Embed Size (px)
Citation preview

American Journal of Medical Genetics 35493-495 (1990)
Letter to the Editor
Autosomal Recessive Form of Mandibular Dysostosis
To the Editor:
In this issue of the American Journal of Medical Ge- netics, Rodriguez and Palacios L19901 report clinical and pathological findings of a new autosomal recessive form of mandibulofacial dysostosis with limb deficiency that is distally postaxial. I recently examined a stillborn female infant, whose pattern of malformation may rep- resent the severe spectrum of the disorder detailed in Rodriguez’ report.
The infant was the product of a 26-week-gestation to a 27-year-old gravida 2 para 1 black woman who denied use of isotretinoin or other vitamin A analogues. The mother developed a urinary tract infection at about the time of conception and was treated with trimethoprim- sulfa. The pregnancy was further complicated by vagi- nal bleeding at 14 weeks of gestation. Subsequent pre- natal ultrasonography showed polyhydramnios, a large cystic hygroma, phocomelia, single kidney, and possible porencephaly or unilateral dilatation of the inferior lat- eral ventricle. There was no family history of consan- guinity. The couple had one normal male child.
The infant was delivered stillborn at 26 weeks gesta- tion. Weight was 480 g, and head circumference was 22 cm (5th centile). Hypertrichosis with a soft cranium, and normal size anterior and posteriar fontanels were noted. Severe mandibulofacial dysostosis included hy- pertelorism, absence of eyelashes, indentation of the lateral inferior border of the left eyelid, malar hypo- plasia, flattened nose with small nostrils, smooth phi- ltrum, microstomia and micrognathia, midline cleft of the soft palate and posterior hard palate, and atresia of both ear canals. Very rudimentary pinnae with central pits had developed inferiorly near the angles of the man- dible. At the normal site of auricle development was a 0.5 cm well-demarcated circular indentation in the skin on the right side and a small circular protuberance of hairless subcutaneous tissue with two pits on the left side. A large cystic hygroma of the neck was present with hypertrichosis extending over the posterior and lateral trunk. Dorsally deviated coccyx and sacrum were widely separated from a patent anus. Severe phocomelia of the upper limbs was accompanied by lower limb defi- ciency with a pterygium extending across the perineum to the knees and arthrogryposis at the hips and knees. Development of fingers and toes was remarkably sym- metric, each consisting of three digits (thumbihallux with two fingers /toes) with well-developed nails. The
Received for publication June 30, 1989.
0 1990 Wiley-Liss, Inc.
right thumb was smaller than the left thumb, and there was mild cutaneous syndactyly of the other two fingers of the left hand and clinodactyly of the corresponding fingers of the right hand. Bilateral single transverse palmar creases were noted. A widened gap between the hallux and second toe was present bilaterally.
Internal examination showed an annular pancreas with obstruction of the distal duodenum and a single horse-shoe kidney with 3 ureters. Externally the heart and great vessels appeared normal. The heart-lung block was lost in transit to a consultant cardiovascular pathology division of another institution. The cerebral hemispheres were symmetrical, but the gyri were poorly formed and the sulci were shallow. Autolysis precluded detailed neuropathologic examination.
Roentgenograms showed severe mandibular hypo- plasia, partial agenesis of the sacrum, and thoracic cage asymmetry (10 ribs on left, 11 ribs on right). Markedly hypoplastic humeri were located subcutaneously on the trunk proximal to the phocomelic upper limbs. There was bilateral aplasia of the radius and ulna. Three metacarpals of the right hand and two metacarpals of the left hand were present with phalanges of the thumb and two fingers on each side.There was marked femoral hypoplasia, to a greater extent on the right, with bowing of the left femur, bilateral tibia1 hypoplasia, and bilat- eral fibular aplasia. Three metatarsals and correspond- ing phalanges of the hallux and 2 toes were present on each foot.
Cytogenetic evaluation of amniocytes and of skin fi- broblasts was normal (46,XX) and did not show prema- ture separation of centromeric heterochromatin.
This stillborn female’s mandibulofacial dysostosis with limb deficiency and internal anomalies is of greater pathologic severity than the pattern of malfor- mations previously described in the postaxial acrofacial dysostosis (Genee-Wiedemann) syndrome [Miller et al., 19791. The presence of relatively well-developed thumbs and halluces render unlikely the diagnosis of a severe form of Nager acrofacial dysostosis [Braga, 1982; Halal et al., 19831.
