Embed Size (px)
Citation preview

ence tomography was performed to evaluate the fovealmorphology in vivo after extraocular muscle surgery.
A 10-year-old female with white hair, pinkish irideswith multiple transillumination defects, and ocular nystag-mus presented for vision rehabilitation. Her skin was milkywhite, without any pigmented nevi or evidence of tanning.The diagnosis of oculocutaneous albinism and legal blind-ness was made at 3 years of age. The patient reportedmoderate photophobia and difficulty after prolonged read-ing. On examination, her near vision decreased over timebecause of fatigue exacerbated by a high-frequency, coarsehorizontal nystagmus with moderate amplitude. Colorvisual acuity using Ishihara plates was intact, near visualacuity was 20/50 at 6 inches, and distance visual acuityusing the Lighthouse Distance Visual Acuity Test was20/400 in both eyes. The patient was unable to maintainfixation on a target and exhibited no null-point.
Bilateral recession of the horizontal muscles decreasedthe amplitude of her nystagmus significantly and improvedher near visual acuity to 20/30. Postoperatively, she wasable to find her centralized null-point with a minimal lefthead tilt and was able to fixate on the aiming beam of anoptical coherence tomography machine. Multiple serialoptical coherence tomography scans in a horizontal andvertical fashion were performed to evaluate the contour ofthe foveal retina in both eyes (Figure 1). No fovealindentation was demonstrated on any cross-sectional im-age. A widespread thickening of the retina occurredthroughout the entire fovea with no difference from thesurrounding macula. The inner retina had a highly reflec-tive signal in red-to-orange colors.
Patients with oculocutaneous albinism have decreasedvisual acuity and absence of the foveal pit, possibly causedby changes in the retinal morphology. Although his-topathologic studies of eyes with oculocutaneous albinismare rare, available serial sections in the anatomical loca-tion of the fovea reveal six to eight layers of ganglion cellswith arcades of capillary vessels2 and fail to disclose thephysiologic foveal pit.3 In our case, serial cross-sectionaloptical coherence tomography scans were also unable todetermine the foveal pit in vivo. The foveal thickness onoptical coherence tomography was greater than 300 �m inour patient with oculocutaneous albinism, compared with150 �m in the normal eye.
The foveal indentation was not apparent and was filledwith hyperreflective tissue, possibly corresponding to mul-tiple ganglion cells layers. Patients with oculocutaneousalbinism frequently report photophobia, because the un-pigmented eye reflects more light. This reduced ability toabsorb light also leads to a greater backscattering of laserlight during optical coherence tomography and possiblyabnormally high signals on the false color image. Theseoptical coherence tomography images assessed the anatom-ical morphology of the fovea in vivo and confirmed fovealhypoplasia in a patient with oculocutaneous albinism.
REFERENCES
1. Summers C, King RA. Ophthalmic features of minimalpigment oculocutaneous albinism. Ophthalmology 1994;101:906–914.
2. Naumann GOH, Lerche W, Schroeder W. Foveolar aplasia intyrosinase-positive oculocutaneous albinisim. Graefes ArchKlin Exp Ophthalmol 1976;200:39–50.
3. Mietz H, Green WR, Wolff SM, Abundo GP. Foveal hyp-oplasia in complete oculocutaneous albinism. A histopatho-logic study. Retina 1992;12:254–260.
