Embed Size (px)
Citation preview

Atrial Natriuretic Peptide Induces Natriuretic PeptideReceptor-cGMP-dependent Protein Kinase Interaction*
Received for publication, April 18, 2003, and in revised form, July 1, 2003Published, JBC Papers in Press, July 10, 2003, DOI 10.1074/jbc.M304098200
Nathan Airhart, Yong-Feng Yang, Charles T. Roberts, Jr., and Michael Silberbach‡
From the Department of Pediatrics and the Heart Research Center, Oregon Health and Science University,Portland, Oregon 97239
Circulating natriuretic peptides such as atrial natri-uretic peptide (ANP) counterbalance the effects of hy-pertension and inhibit cardiac hypertrophy by activat-ing cGMP-dependent protein kinase (PKG). Natriureticpeptide binding to type I receptors (NPRA and NPRB)activates their intrinsic guanylyl cyclase activity, re-sulting in a rapid increase in cytosolic cGMP that sub-sequently activates PKG. Phosphorylation of the recep-tor by an unknown serine/threonine kinase is requiredbefore ligand binding can activate the cyclase. Whilesearching for downstream PKG partners using a yeasttwo-hybrid screen of a human heart cDNA library, weunexpectedly found an upstream association withNPRA. PKG is a serine/threonine kinase capable of phos-phorylating NPRA in vitro; however, regulation ofNPRA by PKG has not been previously reported. Herewe show that PKG is recruited to the plasma membranefollowing ANP treatment, an effect that can be blockedby pharmacological inhibition of PKG activation. Fur-thermore, PKG participates in a ligand-dependent gain-of-function loop that significantly increases the intrin-sic cyclase activity of the receptor. PKG translocation isANP-dependent but not nitric oxide-dependent. Our re-sults suggest that anchoring of PKG to NPRA is a keyevent after ligand binding that determines distal effects.As such, the NPRA-PKG association may represent anovel mechanism for compartmentation of cGMP-medi-ated signaling and regulation of receptor sensitivity.
The natriuretic peptides (NPs)1 are produced by the heart,the vasculature, and the kidney and are an ancient family ofpolypeptide hormones that regulate mammalian blood volumeand blood pressure. More recently, the ability of NPs to mod-ulate cell proliferation (1) and cardiac hypertrophy (2) has beendemonstrated. Physiologically important NP actions includenatriuresis (3), vasodilation (4), and ubiquitous inhibitory ac-tions such as inhibition of smooth muscle proliferation (1, 5),
cardiac fibroblast proliferation (6), cardiomyocyte hypertrophy(2, 7–9), sympathetic tone (10), renin-angiotensin-aldosteroneactivation (11), and hypothalamic-pituitary-adrenal axis sig-naling (12–14). In disease states such as heart failure, NPactions may be limited by resistance to hormone effects (15–17)that are at least in part because of insensitivity of the receptoritself (18).
Natriuretic peptide binding to type I receptors (NPRA andNPRB) on target cells activates their intrinsic guanylyl cyclaseactivity, resulting in a rapid increase in cGMP. DiffusiblecGMP acts as a second messenger primarily by stimulatingPKG (19). PKG is the major mediator of cGMP-induced smoothmuscle relaxation (20). Downstream NP effects that have beendirectly tied to activated PKG include modulation of the L-typecalcium channel (21, 22) and cross-talk with heterologous re-ceptors, such as G protein-coupled receptors (23, 24). Further-more, there is recent evidence that the membrane-bound gua-nylyl cyclase, NPRA, but not soluble cyclases that are activatedby nitric oxide, has potent effects on plasma membrane controlof the calcium ATPase pump (25), suggesting that NO- andNP-mediated effects are compartmentalized in cells.
Although the NPRA cDNA was first cloned more than 13years ago (26), its regulation remains poorly understood. In itsprebound state, the NPRA receptor exists as a homodimer (27),but ligand binding alone is insufficient to induce cyclase activ-ity. Rather, phosphorylation of six serine and threonine resi-dues in the intracellular juxtamembrane-kinase homology do-main makes the receptor susceptible to NP activation (28). Theprotein kinase that mediates receptor phosphorylation isunknown.
We have previously reported that ANP inhibits cardiac hy-pertrophy through cGMP/PKG-mediated activation of the ERKsignaling cascade at the level of MEK (9) but could not demon-strate a direct interaction between PKG and MEK. PKG sub-strates are membrane-bound (29), cytosolic (30), and intranu-clear (31). In an attempt to identify novel proteins that could becandidates for linking PKG to MEK, we used a cytosolic yeasttwo-hybrid system employing PKG as bait. We found that PKGdirectly interacts with NPRA. These results were initially quitesurprising, because PKG was previously thought to regulateonly downstream ANP targets. However, it has been demon-strated previously that PKG is a serine/threonine kinase capa-ble of phosphorylating NPRA in vitro (32). We report for thefirst time the regulation of NPRA by PKG.
EXPERIMENTAL PROCEDURES
Yeast Two-hybrid Studies—Screening for PKG-interacting proteinswas done using a commercially available system (CytoTrap, Strat-agene, La Jolla, CA) that identifies protein-protein interactions in theyeast cytoplasm. Rather than relying on transcriptional activation todetect interactions, the RAS signal transduction cascade is initiated byrecruitment of hSOS to the plasma membrane in a temperature-sensi-tive mutant yeast strain, cdc25H, by virtue of the interaction of its baitfusion partner with a myristoylated prey protein, which allows growth
* This work was supported by grants (to M. S.) from the Friends ofDoernbecher, the Dickinson Family Foundation, and the Glenn andJuanita Struble Research Fund of the Oregon Health and ScienceUniversity Heart Research Center. The costs of publication of thisarticle were defrayed in part by the payment of page charges. Thisarticle must therefore be hereby marked “advertisement” in accordancewith 18 U.S.C. Section 1734 solely to indicate this fact.
