Embed Size (px)
Citation preview

American Journal of Medical Genetics 136A:269–274 (2005)
Clinical ReportAssociation of Adams–Oliver Syndrome With PulmonaryArterio-Venous Malformation in the Same Family:A Further Support to the Vascular HypothesisMauro Maniscalco,1,2* Anna Zedda,1 Stanislao Faraone,1 Guglielmo de Laurentiis,2
Raffaele Verde,1 Valerio Molese,3 Gaetano Lapiccirella,3 and Matteo Sofia2
1Section of Respiratory Medicine, Hospital ‘‘S. Maria della Pieta,’’ Casoria, Naples, Italy2Department of Respiratory Medicine, A.O. Monaldi, University ‘‘Federico II,’’ Naples, Italy3Department of Radiology, Hospital ‘‘S. Maria della Pieta,’’ Casoria, Naples, Italy
Adams–Oliver syndrome (AOS) is a rare diseasecharacterized by congenital scalp defects, term-inal transverse limb defects and cutis marmoratatelangiectatica. A significant incidence of cardiacand vascular malformations has been reported,leading to the hypothesis of a vascular defectearly involved in the pathogenesis. We reporttwo members of the same family with previouslydiagnosed AOS based on clinical phenotype andlater recognized to have pulmonary arterio-venous malformation (PAVM). None of the sub-jects fulfilled current diagnostic criteria ofhereditary hemorrhagic telangiectasia, which isthe most common cause of PAVM. The occurrenceof PAVM in AOS lends support to the hypothesisthat endothelial specific abnormalities could be apatho-physiological mechanism in its develop-ment. Therefore, the role of screening for PAVMin clinical management of subjects with AOS shoulddeserve further studies. � 2005 Wiley-Liss, Inc.
KEY WORDS: Adams–Oliver syndrome; arterio-venous malformation; ortho-deoxia; pulmonary hypertension
INTRODUCTION
Adams–Oliver syndrome (AOS) is a rare syndrome char-acterized by congenital scalp defects and terminal transverselimb defects of variable severity [Adams and Oliver, 1945].Many recent reports of AOS have highlighted the variableexpression of this condition [Farrell et al., 1993; Mempel et al.,1999; Pereira-Da-Silva et al., 2000]. Since the original des-cription, several sporadic, and familial cases of AOS have beenreported with remarkable intrafamilial and interfamilialvariability and autosomal dominant inheritance.
The association of vascular and cardiac malformations inAOS has been increasingly recognized [Toriello et al., 1988;David et al., 1991; Farrell et al., 1993; Frank and Frosch, 1993;Bamforth et al., 1994; Zapata et al., 1995; Lin et al., 1998],suggesting that vascular anomalies may represent an impor-tant component of this syndrome. We report two members of
the same family with AOS (Fig. 1) and pulmonary arterio-venous malformation (PAVM).
CLINICAL REPORT
Patient 1
A 41-year-old male with AOS (II-4, Fig. 1) was admitted toour Department with decreased exercise tolerance following anacute lower airway infection. The diagnosis of AOS had beenmade 2 years earlier at another institution based on char-acteristic clinical features: (a) cutaneous syndactyly of the 2ndand 3rd feet toes (Fig. 2) with radiographs showing absence ofmiddle phalanges of 2–5 toes; (b) venous ectasia of lower limbs,brachydactyly of the hands; (c) diffuse thickening of skulltheca. He also had mental retardation, which is not typical ofAOS (Wechsler Adult Intelligence Scale, verbal IQ 51, nonverbal IQ 57, total IQ 50). The father was diagnosed with AOStransmitted with autosomal dominant inheritance. At the timehe had a normal chromosomal analysis and fragile X syndromewas excluded by DNA studies.
At the time of diagnosis, a chest X-ray showed pulmonaryvascular opacities. Subsequent pulmonary angiographyshowed only dilatation of lobar pulmonary arteries, whichwas inconclusive for pulmonary vascular malformation. Thechest examination, at the time of the recent admission,appeared to be negative except for a slight right diaphragmelevation and increase in vascular shadows close to left hilum(Fig. 5a). Lung volumes showed a moderate restrictive defectwith mild impairment of the lung diffusing capacity to carbonmonoxide. Arterial blood gases at rest revealed mild hypox-emia in comparison to age predicted value, which was onlypartially corrected by breathing oxygen. Contrast echocardio-graphy showed the delayed opacification of left chamber,which was consistent with the presence of intrapulmonaryanatomical shunt. A contrast-enhanced thorax spiral CT scandepicted a large pulmonary arterio-venous communication inleft upper lobe (Fig. 5b,c). The patient refused selectivepulmonary angiography and right heart catheterization.Apart from the pulmonary vascular abnormality, no othercardiovascular alterations were detected. The examinedabdomen and genitalia were normal; liver and the gastro-intestinal-tract (ultrasonographic and gastroscopic studies)did not reveal neither any vascular abnormality nor grossfunctional derangements.