Autosomal Recessive CerebellarAtaxia With Bull’s-eye MacularDystrophyJohannes R. M. Cruysberg, MD, PhD,Kirsti U. Eerola, MD, Hans R. Vrijland, MD,Albert L. Aandekerk, FOPS,Hubertus P. H. Kremer, MD, PhD, andAugust F. Deutman, MD, PhD
PURPOSE: In 1980, we published in the American Journalof Ophthalmology two siblings with hereditary ataxia andatrophic maculopathy. The report is cited in the litera-ture as autosomal dominant cerebellar ataxia with retinaldegeneration. The purpose of the present study is todocument the progression of the neurodegenerative dis-order and to review the diagnosis.DESIGN: Observational case report.METHODS: Twenty years after the original publication,the 52-year-old male patient had new ocular and neuro-logic examinations, fluorescein angiography, moleculargenetic analysis, and biochemical testing.RESULTS: Fluorescein angiography showed marked pro-gression of the macular dystrophy to a bull’s-eye config-uration. Genetic analysis of the patient did not showCAG trinucleotide repeat expansion in the various spino-cerebellar ataxia genes. This excludes the diagnosis ofautosomal dominant cerebellar ataxia with macular de-generation (ADCA type II) with mutation of the spino-cerebellar ataxia 7 gene. Major causes of autosomalrecessive cerebellar ataxia with retinal degeneration,including Friedreich ataxia and congenital disorders ofglycosylation, were also excluded.CONCLUSION: The two patients, previously published inthe American Journal of Ophthalmology by Eerola andcoworkers, did not suffer from presently recognized
Accepted for publication Oct 8, 2001.From the Institutes of Ophthalmology (J.R.M.C., A.L.A., A.F.D.) and
Neurology (H.P.H.K.), University Medical Centre Nijmegen, Nijmegen,The Netherlands, and the Departments of Ophthalmology, St JansGasthuis, Weert, The Netherlands (K.U.E.), and Ignatius Ziekenhuis,Breda, The Netherlands (H.R.V.).
Inquiries to J. R. M. Cruysberg, MD, PhD, Institute of Ophthalmology,University Medical Centre Nijmegen, PO Box 9101, 6500 HB Nijmegen,The Netherlands; fax: �31-24-3540522; e-mail: [email protected]
AMERICAN JOURNAL OF OPHTHALMOLOGY410 MARCH 2002
disorders with cerebellar ataxia and retinal degeneration.The Eerola syndrome probably represents a separateneurodegenerative entity characterized by autosomal re-cessive cerebellar ataxia and progressive macular dystro-phy with a bull’s-eye pattern. (Am J Ophthalmol2002;133:410–413. © 2002 by Elsevier Science Inc.All rights reserved.)
IN 1980, WE PUBLISHED TWO SIBLINGS WITH A NEURODE-
generative disorder characterized by hereditary ataxiaand atrophic maculopathy with bull’s-eye configuration.1One of the patients (patient 1) had the atrophic stage ofbilateral maculopathy, and the other patient (patient 2)showed the beginning of a bull’s-eye maculopathy. At thattime, the disorder was classified as olivo-ponto-cerebellaratrophy with macular degeneration (OPCA III). Althoughautosomal dominant inheritance, usually established inolivo-ponto-cerebellar atrophy with macular degenerationIII, could not be demonstrated with clinical examinationof this family, the report is nevertheless cited in theliterature as autosomal dominant cerebellar ataxia withretinal degeneration (ADCA type II).2 Because of theprogress in molecular genetic research of cerebellar ataxiawith macular degeneration,3,4 we examined one of thepatients again after 20 years to solve the unansweredquestion with respect to classifying the diagnosis of theneurodegenerative disorder in this family.
Patient 1 died at the age of 59 years. Patient 2, a52-year-old man (the 32-year-old man of the previousstudy1), had new ocular and neurologic examinations,fluorescein angiography of the fundus, molecular genetic
analysis, and biochemical testing. Electroretinography wasrefused.
The history of the 52-year-old man confirmed the onsetof gait impairment, speech difficulties, and tremors of thearms and legs since he was 8 years old. He graduallydeteriorated to the extent of requiring a wheel chair at theage of 30 years. Visual acuity had deteriorated since the ageof 30. He could perform transfers from his chair to bed orto the toilet, while he could walk a few meters with help.Other activities of daily living, however, were unimpaired.The disease was similar to that in one of his sisters (patient1 in the original article1). Neither the parents of thepatient, nor anyone else in the extensive family (threebrothers and seven sisters) had a neurologic disorder.