‡ To whom correspondence should be addressed: Doernbecher Chil-dren’s Hospital, 707 S.W. Gaines Rd., Mail Code CDRC-P, Portland, OR97239-2901. Tel.: 503-494-9899; Fax: 503-494-2824; E-mail: [email protected].
1 The abbreviations used are: NP, natriuretic peptide; PKG, cGMP-dependent protein kinase; NO, nitric oxide; ANP, atrial NP; PIPES,1,4-piperazinediethanesulfonic acid; SNP, S-nitroso-N-acetylpenicilla-mine; HEK, human embryonic kidney; MEK, mitogen-activated proteinkinase/extracellular signal-regulated kinase kinase; ERK, extracellularsignal-regulated kinase.
THE JOURNAL OF BIOLOGICAL CHEMISTRY Vol. 278, No. 40, Issue of October 3, pp. 38693–38698, 2003© 2003 by The American Society for Biochemistry and Molecular Biology, Inc. Printed in U.S.A.
This paper is available on line at http://www.jbc.org 38693
by guest on March 19, 2019
http://ww
w.jbc.org/
Dow
nloaded from

at 36 °C. A bovine PKG I� cDNA was cloned in-frame into the hSOSbait plasmid. A human heart cDNA library in the pMyr plasmid (Strat-agene) containing 7.4 � 106 independent clones was cotransfected withthe pPKG-hSOS expression vector into competent cdc25H cells, whichwere then grown for 4 days at 25 °C on minimal glucose plates. Colonieswere isolated and tested for galactose-dependent growth at 36 °C. Plas-mids were extracted from three initially positive colonies, and theinserts were sequenced before retransformation in cdc25H cells to-gether with pPKG-hSOS. Conventional yeast transformation and ma-nipulation protocols were used. Cells were replica-plated onto eitherglucose- or galactose-minimal medium containing relevant amino acids,according to the manufacturer’s protocols.
Immunocytochemistry—HEK-NPRA and HEK293 control cellswere incubated in 4-well chamber slides at a density of �100,000cells/cm2 in Dulbecco’s modified Eagle’s medium with 10% fetal bo-vine serum and 1% penicillin/streptomycin solution (Invitrogen). Af-ter fixation in 3.7% formaldehyde, cells were permeabilized with 0.3%Triton X-100 in phosphate-buffered saline for 10 min and blockedwith 1% horse serum, 0.2% bovine serum albumin in phosphate-buffered saline. Fixed slides were incubated with a primary antibodymixture containing 0.1 �g/ml rabbit anti-PKG I (Stressgen) or rabbitanti-PKA (Santa Cruz Biotechnology) and 0.7 �g/ml mouse anti-FLAG M2 (Sigma). The immunogen used to generate the anti-PKG Iantisera has no homology to PKG II making cross-reactivity ex-tremely unlikely. Cells were blocked for 1 h and then for an additionalhour with fluorescein isothiocyanate or Cy5-conjugated donkey sec-ondary antibodies (Jackson Laboratories); cells were then mountedon coverslips using Gel/Mount mounting medium (Biomedia Corp.).Cardiac cells were treated identically, except fixed cells were incu-bated with a 1:1000 dilution of rhodamine-phalloidin (MolecularProbes, Eugene, OR) for 1 h. Fluorescent imaging was performed witha Leica DMRA upright fluorescent microscope, and images wereacquired with a Hamamatsu ORCA2 CCD camera.
Cardiac Cell Culture—The care of all animals used in this researchwas in accordance with institutional guidelines. Ventricular cardiaccells from 1–2-day-old Harlan Sprague-Dawley rats were prepared asdescribed previously (33). Ventricles were dissected free from atria andquartered. Myocytes were dissociated in trypsin and DNase I andpreplated to remove non-myocyte cells. In all experiments, cells wereplated on gelatin-coated chamber slides (�400,000 cells/chamber) andmaintained overnight at 5% CO2 in Dulbecco’s modified Eagle’s mediumwith 17% Media 199, 10% horse serum, and 5% fetal bovine serum.Cells were incubated in 80% Dulbecco’s modified Eagle’s medium, 20%Media 199 (Invitrogen), and phenylephrine (1 �M) 72 h beforetreatments.
Preparation of Plasma Membranes—Plasma membranes were madeas described previously (26). HEK-NPRA and control cells were platedat equal densities and transfected with 1 �g of the PKG expressionvector using FuGENE (Roche Applied Science). After a 48-h incubation,cells were rinsed with ice-cold phosphate-buffered saline and scrapedinto 0.5 ml of buffer (20 mM HEPES, pH 7.4, 50 mM NaCl, 5 mM EDTA,
10 �g/ml leupeptin, 10 �g/ml aprotinin, and 1 �g/ml pepstatin). Cellswere then mechanically lysed by flushing repeatedly through a 22-gauge needle. The particulate fraction was isolated by centrifugation at5000 � g for 15 min at 4 °C, and the pellet was washed once withscraping buffer prior to experiments.
