Embed Size (px)
Citation preview

August 13 2009MOBIL Summer SchoolLea Thøgersen

Model based on observations and theory. Used to predict and explain new observations
Molecular Modeling Use the computer as a laboratory Do you know any methods? What are they used for?
Today: Molecular Dynamics Experimental observations and simple
physical rules combined to simulate how different atoms move wrt each other.

Topics: Conformational energy, force field and molecular dynamics
Literature: “Part 3” (Chap. 8 Diffraction and Simulation) p.196-200 (first 4 lines), p. 203-207, p. 210-212.
Goal: Obtain basic feeling for the possibilities and limitations of molecular dynamics
Means: active participation from you First session “Conformational Energy and Force Fields”
ends with an exercise Second session “Molecular Dynamics”
includes discussion of a current research study
?


Etot =
Ekin for a molecule e.g. vibration, diffusion coupled with temperature and atom velocities,
but independent of atom positions
Epot for molecule atoms affect each other dependent on atom
type and distance=> Epot coupled with atom positions
conformational energy
Ekin + Epot
{½mv2}
{mgh (gravity) ; ½kx2 (spring)}
?
??
?
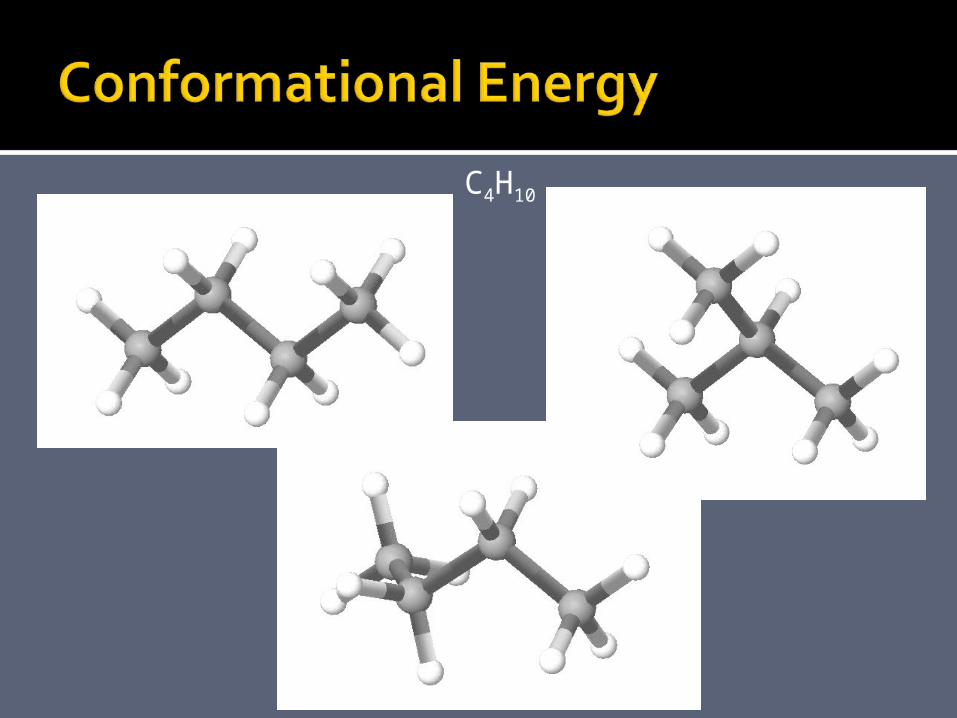
C4H10

Atoms nuclei (protons+neutrons) electrons
Quantum Mechanics: when chemical bonds are formed electrons redistribute on all
atoms in the molecule a carbon (e.g.) would be different from molecule to molecule the distribution of both the electrons and the nuclei in a
molecule determines the conformational energy Experimentally:
atoms of particular type and in particular functional groups behave similar independent of the molecule
IR wave lengths and NMR chemical shifts have characteristic values for certain atom types and groups independent of which molecule they are a part of
Molecular Mechanics: Conformational energy from distribution of only the nuclei Not without problems
??

Energy as function of the relative positions of the atoms => conformational energy
Additive energy contributions Spectroscopy of small molecules suggest that energy
contributions from individual internal coordinates are independent, to a good approximation
Energy function as sum of independent contributions Relative energies instead of absolutes
Easier to define energy penalty than absolute energy Constant contributions can be ignored
Divided in “bonding” and “non-bonding” contributions
1 1
2 2
2 1 2 1
E E S
E E S
E E E E

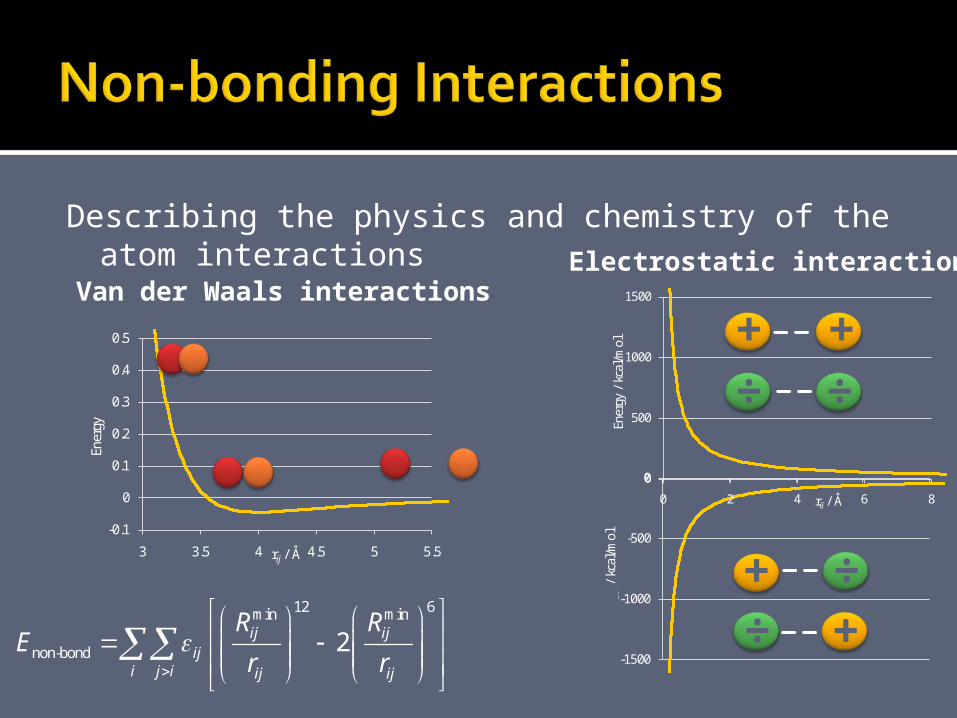
Describing the physics and chemistry of the atom interactions
r or θ
E
2 2bond angle1 1 1bond eq, eq,2 2 2
bonds angles dihedral
1 cos i i i i i i in ii i i n
E k r r k V n
eq
E
φ
bond stretchangle bend
bond rotation=> dihedral
-1500
-1000
-500
0
0 2 4 6 8
Ener
gy /
kca
l/mol
Describing the physics and chemistry of the atom interactions
12 6min min
non-bond0
24
ij ij i jij
i j i i j iij ij ij
R R q qE
r r r
+ +÷ ÷
÷÷+
+-0.1
0
0.1
0.2
0.3
0.4
0.5
3 3.5 4 4.5 5 5.5
Ener
gy
rij / Å
0
500
1000
1500
0 2 4 6 8
Ener
gy /
kca
l/mol
rij / Å
Van der Waals interactionsElectrostatic interactions

