Embed Size (px)
Citation preview

Upc
HZa
b
a
ARRAA
KPCPM
1
relrsdpsPdssnlb
CG
(
h0
Applied Catalysis A: General 490 (2015) 65–70
Contents lists available at ScienceDirect
Applied Catalysis A: General
jou rn al hom ep age: www.elsev ier .com/ locate /apcata
ltradispersed platinum nanoclusters onolydopamine-functionalized carbon nanotubes as an excellentatalyst for methanol oxidation reaction
aoliang Huanga, Zuyu Hea, Xiaomin Lina, Weishuo Ruana, Yingju Liua,b,∗,huohong Yanga,∗
Institute of Biomaterials, Department of Applied Chemistry, College of Sciences, South China Agricultural University, Guangzhou 510642, ChinaState Key Laboratory of Chem/Biosensing & Chemometrics, Hunan University, Changsha 410082, China
r t i c l e i n f o
rticle history:eceived 29 August 2014eceived in revised form 22 October 2014ccepted 10 November 2014
a b s t r a c t
The poor electrocatalytic activity towards methanol oxidation reaction (MOR) and the extremely rarereserves of platinum are considered as two of the most severe obstacles which hinder the commercial-ization of direct methanol fuel cells. In this work, Pt nanoclusters (∼1.81 nm), which are much smallerthan Pt nanoparticles (∼3 nm) in commercial Pt/C, have been uniformly dispersed on polydopamine-
vailable online 15 November 2014
eywords:latinum nanoclustersarbon nanotubeolydopamine
functionalized carbon nanotubes via a simple polyol method. These ultradispersed Pt nanoclusters featurelarge accessible active surface as well as high fraction of surface atoms with low coordination numbers,and therefore not only exhibit excellent mass activity and stability but also show improved specify activityand CO tolerance relative to Pt/C and Pt/CNT catalyst.
© 2014 Elsevier B.V. All rights reserved.
ethanol oxidation reaction. Introduction
Direct methanol fuel cells (DMFC) are expected to become aeliable source of clean energy with broad applications in bothlectric vehicles and portable electronics devices [1,2]. Neverthe-ess, the poor electrocatalytic activity towards methanol oxidationeaction (MOR) and the extremely rare reserves of platinum aretill considered as two of the most severe obstacles which hin-er the commercialization of DMFC [1,3]. To this end, downsizinglatinum particles is one of the most effective and straightforwardolutions. Compared to the state-of-the-art Pt/C catalyst (3–4 nmt nanoparticles deposited on Vulcan XC72) [4], Pt nanoclusters,efined as particles less than 2 nm in size, are reported to exhibiturprisingly higher catalytic activity due to their larger accessibleurface and higher fraction of surface atoms with low coordination
umber [3,5–7]. Evidently, the utilization of Pt nanoclusters as cata-ysts will not only drastically improve electrocatalytic performanceut also considerably maximize the efficiency of per platinum atom.
∗ Corresponding authors at: Institute of Biomaterials, Department of Appliedhemistry, College of Sciences, South China Agricultual University, Wushan,uangzhou 510642, China. Tel.: +86 020 85280325; fax: +86 020 85282366.
E-mail addresses: [email protected] (Y. Liu), [email protected]. Yang).
ttp://dx.doi.org/10.1016/j.apcata.2014.11.014926-860X/© 2014 Elsevier B.V. All rights reserved.
Electrocatalysis by platinum is a typical surface process, that is, theexposed surface and dispersity of Pt play a vital role in its electrocat-alytic performance. Unfortunately, even though many efforts havebeen paid, the synthesis of “clean” and well-dispersed Pt nanoclus-ters as electrocatalyst still remains a great challenge. For example,capping agents (e.g. polymers [8], surfactants [9], ligands [10] anddendrimers [11,12]) which have been widely applied to control thesize of metal particles usually impair their electrocatalytic activityby blocking the active sites [5,13]. As for conventional supportingmaterials (e.g. carbon [14,15], metal oxide [16,17] and metal oxide-carbon [18,19]), Pt particles overgrow and show aggregation to acertain extent, especially in relatively high loading.
Recently, polydopamine (PDA) has emerged as a highly versa-tile mussel-inspired coating and adhesive primer [20]. The PDAfunctionalization shares several advantages to stabilize platinumnanoclusters, including (1) the binding ability with Pt precursor byamino and catechol groups [21]; (2) the �–� interaction betweendopamine (PDA precursor) and graphitized carbon materials suchas carbon nanotube and graphene [22]; (3) scatheless to con-ductive structure of carbon materials [23]; (4) the hydrophilicityand controllable thickness [24]. Carbon nanotube and graphene
both feature excellent electric conductivity and high surface areawhich are vital to electrocatalytic activity of Pt. But, carbon nano-tube, one-dimensional (1D) material, has been reported to forminterconnected conducting network [25], which increases the
6 lysis A
ceTnap
ngodeptcewtpet
2
2
twhLer
2
shdfw
2C
etstwticNw
2
f(sst
6 H. Huang et al. / Applied Cata
ontact between loaded metal and electrolyte as well as thelectron transfer between the current collector and the catalyst.herefore, these provide us a hint that PDA-functionalized carbonanotube (PDA-CNT) could be an ideal substrate for growing andnchoring ultrafine and ultradispersed Pt nanoclusters for high-erformance electrocatalysts towards MOR.
