Embed Size (px)
Citation preview

Biochemische und biophysikalische Untersuchungen an pflanzlichen und bakteriellen Hämoxygenasen
Dissertation zur Erlangung des Grades eines Doktors der Naturwissenschaften
(Doctor rerum naturalium (Dr. rer. nat.)) der Fakultät für Biologie und Biotechnologie
an der Internationalen Graduiertenschule Biowissenschaften der Ruhr-Universität Bochum
angefertigt am Lehrstuhl Biologie der Mikroorganismen
in der Arbeitsgruppe Physiologie der Mikroorganismen
vorgelegt von Björn Gisk
aus Herne
Bochum Oktober 2010
Referentin: Prof. Dr. Nicole Frankenberg-Dinkel
Korreferent: Prof. Dr. Matthias Rögner

Biochemical and biophysical investigations on plant and bacterial heme oxygenases
Dissertation to obtain the degree Doctor rerum naturalium (Dr. rer. nat.)
at the Faculty of Biology and Biotechnology Ruhr-University Bochum
International Graduate School of Biosciences
Ruhr-University Bochum
Department of Microbiology/
Physiology of Microorganisms
Submitted by Björn Gisk
from Herne
Bochum Oktober 2010
First Supervisor: Prof. Dr. Nicole Frankenberg-Dinkel
Second Supervisor: Prof. Dr. Matthias Rögner

Danksagungen
Danksagungen Zuerst möchte ich mich besonders bei meiner Doktormutter Prof. Dr. Nicole Frankenberg-
Dinkel für die äußerst interessante und spannende Aufgabenstellung und die fortwährende
Unterstützung und Diskussionsbereitschaft bedanken. Ich danke ihr für die Einräumung
großer experimenteller Freiheiten und für die Ermöglichung der Teilnahme an internationalen
Konferenzen.
Ich danke ebenfalls Herrn Prof. Dr. Matthias Rögner für die freundliche Übernahme des
Korreferates und für die Ermöglichung der ersten eigenständigen wissenschaftlichen Arbeiten
in seiner Arbeitsgruppe während meiner Bachelorarbeit, wodurch mein wissenschaftliches
Interesse noch mehr denn je geweckt worden ist.
Als nächstes möchte ich mich bei meinen Kooperationspartnern, Prof. Dr. Eckhard Hofmann
aus der Arbeitsgruppe Röntgenstrukturanalyse an Proteinen, PD Dr. Carsten Kötting aus dem
Lehrstuhl Biopyhsik, Prof. Dr. Raphael Stoll aus der Arbeitsgruppe Biomolekulare NRM der
Fakultät für Chemie und Biochemie (alle Ruhr-Universität Bochum) sowie Prof. Dr. Martin
Bröring, Institut für Anorganische und Analytische Chemie der TU Braunschweig, recht
herzlich bedanken, ohne die viele Experimente nicht möglich gewesen wären.
Prof. Dr. Bernhard Grimm (AG Pflanzenphysiologie der Humboldt-Universität zu Berlin)
danke ich für die Messung des Tetrapyrrolgehaltes in Arabidopsis Pflanzen.
Bei Dr. Jessica Wiethaus, Dr. Katalin Barkovits und Dipl. Biol. Andrea W.U. Busch möchte
ich mich besonders bedanken, da sie mir während der letzen Jahre immer mit Rat und Tat zur
Seite standen und durch das freundschaftliche Verhältnis so mancher Tiefpunkt leichter zu
durchschreiten war.
Ebenfalls möchte ich mich bei allen anderen Mitarbeitern der Arbeitsgruppe Physiologie der
Mikroorganismen für die äußerst angenehme Arbeitsatmosphäre bedanken, insbesondere bei
Britta Schubert und Bastian Molitor, die mich mit Humor und Zuverlässigkeit durch die
Promotion begleitet haben. Bei Hanno Boeddinghaus bedanke ich mich für die EDV
Unterstützung.

Danksagungen
Bei Prof. Dr. Richard Vierstra (University of Wisconsin-Madison, USA) bedanke ich mich
für die mir freundlicherweise zur Verfügung gestellten Expressionsklone pET-ho3 und -ho4
und bei Prof. Dr. Takayuki Kohchi (Graduate School of Biostudies, Kyoto University, Japan)
für den Expressionsklon pGEX-mhy1.
Beim DAAD (Deutscher Akademischer Austausch Dienst) bedanke ich mich für die
Finanzierung des einmonatigen Forschungsaufenthaltes in Taiwan/Teipei an der Academia
Sinica am Institut of Plant and Microbial Biology in der Arbeitsgruppe von Prof. Dr. Shih-
Long Tu.
Ich danke zudem meinen Freuden, insbesondere Kathrin, Heiko, Lutz und Stefan, die mir
während des Studiums und der Promotion zur Seite standen.
Nicht zuletzt bedanke ich mich von ganzem Herzen bei meinen Eltern, die mir überhaupt erst
das Studium ermöglicht haben und mich von Anfang bis zum Ende unterstützten.

Meinen Eltern

Inhaltsverzeichnis
I
I Inhaltsverzeichnis
I Inhaltsverzeichnis I
II Abkürzungsverzeichnis III
A Einleitung 1
1. Häm 1
2. Corrole 3
3. Hämoxygenasen 4
3.1 Phylogenetische Verbreitung der Hämoxygenasen 6
3.2 Biologische Relevanz der Hämoxygenasen und der durch den 9 Hämabbau entstandenen Produkte
3.3 Strukturelle Ähnlichkeiten der Hämoxygenasen 13
3.4 Substratbindetasche der Hämoxygenasen 16
3.5 Reaktionsmechanismus der Hämoxygenasen 18
3.6 Hämoxygenasen aus Pseudomonas aeruginosa 22
3.7 Hämoxygenasen aus Arabidopsis thaliana 23
B Aufgabenstellung 25
C Publikationen/Manuskripte 26
1. Characterization of the haem oxygenase protein family in Arabidopsis thaliana 27 reveals a diversity of functions
2. Heme oxygenases from Arabidopsis thaliana reveal different mechanisms of 38 carbon monoxide binding
3. Enzymatic ring opening of an iron corrole by plant-type heme oxygenases: 53 Unexpected substrate and protein selectivities
4. The existence of two heme oxygenases with different regiospecificities is a 67 characteristic trait of pathogenic and non-pathogenic Pseudomonas species
D Diskussion 87
1. Hämoxygenasen aus Arabidopsis thaliana 87
1.1 Hämoxygenasen aus A. thaliana sind nicht im Mitochondrium, 87 sondern im Chloroplast lokalisiert
1.2 HY1, HO3 und HO4 aus A. thaliana sind biochemisch nahezu 88 identisch
1.3 HO2 aus A. thaliana und ihre mögliche Funktion 90
1.4 Arabidopsis Hämoxygenasen unterliegen der Produktinhibierung 94 durch Eisen und BV, jedoch der durch CO nur bedingt
1.5 Arabidopsis Hämoxygenasen sind strukturell ähnlich zu anderen 98 Hämoxygenasen

Inhaltsverzeichnis
II
2. Hämoxygenasen aus Pseudomonaden 100
2.1 Verschiedene Pseudomonas Spezies besitzen alle zwei aktive 100 und regiospezifisch unterschiedliche Hämoxygenasen
3. Corrole als alternative Hämoxygenase-Substrate 104
E Zusammenfassung 107
F Summary 109
G Anhang 111
1. Strukturaufklärung der HO3 aus Arabidopsis thaliana 111
1.1 D 1H-NMR an Arabidopsis HO3 111
1.2 D 1H-15N HSQC-NMR an Arabidopsis HO3 112
1.3 Material und Methoden 112
1.4 Ergebnisse und Diskussion 114
2. Prozentuale Verteilung der Eigenleistung an den Publikationen/Manuskripte 117
3. Konferenzbeiträge 118
4. Lebenslauf 119
5. Erklärung 122
H Literaturverzeichnis 123

Abkürzungsverzeichnis
III
II Abkürzungsverzeichnis
Die Abkürzungen von Nuklein- und Aminosäuren erfolgen nach dem „Nomenclature
Commitee of the International Union of Biochemistry and Molecular Biology“ (NC-IUBMB).
Die Chemikalien werden nach der „International Union of Pure and Applied Chemistry“
(IUPAC) benannt. Die Abkürzungen der Proteine und Gene stellen ihre Namen dar. Des
Weiteren gelten die Abkürzungen für die SI-Einheit („Système International d’unités“). Die
eben genannten Abkürzungen werden im Abkürzungsverzeichnis nicht gesondert aufgeführt.
Å Angström
Abb. Abbildung
Amp Ampicillin
AS Aminosäure
ATP Adenosintriphosphat
BphP Bakteriophytochrom
BphO Hämoxygenase aus Pseudomonas aeruginosa
BR Bilirubin
BV Biliverdin
BvR Biliverdinreduktase
bzw. beziehungsweise
°C grad Celsius
Col Columbia (Ecotyp von Arabidopsis)
Da Dalton
DHBV 15,16-Dihydrobiliverdin
DMSO Dimethylsulfoxid
DNA Desoxyribonukleinsäure
ε molarer Extinktionskoeffizient
EPR elektroparamagnetische Resonanz-Messung
Fd Ferredoxin
Fig. Abbildung (Figure)
FM Frischmaterial
FNR Ferredoxin: NADP+-Oxidoreduktase
FPR Ferredoxin: NADP+-Reduktase
FR Dunkelrotlicht (far-red light)

Abkürzungsverzeichnis
IV
FTIR Fourier-Transformation-Infrarot-Spektroskopie
(fourier transformation infra red spectroscopy)
Fur Eisenaufnahme Regulator
(ferric uptake regulator)
GFP grün fluoreszierendes Protein
(green fluorecent protein)
Glk-6-P-DH Glukose-6-Phosphat-Dehydrogenase
GPC Gelpermeationschromatographie
GST Glutathion-S-Transferase
HSQC Heteronukleare Einzelquanten Koherenz
(Heteronuclear Single Quantum Coherence)
HRM Häm regulatorische Motive
(heme regulatory motifs)
HO Hämoxygenase
HO2, 3, 4 Hämoxygenasen aus Arabidopsis thaliana
HPLC Hochleistungs-Flüssigkeits-Chromatographie
(high performance liquid chromatography)
HY1 Hämoxygenase aus Arabidopsis thaliana
HY2 Ferredoxin-abhängige
Phytochromobilin-Synthase aus Arabidopsis
thaliana
IPTG Isopropyl-1-thio-ß-D-galactosid
Kap. Kapitel
LB Luria Bertani (Medium)
Mr relative molekulare Masse
MME Monomethylester
NADPH Nicotinamid-adenin-dinukleotid-phosphat
(reduziert)
NMR Kernspinresonanz-Spektroskopie
(nuclear magnetic resonance)
PBS Phosphat-gepufferte Salzlösung
(phosphate buffered saline)
PCB Phycocyanobilin
PEB Phycoerythrobilin

Abkürzungsverzeichnis
V
PigA Hämoxygenase aus
Pseudomonas aeruginosa (Eisenmangel
induziert)
PΦB Phytochromobilin
PS Photosystem
Psi pounds per square inch
Phy Phytochrom
PHY Phytochrom Domäne
Proto IX Protoporphyrin IX
R Hellrotlicht (red-light)
rpm Umdrehungen pro Minute
(rounds per minute)
ROS reaktive Sauerstoff Spezies
(reactive oxygen spezies)
RT Raumtemperatur
Tab. Tabelle
TFA Trifluoressigsäure
U Unit
UV/vis Ultraviolett/sichtbar (visible)-Spektroskopie
v/v Volumen pro Volumen (volume per volume)
WT Wildtyp
w/v Gewicht pro Volumen (weight per volume)

Einleitung
1
A Einleitung
1. Häm
Häm ist eines der wichtigsten Bausteine unseres Lebens. Es ist ein essentieller Cofaktor einer
Vielzahl von Enzymen mit unterschiedlichsten Funktionen. Im Hämoglobin der Erythrozyten
dient Häm dem Transport von molekularem Sauerstoff. Im Myoglobin der Muskulatur hat es
eine Funktion als Sauerstoff-Speicher (Hayashi et al., 1973). Durch Oxidoreduktasen, wie
Katalasen und Peroxidasen, die Häm als prosthetische Gruppe besitzen, werden
Redoxreaktionen zur Umsetzung von H2O2 katalysiert (Sorenson&Scandalios, 1980; Alfonso-
Prieto et al., 2009). In Cytochromen ist Häm am Elektronentransport, der Energiegewinnung
und an chemischen Transformationen beteiligt (Wagener et al., 2003). Darüber hinaus ist
Häm in die Synthese von Signalmolekülen involviert, die durch die Stickstoffoxid-Synthase,
Guanylat-Cyclase, Cyclooxygenase und Hydroxylasen produziert werden (Maines, 1997;
Ponka, 1999; Zhao et al., 1999; Wagner et al., 2003). Enzyme die Häm als Cofaktor gebunden
haben, werden auch als Hämproteine bezeichnet.
Häm besitzt neben seinen nützlichen Eigenschaften auch solche, die den Organismus
gefährden können. Ungebundenes Häm wirkt potentiell zytotoxisch. Die toxische Wirkung ist
eine Folge der Fenton-Reaktion. In biologischen Systemen gilt die durch Eisensalze
katalysierte Oxidation organischer Substrate mit Wasserstoffperoxid in saurem Medium als
Hauptquelle reaktiver Sauerstoff-Spezies (reactive oxygen species; ROS)
(Halliwell&Gutteridge, 1984; Everse&Hsia, 1997). Diese ROS können die Zellmembranen,
DNA und Proteine schädigen und spielen eine Rolle bei der Entstehung von oxidativem Stress
(Beri&Chandra, 1993; Jeney et al., 2002; Wagener et al., 2003). Daher ist die Regulierung der
Häm-Homöostase essentiell für jeden Organismus.
Häm zählt zu den Metalloporphyrinen und besteht aus einem planaren Protoporphyrin IX-
Ring. Dieser ist aus vier einzelnen Pyrrolringen (A, B, C und D) aufgebaut, die über meso-
Kohlenstoffbrücken (Methinbrücken) miteinander verbunden sind (Abb. 1). Das
Protoporphyrin IX (Proto IX) wird auch als geschlossenes oder zyklisches Tetrapyrrol
bezeichnet. Entsprechend ihrer Position werden die Methinbrücken des Proto IX mit α, β, γ
und δ benannt. Sie bestehen formal aus einer Einfach- und einer Doppelbindung, wodurch das
Proto IX einen aromatischen Ring mit 18 delokalisierten π-Elektronen bildet. Das ausgeprägte
konjugierte π-Elektronensystem macht das Molekül zu einem guten Cofaktor, da es durch
dieses System in der Lage ist, u. a. Elektronen zu transportieren und Licht im sichtbaren
Bereich zu absorbieren. Proto IX ist ein Schlüsselmolekül in der Biosynthese von zyklischen