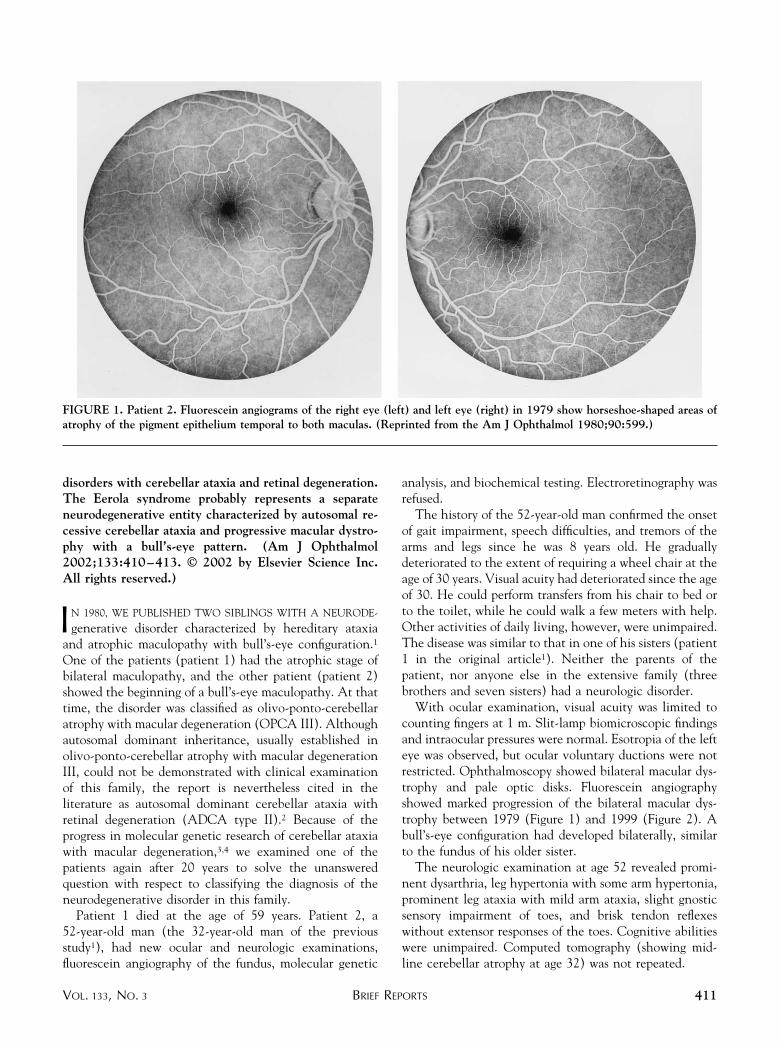
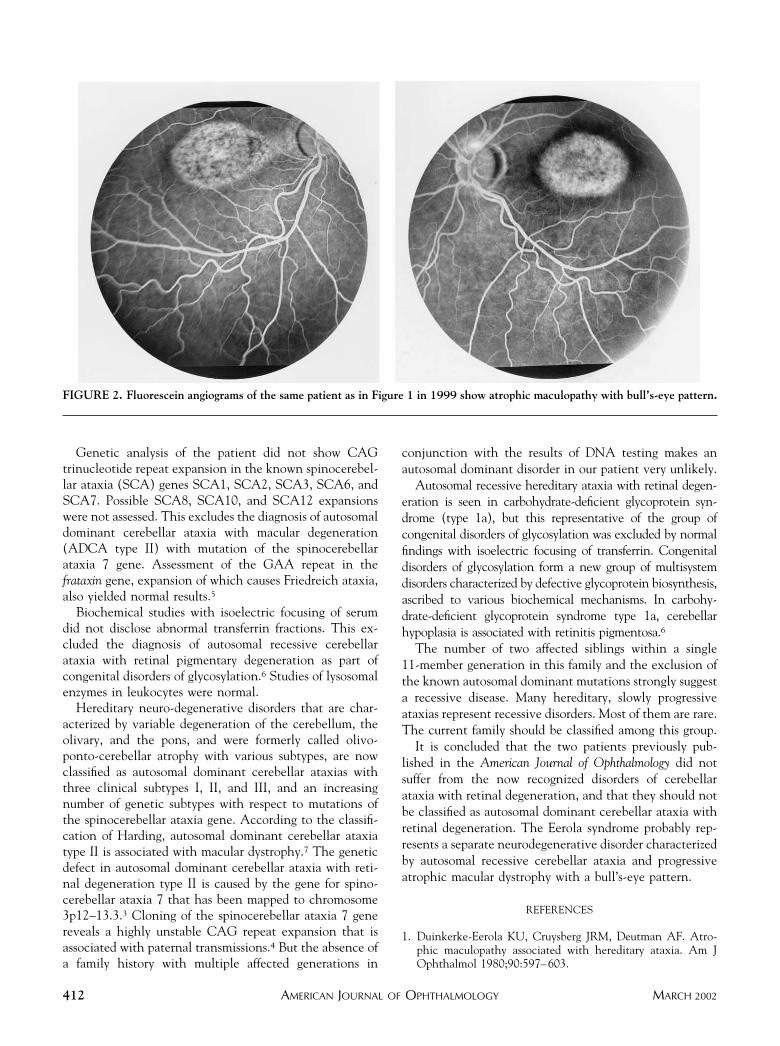
With ocular examination, visual acuity was limited tocounting fingers at 1 m. Slit-lamp biomicroscopic findingsand intraocular pressures were normal. Esotropia of the lefteye was observed, but ocular voluntary ductions were notrestricted. Ophthalmoscopy showed bilateral macular dys-trophy and pale optic disks. Fluorescein angiographyshowed marked progression of the bilateral macular dys-trophy between 1979 (Figure 1) and 1999 (Figure 2). Abull’s-eye configuration had developed bilaterally, similarto the fundus of his older sister.
The neurologic examination at age 52 revealed promi-nent dysarthria, leg hypertonia with some arm hypertonia,prominent leg ataxia with mild arm ataxia, slight gnosticsensory impairment of toes, and brisk tendon reflexeswithout extensor responses of the toes. Cognitive abilitieswere unimpaired. Computed tomography (showing mid-line cerebellar atrophy at age 32) was not repeated.
FIGURE 1. Patient 2. Fluorescein angiograms of the right eye (left) and left eye (right) in 1979 show horseshoe-shaped areas ofatrophy of the pigment epithelium temporal to both maculas. (Reprinted from the Am J Ophthalmol 1980;90:599.)
BRIEF REPORTSVOL. 133, NO. 3 411
Genetic analysis of the patient did not show CAGtrinucleotide repeat expansion in the known spinocerebel-lar ataxia (SCA) genes SCA1, SCA2, SCA3, SCA6, andSCA7. Possible SCA8, SCA10, and SCA12 expansionswere not assessed. This excludes the diagnosis of autosomaldominant cerebellar ataxia with macular degeneration(ADCA type II) with mutation of the spinocerebellarataxia 7 gene. Assessment of the GAA repeat in thefrataxin gene, expansion of which causes Friedreich ataxia,also yielded normal results.5
