Embed Size (px)
Citation preview

Clin Investig (1992) 70:794-801
Guest kecture~ "GeseHschaft fur NephrNogie', 23rd Congress Olinical
Investigator
Autosomal recessive polycystic kidney disease K. Zerres Institut ffir Humangenetik der Universit/it Bonn
© Springer-Verlag 1992
Summary. Autosomal recessive polycystic kidney disease is a rare inherited disorder which usually becomes clinically manifest in early childhood, whereas autosomal dominant polycystic kidney disease usually is a disorder of adult onset. With increasing knowledge and improving diagnostic techniques, it becomes evident that the spectrum of both entities is much more variable than gener- ally known. The presentation of autosomal reces- sive polycystic kidney disease at later ages and sur- vival into adul thood have been reported. The diag- nostic criteria, clinical course, genetics and differ- ential diagnosis of autosomal recessive polycystic kidney disease will be presented.
Key words: Polycystic kidney disease - Autosomal recessive - Congenital hepatic fibrosis - Caroli syn- drome
Autosomal recessive polycystic kidney disease (ARPKD) is a rare inherited disorder which usual- ly becomes clinically manifest in early childhood, whereas autosomal dominant polycystic kidney disease (ADPKD) usually is a disorder of adult onset. With increasing knowledge and improving diagnostic techniques, it has become evident that the spectrum of both entities is much more variable than generally thought. The presentation of A R P K D at later ages and survival into adul thood have been reported. A D P K D seems to be as fre- quent as A R P K D in childhood. In a systematic study on the clinical picture and genetics of cystic kidney diseases in children of the "Arbeitsgemein- schaft ffir P/idiatrische Nephrologie", we have col- lected the data of about 96 children with A R P K D and 105 children with ADPKD.
In the following, the main features of A R P K D will be presented with respect to the differential diagnosis of autosomal dominant polycystic kid-
Abbreviations: ADPKD, ARPKD=autosomal dominant/re- cessive polycystic kidney disease; CHF = congenital hepatic fi- brosis
ney disease and cystic dysplasia, which are summa- rized in Table 1. Many important papers about this subject have been published, which - among others - have been the basis for this review: [1, 3, 5, 6, 8, 10-15, 20, 23, 26-28].
The synonyms include infantile polycystic kid- ney disease, sponge kidney, and polycystic kidneys type Potter I, but the genetic term A R P K D is usu- ally used today.
Pathologic anatomy
Renal involvement is invariably bilateral and large- ly symmetric. The kidneys are usually grossly en- larged but retain a reniform configuration (Fig. 1). In cases with typical manifestations, the capsule surface of the kidney is closely studded with opal- escent dots up to 2 mm. The cut surface discloses that these specks represent the cortical extension of fusiform or cylindrical spaces arranged radially throughout the renal parenchyma from medulla to cortex (Fig. 2). the picture of striation on cut section is therefore produced by diverticular, sac- cular or cystic ectasia of the collecting system, par- ticularly next to the capsule. All cysts are lined
Fig. 1. Autosomal recessive polycystic kidney disease. The cut surface reveals enlarged tubules arranged radially through the renal parenchyma from medulla to cortex. (Courtesy Prof. Dr. R. Waldherr, Heidelberg)
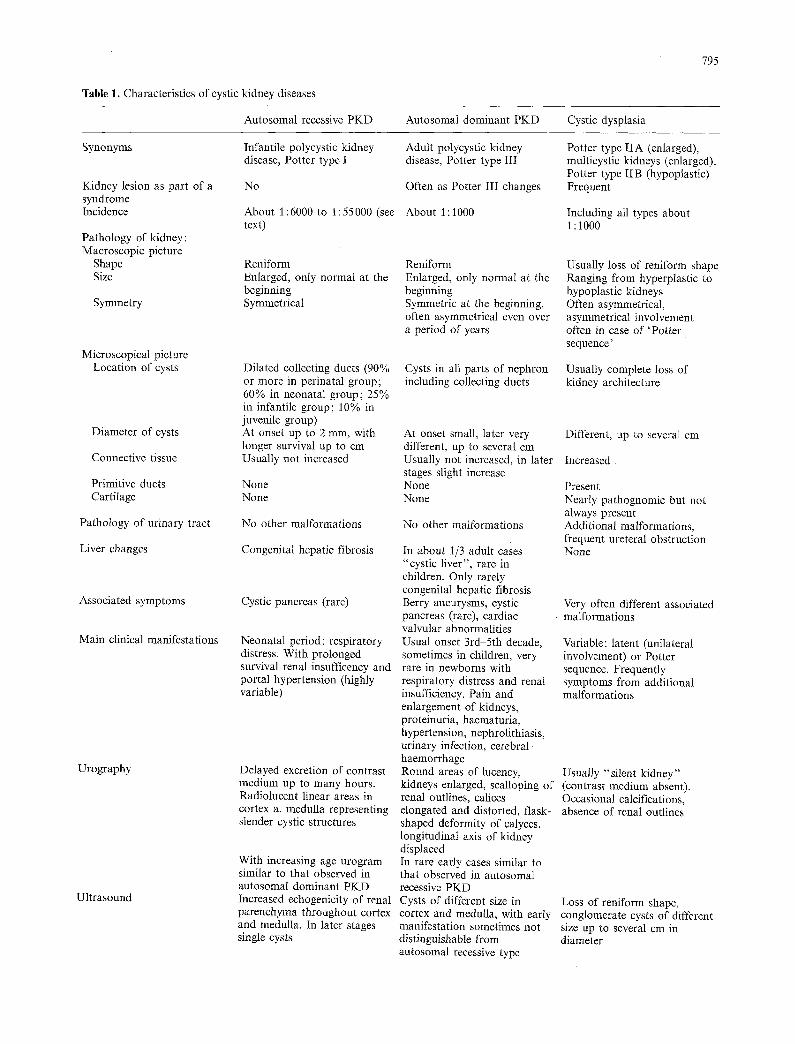
Table 1. Characteristics of cystic kidney diseases
Autosomal recessive PKD Autosomal dominant PKD Cystic dysplasia
795
Synonyms
Kidney lesion as part of a syndrome Incidence
Pathology of kidney: Macroscopic picture
Shape Size
Symmetry
Microscopical picture Location of cysts
Diameter of cysts
Connective tissue
Primitive ducts Cartilage
Pathology of urinary tract
Liver changes
Associated symptoms
Main clinical manifestations
Urography
Ultrasound
Infantile polycystic kidney disease, Potter type I
No
About 1:6000 to 1:55000 (see text)
Reniform Enlarged, only normal at the beginning Symmetrical
Dilated collecting ducts (90% or more in perinatal group; 60% in neonatal group; 25% in infantile group; 10% in juvenile group) At onset up to 2 mm, with longer survival up to cm Usually not increased
None None
No other malformations
Congenital hepatic fibrosis
Cystic pancreas (rare)
Neonatal period: respiratory distress. With prolonged survival renal insufficency and portal hypertension (highly variable)
Delayed excretion of contrast medium up to many hours. Radiolucent linear areas in cortex a. medulla representing slender cystic structures