Einleitung
2
Tetrapyrrolen in allen Organismen. Durch die vier zum Zentrum liegenden Stickstoffatome
der Pyrrolringe können zweiwertige Übergangsmetalle im Proto IX gebunden werden. Im
Falle des Häms wird ein Eisenatom (Fe) gebunden, weshalb Häm auch als
Eisenprotoporphyrin IX bezeichnet wird. Die Insertion des Eisenatoms in das Häm findet bei
allen Lebewesen durch die Ferrochelatase statt (Dailey et al., 2000). In Abhängigkeit des
Valenzelektronen-Status des Eisens kann dieses entweder als Ferro-Form (Fe2+) vorliegen,
dann spricht man von Häm oder als Ferri-Form (Fe3+), welches als Hemin bezeichnet wird.
N
N
N
N
Fe
HOOC HOOC
A
C
B
D
Abbildung 1: Das Häm-b-Molekül. Die vier Pyrrolringe (A, B, C und D) sind über Methinbrücken (α, β γ und δ) miteinander verbunden und weisen Vinyl/Methyl-Seitenketten auf. Dative Bindungen zwischen zwei der Pyrrolstickstoffatome und dem Eisenzentralatom sind als gestrichelte Linien gezeichnet.
Die vier Pyrrolringe des Häms können mit unterschiedlichen Seitenketten substituiert sein.
Daraus ergeben sich verschiedene Variationen des Hämmoleküls (Häm-a, -b, -c, -d, -d1, -o,
-P450, -P460 und Sirohäm) (Ponka, 1999). Als Enzym-Cofaktor sind b- und c-Typ Häme am
weitesten verbreitet. Diese kommen u. a. im Hämoglobin und Myoglobin (b-Typ Häm)
(Tenhunen et al., 1969; Hayashi et al., 1973) und im Cytochrom b6 (c-Typ Häm) vor (de Vitry
et al., 2004).
b-und c-Typ Häme unterscheiden sich dadurch, dass die c-Typ Häme kovalent über die Vinyl-
bzw. Methylreste an die apo-Proteine gebunden werden, b-Typ Häme hingegen nicht kovalent
gebunden in den Proteinen vorliegen.

Einleitung
3
2. Corrole
Hämproteine können nicht nur Häm, sondern auch dem Häm ähnliche Moleküle, wie Corrole
binden.
Corrole (Tetradehydrocorrine) sind chemisch modifizierte Porphyrine, die in der Natur nicht
vorkommen. Sie sind dem Häm strukturell sehr ähnlich. Einer der Unterschiede dieser
tetrapyrrolischen Makrozyklen besteht in dem Fehlen eines meso-Kohlenstoffatoms zwischen
zwei Pyrrolringen (Abb. 2).
NH
NH
HN
NHNN
NNH N
N
HN
N
Porphyrin Corrol Corrin
Abbildung 2: Porphyrinoide Makrozyklen. Das Corrol nimmt eine Position zwischen dem Porphyrin und dem Corrin ein.
Damit stehen die Corrole strukturell zwischen den Porphyrinen und den Corrinen, dem
Grundgerüst des Vitamin B12. Der aromatische Charakter bleibt den Corrolen durch das
18 π-Elektronensystem erhalten (Guilard et al., 2003). Ein weiterer Unterschied zu den
Porphyrinen besteht in der leichten Verzerrung des Corrolring-Rückgrates aus der Ebene der
vier zentralen Stickstoffatome heraus, bedingt durch die Abstoßung der inneren
Wasserstoffatome (Harrison et al., 1971). Aus diesem Grund ist das Corrol, im Vergleich zum
Häm, nicht planar. Ein entscheidender Vorteil der Corrole liegt in der Fähigkeit, Metalle in
formal höheren Oxidationsstufen zu stabilisieren, d. h. ohne Berücksichtigung der
tatsächlichen elektronischen Struktur. Dabei kann ein Corrol z. B. ein Eisen formal als Fe4+
koordinieren (Vogel et al., 1994), wohingegen in den Porphyrinen das Eisen als Fe3+ vorliegt.
Dadurch ergeben sich signifikante Unterschiede in der Reaktivität und Stabilität der Corrole
im Vergleich zum Häm, was Untersuchungen an Corrolen zu einem immer attraktiver
werdenden Bereich der Hämproteinforschung werden lässt. Da die modifizierten
Metalloporphyrine eine ähnliche Struktur aufweisen wie die natürlichen Hämgruppen, können
einem Protein durch eine Substitution der Porphyrine gegen Corrole neue
Reaktionsmöglichkeiten verliehen werden. Dabei sind keine entscheidenden strukturellen
Änderungen an der Metallbindesstelle erforderlich. Beispielsweise konnte durch den Einbau

Einleitung
4
eines Eisenporphyrinoids (Porphycen) an Stelle des Häms in ein Metmyoglobin und
Methämoglobin die Bindungsfähigkeit zu kleinen Molekülen, wie O2, drastisch erhöht werden
(Hayashi et al., 2002). Ein anderes Beispiel ist der Einbau von Corrolen in Myoglobin und
einer Peroxidase, deren Reaktivitäten dadurch verändert werden konnten (Matsuo et al.,
2009).
Durch den Austausch der natürlichen prosthetischen Hämgruppe gegen chemisch modifizierte
Metalloporphyrine in Hämproteine können Erkenntnisse über die physiologischen
Eigenschaften und die Regulation der Hämproteine gewonnen werden. Zudem können die
modifizierten Proteine für biotechnologische Verwendungen genutzt werden.
3. Hämoxygenasen
Nicht nur dem Häm selbst, sondern auch dem Hämabbau kommt eine große Bedeutung in
biologischen Systemen zu. Die Hämspaltung kann neben dem einfachen katabolischen Abbau
auch der Eisengewinnung dienen, die Biosynthese der photosynthetischen und
photosensorischen Phycobiline einleiten und zudem über die Freisetzung von
Kohlenstoffmonoxid (CO) eine Rolle in der Signaltransduktion spielen. Das Schlüsselenzym
der Hämspaltung ist die Hämoxygenase (HO [E.C. 1.14.99.3]). HOs katalysieren die
Umsetzung von Häm zu Biliverdin (BV), Kohlenstoffmonoxid (CO) und Eisen (Fe2+) in
äquimolaren Teilen. Die Sauerstoff-abhängige Reaktion benötigt sieben Elektronen und drei
Sauerstoff-Moleküle (Ortiz de Montellano, 2000; Wilks, 2002; Matsui et al.). Das Substrat
der HOs ist ausschließlich Häm-b (Tenhunen et al., 1969; Matsui et al., 2010).
HOs spalten das Häm regiospezifisch an einer der vier meso-Kohlenstoffbrücken, was zur
Entstehung der vier Biliverdin-Isomere IXα, IXβ, IXγ und IXδ führt (Abb.3). Die meisten
charakterisierten HOs weisen eine Spezifität für den α-meso-Kohlenstoff auf. Hierzu zählen
die HOs aus dem Menschen, aus vielen Gram-positiven und Gram-negativen Bakterien, aus
Hefen und aus allen photosynthetischen Organismen (Cornejo et al., 1998; Muramoto et al.,
2002; Gohya et al., 2006).
HOs, die nicht für den α-meso-Kohlenstoff spezifisch sind, kommen ebenfalls in der Natur
vor und sind in Drosophila melanogasta, im Säuger-Fötus, in der Raubwanze (Rhodnius
prolixus) und in Pseudomonas aeruginosa zu finden.
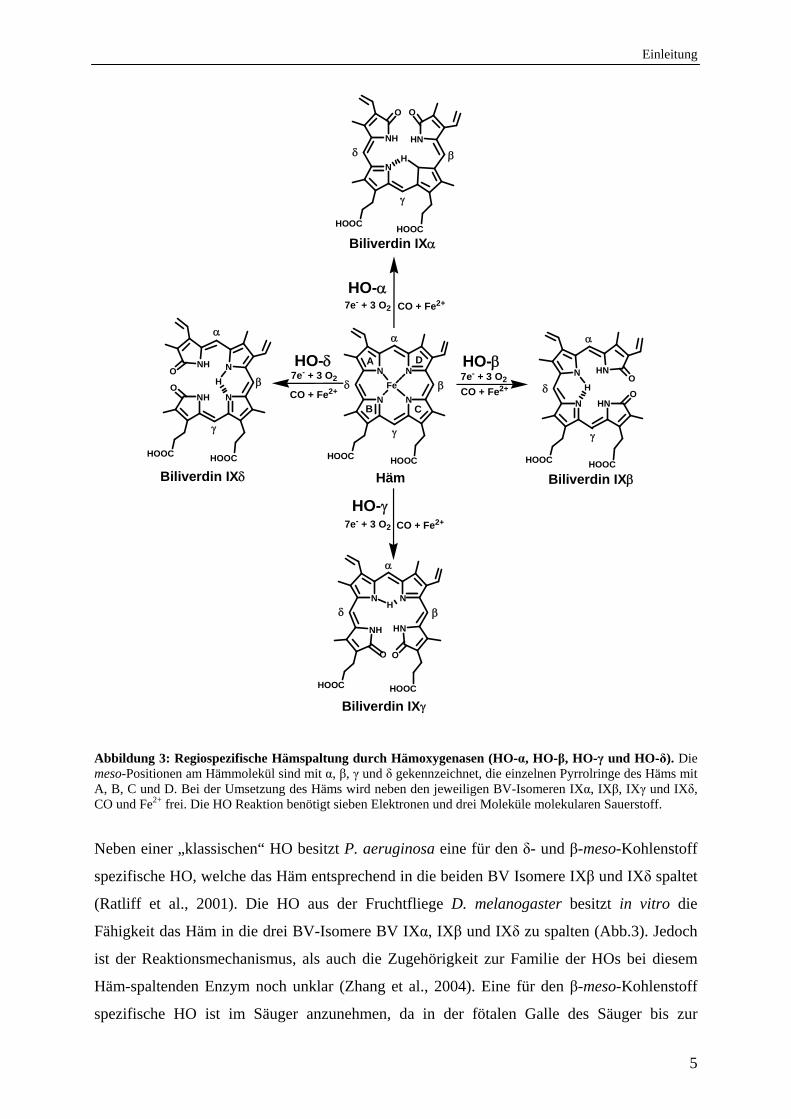
Einleitung
5
N
N
N
N
FeCO + Fe2+
7e- + 3 O2
Biliverdin IX
HOOC HOOC
Häm
NH
NH
N
N
HOOC HOOC
O
O
N
N
HN
HN
HOOC HOOC
O
O
NH
N
HN
HOOCHOOC
O O
H
HH
N
NH
N
HN
HOOC HOOC
O O
H
A
Biliverdin IX Biliverdin IX
Biliverdin IX
7e- + 3 O2
7e- + 3 O2
7e- + 3 O2
CO + Fe2+
CO + Fe2+
CO + Fe2+
HO-
HO-
HO-
HO-
C
D
B
Abbildung 3: Regiospezifische Hämspaltung durch Hämoxygenasen (HO-α, HO-β, HO-γ und HO-δ). Die meso-Positionen am Hämmolekül sind mit α, β, γ und δ gekennzeichnet, die einzelnen Pyrrolringe des Häms mit A, B, C und D. Bei der Umsetzung des Häms wird neben den jeweiligen BV-Isomeren IXα, IXβ, IXγ und IXδ, CO und Fe2+ frei. Die HO Reaktion benötigt sieben Elektronen und drei Moleküle molekularen Sauerstoff.
Neben einer „klassischen“ HO besitzt P. aeruginosa eine für den δ- und β-meso-Kohlenstoff
spezifische HO, welche das Häm entsprechend in die beiden BV Isomere IXβ und IXδ spaltet
(Ratliff et al., 2001). Die HO aus der Fruchtfliege D. melanogaster besitzt in vitro die
Fähigkeit das Häm in die drei BV-Isomere BV IXα, IXβ und IXδ zu spalten (Abb.3). Jedoch
ist der Reaktionsmechanismus, als auch die Zugehörigkeit zur Familie der HOs bei diesem
Häm-spaltenden Enzym noch unklar (Zhang et al., 2004). Eine für den β-meso-Kohlenstoff
spezifische HO ist im Säuger anzunehmen, da in der fötalen Galle des Säuger bis zur