In Vitro Kinase Activity—Kinase activity measurements were per-formed as described (34). Crude membrane lysates were solubilized in0.5 ml of immunoprecipitation buffer (50 mM Tris, 50 mM NaCl, 5 mM
EDTA, 0.05% Nonidet P-40, 10 �g/ml leupeptin, 10 �g/ml aprotinin,and 1 �g/ml pepstatin). Following centrifugation (5000 � g for 15 minat 4 °C), 50 �l of FLAG-M2-conjugated agarose beads (Sigma A220)were added to the supernatant and rocked overnight at 4 °C. Afteradditional washes in 20 mM HEPES, pH 7.4, 50 mM NaCl, 5 mM EDTA,50 �l of kinase buffer (40 mM Tris, pH 7.4, 2 mM MgOAc, 100 �M
isobutylmethylxanthine, 150 �M BPDEtide (PKG-specific substrate,Calbiochem)), 10 �Ci/reaction [�-32P]ATP (specific activity 3,000 Ci/mM,ICN), and 1 �M synthetic protein kinase A inhibitor (Calbiochem) wereadded to the reaction, with or without 5 �M cGMP, and incubated at30 °C for 15 min. 40 �l was then spotted onto Whatman P81 phospho-cellulose paper. After air-drying, papers were washed 5 times with 0.5%phosphoric acid and radioactivity was quantified using a Beckman LS6500 liquid scintillation counter. In other studies, in vitro kinase activ-ity was measured identically except that the immunoprecipitation stepswere omitted.
Guanylyl Cyclase Activity—Measurement of hormone-dependentguanylyl cyclase activity was performed as described (28), except thatcold cGMP was added to the reactions to assure PKG activation incontrol cells. Crude membranes prepared from cells plated at equaldensities were solubilized in 100 �l of assay buffer (25 mM PIPES, pH7.4, 50 mM NaCl, 250 �M isobutylmethylxanthine, 0.1% bovine serumalbumin, 5 mM creatine phosphate, 5 units/assay creatine phosphoki-nase, 100 nM 8-bromo-cGMP, 1 mM GTP, 5.5 �Ci/reaction [�-32P]GTP(specific activity 3000 Ci/mM, ICN), 5 mM MgCl2, 1 mM ATP, 10 �g/mlleupeptin, 10 �g/ml aprotinin, 1 �g/ml pepstatin, 10 mM NaPO4, 0.1 M
NaF, 1 mM Na3VO4, 80 �M �-glycerol phosphate) and treated as de-scribed. Reactions were incubated at 30 °C and terminated by adding500 �l of 110 mM ZnOAc. To precipitate unincorporated [�-32P]GTP, 500�l of 110 mM sodium carbonate was added to each reaction. Following
FIG. 1. Interaction of NPRA and PKG in yeast. Cdc25H cells weretransformed with the indicated plasmids and plated on glucose minimalmedium supplemented with the appropriate amino acids. Three inde-pendent transformant clones harboring prey plasmids encoding NPRA,RIT (a Ras-related GTPase (46)), and an unknown protein (3d) weregrown at 25 °C, indicating successful plasmid co-transformation (leftpanel), replica-plated onto appropriately supplemented galactose min-imal plates, and grown at 36 °C (right panel). Both the NPRA andRIT-containing chimeras grew at 36 °C when co-transformed with thePKG-SOS plasmid, but the RIT chimera also grew with the insertlesspSOS vector, indicating PKG-independent activation. Clone 3d did notgrow at 36 °C.
FIG. 2. PKG is present at the cell membrane. A, PKG kinaseactivity is present in detergent-containing cell membrane lysates fromHEK-NPRA cells but not control cells. NPRA immunoprecipitates oflysates from control cells or cells transfected with a bovine PKG I�expression vector were assayed for PKG activity. Data are expressed aspercent increase from reactions without cGMP (mean � S.E. of threeindependent experiments, each performed in triplicate). B, PKG proteinis present in cell membrane lysates of HEK-NPRA but not control cells(HEK293). HEK-NPRA and control cells were transfected with the PKGexpression vector and then treated for the indicated times with 1 �M
ANP before preparation of detergent-free lysates (upper panel). PKG ispresent in cytosolic lysates of both HEK-NPRA and control cells (middlepanel). NPRA protein is present in cell membrane lysates of HEK-NPRA but not control cells (lower panel). Lysates underwent SDS-PAGE electrophoresis and were immunoblotted with anti-PKG orNPRA antibody (images are representative of three separateexperiments).
NPRA/PKG Interaction38694
by guest on March 19, 2019
http://ww
w.jbc.org/
Dow
nloaded from
centrifugation, the supernatant was added to a chromatography column(Bio-Rad 731–1550) containing 1.0 g of dry neural alumina resin (SigmaA9003) acidified with 5 ml of 1 N perchloric acid. The column waswashed sequentially with 20 ml of 1 N perchloric acid and 20 ml ofwater. [32P]cGMP was eluted into scintillation vials with 10 ml of 200mM ammonium formate and counted using a Beckman LS 6500 liquidscintillation counter.
RESULTS
NPRA and PKG Associate in Yeast—To identify potentialPKG partners, we screened a human heart cDNA library usinga cytosolic yeast two-hybrid system (see “Experimental Proce-dures”). Employing PKG as bait, the single positive clone iden-tified corresponded to an �300-bp cDNA fragment of NPRAencoding the entire C-terminal cyclase domain and part of theso-called hinge region (35) (Fig. 1).
NPRA and PKG Associate in Mammalian Cells—Experi-ments were carried out in HEK293 cells stably transfected withan expression vector encoding FLAG epitope-tagged NPRA (27)(HEK-NPRA). HEK-NPRA cells exhibit a hormone-stimulatedgeneration of cGMP, whereas untransfected HEK293 cells ex-
press minimal endogenous NPRA (28).2 Using FLAG antibody,it was not possible to co-immunoprecipitate levels of PKG fromHEK-NPRA membranes that were detectable by Western im-munoblotting, presumably because detergent solubilizationdisrupted the NPRA-PKG association. Attempts to releaseNPRA from membrane lysates while maintaining PKG associ-ation using a variety of concentrations of both ionic and non-ionic detergents were unsuccessful. However, these same im-munoprecipitates clearly contained low levels of PKG basedupon highly sensitive PKG activity assays (Fig. 2A). BecauseNPRA is a transmembrane protein and membrane localizationof PKG had not been previously reported, we prepared deter-gent-free lysates from crude membranes of HEK-NPRA andcontrol cells that had both been transiently transfected withthe PKG expression vector. PKG protein (Fig. 2B, upper panel)and kinase activity (Fig. 3C) were detectable in the HEK-NPRAmembranes but not the control membrane fraction. Treatment