With increasing age urogram similar to that observed in autosomal dominant PKD Increased echogenicity of renal parenchyma throughout cortex and medulla. In later stages single cysts
Adult polycystic kidney disease, Potter type III
Often as Potter III changes
About 1 : 1000
Reniform Enlarged, only normal at the beginning Symmetric at the beginning, often asymmetrical even over a period of years
Cysts in all parts of nephron including collecting ducts
At onset small, later very different, up to several cm Usually not increased, in later stages slight increase None None
No other malformations
In about 1/3 adult cases "cystic liver", rare in children. Only rarely congenital hepatic fibrosis Berry aneurysms, cystic pancreas (rare), cardiac valvular abnormalities Usual onset 3rd-5th decade, sometimes in children, very rare in newborns with respiratory distress and renal insufficiency. Pain and enlargement of kidneys, proteinuria, haematuria, hypertension, nephrolithiasis, urinary infection, cerebral haemorrhage Round areas of lucency, kidneys enlarged, scalloping of renal outlines, calices elongated and distorted, flask- shaped deformity of calyces, longitudinal axis of kidney displaced In rare early cases similar to that observed in autosomal recessive PKD Cysts of different size in cortex and medulla, with early manifestation sometimes not distinguishable from autosomal recessive type
Potter type IIA (enlarged), multicystic kidneys (enlarged), Potter type IIB (hypoplastic) Frequent
Including all types about i : 1000
Usually loss of reniform shape Ranging from hyperplastic to hypoplastic kidneys Often asymmetrical, asymmetrical involvement often in case of 'Pot ter sequence'
Usually complete loss of kidney architecture
Different, up to several cm
Increased
Present Nearly pathognomic but not always present Additional malformations, frequent ureteral obstruction None
Very often different associated malformations
Variable: latent (unilateral involvement) or Potter sequence. Frequently symptoms from additional malformations
Usually "silent kidney" (contrast medium absent). Occasional calcifications, absence of renal outlines
Loss of reniform shape, conglomerate cysts of different size up to several cm in diameter

796
Table 1 (continued)
Autosomal recessive PKD Autosomal dominant PKD Cystic dysplasia
Risk for siblings 25% 50% (in extremely rare cases of spontaneous mutation no risk)
Risk for children
Manifestation in affected family members
Parental kidneys
Prenatal diagnosis
Potter sequence cases
Below 1% (unless non-affected parent is not related to the affected person, or no case in her/his family) Often similar course in siblings
No alterations
By ultrasound: increased echogenicity, enlargement oligohydromnios. Often visible only in the second half of pregnancy. Biochemical methods not confirmed Rare
50%
Variable, often similar in the same family, Recurrence of early manifestations possible
Demonstration of one affected parent (unless parents are too young do demonstrate cystic changes on ultrasound). Rare cases of spontaneous mutation In extremely rare cases by ultrasound. In informative families by linkage analyses (reliability 95%)
Extremely rare
Unknown, usually below 10% (in rare cases autosomal recessive, autosomal dominant or X-linked inheritance) Usually below /0% (in rare cases of autosomal inheritance up to 50%)
Variable, in same family renal agenesis, dysplasia corticaI cysts and hydronephrosis possible Unilateral agenesis or dysplasia in up to about 10% in one parent
Possible early in pregnancy
Present bilaterally
Fig. 2. Autosomal recessive polycystic kidney disease. Photo- micrograph of kidney in Fig. 1 (x 37.5). (Courtesy Prof. Dr. R. Waldherr, Heidelberg)
Fig. 3. Microscopic appearance of the liver in autosomal reces- sive polycystic kidney disease: "congenital hepatic fibrosis". Increase in number and size of bile ducts and portal fibrosis ( x 56). (Courtesy Prof. Dr. R. Waldherr, Heidelberg)
with a single layer o f cuboidal epithelium, typical for the collection system. The epi thel ium shows no sign o f compress ion . A l though the p r o p o r t i o n o f glomeruli and o ther neph ron segments m a y be slightly reduced, their compos i t ion is largely un- changed. G l o m e r u l a r cysts are not typical for A R P K D . A m a r k e d interstitial pro l i fe ra t ion o f connect ive tissue usually does not occur unless sec-
onda ry s y m p t o m s of an infection exist. The renal pelvis and ureters usual ly a p p e a r normal .