Herein, we fabricated an excellent electrocatalytic active Ptanoclusters (Ptn)/PDA-CNT nanocomposite catalyst via homo-eneously depositing ultradispersed Pt nanoclusters (∼1.81 nm)n polydopamine-functionalized carbon nanotubes. The size andistribution of Pt nanoclusters were identified by transmissionlectron microscope (TEM). X-ray diffraction (XRD) and X-rayhotoelectron spectroscope (XPS) techniques were performed fur-her to crosscheck the tiny size of Pt nanoclusters. Electrochemicalharacterizations showed that the Ptn/PDA-CNT electrocatalystxhibits lower onset potential of methanol and CO oxidation asell as higher mass activity and specific activity in comparison with
hat of Pt/CNT and state-of-the-art 20 wt% Pt/C catalysts. We pro-osed that these improved performance is not simply the result ofnlarged electrochemical active surface area (ECSA), but also dueo the incremental exposure of Pt atoms on the edges and corners.
. Experimentals
.1. Materials
Multi-walled carbon nanotubes (MWNTs, >95% purity, diame-er 20–40 nm) were obtained from Shenzhen Nanotech Port Co. Ltd,hile dopamine hydrochloride and hexachloroplatinic acid hexa-ydrate (H2PtCl6·6H2O) were provided by Aladdin Chemistry Co.,td and Shanghai July Chemical Co., Ltd, respectively. Unless oth-rwise stated, all reagents were of analytical grade and used aseceived without further purification.
.2. Synthesis of PDA coatings modified CNTs
First, 50 mg of MWNTs were dispersed into 45 mL·H2O by ultra-onication for 30 min, followed by the addition of 10 mg dopamineydrochloride. Next, 5 mL 0.01 M KMnO4 aqueous solution wasropwise added, and the solution was stirred at room temperatureor 3 h. The resulting product was separated by filtration, washedith deionized water several times, and then dried at 40 ◦C in oven.
.3. Immobilization of Pt nanoclusters on PDA-functionalizedNTs
In typical procedure, 25 mg of PDA-CNT were dispersed in 50 mLthylene glycol by ultrasonication for 20 min, followed by the addi-ion of an appropriate amount of H2PtCl6. Next, the pH of theolution was adjusted to 8.0 by the addition of 0.1 M NaOH solu-ion. After stirring at room temperature for 20 min, the mixtureas refluxed at 110 ◦C for 2 h. After cooling to room temperature,
he product was washed with ethanol and water for several times,solated with centrifugation, and then dried at 50 ◦C for 24 h. Foromparison, acid treated functionalized CNTs were loaded with PtPs under the same process. The Pt loading of as-prepared catalystas controlled as 20 wt%.
.4. Physical characterization
Powder X-ray diffraction (XRD) measurement was per-ormed using Rigaku D/Max-2200 vpc with Cu K� radiation
� = 0.15406 nm). The XRD measurements were performed by stepcan in 2� range from 10◦ to 70◦ at a scanning speed of 2 s/step (thetep size is 0.02◦/step). The product morphology and microstruc-ure were studied by transmission electron microscopy (TEM; FEI,: General 490 (2015) 65–70
Tecnai 12, 100 kV) at the Instrumental Analysis & Research Centerin South China Agricultural University. The surface properties ofthe samples were analyzed with X-ray photoelectron spectroscopy(XPS, Thermo-VG Scientific, ESCALAB 250) using an Al K� X-raysource in Sun Yat-Sen University (Guangzhou). The peak positionwas calibrated to the C 1s peak at 284.8 eV.
2.5. Electrochemical measurements
The electrochemical measurements were carried out with astandard three-electrode system on an IM6ex electrochemicalworkstation (Zahner, Germany) using a platinum foil and a sat-urated calomel electrode (SCE) as the counter and referenceelectrode, respectively. Unless otherwise stated, all the electrodepotentials are referenced to SCE. To prepare the catalyst suspen-sion, 5 mg catalysts were first dispersed in 1.0 mL doubly distilledwater for 30 min. Then, an aliquot of catalyst suspension was pipet-ted onto the prepolished glassy carbon electrode and dried in roomtemperature, leading to a Pt loading of about 63.7 �g cm−2 for allcatalysts. Finally, 3 �L 0.5 wt% Nafion solution was added to firmlyaffix the electrocatalyst on glassy carbon electrode. The resultingelectrodes were dried overnight before electrochemical measure-ments.
In order to get rid of any effect due to the Nafion film, theworking electrodes were treated by continuous cyclic voltam-metry (CV) between −0.25 and 0.95 V at a scan rate of 50 mV s−1
until a steady CV was obtained in N2-saturated 0.5 M H2SO4 solu-tion. The methanol oxidation experiments were conducted in a2 M CH3OH + 0.1 M H2SO4 electrolyte at a scan rate of 50 mV s−1.Chronoamperometry (CA) curves were recorded at 0.65 V in asolution of 2 M CH3OH + 0.5 M H2SO4 for 1200 s to investigatethe long-term performance of as-prepared electrocatalysts. ForCO stripping voltammetry, CO gas was purged into N2-saturated0.5 M H2SO4 at a position close to the working electrode for20 min. After the excess CO was purged out with N2 for 20 min,the CO stripping voltammetry was performed at a scan rate of10 mV s−1.
