la
descripción, identificación y cuantificación, desde una perspectiva holística de todos los
metabolitos, considerados como los productos finales de la expresión génica.
Al igual que el transcriptoma y el proteoma, el metaboloma es dinámico, experimenta cambios
muy rápidos que proporciona una visión del estado celular. Sin embargo, al contrario que los
genes y las proteínas, cuyas funciones están sujetas a la regulación epigenética y a las
modificaciones postraduccionales, respectivamente, los metabolitos actúan como indicadores
directos de la actividad bioquímica y son, por tanto, fácilmente correlacionados con el
fenotipo (Sheth et al., 2014). Por dicho motivo, la metabolómica está llamada a complementar
la información bioquímica obtenida de los genes y las proteínas, facilitando las
reconstrucciones genómicas actuales del metabolismo y mejorando nuestra comprensión de
la biología celular y fisiología de diferentes sistemas biológicos.
La plasticidad vegetal que permite la adaptación de estos organismos sésiles al medio y a los
cambios ambientales origina una elevada diversidad físico-química en las estructuras

pág. 10
moleculares y la naturaleza química de los metabolitos, especialmente los denominados
metabolitos secundarios.
Se ha estimado que existen aproximadamente 200.000 metabolitos en plantas. Cada planta
puede producir entre 5.000-25.000 metabolitos diferentes (Trethewey, 2004). Además de los
carbohidratos, ácidos grasos y proteínas, se han identificado más de 100.000 metabolitos
secundarios, como los isoprenoides, fenilpropanoides y alcaloides. La biosíntesis y la
acumulación de los metabolitos secundarios muestran una gran especificidad espacio-
temporal y son, en ocasiones, compuestos únicos en ciertas especies vegetales.
Al contrario que las proteínas, la estructura química no puede ser predicha basándonos en la
secuencia del genoma, haciendo incluso más difícil la determinación. Tal complejidad es un
reto analítico enorme, necesitándose poderosas herramientas para la separación y la
caracterización de las muestras.
Ilustración 1. Flujo de trabajo de un análisis metabolómico. En un procedimiento de metabolómica no
dirigida, los metabolitos de las muestras biológicas son previamente aislados. Subsecuentemente, se
obtienen los datos espectrales a partir de técnicas de NMR, LC-MS y GC-MS. Una vez adquiridos los datos,
estos son procesados empleando diversos softwares bioinformáticos para alcanzar la identificación del mayor
número posible de compuestos y finalmente realizar una interpretación biológica adecuada. Esquema
adaptado de Alonso et al, 2015.

pág. 11
Son múltiples las metodologías que se emplean para la detección, cuantificación y
elucidación de la estructura de los metabolitos.
El flujo de trabajo de un análisis metabolómico (Ilustración 1) comprende al menos 5 etapas
fundamentales; (1) la preparación de la muestra, (2) el empleo de métodos analíticos para la
adquisición de los datos, (3) el análisis de los datos y (4) la identificación y cuantificación de
los datos empleando análisis estadísticos y bioinformáticos para alcanzar (5) una
aproximación o interpretación biológica de los resultados.
En el intento de identificar las diferencias fenotípicas observables para su correlación con el
fenotipo molecular y metabolómico se ha realizado un análisis fenotípico, mediante la
caracterización morfométrica de los frutos (Porter et al., 1946). Se ha realizado un estudio
fenotípico y metabolómico, empleando desde métodos bioquímicos clásicos, para la
determinación de compuestos fenólicos (Ainsworth et al., 2007), azúcares (Miller, 1959) y
aminoácidos (Smith y Agiza, 1951), hasta técnicas que permiten una aproximación holística
(espectroscopía del infrarrojo cercano (NIRS), resonancia magnética nuclear (NMR) y
espectrometría de masas (MS)).
La espectroscopía del infrarrojo cercano es un método no destructivo, simple, fiable y
preciso, que permite el completo análisis químico en un único experimento, incluyendo fibra,
almidón, proteína y ácidos grasos en productos agrícolas tales como granos y aceites de
semillas (Valero et al., 2010). Su uso generalizado se debe principalmente a que permite
realizar análisis cualitativos y cuantitativos de componentes en muestras, con un mínimo de
preparación, empleándose en programas de reforestación para la selección de los individuos
más aptos (Schimleck et al., 2000), diferenciación de especies (Adedipe et al., 2008) y la
determinación del contenido lipídico (Sousa-Correia et al., 2007).
Se basa en la absorbancia que presentan los diferentes compuestos orgánicos en ciertas
regiones de longitud de onda (entre 800 y 2500 nm). Esta técnica combina la espectroscopía,
la estadística y la computación y genera modelos matemáticos que relacionan la composición
química (presencia de grupos químicos activos) con cambios de energía en la región
correspondiente al rango infrarrojo cercano (Vásquez et al., 2004).
La resonancia magnética nuclear es una alternativa a las tecnologías basadas en
espectrometría de masas para análisis metabolómicos (Ratcliffe y Shachar-Hill, 2001). Es una

pág. 12
técnica de espectroscopía rápida y altamente reproducible que se basa en la energía de
absorción y reemisión de los núcleos atómicos debido a las variaciones en el campo magnético
externo (Bothwell y Griffin, 2011). Se genera diferentes tipos de información metabolómica
dependiendo del núcleo atómico al cual va dirigido la aplicación de los campos magnéticos.
Esta tecnología se fundamenta en la composición uniforme de los metabolitos, constituidos
básicamente por hidrógeno, carbono, nitrógeno, oxígeno y fosforo. Sin embargo, el hidrógeno
es el núcleo normalmente elegido en el análisis de muestras de origen biológico (1H-NMR),
debido a su abundancia natural en las muestras biológicas. Pero, a pesar de ser menos
frecuente, también se emplean en NMR otros átomos como el carbono (13C-NMR) y el fosforo
(31P-NMR), aportando una información adicional de los diversos tipos de metabolitos (Reo,
2002).
Además, NMR no requiere de pasos previos de separación o preparación de las muestras para
la detección de metabolitos, siendo incluso posible empleando extractos crudos. Debido a
esto, el espectro de NMR da información cuantitativa muy detallada que es altamente
reproducible (Last et al., 2007). Los datos espectrales resultantes en NMR no solo permiten la
cuantificación de la concentración de metabolitos sino que también aportan información
sobre la estructura química. Los áreas de los picos en los espectros generados por cada
molécula son empleados como medida indirecta de la cantidad de los metabolitos en la
muestra, mientras que el patrón de picos formado en el espectro informa de las propiedades
físicas de la molécula, lo que permite la identificación del tipo de metabolito.
La ausencia de derivatización y su naturaleza no destructiva hacen posible la recuperación de
muestra analizada después del análisis para su empleo en análisis posteriores. Sin embargo,
NMR es mucho menos sensible que los métodos de MS y la resolución de los espectros es más
pobre, razones por las cuales NMR es menos empleado en la metabolómica de plantas que
MS (Last et al., 2007). A pesar de ello, NMR es un buen complemento para el perfil obtenido
con MS y es un método útil para la obtención de la huella metabólica (Krishnan et al., 2005).
La espectrometría de masas es una técnica analítica que permite la adquisición de
datos espectrales en función de la relación masa/carga (m/z) y la intensidad relativa de los
elementos analizados. Para que el espectrómetro pueda generar los picos de señal de cada
metabolito, las muestras biológicas deben ser previamente ionizadas. Los compuestos
ionizados resultantes de cada molécula generarán diferentes patrones de picos que definen

pág. 13
la huella o el perfil de la molécula original. En metabolómica, generalmente se requieren de
un paso previo de separación. Este paso reduce la elevada complejidad de la muestra biológica
y permite dirigir el análisis a un conjunto determinado de compuestos. Las columnas de
cromatografía de líquidos y gases (LC y GC, respectivamente) son las técnicas de separación
más comúnmente empleadas (Theodoridis et al., 2011). Dicha técnica de separación
cromatográfica está basada en la interacción de los diferentes metabolitos contenidos en la
muestra con los materiales adsorbentes en la columna cromatográfica. De este modo, los
metabolitos con diferentes propiedades químicas poseerán distintos tiempos de retención
(tiempo necesario para pasar a través de la columna). El tiempo requerido por cada
metabolito, se emplea junto a los valores de la relación masa/carga que se obtendrán en el
espectrómetro de masas generando la información necesaria para la identificación de los
metabolitos. Así, se combinan ambas técnicas, denominándose LC-MS y GC-MS.
Para la realización de este trabajo de investigación, desde un punto de vista
interdisciplinar, se ha requerido de conceptos científicos y aproximaciones experimentales del
campo de la biología y la química (desde aspectos fisiológicos del organismo de estudio y
propiedades físicas y químicas de los componentes moleculares) para alcanzar la comprensión
de los mecanismos moleculares en los sistemas biológicos complejos.
Se debe destacar la naturaleza novedosa de esta aproximación al perfil metabolómico del
fruto de encina u otras especies forestales. Esto queda reflejado debido a la escasa bibliografía
presente. De hecho, la mayoría de los trabajos que han enfrentado sus problemas con este
tipo de aproximación han sido referentes a la determinación del perfil metabolómico de
respuesta a distintos estreses. En ellos, han empleado 1H-NMR para la determinación del perfil
en la respuesta a heridas en el proceso de germinación de semillas de Quercus ilex (Sardans
et al., 2014), GC-MS en respuesta a estrés biótico (Adjami et al., 2016, Kersten et al., 2013) o
espectroscopía FT-IR y GC en respuesta a la desecación (Connor et al, 2003). En definitiva, no
se ha realizado la determinación del perfil de metabolitos para el estudio de la variabilidad
poblacional como se ha pretendido realizar en este trabajo.
2. Materiales y Métodos
Se han empleado una serie de aproximaciones metodológicas, incluyendo técnicas no
destructivas (NIRS y NMR) y técnicas in vitro de los extractos de harina de bellota (ensayos
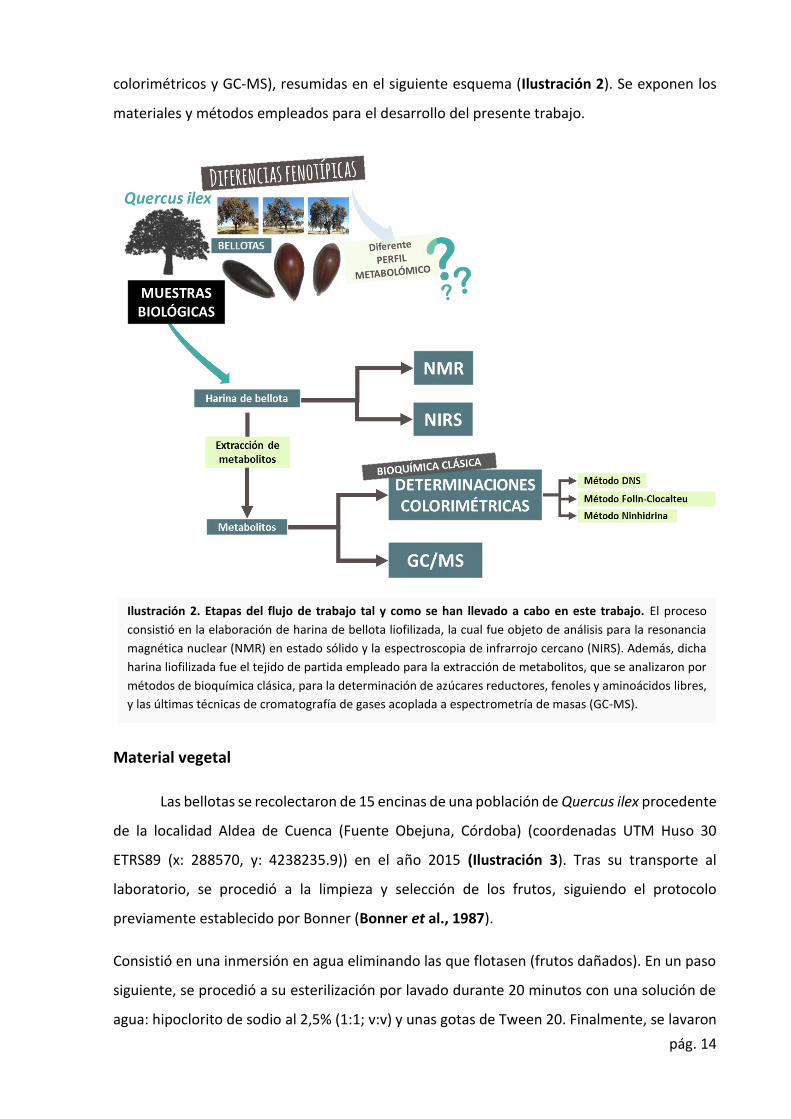
pág. 14
colorimétricos y GC-MS), resumidas en el siguiente esquema (Ilustración 2). Se exponen los
materiales y métodos empleados para el desarrollo del presente trabajo.
Material vegetal
Las bellotas se recolectaron de 15 encinas de una población de Quercus ilex procedente
de la localidad Aldea de Cuenca (Fuente Obejuna, Córdoba) (coordenadas UTM Huso 30
ETRS89 (x: 288570, y: 4238235.9)) en el año 2015 (Ilustración 3). Tras su transporte al
laboratorio, se procedió a la limpieza y selección de los frutos, siguiendo el protocolo
previamente establecido por Bonner (Bonner et al., 1987).
Consistió en una inmersión en agua eliminando las que flotasen (frutos dañados). En un paso
siguiente, se procedió a su esterilización por lavado durante 20 minutos con una solución de
agua: hipoclorito de sodio al 2,5% (1:1; v:v) y unas gotas de Tween 20. Finalmente, se lavaron
Ilustración 2. Etapas del flujo de trabajo tal y como se han llevado a cabo en este trabajo. El proceso
consistió en la elaboración de harina de bellota liofilizada, la cual fue objeto de análisis para la resonancia
magnética nuclear (NMR) en estado sólido y la espectroscopia de infrarrojo cercano (NIRS). Además, dicha
harina liofilizada fue el tejido de partida empleado para la extracción de metabolitos, que se analizaron por
métodos de bioquímica clásica, para la determinación de azúcares reductores, fenoles y aminoácidos libres,
y las últimas técnicas de cromatografía de gases acoplada a espectrometría de masas (GC-MS).