A R P K D is invar iably associated with a gener- alized por ta l and in ter lobular fibrosis o f the liver a ccompan ied by bil iary duct hyperp las ia and small, distal por ta l vein branches (Fig. 3). The in- t rahepat ic por ta l invo lvement results in a presinu- soidal b lock with signs of por ta l hyper tension. AI-
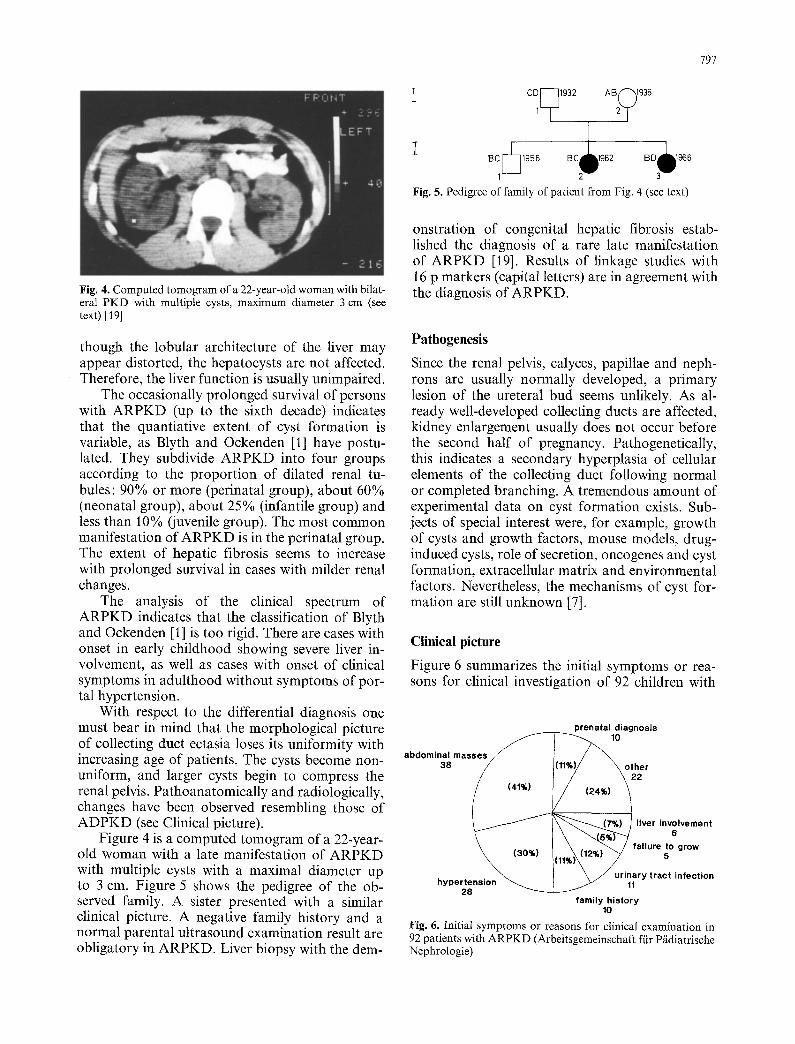
Fig. 4. Computed tomogram of a 22-year-old woman with bilat- eral PKD with multiple cysts, maximum diameter 3 cm (see text) [19]
though the lobular architecture of the liver may appear distorted, the hepatocysts are not affected. Therefore, the liver function is usually unimpaired.
The occasionally prolonged survival of persons with ARPKD (up to the sixth decade) indicates that the quantiative extent of cyst formation is variable, as Blyth and Ockenden [1] have postu- lated. They subdivide ARPKD into four groups according to the proportion of dilated renal tu- bules: 90% or more (perinatal group), about 60% (neonatal group), about 25% (infantile group) and less than 10% (juvenile group). The most common manifestation of ARPKD is in the perinatal group. The extent of hepatic fibrosis seems to increase with prolonged survival in cases with milder renal changes.
The analysis of the clinical spectrum of ARPKD indicates that the classification of Blyth and Ockenden [1] is too rigid. There are cases with onset in early childhood showing severe liver in- volvement, as well as cases with onset of clinical symptoms in adulthood without symptoms of por- tal hypertension.
With respect to the differential diagnosis one must bear in mind that the morphological picture of collecting duct ectasia loses its uniformity with increasing age of patients. The cysts become non- uniform, and larger cysts begin to compress the renal pelvis. Pathoanatomically and radiologically, changes have been observed resembling those of ADPKD (see Clinical picture).