3. Results and discussion
3.1. The morphology characterization of the Ptn/PDA-CNT
Fig. 1A shows the XRD patterns of PDA-CNT and raw CNT. Thestrong diffraction peak at 26.6◦ and the other four weak peakscan be well indexed to the diffraction from the correspondingcrystallographic planes of graphitic carbon nanotubes (JCPDS Cardno. 26-1079). No differences can be found between PDA-CNT andraw CNT in XRD patterns, indicating the graphite structure of CNTretains after functionalization of PDA. Fig. 1B shows the XPS surveyspectra of Ptn/PDA-CNT and Pt/CNT. Apart from the similar C, O andPt signals of Pt/CNT, additional N element (2.87 at%) which comesfrom nitrogen-containing functional groups of polydopamine wasalso detected in Ptn/PDA-CNT. Moreover, as shown in Fig. 1 C, theC 1s peak can be deconvolved into sp2-hybridized C C (284.8 eV),C N (285.4 eV), C O and C N (286.3 eV) as well as C O (288.2 eV).In Fig. 1D, the peaks at the binding energy of 398.8 and 400.4 eVcorrespond to pyridinic and pyrrolic N species, respectively [26,27],which are consistent with two reported structures of polydopaminein PDA sub-micrometer spheres [28]. Additionally, in Fig. 1E, the O1s peak of C O (531.4 eV) and C O (532.6 eV) can be ascribed tothe o-quinone and catechol moieties in PDA.
Then, the morphology of Ptn/PDA-CNT was examined by TEM.As displayed in Fig. 2A, ultrafine Pt nanoclusters are evenly dis-persed on the surface of Ptn/PDA-CNT without aggregation. Themagnified TEM image of Ptn/PDA-CNT clearly shows separated
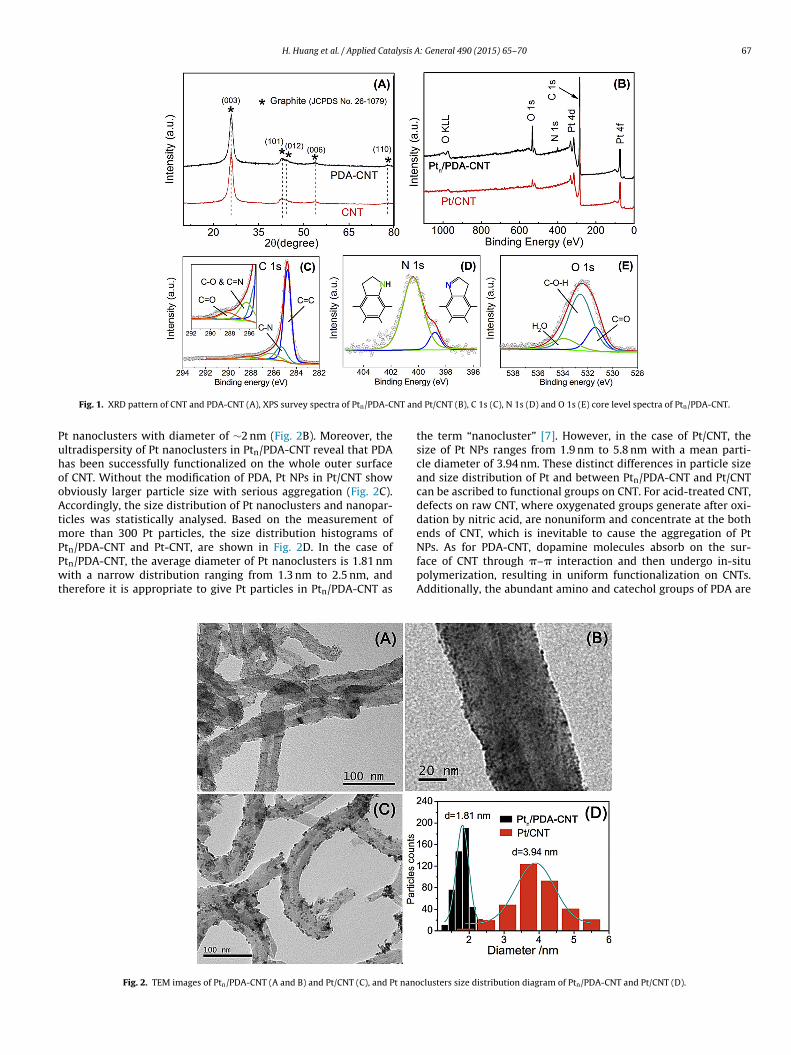
H. Huang et al. / Applied Catalysis A: General 490 (2015) 65–70 67
NT an
PuhooAtmPPwt
Fig. 1. XRD pattern of CNT and PDA-CNT (A), XPS survey spectra of Ptn/PDA-C
t nanoclusters with diameter of ∼2 nm (Fig. 2B). Moreover, theltradispersity of Pt nanoclusters in Ptn/PDA-CNT reveal that PDAas been successfully functionalized on the whole outer surfacef CNT. Without the modification of PDA, Pt NPs in Pt/CNT showbviously larger particle size with serious aggregation (Fig. 2C).ccordingly, the size distribution of Pt nanoclusters and nanopar-
icles was statistically analysed. Based on the measurement ofore than 300 Pt particles, the size distribution histograms of
tn/PDA-CNT and Pt-CNT, are shown in Fig. 2D. In the case oftn/PDA-CNT, the average diameter of Pt nanoclusters is 1.81 nmith a narrow distribution ranging from 1.3 nm to 2.5 nm, and
herefore it is appropriate to give Pt particles in Ptn/PDA-CNT as
Fig. 2. TEM images of Ptn/PDA-CNT (A and B) and Pt/CNT (C), and Pt nano
d Pt/CNT (B), C 1s (C), N 1s (D) and O 1s (E) core level spectra of Ptn/PDA-CNT.