pág. 15
abundantemente con agua, secadas a temperatura ambiente y almacenadas a 4 oC en bolsas
de polietileno herméticamente cerradas (Caliskan., 2014, Corbineau et al., 2014).
Morfometría y contenido hídrico
Se determinaron los siguientes parámetros morfométricos de los frutos: longitud,
diámetro máximo, volumen, peso fresco y peso seco. El muestreo se llevó a cabo en 10
bellotas con un tamaño homogéneo y en buen estado. A cada bellota se le realizó una
fotografía sobre papel milimetrado y a partir de ésta se determinó la longitud y diámetro
máximo. La medida del volumen se obtuvo a partir del agua desplazada tras la inmersión del
fruto en una probeta de 20 mL. El peso de los frutos se determinó con una balanza analítica
(Mettler AJ150). Se midió el peso seco y el peso seco tras la eliminación del pericarpo de los
frutos. El peso seco fue medido tras 96 horas en estufa a 65 oC. El contenido hídrico, en tanto
por ciento, se obtuvo según la fórmula CH = (Peso fresco - Peso seco)/(Peso seco) x 100).
Las características morfométricas de longitud, diámetro máximo, volumen, peso fresco
y peso seco y la realización de un análisis estadístico multivariante de componentes
principales (PCA) permitieron la selección tres bellotas que correspondían a los tres grupos
más diferenciados (Ilustración 4).
Ilustración 3. Área de muestreo. Aldea de Cuenca (Fuente Obejuna, Provincia de Córdoba). Se enumeran las
15 encinas muestreadas. Las encinas seleccionadas fueron las encinas correspondientes a los números 3, 7
y 14.

pág. 16
Las características morfométricas de los frutos de las encinas seleccionadas se muestran en la
Tabla 1.
Tabla 1. Parámetros morfométricos. Se muestran los resultados de peso fresco (g), peso seco (g), contenido
hídrico (%), longitud (mm), diámetro máximo (mm) y volumen (mL) en valores medios ± error estándar. Se
realizó test de Friedman (no paramétricas) y test de ANOVA (paramétricas). Todas las variables presentaban
diferencias significativas. Las medidas de longitud, diámetro máximo y volumen se realizaron sobre los frutos
completos. El peso seco y l peso fresco se obtuvo tras la eliminación del pericarpo. Abreviaturas: E3 (Encina
3), E7 (Encina 7), E14 (Encina 14)
ID Peso Fresco (g) Peso Seco (g) Contenido
Hídrico (%) Longitud
(mm) Diámetro
máximo (mm) Volumen (mL)
E3 3.4 ± 0.3 2.1 ± 0.2 37.7 ± 2.6 34.6 ± 2.4 17.2 ± 1.4 3.8 ± 0.4
E7 4.5 ± 0.5 2.7 ± 0.3 40.8 ± 1.9 44.7 ± 1.3 15.0 ± 1.6 5.0 ± 0.7
E14 8.4 ± 0.9 5.2 ± 0.6 39.0 ± 2.8 47.1 ± 1.3 23.2 ± 1.3 8.9 ± 0.9
Ilustración 4. Selección según características morfométricas de los frutos. Las encinas están numeradas de
1 a 15. Se realizó la caracterización según volumen (mL), diámetro máximo (mm), longitud (mm), contenido
hídrico (%), peso seco (g) y peso fresco (g). A partir de las variables nombradas se realizó una prueba de
estadística multivariante, análisis de componentes principales (PCA), lo que nos distribuyó los distintos frutos
según dichas características. Las encinas seleccionadas fueron las encinas correspondientes a los números 3,
7 y 14.
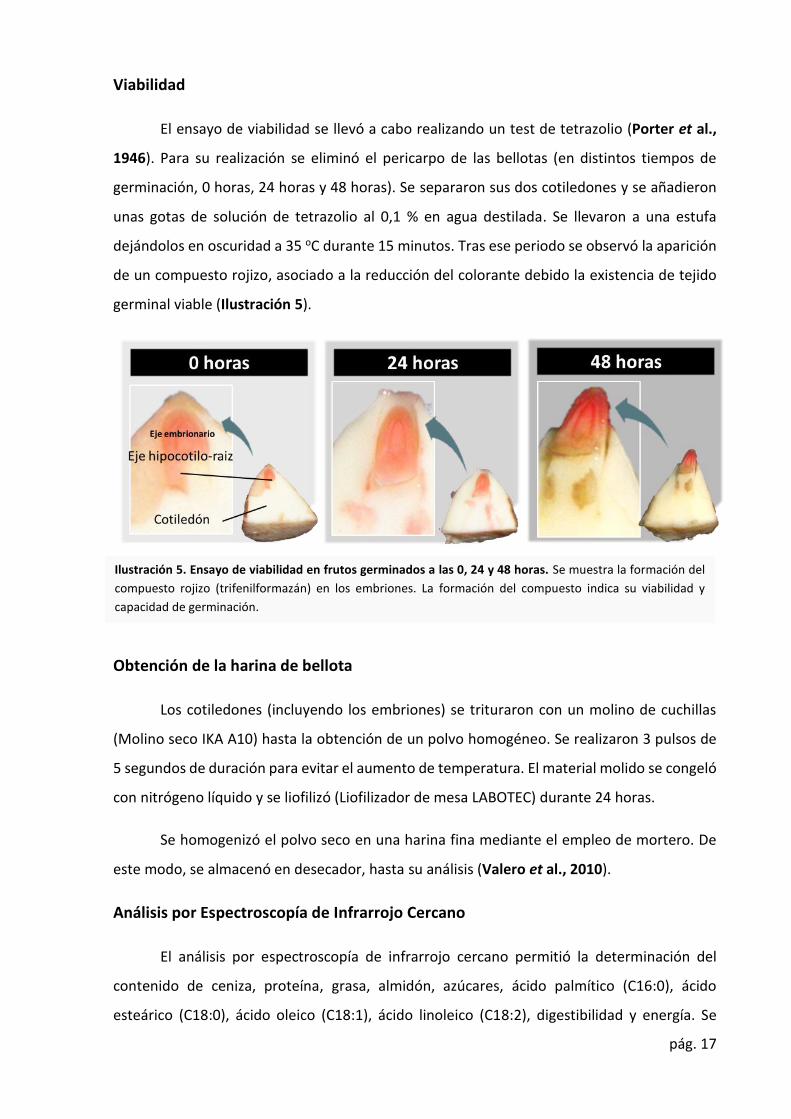
pág. 17
Viabilidad
El ensayo de viabilidad se llevó a cabo realizando un test de tetrazolio (Porter et al.,
1946). Para su realización se eliminó el pericarpo de las bellotas (en distintos tiempos de
germinación, 0 horas, 24 horas y 48 horas). Se separaron sus dos cotiledones y se añadieron
unas gotas de solución de tetrazolio al 0,1 % en agua destilada. Se llevaron a una estufa
dejándolos en oscuridad a 35 oC durante 15 minutos. Tras ese periodo se observó la aparición
de un compuesto rojizo, asociado a la reducción del colorante debido la existencia de tejido
germinal viable (Ilustración 5).
Obtención de la harina de bellota
Los cotiledones (incluyendo los embriones) se trituraron con un molino de cuchillas
(Molino seco IKA A10) hasta la obtención de un polvo homogéneo. Se realizaron 3 pulsos de
5 segundos de duración para evitar el aumento de temperatura. El material molido se congeló
con nitrógeno líquido y se liofilizó (Liofilizador de mesa LABOTEC) durante 24 horas.
Se homogenizó el polvo seco en una harina fina mediante el empleo de mortero. De
este modo, se almacenó en desecador, hasta su análisis (Valero et al., 2010).
Análisis por Espectroscopía de Infrarrojo Cercano
El análisis por espectroscopía de infrarrojo cercano permitió la determinación del
contenido de ceniza, proteína, grasa, almidón, azúcares, ácido palmítico (C16:0), ácido
esteárico (C18:0), ácido oleico (C18:1), ácido linoleico (C18:2), digestibilidad y energía. Se
Ilustración 5. Ensayo de viabilidad en frutos germinados a las 0, 24 y 48 horas. Se muestra la formación del
compuesto rojizo (trifenilformazán) en los embriones. La formación del compuesto indica su viabilidad y
capacidad de germinación.