Constants in the energy expression should be determinedex.
Based on experimental observations and QM computations.
Hard and tedious work to construct a good and general force field.
? 2bond1bond eq,2
bonds
i i ii
E k r r
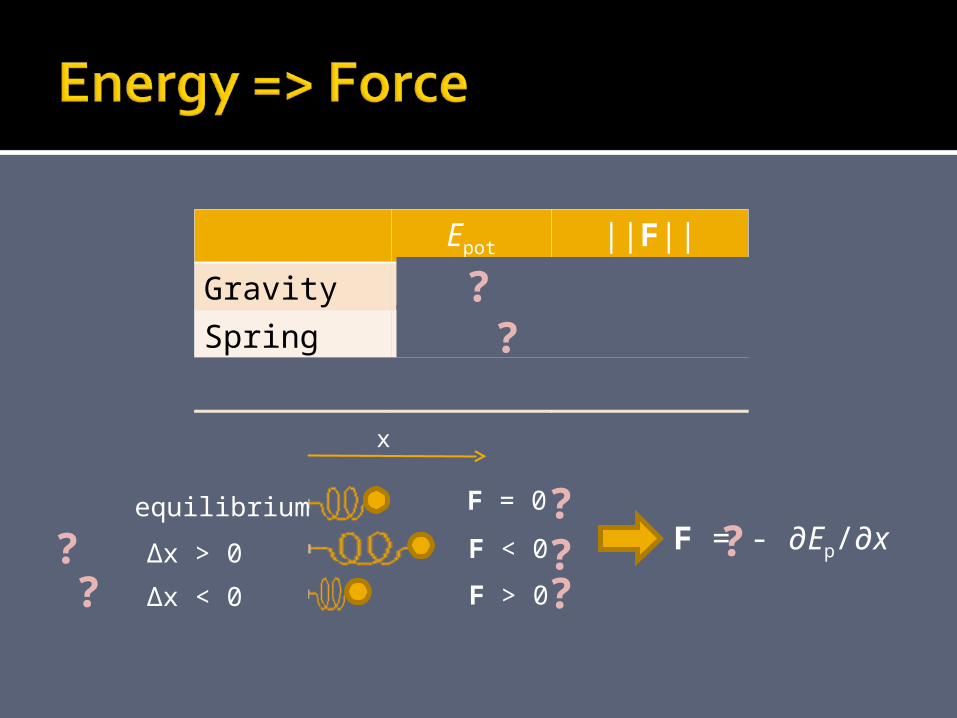
Epot ||F||
Gravity mgh mg
Spring ½k(Δx)2 k Δx
Generally Ep ∂Ep/∂x
x
equilibrium
∆x > 0
∆x < 0
F = 0
F < 0
F > 0
F = - ∂Ep/∂x??
???
?
??
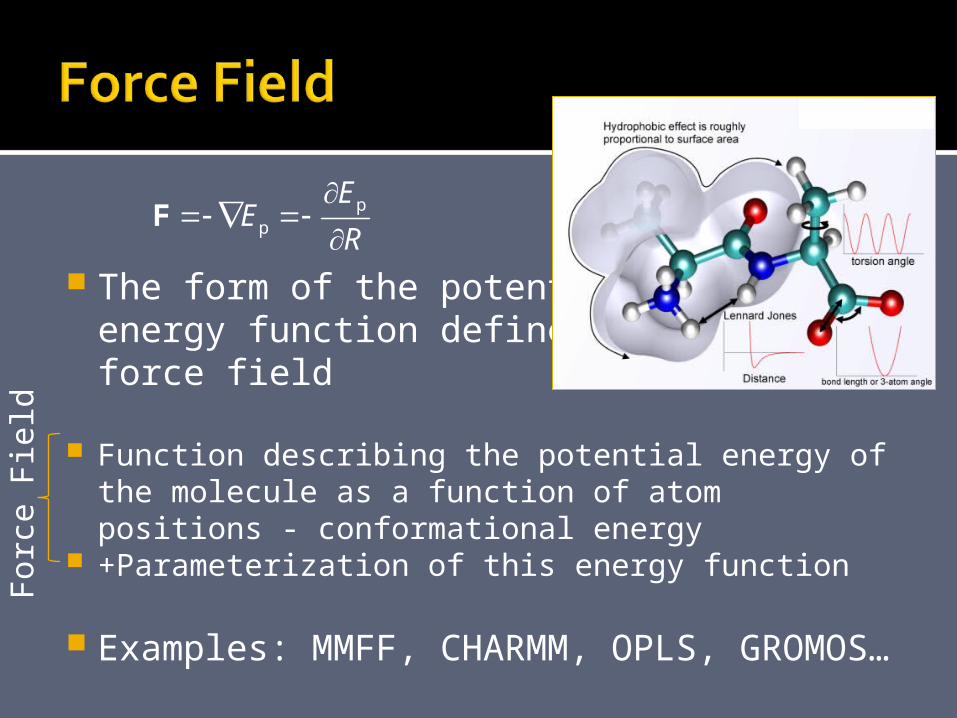
The form of the potentialenergy function defines a force field
Function describing the potential energy of the molecule as a function of atom positions - conformational energy
+Parameterization of this energy function
Examples: MMFF, CHARMM, OPLS, GROMOS…
pp
EE
R
F
Forc
e F
ield
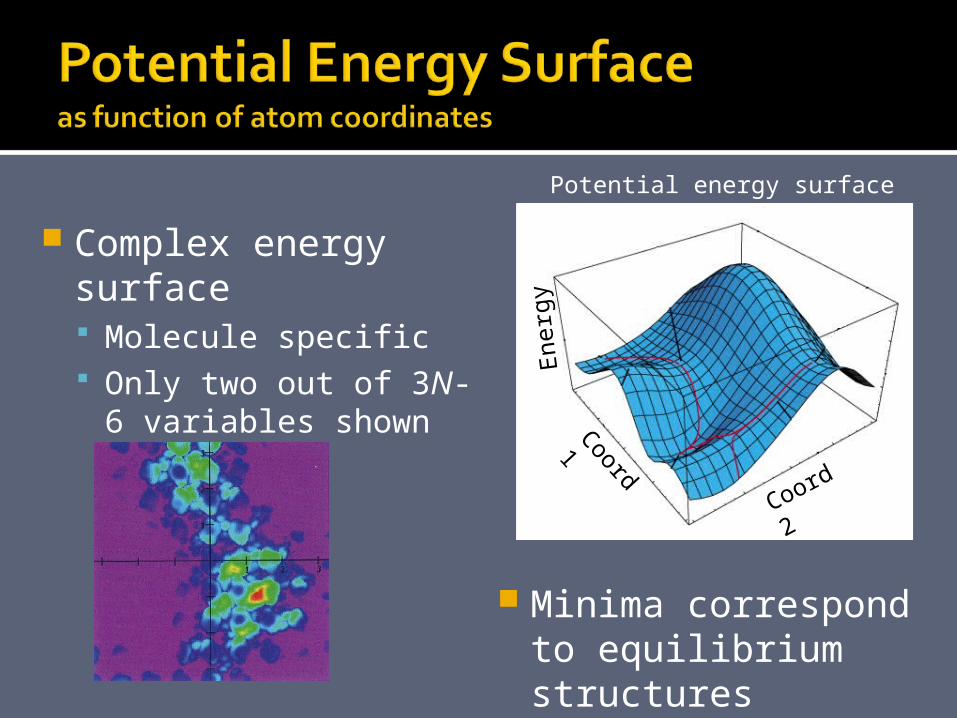
Complex energy surface Molecule specific Only two out of 3N-6
variables shown here.
Potential energy surface
Energ
y
Coord
1
Coord
2
Minima correspond to equilibrium structures