2 M. Chinkers, personal communication.
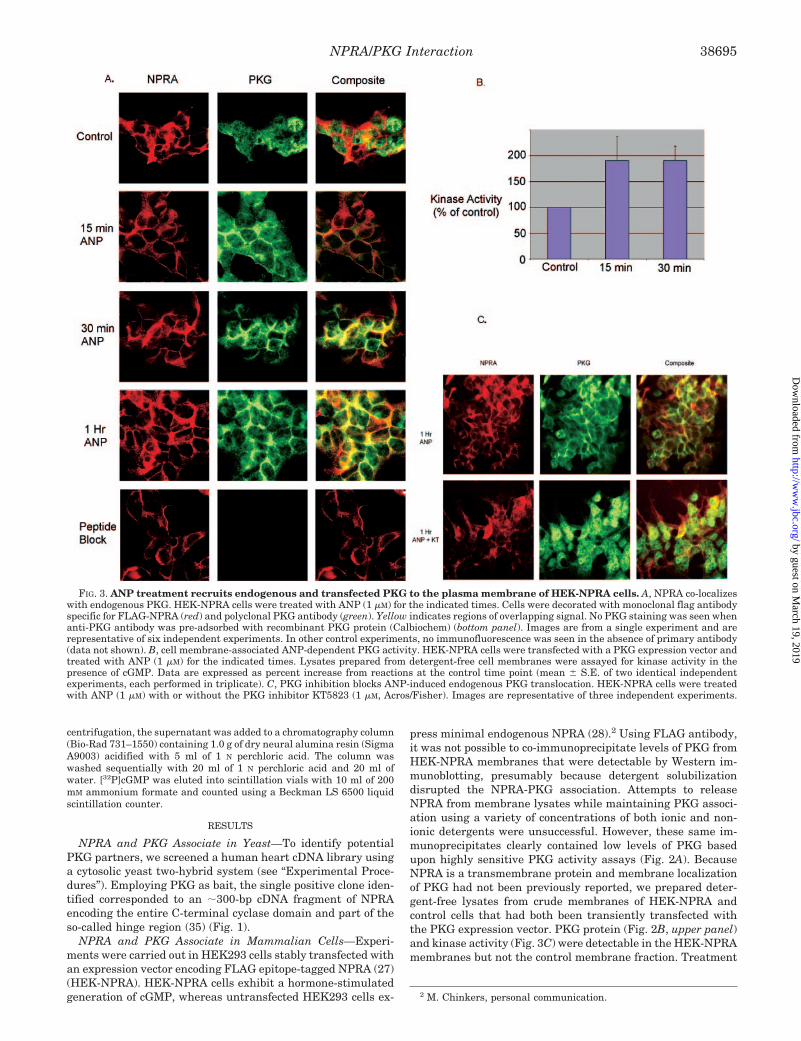
FIG. 3. ANP treatment recruits endogenous and transfected PKG to the plasma membrane of HEK-NPRA cells. A, NPRA co-localizeswith endogenous PKG. HEK-NPRA cells were treated with ANP (1 �M) for the indicated times. Cells were decorated with monoclonal flag antibodyspecific for FLAG-NPRA (red) and polyclonal PKG antibody (green). Yellow indicates regions of overlapping signal. No PKG staining was seen whenanti-PKG antibody was pre-adsorbed with recombinant PKG protein (Calbiochem) (bottom panel). Images are from a single experiment and arerepresentative of six independent experiments. In other control experiments, no immunofluorescence was seen in the absence of primary antibody(data not shown). B, cell membrane-associated ANP-dependent PKG activity. HEK-NPRA cells were transfected with a PKG expression vector andtreated with ANP (1 �M) for the indicated times. Lysates prepared from detergent-free cell membranes were assayed for kinase activity in thepresence of cGMP. Data are expressed as percent increase from reactions at the control time point (mean � S.E. of two identical independentexperiments, each performed in triplicate). C, PKG inhibition blocks ANP-induced endogenous PKG translocation. HEK-NPRA cells were treatedwith ANP (1 �M) with or without the PKG inhibitor KT5823 (1 �M, Acros/Fisher). Images are representative of three independent experiments.
NPRA/PKG Interaction 38695
by guest on March 19, 2019
http://ww
w.jbc.org/
Dow
nloaded from
with ANP for 15 and 60 min, but not 5 min, demonstrated anapparent increase in membrane-associated PKG protein in theHEK-NPRA membranes only (Fig. 2B). PKG protein was pres-ent equally in cytosolic fractions of HEK-NPRA and controlcells (Fig. 2B, middle panel), and NPRA protein was observedonly in the HEK-NPRA membranes (Fig. 2B, lower panel). Inadditional experiments, ANP treatment for either 15 or 30 minresulted in a significant increase in PKG activity in crudemembrane preparations of HEK-NPRA cells compared withuntreated cells (Fig. 3B). Taken together, these data suggestthat PKG is prebound to unliganded receptor and that ANPbinding to NPRA recruits additional PKG to the cellmembrane.