Figure 4 is a computed tomogram of a 22-year- old woman with a late manifestation of ARPKD with multiple cysts with a maximal diameter up to 3 cm. Figure 5 shows the pedigree of the ob- served family. A sister presented with a similar clinical picture. A negative family history and a normal parental ultrasound examination result are obligatory in ARPKD. Liver biopsy with the dem-
797
°o,++ 1 - - 2 + 3-
Fig. 5. Pedigree of family of patient from Fig. 4 (see text)
onstration of congenital hepatic fibrosis estab- lished the diagnosis of a rare late manifestation of ARPKD [19]. Results of linkage studies with 16 p markers (capital letters) are in agreement with the diagnosis of ARPKD.
Pathogenesis
Since the renal pelvis, calyces, papillae and neph- rons are usually normally developed, a primary lesion of the ureteral bud seems unlikely. As al- ready well-developed collecting ducts are affected, kidney enlargement usually does not occur before the second half of pregnancy. Pathogenetically, this indicates a secondary hyperplasia of cellular elements of the collecting duct following normal or completed branching. A tremendous amount of experimental data on cyst formation exists. Sub- jects of special interest were, for example, growth of cysts and growth factors, mouse models, drug- induced cysts, role of secretion, oncogenes and cyst formation, extracellular matrix and environmental factors. Nevertheless, the mechanisms of cyst for- mation are still unknown [7].
Clinical picture
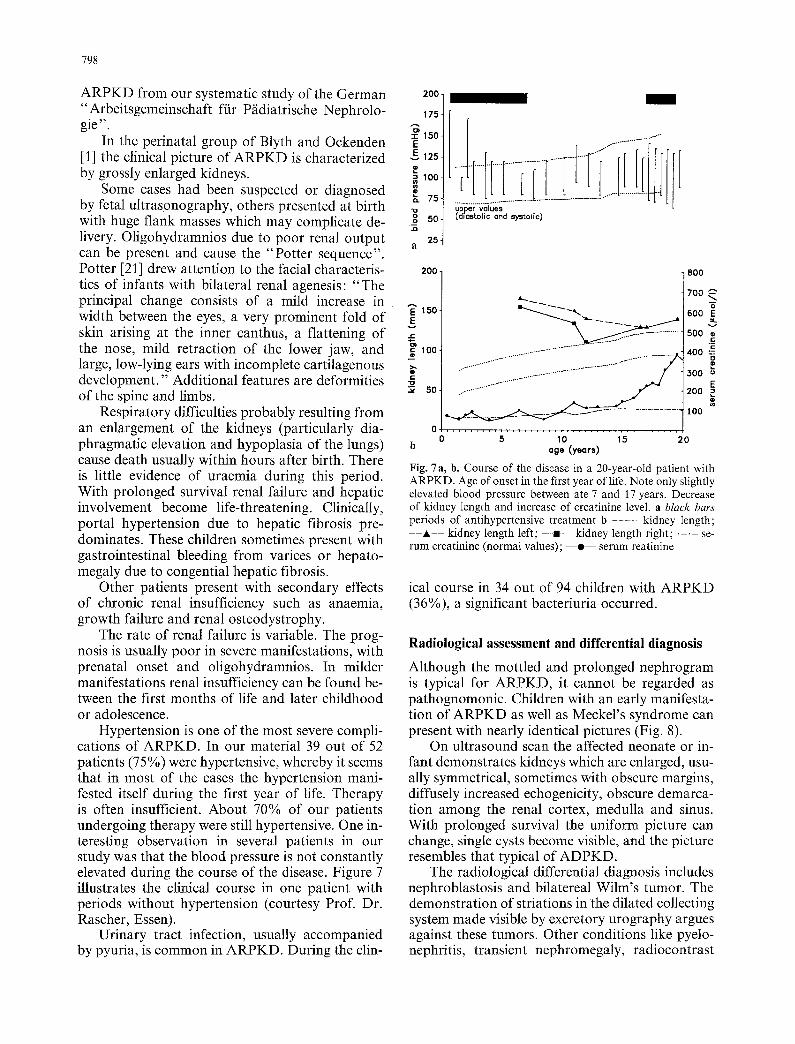
Figure 6 summarizes the initial symptoms or rea- sons for clinical investigation of 92 children with
prenatal diagnosis
abdominal masses 38 other
liver involvement 6
failure to grow 5
urinary tract infection hypertension 11
28 family history
10
Fig. 6. Initial symptoms ol- reasons for clinical examination in 92 patients with ARPKD (Arbeitsgemeinschaft f/Jr P/idiatrische Nephrologie)
798
ARPKD from our systematic study of the German "Arbeitsgemeinschaft ffir P/idiatrische Nephrolo- gie"
In the perinatal group of Blyth and Ockenden [1] the clinical picture of ARPKD is characterized by grossly enlarged kidneys.
Some cases had been suspected or diagnosed by fetal ultrasonography, others presented at birth with huge flank masses which may complicate de- livery. Oligohydramnios due to poor renal output can be present and cause the "Potter sequence". Potter [21] drew attention to the facial characteris- tics of infants with bilateral renal agenesis: "The principal change consists of a mild increase in width between the eyes, a very prominent fold of skin arising at the inner canthus, a flattening of the nose, mild retraction of the lower jaw, and large, low-lying ears with incomplete cartilagenous development." Additional features are deformities of the spine and limbs.
Respiratory difficulties probably resulting from an enlargement of the kidneys (particularly dia- phragmatic elevation and hypoplasia of the lungs) cause death usually within hours after birth. There is little evidence of uraemia during this period. With prolonged survival renal failure and hepatic involvement become life-threatening. Clinically, portal hypertension due to hepatic fibrosis pre- dominates. These children sometimes present with gastrointestinal bleeding from varices or hepato- megaly due to congential hepatic fibrosis.