His 12-year-old son (patient 2 of the present report) (III-3,Fig. 1) had similar cutis marmorata, distal digital hypoplasia,and lower limb venous ectasia (Fig. 3). Another child, adaughter (III-1, Fig. 1), who had been the first to receiveformal AOS diagnosis, had aplasia cutis congenita associatedalso with skull defect, distal digital hypoplasia, and cutis
*Correspondence to: Mauro Maniscalco, MD, L.go delle Mimose1, 80131 Napoli, Italy. E-mail: [email protected]
Received 14 January 2005; Accepted 9 May 2005
DOI 10.1002/ajmg.a.30828
� 2005 Wiley-Liss, Inc.
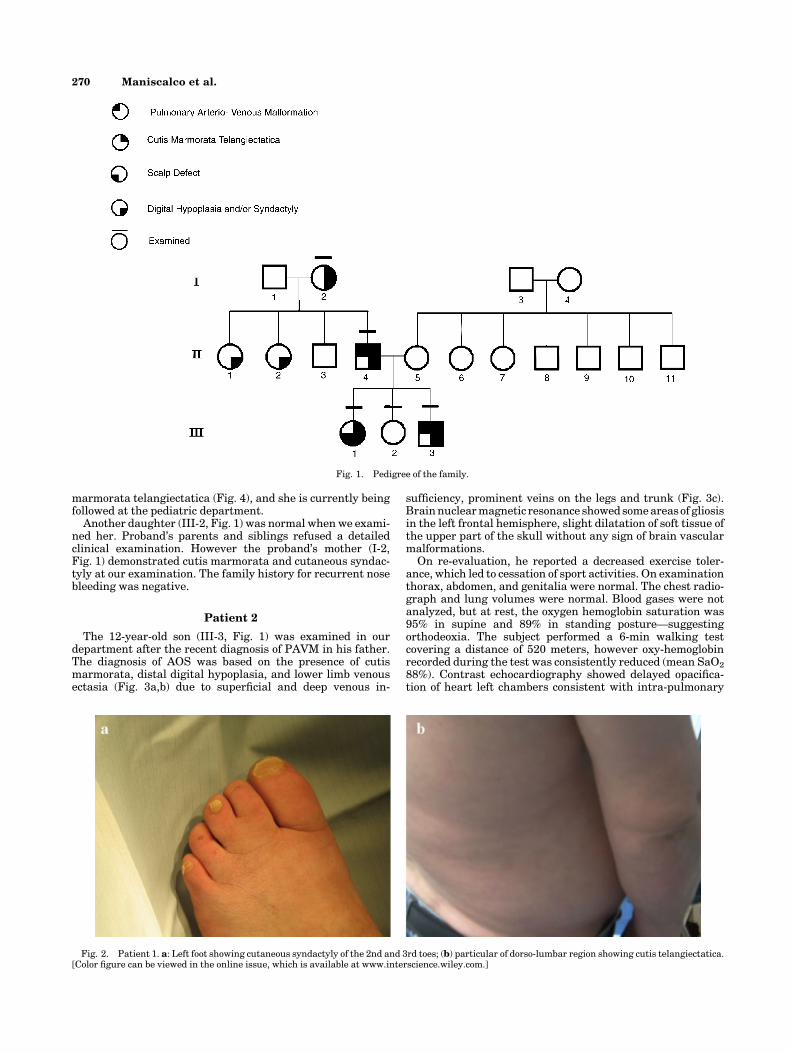
marmorata telangiectatica (Fig. 4), and she is currently beingfollowed at the pediatric department.
Another daughter (III-2, Fig. 1) was normal when we exami-ned her. Proband’s parents and siblings refused a detailedclinical examination. However the proband’s mother (I-2,Fig. 1) demonstrated cutis marmorata and cutaneous syndac-tyly at our examination. The family history for recurrent nosebleeding was negative.