Biochemical studies with isoelectric focusing of serumdid not disclose abnormal transferrin fractions. This ex-cluded the diagnosis of autosomal recessive cerebellarataxia with retinal pigmentary degeneration as part ofcongenital disorders of glycosylation.6 Studies of lysosomalenzymes in leukocytes were normal.
Hereditary neuro-degenerative disorders that are char-acterized by variable degeneration of the cerebellum, theolivary, and the pons, and were formerly called olivo-ponto-cerebellar atrophy with various subtypes, are nowclassified as autosomal dominant cerebellar ataxias withthree clinical subtypes I, II, and III, and an increasingnumber of genetic subtypes with respect to mutations ofthe spinocerebellar ataxia gene. According to the classifi-cation of Harding, autosomal dominant cerebellar ataxiatype II is associated with macular dystrophy.7 The geneticdefect in autosomal dominant cerebellar ataxia with reti-nal degeneration type II is caused by the gene for spino-cerebellar ataxia 7 that has been mapped to chromosome3p12–13.3.3 Cloning of the spinocerebellar ataxia 7 genereveals a highly unstable CAG repeat expansion that isassociated with paternal transmissions.4 But the absence ofa family history with multiple affected generations in
conjunction with the results of DNA testing makes anautosomal dominant disorder in our patient very unlikely.