Q1 Bond Stretch: Which of the three lines represent the stretching of the double C=C bond in propene and why?
Q2 Bond Rotation: Which line represents the single bond, which represents the double bond and why?How many interactions contribute in fact to the rotation around the single and the double bonds?
Q3 vdW Interactions: Which line represents the H-H interaction, which represents the C-H interaction and why?
Q4: What constitutes a force field, and why does it make sense to call it a ”force field”?
Number 2. The equilibrium is found for a shorter distance (than for the solid line), and the graph is steeper, meaning the force constant is higher, meaning the bond is stronger.
Number 1 = single bond, number 2 = double bond. Number 1 has three minima (characteristic of an sp3 bond) and a low rotation barrier. Number 2 has two minima (characteristic of an sp2 bond) and a high rotation barrier.The double bond rotation has four contributions (5-1-2-6, 5-1-2-3, 4-1-2-6, 4-1-2-3)The single bond rotation has six contributions (6-2-3-{7,8,9} and 1-2-3-{7,8,9})
Number 1 = H-H interaction, number 2 = C-H interaction. Hydrogen is a smaller atom than carbon, and therefore the minimum vdW distance is smaller for H-H than for H-C.
A force field consists of a potential energy function and the parameters for the function.It is called a force field since the first derivative of the potential energy wrt the position of an atom gives the force acting on this atom from the rest of the atoms in the system.