Patient 2
The 12-year-old son (III-3, Fig. 1) was examined in ourdepartment after the recent diagnosis of PAVM in his father.The diagnosis of AOS was based on the presence of cutismarmorata, distal digital hypoplasia, and lower limb venousectasia (Fig. 3a,b) due to superficial and deep venous in-
sufficiency, prominent veins on the legs and trunk (Fig. 3c).Brain nuclear magnetic resonance showed some areas of gliosisin the left frontal hemisphere, slight dilatation of soft tissue ofthe upper part of the skull without any sign of brain vascularmalformations.
On re-evaluation, he reported a decreased exercise toler-ance, which led to cessation of sport activities. On examinationthorax, abdomen, and genitalia were normal. The chest radio-graph and lung volumes were normal. Blood gases were notanalyzed, but at rest, the oxygen hemoglobin saturation was95% in supine and 89% in standing posture—suggestingorthodeoxia. The subject performed a 6-min walking testcovering a distance of 520 meters, however oxy-hemoglobinrecorded during the test was consistently reduced (mean SaO2
88%). Contrast echocardiography showed delayed opacifica-tion of heart left chambers consistent with intra-pulmonary
Fig. 1. Pedigree of the family.
Fig. 2. Patient 1. a: Left foot showing cutaneous syndactyly of the 2nd and 3rd toes; (b) particular of dorso-lumbar region showing cutis telangiectatica.[Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
270 Maniscalco et al.

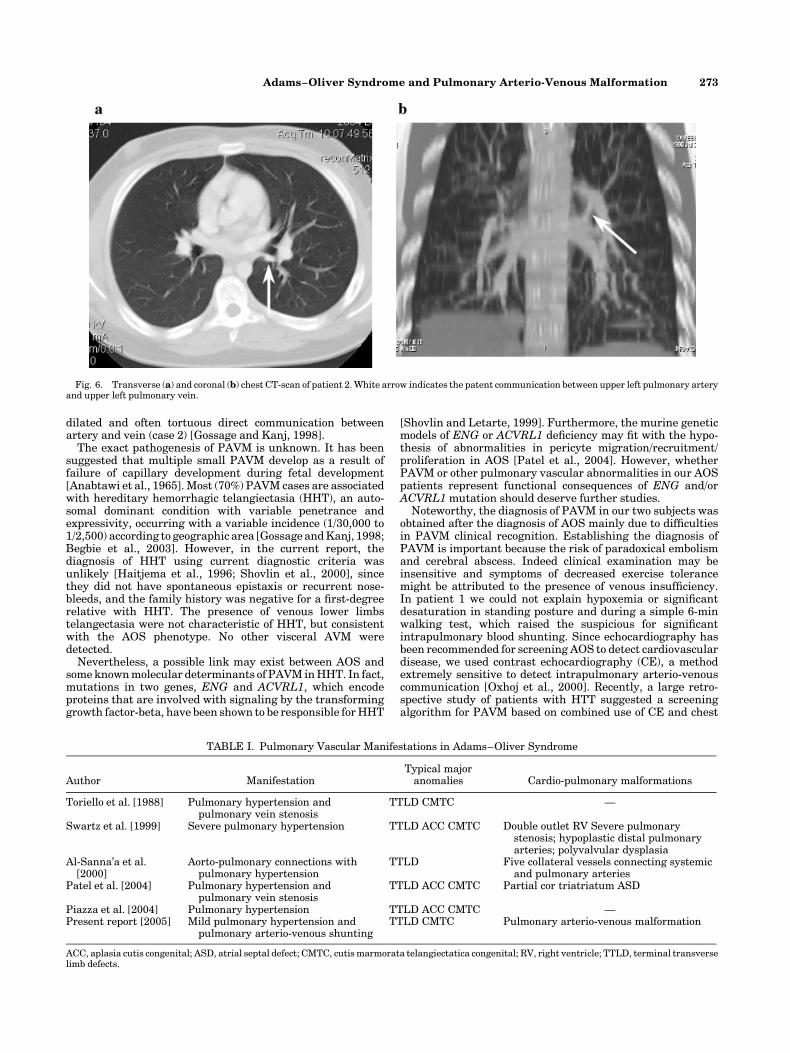
shunting. Spiral CT scan image reconstruction demonstrated alarge connection between upper lobar left pulmonary arteryand the lower left pulmonary vein (Fig. 6a,b). No othercardiovascular abnormalities were detectable and gastro-intestinal ultrasonography did not detect vascular or grossfunctional derangements. Furthermore complete blood countand serum iron were normal.