the term “nanocluster” [7]. However, in the case of Pt/CNT, thesize of Pt NPs ranges from 1.9 nm to 5.8 nm with a mean parti-cle diameter of 3.94 nm. These distinct differences in particle sizeand size distribution of Pt and between Ptn/PDA-CNT and Pt/CNTcan be ascribed to functional groups on CNT. For acid-treated CNT,defects on raw CNT, where oxygenated groups generate after oxi-dation by nitric acid, are nonuniform and concentrate at the bothends of CNT, which is inevitable to cause the aggregation of Pt
NPs. As for PDA-CNT, dopamine molecules absorb on the sur-face of CNT through �–� interaction and then undergo in-situpolymerization, resulting in uniform functionalization on CNTs.Additionally, the abundant amino and catechol groups of PDA areclusters size distribution diagram of Ptn/PDA-CNT and Pt/CNT (D).
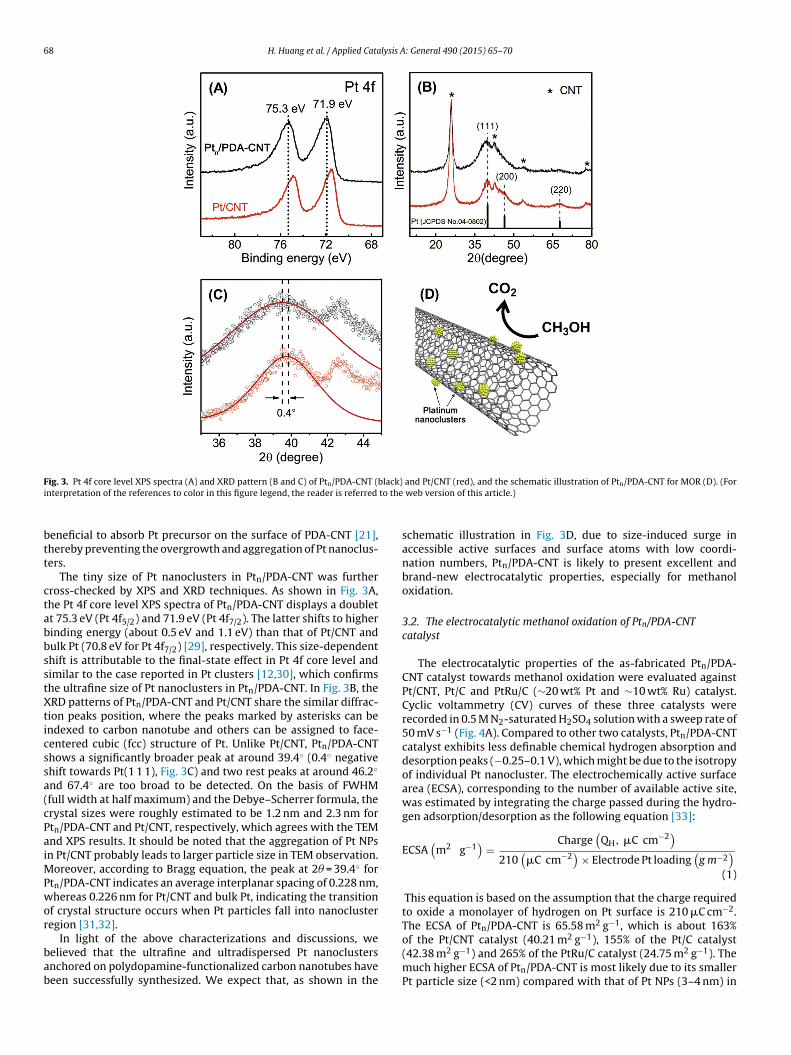
68 H. Huang et al. / Applied Catalysis A: General 490 (2015) 65–70
F black)i to the
btt
ctabbsstXticssa(cPaiMPwor
bab
ig. 3. Pt 4f core level XPS spectra (A) and XRD pattern (B and C) of Ptn/PDA-CNT (nterpretation of the references to color in this figure legend, the reader is referred
eneficial to absorb Pt precursor on the surface of PDA-CNT [21],hereby preventing the overgrowth and aggregation of Pt nanoclus-ers.