The normal chromosomes, severity of the mandi- bulofacial dysostosis, hypertrichosis, and digital find- ings are atypical for Roberts SC (pseudothalidomide) syndrome. Roberts syndrome is characterized by preax- ial digital deficiency.
This stillborn fetus did not have the lateral facial clefting characteristic of the syndrome of acrofacial dys- ostosis with severe facial clefting [Kawira et al., 19841. The infant’s postaxial pattern of digital deficiency, the presence of metatarsals, and the specific internal anom- alies are additional differentiating findings.
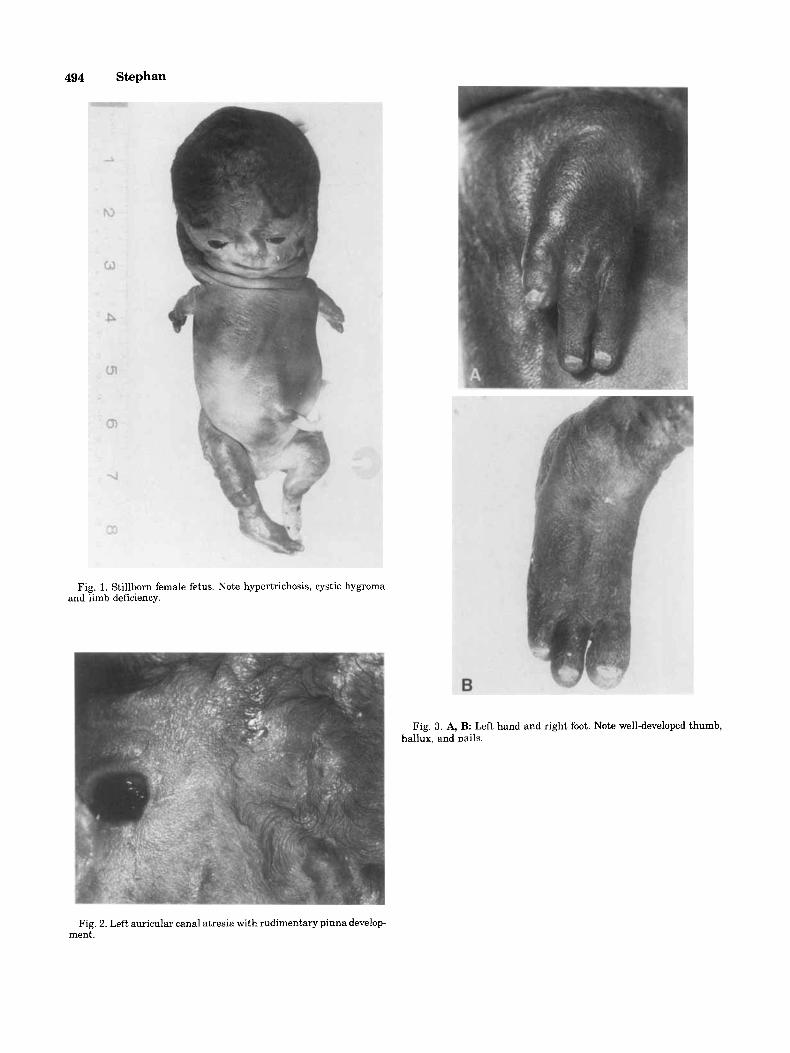
494 Stephan
Fig. 1. Stillborn female fetus. Note hypertrichosis, cystic hygroma and limb deficiency.
Fig. 3. A, B: Left hand and right foot. Note well-developed thumb, hallux. and nails.
Fig. 2. Left auricular canal atresia with rudimentary pinna develop- ment.