DISCUSSION
AOS is usually diagnosed when both terminal transverselimb and scalp defects present concomitantly [Adams andOliver, 1945; Kuster et al., 1988; Whitley and Gorlin, 1991].Patients with isolated limb or scalp defects, however, may alsobe considered to have AOS if both defects are present in familymembers [Whitley and Gorlin, 1991]. In the reported family,intra-familial variability with autosomal dominant inheri-tance was observed. The younger child (III-1, Fig. 1), showedpartial skull defect, distal digital hypoplasia, and cutismarmorata telangiectatica.
This is the first report of PAVM in familial AOS, which webelieve further supports the hypothesis for a causal role of avascular defect in AOS pathogenesis. Cardiovascular malfor-mations have been reported in about 20% of AOS patients, witha predominance of obstructive defects of the left heart [Linet al., 1998]. We believe that the PAVMs in our patients werecongenital. Acquired causes related to liver or lung chronicdisease were carefully excluded. The occurrence of PAVMs in
two members of the same family suggests a common under-lying mechanism.
Pulmonary vascular abnormalities have been described inAOS [Toriello et al., 1988; Chitayat et al., 1992; Fryns et al.,1996; Swartz et al., 1999], including pulmonary vein stenosisand pulmonary hypertension, or severe infantile unexplainedpulmonary hypertension (Table I). Abnormal pulmonary vas-culature with persistence of primitive systemic, pulmonaryartery vascular connection, supra-systemic pulmonary arterypressures has been reported in another young patient withterminal transverse limb defects typically seen in AOS butwith no scalp defect [Al-Sanna’a et al., 2000]. It has beenhypothesized that the limb defects in this syndrome are theresult of vascular disruption due to hemodynamically unstablevessels during the period of 6–8th week of embryonic life ordue to an abnormality of a small vessel structure as suggestedearlier [Swartz et al., 1999]. More recently other two cases withadditional features including intrauterine growth retardation,cutis marmorata telangiectatica congenital, pulmonary hyper-tension, and osteopenia were reported [Patel et al., 2004].Based on a histopathologic examination of one of these pati-ents, abnormal pericyte recruitment/coverage of the vascula-ture as major pathophysiologic mechanism in AOS wasinvoked. Accordingly, our cases showed PAVMs caused byabnormal communications between pulmonary arteries andpulmonary veins, which are most commonly congenital innature, demonstrating two of three typical appearancesdescribed in the literature as a large, single sac (case 1), or a
Fig. 3. Patient 2 at age 12 years. a: Right foot showing cutaneous syndactyly of 2nd and 3rd toes; (b) radiographs of the feet showing digital hypoplasia offirst metatarsal of left foot; (c) particular of dorso-lumbar region showing cutis telangiectatica. [Color figure can be viewed in the online issue, which isavailable at www.interscience.wiley.com.]