Other patients present with secondary effects of chronic renal insufficiency such as anaemia, growth failure and renal osteodystrophy.
The rate of renal failure is variable. The prog- nosis is usually poor in severe manifestations, with prenatal onset and oligohydramnios. In milder manifestations renal insufficiency can be found be- tween the first months of life and later childhood or adolescence.
Hypertension is one of the most severe compli- cations of ARPKD. In our material 39 out of 52 patients (75%) were hypertensive, whereby it seems that in most of the cases the hypertension mani- fested itself during the first year of life. Therapy is often insufficient. About 70% of our patients undergoing therapy were still hypertensive. One in- teresting observation in several patients in our study was that the blood pressure is not constantly elevated during the course of the disease. Figure 7 illustrates the clinical course in one patient with periods without hypertension (courtesy Prof. Dr. Rascher, Essen).
Urinary tract infection, usually accompanied by pyuria, is common in ARPKD. During the clin-
200
175.
150. E
,~fi 125-
~100 .
75.
50- x I
25- a
/
................................ iiii!iiii!ii i! ilil iiiiilil iilili ilill upper values [d;astolic and systogc)
200
A E 150 E
~ 100
c " o "~ 5o
0
800
700 -~ o
600 E
5oo
400 :~ P
300 E
200 p
I00
5 10 15 20 b age (years)
Fig. 7 a, b. Course of the disease in a 20-year-old patient with ARPKD. Age of onset in the first year of life. Note only slightly elevated blood pressure between ate 7 and 17 years. Decrease of kidney length and increase of creatinine level, a black bars periods of antihypertensive treatment b . . . . kidney length;
- A - - kidney length left; - - a - - kidney length right; . . . . se- rum creatinine (normal values); - - e - - serum reatinine
ical course in 34 out of 94 children with ARPKD (36%), a significant bacteriuria occurred.
Radiological assessment and differential diagnosis
Although the mottled and prolonged nephrogram is typical for ARPKD, it cannot be regarded as pathognomonic. Children with an early manifesta- tion of ARPKD as well as Meckel's syndrome can present with nearly identical pictures (Fig. 8).
On ultrasound scan the affected neonate or in- fant demonstrates kidneys which are enlarged, usu- ally symmetrical, sometimes with obscure margins, diffusely increased echogenicity, obscure demarca- tion among the renal cortex, medulla and sinus. With prolonged survival the uniform picture can change, single cysts become visible, and the picture resembles that typical of ADPKD.
The radiological differential diagnosis includes nephroblastosis and bilatereal Wilm's tumor. The demonstration of striations in the dilated collecting system made visible by excretory urography argues against these tumors. Other conditions like pyelo- nephritis, transient nephromegaly, radiocontrast
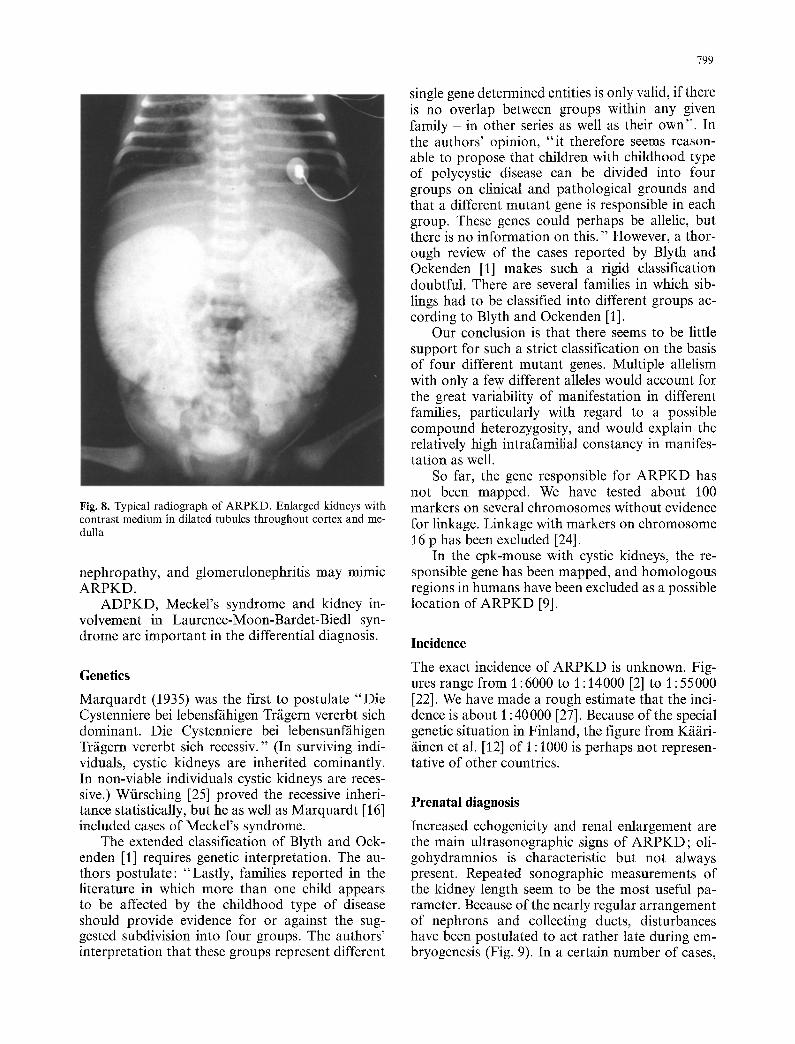
Fig. 8. Typical radiograph of ARPKD. Enlarged kidneys with contrast medium in dilated tubules throughout cortex and me- dulla
nephropathy, and glomerulonephritis may mimic ARPKD.