We next used immunofluorescence microscopy to verify theANP-dependent co-localization of NPRA and endogenous PKG.After ANP treatment for 15, 30, and 60 min, we observedmarkedly increased staining of PKG at the membrane in HEK-NPRA cells (Fig. 3A) but not in control cells (data not shown).Furthermore, the PKG inhibitor KT5823 completely blockedthe ANP-induced translocation of PKG to the plasma mem-brane (Fig. 3C), suggesting that activation of PKG is necessary
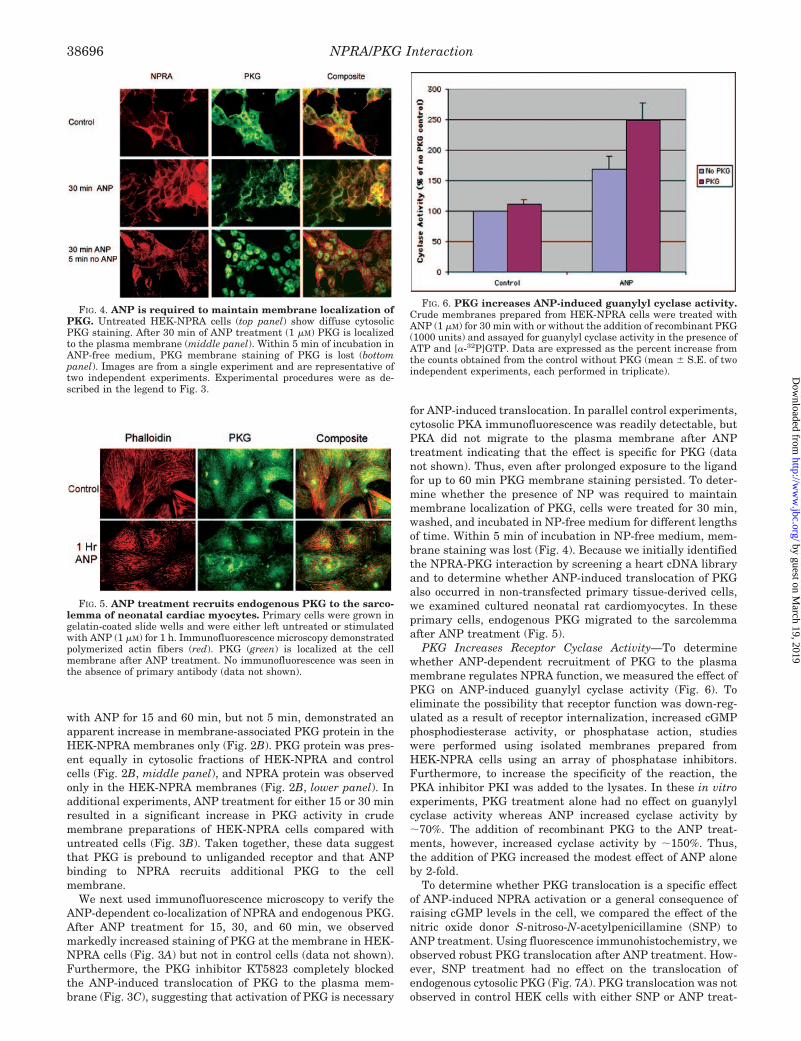
for ANP-induced translocation. In parallel control experiments,cytosolic PKA immunofluorescence was readily detectable, butPKA did not migrate to the plasma membrane after ANPtreatment indicating that the effect is specific for PKG (datanot shown). Thus, even after prolonged exposure to the ligandfor up to 60 min PKG membrane staining persisted. To deter-mine whether the presence of NP was required to maintainmembrane localization of PKG, cells were treated for 30 min,washed, and incubated in NP-free medium for different lengthsof time. Within 5 min of incubation in NP-free medium, mem-brane staining was lost (Fig. 4). Because we initially identifiedthe NPRA-PKG interaction by screening a heart cDNA libraryand to determine whether ANP-induced translocation of PKGalso occurred in non-transfected primary tissue-derived cells,we examined cultured neonatal rat cardiomyocytes. In theseprimary cells, endogenous PKG migrated to the sarcolemmaafter ANP treatment (Fig. 5).
PKG Increases Receptor Cyclase Activity—To determinewhether ANP-dependent recruitment of PKG to the plasmamembrane regulates NPRA function, we measured the effect ofPKG on ANP-induced guanylyl cyclase activity (Fig. 6). Toeliminate the possibility that receptor function was down-reg-ulated as a result of receptor internalization, increased cGMPphosphodiesterase activity, or phosphatase action, studieswere performed using isolated membranes prepared fromHEK-NPRA cells using an array of phosphatase inhibitors.Furthermore, to increase the specificity of the reaction, thePKA inhibitor PKI was added to the lysates. In these in vitroexperiments, PKG treatment alone had no effect on guanylylcyclase activity whereas ANP increased cyclase activity by�70%. The addition of recombinant PKG to the ANP treat-ments, however, increased cyclase activity by �150%. Thus,the addition of PKG increased the modest effect of ANP aloneby 2-fold.
To determine whether PKG translocation is a specific effectof ANP-induced NPRA activation or a general consequence ofraising cGMP levels in the cell, we compared the effect of thenitric oxide donor S-nitroso-N-acetylpenicillamine (SNP) toANP treatment. Using fluorescence immunohistochemistry, weobserved robust PKG translocation after ANP treatment. How-ever, SNP treatment had no effect on the translocation ofendogenous cytosolic PKG (Fig. 7A). PKG translocation was notobserved in control HEK cells with either SNP or ANP treat-
FIG. 4. ANP is required to maintain membrane localization ofPKG. Untreated HEK-NPRA cells (top panel) show diffuse cytosolicPKG staining. After 30 min of ANP treatment (1 �M) PKG is localizedto the plasma membrane (middle panel). Within 5 min of incubation inANP-free medium, PKG membrane staining of PKG is lost (bottompanel). Images are from a single experiment and are representative oftwo independent experiments. Experimental procedures were as de-scribed in the legend to Fig. 3.