Einleitung
6
zwanzigsten Woche bis zu 87 % BV IXβ feststellbar sind. Erst im adulten Säuger ist mit
95-97 % fast ausschließlich BV IXα nachweisbar (Yamaguchi et al., 1994; Shalloe et al.,
1996). In der Raubwanze konnte eine für den γ-meso-Kohlenstoff spezifische HO identifiziert
werden (Paiva-Silva et al., 2006).
Die durch die HOs entstandenen Biliverdine werden als offenkettige Tetrapyrrole (Biline)
bezeichnet (Bonnett&McDonagh, 1973) und erfüllen verschiedenste Funktionen in den
unterschiedlichsten Organismen (Kap. 3.2).
3.1 Phylogenetische Verbreitung der Hämoxygenasen
Die erste HO wurde 1968 von Tenhunen et al. im Menschen beschrieben und zunächst der
Cytochrom P450-Familie zugeordnet (Tenhunen et al., 1969). Erst nach der biochemischen
Charakterisierung im Jahre 1978 wurde sie einer eigenen Enzymklasse zugeordnet
(Yoshida&Kikuchi, 1978). Diese Zuordnung war notwendig, da sich die HO-Reaktion von
der Reaktion anderer Häm-abhängiger Enzyme, wie der Cytochrom P450-Familie (Poulos,
1988; Sevrioukova&Peterson, 1995), den Katalasen und Peroxidasen, deutlich unterscheidet
(Alfonso-Prieto et al., 2009). HOs sind nach der Definition keine Hämproteine, formen jedoch
einen HO:Häm-Komplex im Verhältnis 1:1 (Yoshida&Kikuchi, 1978, 1979).
HOs sind weit verbreitete Enzyme und wurden nicht nur in Säugern gefunden und
charakterisiert (Tenhunen et al., 1969; Wilks et al., 1995), sondern auch in höheren Pflanzen
(Davis et al., 2001), Insekten (Paiva-Silva et al., 2006), pathogenen- (Ochsner&Vasil, 1996;
Ratliff et al., 2001) und nicht pathogenen Bakterien (Wilks&Schmitt, 1998), Hefen (Kim et
al., 2006) sowie in Algen (Richaud&Zabulon, 1997; Douglas&Penny, 1999) und
Cyanobakterien (Cornejo&Beale, 1988; Cornejo&Beale, 1997; Richaud&Zabulon, 1997;
Zhang et al., 2005). Lediglich in Archae wurden bisher noch keine HOs gefunden.
Einige Organismen verfügen über mehr als eine HO. So weist der Mensch drei HO-Isoformen
auf (hHO-1 bis hHO-3). Die hHO-1 und hHO-2 sind zu ~ 42 % relativ identisch. Die hHO-3
weist deutlich weniger Homologie zu den beiden anderen HOs auf, allerdings ist sie der
hHO-2 phylogenetisch näherstehend. Die drei Enzyme sind Produkte verschiedener Gene und
differieren im Expressionsmuster, in der Gewebelokalisation sowie in ihrer Regulation
(Tenhunen et al., 1969; Maines, 1988; McCoubrey et al., 1997; Otterbein&Choi, 2000;
Wagener et al., 2003). In höheren Pflanzen sind multiple Gene der HO-Familie in einer
Vielzahl von Pflanzen bekannt, darunter in Apfel, Hirse, Erbse, Mais, Sojabone, Tomate und
Arabidopsis thaliana (Muramoto et al., 1999; Davis et al., 2001; Terry et al., 2002; Gohya et
al., 2006; Linley et al., 2006). Im cyanobakteriellem Genom von Synechocystis sp. PCC 6803

Einleitung
7
wurden zwei für HOs kodierende Gene gefunden, Syho-1 und Syho-2, deren Produkte eine
Aminosäuresequenzhomologie von 51 % zu einander aufweisen (Cornejo et al., 1998; Zhang
et al., 2005). Beide Proteine, SyHO-1 und SyHO-2, zeigen in vitro eine HO-Aktivität. Die für
HO-1 und HO-2 kodierenden Gene wurden auch in weiteren sequenzierten Cyanobakterien
wie Anabaena sp. PCC7120 und Thermosynechococcus elongatus identifiziert (Hess et al.,
2001; Migita et al., 2003). Das Bakterium P. aeruginosa zählt ebenfalls zu den Organismen,
die nicht nur eine HO besitzen. Neben pigA ist in dem Genom noch das Hämoxygenase-Gen
bphO kodiert. Das Besondere an den beiden HOs ist ihre unterschiedliche Regiospezifität.
Während PigA das Häm in die beiden BV Isomere BV IXδ und BV IXβ spaltet, spaltet BphO
das Häm zu BV IXα (Ratliff et al., 2001; Wegele et al., 2004).
HOs sind lösliche, cytosolische Proteine mit einer relativen molekularen Masse von 21 kDa
bis 27 kDa. Eine Ausnahme stellen die Säuger HOs dar, welche über eine zusätzliche
Membranbindedomäne an mikrosomalen Membranen verankert sind (Tenhunen et al., 1969;
Maines, 1988; McCoubrey et al., 1997; Otterbein&Choi, 2000; Wagener et al., 2003).
Entsprechend weisen Säuger HOs eine höhere relative molekulare Masse von 32 kDa bis 36
kDa auf.
HOs in photosynthetischen Organismen sind im Nukleus der Zelle kodiert und in den
Plastiden lokalisiert. Um in den Chloroplasten zu gelangen weisen sie deshalb eine
N-terminale Transit-Peptid-Lokalisationssequenz für das Organell auf. Diese N-terminale
Sequenz wurde bei pflanzlichen HOs (Terry et al., 1993; Muramoto et al., 1999) und bei HOs
aus Algen (Richaud&Zabulon, 1997; Douglas&Penny, 1999) gefunden.
Sequenzanalysen verschiedener HOs geben einen Einblick in die enge phylogenetische
Verwandtschaft dieser Enzyme, die in Abbildung 4 dargestellt ist. Dabei sind die HOs in zehn
Familien eingeteilt, die sich auf Grund eines Aminosäuresequenzvergleiches ergeben.

Einleitung
8
Abbildung 4: Phylogenetischer Stammbaum von ausgewälten HOs. Ein Sequenzalignment mit dem ClustalW Algorithmus wurde verwendet um den phylogenetischen Stammbau mit WH8102, Synechococcus sp. WH8102; PCC6803, Synechocystis sp. PCC6803; Hs, Homo sapiens; Cd, Corynebacterium diphteriae; Sc, Saccharomyces cervisiae; Ca, Candida albicans; At, Arabidopsis thaliana; Gm, Glycine max; Sb, Sorghum bicolor; Sa, Staphylococcus aureus; PA, Pseudomonas aeruginosa (Stamm PAO1 bzw. PA14); Pf, Pseudomonas fluorescens 5; Pp, Pseudomonas putida KT2440, Ps, Pseudomonas syringae DC3000; Nm, Neisseria meningitidis; Dr, Deinococcus radiodurans HOs zu erstellen. Rechts neben den einzelnen Organismen sind die jeweiligen HO Familien zusammengefasst. Die Familie der Isd-Proteine ist ebenfalls aufgeführt, obwohl sie nicht zu den HOs, sondern zu den Monooxygenasen zählt.
Sa_IsdG
Sa_IsdI
At_HY1
At_HO3
Gm_HO1
Gm_HO3
Sb_HO1
At_HO4
At_HO2
Sb_HO2
WH8102 HO1
PCC6803 HO1
PCC6803 HO2
Hs_HO-1
Hs_HO-3
Hs_HO-2
Cd_HmuO
PAO1_PigA
PA14_PigA
Pf_PigA
Pp_PigA
Ps_PigA
Nm_HemO
PAO1_BphO
Dr_BphO
Cyanobakterielle HO1 Familie
Säuger HO1 Familie
Säuger HO2 Fam.
Pflanzliche HO1 Familie
Pflanzliche HO2 Familie
Isd Familie
PigA Familie
BphO Familie PA14_BphO
Sc_HmX1
Ca_CaHmX1
Hefen HO1 Familie
Cyanobakt. HO2 Familie
HmuO Familie

Einleitung
9
Eine weitere Klasse Häm abbauender Enzyme stellt die Isd-Familie dar, welche u. a. in den
Gram-positiven pathogenen Bakterien Staphylococcus aureus (Skaar et al., 2003) und
Bacillus anthracis (Skaar et al., 2006) beschrieben wurde. Ursprünglich als HOs klassifiziert,
zählen die Isd-Proteine auf Grund ihrer Struktur jedoch zu den Monooxygenasen. Dem
entsprechend ergibt sich eine Reihe von Unterschieden im Vergleich zu den HOs. Isd-
Proteine weisen mit ~ 13 kDa eine deutlich geringere relative molekulare Masse als HOs auf
und es besteht nur eine sehr geringe Homologie zu den bakteriellen HOs. Des Weiteren
spalten Isd-Proteine das Häm nicht zu einem BV-Isomer, sondern zu einem dem BV
ähnlichen Produkt, dem oxo-Bilirubin Chromophor Staphylobilin (Reniere et al., 2010). Der
hierbei zugrunde liegende Reaktionsmechanismus ist noch nicht geklärt. Die unter
Eisenmangelbedingungen produzierten Isd-Proteine dienen der Eisenversorgung des
Bakteriums.
3.2 Biologische Relevanz der Hämoxygenasen und der durch den Hämabbau entstandenen Produkte
Die Stoffwechselfunktionen von HOs sind äußerst mannigfaltig. HOs erfüllen dabei katabole
oder anabole Funktionen, je nachdem, ob sie lediglich überschüssiges Häm abbauen oder die
Nutzung der freigesetzten Hämspaltprodukte BV, Eisen und CO ermöglichen.
Die Funktion des COs
CO ist ein Signalmolekül in den verschiedensten Organismen und wird u. a. durch HOs beim
Hämabbau generiert.
In Säugern wird CO durch die konstitutiv exprimierte hHO-2 generiert (Yoshida&Kikuchi,
1978; Maines, 1997), welches als physiologisches Signalmolekül u. a. die Aktivierung der
Guanylatzyklase bewirkt (Kharitonov et al., 1995). Die hHO-2 weist die höchste Aktivität im
Gehirn und im Hoden auf (Maines, 1997). 2009 wurde eine weitere entscheidende Funktion
der hHO-2 durch Yi und Kollegen gefunden (Yi et al., 2009). Die hHO-2 verfügt über drei
Häm regulatorische Motive (heme regulatory motifs (HRM)), die jeweils aus einem Cystein
und einem Prolin bestehen. In Abhängigkeit vom Redoxzustand der Zelle findet ein
Thiol/Disulfid Wechsel dieser HRM statt, wodurch die Affinität der HO zum Substrat Häm
beeinflusst wird. Dadurch wird die hHO-2 katalysierte Freisetzung des Signalmoleküls CO
redoxabhängig reguliert (Yi&Ragsdale, 2007).

Einleitung
10
In Pflanzen ist das durch HOs freigesetzte CO an der Auxin-induzierten
Adventivwurzelbildung und an der Eisen-Homöostase beteiligt (Guo et al., 2008; Xuan et al.,
2008).
Die Funktion des Eisens
Eisen ist ein essentieller Baustein und zugleich ein limitierender Faktor bei der Entwicklung
allen Lebens. Daher ist es für Organismen wesentlich die Eisenversorgung zu sichern. So
stellt Häm, welches z.B. beim Abbau von Hämproteinen frei wird oder aber auch durch
pathogene Bakterien vom Wirt aufgenommen wird, eine durch HOs nutzbare Eisenquelle dar.
Die bakteriellen HOs HmuO aus Corynebacterium diphtheriae und HemO aus Neisseria
meningitidis sind maßgeblich an der Eisengewinnung aus Häm und Hämoglobin beteiligt und
beugen durch den Hämabbau der Hämtoxizität vor (Schmitt, 1997a, 1997b; Zhu et al., 2000).
In opportunistisch pathogenen Bakterien, wie P. aeruginosa, wird das Häm durch PigA zur
Eisengewinnung genutzt (Zhu et al., 2000; Ratliff et al., 2001). Bei Algen, wie Rhodella
violacae, und Hefen, wie Saccharomyces cerevisiae und Candida albicans, dient der
Hämabbau durch HOs ebenfalls der Versorgung mit extrazellulärem Eisen
(Richaud&Zabulon, 1997; Kim et al., 2006).
Die Funktion des Biliverdins
BV übernimmt die meisten biologischen Funktionen von den bei der Hämspaltung gebildeten
Produkten. Dies ist vor allem dadurch begründet, dass BV das erste Molekül der Biosynthese
aller weiteren offenkettigen Tetrapyrrole darstellt.
Im Säuger wird das BV durch die unter Stressbedingungen und durch chemische Reagenzien
induzierte hHO-1 gebildet (Poss&Tonegawa, 1997a, 1997b; Otterbein&Choi, 2000) und
übernimmt zwei wesentliche Funktionen. Zum einen ist BV ein Abbauprodukt des Häms aus
Hämoglobin der gealterten Erythrozyten und zum anderen wird das durch die Hämspaltung
gebildete BV IXα weiter durch die Biliverdin-Reduktase (BvR) zum Bilirubin (BR) reduziert
(Maines&Trakshel, 1993) (Abb. 5). BV und BR wirken im Körper als physiologische
Antioxidantien und können Zell- und Gewebsschädigungen vorbeugen (Stocker et al., 1987a,
1987b; Baranano et al., 2002).
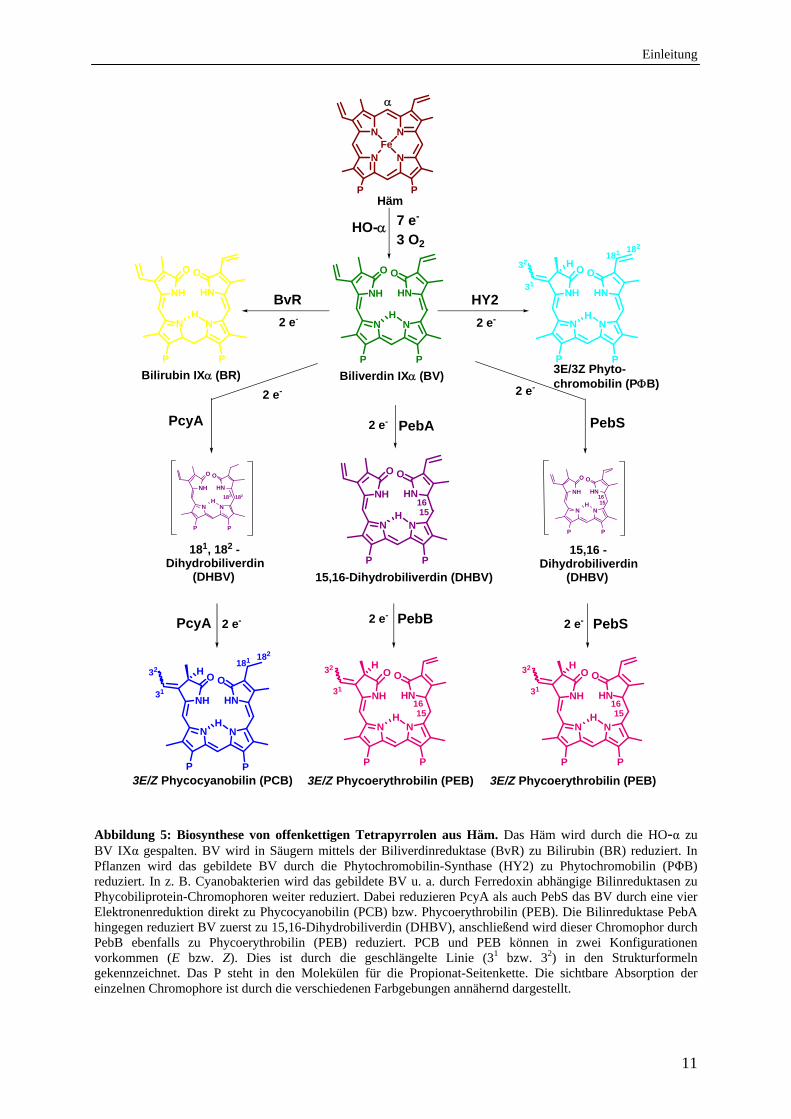
Einleitung
11
N
HNNH
O
N
PP
O
H
N
HNNH
O
N
PP
O
H1615
N
HNNH
O
N
PP
OH
H
32
31
Biliverdin IX(BV)
15,16-Dihydrobiliverdin (DHBV)
181, 182 -Dihydrobiliverdin (DHBV)
3E/Z Phycocyanobilin (PCB)
N
HNNH
O
N
PP
O
H
H
1615
182
181
N
HNNH
O
N
PP
O
H1615
N
HNNH
O
N
PP
O
H181 182
31
32
N
HNNH
O
N
PP
O
H
H
1615
31
32
2 e-
PcyA
PcyA 2 e-
2 e- PebS
PebS2 e-
PebA
PebB2 e-
2 e-
15,16 -Dihydrobiliverdin (DHBV)
BvR
2 e-N
HNNH
O
N
PP
OH
H
32
31
182
181
3E/3Z Phyto-chromobilin (PB)
2 e-
HY2
N
HNNH
O
N
PP
O
H
Bilirubin IX(BR)
N
N
N
N
P P
Fe
HO- 7 e-
3 O2
Häm
3E/Z Phycoerythrobilin (PEB) 3E/Z Phycoerythrobilin (PEB)
Abbildung 5: Biosynthese von offenkettigen Tetrapyrrolen aus Häm. Das Häm wird durch die HO-α zu BV IXα gespalten. BV wird in Säugern mittels der Biliverdinreduktase (BvR) zu Bilirubin (BR) reduziert. In Pflanzen wird das gebildete BV durch die Phytochromobilin-Synthase (HY2) zu Phytochromobilin (PΦB) reduziert. In z. B. Cyanobakterien wird das gebildete BV u. a. durch Ferredoxin abhängige Bilinreduktasen zu Phycobiliprotein-Chromophoren weiter reduziert. Dabei reduzieren PcyA als auch PebS das BV durch eine vier Elektronenreduktion direkt zu Phycocyanobilin (PCB) bzw. Phycoerythrobilin (PEB). Die Bilinreduktase PebA hingegen reduziert BV zuerst zu 15,16-Dihydrobiliverdin (DHBV), anschließend wird dieser Chromophor durch PebB ebenfalls zu Phycoerythrobilin (PEB) reduziert. PCB und PEB können in zwei Konfigurationen vorkommen (E bzw. Z). Dies ist durch die geschlängelte Linie (31 bzw. 32) in den Strukturformeln gekennzeichnet. Das P steht in den Molekülen für die Propionat-Seitenkette. Die sichtbare Absorption der einzelnen Chromophore ist durch die verschiedenen Farbgebungen annähernd dargestellt.