The tiny size of Pt nanoclusters in Ptn/PDA-CNT was furtherross-checked by XPS and XRD techniques. As shown in Fig. 3A,he Pt 4f core level XPS spectra of Ptn/PDA-CNT displays a doublett 75.3 eV (Pt 4f5/2) and 71.9 eV (Pt 4f7/2). The latter shifts to higherinding energy (about 0.5 eV and 1.1 eV) than that of Pt/CNT andulk Pt (70.8 eV for Pt 4f7/2) [29], respectively. This size-dependenthift is attributable to the final-state effect in Pt 4f core level andimilar to the case reported in Pt clusters [12,30], which confirmshe ultrafine size of Pt nanoclusters in Ptn/PDA-CNT. In Fig. 3B, theRD patterns of Ptn/PDA-CNT and Pt/CNT share the similar diffrac-
ion peaks position, where the peaks marked by asterisks can bendexed to carbon nanotube and others can be assigned to face-entered cubic (fcc) structure of Pt. Unlike Pt/CNT, Ptn/PDA-CNThows a significantly broader peak at around 39.4◦ (0.4◦ negativehift towards Pt(1 1 1), Fig. 3C) and two rest peaks at around 46.2◦
nd 67.4◦ are too broad to be detected. On the basis of FWHMfull width at half maximum) and the Debye–Scherrer formula, therystal sizes were roughly estimated to be 1.2 nm and 2.3 nm fortn/PDA-CNT and Pt/CNT, respectively, which agrees with the TEMnd XPS results. It should be noted that the aggregation of Pt NPsn Pt/CNT probably leads to larger particle size in TEM observation.
oreover, according to Bragg equation, the peak at 2� = 39.4◦ fortn/PDA-CNT indicates an average interplanar spacing of 0.228 nm,hereas 0.226 nm for Pt/CNT and bulk Pt, indicating the transition
f crystal structure occurs when Pt particles fall into nanoclusteregion [31,32].
In light of the above characterizations and discussions, weelieved that the ultrafine and ultradispersed Pt nanoclustersnchored on polydopamine-functionalized carbon nanotubes haveeen successfully synthesized. We expect that, as shown in the
and Pt/CNT (red), and the schematic illustration of Ptn/PDA-CNT for MOR (D). (For web version of this article.)
schematic illustration in Fig. 3D, due to size-induced surge inaccessible active surfaces and surface atoms with low coordi-nation numbers, Ptn/PDA-CNT is likely to present excellent andbrand-new electrocatalytic properties, especially for methanoloxidation.
3.2. The electrocatalytic methanol oxidation of Ptn/PDA-CNTcatalyst
The electrocatalytic properties of the as-fabricated Ptn/PDA-CNT catalyst towards methanol oxidation were evaluated againstPt/CNT, Pt/C and PtRu/C (∼20 wt% Pt and ∼10 wt% Ru) catalyst.Cyclic voltammetry (CV) curves of these three catalysts wererecorded in 0.5 M N2-saturated H2SO4 solution with a sweep rate of50 mV s−1 (Fig. 4A). Compared to other two catalysts, Ptn/PDA-CNTcatalyst exhibits less definable chemical hydrogen absorption anddesorption peaks (−0.25–0.1 V), which might be due to the isotropyof individual Pt nanocluster. The electrochemically active surfacearea (ECSA), corresponding to the number of available active site,was estimated by integrating the charge passed during the hydro-gen adsorption/desorption as the following equation [33]:
ECSA(
m2 g−1)
=Charge
(QH, �C cm−2
)
210(
�C cm−2)
× Electrode Pt loading(
g m−2)
(1)
This equation is based on the assumption that the charge requiredto oxide a monolayer of hydrogen on Pt surface is 210 �C cm−2.The ECSA of Ptn/PDA-CNT is 65.58 m2 g−1, which is about 163%
of the Pt/CNT catalyst (40.21 m2 g−1), 155% of the Pt/C catalyst(42.38 m2 g−1) and 265% of the PtRu/C catalyst (24.75 m2 g−1). Themuch higher ECSA of Ptn/PDA-CNT is most likely due to its smallerPt particle size (<2 nm) compared with that of Pt NPs (3–4 nm) in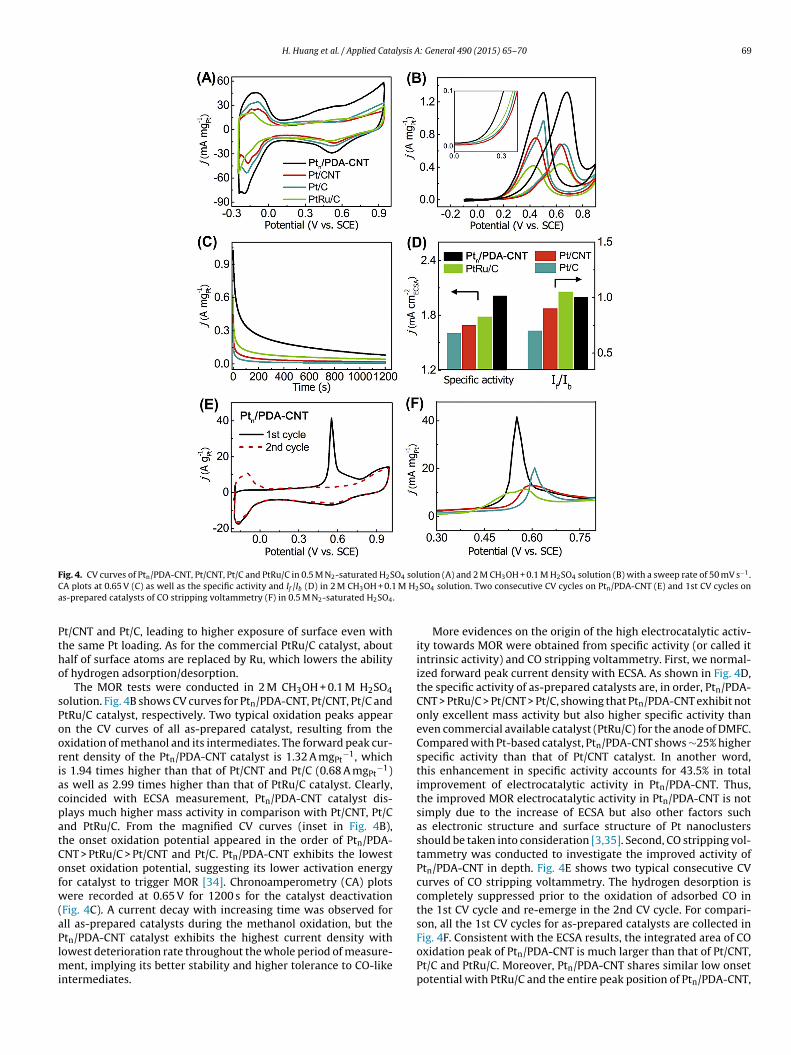
H. Huang et al. / Applied Catalysis A: General 490 (2015) 65–70 69
F O4 sol −1
C M H2
a .
Ptho
sPooriacpatCofw(aPlmi
ig. 4. CV curves of Ptn/PDA-CNT, Pt/CNT, Pt/C and PtRu/C in 0.5 M N2-saturated H2SA plots at 0.65 V (C) as well as the specific activity and If /Ib (D) in 2 M CH3OH + 0.1s-prepared catalysts of CO stripping voltammetry (F) in 0.5 M N2-saturated H2SO4
t/CNT and Pt/C, leading to higher exposure of surface even withhe same Pt loading. As for the commercial PtRu/C catalyst, aboutalf of surface atoms are replaced by Ru, which lowers the abilityf hydrogen adsorption/desorption.
The MOR tests were conducted in 2 M CH3OH + 0.1 M H2SO4olution. Fig. 4B shows CV curves for Ptn/PDA-CNT, Pt/CNT, Pt/C andtRu/C catalyst, respectively. Two typical oxidation peaks appearn the CV curves of all as-prepared catalyst, resulting from thexidation of methanol and its intermediates. The forward peak cur-ent density of the Ptn/PDA-CNT catalyst is 1.32 A mgPt
−1, whichs 1.94 times higher than that of Pt/CNT and Pt/C (0.68 A mgPt
−1)s well as 2.99 times higher than that of PtRu/C catalyst. Clearly,oincided with ECSA measurement, Ptn/PDA-CNT catalyst dis-lays much higher mass activity in comparison with Pt/CNT, Pt/Cnd PtRu/C. From the magnified CV curves (inset in Fig. 4B),he onset oxidation potential appeared in the order of Ptn/PDA-NT > PtRu/C > Pt/CNT and Pt/C. Ptn/PDA-CNT exhibits the lowestnset oxidation potential, suggesting its lower activation energyor catalyst to trigger MOR [34]. Chronoamperometry (CA) plotsere recorded at 0.65 V for 1200 s for the catalyst deactivation
Fig. 4C). A current decay with increasing time was observed forll as-prepared catalysts during the methanol oxidation, but the
tn/PDA-CNT catalyst exhibits the highest current density withowest deterioration rate throughout the whole period of measure-ent, implying its better stability and higher tolerance to CO-likentermediates.
