Embed Size (px)
Citation preview

American Journal of Medical Genetics 27:651-660 (1987)
Apparently New Autosomal Recessive Syndrome of Mental Retardation, Dista1 Limb Deficiencies, Oral Involvement, and Possible Renal Defect
M. Buttiens and J.P Fryns
Center for Human Genetics, University of Leuven, Leuven, Belgium
This paper illustrates a syndrome of distal limb deficiency and oral defects in two sibs, a iiioderately mentally retarded man and his mildly retarded sister. Both have rnicroretrognathia, rnicrostomia, normal tongue, and symmetric severe limb defi- ciencies. This seems to be a previously undcscribed syndrome. The nosology of the different orofacial syndroines associated with distal limb deficiencies is discussed.
Key words: limb deficiency-mental retardation syndrome, microretrognathia, microstomia
INTRODUCTION
The sporadic association of peromelia and micrognathia, with or without cranial nerve palsies and/or tongue hypoplasia, has been described under severa1 names: Hanhart syndrome, aglossia-adactylia syndrome, hypoglossia-hypodactylia syn- drome, ankyloglossia superior or glossapalatine-ankylosis syndrome, Mobius syn- drome and Poland-Mobius syndrome [Herrmann et al, 19761. A complex nosological classification has remained problematic : some authors consider these syndromes as isolated entities with specific characteristics; others have introduced terms such as “community of face-limb malformation syndromes” [Kaplan et al, 19761, “oroman- dibular limb hypogenesis syndrome” [Goodman and Gorlin, 19771, and “ facial-limb disruptive spectrum” [Smith and Jones, 19821, referring to the striking overlap between these “entities.” Herrmann et al [1976] divided them into two groups, Hanhart syndrome and Poland-Mobius syndrome.
Received for publication August 23, 1986; revision received January 5, 1987.
Address reprint requests to J.P. Fryns, Center for Hurnan Genetics, U.Z. Gasthuisberg, Herestraat 49, B-3000 Leuven, Belgium.
O 1987 Alan R. Liss, Inc.

652 Buttiens and Fryns
Fig. 1. The typical craniofacial niorphology (patient i ) , with midfacial hypoplasia and micrognathia
As far as we know, al1 reported cases have been sporadic, and consanguinity was rarely documented. In this report, we describe a moderately mentally retarded brother and his mildly mentally retarded sister with micrognathia and severe symme- tric limb defects without tongue abnormalities or cranial nerve palsy .
CLlNlCAL REPORTS Patient 1
C.A., a 21-year-old man, was examined during a follow-up hospitalization in the Department of Nephrology. At age 16 years, evaluation of persistent proteinuria documented an oligomeganephronia. He is moderately mentally retarded (IQ 45; Terman scale). At birth the combination of severe oral and limb defects was noted, and he was admitted to an institution for the mentally retarded from age 5 months. He is the second child of normal Greek parents. As far as we could determine, there were no familia1 antecedents of consanguinity, congenital malformations, or mental retardation.