Einleitung
12
Aber auch in Pflanzenzellen schützt BV vor Zellschädigungen die durch oxidativen Stress,
vornehmlich durch ROS, hervorgerufen werden. Es wurde für einige pflanzliche HOs bereits
gezeigt, dass sie durch die Bildung von BV an der Beseitigung der ROS beteiligt sind.
So hat die HO1 in der Sojabohne beispielsweise eine wichtige Funktion für den antioxidativen
Schutz der Zelle gegen Cadmium-induzierten oxidativen Stress (Balestrasse et al., 2005) und
gegen durch UV-B erzeugten Stress. Dabei wird die Expression der HO1 mit zunehmendem
Stress in der Pflanze erhöht. Dadurch erfolgt letztlich ein Anstieg des Antioxidant BV in der
Pflanzenzelle, was den Schutz vor oxidativen Schaden steigert (Yannarelli et al., 2006).
In Reptilien, Fischen und Eierschalen von Vögeln dient das HO Produkt BV IXα der
Pigmentierung (Terry et al., 2002). Insekten nutzen BV IXγ zur Pigmentierung der Flügel
(Paiva-Silva et al., 2006).
In Pflanzen wird das gebildete BV IXα durch die Phytochromobilin-Synthase (HY2) zum
Phytochrom-Chromophor Phytochromobilin (PΦB) reduziert (Abb.5). PΦB wird als
Chromophor der Photorezeptoren der Phytochromfamilie verwendet (light-sensing) (Terry et
al., 1995; Frankenberg et al., 2001; Kohchi et al., 2001).
In Cyanobakterien und Algen ist das durch die HOs gebildete BV IXα die Vorstufe für die
Phycobiline, die als Chromophore in den Phycobilisomen zur photosynthetischen
Lichtsammlung (light-harvesting) beitragen (Beal, 1993) (Abb. 5).
Dabei wird in dem Cyanobakterium Anabaena sp. PCC7120 das BV IXα durch die
Phycocyanobilin: Ferredoxin Oxidoreduktase (PcyA) zu Phycocyanobilin (PCB) reduziert.
Diese Reduktion benötigt vier Elektronen (Frankenberg&Lagarias, 2003).
In den Cyanobakterien Fremyella diplosiphon (Calothrix oder Tolypothrix sp. PCC 7601) und
Synechococcus sp. WH8020 wurden neben PcyA zwei weitere Enzyme gefunden, die für die
Biosynthese von Phycoerythrobilin (PEB) benötigt werden.
Die erste Reduktion erfolgt durch die ebenfalls Ferredoxin-abhängige Bilinreduktase PebA
(15,16-Dihydrobiliverdin: Ferredoxin Oxidoreduktase). Bei dieser Reduktion werden, wie bei
der HY2, zwei Elektronen benötigt. Das Reaktionsprodukt von PebA, das 15,16-
Dihydrobiliverdin (DHBV), wird anschließend durch PebB (PEB: Ferredoxin
Oxidoreduktase) unter Verwendung zwei weiterer Elektronen zum PEB reduziert
(Dammeyer&Frankenberg-Dinkel, 2006, 2008). Interessanterweise konnte im
Prochlorococcus Myovirus P-SSM2 ein Enzym gefunden werden, das, ähnlich dem PcyA,
eine vier Elektronen-Reduktion durchführt. Dieses Enzym, die Phycoerythrobilin-Synthase
(PebS), synthetisiert das PEB direkt aus BV IX (Dammeyer et al., 2008). Alle
synthetisierten Chromophore werden anschließend kovalent an ihr jeweiliges Phycobiliprotein

Einleitung
13
gebunden. Die Bindung des Chromophors an das jeweilige apo-Protein kann autokatalytisch
erfolgen, wie es bei PΦB und dem Phytochrom vorkommt (Parks&Quail, 1991; Terry et al.,
1993) oder durch eine Lyase vermittelte Reaktion. Lyasen sind Enzyme, die den jeweiligen
Chromophor in der richtigen Konformation an das jeweilige Phycobiliprotein binden, so dass
die Lichtsammlung gewährleistet ist (Schluchter et al., 2010).
Die unterschiedliche Farbigkeit (Abb. 5) der offenkettigen Tetrapyrrolen entsteht durch die
Veränderung des konjugierten π-Elektronensystems, welches ebenfalls einen entscheidenden
Einfluss auf die Absorptionsmaxima der jeweiligen Biline hat. Darüber hinaus bewirkt die
Protonierung des basischen Pyrrolstickstoffs eine Rotverschiebung und Erhöhung des
langwelligen Absorptionsmaximums (Gossauer&Hirsch, 1974; Brandlmeier et al., 1981). Die
Konjugation der einzelnen Doppelbindung, bzw. die Delokalisation über mehrere
Pyrrolringsysteme hinweg, erfolgt über Methinbrücken zwischen den Ringen. Eine Torsion
der Ringe zueinander beeinflusst die Konjugation des Gesamtsystems. Ein Torsionswinkel Θ
von 90° um die formale Einfachbindung bewirkt eine vollständige Entkopplung der
jeweiligen Subchromophore links und rechts der Brücke. Eine Verringerung des
Torsionswinkels ruft eine graduell zunehmende Konjugation hervor, was in der bathochromen
Verschiebung der Absorptionsmaxima zu beobachten ist (Falk, 1989).
Durch all diese beschriebenen unterschiedlichsten Prozesse, in denen HOs maßgeblich
involviert sind, sind diese Enzyme von besonderer Wichtigkeit in den verschiedensten
Organismen und nehmen einen immer größer werdenden Stellenwert in der
Grundlagenforschung ein.
3.3 Strukturelle Ähnlichkeiten der Hämoxygenasen
Bis zum heutigen Zeitpunkt wurden Kristallstrukturen von sieben verschiedenen HOs und
zwei Mitgliedern der Isd-Familie bestimmt. Darunter sind die lösliche menschliche hHO-1
ohne Membranbindedomäne (Schuller et al., 1999), eine HO aus der Ratte (Sugishima et al.,
2000), drei bakterielle HOs (N. meningitidis (Schuller et al., 2001), HmuO aus C. diphteriae
(Hirotsu et al., 2003) und PigA aus P. aeruginosa (Friedman et al., 2004)) und zwei
Isoformen cyanobakterieller HOs aus Synechosystis sp. PCC6803 SyHO-1 (Sugishima et al.,
2004) sowie SyHO-2 (Sugishima et al., 2005). Aus der Isd-Familie wurden bisher IsdG und
IsdI kristallisiert und ihre Struktur bestimmt (Wu et al., 2005; Lee et al., 2008; Reniere et al.,
2010). Durch NMR Daten konnten ergänzende Informationen über die Orientierung des
Substrates Häm und des Produktes BV in den jeweiligen untersuchten HOs gewonnen werden
(Hernandez et al., 1994; Wegele et al., 2004).

Einleitung
14
Die Säuger HOs weisen mit ~ 28 kDa (ohne die Membranbindedomäne) eine etwas größere
relative molekulare Masse auf, als die bakteriellen HOs (24-26 kDa). Die Monooxygenasen
der Isd-Familie sind mit nur ~ 13 kDa ungewöhnlich klein. Hingegen sind die Strukturen der
HOs, mit Ausnahme der Proteine der Isd-Familie, aus den verschiedenen Organismen sehr
ähnlich.
Trotz der geringen Sequenzähnlichkeit der HOs aus den verschiedenen Organismen besitzen
alle bis dato kritallisierten HOs eine charakteristische α-helikale Konformation, die aus acht
rechtsgängigen α-Helices besteht (Abb. 6 (hHO-1 und PigA)). Dadurch weisen die HOs eine
kompakt gefaltete Domäne auf. Bis auf die homodimere Hämoxygenase SyHO-2 sind alle
anderen untersuchten HOs monomere Proteine. Das Hämmolekül liegt in der Substrattasche
(aktiven Tasche) der HOs, wie in einem „Sandwich“, zwischen der proximalen und der
distalen Helix.
Abbildung 6: Darstellung zweier HOs und eines Isd-Proteins als Bändermodell. Gezeigt sind die menschliche HO1 (hHO-1) (PDB code 1N45), die bakterielle HO aus P. aeruginosa (PigA) (PDB code 1SK7) und die Monooxygenase IsdI aus S. aureus (PDB code 3LGN). Das Hämmolekül sowie das neutrale Imidazol des Histidin-Restes, welches das Eisen des Häms koordiniert, sind in der „Stick“-Darstellung gezeigt. Das Hämmolekül ist mit: C= grün, N= blau, O= rot und Fe= braun, dargestellt.