ADPKD, Meckel's syndrome and kidney in- volvement in Laurence-Moon-Bardet-Biedl syn- drome are important in the differential diagnosis.
Genetics
Marquardt (1935) was the first to postulate "Die Cystenniere bei lebensf/ihigen Tr/igern vererbt sich dominant. Die Cystenniere bei lebensunf/ihigen Trfigern vererbt sich recessiv." (In surviving indi- viduals, cystic kidneys are inherited cominantly. In non-viable individuals cystic kidneys are reces- sive.) Wfirsching [25] proved the recessive inheri- tance statistically, but he as well as Marquardt [16] included cases of Meckel's syndrome.
The extended classification of Blyth and Ock- enden [1] requires genetic interpretation. The au- thors postulate: "Lastly, families reported in the literature in which more than one child appears to be affected by the childhood type of disease should provide evidence for or against the sug- gested subdivision into four groups. The authors' interpretation that these groups represent different
799
single gene determined entities is only valid, if there is no overlap between groups within any given family - in other series as well as their own". In the authors' opinion, "i t therefore seems reason- able to propose that children with childhood type of polycystic disease can be divided into four groups on clinical and pathological grounds and that a different mutant gene is responsible in each group. These genes could perhaps be allelic, but there is no information on this." However, a thor- ough review of the cases reported by Blyth and Ockenden [1] makes such a rigid classification doubtful. There are several families in which sib- lings had to be classified into different groups ac- cording to Blyth and Ockenden [1].
Our conclusion is that there seems to be little support for such a strict classification on the basis of four different mutant genes. Multiple allelism with only a few different alleles would account for the great variability of manifestation in different families, particularly with regard to a possible compound heterozygosity, and would explain the relatively high intrafamilial constancy in manifes- tation as well.
So far, the gene responsible for ARPKD has not been mapped. We have tested about 100 markers on several chromosomes without evidence for linkage. Linkage with markers on chromosome 16 p has been excluded [24].
In the cpk-mouse with cystic kidneys, the re- sponsible gene has been mapped, and homologous regions in humans have been excluded as a possible location of ARPKD [9].
Incidence
The exact incidence of ARPKD is unknown. Fig- ures range from 1:6000 to 1 : 14000 [2] to 1 : 55000 [22]. We have made a rough estimate that the inci- dence is about 1:40000 [27]. Because of the special genetic situation in Finland, the figure from K/i/iri- /linen et al. [12] of 1 : 1000 is perhaps not represen- tative of other countries.
Prenatal diagnosis
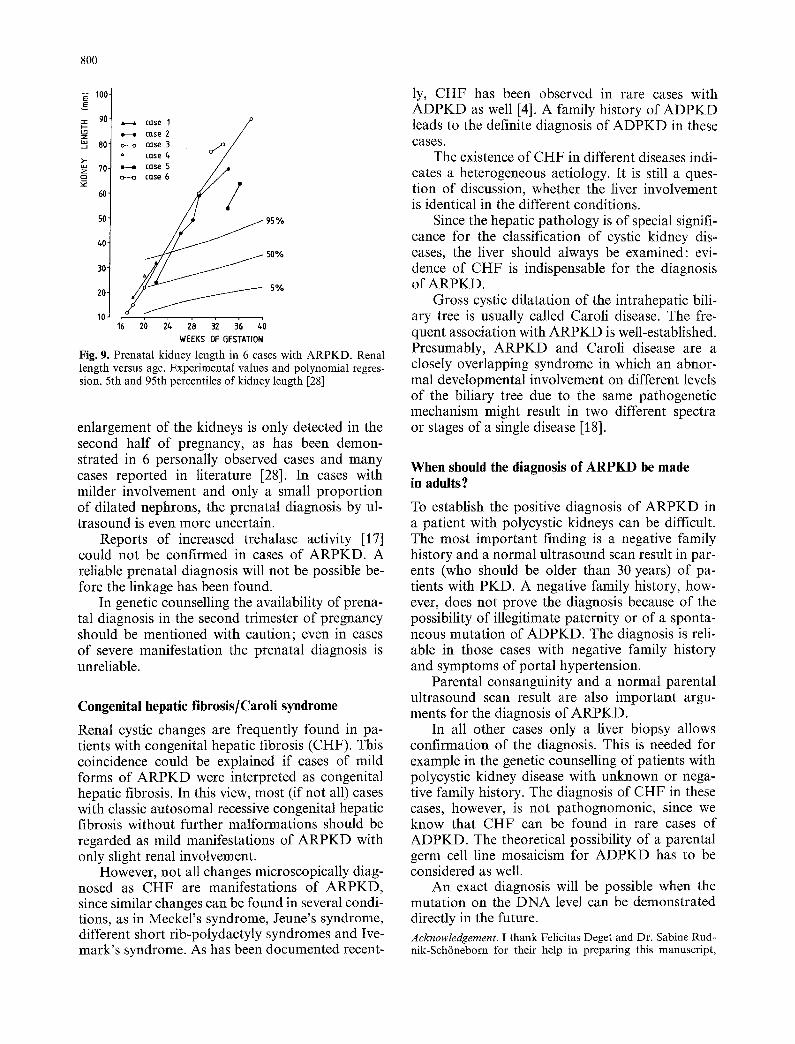
Increased echogenicity and renal enlargement are the main ultrasonographic signs of ARPKD; oli- gohydramnios is characteristic but not always present. Repeated sonographic measurements of the kidney length seem to be the most useful pa- rameter. Because of the nearly regular arrangement of nephrons and collecting ducts, disturbances have been postulated to act rather late during em- bryogenesis (Fig. 9). In a certain number of cases,
800
"~ 100 E
90- ~ case 1 / P H case Z /
80" ~ case 3 o./-°,// case /.