A Virtual Experiment
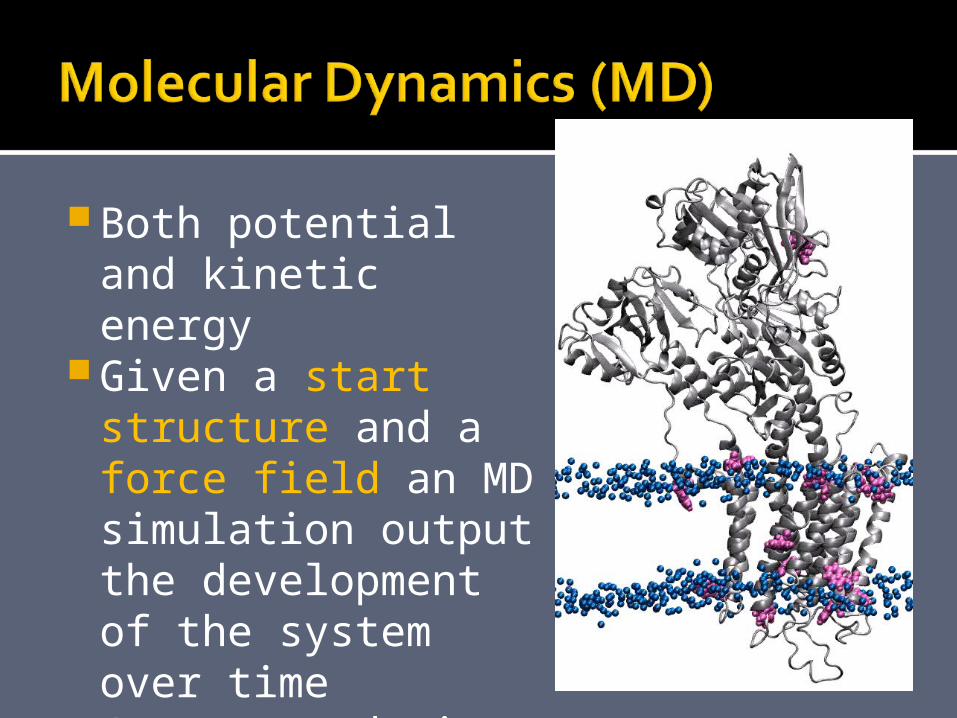
Both potential and kinetic energy
Given a start structure and a force field an MD simulation output the development of the system over time (nanosecond time scale)
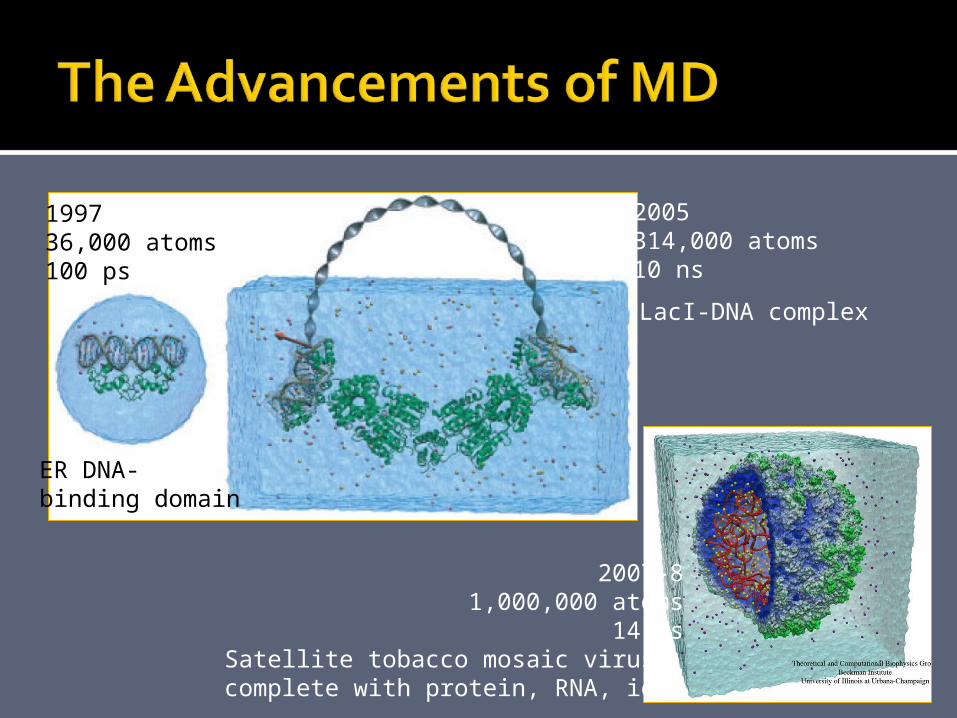
199736,000 atoms100 ps
2005314,000 atoms10 ns
2007-81,000,000 atoms
14 nsSatellite tobacco mosaic virus,
complete with protein, RNA, ions
ER DNA-binding domain
LacI-DNA complex
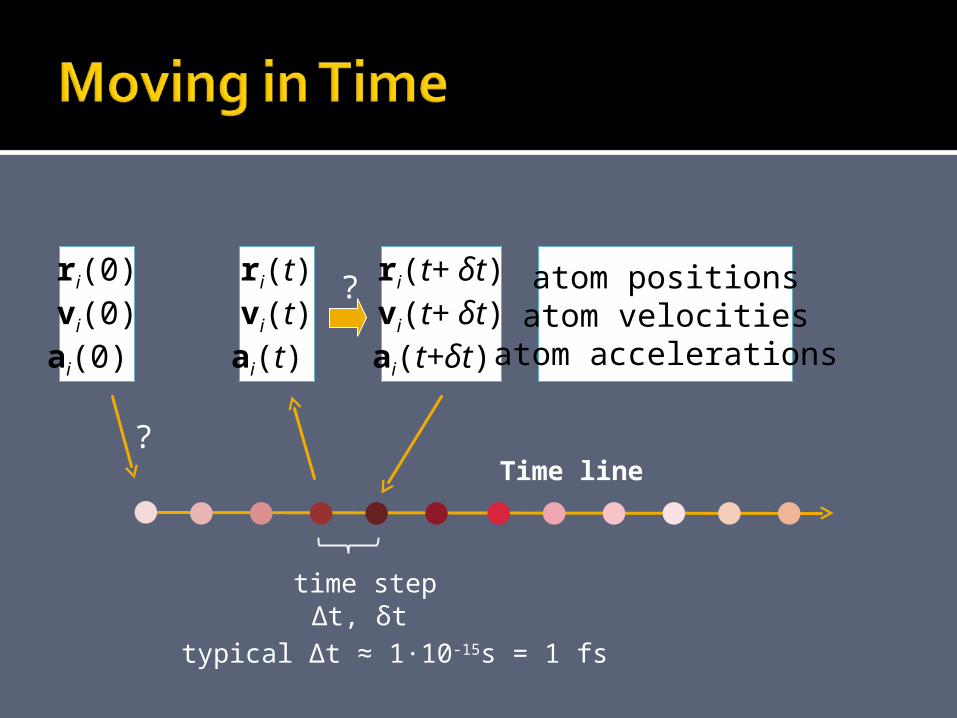
ri(t)vi(t)ai(t)
Time line
time step∆t, δt
typical ∆t ≈ 1·10-15s = 1 fs
ri(t+ δt)vi(t+ δt)ai(t+δt)
atom positionsatom velocities
atom accelerations
ri(0)vi(0)ai(0)
?
?

Find initial coordinates r(t=0) for all atoms in the system For proteins an X-ray or NMR structure is
used or modified Water and lipid can be found pre-
equilibrated from the modeling software or on the web
Smaller molecules can be sketched naively and pre-optimized within the modeling software
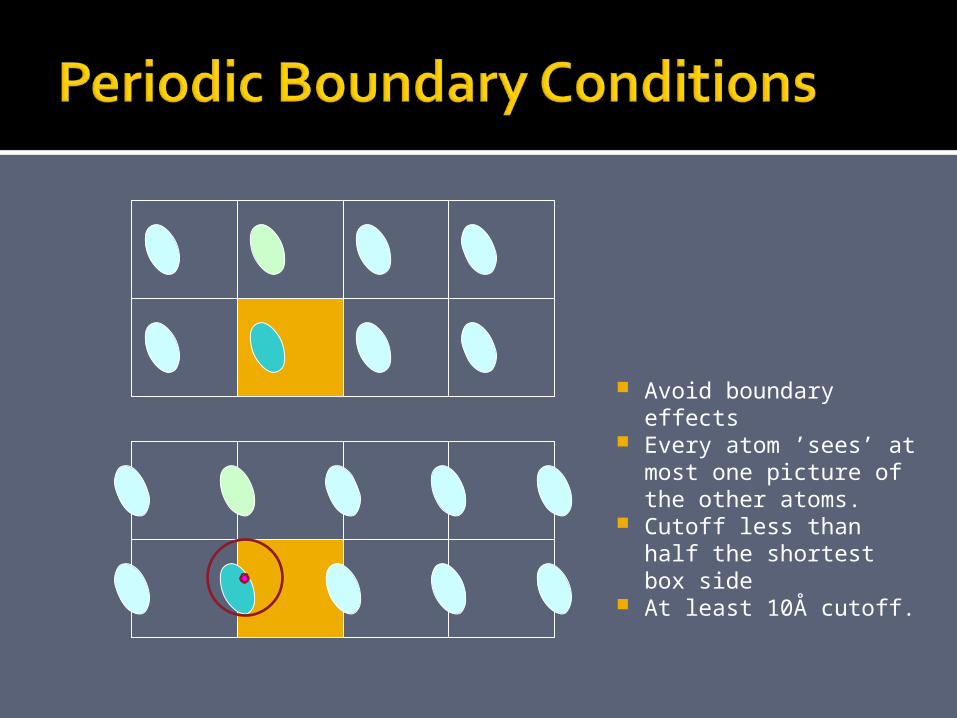
Avoid boundary effects
Every atom ’sees’ at most one picture of the other atoms.
Cutoff less than half the shortest box side
At least 10Å cutoff.