Einleitung
15
Das Eisenzentralatom des Häms wird in allen HOs über einen N-terminalen konservierten
Histidin-Rest koordiniert (Kap. 3.4). Die Carboxylgruppen der Propionatseitenketten des
Häms sind an der molekularen Oberfläche exponiert. Eine weitere strukturelle Gemeinsamkeit
aller HOs besteht in der Verformung der distalen Helix. Diese weist eine ~ 50° Beugung
oberhalb der Hämoberfläche auf. Diese Beugung besteht aus einer sehr Glycin-reichen
Sequenz. Diese Aminosäuresequenz in der distalen Helix, die auch als G-X-X-X-G Motiv
bezeichnet wird, ist nur bei Säugern und Bakterien konserviert, pflanzlichen HOs und den
Monooxygenasen der Isd-Familie fehlt dieses Motiv.
Durch die Glycin-Reste ist die distale Helix sehr flexibel und äußerst wichtig für die
Hämbindung, die katalytische Aktivität des Proteins und für die Freisetzung des BV (Wilks,
2002; Colas&Ortiz de Montellano, 2003).
Anhand der Beugung der distalen Helix und der Positionierung des Häms in der
Substrattasche werden bei den HOs drei Typen von Konformationen unterschieden. Die
”offene” Konformation wurde für die menschliche hHO-1 (Schuller et al., 1999), die
”geschlossene” für HemO aus N. meningitidis (Schuller et al., 2001), HmuO aus
C. diphtheriae (Hirotsu et al., 2003) sowie PigA aus P. aeruginosa (Friedman et al., 2004)
und die ”mehr geschlossene” für HO-1 aus der Ratte (Sugishima et al., 2000) beschrieben.
Die ”offene” und die ”geschlossene” Konformation sind auf Grund der stabilisierenden
Wasserstoffbrücken zwischen einem Glycin-Rest der distalen Helix und dem
Eisenzentralatom des Häm stabiler als die ”mehr geschlossene” Konformation. Ebenfalls
weisen alle HOs an der molekularen Oberfläche in der Nähe der Substrattasche ein positives
elektrostatisches Potential auf. Diese positive Ladung ist für den Komplex mit dem
Elektronendonator von Vorteil. Die Cytochrom P450-Reduktase als physiologischer
Elektronendonator für die hHO-1 oder das reduzierte Ferredoxin für die bakteriellen HOs
weisen eine überwiegend negative Ladung an der Oberfläche auf, so dass die
Komplexbildung zwischen der HO und dem notwendigen Elektronenübertäger gewährleistet
ist (Wang et al., 1997; Sugishima et al., 2004). Ebenfalls wichtig für die Interaktion der HO
mit dem Elektronenüberträger ist ein Netzwerk aus ionischen Wechselwirkungen zwischen
den beiden Proteinen (Akashi et al., 1999).
Die Monooxygenasen der Isd-Familie sind, im Gegensatz zu den HOs, Homodimere
bestehend aus α-Helices und β-Faltblättern (Abb. 6). Sie weisen eine hohe strukturelle
Ähnlichkeit zur Monooxygenase aus Streptomyces coelicolor (Sciara et al., 2003) auf. Die
IsdI-Monomere sind jeweils C-terminal über antiparallele β-Faltblätter in einem Homodimer
organisiert. Dabei wird ein β-Fass im dimeren Zwischenraum ausgebildet, ähnlich wie bei

Einleitung
16
Proteinen der Ferredoxin-Superfamilie (Wu et al., 2005). Jede monomere Einheit bindet
jeweils ein Hämmolekül, welches, ähnlich wie bei den HOs, über einen Histidin-Rest
koordiniert wird.
3.4 Substratbindetasche der Hämoxygenasen
Auf Grund der unterschiedlichen Regiospezifität der HOs werden im folgenden Kapitel eine
für den α-meso-Kohlenstoff spezifische menschliche HO (hHO-1) mit einer für den δ- und β-
meso-Kohlenstoff spezifischen HO (PigA aus P. aeruginosa) genauer miteinander verglichen,
um die Regiospezifität der Enzyme zu erklären (Abb. 7).
Wie in Kapitel 3.3 beschrieben befindet sich das Häm in der aktiven Tasche zwischen distaler
und proximaler Helix. In allen bekannten HOs wird das Eisenzentralatom des Häms über die
neutrale Imidazolgruppe eines konservierten Histidin-Restes der proximalen Helix
koordiniert. Dieses wurde mittels Röntgenstrukturanalyse, elektroparamagnetischen
Resonanz-Messungen (EPR), Resonanz-Raman-Messungen, Kernspinresonanz-Spektroskopie
(Nuclear Magnetic Resonance (NMR)) und Mutagenesestudien untersucht (Wilks et al., 1995;
Chu et al., 1999). Durch die Koordination des konservierten Histidin-Restes liegt das Eisen in
den HOs als fünf-fach koordiniertes Eisen im Grundzustand vor. Der sechste
Koordinationspartner des Eisenszentralatoms des Häm ist häufig ein diatomisches Gas
(vorzugsweise O2) (Sugishima et al., 2002) oder aber ein Wassermolekül, welches sich in der
aktiven Tasche des Proteins befindet (Sugishima et al., 2000). Die Orientierung des
Imidazolrings des Histidin-Restes zum Häm ist sowohl bei hHO-1, als auch PigA dieselbe.
Der Abstand zwischen dem Stickstoff des Imidazolrings vom Histidin-Rest zum
Eisenzentralatom des Häms kann annähernd als halber Vektor der N-Fe-N Bindung im
Pyrrolring bezeichnet werden (Friedman et al., 2004). Ein Netzwerk aus
Wasserstoffbrückenbindungen, das innerhalb der distalen Substrattasche zwischen den
Seitenketten des Proteins und des umgebenden Solvens vorhanden ist, gewährleistet eine
angemessene Protonenabgabe von Wassermolekülen zum distalen Sauerstoff-Atom des
Eisen-gebundenen O2-Liganden. Das starre Netzwerk der über die Wasserstoffe des Proteins
gebundenen Wassermoleküle stabilisiert darüber hinaus die aktive Ferrihydroperoxy-Spezie
[(Fe3+)-O-OH] in der aktiven HO. Diese Gemeinsamkeit des über Wasserstoffbindungen
gebundenen Wassers in der Substrattasche weisen alle bisher strukturell charakterisierten HOs
auf, was auf den gleichen katalytischen Mechanismus aller HOs hindeutet.

Einleitung
17
Abbildung 7: Substrattasche der menschlichen hHO-1 sowie der PigA aus P. aeruginosa. Die Darstellung und Farbgebung entspricht der aus Abb. 6. Das Häm befindet sich zwischen der proximalen- und distalen Helix. Die entscheidenden meso-Kohlenstoffatome des Häms sind bezeichnet.
Die Orientierung des Häms in der aktiven Tasche der jeweiligen HOs ist allerdings recht
unterschiedlich. Das Häm kann z. B. in der hHO-1 durch eine Drehung (Spiegelung) entlang
der α/γ-meso-Achse um 180° in zwei Orientierungen in der Substrattasche vorliegen
(Hernandez et al., 1994). Die Reaktionsprodukte unterscheiden sich dabei allerdings nicht. Es
wird das BV IXα Isomer gebildet. In PigA hingegen ist das Häm horizontal um ~ 110°, im
Vergleich zur hHO-1, in der Substrattasche gedreht (Abb. 7).
Durch diese Rotation ist der δ-meso-Kohlenstoff des Häms näher an den in der Hämspaltung
involvierten Aminosäure-Reste positioniert, wodurch die Regiospezifität für das BV IXδ
Isomer zu erklären ist. Die für die Hämspaltung notwendigen Aminosäuren der HOs befinden
sich im hinteren Teil der Substrattasche. Dieser strukturelle Unterschied resultiert in
verschiedenen Interaktionen zwischen dem Protein und den Propionatseitenketten des Häms.
Durch Mutationen in der distalen Helix der hHO-1 konnte diese für den α-meso-Kohlenstoff
spezifische HO in eine δ-meso-Kohlenstoff spezifische HO umgewandelt werden. Dabei ist
das Häm, wie in der Hämoxygenase PigA, rotiert (Zhou et al., 2000).

Einleitung
18
Ebenfalls konnten durch die PigA Kristallstruktur und durch gezielte Punktmutationen in
diesem Protein die für die Regiospezifität essentiellen Aminosäure-Reste ausfindig gemacht
werden (Fujii et al., 2004). Durch zwei Lysin-Reste und ein Phenylalanin-Rest ist das Häm in
der PigA so spezifisch koordiniert, dass das Häm an dem δ-meso-Kohlenstoff gespalten wird
(Fujii et al., 2004). Anhand der Substrattasche der PigA ist allerdings auch gut zu erkennen,
dass eine Drehung des Häms entlang der α/γ-meso-Achse um ~ 180° den β-meso-Kohlenstoff
in den hinteren Teil der aktiven Tasche kommen lässt, wodurch das Häm dann zum BV IXβ
Isomer gespalten werden kann. Diese Beobachtungen wurden auch bei in vitro Experimenten
konstatiert (Ratliff et al., 2001; Wegele et al., 2004). Dabei entsteht bei der Hämspaltung
durch PigA sowohl BV IXδ als auch BV IXβ im Verhältnis 70:30 (Caignan et al., 2002).
3.5 Reaktionsmechanismus der Hämoxygenasen
Obwohl die HOs in sehr verschiedenen Organismen vorkommen und die Hämabbauprodukte
eine sehr unterschiedliche Funktion erfüllen, ist den HOs aus diversen Organismen (Säuger,
Pflanzen und Bakterien) auf Grund hoher struktureller Ähnlichkeiten derselbe
Reaktionsmechanismus der Hämspaltung abzuleiten. Die Monooxygenasen IsdG und IsdI der
Isd-Familie stellen dabei eine Ausnahme dar. Dieser Reaktionsmechanismus wurde noch
nicht untersucht.
Der Reaktionsmechanismus der HOs konnte in den letzen Jahren anhand von verschiedenen
Kristallstrukturen sowie durch UV/vis-Spektroskopie, EPR- und Resonanz-Raman-
Messungen detailliert verfolgt und aufgeklärt werden. Eine der ersten kristallisierten (Abb. 6)
und am besten biochemisch charakterisierten HOs ist die hHO-1 aus dem Menschen an der
die Reaktion des Hämabbaus im Folgenden beschrieben wird.
Die benötigten Elektronen für die Hämspaltung werden von allen aktiven Säuger HOs in vivo
und von Bakterien in vitro vom NADPH (Nikotinamid-adenin-dinukleotid-hydrophosphat)
und der NADPH-Cytochrom P450-Reduktase, die als Elektronendonator fungiert, bezogen
(Yoshida&Kikuchi, 1978; Maines, 1988; Sono et al., 1996; Zhu et al., 2000; Ratliff et al.,
2001). In dem opportunistisch pathogenen Bakterium P. aeruginosa wurde ein der
Ferredoxin-NADPH-Oxidoreduktase (FNR) ähnliches Protein (FPR: Ferredoxin-NADP+-
Reduktase) gefunden, was als natürlicher Elektronendonator fungiert (Wang et al., 2007). In
photosynthtetischen Organismen (einschließlich photosynthetischen Bakterien) konnte durch
in vitro Experimente reduziertes Ferredoxin als Elektronendonator nachgewiesen werden
(Cornejo et al., 1998; Muramoto et al., 2002). Interessanterweise verläuft die Katalyse in
Anwesenheit eines zweiten, zusätzlichen Reduktionsmittels, wie Ascorbat oder Trolox, bei
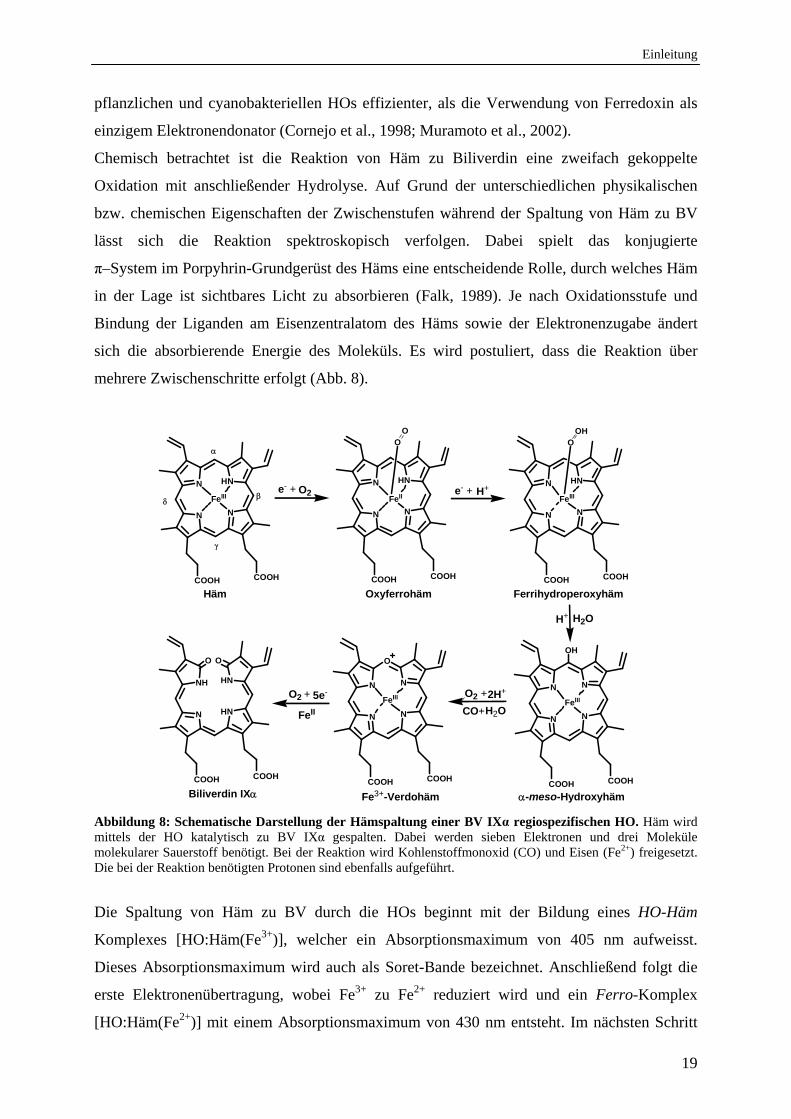
Einleitung
19
pflanzlichen und cyanobakteriellen HOs effizienter, als die Verwendung von Ferredoxin als
einzigem Elektronendonator (Cornejo et al., 1998; Muramoto et al., 2002).
Chemisch betrachtet ist die Reaktion von Häm zu Biliverdin eine zweifach gekoppelte
Oxidation mit anschließender Hydrolyse. Auf Grund der unterschiedlichen physikalischen
bzw. chemischen Eigenschaften der Zwischenstufen während der Spaltung von Häm zu BV
lässt sich die Reaktion spektroskopisch verfolgen. Dabei spielt das konjugierte
π–System im Porpyhrin-Grundgerüst des Häms eine entscheidende Rolle, durch welches Häm
in der Lage ist sichtbares Licht zu absorbieren (Falk, 1989). Je nach Oxidationsstufe und
Bindung der Liganden am Eisenzentralatom des Häms sowie der Elektronenzugabe ändert
sich die absorbierende Energie des Moleküls. Es wird postuliert, dass die Reaktion über
mehrere Zwischenschritte erfolgt (Abb. 8).
O
N HN
N N
FeIII
COOHCOOH
N N
N N
FeIII
COOHCOOH
OH
O2
-meso-Hydroxyhäm
N
O
N
N N
FeIII
COOHCOOH
NH HN
N HN
COOHCOOH
Fe3+-Verdohäm
O2 +
FeII
O
Biliverdin IX
N HN
N N
FeII
COOHCOOH
O2 +
OO
e- +N HN
N N
FeIII
COOHCOOH
e- +
CO+
2H+
H2O
5e-
H+
Häm Oxyferrohäm Ferrihydroperoxyhäm
OOH
H+ H2O
Abbildung 8: Schematische Darstellung der Hämspaltung einer BV IXα regiospezifischen HO. Häm wird mittels der HO katalytisch zu BV IXα gespalten. Dabei werden sieben Elektronen und drei Moleküle molekularer Sauerstoff benötigt. Bei der Reaktion wird Kohlenstoffmonoxid (CO) und Eisen (Fe2+) freigesetzt. Die bei der Reaktion benötigten Protonen sind ebenfalls aufgeführt.
Die Spaltung von Häm zu BV durch die HOs beginnt mit der Bildung eines HO-Häm
Komplexes [HO:Häm(Fe3+)], welcher ein Absorptionsmaximum von 405 nm aufweisst.
Dieses Absorptionsmaximum wird auch als Soret-Bande bezeichnet. Anschließend folgt die
erste Elektronenübertragung, wobei Fe3+ zu Fe2+ reduziert wird und ein Ferro-Komplex
[HO:Häm(Fe2+)] mit einem Absorptionsmaximum von 430 nm entsteht. Im nächsten Schritt