ution (A) and 2 M CH3OH + 0.1 M H2SO4 solution (B) with a sweep rate of 50 mV s .SO4 solution. Two consecutive CV cycles on Ptn/PDA-CNT (E) and 1st CV cycles on
More evidences on the origin of the high electrocatalytic activ-ity towards MOR were obtained from specific activity (or called itintrinsic activity) and CO stripping voltammetry. First, we normal-ized forward peak current density with ECSA. As shown in Fig. 4D,the specific activity of as-prepared catalysts are, in order, Ptn/PDA-CNT > PtRu/C > Pt/CNT > Pt/C, showing that Ptn/PDA-CNT exhibit notonly excellent mass activity but also higher specific activity thaneven commercial available catalyst (PtRu/C) for the anode of DMFC.Compared with Pt-based catalyst, Ptn/PDA-CNT shows ∼25% higherspecific activity than that of Pt/CNT catalyst. In another word,this enhancement in specific activity accounts for 43.5% in totalimprovement of electrocatalytic activity in Ptn/PDA-CNT. Thus,the improved MOR electrocatalytic activity in Ptn/PDA-CNT is notsimply due to the increase of ECSA but also other factors suchas electronic structure and surface structure of Pt nanoclustersshould be taken into consideration [3,35]. Second, CO stripping vol-tammetry was conducted to investigate the improved activity ofPtn/PDA-CNT in depth. Fig. 4E shows two typical consecutive CVcurves of CO stripping voltammetry. The hydrogen desorption iscompletely suppressed prior to the oxidation of adsorbed CO inthe 1st CV cycle and re-emerge in the 2nd CV cycle. For compari-son, all the 1st CV cycles for as-prepared catalysts are collected in
Fig. 4F. Consistent with the ECSA results, the integrated area of COoxidation peak of Ptn/PDA-CNT is much larger than that of Pt/CNT,Pt/C and PtRu/C. Moreover, Ptn/PDA-CNT shares similar low onsetpotential with PtRu/C and the entire peak position of Ptn/PDA-CNT,
7 lysis A
bsitd
tccwottfnTntc
4
ntPhufnaed
A
tPFdPovC
R
[
[
[
[
[[
[
[
[
[
[
[
[
[
[[
[
[[[
[
[[
[
[C 116 (2012) 16969–16978.
0 H. Huang et al. / Applied Cata
oth the onset and peak potential, displays a significant negativehift towards that of Pt/CNT and Pt/C, indicating the Ptn/PDA-CNTs capable to significantly decrease the over-potential of CO oxida-ion. These results agree with the ratio of the forward peak currentensity (If) to the backward peak current density (Ib) in Fig. 4D.
Overall, the above results indicate the catalysis of Pt nanoclus-ers towards methanol and poisoning intermediate in Ptn/PDA-CNTatalyst behaves different from that of Pt/CNT and commercial Pt/Catalyst. It should be noted that transition metal oxide or Pt alloy,hich can alter the surface environment and electronic structure
f Pt, is not involved in present study. The possible mechanism forhe improved electrocatalytic properties of Ptn/PDA-CNT might beypical surface effect in nanoscale. According to Long’s work [32],rom 2.9 nm Pt NPs to 1.8 nm Pt nanoclusters, Pt-Pt coordinationumber significantly decrease from ∼9.8 to ∼7.4 (12 for bulk Pt).hese incremental fraction surface atoms with lower coordinationumber are widely reported to exhibit higher catalytic activity thanhe common surface atoms and serve as the active sites for breakinghemical bonds [36,37].
. Conclusions
We fabricated an excellent electrocatalytic active Ptn/PDA-CNTanocomposite catalyst by depositing ultradispersed Pt nanoclus-ers on PDA-CNT. Owing to the mussel-inspired polydopamine,t nanoclusters (∼1.81 nm) with narrow size distribution wereomogeneously anchored on PDA-CNT without aggregation. Theseltradispersed Pt nanoclusters feature large accessible active sur-ace and high fraction of surface atoms with low coordinationumbers and therefore not only exhibit excellent mass activitynd stability but also show improved specify activity and CO tol-rance. This present strategy may provide a promising route forevelopment of next-generation anode catalyst for DMFCs.
cknowledgements
This work was supported by the National Science Founda-ion of China (21105030, 21475047), the Science and Technologylanning Project of Guangdong Province (2013B02080007), theoundation for High-level Talents in Higher Education of Guang-ong Province, the Student Innovative Project of Guangdongrovince (201410564196), the Open Fund of State Key Laboratoryf Chem/Biosensing & Chemometrics (2012013), the Student Inno-ative Project of Guangdong Province and the 211 Project of Southhina Agricultural University (2009B010100001).
eferences
[1] M.K. Debe, Nature 486 (2012) 43–51.
[
[[
: General 490 (2015) 65–70
[2] M. Winter, R.J. Brodd, Chem. Rev. 104 (2004) 4245–4270.[3] S. Sun, G. Zhang, N. Gauquelin, N. Chen, J. Zhou, S. Yang, W. Chen, X. Meng, D.
Geng, M. Banis, R. Li, S. Ye, S. Knights, G.A. Botton, T.K. Sham, X.L. Sun, Sci. Rep.3 (2013) 1775.
[4] W. Vogel, J. Phys. Chem. C 112 (2008) 13475–13482.[5] X. Chen, G. Wu, J. Chen, X. Chen, Z. Xie, X. Wang, J. Am. Chem. Soc. 133 (2011)
3693–3695.[6] X. Yang, A. Wang, B. Qiao, J. Li, J. Liu, T. Zhang, Acc. Chem. Res. 46 (2013)
1740–1748.[7] Y. Lu, W. Chen, Chem. Soc. Rev. 41 (2012) 3594–3623.[8] Z. Shen, H. Duan, H. Frey, Adv. Mater. 19 (2007) 349–352.[9] X. Yuan, Z. Luo, Q. Zhang, X. Zhang, Y. Zheng, J.Y. Lee, J. Xie, ACS Nano 5 (2011)
8800–8808.10] Y. Li, O. Zaluzhna, B. Xu, Y. Gao, J.M. Modest, Y.J. Tong, J. Am. Chem. Soc. 133
(2011) 2092–2095.11] Y. Huang, H. Huang, Y. Liu, Y. Xie, Z. Liang, C. Liu, J. Power Sources 201 (2012)
81–87.12] K. Yamamoto, T. Imaoka, W.J. Chun, O. Enoki, H. Katoh, M. Takenaga, A. Sonoi,
Nat. Chem. 1 (2009) 397–402.13] D. Li, C. Wang, D. Tripkovic, S. Sun, N.M. Markovic, V.R. Stamenkovic, ACS Catal.