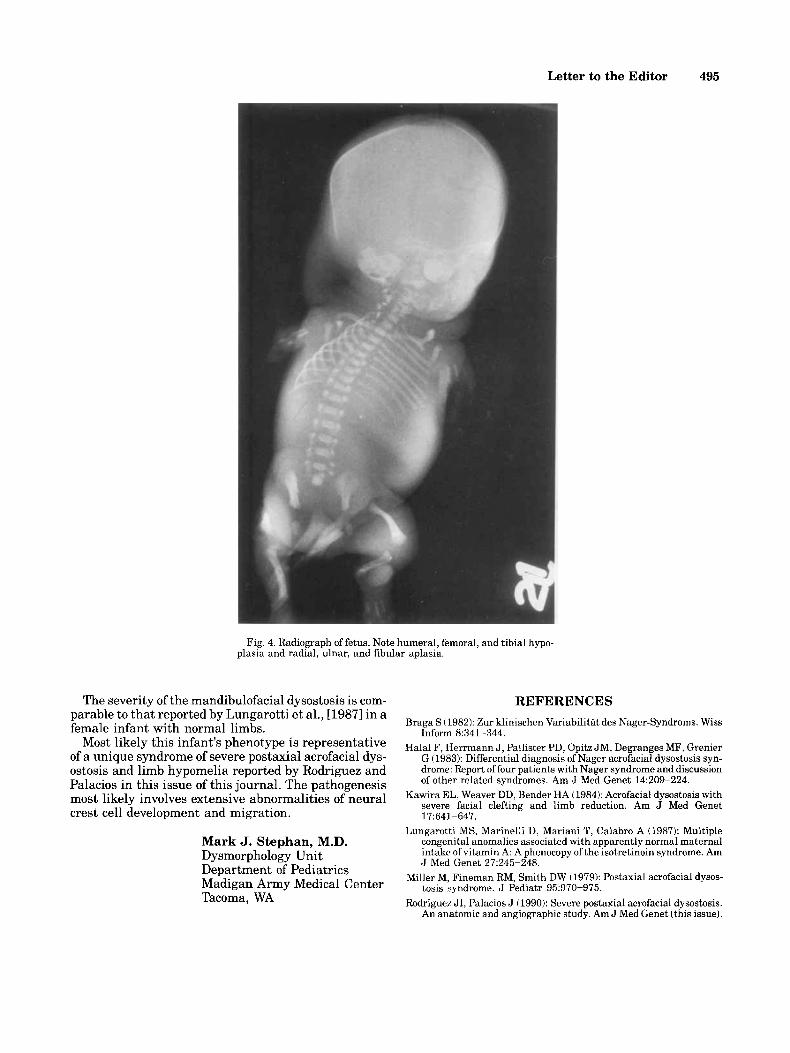
Letter to the Editor 495
Fig. 4. Radiograph of fetus. Note humeral, femoral, and tibia1 hypo- plasia and radial, ulnar, and fibular aplasia.
The severity of the mandibulofacial dysostosis is com- parable to that reported by Lungarotti et al., [19871 in a female infant with normal limbs.
Most likely this infant’s phenotype is representative of a unique syndrome of severe postaxial acrofacial dys- ostosis and limb hypomelia reported by Rodriguez and Palacios in this issue of this journal. The pathogenesis most likely involves extensive abnormalities of neural crest cell development and migration.
Mark J. Stephan, M.D. Dysmorphology Unit Department of Pediatrics Madigan Army Medical Center Tacoma, WA
REFERENCES Braga S (1982): Zur klinischen Variabilitat des Nager-Syndroms. Wiss
Inform 8:341-344. Halal F, Herrmann J , Pallister PD, Opitz JM, Degranges MF, Grenier
G (1983): Differential diagnosis of Nager acrofacial dysostosis syn- drome: Report of four patients with Nager syndrome and discussion of other related syndromes. Am J Med Genet 14:209-224.
Kawira EL, Weaver DD, Bender HA (1984): Acrofacial dysostosis with severe facial clefting and limb reduction. Am J Med Genet 17:641-647.
Lungarotti MS, Marinelli D, Mariani T, Calabro A (1987): Multiple congenital anomalies associated with apparently normal maternal intake of vitamin A: A phenocopy of the isotretinoin syndrome. Am J Med Genet 27945-248.
Miller M, Fineman RM, Smith DW (1979): Postaxial acrofacial dysos- tosis syndrome. J Pediatr 95:970-975.
Rodriguez JI, Palacios J (1990): Severe postaxial acrofacial dysostosis. An anatomic and angiographic study. Am J Med Genet (this issue).



![Autosomal recessive ichthyosis with limb reduction defect ... · including autosomal dominant, autosomal recessive and X-linked inheritance [1,2]. Associated cutaneous and extracutaneous](https://img.pdfslide.net/doc/110x75/5ec8c9b91adfdf12ab3e663c/autosomal-recessive-ichthyosis-with-limb-reduction-defect-including-autosomal.jpg)

