On clinical examination, we found peculiar facial anomalies associated with severe limb defects. His height was 163 cm, weight 55 kg, and head circumference (OFC) 55 cm. He had severe micrognathia (Fig. 1), normal ocular motility, high nasal bridge, maxillary hypoplasia, a relatively small mouth, highly arched palate, and narrow upper dental arch but no oligodentia or hypoplasia of the tongue. Severe limb defects were present (Fig. 2), with deficiency of the dista1 two-thirds of the forearms and similar transverse defects of feet (with absence of toes). Secondary sexual development was normal, but the left testis was undescended. There was no evidence of cardiac or intestinal malformations. Ophthalmological examination showed
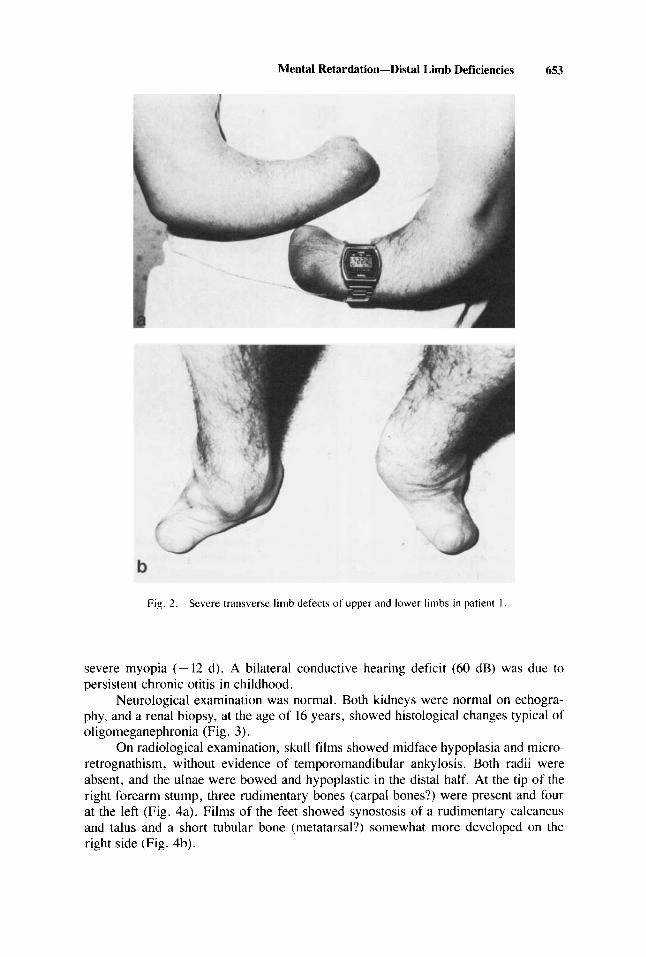
Mental Retardation-Dista1 Lirnb Deficiencies 653
Fig. 2. Scvcre transverse limb defects of upper and lower limbc i n patient 1
severe myopia (-12 d). A bilateral conductive hearing deficit (60 dB) was due to persistent chronic otitis in childhood.
Neurological examination was normal. Both kidneys were normal on echogra- phy, and a renal biopsy, at the age of 16 years, showed histological changes typical of oligomeganephronia (Fig. 3).
On radiological examination, skull films showed midface hypoplasia and micro- retrognathism, without evidence of temporomandibular ankylosis. Both radii were absent, and the ulnae were bowed and hypoplastic in the dista1 half. At the tip of the right forearm stump, three rudimentary bones (carpa1 bones?) were present and four at the left (Fig. 4a). Films of the feet showed synostosis of a rudimentary calcaneus and talus and a short tubular bone (metatarsal?) somewhat more developed on the right side (Fig. 4b).
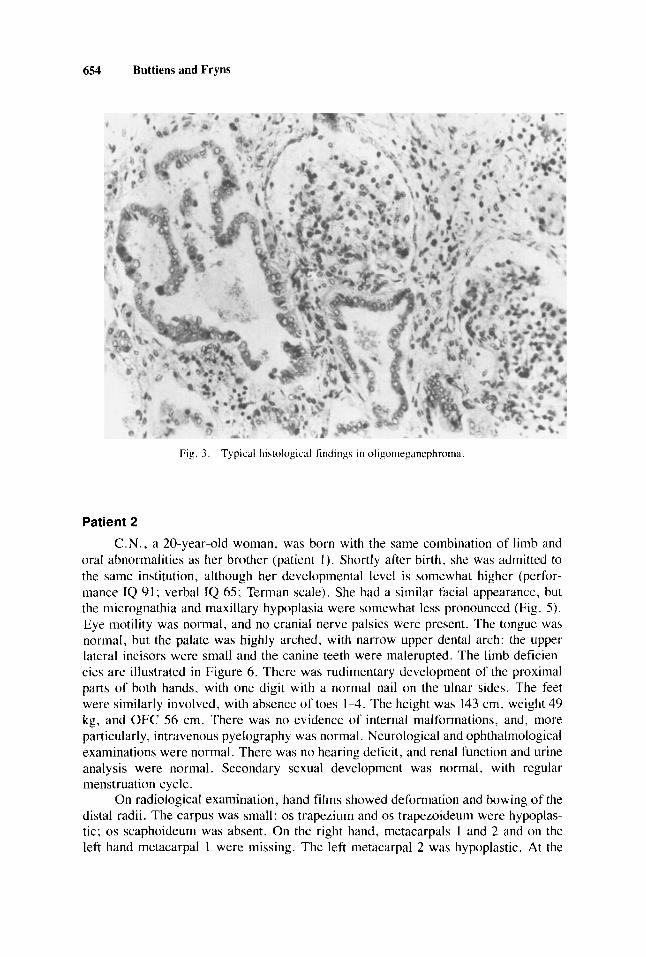
654 Buttiens and Fryns
Fig. 3 . Typical histological tindings in oligoriieganephroina
Patient 2
C.N., a 20-year-old woman, was born with the same combination of limb and oral abnormaiities as her brother (patient 1 ) . Shortly after birth, she was admitted to thc same institution, although her developmental ievel is somewhat higher (perfor- mance IQ 91; verbal IQ 65; Terman scale). She had a similar facial appearance, but the rnicrognathia and maxiilary hypoplasia were soniewhat lcss pronounced (Fig. 5 ) . Eye iriotility was normal, and no cranial nerve palsies were present. The tongue was normal, but thc palate was highly arched, with narrow upper dental arch: the upper lateral incisors were small and the canine teeth were malerupted. The iimb deficien- cies are illustrated in Figure 6 . There was rudinientary development of the proximal parts of both hands, with one digit with a normal nail on the ulnar sides. The feet were siinilarly involved, with absence of toes 1-4. The height was 143 cm, weight 49 kg, and OFC 56 cm. There was no evidence of interna1 malformations, and, more particular1 y, intravenous py elography was normal. Ncurological and ophthalmological examinations were normal. There was no hearing deficit. and renal function and urine analysis were normal. Secondary sexual development was normal, with regular menstruation cycle.