~- iii.--i case S J ~ll 70-
c::) 60- ~ CQSe 6 / /
~0
30 ~ 50%
20 / ~ 5%
10 1; 20 2/* i8 32 3'6 ~'0 WEEKS OF GESTATION
Fig. 9. Prenatal kidney length in 6 cases with ARPKD. Renal length versus age. Experimental values and polynomial regres- sion, 5th and 95th percentiles of kidney length [28]
enlargement of the kidneys is only detected in the second half of pregnancy, as has been demon- strated in 6 personally observed cases and many cases reported in literature [28]. In cases with milder involvement and only a small proportion of dilated nephrons, the prenatal diagnosis by ul- trasound is even more uncertain.
Reports of increased trehalase activity [17] could not be confirmed in cases of ARPKD. A reliable prenatal diagnosis will not be possible be- fore the linkage has been found.
In genetic counselling the availability of prena- tal diagnosis in the second trimester of pregnancy should be mentioned with caution; even in cases of severe manifestation the prenatal diagnosis is unreliable.
Congenital hepatic fibrosis/Caroli syndrome
Renal cystic changes are frequently found in pa- tients with congenital hepatic fibrosis (CHF). This coincidence could be explained if cases of mild forms of ARPKD were interpreted as congenital hepatic fibrosis. In this view, most (if not all) cases with classic autosomal recessive congenital hepatic fibrosis without further malformations should be regarded as mild manifestations of ARPKD with only slight renal involvement.
However, not all changes microscopically diag- nosed as CHF are manifestations of ARPKD, since similar changes can be found in several condi- tions, as in Meckel's syndrome, Jeune's syndrome, different short rib-polydactyly syndromes and Ive- mark's syndrome. As has been documented recent-
ly, CHF has been observed in rare cases with ADPKD as well [4]. A family history of ADPKD leads to the definite diagnosis of ADPKD in these cases.
The existence of CHF in different diseases indi- cates a heterogeneous aetiology. It is still a ques- tion of discussion, whether the liver involvement is identical in the different conditions.
Since the hepatic pathology is of special signifi- cance for the classification of cystic kidney dis- eases, the liver should always be examined: evi- dence of CHF is indispensable for the diagnosis of ARPKD.
Gross cystic dilatation of the intrahepatic bili- ary tree is usually called Caroli disease. The fre- quent association with ARPKD is well-established. Presumably, ARPKD and Caroli disease are a closely overlapping syndrome in which an abnor- mal developmental involvement on different levels of the biliary tree due to the same pathogenetic mechanism might result in two different spectra or stages of a single disease [18].
When should the diagnosis of ARPKD be made in adults?
To establish the positive diagnosis of ARPKD in a patient with polycystic kidneys can be difficult. The most important finding is a negative family history and a normal ultrasound scan result in par- ents (who should be older than 30 years) of pa- tients with PKD. A negative family history, how- ever, does not prove the diagnosis because of the possibility of illegitimate paternity or of a sponta- neous mutation of ADPKD. The diagnosis is reli- able in those cases with negative family history and symptoms of portal hypertension.
Parental consanguinity and a normal parental ultrasound scan result are also important argu- ments for the diagnosis of ARPKD.
In all other cases only a liver biopsy allows confirmation of the diagnosis. This is needed for example in the genetic counselling of patients with polycystic kidney disease with unknown or nega- tive family history. The diagnosis of CHF in these cases, however, is not pathognomonic, since we know that CHF can be found in rare cases of ADPKD. The theoretical possibility of a parental germ cell line mosaicism for ADPKD has to be considered as well.
An exact diagnosis will be possible when the mutation on the DNA level can be demonstrated directly in the future. Acknowledgement. I thank Felicitas Deget and Dr. Sabine Rud- nik-Sch6neborn for their help in preparing this manuscript,

801
as well as Prof. R. Waldherr, Prof. W. Rascher and Priv.-Doz. H.P.H. Neumann for their kind support.