Autosomal recessive hereditary ataxia with retinal degen-eration is seen in carbohydrate-deficient glycoprotein syn-drome (type 1a), but this representative of the group ofcongenital disorders of glycosylation was excluded by normalfindings with isoelectric focusing of transferrin. Congenitaldisorders of glycosylation form a new group of multisystemdisorders characterized by defective glycoprotein biosynthesis,ascribed to various biochemical mechanisms. In carbohy-drate-deficient glycoprotein syndrome type 1a, cerebellarhypoplasia is associated with retinitis pigmentosa.6
The number of two affected siblings within a single11-member generation in this family and the exclusion ofthe known autosomal dominant mutations strongly suggesta recessive disease. Many hereditary, slowly progressiveataxias represent recessive disorders. Most of them are rare.The current family should be classified among this group.
It is concluded that the two patients previously pub-lished in the American Journal of Ophthalmology did notsuffer from the now recognized disorders of cerebellarataxia with retinal degeneration, and that they should notbe classified as autosomal dominant cerebellar ataxia withretinal degeneration. The Eerola syndrome probably rep-resents a separate neurodegenerative disorder characterizedby autosomal recessive cerebellar ataxia and progressiveatrophic macular dystrophy with a bull’s-eye pattern.
REFERENCES
1. Duinkerke-Eerola KU, Cruysberg JRM, Deutman AF. Atro-phic maculopathy associated with hereditary ataxia. Am JOphthalmol 1980;90:597–603.
FIGURE 2. Fluorescein angiograms of the same patient as in Figure 1 in 1999 show atrophic maculopathy with bull’s-eye pattern.
AMERICAN JOURNAL OF OPHTHALMOLOGY412 MARCH 2002
2. OMIM Online Mendelian Inheritance in Man. http://www.ncbi.nlm.nih.gov/Omim/. June 2001.
3. David G, Giunti P, Abbas N, et al. The gene for autosomaldominant cerebellar ataxia type II is located in a 5-cM regionin 3p12-p13: genetic and physical mapping of the SCA7 locus.Am J Hum Genet 1996;59:1328–1336.
4. David G, Abbas N, Stevanin G, et al. Cloning of the SCA7gene reveals a highly unstable CAG repeat expansion. NatGenet 1997;17:65–70.
5. Campuzano V, Montermini L, Molto MD, et al. Friedreich’sataxia: autosomal recessive disease caused by an intonic GAAtriplet repeat expansion. Science 1996;271:1423–1427.
6. de Lonlay P, Seta N, Barrot S, et al. A broad spectrum ofclinical presentations in congenital disorders of glycosylationI: a series of 26 cases. J Med Genet 2001;38:14–19.