On radiological examination, hand fiims showed deformation and bowing of the dista1 radii. The carpus was smali: os trapezium and os trapezoideum were hypoplas- tic; os scaphoideum was absent. On the right hand, metacarpals 1 and 2 and on the left hand metacarpal 1 were missing. The left metacarpal 2 was hypoplastic. At the
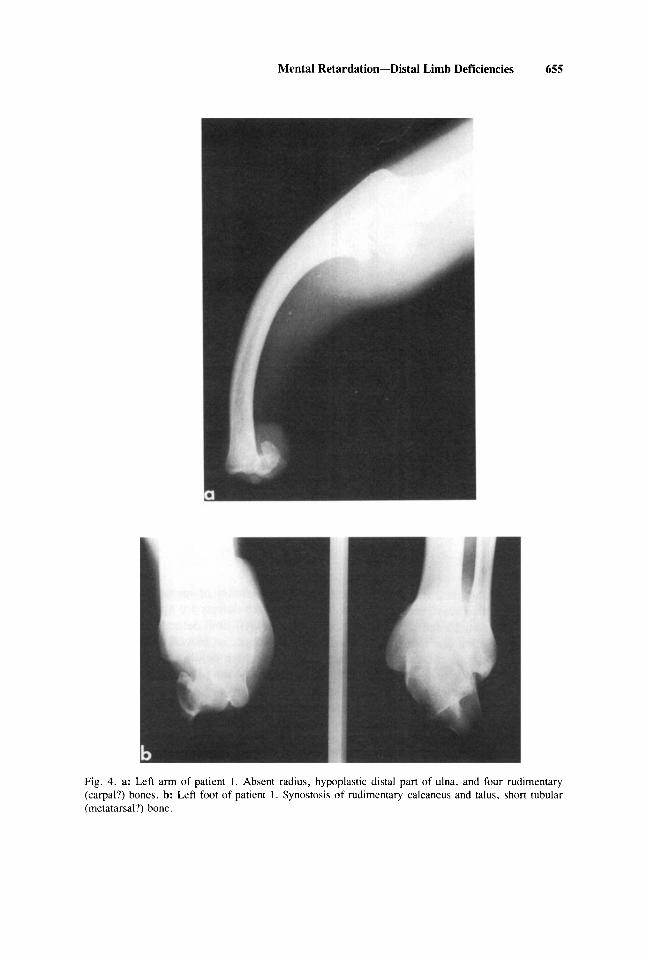
Mental Retardation-Dista1 Limb Deficiencies 655
Fig. 4. a: Left arm of patient 1. Absent radius, hypoplastic dista1 part of ulna, and four rudimentary (carpal?) bonec. b: Left foot of patient 1. Synostosic of rudimentary calcaneus and talus, short tubular (metatarsal?) bone.

656 Buttiens and Fryns
Fig. 5 . Sirnilar craniofacial features in patient 2 (aister ofpatient 1 ) .