Epot ||F||
Spring ½k(Δx)2 k Δx
Generally Ep ∂Ep/∂xx
equilibrium
∆x > 0
∆x < 0
F = 0
F < 0
F > 0
F = - ∂Ep/∂x = -G
F = m a
r(t=0) => F(r(t=0)) => a(t=0)
?
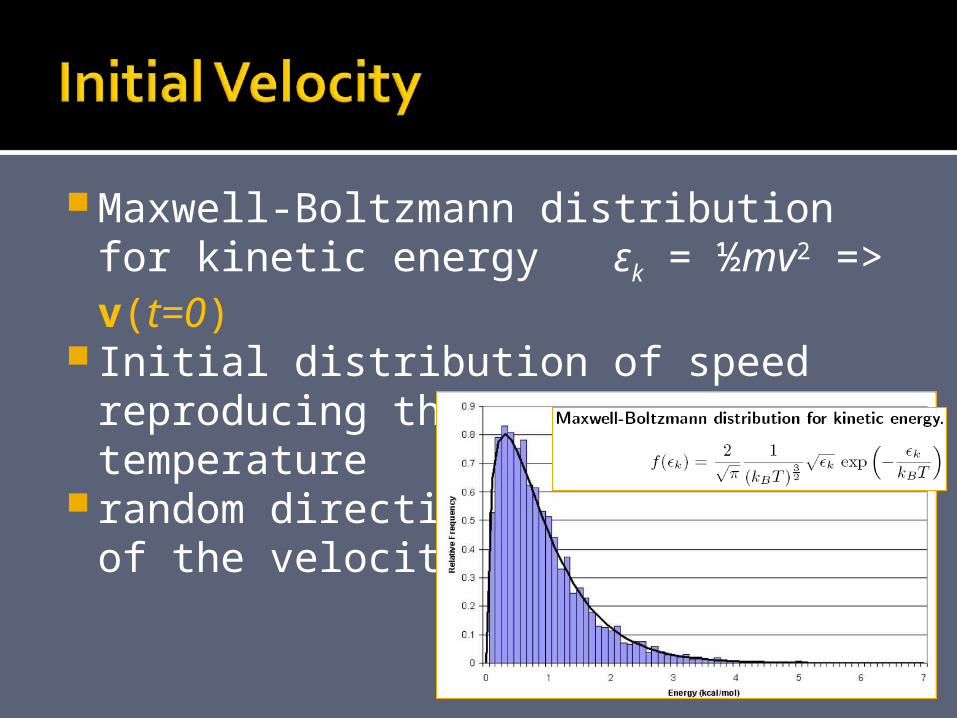
Maxwell-Boltzmann distribution for kinetic energy εk = ½mv2 => v(t=0)
Initial distribution of speed reproducing the requested temperature
random directionsof the velocities

212
1
12
( )
i i
i i i
i i i p im t t
i i i i
t t t t t t t
t t t t m E
t t t t t t t
r
r r v a
a F r r
v v a a
Time line
time step∆t, δt
typical ∆t ≈ 1·10-15s = 1 fs
ri(t)
ai(t)
vi(t)
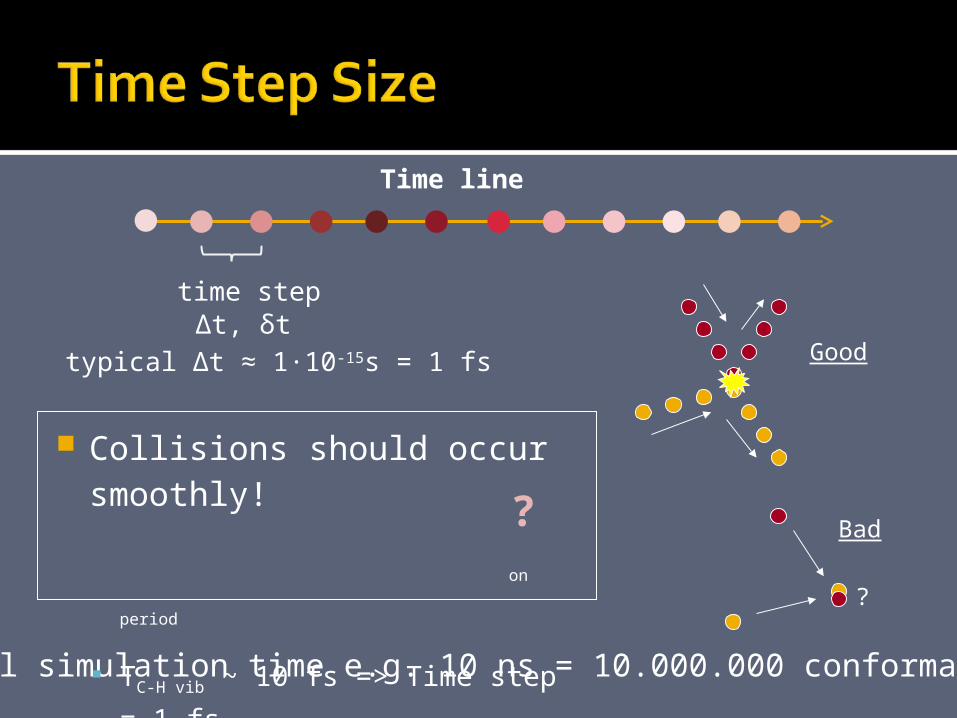
Good
?
Bad
Total simulation time e.g. 10 ns = 10.000.000 conformations
Collisions should occur smoothly!
Time step ~ 1/10 Tfast motion period
TC-H vib ~ 10 fs => Time step = 1
fs
Time line
time step∆t, δt
typical ∆t ≈ 1·10-15s = 1 fs
?

Build the system Clean pdb-structure for unwanted atoms Add missing atoms Add the environment Make a structure file describing connections
Minimization of the system Some 2000 steps, gradient < 5 or so To remove clashes
Equilibration of the system Maybe constraining some atoms to their initial position too keep overall
structure Maybe starting from low temperature, and slowly increasing it to the
wanted Maybe letting the volume adjust properly to the size of the system Energy and RMSD should level out
Production run Constant temp, vol, pressure?