pág. 18
empleó un espectrofotómetro Foss-NIRSystems 6500 System II (Foss-NIRSystems Inc., Silver
Spring) (Unidad de Espectroscopía NIR/MIR en el Servicio central de apoyo a la Investigación
(SCAI) de la Universidad de Córdoba (http://www.uco.es/servicios/scai/nir.html) equipado
con un módulo transportador rectangular de muestra de ¼ y dos detectores que analizan dos
regiones del espectro (visible e infrarrojo cercano). Los datos espectrales se adquirieron cada
2 nm y se registraron y analizando con el software WinISI 1.50 (Infrasoft International, Port
Matilda, PA, USA). Para la cuantificación de los compuestos se emplearon algoritmos tal y
como se describe en el trabajo de Valero (Valero et al., 2010).
El análisis estadístico de los resultados obtenidos en NIRS se llevó a cabo empleando
el software estadístico R (versión 3.2.5, https://cran.r-project.org/src/base/R-3/). Se
realizaron los análisis de estadística descriptiva de los distintos compuestos identificados
(media y error estándar). Se realizaron los correspondientes test de normalidad de las
distribuciones de las distintas variables mediante el test de Shaphiro (p>0,05, se aceptaría la
hipótesis nula, por tanto, se consideran como variables paramétricas). Aquellas variables que
siguieron una distribución normal, se sometieron a un test ANOVA de un factor, para
establecer si las diferencias mostradas serían estadísticamente significativas (p>0,05 se
aceptaría la hipótesis nula, que indicaría igualdad de las medias, es decir, no existen
diferencias estadísticamente significativas entre los grupos). Aquellas variables que no
siguieron una distribución normal, se sometieron a un test de Friedman (p>0,05 se aceptaría
la hipótesis nula, que indicaría igualdad de las medias, es decir, no existen diferencias
estadísticamente significativas entre los grupos).
Se realizaron las respectivas correlaciones de Pearson para el estudio de las posibles
relaciones entre las variables obtenidas con NIRS y las características morfométricas, cuya
representación gráfica se mostrará mediante un gráfico de correlaciones en el
correspondiente apartado de los resultados y discusión.
Análisis por Resonancia Magnética Nuclear
Se obtuvieron los espectros de 13C-NMR en un espectrómetro de NMR Bruker Avance
III HD 400 WB en la Unidad de Resonancia Magnética Nuclear en el SCAI de la Universidad de
Córdoba (http://www.uco.es/servicios/scai/rmn.html). Para la obtención de los diversos
valores del espectro se empleó una secuencia de pulso CP/MAS (Solid-state Cross-Polarization
Magic Angle Spinning) con un tiempo de repetición de 4 segundos y un tiempo de contacto

pág. 19
de 1,9 microsegundos. La muestra de harina liofilizada fue sometida a una frecuencia de giro
de 13 KHz. Se empleó la referencia de TMS (tetrametilsilano) a 0 ppm. Para el correspondiente
análisis de los resultados y visualización de los espectros se empleó el software Dmfit (Massiot
et al., 2002) y para la identificación de las distintas regiones del espectro se emplearon una
serie de referencias que se especificarán en el apartado de los resultados y discusión.
Extracción de metabolitos
Se llevó a cabo la extracción de metabolitos mediante un procedimiento clásico de
dobles solventes y siguiendo la metodología descrita por el Dr. Luis Valledor (Universidad de
Oviedo) (Valledor et al., 2014) (Ilustración 6).
Ilustración 6. Procedimiento experimental para la extracción de metabolitos. 1. Homogeneización harina
de bellota. 2. Primera extracción con solución de extracción de metabolitos [Cloroformo: Metanol: Agua
(2.5:1:0.5)]. 3. Segunda extracción con solución de extracción de metabolitos. Tratamiento pellet con
metanol al 100 % con 0.75 % de β-mercaptoetanol. 4. Sobrenadante resultante que contiene los metabolitos.
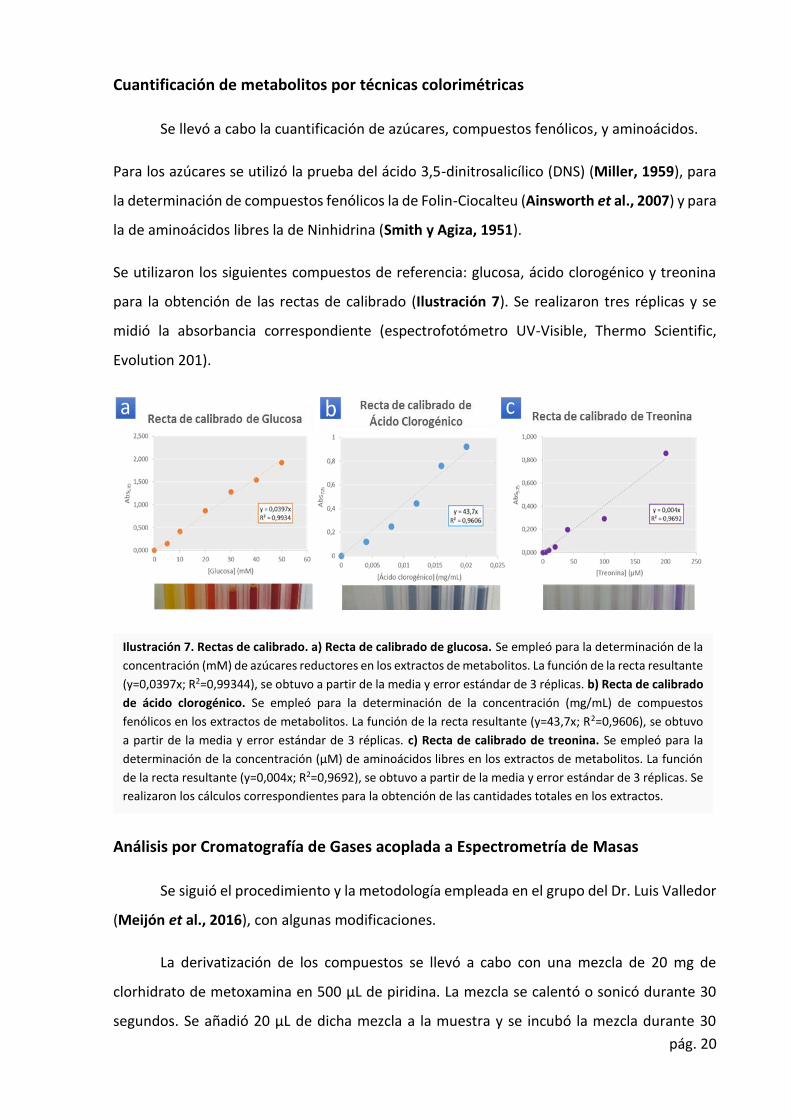
pág. 20
Cuantificación de metabolitos por técnicas colorimétricas
Se llevó a cabo la cuantificación de azúcares, compuestos fenólicos, y aminoácidos.
Para los azúcares se utilizó la prueba del ácido 3,5-dinitrosalicílico (DNS) (Miller, 1959), para
la determinación de compuestos fenólicos la de Folin-Ciocalteu (Ainsworth et al., 2007) y para
la de aminoácidos libres la de Ninhidrina (Smith y Agiza, 1951).
Se utilizaron los siguientes compuestos de referencia: glucosa, ácido clorogénico y treonina
para la obtención de las rectas de calibrado (Ilustración 7). Se realizaron tres réplicas y se
midió la absorbancia correspondiente (espectrofotómetro UV-Visible, Thermo Scientific,
Evolution 201).
Análisis por Cromatografía de Gases acoplada a Espectrometría de Masas
Se siguió el procedimiento y la metodología empleada en el grupo del Dr. Luis Valledor
(Meijón et al., 2016), con algunas modificaciones.
La derivatización de los compuestos se llevó a cabo con una mezcla de 20 mg de
clorhidrato de metoxamina en 500 µL de piridina. La mezcla se calentó o sonicó durante 30
segundos. Se añadió 20 µL de dicha mezcla a la muestra y se incubó la mezcla durante 30
Ilustración 7. Rectas de calibrado. a) Recta de calibrado de glucosa. Se empleó para la determinación de la
concentración (mM) de azúcares reductores en los extractos de metabolitos. La función de la recta resultante
(y=0,0397x; R2=0,99344), se obtuvo a partir de la media y error estándar de 3 réplicas. b) Recta de calibrado
de ácido clorogénico. Se empleó para la determinación de la concentración (mg/mL) de compuestos
fenólicos en los extractos de metabolitos. La función de la recta resultante (y=43,7x; R2=0,9606), se obtuvo
a partir de la media y error estándar de 3 réplicas. c) Recta de calibrado de treonina. Se empleó para la
determinación de la concentración (µM) de aminoácidos libres en los extractos de metabolitos. La función
de la recta resultante (y=0,004x; R2=0,9692), se obtuvo a partir de la media y error estándar de 3 réplicas. Se
realizaron los cálculos correspondientes para la obtención de las cantidades totales en los extractos.

pág. 21
minutos a 37 oC en un agitador térmico. Para la silación de los metabolitos se añadió 70 µL de
MSTFA a las muestras y se incubaron durante 30 minutos a 37 oC en un agitador térmico. Tras
este tiempo, se centrifugó durante 3 minutos a 20.000 g, transfiriendo el sobrenadante a los
microviales de GC.
Las medidas de GC-MS se realizaron siguiendo el procedimiento (Furuhashi et al.,
2012) en un instrumento de triple cuadrupolo (TSQ Quantum GC, Thermo). Se inyectó 1 µL de
muestra y se realizó la separación por cromatografía de gases mediante una columna capilar
HP-5MS (30m x 0,25 mm x 0,25 mm, Agilent Technologies). Las temperaturas del horno fueron
incrementando desde 80 oC a 200 oC, (3 oC/min), luego, fue incrementando desde 200 oC a
250 oC durante 3 minutos (10 oC/min) y por último, 250 oC/min hasta 280 oC durante 4 minutos
(30 oC/min). Después de la separación, las condiciones de temperatura fueron 25 oC durante
4 minutos. La temperatura del inyector fue 265 oC. El caudal del proceso cromatográfico fue
de 1 mL/ min. El intervalo de tiempo de detección fue de 7,10 a 48,5 minutos. El análisis de
masas (m/z) se llevó a cabo en un equipo de impacto electrónico (EI) a 70 eV en intervalos de
escaneado de 40-600 m/z.
Se realizó el análisis y procesamiento de los datos obtenidos por GC-MS empleando el
software MZmine 2.20 (Pluskal et al, 2010), con el cual se señalaron los picos y se registraron
los tiempos de retención, las masas espectrales características y las intensidades de cada uno.
Para la identificación de los metabolitos se emplearon diversas bases de datos (Alkane, Fiehn
library 1 y 2, Gölm Metabolome Database, GC-TSQ y MoSys) y el software NIST.MS Search
(versión 2.01, http://chemdata.nist.gov/mass-spc/ms-search/).
3. Presentación de los resultados y discusión
Mediante la integración de técnicas de bioquímica clásica de cuantificación de
metabolitos y las modernas aproximaciones holísticas de metabolómica, GC acoplado a MS,
NIRS y RMN, se ha pretendido establecer el perfil de metabolitos o metaboloma del fruto de
la encina (Quercus ilex subsp. Ballota [Desf.] Samp), la bellota. Dicho trabajo, de carácter
marcadamente metodológico tiene un doble objetivo y proyección futura, la caracterización
o catalogación de la variabilidad poblacional y la búsqueda de compuestos con posible
actividad biológica. Se emplearon, para ello, frutos maduros de una única población,
localizada en Aldea de Cuenca, Córdoba. Para cada una de las muestras se determinaron los
parámetros morfométricos (longitud, diámetro máximo, volumen, peso fresco y peso seco),