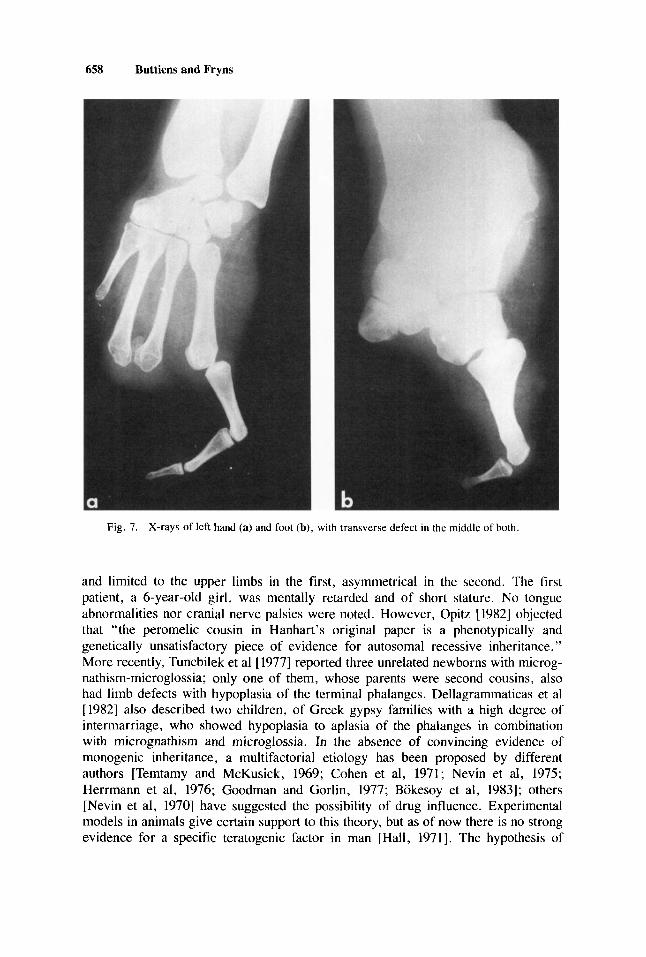
medial side of both distal fourth metacarpals, a small, rudimentary, accessory bone was present. Both fifth metacarpals and phalanges were normal (Fig. 7a). Foot filrns showed normal tal¡ and calcanei. Except for os cuboideum and os navicuiare, al1 other midfoot bones were absent. The base of metatarsal 1 was a rudiment; metatar- sals 2-4 were missing. The fifth metatarsai was well formed, with a rather large base and only two small phalanges (Fig. 7b).
DISCUSSION
These sibs present the hitherto unreported association of mental retardation, distal limb deficiencies, and oral involvement. The limb defects are not a total distal transverse peromelia but rather a paraxial radiai/preaxial limb defect. Both patients have micrognathia and microstomia, with normal tongue, as is well documented in the so called oromandibular malformations-transverse limb defect syndrornes. Over the past years, increasing confusion has developed concerning the classification and the etiology of these syndromes. Hall [1971] proposed five types of orornandibular and limb hypogenesis syndromes in which hypoglossia was the common feature. In a fifth category of “miscellaneous syndromes, ” he classified the Charlie-M syndrome, Pierre-Robin syndrome, Mobius syndrome, Hanhart syndrome, and amniotic band syndrome. In a thorough review of the literature, Herrmann et al [ 19761 distinguished two subgroups, ie, Hanhart syndrome and Poland-Mobius syndrome, and gave a detailed analysis of the phenotypic features and the etiologic aspects. These two sibs do not fa11 into any of these proposed classifications. Their symmetric limb involve- ment is clearly distinct and different from those reported until now. They seem to be examples of an apparently new autosomal recessive syndrome. Furthermore, there is
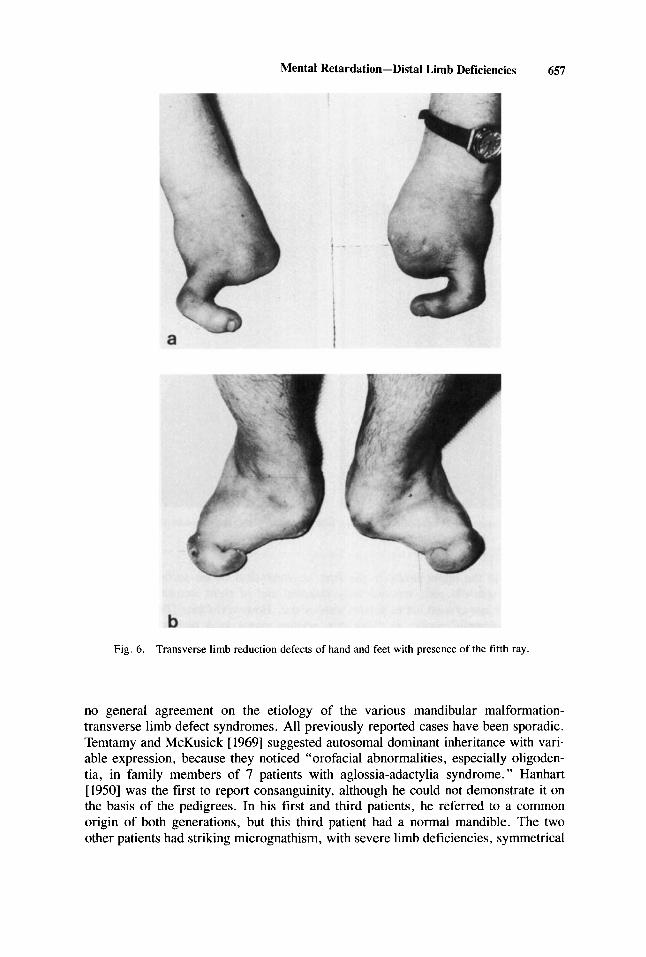
Mental Retardation-Distai Limb Deficiencies 657
Fig. 6. Transverse limb reduction defects of hand and feet with presence of the tifth ray.
no general agreement on the etiology of the various mandibular malformation- transverse limb defect syndromes. Al1 previously reported cases have been sporadic. Temtamy and McKusick [ 19691 suggested autosomal dominant inheritance with vari- able expression, because they noticed “orofacial abnormalities, especially oligoden- tia, in family members of 7 patients with aglossia-adactylia syndrome. ” Hanhart [1950] was the first to report consanguinity, although he could not demonstrate it on the basis of the pedigrees. In his first and third patients, he referred to a common origin of both generations, but this third patient had a normal mandible. The two other patients had striking micrognathism, with severe limb deficiencies, symmetrical
658 Buttiens and Fryns
Fig. 7. X-rays of left hand (a) and foot (b), with transverse defect in the middle of both.