References
1. Blyth H, Ockenden BG (1971) Polycystic disease of kidneys and liver presenting in childhood. J Med Genet 8:257-284
2. Bosniak MA, Ambos MA (1975) Polycystic kidney disease. Semin Roentgenol 10:133-143
3. Chilton SJ, Cremin BJ (1981) The spectrum of polycystic disease in children. Pediatr Radiol 11:9-15
4. Cobben JM, Breuning MH, Schoots C, TenKate LP, Zerres K (1990) Congenital hepatic fibrosis in autosomal dominant polycystic kidney disease. Kidney Int 38 : 880-885
5. Cole B (1990) Autosomal recessive polycystic kidney dis- ease. In: Gardner KD, Bernstein J (eds) The cystic kidney. Kluwer, Dordrecht, p 295
6. Cole BR, Conley SB, Stapleton FB (1987) Polycystic kidney disease in the first year of life. J Pediatr 111:693-699
7. Gabow PA (1991) Polycystic kidney disease: clues to patho- genesis. Kidney Int 40: 989-996
8. Gang GL, Herrin JT (1986) Infantile polycystic disease of the liver and kidneys. Clin Nephrol 25 : 28-36
9. Guay-Woodford LM, D'Eustachio P, Miicher G, Uhlhaas S, Zerres K (1990) Testing the hypothesis that murine con- genital polycystic kidney disease (cpk) and human autoso- real recessive polycystic kidney disease (ARPKD) affect ho- mologous genes. JASN 1 : 300A
10. Kfifiri/iinen H (1987) Polycystic kidney disease in children: a genetic and epidemiological study of 82 Finnish patients. J Med Genet 24:474-481
11. K~i~irifiinen H, J/i/iskefiinen J, Kirisaari L, Koskimies O, Norio R (1988) Dominant and recessive polycystic kidney disease in children: classification by intravenous pyelogra- phy, ultrasound and computed tomography. Pediatr Radiol 18:45-50
12, K/i/iri/iinen H, Koskimies O, Norio R (1988) Dominant and recessive polycystic kidney disease in children: evaluation of clinical features and laboratory data. Pediatr Nephrol 2: 296-302
13. Kaplan BS, Fay J, Shah V, Dillon MJ, Barrett TM (1989) Autosomal recessive polycystic kidney disease. Pediatr Nephrol 3 : 43-49
14. Liebermann E, Salinas-Madrigal L, Gwinn JL, Brennan LP, Fine RN (1971) Infantile polycystic disease of the kidneys and liver. Medicine 50 : 277-318
15. Lundin PM, Olow I (1961) Polycystic kidneys in newborns, infants and children, a clinical and pathological study. Acta Paediatr 50:185-200
16. Marquardt W (1935) Cystennieren, Cystenleber und Cys- tenpancreas bei zwei Geschwistern. Thesis, University of Tfibingen
17. Morin PR, Potier M, Dallaire L, Melancon SB, Boisvert J (1981) Prenatal detection of the autosomat dominant re- cessive type of polycystic kidney disease by trehalase assay in amniotic fluid. Prenat Diagn I : 75-79
18. Nakanuma Y, Terada T, Ohtal G, Kurachi M, Matsubara F (1982) Caroli's disease in congenital hepatic fibrosis and infantile polycystic disease. Liver 2:346354
19. Neumann HPH, Zerres K, Fischer CL, Wolff G, Schaefer HE, Gal A, Wirth B, Kr6pelin T, Haag K, Schollmeyer P (1988) Late manifestation of autosomal-recessive polycys- tic kidney disease in two sisters. Am J Nephrol 8:194-197
20. Osathanondh V, Potter EL (1964) Pathogenesis of polycys- tic kidneys. Type I due to hyperplasia of interstitial portions of collecting tubules. Arch Pathol 77: 466-473
21. Potter EL (1946) Facial characteristics of infants with bilat- eral renal agenesis. Am J Obstet Gynecol 51 : 885-888
22. Potter EL (1972) Normal and abnormal development of the kidney. Year Book Medical , Chicago
23. Rall JE, Odel HM (1949) Congential polycystic disease of the kidney: review of the literature, and data on 207 cases. Am J Med Sci 218:399-407
24. Wirth B, Zerres K, Fischbach M, Claus D, Neumann HPH, Lennert T, Brodehl J, Neugebauer M, Miiller-Wiefel DE, Geisert J, Gal A (1987) Autosomal recessive and dominant polycystic kidney disease are not allelic. Hum Genet 77:221-222
25. Wfirsching F (1957) Zur Erbpathologie und Ph/inogenese der Cystenieren. Thesis, University of Munich
26. Zerres K (1987) Genetics of cystic kidney diseases. Pediatr Nephrol 1 : 397-404
27. Zerres K, V61pel MC, Weil3 H (1984) Cystic kidneys. Genet- ics, pathologic anatomy, clinical picture, and prenatal diag- nosis. Hum Genet 68:104-135
28. Zerres K, Hansmann M, Mallmann R, Gembruch U (1988) Autosomal recessive polycystic kidney disease. Problems of prenatal diagnosis. Prenat Diagn 8:215-229
Prof. Dr. med. K. Zerres Institut fiir Humangenetik der Universitfit Bonn Wilhelmstrasse 31 W-5300 Bonn 3, FRG


![Autosomal recessive ichthyosis with limb reduction defect ... · including autosomal dominant, autosomal recessive and X-linked inheritance [1,2]. Associated cutaneous and extracutaneous](https://img.pdfslide.net/doc/110x75/5ec8c9b91adfdf12ab3e663c/autosomal-recessive-ichthyosis-with-limb-reduction-defect-including-autosomal.jpg)
![Clinical manifestations of autosomal recessive polycystic kidney ... · viduals to survive the perinatal period [ 8, 10]. Pulmonaryhypoplasia,aserio uscomplicationthatgenerally occurs](https://img.pdfslide.net/doc/110x75/5f09f6827e708231d4295907/clinical-manifestations-of-autosomal-recessive-polycystic-kidney-viduals-to.jpg)

