7. Harding AE. Clinical features and classification of inheritedataxias. Adv Neurol 1993;61:1–14.
Scanning Laser OphthalmoscopeFindings in Acute MacularNeuroretinopathyArnd Gandorfer, MD, and Michael W. Ulbig, MD
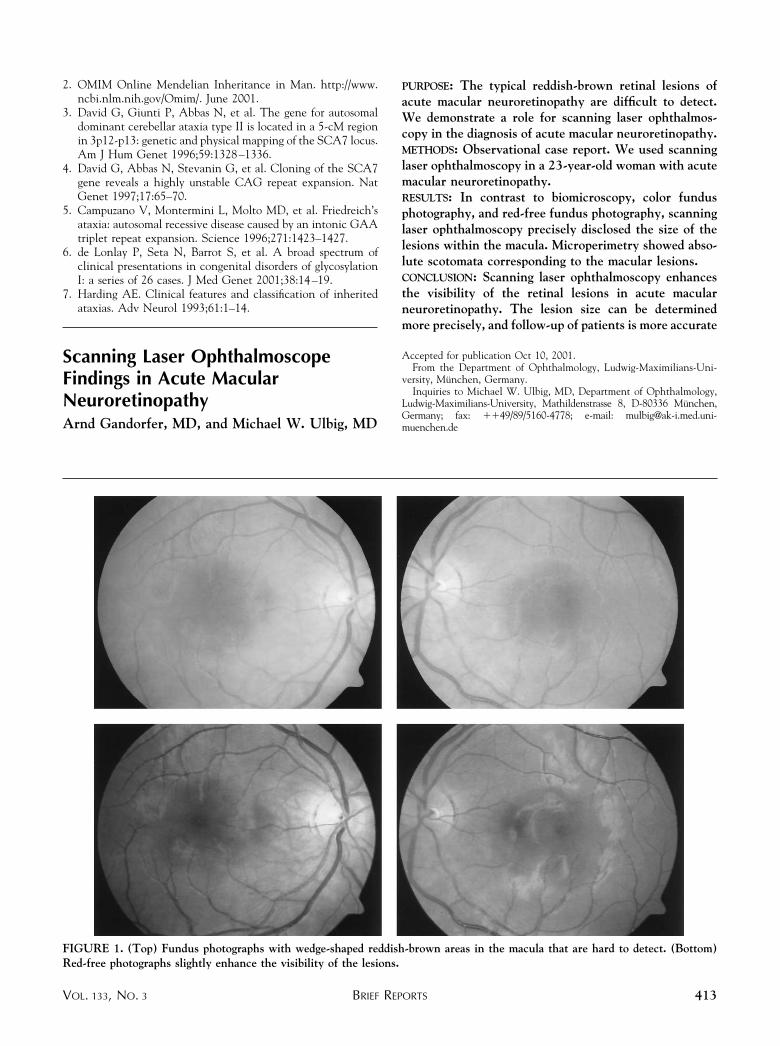
PURPOSE: The typical reddish-brown retinal lesions ofacute macular neuroretinopathy are difficult to detect.We demonstrate a role for scanning laser ophthalmos-copy in the diagnosis of acute macular neuroretinopathy.METHODS: Observational case report. We used scanninglaser ophthalmoscopy in a 23-year-old woman with acutemacular neuroretinopathy.RESULTS: In contrast to biomicroscopy, color fundusphotography, and red-free fundus photography, scanninglaser ophthalmoscopy precisely disclosed the size of thelesions within the macula. Microperimetry showed abso-lute scotomata corresponding to the macular lesions.CONCLUSION: Scanning laser ophthalmoscopy enhancesthe visibility of the retinal lesions in acute macularneuroretinopathy. The lesion size can be determinedmore precisely, and follow-up of patients is more accurate
Accepted for publication Oct 10, 2001.From the Department of Ophthalmology, Ludwig-Maximilians-Uni-
versity, Munchen, Germany.Inquiries to Michael W. Ulbig, MD, Department of Ophthalmology,
Ludwig-Maximilians-University, Mathildenstrasse 8, D-80336 Munchen,Germany; fax: ��49/89/5160-4778; e-mail: [email protected]
FIGURE 1. (Top) Fundus photographs with wedge-shaped reddish-brown areas in the macula that are hard to detect. (Bottom)Red-free photographs slightly enhance the visibility of the lesions.
BRIEF REPORTSVOL. 133, NO. 3 413




![Ataxia telangiectasia: a reviewataxia, oculocutaneous telangiectasia and frequent pul-monary infection [1]. Definition A-T is an autosomal recessive cerebellar ataxia [2]. It has also](https://img.pdfslide.net/doc/110x75/60c0274fdc425b48211dfd10/ataxia-telangiectasia-a-review-ataxia-oculocutaneous-telangiectasia-and-frequent.jpg)
