Einleitung
20
erfolgt die schnelle Bindung von molekularem Sauerstoff an das reduzierte Eisen des Ferro-
Komplexes. Es entsteht ein metastabiler Oxyferro-Komplex [HO:Häm(Fe2+)-O2], der ein
Absorptionsmaximun von 410 nm besitzt (Yoshida&Kikuchi, 1978; Yoshida et al., 1980).
Der Oxyferro-Komplex besitzt ebenfalls zwei weitere Absorptionsmaxima bei 540 nm und
569 nm, die als Q-Banden bezeichnet werden. Durch eine weitere Ein-Elektronen-Reduktion
und einer Protonierung (das Proton stammt aus dem Wasser der Substrattasche, welches sich
in der Nähe der distalen Helix befindet) entsteht ein Ferrihydroperoxy-Komplex
[HO:Häm(Fe3+)-O-OH] (Wilks&Ortiz de Montellano, 1993; Davydov et al., 2002).
Anschließend bindet das terminale Sauerstoff-Atom des Hydroperoxid-Intermediates an das
α-meso-Kohlenstoffatom des Porphyrinrings und es entsteht das α-meso-Hydroxyhäm
(Wilks&Ortiz de Montellano, 1993). Das Eisenatom des gebildeten α-meso-Hydroxyhäms
befindet sich hierbei in einem fünf-fach koordinierten high-spin Zustand und liegt als
Oxophlorin-Resonanz vor, die auch als Fe2+-Porphyrin π-neutrales-Radikal bezeichnet wird
(Matera et al., 1996).
Im nächsten Schritt der enzymatischen Spaltung des Häms wird, ausgehend vom α-meso-
Hydroxyhäm, Verdohäm gebildet, wobei Kohlenstoffmonoxid (CO) freigesetzt wird. Dieser
Schritt benötigt ebenfalls molekularen Sauerstoff und zwei Protonen. Über den
Reaktionsmechanismus in diesem Schritt gibt es verschiedene Theorien. So wird zum einen
postuliert, dass ein O2-Molekül ausreicht, um das Fe3+-Verdohäm zu bilden und darauf
folgend ein Elektron zur Reduktion des Fe3+- zu Fe2+-Verdohäm benötigt wird (Liu et al.,
1997). Demgegenüber wird davon ausgegangen, dass sowohl das O2-Molekül als auch ein
Elektron gleichzeitig bei dieser Reaktion vorhanden sein müssen (Matera et al., 1996). Eine
weitere Hypothese ist, dass das O2-Molekül alleine ausreicht, um das Fe2+-Verdohäm zu
bilden (Sakamoto et al., 1999). Einigkeit besteht darin, dass das O2-Molekül schneller mit
dem Porphyrin Makrozyklus reagiert, als mit dem Eisenzentralatom. Des Weiteren ist
bekannt, dass diese Stufe im Hämabbau nicht durch frei werdendes CO inhibiert werden kann
und dass hierbei nun alle vier Vorstufen der BV Isomere entstehen können (Zhang et al.,
2003). Der nun gebildete Ferro-Verdohäm Komplex liegt in den HOs als sechs-fach
koordinierter high-spin Zustand mit einem proximalen Imidazol Liganden (Histidin-Rest) am
Protein und als axial koordiniertes Hydroxid vor (Takahashi et al., 1997). Die positive Ladung
des Verdohäm-Rings lässt einen partiellen Fe3+ Charakter im Fe2+-Verdohäm-HO Komplex
zu, wodurch Liganden, die sonst nur im Fe3+ Oxidationszustand gebunden werden können
(z. B. Cyanide und Azide) binden und die weitere Reaktion der HO inhibieren können. Das
Absorptionsmaximum von Verdohäm liegt bei 630 nm. Der letzte Schritt vom Verdohäm zum

Einleitung
21
Biliverdin ist noch nicht sehr ausführlich untersucht worden. In Anwesenheit von
molekularem Sauerstoff und Elektronen entsteht Eisen-Biliverdin [(Fe3+)-BV], das ein
Absorptionsmaximum von 670 nm besitzt. Es wird davon ausgegangen, dass der
Verdohämring durch Oxidation zu BV geöffnet wird. Dem zu Folge ist ein hydrolytischer
Mechanismus für die Ringöffnung ausgeschlossen. Für die Freisetzung des BVs muss Fe3+
aus dem Eisen-Biliverdin-Komplex zu Fe2+ reduziert werden (Yoshida et al., 1980). Die
Freisetzung des BVs ist der bisher am wenigsten charakterisierte, aber auch gleichzeitig der
Geschwindigkeitsbestimmende Schritt dieser Reaktion (Liu&Ortiz de Montellano, 2000). In
vitro Experimente mit der Säuger hHO-1 haben gezeigt, dass durch Zugabe von BvR die
Reaktionsgeschwindigkeit des Hämabbaus gesteigert und dadurch eine schnellere Freisetzung
des BVs aus der HO gewährleistet wird (Maines&Trakshel, 1993).
Dabei ist der Reaktionsmechanismus aller Hämproteine (z. B. Katalasen) bis zum Oxyferro-
Komplex [HO:Häm(Fe2+)-O2] identisch. Erst an dieser Stelle des Reaktionsmechanismus
unterscheiden sich die HOs im Vergleich zu anderen Hämproteinen (z. B. Katalasen)
(Alfonso-Prieto et al., 2009). Dieser Oxyferro-Komplex ist für die darauf folgende
Hydroxylierung des α-meso-Hydroxyhäms ein kritischer Zustand. Kommt zu diesem
metastabilen Oxyferro-Komplex [HO:Häm(Fe2+)-O2] ein weiteres Elektron hinzu, so entsteht
daraus ein aktives Peroxidintermediat. Die HOs spalten die [O-O]-Bindung im Oxyferro-
Komplex heterolytisch, wobei ein Ferryl-Intermediat [(Fe-O)3+] entsteht, welches schneller
gebildet wird, als das aktive Peroxidintermediat. Würde bei der Reaktion Wasserstoffperoxid
(H2O2) entstehen und gleichzeitig Sauerstoff anwesend sein, so würde das α-meso-
Hydroxyhäm sofort gebildet und zu Verdohäm umgewandelt werden. H2O2 ist äquivalent zu
einem Molekül O2 und zwei Elektronen in der Reaktion von Häm zu Verdohäm, aber es kann
nicht die weitere Reaktion von Verdohäm zu Biliverdin katalysieren. Darüber hinaus kann es
bei einem Überschuss an H2O2 dazu kommen, dass das Häm unspezifisch gespalten wird und
Mono- oder Dipyrrole entstehen (Wilks&Ortiz de Montellano, 1993). Um zu gewährleisten,
dass die in vitro untersuchten Proteine tatsächlich aktive HOs mit katalytischer Aktivität sind,
ist es essentiell Katalasen in die Reaktionsansätze hinzuzugeben. Damit kann sichergestellt
werden, dass die Umsetzung des Häms zu BV nicht chemisch (durch evtl. entstandenes
H2O2), sondern enzymatisch geschieht. Durch Zugabe von Katalase wird H2O2 zu H2O und O2
umgesetzt und das Häm kann nicht unspezifisch gespalten werden.

Einleitung
22
3.6 Hämoxygenasen aus Pseudomonas aeruginosa
Pseudomonas aeruginosa ist ein Gram-negatives opportunistisch pathogenes Bakterium, das
in den unterschiedlichsten Habitaten zu finden ist. Dieses Bakterium kann verschiedene
Infektionen auslösen, speziell bei immunsupprimierten Personen (Gordon et al., 1998). Eine
der schwerwiegendsten Infektionen, die durch P. aeruginosa verursacht werden, ist eine
Lungenentzündung bei Patienten mit zystischer Fibrose (Heilesen et al., 1983). Damit sich das
Bakterium im Schleim der erkrankten Patienten ansiedeln kann, produziert es eine Vielzahl an
Virulenzfaktoren. Zu diesen Faktoren kann auch die Hämoxygenase PigA gezählt werden
(Ratliff et al., 2001). Das pigA Gen (Pseudomonas iron induced gene) wurde bei der Suche
nach Eisen-regulierten Genen in einem Gencluster mit vier weiteren Genen, pigA-E,
(Ochsner&Vasil, 1996) gefunden. Es konnte gezeigt werden, dass PigA an der Hämspaltung
von extrazellulärem Häm beteiligt ist (Bhakta&Wilks, 2006). Die Hauptaufgabe der PigA
liegt daher wahrscheinlich in der Eisengewinnung aus dem Häm des Wirtsorganismus unter
Eisen-limitierten Bedingungen. PigA aus P. aeruginosa spaltet das Häm in vitro an der
δ- sowie β-meso-Kohlenstoffbrücke (Ratliff et al., 2001), wodurch die BV Isomere BV IXδ
und BV IXβ im Verhältnis 70:30 entstehen (Caignan et al., 2002). Die Funktion dieser
gebildeten Isomere ist noch unklar.
Interessanterweise besitzt P. aeruginosa neben PigA eine zweite HO, die BphO. Somit ist
P. aeruginosa das bis heute einzig bekannte Bakterium, das zwei regiospezifisch
unterschiedliche HOs besitzt. BphO ist eine „klassische“ HO, die für den α-meso-Kohlenstoff
spezifisch ist und das Häm zu BV IXα spaltet (Wegele et al., 2004). Das für diese HO
kodierende Gen bphO ist mit einem für ein bakterielles Phytochrom kodierenden Gen bphP
(bacterial phytochrome) stromaufwärts in einem Operon organisiert und wird zusammen
transkribiert (Tasler et al., 2005; Barkovits et al., 2008). Biochemische Analysen konnten
zeigen, dass BphO durch die Spaltung von Häm zu BV IXα den natürlichen Chromophor für
das bakterielle Phytochrom, einem putativen Rotlichtsensor, liefert (Wegele et al., 2004).
Daher wird diese HO in die Familie der bakteriellen Phytochrom Hämoxygenasen (BphO,
Bacterial phytochrome heme oxygenase) eingeordnet und bildet zusammen mit BphP eine
physiologisch funktionelle Einheit, das Phytochrom-System. Zusammenfassend kann die
Funktion der HOs der PigA-Familie aus pathogenen Bakterien als katabolisch beschrieben
werden. Diese HOs bauen das Häm ab, um den Organismus mit Eisen zu versorgen. Die HOs
der BphO-Familie weisen hingegen eine anabole Funktion auf. Ihre Hämspaltung dient der
Phytochrom-Chromophorsynthese.

Einleitung
23
3.7 Hämoxygenasen aus Arabidopsis thaliana
Arabidopsis thaliana, Acker-Schmalwand oder auch Schotenkresse genannt, ist ein einjährig
blühendes Wildkraut aus der Familie der Kreuzblütler (Brassicaceae) (Laibach, 1907). Sie
weist eine hohe Fruchtbarkeit auf, besitzt einen kurzen Generationszyklus von nur acht
Wochen (von der Keimung des Samens bis zur Reife der Samen) und ist bei einer geringen
Raumbeanspruchung leicht zu kultivieren (Laibach, 1943). Im Jahre 2000 wurde die
vollständige Nukleotidsequenz des Genoms analysiert (The-Arabidopsis-Genome-Initiative,
2000). Das fünf Chromosomen umfassende Genom, welches ~ 28.000 Gene aufweist, ist
heute ausführlich kartiert. Obwohl A. thaliana keine große wirtschaftliche Bedeutung
zukommt, können auf Grund der engen Verwandtschaft mit Kulturarten, wie Kohl (Brassica
oleracea) oder Raps (Brassica napus), die für Arabidopsis erarbeiteten Informationen über
funktionale Zusammenhänge zwischen Genotyp, Umwelt und Phänotyp auf Nutzpflanzen
übertragen werden.
A. thaliana war die erste höhere Pflanze in der eine Hämoxygenase (HY1) beschrieben wurde
(Davis et al., 1999; Muramoto et al., 1999). Eine HO defiziente hy1 Mutante von Arabidopsis
wurde ursprünglich durch einen genetischen screen von Mutanten mit langen Hypocotylen in
weißem Licht entdeckt, die keine Veränderung bei einem Lichtwechsel von hell- zu dunkel-
rotem Licht zeigten (Koornneef et al., 1980). Es stellte sich jedoch heraus, dass diese Pflanze
drei weitere kodierende Gene (HO2, HO3 und HO4) für HOs besitzt (Davis et al., 2001; Terry
et al., 2002). A. thaliana ist der erste bekannte Organismus, der vier putative HOs (HY1,
HO2, HO3 und HO4) besitzt. Die HOs sind alle im Zellkern kodiert und haben eine
N-terminale Transit-Peptid-Lokalisationssequenz für Plastiden (Davis et al., 2001). Die
Lokalisation für HY1 im Stroma des Chloroplasten konnte durch Verwendung des grün
fluoreszierenden Proteins (GFP) und Immunoblot Untersuchungen demonstriert werden
(Muramoto et al., 1999).
Die HOs aus A. thaliana können in zwei Unterfamilien eingeteilt werden (HO1- und HO2-
Unterfamilie) (Davis et al., 2001). Die HO1-Unterfamilie besteht aus HY1, HO3 und HO4. In
die HO2-Unterfamile wird nur die HO2 gefasst. Diese Unterscheidungen wurden auf Grund
der Aminosäuresequenzen der einzelnen HOs getroffen (Teil D Kap. 1.3). Die HO2-
Unterfamilie weist einen Bereich reich an kleinen Aminosäure-Resten, wie Glycin, Alanin,
Valin und Serin auf (Spacer-Region), gefolgt von einer weiteren auffälligen Region, die reich
an Glutamat- und Aspartat-Resten ist. Eine zusätzliche Unterscheidung ergibt sich aus der
potentiellen Koordination des Substrats Häm im Protein. Genau wie bei bakteriellen oder
Säuger HOs weisen alle Mitglieder der HO1-Unterfamilie aus Arabidopsis zur Koordination