pág. 22
la viabilidad, se molió para obtener harina, se liofilizó y utilizó como material de partida para
sus correspondientes análisis con espectroscopía de infrarrojo cercano (NIRS) y de resonancia
magnética nuclear (NMR). A partir de dicha harina, se realizó la extracción de metabolitos
siguiendo un protocolo clásico de extracción, basado en el empleo de cloroformo: metanol:
agua. Los extractos se analizaron por técnicas colorimétricas para la determinación de
compuestos fenólicos, aminoácidos libres y azúcares, y, por último, se analizaron con
cromatografía de gases acoplado a espectrometría de masas (GC-MS).
Análisis por métodos colorimétricos
En los extractos de metabolitos se determinaron el contenido (µg equivalentes de
compuesto de referencia /g de harina) en fenólicos, aminoácidos libres y azúcares (Tabla 2).
Los compuestos mayoritarios fueron los azúcares, con un rango de 2270.4 a 995.7 µg
equivalentes de glucosa por g de harina liofilizada, seguido de los compuestos fenólicos con
un rango de 518.0 a 296.0 µg equivalentes de ácido clorogénico por g de harina liofilizada y
por último, los aminoácidos libres con un rango de 400.4 µg equivalentes de treonina por g de
harina liofilizada a valores en torno al límite de detección.
Existieron diferencias significativas (p ≤ 0.05) entre el contenido de aminoácidos libres
de los tres individuos analizados, con valores superiores para E7. No existieron diferencias
significativas (p ≥ 0.05) entre el contenido de azúcares y compuestos fenólicos de los tres
individuos analizados, obteniéndose mayores valores del contenido de azúcares para E14 y de
compuestos fenólicos para E3. Valores similares de compuestos fenólicos han sido
Tabla 2. Contenido en compuestos fenólicos, aminoácidos libres y azúcares en los extractos de harina de bellota. Se muestran los resultados en valores medios ± error estándar correspondiente a tres réplicas biológicas por individuo, E3, E7 y E14. L.d. Valores en torno al límite de detección. El análisis estadístico de los datos se llevó a cabo mediante test paramétricos (test de ANOVA) y test no paramétricos (test de Friedman) para determinar la existencia de diferencias estadísticamente significativas. a. Indica variable con diferencias estadísticamente significativas entre individuos (p ≤ 0.05). Unidades de compuestos fenólicos (µg equivalentes de ácido clorogénico s por g harina liofilizada), aminoácidos libres (µg equivalentes de treonina por g harina liofilizada) y azúcares (µg equivalentes de glucosa por g harina liofilizad)
ID Fenólicos
(µg equ. A. Clorogénico /g Harina) Aminoácidos libres
(µg equ. Treonina /g Harina) Azúcares
(µg equ. Glucosa /g Harina)
E3 518.0 ± 74.0 43.9 ± 59.6a 2259.1 ± 345.7
E7 296.0 ± 185.0 400.4 ± 120.9a 995.7 ± 408.9
E14 444.0 ± 74.0 L.d.a 2270.4 ± 469.5
pág. 23
previamente referenciados (Cantos et al., 2003). Por el contrario, otros autores han señalado
valores muy superiores de azúcares y aminoácidos libres (Midilli et al., 2008 y Özcan, 2006).
Estas diferencias pudieran deberse al protocolo de extracción, al método de análisis, al
material de partida o incluso a errores en la determinación.
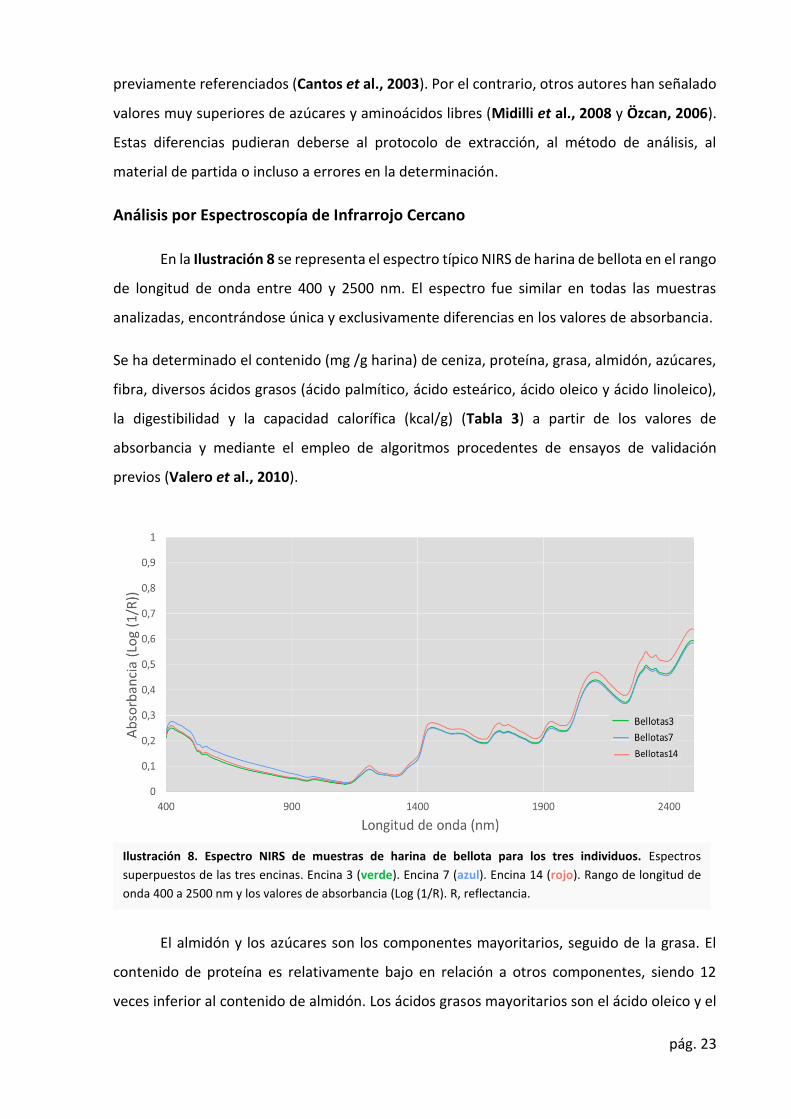
Análisis por Espectroscopía de Infrarrojo Cercano
En la Ilustración 8 se representa el espectro típico NIRS de harina de bellota en el rango
de longitud de onda entre 400 y 2500 nm. El espectro fue similar en todas las muestras
analizadas, encontrándose única y exclusivamente diferencias en los valores de absorbancia.
Se ha determinado el contenido (mg /g harina) de ceniza, proteína, grasa, almidón, azúcares,
fibra, diversos ácidos grasos (ácido palmítico, ácido esteárico, ácido oleico y ácido linoleico),
la digestibilidad y la capacidad calorífica (kcal/g) (Tabla 3) a partir de los valores de
absorbancia y mediante el empleo de algoritmos procedentes de ensayos de validación
previos (Valero et al., 2010).
El almidón y los azúcares son los componentes mayoritarios, seguido de la grasa. El
contenido de proteína es relativamente bajo en relación a otros componentes, siendo 12
veces inferior al contenido de almidón. Los ácidos grasos mayoritarios son el ácido oleico y el
Ilustración 8. Espectro NIRS de muestras de harina de bellota para los tres individuos. Espectros
superpuestos de las tres encinas. Encina 3 (verde). Encina 7 (azul). Encina 14 (rojo). Rango de longitud de
onda 400 a 2500 nm y los valores de absorbancia (Log (1/R). R, reflectancia.
pág. 24
ácido palmítico. Le siguen el contenido de ácido linoleico y de ácido esteárico, como
minoritarios.
A tenor de los valores de azúcares encontrados se pudo concluir que su contenido fue
similar para los tres individuos analizados, algo que contradijo lo datos obtenidos por el
método colorimétrico. Se obtuvieron valores 50 veces menores con los métodos colorimétrico
que con NIRS. Se podría establecer como anómalo el resultado obtenido en E7 con el método
colorimétrico. Esto puede indicar un error en el proceso de extracción o en la determinación.
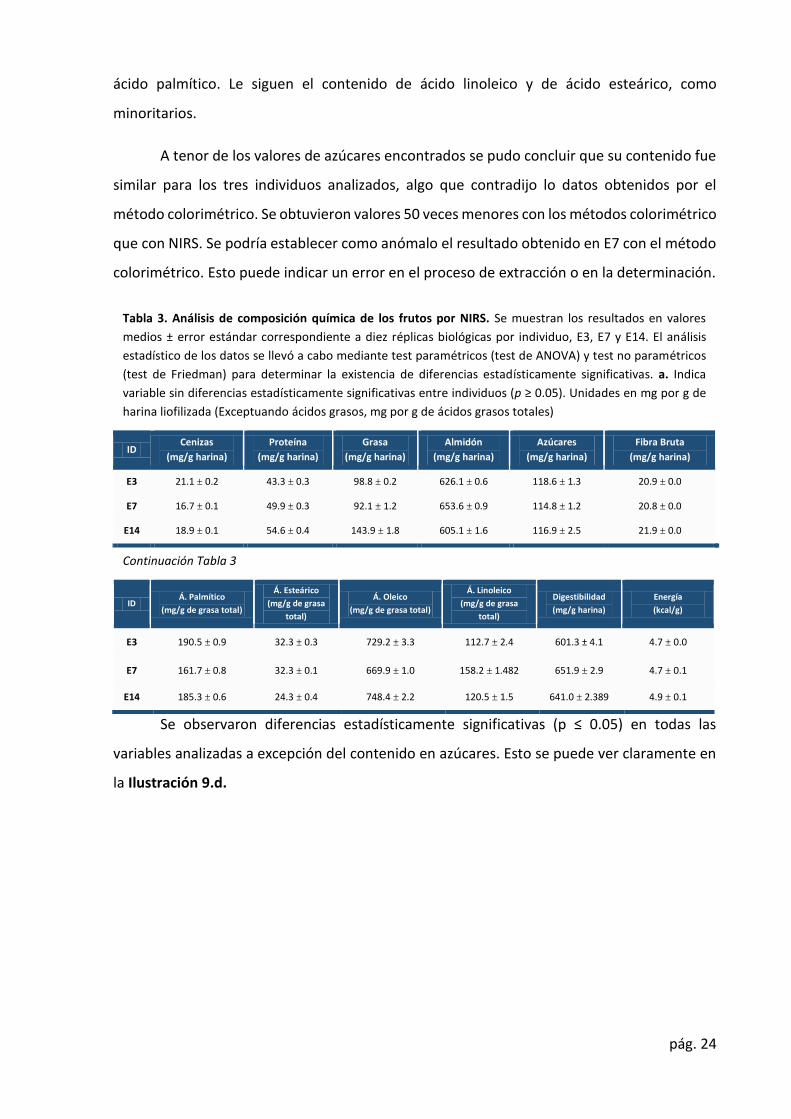
Tabla 3. Análisis de composición química de los frutos por NIRS. Se muestran los resultados en valores
medios ± error estándar correspondiente a diez réplicas biológicas por individuo, E3, E7 y E14. El análisis
estadístico de los datos se llevó a cabo mediante test paramétricos (test de ANOVA) y test no paramétricos
(test de Friedman) para determinar la existencia de diferencias estadísticamente significativas. a. Indica
variable sin diferencias estadísticamente significativas entre individuos (p ≥ 0.05). Unidades en mg por g de
harina liofilizada (Exceptuando ácidos grasos, mg por g de ácidos grasos totales)
ID Cenizas (mg/g harina)
Proteína (mg/g harina)
Grasa (mg/g harina)
Almidón (mg/g harina)
Azúcares (mg/g harina)
Fibra Bruta (mg/g harina)
E3 21.1 ± 0.2 43.3 ± 0.3 98.8 ± 0.2 626.1 ± 0.6 118.6 ± 1.3 20.9 ± 0.0 E7 16.7 ± 0.1 49.9 ± 0.3 92.1 ± 1.2 653.6 ± 0.9 114.8 ± 1.2 20.8 ± 0.0
E14 18.9 ± 0.1 54.6 ± 0.4 143.9 ± 1.8 605.1 ± 1.6 116.9 ± 2.5 21.9 ± 0.0
Continuación Tabla 3
ID Á. Palmítico (mg/g de grasa total)
Á. Esteárico (mg/g de grasa
total) Á. Oleico
(mg/g de grasa total) Á. Linoleico
(mg/g de grasa
total) Digestibilidad
(mg/g harina) Energía (kcal/g)
E3 190.5 ± 0.9 32.3 ± 0.3 729.2 ± 3.3 112.7 ± 2.4 601.3 ± 4.1 4.7 ± 0.0
E7 161.7 ± 0.8 32.3 ± 0.1 669.9 ± 1.0 158.2 ± 1.482 651.9 ± 2.9 4.7 ± 0.1 E14 185.3 ± 0.6 24.3 ± 0.4 748.4 ± 2.2 120.5 ± 1.5 641.0 ± 2.389 4.9 ± 0.1
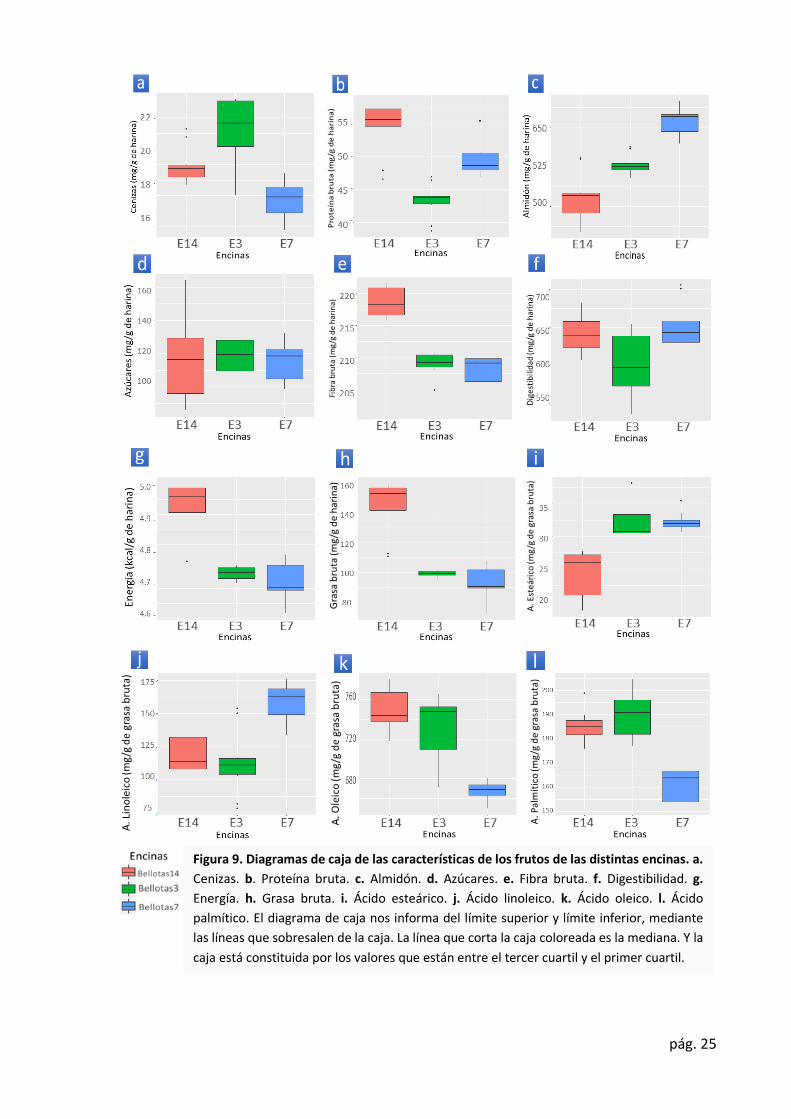
Se observaron diferencias estadísticamente significativas (p ≤ 0.05) en todas las
variables analizadas a excepción del contenido en azúcares. Esto se puede ver claramente en
la Ilustración 9.d.
pág. 25
Figura 9. Diagramas de caja de las características de los frutos de las distintas encinas. a.
Cenizas. b. Proteína bruta. c. Almidón. d. Azúcares. e. Fibra bruta. f. Digestibilidad. g.
Energía. h. Grasa bruta. i. Ácido esteárico. j. Ácido linoleico. k. Ácido oleico. l. Ácido
palmítico. El diagrama de caja nos informa del límite superior y límite inferior, mediante
las líneas que sobresalen de la caja. La línea que corta la caja coloreada es la mediana. Y la
caja está constituida por los valores que están entre el tercer cuartil y el primer cuartil.