2 (2012) 1358–1362.14] S. Mayavan, J.B. Sim, S.M. Choi, J. Mater. Chem. 22 (2012) 6953–6958.15] S. Mayavan, H.S. Jang, M.J. Lee, S.H. Choi, S.M. Choi, J. Mater. Chem. A 1 (2013)
3489–3494.16] C. Zhang, H. Yu, Y. Li, Y. Gao, Y. Zhao, W. Song, Z. Shao, B. Yi, ChemSusChem 6
(2013) 659–666.17] V.T. Thanh Ho, K.C. Pillai, H.L. Chou, C.J. Pan, J. Rick, W.N. Su, B.J. Hwang, J.F. Lee,
H.S. Sheu, W.T. Chuang, Energy Environ. Sci. 4 (2011) 4194–4200.18] L. Zhang, L. Wang, C.M.B. Holt, B. Zahiri, Z. Li, K. Malek, T. Navessin, M.H. Eiker-
ling, D. Mitlin, Energy Environ. Sci. 5 (2012) 6156–6172.19] H. Huang, Y. Liu, Q. Gao, W. Ruan, X. Lin, X. Li, ACS Appl. Mater. Interfaces 6
(2014) 10258–10264.20] H. Lee, S.M. Dellatore, W.M. Miller, P.B. Messersmith, Science 318 (2007)
426–430.21] M. Lin, H. Huang, Y. Liu, C. Liang, S. Fei, X. Chen, C. Ni, Nanotechnology 24 (2013)
065501.22] J. Zhao, W. Zhang, P. Sherrell, J.M. Razal, X. Huang, A.I. Minett, J. Chen, ACS Appl.
Mater. Interfaces 4 (2012) 44–48.23] B. Fei, B. Qian, Z. Yang, R. Wang, W. Liu, C.L. Mak, J.H. Xin, Carbon 46 (2008)
1795–1797.24] H. Hu, B. Yu, Q. Ye, Y. Gu, F. Zhou, Carbon 48 (2010) 2347–2353.25] Y. Xiao, W. Jiang, S. Wan, X. Zhang, J. Hu, Z. Wei, L. Wan, J. Mater. Chem. A 1
(2013) 7463–7468.26] P. Chen, J. Yang, S. Li, Z. Wang, T. Xiao, Y. Qian, S. Yu, Nano Energy 2 (2013)
249–256.27] G. Wang, L. Jia, Y. Zhu, B. Hou, D. Li, Y. Sun, RSC Adv. 2 (2012) 11249–11252.28] K. Ai, Y. Liu, C. Ruan, L. Lu, G.M. Lu, Adv. Mater. 25 (2013) 998–1003.29] B.V. Crist, Handbook of Monochromatic XPS Spectra: The Elements and Native
Oxides, John Wiley & Sons Ltd, Chichester, 2000.30] Y. Sun, Y. Wang, J.S. Pan, L. Wang, C.Q. Sun, J. Phys. Chem. C 113 (2009)
14696–14701.31] Z. Wu, J. Chen, R. Jin, Adv. Funct. Mater. 21 (2011) 177–183.32] L. Li, L. Wang, D.D. Johnson, Z. Zhang, S.I. Sanchez, J.H. Kang, R.G. Nuzzo, Q.
Wang, A.I. Frenkel, J. Li, J. Ciston, E.A. Stach, J.C. Yang, J. Am. Chem. Soc. 135(2013) 13062–13072.
33] S. Sun, G. Zhang, D. Geng, Y. Chen, R. Li, M. Cai, X. Sun, Angew. Chem. Int. Ed. 50(2011) 422–426.
34] T. Chen, T.M. Luo, Y. Yang, Y. Wei, K. Wang, T. Lin, T. Wen, C.H. Lee, J. Phys. Chem.
35] J.N. Tiwari, K. Nath, S. Kumar, R.N. Tiwari, K.C. Kemp, N.H. Le, D.H. Youn, J.S. Lee,K.S. Kim, Nat. Commun. 4 (2013) 2221.
36] N. Tian, Z. Zhou, S. Sun, Y. Ding, Z. Wang, Science 316 (2007) 732–735.37] B. Wu, N. Zheng, Nano Today 8 (2013) 168–197.



![Applied Catalysis B: Environmental652 K. Deplanche et al. / Applied Catalysis B: Environmental 147 (2014) 651–665 [4]. This microbial ability can be harnessed for biotechnological](https://img.pdfslide.net/doc/110x75/60b11e48affdb175503a2f34/applied-catalysis-b-environmental-652-k-deplanche-et-al-applied-catalysis-b.jpg)

