Einleitung
24
des Eisenzentralatoms des Häms ein Histidin-Rest auf, hingegen besitzt die HO2 an dieser
Stelle einen Arginin-Rest (Abb. 10). Der Austausch des Histidin-Restes zu einem Arginin-
Rest ist für die Enzymaktivität bei den Säuger-orthologen HOs von großer Bedeutung. Diese
sind durch den Aminosäureaustausch nicht mehr in der Lage das Häm zu binden und
umszusetzen (Davis et al., 2001; Ortiz de Montellano&Wilks, 2001).
Durch einen Aminosäuresequenzvergleich der Arabidopsis HY1 zu den anderen drei HOs
konnten folgende Homologien bestimmt werden. Die HY1 weist mit 85 % Identität und 91 %
Ähnlichkeit die höchste Homologie zur HO3 auf. Die Homologien der HY1 zur HO4 mit
69 % Identität und 78 % Ähnlichkeit sowie zur HO2 mit 43 % Identität und 66 % Ähnlichkeit
sind deutlich niedrigerer.
Biochemisch charakterisiert wurde bisher nur HY1. Es konnte gezeigt werden, dass HY1 in
vitro Häm zu BV IXα, CO und Eisen umsetzt und ähnliche biochemische Parameter anderer
untersuchter HOs zeigt. Das Protein ist Ferredoxin-abhängig, und die Aktivität kann durch
Zugabe von Ascorbat um das zehn-fache gesteigert werden. Auch benötigt das Protein in vitro
einen Eisenchelator um das gebildete BV freizusetzen (Muramoto et al., 2002). Dieses wurde
bisher auch für bakterielle HOs gezeigt (Wegele et al., 2004). Dass die HY1 Ferredoxin-
abhängig ist, konnte biochemisch durch Zugabe von isolierten Spinat-Thylakoiden in vitro
demonstriert werden (Muramoto et al., 2002).
Für alle vier putativen Arabidopsis HOs (HY1, HO2, HO3 und HO4) wird eine Beteiligung in
der Biosynthese von Phytochrom-Chromophoren postuliert (Emborg et al., 2006). Dabei
scheinen HY1 und HO2 einen deutlich größeren Einfluss auf die Synthese von BV zu haben
als HO3 und HO4 (Davis et al., 1999; Terry&Kendrick, 1999; Emborg et al., 2006). HY1 ist
die dominante Isoform für die holo-Phytochromassemblierung, aber HO3 und HO4 in
Abwesenheit von HY1 scheinen ebenfalls wichtig dafür zu sein (Emborg et al., 2006). Die
genauen Funktionen, vor allem von HO2, die einen leichten Phänotyp in der ho2 Pflanze zeigt
(Teil D Kap. 1.3) und von HO3 und HO4, sind noch unklar. Es stellt sich somit die Frage,
warum A. thaliana über vier HOs verfügt.

Aufgabenstellung
25
B Aufgabenstellung
Die Ziele dieser Arbeit gliederten sich in drei verschiedene Hauptprojekte.
Das erste Ziel bestand in der biochemischen Charakterisierung der bis dato noch nicht
charakterisierten drei putativen HOs (HO2, HO3 und HO4) aus Arabidopsis thaliana im
Vergleich zur HY1. Durch UV/vis-spektroskopische Untersuchungen sollten u. a. die
Aktivität und die Verwendung des natürlichen Elektronendonators nachgewiesen werden
sowie die HOs mittels enzymologischer Parameter detailiert beschrieben werden. Das
jeweilige Reaktionsprodukt der verschiedenen HOs sollte mittels HPLC (high preasure liquid
chromatogrphy) charakterisiert werden. Durch Gelfiltrationsexperimente sollte der
Oligiomerisierungsgrad der Proteine untersucht werden. Die Strukturaufklärung einer aktiven
pflanzlichen HO sollte mit Hilfe der Röntgenstrukturanalyse und Kernspinresonanz-
spektroskopie (Nuclear Magnetic Resonance (NMR)) geschehen. Ebenfalls sollten durch die
NMR die an der Hämbindung beteiligten Aminosäure-Reste ausfindig gemacht werden.
Durch den Einsatz von Flash-Photolyse- und FTIR-Techniken (Fourier-Transformation
Infrarot-Spektroskopie) sollten die Bindungseigenschaften der pflanzlichen HOs zum
Kohlenstoffmonoxid (CO) untersucht werden.
Im zweiten Teilprojekt sollte geklärt werden, ob Pseudomonas aeruginosa der einzige
beschriebene Organismus ist, der zwei aktive HOs (BphO und PigA) mit verschiedener
Regiospezifität besitzt oder ob auch andere Pseudomonaden zwei regiospezifisch
unterschiedliche HOs aufweisen. Dazu sollten HOs verschiedener Pseudomonas Spezies
biochemisch charakterisiert und ihre jeweiligen Reaktionsprodukte analysiert werden.
Das dritte Ziel dieser Arbeit bestand im erstmaligen Einbau und der Charakterisierung eines
Metalloporphyrins (Eisencorrol (Fe-CH(cor)) in HOs. Dazu sollten zwei verschiedene
regioisomere Eisencorrole, anstatt des Häms, in die aktiven Arabidopsis HOs, als auch in
beide regiospezifisch unterschiedlichen HOs aus P. aeruginosa eingebaut und auf ihre
Aktivitäten hin untersucht werden. Dabei sollten die oben bereits aufgeführten Methoden
verwendet werden. Der Einbau der Eisencorrole in die verschiedenen HOs sollte der weiteren
Charakterisierung der Proteine dienen und einen ersten Ansatz über Eisencorrole in HOs für
zukünftige biotechnologische Anwendungen liefern.

Publikationen/Manuskripte
26
C Publikationen/Manuskripte
1. Characterization of the haem oxygenase protein family in Arabidopsis
thaliana reveals a diversity of functions
Gisk, B., Yasui, Y., Kohchi, T. & Frankenberg-Dinkel, N. (2010)
Biochemical Journal 425:425-434
2. Heme oxygenases from Arabidopsis thaliana reveal different mechanisms
of carbon monoxide binding
Gisk, B., Frankenberg-Dinkel, N. & Koetting, C.
(Manuskript bei “Biochemical and Biophysical Research Communications” eingereicht)
3. Enzymatic ring opening of an iron corrole by plant-type
heme oxygenases: Unexpected substrate and protein selectivities
Gisk, B., Brégier, F., Krueger, R. A., Broering, M. & Frankenberg-Dinkel, N.
(Manuskript bei “Biochemistry” in Revision)
4. The existence of two heme oxygenases with different regiospecificities is a
characteristic trait of pathogenic and non-pathogenic Pseudomonas
species
Gisk, B., Aras, M., Wiethaus, J. & Frankenberg-Dinkel, N.
(Manuskript in Vorbereitung)

Publikationen/Manuskripte
27
1. Characterization of the haem oxygenase protein family
in Arabidopsis thaliana reveals a diversity of functions
Gisk, B., Yasui, Y., Kohchi, T. & Frankenberg-Dinkel, N. (2010)
Biochemical Journal 425: 425-434

Biochem. J. (2010) 425, 425–434 (Printed in Great Britain) doi:10.1042/BJ20090775 425
Characterization of the haem oxygenase protein family in Arabidopsisthaliana reveals a diversity of functionsBjoern GISK*, Yukiko YASUI†, Takayuki KOHCHI† and Nicole FRANKENBERG-DINKEL*1
*Physiology of Microorganisms, Ruhr-University Bochum, Universitaetsstr. 150, 44780 Bochum, Germany, and †Graduate School of Biostudies, Kyoto University,Kyoto 606-8502, Japan
HOs (haem oxygenases) catalyse the oxidative cleavage of haemto BV (biliverdin), iron and carbon monoxide. In plants, theproduct of the reaction is BV IXα, the precursor of the PHY(phytochrome) chromophore and is thus essential for properphotomorphogenesis. Arabidopsis thaliana contains one majorbiochemically characterized HO (HY1) and three additionalputative HOs (HO2, HO3 and HO4). All four proteins are encodedin the nucleus but contain chloroplast translocation sequencesat their N-termini. The transit peptides of all four proteins aresufficient for chloroplast translocalization as shown by GFP(green fluorescent protein) reporter gene fusions. Overall, all fourproteins can be divided into two subfamilies: HO1 and HO2.Here we show that all members of the HO1 subfamily (HY1,HO3 and HO4) are active monomeric HOs and can convert haemto BV IXα using spinach Fd (ferredoxin) as an electron donor.Addition of a second electron donor, such as ascorbate, led to
a 10-fold increase in the haem conversion rate. Furthermore,haem turnover is also promoted by light when spinach thylakoidsare present. All HO1 family members displayed similar kineticparameters indicating they all have a possible involvement in PHYchromophore biosynthesis. HO2 did not yield sufficient amountsof soluble protein and therefore required the construction of asynthetic gene adapted to the codon usage of Escherichia coli.HO2 is unable to bind or degrade haem and therefore it is not ahaem oxygenase. However, HO2 shows strong binding of proto IX(protoporphyrin IX), a precursor for both haem and chlorophyllbiosynthesis. A possible function of HO2 in the regulation oftetrapyrrole metabolism is discussed.
Key words: Arabidopsis thaliana, biliverdin (BV), haemoxygenase (HO), HO2, protoporphyrin IX (proto IX).
INTRODUCTION
Light is one of the most essential factors for plant growth anddevelopment. In order to sense light, a variety of photoreceptorsspanning nearly the entire visible and UV region of the spectrumhave evolved. Sensing of red/far-red light is mediated via PHYs(phytochromes) which are able to photoisomerize between twostable conformations, the red-light-absorbing Pr-form and the far-red-light-absorbing Pfr-form. This photoconversion between thetwo spectrally distinct forms is mediated by a covalently boundopen-chain tetrapyrrole (bilin), which is PB (phytochromobilin)in plants. In Arabidopsis thaliana the PHY superfamily consistsof five members (PHYA–E) which have different functions ingrowth and development [1].
The biosynthesis of the apoPHY occurs in the cytosol, whereasthe biosynthesis of PB takes place in the chloroplast [1]. Anearly precursor of PB is proto IX (protoporphyrin IX), which islocated at the branch point of haem and chlorophyll biosynthesis[2]. At this point Mg2+ is inserted by magnesium chelatase toultimately yield chlorophyll, whereas ferrochelatase inserts Fe2+
to generate haem, the direct precursor of all bilins. Cleavageof haem by HO (haem oxygenase; E.C.1.14.99.3) subsequentlyyields the first open-chain product BV (biliverdin) IXα. Haemdegradation requires three molecules of molecular oxygen andseven electrons and results in formation of BV IXα, carbonmonoxide and Fe2+ via the intermediates α-meso-hydroxyhaem,α-verdohaem and the iron(III)–BV IXα complex (Figure 1) [3].BV IXα is subsequently reduced by PB synthase HY2, a
member of the Fd (ferredoxin)-dependent bilin reductases, toyield 3Z-PB [4,5]. Next, PB is transported out of the plastidand assembles with the apoPHY in an autocatalytic reaction toform functional holoPHY. 3Z-PB is probably isomerized to the3E-isomer prior to the assembly with apoPHY in the cytosol.Whether the 3Z- or the 3E-isomer (or both) is transported out ofthe chloroplast is currently unknown. Whereas there is a singlegene encoding PB synthase in Arabidopsis thaliana [4], fourputative HO genes have been identified [6].
The four HOs of Arabidopsis cluster into two subfamilies: theHO1 subfamily and the HO2 subfamily. Whereas HY1 (HO1),HO3 and HO4 belong to the HO1 subfamily, HO2 is the onlymember of the HO2 subfamily. Both HO subfamilies are alsopresent in other plant species, such as soybean, tomato, sorghum,rice and pine [6,7]. The main difference between these twosubfamilies is an inserted spacer sequence in the HO2 subfamily,of approx. 34–55 amino acid residues, which is rich in glutamate,aspartate and glycine residues. Furthermore, all members of theHO2 family lack the conserved histidine residue in the activesite, which is involved in haem–iron co-ordination in the HO1family [6]. However, phenotypic analysis of the ho2-1 mutant inArabidopsis suggested that HO2 does also contribute to properphotomorphogenesis [6]. Some of the observed phenotypesresemble that of the hy1-100 mutant, but to a lesser extent. Pheno-types observed include hypocotyl elongation under red and far-red light, smaller rosette leaves and chlorosis. Moreover, a slowerdegradation rate after red light irradiation of PHYA was observedindicating a mixed (apo- and holo-) PHYA pool in the ho2-1
Abbreviations used: BV, biliverdin; CaMV, cauliflower mosaic virus; DCIP, dichloroindophenol; DCMU, dichlorophenyldimethylurea; Fd, ferredoxin;GFP, green fluorescent protein; GST, glutathione transferase; HO, haem oxygenase; IPTG, isopropyl β-D-thiogalactoside; PHY, phytochrome; proto IX,protoporphyrin IX; PSI, photosystem I; PB, phytochromobilin; TFA, trifluoroacetic acid; YFP, yellow fluorescent protein.
1 To whom correspondence should be addressed (email [email protected]).The synthetic DNA sequence for HO2 has been deposited in GenbankTM under the accession number FJ854354.
c© The Authors Journal compilation c© 2010 Biochemical Society
www.biochemj.org
Bio
chem
ical
Jo
urn
al