Adams–Oliver Syndrome and Pulmonary Arterio-Venous Malformation 271
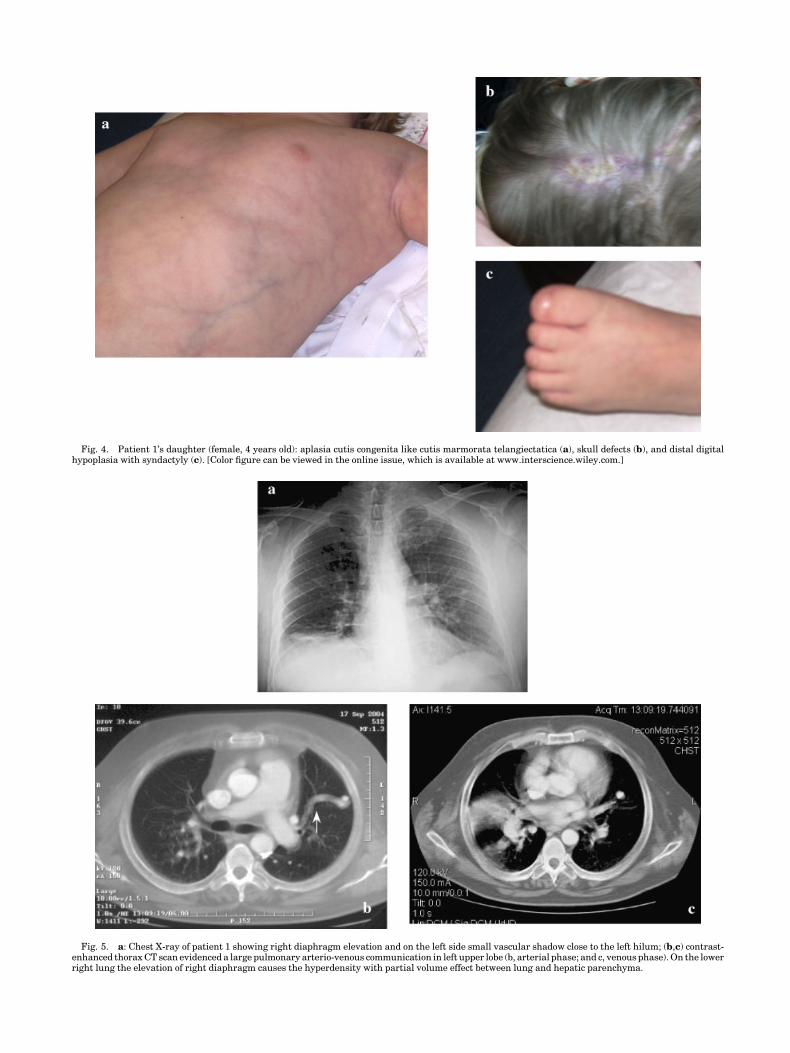
Fig. 4. Patient 1’s daughter (female, 4 years old): aplasia cutis congenita like cutis marmorata telangiectatica (a), skull defects (b), and distal digitalhypoplasia with syndactyly (c). [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
Fig. 5. a: Chest X-ray of patient 1 showing right diaphragm elevation and on the left side small vascular shadow close to the left hilum; (b,c) contrast-enhanced thorax CT scan evidenced a large pulmonary arterio-venous communication in left upper lobe (b, arterial phase; and c, venous phase). On the lowerright lung the elevation of right diaphragm causes the hyperdensity with partial volume effect between lung and hepatic parenchyma.
dilated and often tortuous direct communication betweenartery and vein (case 2) [Gossage and Kanj, 1998].
The exact pathogenesis of PAVM is unknown. It has beensuggested that multiple small PAVM develop as a result offailure of capillary development during fetal development[Anabtawi et al., 1965]. Most (70%) PAVM cases are associatedwith hereditary hemorrhagic telangiectasia (HHT), an auto-somal dominant condition with variable penetrance andexpressivity, occurring with a variable incidence (1/30,000 to1/2,500) according to geographic area [Gossage and Kanj, 1998;Begbie et al., 2003]. However, in the current report, thediagnosis of HHT using current diagnostic criteria wasunlikely [Haitjema et al., 1996; Shovlin et al., 2000], sincethey did not have spontaneous epistaxis or recurrent nose-bleeds, and the family history was negative for a first-degreerelative with HHT. The presence of venous lower limbstelangectasia were not characteristic of HHT, but consistentwith the AOS phenotype. No other visceral AVM weredetected.
Nevertheless, a possible link may exist between AOS andsome known molecular determinants of PAVM in HHT. In fact,mutations in two genes, ENG and ACVRL1, which encodeproteins that are involved with signaling by the transforminggrowth factor-beta, have been shown to be responsible for HHT
[Shovlin and Letarte, 1999]. Furthermore, the murine geneticmodels of ENG or ACVRL1 deficiency may fit with the hypo-thesis of abnormalities in pericyte migration/recruitment/proliferation in AOS [Patel et al., 2004]. However, whetherPAVM or other pulmonary vascular abnormalities in our AOSpatients represent functional consequences of ENG and/orACVRL1 mutation should deserve further studies.