Experimenting with different setups to see what happens – is the system stable? Mutations, temperature, pressure,
environment.... Test out hypotheses based on experiment
Detailed information at the atomic level Free energy differences – site-directed
mutagenesis Other thermodynamics stuff Poke it / steer it

X-ray, NMR and various biophysical studies and mutation studies and more? Model the hypothesis, does the modelled response fit the
experiment? If so, both the experiment and simulation conclusion is strengthen and a higher level of understanding is gained
Shortcomings of MD: Timescale - ns is very short – no conformational changes System size – the dimensions of the model are less than
nm No electrons – polarization cannot be described
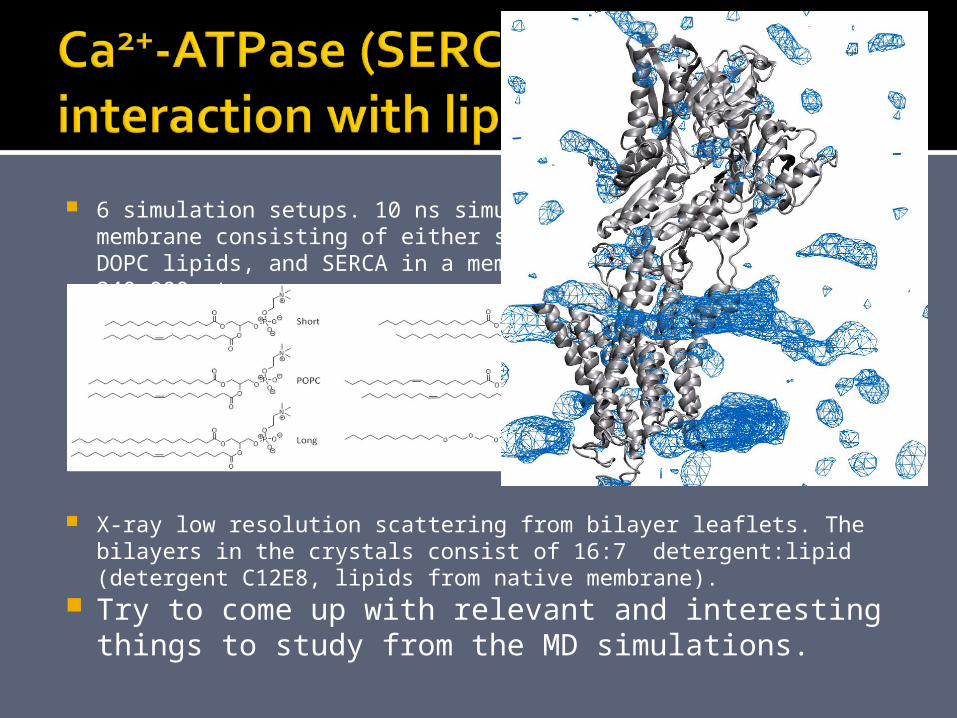
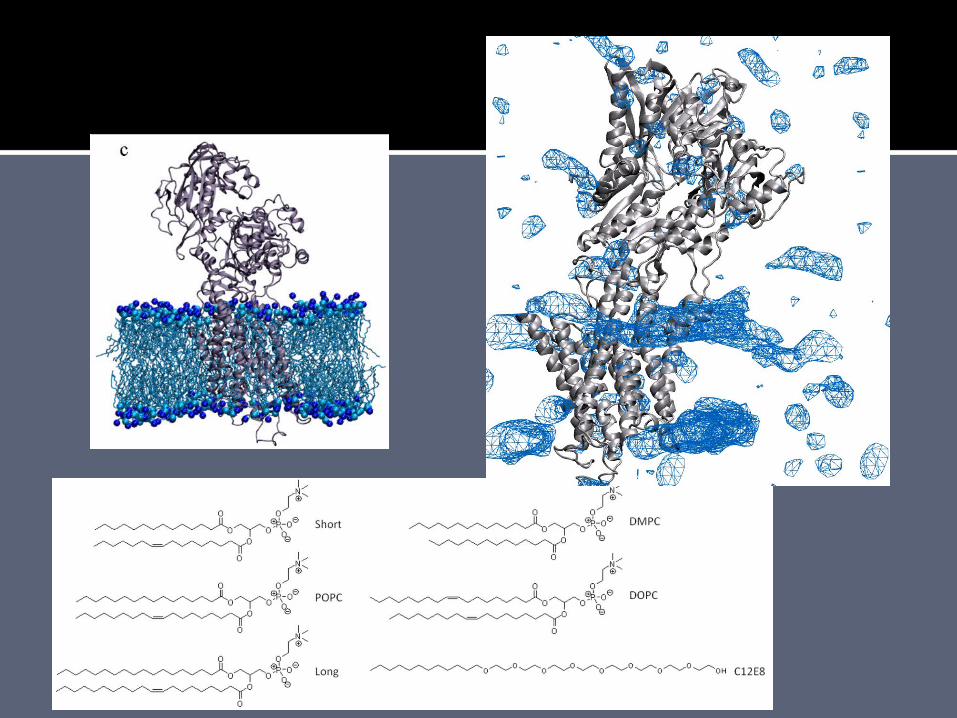
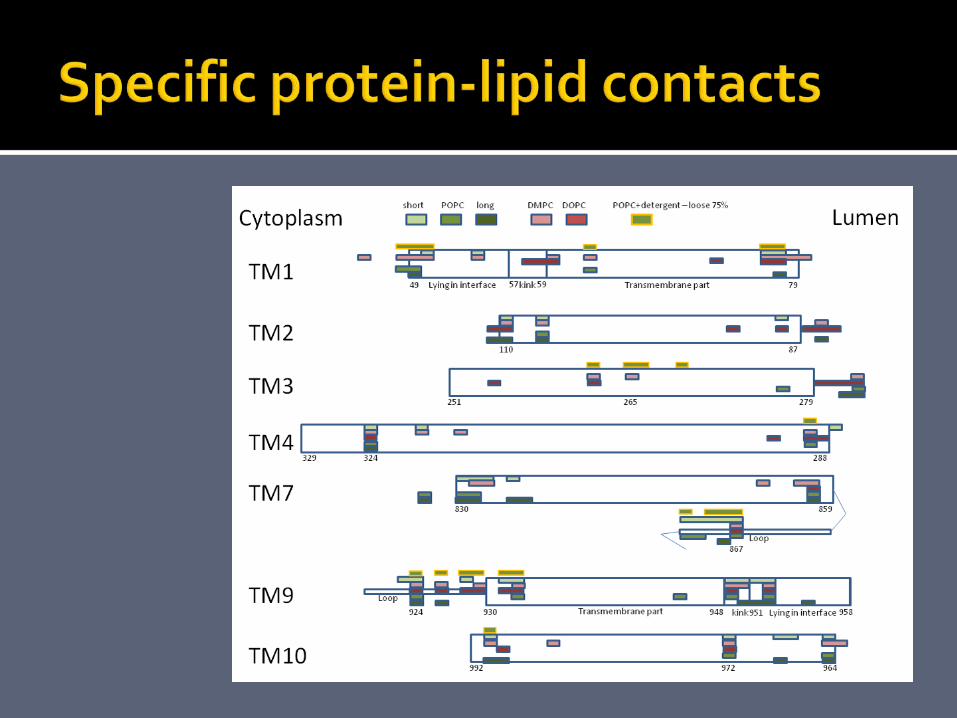