FIG. 5. ANP treatment recruits endogenous PKG to the sarco-lemma of neonatal cardiac myocytes. Primary cells were grown ingelatin-coated slide wells and were either left untreated or stimulatedwith ANP (1 �M) for 1 h. Immunofluorescence microscopy demonstratedpolymerized actin fibers (red). PKG (green) is localized at the cellmembrane after ANP treatment. No immunofluorescence was seen inthe absence of primary antibody (data not shown).
FIG. 6. PKG increases ANP-induced guanylyl cyclase activity.Crude membranes prepared from HEK-NPRA cells were treated withANP (1 �M) for 30 min with or without the addition of recombinant PKG(1000 units) and assayed for guanylyl cyclase activity in the presence ofATP and [�-32P]GTP. Data are expressed as the percent increase fromthe counts obtained from the control without PKG (mean � S.E. of twoindependent experiments, each performed in triplicate).
NPRA/PKG Interaction38696
by guest on March 19, 2019
http://ww
w.jbc.org/
Dow
nloaded from
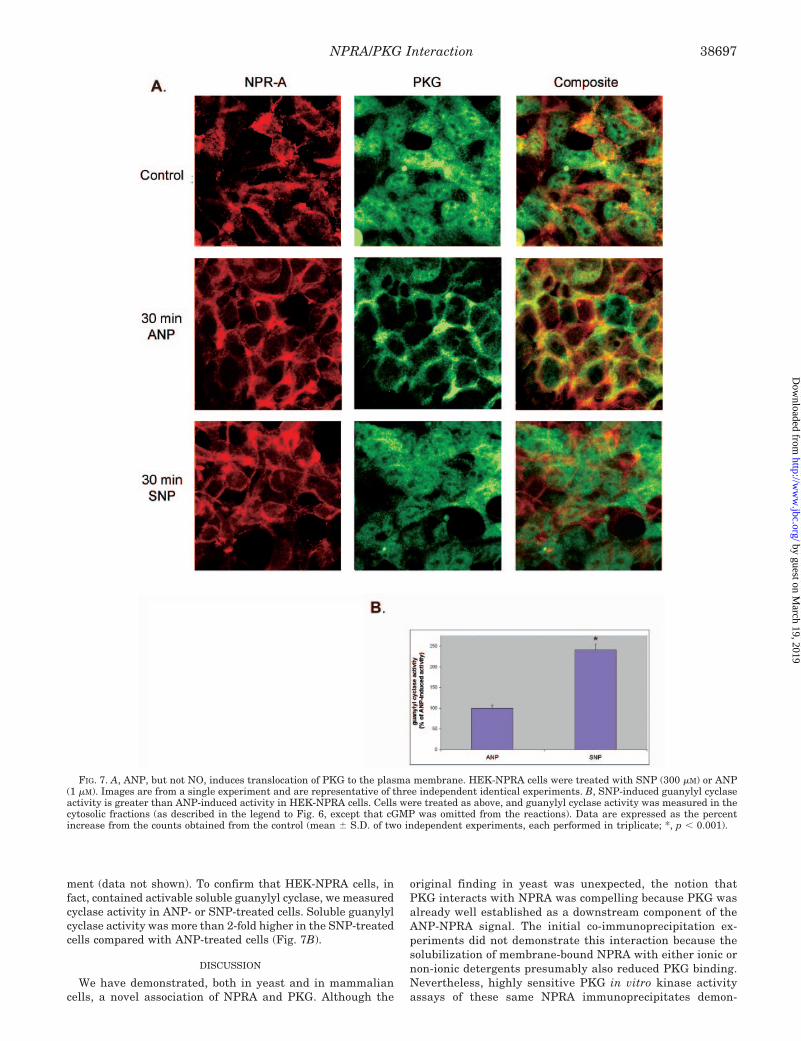
ment (data not shown). To confirm that HEK-NPRA cells, infact, contained activable soluble guanylyl cyclase, we measuredcyclase activity in ANP- or SNP-treated cells. Soluble guanylylcyclase activity was more than 2-fold higher in the SNP-treatedcells compared with ANP-treated cells (Fig. 7B).
DISCUSSION
We have demonstrated, both in yeast and in mammaliancells, a novel association of NPRA and PKG. Although the
original finding in yeast was unexpected, the notion thatPKG interacts with NPRA was compelling because PKG wasalready well established as a downstream component of theANP-NPRA signal. The initial co-immunoprecipitation ex-periments did not demonstrate this interaction because thesolubilization of membrane-bound NPRA with either ionic ornon-ionic detergents presumably also reduced PKG binding.Nevertheless, highly sensitive PKG in vitro kinase activityassays of these same NPRA immunoprecipitates demon-
FIG. 7. A, ANP, but not NO, induces translocation of PKG to the plasma membrane. HEK-NPRA cells were treated with SNP (300 �M) or ANP(1 �M). Images are from a single experiment and are representative of three independent identical experiments. B, SNP-induced guanylyl cyclaseactivity is greater than ANP-induced activity in HEK-NPRA cells. Cells were treated as above, and guanylyl cyclase activity was measured in thecytosolic fractions (as described in the legend to Fig. 6, except that cGMP was omitted from the reactions). Data are expressed as the percentincrease from the counts obtained from the control (mean � S.D. of two independent experiments, each performed in triplicate; *, p � 0.001).
NPRA/PKG Interaction 38697
by guest on March 19, 2019
http://ww
w.jbc.org/
Dow
nloaded from

strated that they contained PKG activity. Additionally, wehave shown that PKG is found in membrane extracts fromcells that had been stably transfected with an NPRA expres-sion vector but not from non-expressing control cells. Finally,ANP-dependent co-localization of NPRA and PKG at theplasma membrane was subsequently corroborated by immu-nofluorescence microscopy.