Noteworthy, the diagnosis of PAVM in our two subjects wasobtained after the diagnosis of AOS mainly due to difficultiesin PAVM clinical recognition. Establishing the diagnosis ofPAVM is important because the risk of paradoxical embolismand cerebral abscess. Indeed clinical examination may beinsensitive and symptoms of decreased exercise tolerancemight be attributed to the presence of venous insufficiency.In patient 1 we could not explain hypoxemia or significantdesaturation in standing posture and during a simple 6-minwalking test, which raised the suspicious for significantintrapulmonary blood shunting. Since echocardiography hasbeen recommended for screening AOS to detect cardiovasculardisease, we used contrast echocardiography (CE), a methodextremely sensitive to detect intrapulmonary arterio-venouscommunication [Oxhoj et al., 2000]. Recently, a large retro-spective study of patients with HTT suggested a screeningalgorithm for PAVM based on combined use of CE and chest
TABLE I. Pulmonary Vascular Manifestations in Adams–Oliver Syndrome
Author ManifestationTypical major
anomalies Cardio-pulmonary malformations
Toriello et al. [1988] Pulmonary hypertension andpulmonary vein stenosis
TTLD CMTC —
Swartz et al. [1999] Severe pulmonary hypertension TTLD ACC CMTC Double outlet RV Severe pulmonarystenosis; hypoplastic distal pulmonaryarteries; polyvalvular dysplasia
Al-Sanna’a et al.[2000]
Aorto-pulmonary connections withpulmonary hypertension
TTLD Five collateral vessels connecting systemicand pulmonary arteries
Patel et al. [2004] Pulmonary hypertension andpulmonary vein stenosis
TTLD ACC CMTC Partial cor triatriatum ASD
Piazza et al. [2004] Pulmonary hypertension TTLD ACC CMTC —Present report [2005] Mild pulmonary hypertension and
pulmonary arterio-venous shuntingTTLD CMTC Pulmonary arterio-venous malformation
ACC, aplasia cutis congenital; ASD, atrial septal defect; CMTC, cutis marmorata telangiectatica congenital; RV, right ventricle; TTLD, terminal transverselimb defects.
Fig. 6. Transverse (a) and coronal (b) chest CT-scan of patient 2. White arrow indicates the patent communication between upper left pulmonary arteryand upper left pulmonary vein.
Adams–Oliver Syndrome and Pulmonary Arterio-Venous Malformation 273

radiograph, followed by chest CT [Cottin et al., 2004]. However,further studies in larger number of subjects are needed toestablish the role of chest X-ray, oximetry, and CE for PAVMscreening in AOS.
The reported association of PAVM in an AOS family does notseem coincidental, suggesting a pathogenic role of abnormalendothelium pathway signaling in the development of thesyndrome. Moreover, since PAVM may cause potentiallyhazardous complications, screening for PAVMs among AOSpatients and their relatives warrants further studies.
ACKNOWLEDGMENTS
We thank Dr. Luigi Pianese for manuscript revision andTanja Sobko for technical assistance.
REFERENCES
Adams FH, Oliver C. 1945. Hereditary deformities in man due to arresteddevelopment. J Hered 36:3–7.
Al-Sanna’a N, Adatia I, Teebi AS. 2000. Transverse limb defects associatedwith aorto-pulmonary vascular abnormalities: Vascular disruptionsequence or atypical presentation of Adams–Oliver syndrome? Am JMed Genet 94:400–404.
Anabtawi IN, Ellison RG, Ellison LT. 1965. Pulmonary arteriovenousaneurysms and fistulas. Anatomical variations, embryology, and classi-fication. Ann Thorac Surg 122:277–285.
Bamforth JS, Kaurah P, Byrne J, Ferreira P. 1994. Adams Oliver syndrome:A family with extreme variability in clinical expression. Am J Med Genet49:393–396.
Begbie ME, Wallace GM, Shovlin CL. 2003. Hereditary haemorrhagictelangiectasia (Osler–Weber–Rendu syndrome): A view from the 21stcentury. Postgrad Med J 79:18–24.
Chitayat D, Meunier C, Hodgkinson KA, Robb L, Azouz M. 1992. Acrania:A manifestation of the Adams–Oliver syndrome. Am J Med Genet 44:562–566.
Cottin V, Plauchu H, Bayle JY, Barthelet M, Revel D, Cordier JF. 2004.Pulmonary arteriovenous malformations in patients with hereditaryhemorrhagic telangiectasia. Am J Respir Crit Care Med 169:994–1000.
David A, Roze JC, Melon-David V. 1991. Adams–Oliver syndromeassociated with congenital heart defect: Not a coincidence. Am J MedGenet 40:126–127.