pág. 26
Los resultados obtenidos en mg /g harina son similares a los obtenidos por Valero,
confirmando la robustez y precisión de la técnica. Los rangos de valores encontrados fueron
de 16.7 (E7) a 21.1 (E3) para cenizas, de 43.3 (E3) a 54.6 (E14) para proteína, de 605.1 (E14) a
653.6 (E7) para almidón, de 114,8 (E7) a 118.6 (E3) para azúcares, de 92.1 (E7) a 143.9 (E14)
para grasa, y de 161.7 (E7) a 190.5 (E14) para fibra. Estos valores no se alejan de los obtenidos
por Rodríguez (Rodríguez et al., 2009). Los valores de ácidos grasos obtenidos y expresados
como mg /g de grasa total fueron de 161.7 (E7) 190.5 (E3) para ácido palmítico, de 24.3 (E7) a
32.3 (E14) para ácido esteárico, de 112.7 (E3) a 158.2 (E14) para ácido linoleico y de 63.0 (E7)
a 74.8 (E14) para ácido oleico. Los valores de digestibilidad fueron de 601.3 (E13) a 651.9 (E7)
mg /g harina. Los valores de capacidad calorífica fueron de 4.7 (E7) a 4.9 (E14) kcal/g harina.
Análisis por Resonancia Magnética Nuclear
La espectroscopía 13C CP-MAS NMR se ha empleado ampliamente para la
caracterización de materiales sólidos naturales como semillas (O´Donnell et al., 1981).
Mediante el empleo de esta técnica se ha obtenido información sobre las resonancias de los
carbonos de diferentes familias de componentes, entre ellos proteínas, carbohidratos y ácidos
grasos, lo que permite su análisis cualitativo y cuantitativo. El espectros de 13C NMR de la
harina de bellota presentó unas características espectrales que son típicas de otros materiales
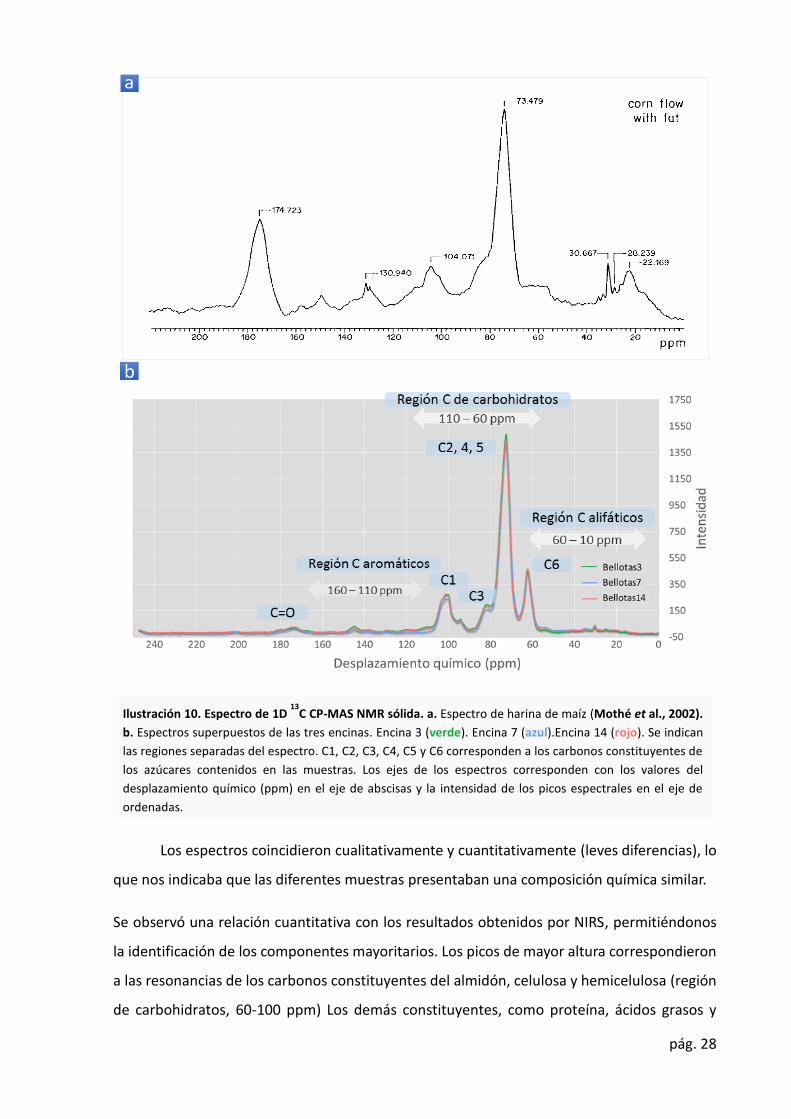
vegetales (Ilustración 10.a) (Mothé et al., 2002). En la Ilustración 10.b se representa el
espectro típico NMR de la harina de bellota en el desplazamiento químico entre 0 y 250 ppm.
De acuerdo a trabajos previos que han empleado esta técnica para el estudio de
materiales biológicos similares (Wooten, 1995) y asumiendo que nuestra muestra cumple un
patrón similar, el espectro pudo ser convenientemente dividido en cuatro regiones: la región
de carbonos alifáticos, 10-60 ppm; la región de carbonos de carbohidratos, 60-110 ppm; la
región de carbonos aromáticos, 110-165 ppm; y la región de carbonos carboxílicos, 165-190
ppm. Las divisiones del espectro se indican en la Ilustración 10.b.
La región alifática contiene las resonancias correspondientes a los carbonos de cadenas
alifáticas de proteínas, aminoácidos, ácidos orgánicos, lípidos, alcaloides, productos naturales
y aceites. También corresponden a resonancias de carbonos de grupos metilos pertenecientes
a las cadenas laterales acetilo de la hemicelulosa y algunos azúcares.

pág. 27
La región de carbohidratos exhibe predominantemente resonancias de carbonos unidos a
oxígeno. Los polisacáridos (celulosa, hemicelulosa, almidón y pectina) son los componentes
mayoritarios en esta categoría. Monosacáridos (principalmente glucosa y fructosa), alcaloides,
y ácidos orgánicos también poseen resonancias en dicha región.
La región aromática posee resonancias correspondientes a carbonos de proteínas y cadenas
laterales de aminoácidos, alcaloides y fenólicos como lignina o ácido clorogénico.
La región carboxílica contiene las resonancias de los carbonos carboxilo y carbonilo
pertenecientes a pectinas, ácidos orgánicos, aminoácidos, el esqueleto carbonado de
proteínas y los grupos carboxilos de las cadenas laterales, lignina, ácido clorogénico, etc. Los
grupos carbonilos peptídicos de proteínas, los carbonos carboxílicos de pectinas y ácidos
orgánicos, también, se localizan en dicha región.
De este modo, a partir del espectro 1D 13C CP-MAS de Arabidopsis thaliana (Wheeler
et al., 2015) se han identificado 9 picos en las 4 regiones establecidas del espectro. El primer
pico (173 ppm) se identificó en la región de carbonos carboxílicos (mayores valores de
desplazamiento químico del espectro). Constituido por las resonancias de los grupos carbonilo
de residuos de ácido galacturónico (principalmente de hemicelulosa, constituyente de la
fibra), de lípidos y proteínas. Seguidamente, se detectó un pico a 144 ppm. Este pico localizado
en la región de carbonos aromáticos está constituido por las resonancias de los carbonos
aromáticos de proteínas (aminoácidos aromáticos) y lignina. Desde el tercer al séptimo pico
identificado se localizaban las resonancias de los carbonos pertenecientes a carbohidratos
(60-110 ppm). El tercer pico (101 ppm) correspondió a las resonancias de los C1 de celulosa y
hemicelulosa y el cuarto pico (94 ppm) a las resonancias del C1 del almidón. El quinto pico que
se identificó (81.6 ppm) estaba constituido por las resonancias de los C4 de celulosa,
hemicelulosa y almidón. El sexto pico (72.5 ppm) correspondió a las resonancias de los C2, C3
y C5 de celulosa, hemicelulosa y almidón. Y el séptimo pico (62 ppm) perteneciente a la región
de carbonos de carbohidratos, correspondió las resonancias del C6 de celulosa, hemicelulosa
y almidón. El octavo pico identificado (55 ppm) se localizó en la región de carbonos alifáticos,
correspondió a las resonancias de los grupos OCH3 de la lignina. Y el último pico identificado
(31 ppm) correspondió a los grupos (CH2)n y CH3 de lípidos y proteínas.
pág. 28
Los espectros coincidieron cualitativamente y cuantitativamente (leves diferencias), lo
que nos indicaba que las diferentes muestras presentaban una composición química similar.
Se observó una relación cuantitativa con los resultados obtenidos por NIRS, permitiéndonos
la identificación de los componentes mayoritarios. Los picos de mayor altura correspondieron
a las resonancias de los carbonos constituyentes del almidón, celulosa y hemicelulosa (región
de carbohidratos, 60-100 ppm) Los demás constituyentes, como proteína, ácidos grasos y
Ilustración 10. Espectro de 1D 13
C CP-MAS NMR sólida. a. Espectro de harina de maíz (Mothé et al., 2002).
b. Espectros superpuestos de las tres encinas. Encina 3 (verde). Encina 7 (azul).Encina 14 (rojo). Se indican
las regiones separadas del espectro. C1, C2, C3, C4, C5 y C6 corresponden a los carbonos constituyentes de
los azúcares contenidos en las muestras. Los ejes de los espectros corresponden con los valores del
desplazamiento químico (ppm) en el eje de abscisas y la intensidad de los picos espectrales en el eje de
ordenadas.