6 simulation setups. 10 ns simulations of SERCA in a membrane consisting of either short, POPC, long, DMPC, or DOPC lipids, and SERCA in a membrane of 2:1 C12E8:POPC. 200-240.000 atoms.
X-ray low resolution scattering from bilayer leaflets. The bilayers in the crystals consist of 16:7 detergent:lipid (detergent C12E8, lipids from native membrane).
Try to come up with relevant and interesting things to study from the MD simulations.
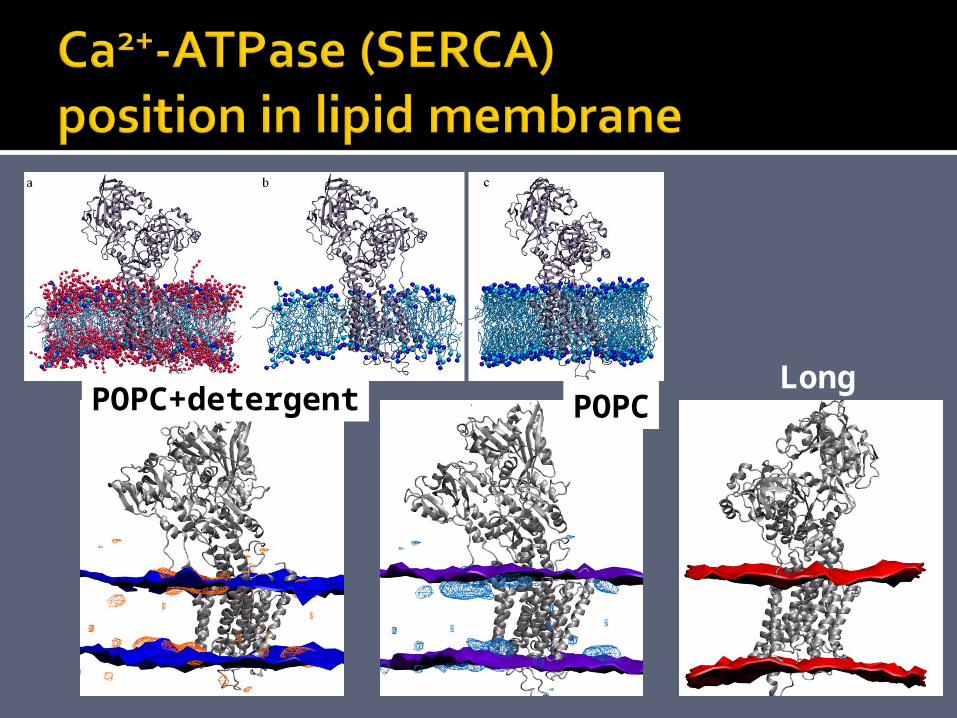
LongPOPCPOPC+detergent
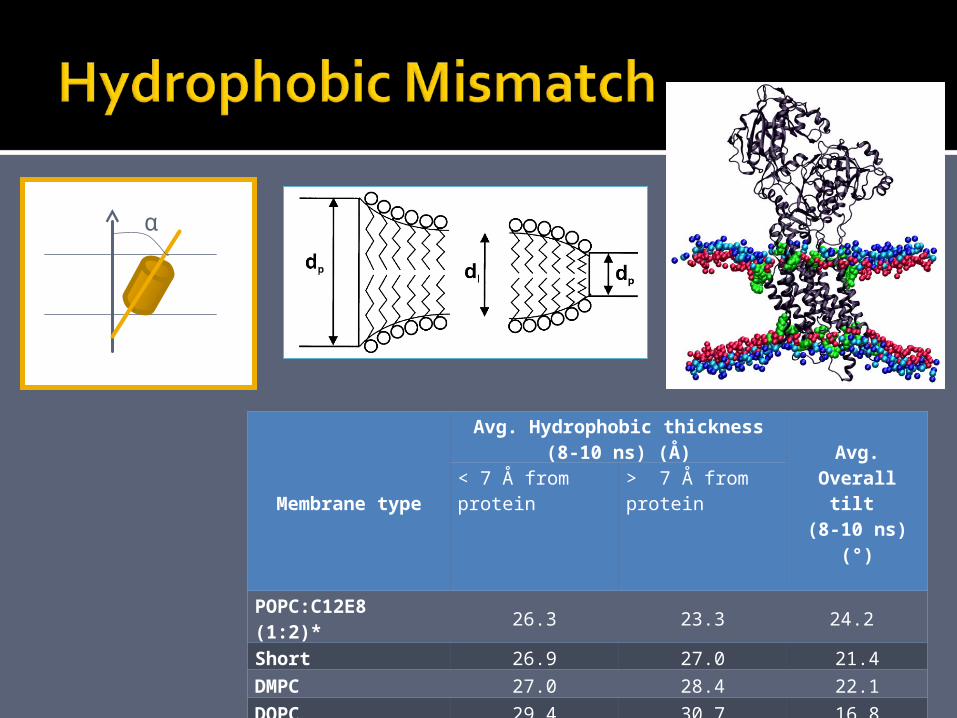
Membrane typeAvg. Hydrophobic thickness (8-10 ns) (Å)
Avg. Overall tilt (8-10 ns) (°)
< 7 Å from protein > 7 Å from protein
POPC:C12E8 (1:2)* 26.3 23.3 24.2 Short 26.9 27.0 21.4DMPC 27.0 28.4 22.1DOPC 29.4 30.7 16.8POPC 29.5 30.7 18.7Long 32.7 34.3 17.5purePOPC 31.3 -purePOPC:C12E8 (1:2)* 24.0 -
α