Farrell SA, Warda LJ, LaFlair P, Szymonowicz W. 1993. Adams–Oliversyndrome: A case with juvenile chronic myelogenous leukemia andchylothorax. Am J Med Genet 47:1175–1179.
Frank RA, Frosch PJ. 1993. Adams–Oliver syndrome: Cutis marmoratateleangiectatica congenita with multiple anomalies. Dermatology 187:205–208.
Fryns JP, Legius E, Demaerel P, van den Berghe H. 1996. Congenital scalpdefect, distal limb reduction anomalies, right spastic hemiplegia and
hypoplasia of the left arteria cerebri media. Further evidence thatinterruption of early embryonic blood supply may result in Adams–Oliver (plus) syndrome. Clin Genet 50:505–509.
Gossage JR, Kanj G. 1998. Pulmonary arteriovenous malformations. A stateof the art review. Am J Respir Crit Care Med 158:643–661.
Haitjema T, Westermann CJ, Overtoom TT, Timmer R, Disch F, Mauser H,Lammers JW. 1996. Hereditary hemorrhagic telangiectasia (Osler–Weber–Rendu disease): New insights in pathogenesis, complications,and treatment. Arch Intern Med 156:714–719.
Kuster W, Lenz W, Kaariainen H, Majewski F. 1988. Congenital scalpdefects with distal limb anomalies (Adams–Oliver syndrome): Report often cases and review of the literature. Am J Med Genet 31:99–115.
Lin AE, Westgate MN, van der Velde ME, Lacro RV, Holmes LB. 1998.Adams–Oliver syndrome associated with cardiovascular malforma-tions. Clin Dysmorphol 7:235–241.
Mempel M, Abeck D, Lange I, Strom K, Caliebe A, Beham A, Kautza M,Worret WI, Neubauer BA, Ring J, Schroeder H, Folster-Holst R. 1999.The wide spectrum of clinical expression in Adams–Oliver syndrome:A report of two cases. Br J Dermatol 140:1157–1160.
Oxhoj H, Kjeldsen AD, Nielsen G. 2000. Screening for pulmonary arterio-venous malformations: Contrast echocardiography versus pulse oxime-try. Scand Cardiovasc J 34:281–285.
Patel MS, Taylor GP, Bharya S, Al-Sanna’a N, Adatia I, Chitayat D, SuzanneLewis ME, Human DG. 2004. Abnormal pericyte recruitment as a causefor pulmonary hypertension in Adams–Oliver syndrome. Am J MedGenet 129:294–299.
Pereira-Da-Silva L, Leal F, Santos GC, Videira Amaral JM, Feijoo MJ.2000. Clinical evidence of vascular abnormalities at birth in Adams–Oliver syndrome: Report of two further cases. Am J Med Genet 94:75–76.
Piazza AJ, Blackston D, Sola A. 2004. A case of Adams–Oliver sindrome withassociated brain and pulmonary involvement: Further evidence of vas-cular pathology? Am J Med Genet 130:172–175.
Shovlin CL, Letarte M. 1999. Hereditary haemorrhagic telangiectasia andpulmonary arteriovenous malformations: Issues in clinical managementand review of pathogenic mechanisms. Thorax 54:714–729.
Shovlin CL, Guttmacher AE, Buscarini E, Faughnan ME, Hyland RH,Westermann CJ, Kjeldsen AD, Plauchu H. 2000. Diagnostic criteriafor hereditary hemorrhagic telangiectasia (Rendu–Osler–Weber syn-drome). Am J Med Genet 91:66–67.
Swartz EN, Sanatani S, Sandor GG, Schreiber RA. 1999. Vascularabnormalities in Adams–Oliver syndrome: Cause or effect? Am J MedGenet 82:49–52.
Toriello HV, Graff RG, Florentine MF, Lacina S, Moore WD. 1988. Scalp andlimb defects with cutis marmorata telangiectatica congenita: Adams–Oliver syndrome? Am J Med Genet 29:269–276.
Whitley CB, Gorlin RJ. 1991. Adams–Oliver syndrome revisited. Am J MedGenet 40:319–326.
Zapata HH, Sletten LJ, Pierpont ME. 1995. Congenital cardiac malfor-mations in Adams–Oliver syndrome. Clin Genet 47:80–84.
274 Maniscalco et al.





