pág. 29
fibra, estarían en menor cantidad, como indican las menores alturas de los picos en las
regiones de resonancia de los grupos carbonilos (C=O), carbonos aromáticos y carbonos
alifáticos. Esta distribución coincidió con los resultados obtenidos con NIRS, y como se
publican en otros trabajos sobre la composición de harina de bellota (Silva et al., 2016).
Al contrario que NIRS, NMR no nos ha permitido realizar un análisis individualizado de
compuestos específicos. Esto impidió establecer que individuo posee una mayor
concentración de un determinado componente, puesto que un pico estaba constituido por las
resonancias de carbonos de distintos compuestos. Esto se observó, en los picos de las
resonancias de los carbonos de carbohidratos, que constituyeron la suma del contenido en
almidón, celulosa y hemicelulosa. Así, se observó que la E7 presentó menores alturas de los
picos, lo que indicó una menor concentración de almidón, celulosa y hemicelulosa. Sin
embargo, los resultados de NIRS, nos indicaron lo contrario (E3 (62,6) E7 (65,4) y E14 (60,5)).
Los resultados obtenidos con 1D 13C CP-MAS NMR nos permitieron identificar leves
diferencias entre los diferentes individuos analizados. Se observó que las E3 y E7 presentaron
picos de mayor altura en la región de las resonancias de carbonos de carbohidratos en el
espectro de NMR (Ilustración 10). En cambio, E14, que poseyeron un mayor contenido de
grasa y proteína, presentaron picos de mayor altura en las regiones de las resonancias de los
carbonos de los grupos carbonilo (enlaces peptídicos) y de cadena alifáticas (de ácidos grasos
y proteínas).
Para una estimación cuantitativa de compuestos o familias de compuestos sería
necesario la generación de ecuaciones de predicción, como se emplea en NIRS, algo que no se
ha llevado a cabo con muestras de harina de bellotas o de semillas.
Análisis por Cromatografía de Gases acoplada a Espectrometría de Masas
Los métodos colorimétricos, NIRS y 1D 13C CP-MAS NMR analizados previamente, no
nos han aportado información sobre compuestos individuales. Para poder identificar
metabolitos se necesitaron de una etapa de purificación de los mismos, tal y como la
cromatografía de gases o líquida que puede acoplarse a una posterior identificación y
cuantificación por espectrometría de masas. Empleando los extractos de las harinas, en el
presente trabajo se realizó un análisis por GC-MS. A partir de los espectros m/z y los tiempos
de retención y el posterior búsqueda en las bases de datos (Alkane, Fiehn library 1 y 2, Gölm
pág. 30
Metabolome Database, GC-TSQ y MoSys) se identificaron algunos de los compuestos. No se
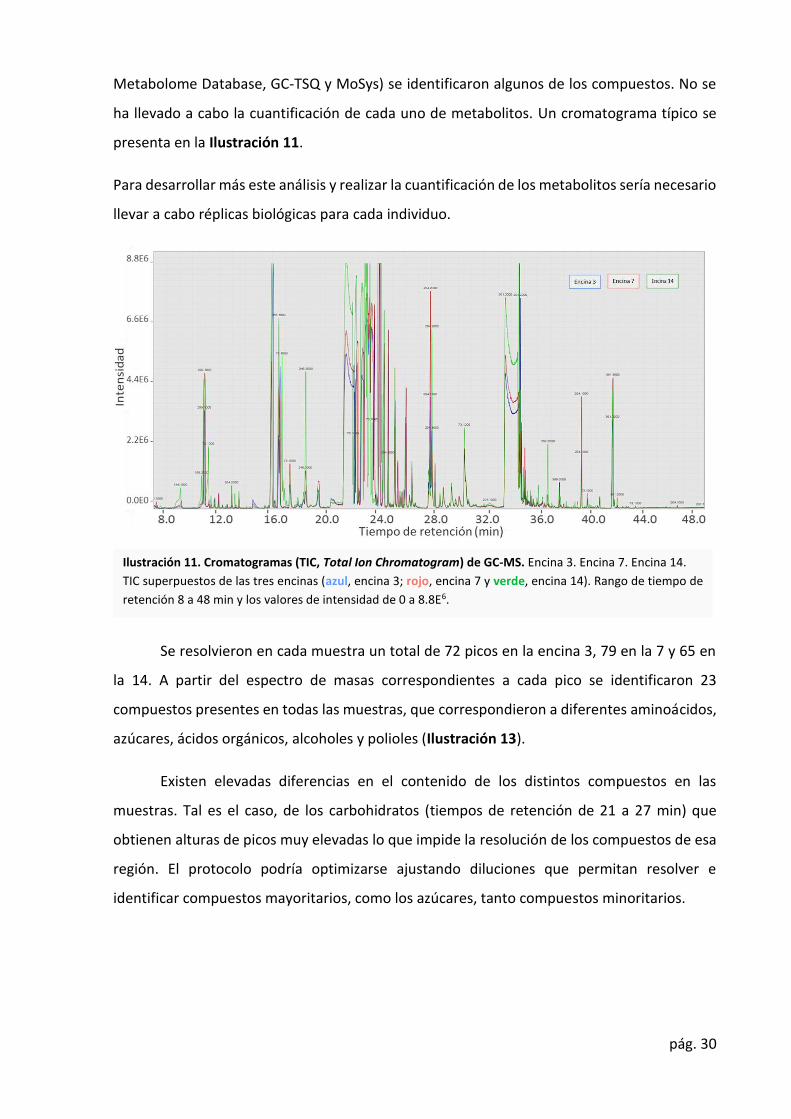
ha llevado a cabo la cuantificación de cada uno de metabolitos. Un cromatograma típico se
presenta en la Ilustración 11.
Para desarrollar más este análisis y realizar la cuantificación de los metabolitos sería necesario
llevar a cabo réplicas biológicas para cada individuo.
Se resolvieron en cada muestra un total de 72 picos en la encina 3, 79 en la 7 y 65 en
la 14. A partir del espectro de masas correspondientes a cada pico se identificaron 23
compuestos presentes en todas las muestras, que correspondieron a diferentes aminoácidos,
azúcares, ácidos orgánicos, alcoholes y polioles (Ilustración 13).
Existen elevadas diferencias en el contenido de los distintos compuestos en las
muestras. Tal es el caso, de los carbohidratos (tiempos de retención de 21 a 27 min) que
obtienen alturas de picos muy elevadas lo que impide la resolución de los compuestos de esa
región. El protocolo podría optimizarse ajustando diluciones que permitan resolver e
identificar compuestos mayoritarios, como los azúcares, tanto compuestos minoritarios.
Ilustración 11. Cromatogramas (TIC, Total Ion Chromatogram) de GC-MS. Encina 3. Encina 7. Encina 14.
TIC superpuestos de las tres encinas (azul, encina 3; rojo, encina 7 y verde, encina 14). Rango de tiempo de
retención 8 a 48 min y los valores de intensidad de 0 a 8.8E6.

pág. 31
Se tomaron las alturas de pico como valor de cantidad en la muestra. Atendiendo a la
altura de pico los metabolitos mayoritarios fueron los azúcares. En la Ilustración 12 se
muestran las concentraciones relativas de los picos de los metabolitos identificados
considerando la altura de los picos como indicador de concentración de cada compuesto en
la muestra. De acuerdo, a esta asunción se establece una mayor concentración de metabolitos
en la E14. De hecho, en el cromatograma (Ilustración 11) se observa una mayor altura de los
picos correspondientes a la E14.
Todas las técnicas empleadas han dado información sobre la información química de los
frutos.
Ilustración 12. Heatmap de los metabolitos identificados en los extractos de los frutos de las tres encinas.
Los valores de la escala van de 1 a 0. Los valores de cada metabolito en función de la escala se han obtenido
debido a la normalización al dividir todos los valores de altura de pico con el valor mayor, siendo más
próximos a 1 los de mayor valor y cercanos a 0 los de menor valor.
Ilustración 13. Metabolitos identificados. Se presenta los 23 metabolitos identificados inequívocamente. Se
han seleccionado aquellos metabolitos identificados presentes en las tres muestras. Los metabolitos fueron
identificados tras la comparación con las bases de datos (Alkane, Fiehn library 1 y 2, Gölm Metabolome
Database, GC-TSQ y MoSys).

pág. 32
Relación entre la composición química y las características morfométricas
Se llevó a cabo un análisis estadístico para identificar la relación entre la variabilidad
en composición química y metaboloma y las características morfométricas de los frutos de las
encinas.
Autores como Valero establecieron correlaciones entre la composición química con las
características morfométricas de longitud, peso y diámetro máximo (Valero et al., 2010 y
Valero et al., 2011). Se ha realizado una matriz de correlaciones para mostrar las posibles
correlaciones (Ilustración 14)
Analizando la matriz de correlaciones, se arrojan resultados que confirman las
hipótesis previamente establecidas en los trabajos de Valero. Existió correlación positiva entre
volumen y la cantidad de proteína de las bellotas (r2= 0.75). También se observó dicha
correlación positiva con la cantidad de fibra, grasa y energía (r2= 0.62, r2= 0.60, r2= 0.61). Por
el contrario, se observó una correlación negativa entre el diámetro de los frutos y la cantidad
de almidón (r2= -0.68). No se observó correlación entre el volumen y el contenido en azúcares
(r2= 0.06) y el contenido hídrico de los frutos (r2= 0.03).
La existencia de correlaciones entre la composición química y las características
morfométricas, nos permiten seleccionar aquellos frutos con una determinada composición
química, mediante el análisis de sus características fenotípicas y sin necesidad del empleo de
métodos para su cuantificación.
Ilustración 14. Matriz de
correlaciones entre las
características morfométricas y
los datos de composición
química obtenidos mediante
NIRS. Los valores de la escala van
de 1 a -1. Los valores de r
próximos a 1 indican una
correlación positiva (azul). Los
valores de r próximos a -1 indican
una correlación negativa (rojo).
Los valores de r próximos a 0
indican ausencia de correlación.
Los valores de r fueron obtenidos
mediante el test de correlación
de Pearson entre las variables.