We have also demonstrated ligand-dependent “feed-forward”regulation of NPRA by PKG. The requirement for an NPRAkinase was first suggested by Potter and Hunter (28), whodemonstrated that phosphorylation of serine/threonine resi-dues in the NPRA kinase-like domain was essential for recep-tor function (18). Our studies suggest that prebound PKG is theNPRA kinase and that ANP binding is necessary and sufficientfor both recruitment to and maintenance of membrane-boundPKG. Our findings may also be of relevance to brain natriureticpeptide because it is nearly identical in structure to ANP, andboth ANP and brain natriuretic peptide preferentially bindwith high affinity to NPRA (36). Finally, this ligand-dependentprocess appears to regulate the intrinsic guanylyl cyclase ac-tivity of NPRA. Thus, NPRA-PKG interaction may play a rolein determining receptor sensitivity.
We have demonstrated that ANP, but not the NO donor SNP,induces PKG translocation. These data suggest that PKGmembrane recruitment is an effect of NPRA activation and nota general consequence of raising cGMP levels. Thus, solubleand particulate guanylyl cyclase appear to be compartmental-ized with respect to PKG migration. How this observationrelates to the regulation of diverse downstream PKG actionswill require additional study. It is well established that bothnitric oxide and NPs signal through cGMP generated by solubleand membrane-bound guanylyl cyclases, respectively. Most ofthe downstream actions of GMP (37) occur through its bindingand subsequent activation of PKG, but cGMP also directlyregulates ion channels (38–41) and phosphodiesterases (42).There is recent evidence that particulate, but not soluble, cy-clases have potent effects on plasma membrane control of cal-cium homeostasis (25). Indeed, whereas it is generally believedthat such spatially and temporally distinct cGMP signals cancoexist within cells, a precise mechanism for differentiatingNP- versus NO-specified cellular events has never been pro-posed. Our data support the speculation that NP binding re-cruits PKG to a membrane pool that may segregate it fromsoluble guanylyl cyclase-generated cGMP and may simulta-neously promote PKG activation via receptor cyclase-generatedcGMP through NP binding. The existence of locally increasedcGMP at the plasma membrane or elsewhere may explain earlyobservations that despite extremely low cGMP concentrationsin whole-cell lysates, cGMP-targeted ligands have potent andimportant biological effects.
Manipulation of the NPRA-PKG interaction may favor en-hanced ANP signaling in disease states. Although ANP is apotent vasodilator and diuretic in normal subjects, its benefi-cial effects are blunted in heart failure (15), despite increasedcirculating ANP levels (43). Therefore, ANP-dependent recep-tor desensitization (16, 17, 44) may limit the therapeutic utilityof natriuretic peptide-based therapies. In fact, homologous de-sensitization of NPRA may be triggered specifically by a reduc-tion in NPRA kinase activity (45). Although our results indi-cate that the recruitment of PKG to the plasma membraneamplifies cyclase activity, further studies will be required todefine the critical sequence or motif of the NPRA receptorresponsible for PKG translocation and to determine whetherexclusion of PKG from its NPRA-binding sites results indesensitization.
Acknowledgments—We thank Michael Chinkers (University ofSouthern Alabama) for the HEK-NPRA cells, Thomas Lincoln (Univer-sity of Alabama) for the PKG I� expression vector, and Philip Stork,Richard Goodman, and Elizabeth Wilson (Oregon Health and ScienceUniversity) for helpful discussions.
REFERENCES
1. Morishita, R., Gibbons, G. H., Pratt, R. E., Tomita, N., Kaneda, Y., Ogihara, T.,and Dzau, V. J. (1994) J. Clin. Investig. 94, 824–829
2. Kishimoto, I., Rossi, K., and Garbers, D. L. (2001) Proc. Natl. Acad. Sci.U. S. A. 98, 2703–2706
3. Kishimoto, I., Dubois, S. K., and Garbers, D. L. (1996) Proc. Natl. Acad. Sci.U. S. A. 93, 6215–6219
4. Currie, M. G., Geller, D. M., Cole, B. R., Boylan, J. G., YuSheng, W., Holmberg,S. W., and Needleman, P. (1983) Science 221, 71–73
5. Appel, R. G. (1992) Am. J. Physiol. 262, F911–F9186. Cao, L., and Gardner, D. G. (1995) Hypertension 25, 227–2347. Knowles, J. W., Esposito, G., Mao, L., Hagaman, J. R., Fox, J. E., Smithies, O.,
Rockman, H. A., and Maeda, N. (2001) J. Clin. Investig. 107, 975–9848. Fiedler, B., Lohmann, S. M., Smolenski, A., Linnemuller, S., Pieske, B.,
Schroder, F., Molkentin, J. D., Drexler, H., and Wollert, K. C. (2002) Proc.Natl. Acad. Sci. U. S. A. 99, 11363–11368
9. Silberbach, M., Gorenc, T., Hershberger, R. E., Stork, P. J., Steyger, P. S., andRoberts, C. T., Jr. (1999) J. Biol. Chem. 274, 24858–24864
10. Schultz, H. D., Gardner, D. G., Deschepper, C. F., Coleridge, H. M., andColeridge, J. C. (1988) Am. J. Physiol. 255, R6–R13
11. Olson, L. J., Lowe, D. G., and Drewett, J. G. (1996) Mol. Pharmacol. 50,430–435
12. Wiedemann, K., Jahn, H., and Kellner, M. (2000) Exp. Clin. Endocrinol.Diabetes 108, 5–13
13. Levin, E. R., Gardner, D. G., and Samson, W. K. (1998) N. Engl. J. Med. 339,321–328
14. Drewett, J. G., and Garbers, D. L. (1994) Endocr. Rev. 15, 135–16215. Cody, R. J., Atlas, S. A., and Laragh, J. H. (1988) Eur. Heart J. 9, 29–3316. Ku, D. D., Guo, L., Dai, J., Acuff, C. G., and Steinhelper, M. E. (1996) Am. J.