From Theoretical and Computational Biophysics Group, University of Illinois at Urbana-Champaignhttp://www.ks.uiuc.edu/Gallery/
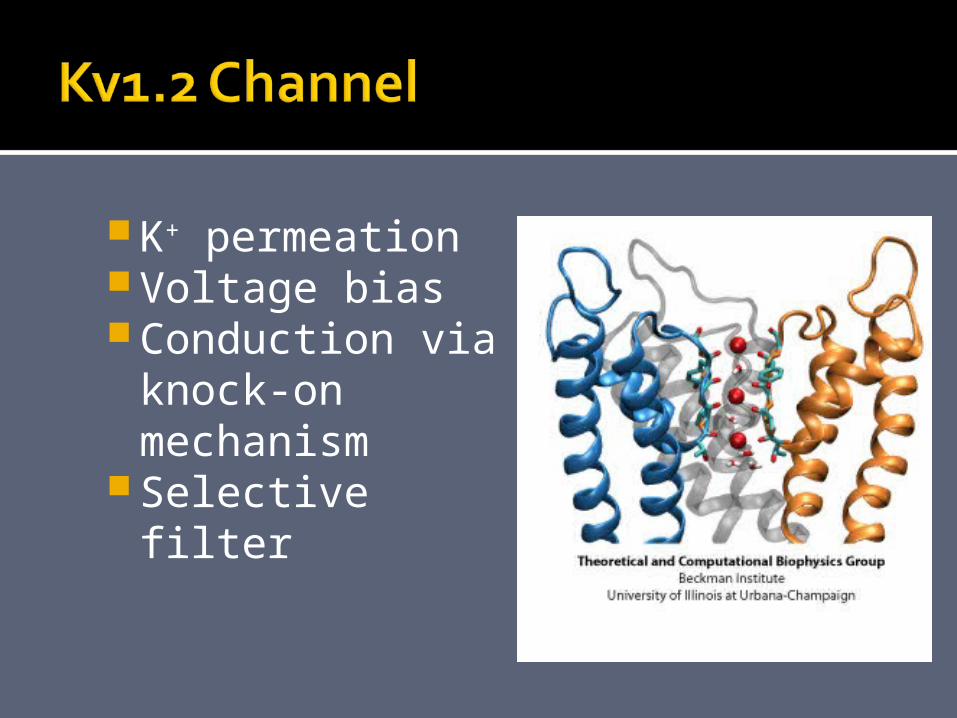
K+ permeationVoltage biasConduction via
knock-on mechanism
Selective filter
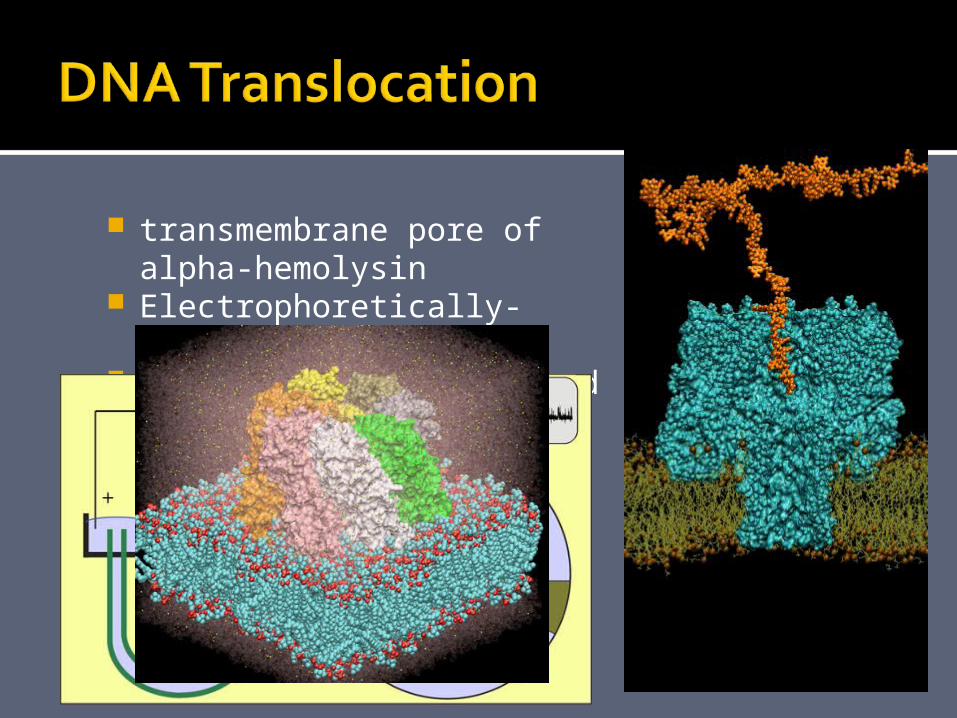
transmembrane pore of alpha-hemolysin
Electrophoretically-driven 58-nucleotide DNA strand
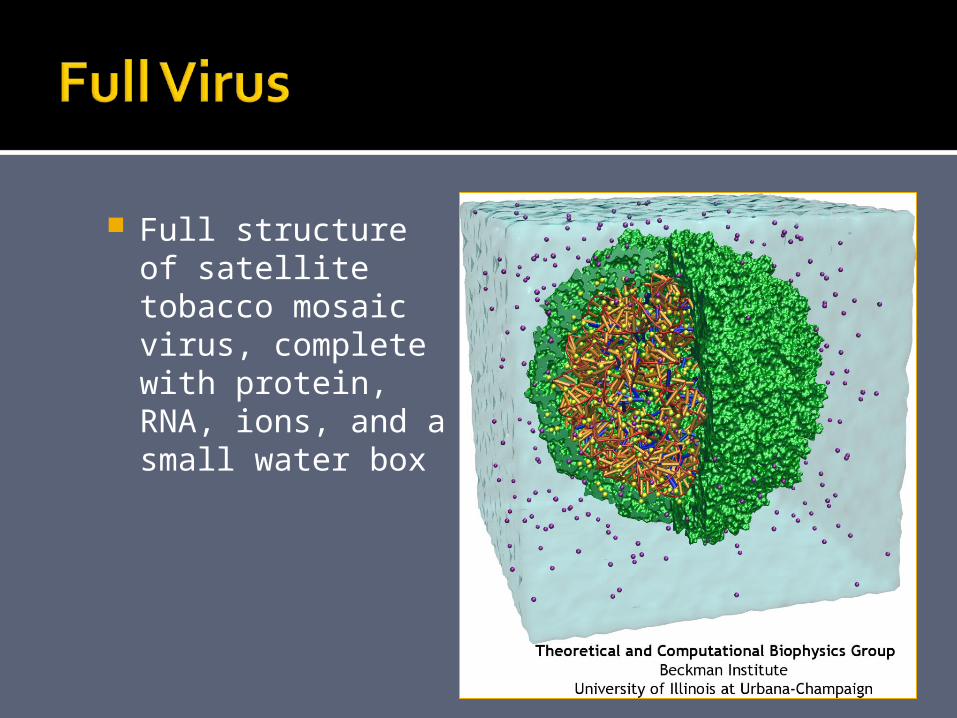
Full structure of satellite tobacco mosaic virus, complete with protein, RNA, ions, and a small water box






















![Mobil İnternet & Mobil Reklam Teknolojileri [Mobil 13]](https://img.pdfslide.net/doc/110x75/548c5a73b47959a8778b4989/mobil-internet-mobil-reklam-teknolojileri-mobil-13.jpg)

![Showrooming: Mağazacılık Öldü Yaşasın Mobil! [Mobil 13]](https://img.pdfslide.net/doc/110x75/5491ec65b479597e6a8b57e5/showrooming-magazacilik-oeldue-yasasin-mobil-mobil-13.jpg)
![Mobil Mecralarda Prime-Time [Mobil 13]](https://img.pdfslide.net/doc/110x75/5491ec16ac79595e288b4664/mobil-mecralarda-prime-time-mobil-13.jpg)