pág. 33
Valoración de las diferentes técnicas empleadas
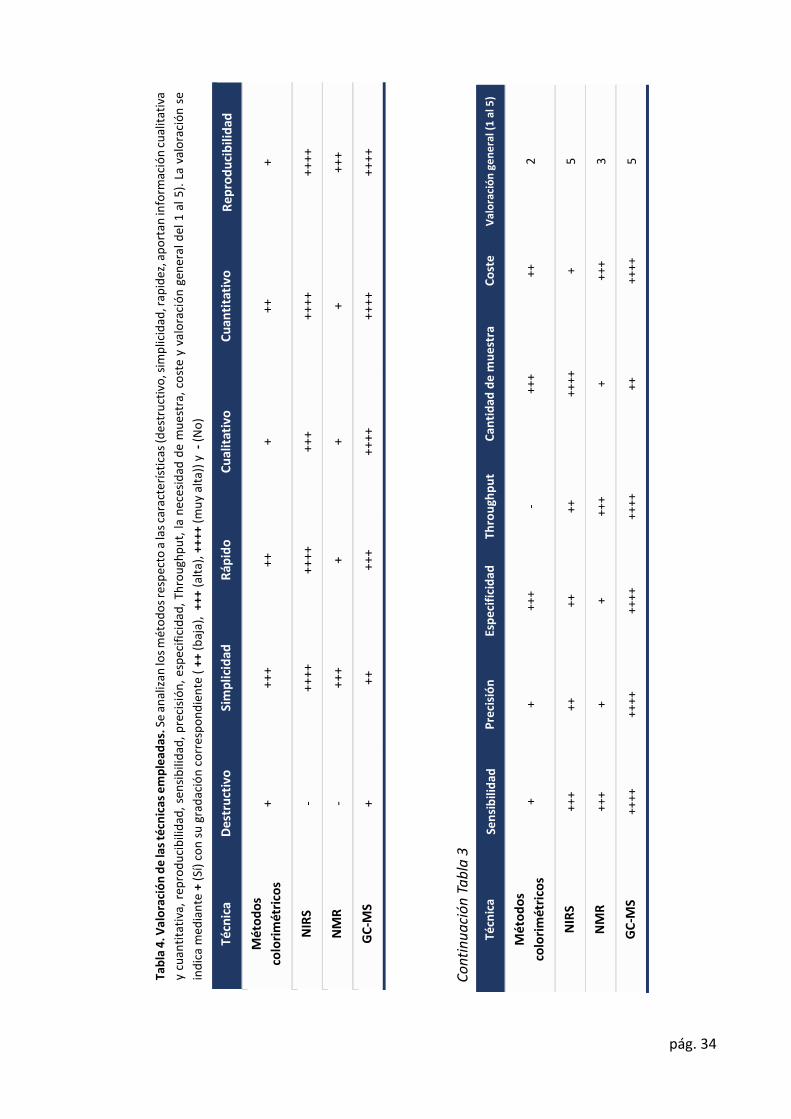
En esta sección se van a valoraron las diferentes técnicas empleadas (Tabla 4).
Individualmente, ninguna de las técnicas permite, por sí sola, abordar los objetivos de estudios
de variabilidad, catalogación y búsqueda de principios bioactivos. Cada técnica aporta
información que puede complementarse para adquirir conocimiento de la composición, el
perfil de metabolitos y la variabilidad intrapoblacional.
NIRS nos aporta información validada sobre la composición química, que nos permiten
identificar diferencias entre individuos. Sin embargo, no nos permite identificar compuestos
que no se ha establecido previamente en las ecuaciones de calibrado.
Con GC-MS podemos adquirir información tanto de compuestos individuales, como la
detección de variabilidad entre individuos. Pero, debido a que el contenido de metabolitos
secundarios es menor que el de metabolitos primarios no es una técnica que nos permita
identificar una gran mayoría de metabolitos secundarios, que son los que realmente definen
el fenotipo. A su vez, la complejidad de los compuestos, que pueden llegar a diferenciarse en
un único grupo funcional, dificulta su identificación. El incremento del material de partida, no
es una solución válida, como puede observarse en la región saturada del cromatograma
debido al elevado contenido en azúcares (Ilustración 11). Por ello, para su detección e
identificación se requieren de técnicas como LC-Orbitrap-MS (Meijón et al., 2016). Por lo que
la combinación de ambas técnicas (GC-MS y LC-Orbitrap-MS) sería una buena opción para la
metabolómica no dirigida y alcanzar una máxima cobertura del metaboloma.
Además, de estas dos técnicas, el empleo de NMR, como paso previo a la extracción de
metabolitos, sería ideal. Mediante NMR en estado sólido, podríamos identificar aquellas
muestras más diferenciadas e identificar cual es la familia de compuestos que origina dicha
variabilidad. Es decir, nos permitiría una metabolómica más dirigida.
pág. 34
Tab
la 4
. Va
lora
ció
n d
e la
s té
cnic
as e
mp
lead
as. S
e an
aliz
an lo
s m
éto
do
s re
spec
to a
las
cara
cter
ísti
cas
(de
stru
ctiv
o, s
imp
licid
ad, r
apid
ez, a
po
rtan
info
rmac
ión
cu
alit
ativ
a
y cu
anti
tati
va,
rep
rod
uci
bili
dad
, se
nsi
bili
dad
, p
reci
sió
n,
esp
ecif
icid
ad,
Thro
ugh
pu
t, l
a n
eces
idad
de
mu
est
ra,
cost
e y
valo
raci
ón
gen
eral
del
1 a
l 5
). L
a va
lora
ció
n s
e
ind
ica
med
ian
te +
(Sí
) co
n s
u g
rad
ació
n c
orr
esp
on
die
nte
( +
+ (b
aja)
, ++
+ (a
lta)
, ++
++ (
mu
y al
ta))
y -
(N
o)
Técn
ica
Des
tru
ctiv
o
Sim
plic
idad
R
ápid
o
Cu
alit
ativ
o
Cu
anti
tati
vo
Re
pro
du
cib
ilid
ad
Mét
od
os
colo
rim
étri
cos
+ ++
+ ++
+
++
+
NIR
S -
++++
++
++
+++
++++
++
++
NM
R
- ++
+ +
+ +
+++
GC
-MS
+ ++
++
+ ++
++
++++
++
++
Co
ntin
ua
ció
n T
ab
la 3
Técn
ica
Sen
sib
ilid
ad
Pre
cisi
ón
Es
pe
cifi
cid
ad
Thro
ugh
pu
t
Can
tid
ad d
e m
ue
stra
C
ost
e
Val
ora
ció
n g
en
era
l (1
al 5
)
Mét
od
os
colo
rim
étri
cos
+
+
+++
- ++
+
++
2
NIR
S ++
+
++
++
++
++++
+
5
NM
R
+++
+
+
+++
+
+++
3
GC
-MS
++++
++++
++++
++++
++
++++
5

pág. 35
4. Conclusiones
A partir del análisis del perfil de metabolitos del fruto de encina empleando las técnicas
colorimétricas, NIRS, NMR y GC-MS, se puede concluir:
i. Los componentes mayoritarios en el fruto de encina fueron almidón, azúcares, ácidos
grasos y proteínas.
ii. El análisis in vitro de los extractos permitió cuantificar los valores de aminoácidos
libres, azúcares y compuestos fenólicos mediante ensayos colorimétricos.
iii. NIRS permitió la determinación de los valores de ceniza, proteína, grasa, almidón,
azúcares, fibra, diversos ácidos grasos, la digestibilidad y capacidad calorífica de los
frutos. Y con 13C NMR se establecieron las zonas del espectro que contenían las
resonancias de los carbonos de los constituyentes mayoritarios.
iv. Con GC-MS se identificaron 23 metabolitos (diferentes aminoácidos, azúcares, ácidos
orgánicos, alcoholes y polioles).
v. Los resultados de composición química y las características morfométricas permitieron
establecer diferencias entre los individuos de la población.
vi. Se observaron correlaciones positivas y negativas entre la composición química y las
características morfométricas.
vii. Tras la evaluación de las diversas técnicas empleadas se pudo establecer que las
técnicas que generan mejor información para la descripción del perfil de metabolitos
del fruto de encina son NIRS y GC-MS.
Conclusions
Considering the analysis of the metabolites profile obtained by means of colorimetric
assays, NIRS, NMR and GC-MS could be concluded:
i. Starch, sugars, fatty acids and proteins have been the mayor acorn components.
ii. In vitro acorn flour extract analysis allowed to obtain values of free amino acids, sugars
and phenolic compounds.
iii. Non-destructive techniques such as NIRS analyzed ash, protein, fat, starch, sugars,
different fatty acids, digestibility and energy. The 13C NMR spectra of acorn flour has
exhibited the same spectral characteristic found in other plant materials. The detected
regions contain carbons resonances due to the carbons in mayor acorn components.

pág. 36
iv. GC-MS enabled to detection 23 metabolites (amino acids, sugars, fatty acids, alcohols
and polyols).
v. Differences in intrapopulation acorn chemical composition and morphometry have
been detected. Allowing to state that there are differences among Holm oak trees.
vi. Positive and negative correlations between chemical composition and morphometry
has been described.
vii. Analyzing the previous results, it could be stated that NIRS and GC-MS are the best
option to study Holm oak acorn metabolites profiles.
5. Bibliografía
1. Sghaier-Hammami B., Valero-Galvàn J., Romero-Rodríguez M. C., Navarro-Cerrillo R. M.,
Abdelly C., Jorrín-Novo J. (2013). Physiological and proteomics analyses of Holm oak
(Quercus ilex subsp. ballota [Desf.] Samp.) responses to Phytophthora cinnamomi. Plant
Physiology and Biochemistry 71:191–202
2. Romero-Rodríguez MC., Pascual J., Valledor L., Jorrín-Novo J. (2014) Improving the quality
of protein identification in non-model species. Characterization of Quercus ilex seed and
Pinus radiate needle proteomes by using SEQUEST and custom databases. Journal of
Proteomics 105:85-91
3. Jorrín-Novo J., Navarro-Cerrillo R.M. (2014) Variabilidad y respuesta a distintos estreses
en poblaciones de encina (Quercus ilex L.) en Andalucía mediante una aproximación
proteómica. Ecosistemas 23:99-107
4. Simova-Stoilova L. P., Romero-Rodríguez M. C., Sánchez-Lucas R., Navarro-Cerrillo R. M.,
Medina-Aunon J. A. and. Jorrín-Novo J. (2015) DE proteomics analysis of drought treated
seedlings of Quercus ilex supports a root active strategy for metabolic adaptation in
response to water shortage. Frontiers in Plant Science 6:627
5. Castro-Díez P., Villar-Salvador P., Pérez-Rontomé C., Maestro-Martínez M., Montserrat-
Martí G. (1997) Leaf morphology and leaf chemical composition in three Quercus
(Fagaceae) species along a rainfall gradient in NE Spain. Trees-Structure and Function
11:127–34.
6. Valero-Galván J., Jorrín-Novo J., Cabrera A., Ariza D., García-Olmo J., Cerrillo R. (2010)
Population variability based on the morphometry and chemical composition of the acorn
in Holm oak (Quercus ilex subsp. ballota [Desf.] Samp.). European Journal of Forest
Research 131:893-904