and lirnited to the upper iimbs in the first, asymmetrical in the second. The first patient, a 6-year-old girl, was mentaily retarded and of short stature. No tongue abnormalities nor cranial nerve palsies were noted. However, Opitz [ 19821 objected that “the peromelic cousin in Hanhart’s original paper is a phenotypically and geneticaliy unsatisfactory piece of evidence for autosomal recessive inheritance. ” More recently, Tuncbiiek et al [ 19771 reported three unrelated newborns with microg- nathism-microglossia; only one of them, whose parents were second cousins, also had limb defects with hypoplasia of the terminal phalanges. Dellagrammaticas et al [1982] also described two chiidren, of Greek gypsy families with a high degree of intermarriage, who showed hypoplasia to apiasia of the phalanges in combination with micrognathism and microgiossia. In the absence of convincing evidence of monogenic inheritance, a multifactorial etiology has been proposed by different authors [Temtamy and McKusick, 1969; Cohen et al, 1971; Nevin et al, 1975; Herrmann et al, 1976; Goodrnan and Gorlin, 1977; Bokesoy et al, 19831; others [Nevin et al, 19701 have suggested the possibility of drug influence. Experimental models in anirnals give certain support to this theory, but as of now there is no strong evidence for a specific teratogenic factor in man [Hall, 19711. The hypothesis of

Mental Retardation-Dista1 Limb Deficiencies 659
vascular accidents in early developmental stages has also been proposed [Kaplan et al, 1976; Johnson and Robinow, 19791, and the resemblance to some cases of amniotic band syndrome has lead some authors to suggest an identical mechanism in the oromandibular limb hypogenesis syndromes.
In addition to the symmetrical dista1 limb deficiencies and oral defects, both sibs are mentally retarded. In the male, additional features are severe myopia and oligomeganephronia. These symptoms are not present in the female sib, who is of short stature with disproportionately large head. Although present in only one of the two sibs, mental retardation, myopia, oligomeganephronia, and short stature may be important but variable components of this syndrome. Renal malformations are known to be associated frequently with other organ malformations, particularly when the renal malformation is bilateral. Malformations of the lower limbs seem to be more frequently associated with kidney malformations than are malformations of the upper limbs [Temtamy and McKusick, 19781. Garner and Bixler [ 19691 mentioned renal malformations (agenesis of the right iudney) in one of his patients, but this patient with hypoplastic thumb, equinovarus position of the feet without other iimb defects, and mental retardation with microgyria doesn’t fit well into the group of syndromes discussed above. As far as we know, oligomeganephronia, a congenital renal devel- opmental defect, has never been reported in association with limb deficiencies.