Physiol. 271, H2368–H237617. Tsutamoto, T., Kanamori, T., Morigami, N., Sugimoto, Y., Yamaoka, O., and
Kinoshita, M. (1993) Circulation 87, 70–7518. Potter, L. R., and Hunter, T. (1999) Mol. Biol. Cell 10, 1811–182019. Silberbach, M., and Roberts, C. T., Jr. (2001) Cell. Signal. 13, 221–23120. Pfeifer, A., Klatt, P., Massberg, S., Ny, L., Sausbier, M., Hirneiss, C., Wang,
G. X., Korth, M., Aszodi, A., Andersson, K. E., Krombach, F., Mayerhofer,A., Ruth, P., Fassler, R., and Hofmann, F. (1998) EMBO J. 17, 3045–3051
21. Nara, M., Dhulipala, P. D., Ji, G. J., Kamasani, U. R., Wang, Y. X., Matalon,S., and Kotlikoff, M. I. (2000) Am. J. Physiol. 279, C1938–C1945
22. Tohse, N., Nakaya, H., Takeda, Y., and Kanno, M. (1995) Br. J. Pharmacol.114, 1076–1082
23. Yamamoto, S., Yan, F., Zhou, H., and Tai, H. H. (2001) Arch. Biochem.Biophys. 393, 97–105
24. Pedram, A., Razandi, M., Kehrl, J., and Levin, E. R. (2000) J. Biol. Chem. 275,7365–7372
25. Zolle, O., Lawrie, A. M., and Simpson, A. W. M. (2000) J. Biol. Chem. 275,25892–25899
26. Chinkers, M., Garbers, D. L., Chang, M. S., Lowe, D. G., Chin, H. M., Goeddel,D. V., and Schulz, S. (1989) Nature 338, 78–83
27. Chinkers, M., and Wilson, E. M. (1992) J. Biol. Chem. 267, 18589–1859728. Potter, L. R., and Hunter, T. (1998) Mol. Cell. Biol. 18, 2164–217229. Jiang, L. H., Gawler, D. J., Hodson, N., Milligan, C. J., Pearson, H. A., Porter,
V., and Wray, D. (2000) J. Biol. Chem. 275, 6135–614330. Lincoln, T. M., Dey, N., and Sellak, H. (2001) J. Appl. Physiol. 91, 1421–143031. Gudi, T., Lohmann, S. M., and Pilz, R. B. (1997) Mol. Cell. Biol. 17, 5244–525432. Larose, L., Rondeau, J. J., Ong, H., and De Lean, A. (1992) Mol. Cell. Biochem.
115, 203–21133. Simpson, P. (1985) Circ. Res. 56, 884–89434. Patel, A. I., and Diamond, J. (1997) J. Pharmacol. Exp. Ther. 283, 885–89335. Potter, L. R., and Hunter, T. (2001) J. Biol. Chem. 276, 6057–606036. Schulz, S., Singh, S., Bellet, R. A., Singh, G., Tubb, D. J., Chin, H., and
Garbers, D. L. (1989) Cell 58, 1155–116237. Eigenthaler, M., Lohmann, S. M., Walter, U., and Pilz, R. B. (1999) Rev.
Physiol. Biochem. Pharmacol. 135, 173–20938. Mery, P. F., Lohmann, S. M., Walter, U., and Fischmeister, R. (1991) Proc.
Natl. Acad. Sci. U. S. A. 88, 1197–120139. Delay, R. J., Dubin, A. E., and Dionne, V. E. (1997) J. Membr. Biol. 159, 53–6040. Anthony, T. L., Brooks, H. L., Boassa, D., Leonov, S., Yanochko, G. M., Regan,
J. W., and Yool, A. J. (2000) Mol. Pharmacol. 57, 576–58841. Hagen, V., Dzeja, C., Bendig, J., Baeger, I., and Kaupp, U. B. (1998) J.
Photochem. Photobiol. B Biol. 42, 71–7842. Francis, S. H., Colbran, J. L., McAllister-Lucas, L. M., and Corbin, J. D. (1994)
J. Biol. Chem. 269, 22477–2248043. Burnett, J. C., Jr., Kao, P. C., Hu, D. C., Heser, D. W., Heublein, D., Granger,
J. P., Opgenorth, T. J., and Reeder, G. S. (1986) Science 231, 1145–114744. Matsumoto, T., Wada, A., Tsutamoto, T., Omura, T., Yokohama, H., Ohnishi,
M., Nakae, I., Takahashi, M., and Kinoshita, M. (1999) Am. J. Physiol. 276,H1935–H1942
45. Joubert, S., Labrecque, J., and De Lean, A. (2001) Biochemistry 40,11096–11105
46. Spencer, M. L., Shao, H., and Andres, D. A. (2002) J. Biol. Chem. 277,20160–20168
NPRA/PKG Interaction38698
by guest on March 19, 2019
http://ww
w.jbc.org/
Dow
nloaded from

Nathan Airhart, Yong-Feng Yang, Charles T. Roberts Jr. and Michael SilberbachProtein Kinase Interaction
Atrial Natriuretic Peptide Induces Natriuretic Peptide Receptor-cGMP-dependent
doi: 10.1074/jbc.M304098200 originally published online July 10, 20032003, 278:38693-38698.J. Biol. Chem.
10.1074/jbc.M304098200Access the most updated version of this article at doi:
Alerts:
When a correction for this article is posted•
When this article is cited•
to choose from all of JBC's e-mail alertsClick here
http://www.jbc.org/content/278/40/38693.full.html#ref-list-1
This article cites 46 references, 24 of which can be accessed free at
by guest on March 19, 2019
http://ww
w.jbc.org/
Dow
nloaded from





