pág. 37
7. Valero-Galván J., Valledor L. Navarro Cerrillo R. M., Gil Pelegrín E., Jorrín-Novo J V. (2011)
Studies of variability in Holm oak (Quercus ilex subsp. ballota[Desf.] Samp.) through
acorn protein profile analysis. Journal of Proteomics 74:1244 – 1255
8. Jimenez P, de Heredia UL, Collada C, Lorenzo Z, Gil L. (2004) High variability of chloroplast
DNA in three Mediterranean evergreen oaks indicates complex evolutionary history.
Heredity 93:510–515.
9. Lumaret R, Jabbour-Zahab R. (2009) Ancient and current gene flow between two
distantly related Mediterranean oak species, Quercus suber and Q. ilex. Annals of Botany
104:725–736.
10. Jorge I., Navarro Cerrillo RM., Lenz C, Ariza D., Porras C., Jorrin J. (2005) The Holm oak leaf
proteome: analytical and biological variability in the protein expression level assessed
by 2-DE and protein identification tandem mass spectrometry de novo sequencing and
sequence similarity searching. Proteomics 5:222–34.
11. Cantos E., Espín J.C., López-Bote C., De la Hoz L., Ordoñez J. and Tomás-Barberán F. A.
(2003) Phenolic Compounds and Fatty Acids from Acorns (Quercus spp.), the Main
Dietary Constituent of Free-Ranged Iberian Pigs. Journal of Agricultural and Food
Chemistry 51:6248−6255
12. Karioti A., Sokovic M., Ciric A., Koukoulitsa C., Bilia A. and Skaltsa H. (2011) Antimicrobial
properties of Quercus ilex L. proanthocyanidin dimers and simple phenolics: Evaluation
of their synergistic activity with conventional antimicrobials and prediction of
their pharmacokinetic profile. Journal of Agricultural and Food Chemistry 59:6412–6422
13. Yarnes C.T., Boecklen W.J., Tuominen K. and Salminen J. (2006) Defining phytochemical
phenotypes:size and shape analysis of phenolic compounds in oaks (Fagaceae, Quercus)
of the Chihuahuan Desert. Canadian Journal Botanical 84:1233-1248
14. Güllüce M., Adıgüzel A., Ögütçü H., Tengül M., Karaman I. and Tahin F. (2004)
Antimicrobial effects of Quercus ilex L. extract. Phytotherapy Research 18:208-211
15. Scherling C., Roscher C., Giavalisco P., Detlef Schulze E. and Weckwerth W. (2010)
Metabolomics unravel contrasting effects of biodiversity on the performance of
individual plant species. PLoS ONE 5(9):e12569
16. Oliver S. G., Winson M. K., Kell D. B., Baganz F. (1998) Systematic functional analysis of
the yeast genome. Trends in Biotechnology 16:373–378
17. Fiehn O. (2002) Metabolomics – the link between genotypes and phenotypes. Plant
Molecular Biology 48:155–171

pág. 38
18. Sheth B., Thaker V. (2014) Plant systems biology: insights, advances and challenges.
Planta 240:33-54
19. Patti G.J., Yanes O. et al. (2012) Innovation: Metabolomics: the apogee of the omics
trilogy. Nature Reviews Molecular Cell Biology 13:263-269
20. Weckwerth W. (2011) Green systems biology - From single genomes, proteomes and
metabolomes to ecosystems research and biotechnology. Journal of Proteomics 75:284-
305
21. Trethewey R.N (2004) Metabolite profiling as an aid to metabolic engineering in plants.
Current Opinion in Plant Biology 7:196-201
22. Alonso A. Marsal S. Julià A. (2015) Analytical methods in untargeted metabolomics: state
of the art in 2015. Frontiers in Bioengineering and Biotechnology 3
23. Ainsworth E.A. and Gillespie K.M (2007) Estimation of total phenolic content and other
oxidation substrates in plant tissues using Folin–Ciocalteu reagent. Nature protocols
2:875-877
24. Miller, G.L., (1959) Use of dinitrosalicylic acid reagent for determination of reducing
sugar. Analytical Chemistry 31:426
25. Smith A. M. and Agiza A. H. (1951) The determination of amino acids colorimetrically the
Ninhydrin reaction. The Wdinburgh and Eastof Scotland college of agriculture 6:623-627
26. Porter R. H., Durrell M., and Romm H. J. (1946) The use of 2,3,5-tiriphenyl-
tetrazoliumchloride as a measure of seed germinability Plant physiology 1384:149-159
27. Schimleck, LR, Raymond, CA, Beadle, CL, Downes, GM, Kube, PD & French, J (2000)
Applications of NIR spectroscopy to forest research. Appita Journal: Journal of the
Technical Association of the Australian and New Zealand Pulp and Paper Industry 53:458-
464.
28. Adedipe O. E., Dawson-Andoh B., Slahor J. and Osborn L. (2008) Classification of red oak
(Quercus rubra) and white oak (Quercus alba) wood using a near infrared spectrometer
and soft independent modelling of class analogies. Journal of Near Infrared Spectroscopy
16:49-57
29. Sousa-Correia C., Alves A., Rodrigues J., Ferreira-Dias S., Abreu J.M, Maxted N., Ford-Lloyd
B. and Schwanninger M. (2007) Oil content estimation of individual kernels of Quercus
ilex subsp. Rotundifolia [(Lam) O. Schwarz] acorns by Fourier transform near infrared
spectroscopy and partial least squares regression. Journal of Near Infrared Spectroscopy
5:247–260

pág. 39
30. Vásquez D. R., Abadía B., and Arreaza L. C. (2004) Near Infrared Reflectance Spectroscopy
(NIRS) applied to nutritional characterization of Guinea grass and corn grain. Revista
Corpoica 5
31. Theodoridis G. A., Gikab H. G., Want E. J. and Wilson I. D. (2011) Liquid chromatography-
mass spectrometry based global metabolite profiling: A review. Analytica Chimica Acta
711:7–16
32. Ratcliffe R G. and Shachar-Hill Y. (2011) Probing plant metabolism with NMR. Annual
Review of Plant Physiology and Plant Molecular Biology 52:499-526
33. Bothwell J.H., Griffin J.L. (2011) An introduction to biological nuclear magnetic resonance
spectroscopy. Biological Review 86:493-510.
34. Reo N. V. (2002) NMR-Based metabolomics. Drug and chemical toxicology 25:375–382
35. Last R. L., Jones A. D. and Shachar-Hill Y. (2007) Towards the plant metabolome and
beyond Nature Reviews 8:167-174
36. Krishnan P., Kruger N. J. and. Ratcliffe R. G, (2005) Metabolite fingerprinting and profiling
in plants using NMR. Journal of Experimental Botany 56:255-265
37. Sardans J., Gargallo-Garriga A., Perez-Trujillo M., Parella T.J., Seco, R., Filella I. and
Peñuelas J. (2014) Metabolic responses of Quercus ilex seedlings to wounding analysed
with nuclear magnetic resonance profiling. Plant Biology 16:395-403
38. Adjami Y., Ghanem R., Daas H., Laid Ouakid M., Pujade Villar J., Bairi A. (2016) Influence of
carpophagous attack on metabolites of cork oak (Quercus suber) acorns. Turkish Journal
of Forestry 17:51-57
39. Kersten B., Ghirardo A., Schnitzler J.P., Kanawati B., Schmitt-Kopplin P, Fladung M.,
Schroeder H. (2013) Integrated transcriptomics and metabolomics decipher differences
in the resistance of pedunculate oak to the herbivore Tortrix viridana L. BCM Genomics
4:737
40. Connor K.F. and Sowa S. (2003) Effects of desiccation on the physiology and biochemistry
of Quercus alba acorns. Tree Physiology 23:1147-1152
41. Bonner, F.T.; Vozzo, John A. (1987) Seed Biology and Technology of Quercus. Gen. Tech.
Rep. SO-66. New Orleans, LA: U.S. Dept of Agriculture, Forest Service, Southern Forest
Experiment Station. 21 p.
42. Caliskan S. (2014) Germination and seedling growth of holm oak (Quercus ilex L.): effects
of provenance, temperature, and radicle pruning. iForest - Biogeosciences and Forestry
7:103-109

pág. 40
43. Corbineau F., Xia Q., Bailly C. and El-Maarouf-Bouteau H. (2014) Ethylene, a key factor in
the regulation of seed dormancy. Frontiers in Plant Science 5:539
44. Porter R.H., Durrell M. and Romm H. J (1946) The use of 2,3,5-Triphenyl-
Tetrazoliumchloride as a measure of seed germinability. Plant Physiology 86:149-159
45. Massiot D., Fayon F., Capron M., King I., Le Calvé S., Alonso B., Durand J.O., Bujoli B., Gan
Z., Hoatson G. (2002) Modelling one and two-dimensional solid-state NMR spectra.
Magnetic Resonance in Chemistry 40:70-76
46. Valledor L., Escandón M., Meijón M., Nukarinen E., Cañal M.J. and Weckwerth W. (2014)
A universal protocol for the combined isolation of metabolites, DNA, long RNAs, small
RNAs, and proteins from plants and microorganisms The Plant Journal 79:173–180
47. Meijón M., Feito I., Oravec M., Delatorre C., Weckwerth W., Majada J. and Valledor L.
(2016) Exploring natural variation of Pinus pinaster Aiton using metabolomics: Is it
possible to identify the region of origin of a pine from its metabolites? Molecular Ecology
25:959–976
48. Furuhashi T., Fragner L., Furuhashi K., Valledor L., Sun X. and Weckwerth W. (2012)
Metabolite changes with induction of Cuscuta haustorium and translocation from host
plants Journal of Plant Interactions 7:84-93
49. Pluskal T., Castillo S., Villar-Briones A., Orešič M. (2010) MZmine 2: Modular Framework
for Processing, Visualizing, and Analyzing Mass Spectrometry Based Molecular Profile
Data. BMC Bioinformatics 11:395
50. Midilli M., Muglah Ö.H., Altintas L., Erol H. and Cakir S. (2008) Shelled acorn seed (Quercus
cerris) as a diet ingredient on the performance of growing Japanese Quail. South African
Journal of Animal Science 1:38-42
51. Özcan T. (2006) Total protein and amino acid compositions in the acorns of Turkish
Quercus L. taxa. Genetic Resources and Crop Evolution 53:419–429
52. Rodríguez-Estévez V., García A, Gómez A.G. (2009) Characteristics of the acorns selected
by free range Iberian pigs during the montanera season. Livestock Science 122:169-176.
53. O'Donnell D.J., Ackerman J, J., Maciel G.E. (1981) Comparative study of whole seed
protein and starch content via cross polarization-magic angle spinning carbon-13 nuclear
magnetic resonance spectroscopy. Journal of agricultural and food chemistry 3:514-518.
54. Mothé C. G., Machado M. G. S., Tavares M. I. B. (2002) Solid-State 13C-NMR and Study of
the Subproducts Obtained from Corn Industry. Journal of Applied Polymer Science
84:1680–1685

pág. 41
55. Wooten J. B. (1995) 13C CPMAS NMR of Bright and Burley Tobaccos. Journal of agricultural
and food chemistry 43:2858-2868
56. Wheeler H.L., Soong R., Courtier-Murias D., Botana A., Fortier-Mcgill B., Maas W.E, Fey M.,
Hutchins H., Krishnamurthy S., Kumar R., Monette M., Stronks H.J., Campbell M.M. and
Simpson A. (2015) Comprehensive multiphase NMR: a promising technology to study
plants in their native state. Magnetic Resonance in Chemistry 53:735-744
57. Silva S., Costa M. E., Borges A., Carvalho A. P., Monteiro M. J., Pintado M. E. (2016)
Nutritional characterization of acorn flour (a traditional component of the
Mediterranean gastronomical folklore). Food Measure 10:584-589

![Estabelecimento do perfil metabolómico volátil da urina e · Na urina o grupo de pacientes com cancro da mama foi maioritariamente caracterizado pelo metabolito 1-[2-(Isobutiriloxi)-1-metiletil]-2,2-dimetilpropil](https://img.pdfslide.net/doc/110x75/5bfed70f09d3f2641b8bb8b4/estabelecimento-do-perfil-metabolomico-volatil-da-urina-e-na-urina-o-grupo.jpg)



