REFERENCES
Bokesoy 1, Aksüyek C, Denitz E (1983): Oromandibular limb hypogenesisíHanhart’s syndrome: Posible drug influence on the malformation. Clin Genct 24:47-49.
Cohen MM, Pantke H, Siris E (1971): Nosologic and genetic considerations in the aglossyadactyly syndrome. In Bergsma D, McKusick V, Jorgcnson R, Hussels 1 (eds): “The Clinical Delineation
Part XI, Orofacial Structurcs.” Ncw York: Alan R. L i s , Inc., for The National rch of Dirnes. BD:OAS V11(7):237-240.
Dellagrammaticas H, Traki M, Kapiki A, Sianidou L, Philippidis P, Papas C, Bartsocas C (1982): Hanhart syndrome: Possibility of autosonial recessive inheritance. In Papadatos CJ, Bartsocas CS (eds): “Skeletal Dysplasias.” New York: Alan R . Liss, Inc.. pp 299-304.
Garner LD, Bixler D (1969): Micrognathia, an ociatcd dcfect of Hanhart’s syndrome, types 11 and 111. Oral Surg 27:601-606.
Goodrnan R, Gorlin RJ (1977): “Atlas of the Face i n Genetic Disorders, 2nd ed.” St. Louis: C.V. Mosby Co.
Hall BD (1971): Aglossia-adactylia. In Bergsma D, McKusick V, Jorgenson R, Hussels 1 (cds): “The Clinical Delineation of Birth Defccts: Part XI. Orofacial Structures.” New York: Alan R. Lisc, lnc., for The National Foundation-March of Dimes. BD:OAS VII(7):233-236.
Hanhart E (1950): Über die Kombination von Perornelie mit Mikrognathie, ein neucs Syndrom beim Menschen, entsprechend der Akroteriasis congenita von Wriedt und Mohr beim Rinde. Arch Klaus-Stift Vererb Forcch 25:53 1-544.
Herrmann 1, Pallicter PD, Gilbert EF, Viseskul C, Bersu E, Pettersen JC, Opitz JM (1976): Studies oí’ malformation syndromes of man XXXXI B: Nosologic studies in the Hanhart and the Mobius syndrorne. Eur J Pediatr 122: 19-55.
Johnson GF, Robinow M (1979): Aglossia-adactylia. Radiology 128: 127-132. Kaplan P, Cummings C , Fraser FC (1976): A “community” of face-limb malformation syndromes. J
Nevin NC, Burrows D, Allen G, Kernohan DC (1975): Aglossia-adactylia syndrome. J Med Genet
Nevin NC, Dodge JA, Kernohan DC (1970): Aglossia-adactylia syndrome. Oral Surg Opitz JM (1982): Comment. In Papadatos CJ, Bartsocas CS (eds): “Skeletal Dyspla.
Pediatr 89:241-247.
12 89-93.
Alan R. Liss. Inc., p 305.

660 Buttiens and Fryns
Smith DW. Jones KL (1982): Facial-limb disruptive spectrum. In: “Recognizable Patterns of Human Malformation. Genetics: Embryologic and Clinical Aspects, 3rd Ed.” Philadelphia: WB Saunders
Temtamy S, McKusick VA (1969): Synopsis of hand rnalformations with particular emphasis on genetic factors. In Bergsrna D, McKusick V, Hall J, Scott C (eds): “The Clinical Delineation of Birth Defects: Part 111, Limb Malformations.” New York: Alan R. Liss, he . , for The National Foundation-March of Dimes. BD:OAS V(3): 125-184.
Temtamy SA, McKucick VA (1978): “The Genetics of Hand Malforniations.” New York: Alan R. L i s , Inc., for The National Foundation-March of Dimes. BD:OAS XIV(3).
Tuncbilek E, Yalcin C , Atasu M (1977): Aglossia-adactylia syndrome (special emphasis on the inheri- tance pattern). Clin Genet 11:421-423.
CO., pp 501-503.
Edited by John M. Opitz and James F. Reynolds





