426 B. Gisk and others
Figure 1 Haem degradation as catalysed by HO
Most HOs cleave haem (heme) at the α-meso carbon position yielding biliverdin IXα. Thisreaction requires seven electrons and three molecules of molecular oxygen. Carbon monoxideand iron are released.
mutant [6]. Therefore, loss of HO2 might limit the availabilityof PB.
All four Arabidopsis HO genes are transcribed, but withdifferent expression levels. Whereas HY1 shows the highestexpression level followed by HO2, the expression of HO3 andHO4 is detectable, but extremely low [8,9]. In addition, geneexpression profiling revealed that HO2 expression is slightlyinduced by light at the onset of greening, whereas HY1, HO3and HO4 are constitutively expressed [9]. Whereas the mainfunction of plant HOs seems to be the biosynthesis of the PHYchromophore, mammalian HOs are involved in iron utilizationand the oxidative stress response, as well as showing potentanti-inflammatory effects [10–12]. In mammals, three isoformsexist, HO-1, HO-2 and HO-3. HO-3 is an isoform with lowcatalytic activity and unknown physiological function [13]. HO-2, on the other hand, is constitutively expressed and thought tobe mostly involved in signalling pathways. HO-1 expression isinduced by oxidative stress and the HO reaction product BV, or thesubsequent reduction product, bilirubin has anti-oxidant function[14]. Reports on a HO from soybean also suggest a function in theoxidative stress response in plants [15,16]. However, mammalianand plant HOs seem to differ in the use of the required reductant.Mammalian HOs were shown to use NADPH–cytochrome P450reductase (NADPH–cytochrome c reductase) [17–19] as thepreferred reductant, whereas plant, cyanobacterial and bacterialHOs seem to prefer Fd [20–23]. In addition, plant HOs appear torequire a second reductant, such as ascorbate, for full activity [21].
The most common open-chain product of the HO reaction isBV IXα, resulting from the oxidative cleavage of haem at theα-meso carbon bridge. However, several previous reports provethe existence of HOs with alternative cleavage sites resulting in thethree other BV IX isomers, β, γ and δ [24,25]. The regiospecificityof these HOs cannot be predicted from the primary amino acidsequence or crystal structures, but rather seem to be due to adifferent orientation of the haem substrate in the active site pocketas shown for the β- and δ-specific HO (PigA) from Pseudomonasaeruginosa [26,27].
Until now, HY1 is the only biochemically characterized HOfrom Arabidopsis [21]. The functionality of HO3 and HO4 wasonly indirectly proven by co-expression of the HOs with theapoPHY from Deinococcus radiodurans (DrBphP), which resul-ted in the binding of the HO reaction product BV to the PHY [8].Whether HO2 is a true HO has not been biochemically addressedat all due to low expression rates and solubility issues. However,the phenotype at least points towards a function within tetrapyrrolemetabolism. In the present study we show biochemical data which
gives new insights into the function of all four Arabidopsis HOs.For the first time we were able to obtain sufficient amounts ofrecombinant HO2 by using a codon-optimized synthetic gene.Interestingly, HO2 is not able to bind or convert haem and istherefore not a true HO. However, its strong binding of proto IXpoints towards a role for HO2 in tetrapyrrole metabolism.
EXPERIMENTAL
Chemicals
All chemicals were purchased from Sigma–Aldrich and met orexceeded the American Chemical Society specifications. HPLC-grade acetone and 80% (v/v) formic acid were purchased fromMallinckrodt Baker.
Subcellular localization experiments with GFP (green fluorescentprotein) fusions
The coding regions for the putative transit peptide for HY1(HO1), HO2, HO3, and HO4 were amplified from Arabidopsis(Columbia) cDNA with the following primers: for HY1, 5′-CG-GGATCCCACCATGGCGTATTTAGCTCCGA-3′ and 5′-CC-GCTCGAGGCAGTAGTAGCCGCAACCA-3′; for HO2, 5′-CG-GGATCCCACCATGGCTTCTCTTCTCAGGC-3′ and 5′-CCG-CTCGAGTGTGAGGCCTTTTGTTGTGA-3′; for HO3, 5′-CG-GGATCCCACCATGGCTACAACAAGACTTAAC-3′ and 5′-CCGCTCGAGATAGCCGCCGCAGTCACC-3′; and for HO4,5′-CGGGATCCCACCATGGCTACATCAAGACTTAATGC-3′
and 5′-CCGCTCGAGGCCACAACATTCACCACCA-3′. Theamplified DNA fragments were digested with BamHI and XhoIand then cloned into the corresponding sites of pENTRTM1Avector (Invitrogen) to produce the entry clones. The entry clonefor intact GFP was prepared by self-ligation of pENTRTM1A,digested with XmnI and EcoRV. After verification of theorientation and the nucleotide sequences, a recombinationreaction was performed between the entry clones and thedestination vector pGWB5 [28] using LR Clonase (Invitrogen)to generate an in-frame fusion with the GFP reporter gene underthe CaMV (cauliflower mosaic virus) 35S promoter. The plasmidpGWB42-MIRO1TM for the mitochondrial marker YFP (yellowfluorescent protein)–MIRO1 [29] was obtained from Dr Yamaoka(University of Tokyo, Japan). The transient assays of fluorescentprotein expression in Nicotiana benthamiana were performed asdescribed in [30] by using Agrobacterium tumefaciens C58C1pGV2260. After 36 h of co-cultivation in the dark, the signals forGFP and chloroplast autofluorescence were examined under aconfocal microscope (FluoView 1000, Olympus).
Construction of the HO2 expression vector
A synthetic gene encoding the mature (without the predictedtransit peptide sequence) HO2 was adapted to the codon biasof Escherichia coli genes to provide a successful expression. Thesynthetic gene, HO2syn, of 738 bp was assembled from syntheticoligonucleotides (Geneart GmbH) with addition of a 5′ BamHIand a 3′ XhoI restriction site respectively. After cleavage with therespective enzymes the artificial gene was cloned into pGEX-6P-1to generate pGEX-mHO2 (GE Healthcare).
Expression and purification of recombinant HOs
HY1 was produced and purified using the plasmid pGEX-mHY1as described previously [31] with minor modifications. Thepurified mature (without the predicted transit peptide) HY1 was
c© The Authors Journal compilation c© 2010 Biochemical Society

Arabidopsis haem oxygenases 427
dialysed against 100 mM potassium phosphate buffer, pH 7.2.The same protocol was used for the expression and purificationof mature HO2syn, except that its GST (glutathione transferase)-fusion has a recognition sequence for the PreScission® proteaseinstead of for thrombin.
The pET24a-mHO3 and pET24a-mHO4 plasmids forexpressing recombinant C-terminally His6-tagged mature HO3and HO4 production were a gift from by Dr R.D.Vierstra(University of Wisconsin, Madison, U.S.A.). An overview of theemployed constructs is given in Figure 3(A).
For production of recombinant HO3 and HO4, 2 litres ofLB (Luria–Bertani) media containing 50 mg/ml kanamycin wasinoculated at a dilution of 1:100 from an overnight cultureof E. coli strain BL21 (DE3) carrying the respective plasmidconstructs and were grown at 37 C to an approx. D578 of0.6–0.8. After a temperature shift to 17 C, protein productionwas induced by the addition of 125 μM IPTG (isopropyl β-D-thiogalactoside), and cells were cultured for additional 15 hwith shaking at 180 rev./min. Afterwards cells were harvestedby centrifugation (13000 g for 15 min) and stored at −20 C.Cell lysis and disruption were performed as described previously[31]. The supernatant was loaded on a Talon®-cobalt-affinitychromatography column (Clontech) and subsequent washing andelution steps were performed according to the manufacturer’sinstructions. Proteins were eluted with a buffer containing 75 mMimidazole. The protein solutions were pooled and dialysed againstreaction buffer (100 mM potassium phosphate buffer, pH 7.2).
Analysis of the quaternary structure using gel-permeationchromatography
The oligomerization state of the four enzymes was investigatedusing analytical gel-permeation chromatography. A SuperdexTM
75 HR10/30 column (GE Healthcare), equilibrated with 100 mMpotassium phosphate, pH 7.2, at a flow rate of 0.5 ml/min, wasemployed. Protein was monitored by absorbance at 280 nmusing the UV-900 detector of an AEKTA-explorer system(GE Healthcare). Standard proteins, with known molecularmass (in kDa: apoferritin, 443.0; β-amylase, 200.0; alcoholdehydrogenase, 150.0; albumin, 66.0; carboanhydrase, 29.0; andcytochrome c, 12.4), were used to generate a calibration curvefrom which the quaternary structure was calculated.
Reconstitution and titration of HOs with haem
Crystalline haemin (15 mg) was dissolved in 5 ml of DMSOto generate a 4.6 mM haemin solution. Haemin was added tothe purified HOs to give a final 2:1 haem/protein ratio. Thesample was subsequently applied to a hydroxylapatite column(1.0 cm × 2.0 cm; resin from Fluka) which was equilibratedwith 10 mM potassium phosphate buffer, pH 7.2. The columnwas then washed with the same buffer until no more freehaem could be detected spectrophotometrically, and the proteinwas eluted with 100 mM potassium phosphate buffer, pH 7.2.The fractions containing detectable haem–HO complex (at A280
and A405) were pooled and concentrated using a Vivaspin®
(Vivascience) concentrator at 40000 g. The concentration of therecombinantly produced HOs was determined using the molarextinction coefficient (ε280) calculated from the deduced aminoacid composition [32].
Titration of HOs with haem was monitored by absorptionspectroscopy. Aliquots of haem (0.58–10.0 μM) were added to thecuvette containing 5 μM HO at 25 C and spectra were recorded5 min after haem addition. The same haem concentrations wereadded to a sample cuvette containing only buffer as a control. The
Kd value was obtained from the difference between spectra of freehaem and protein-bound haem at 405 nm.
Determination of the extinction coefficient for the haem–HOcomplexes
The millimolar extinction coefficient at 405 nm for the haem–HO complex was determined using the pyridine haemochromemethod [33]. The isolated haem–HO complex (10 μM) wasconverted to a pyridine-haemochrome in a 500 μl sample volumeby addition of 62.5 μl of pyridine and 62.5 μl of 0.5 M NaOHsolution. The spectrum of the oxidized pyridine haemochromewas recorded. After addition of an excess of dithionitethe spectrum of the reduced ferrous pyridine haemochromewas recorded. The difference spectrum of the reduced andoxidized form was calculated at 557 nm. The concentration ofhaem was determined from the extinction coefficient (ε405 =34.530 mM−1 · cm−1).
HO assay
HO activity was measured in 500 μl (final volume) of a solutioncontaining 10 μM HO, 10 μM haem, 0.15 mg/ml BSA, 4.6 μMFd (spinach), 0.025 unit/ml spinach Fd-NADP+ oxidoreductaseand 10 μM catalase from Aspergillus niger in 100 mM potassiumphosphate buffer, pH 7.2.
The reaction was started by addition of an NADPH-regenerating system containing 6.5 mM glucose 6-phosphate,0.82 mM NADP+, and 1.1 units/ml glucose-6-phosphatedehydrogenase. Spectral changes between 300 and 900 nm weremonitored for 30 min at 25 C using an Agilent Technologies8453 UV–visible spectroscopy system. Sodium ascorbate and/ordesferrioxamine were added to a final concentration of 5 mMunless otherwise noted. The rate of BV IXα formation at 25 Cwas calculated using the absorbance change at 670 nm. Theconcentration of BV IXα was estimated using an extinctioncoefficient at 670 nm of 6.25 mM−1 · cm−1 in 100 mM potassiumphosphate buffer, pH 7.2, determined from published values ofother solvents [34]. Km values were calculated for the HOs usingEadie–Hofstee and Lineweaver–Burk plots (the standard assayswith 10 μM HO, and various haem concentrations between 1 μMand 100 μM, were used). Temperature dependence of the HOreaction was measured between 10 C and 45 C. The activationenergy (Ea) was calculated based on an Arrhenius plot. The pHdependence of the reaction was monitored between pH 5.0 and9.0 in 100 mM potassium phosphate buffer and for all assays thestandard conditions as described above were used.
Isolation of thylakoid membrane and HO assay
Fresh spinach leaves (25 g) were added to 35 ml of digestionbuffer [50 mM Tris/HCl, pH 7, containing 0.4 M sucrose, 20 mMKCl and 0.5 mM KH2PO4) and homogenized for 5 min. Thehomogenized leaves were then filtered through a fleece clothand the filtrate centrifuged at 40000 g for 5 min, The pelletwas resuspended in 10 ml of digestion buffer. After a secondcentrifugation step at 40000 g for 5 min, the pellet wasresuspended in 5 ml buffer (100 mM Hepes, pH 8.0, containing0.66 M sucrose, 40 mM KCl, 3 mM KH2PO4 and 4 mM MgCl2)and 5 ml of H2O. The chlorophyll content of these crudemembranes was calculated as described previously [35]. Forthe HO assay, 10 μM HO–haem complex was incubated with0.15 mg/ml BSA, 4.6 μM Fd (spinach), 5 mM sodium ascorbate,5 mM tiron and isolated thylakoid membranes (containing 10 μg
c© The Authors Journal compilation c© 2010 Biochemical Society

428 B. Gisk and others
of chlorophyll) in 100 mM potassium phosphate buffer, pH 7.2,either in the dark or for 100 s in the dark followed by irradiationfor 200 s with 460 W · m−2 of actinic white light. Absorbancewas measured at 680 nm and HO activity was calculated by therate of absorbance change at this wavelength. The reaction wasinhibited with 50 μM DCMU (dichlorophenyldimethylurea) or50 μM DCMU and 200 μM reduced DCIP (dichloroindophenol).DCIP was reduced using 0.1 mM HCl and 5 mM ascorbate priorto use.
HPLC analysis of HO reaction products
HO assay mixtures were acidified with 10-fold 0.1% TFA(trifluoroacetic acid) and loaded on to a Waters C18- Sep-Paklight cartridge which had been preconditioned twice with thefollowing sequence of solutions: 3 ml of acetonitrile, to wetthe Sep-Pak; 3 ml of MilliQ water; 3 ml of 0.1% TFA; and 3 ml of10% (v/v) methanol in 0.1% TFA. After the sample was loadedon to the Sep-Pak, it was washed with 6 ml of 0.1% TFA, and 6 mlof a methanol/0.1% TFA solution (4:1; v/v). The tetrapyrroleswere then eluted from the Sep-Pak using 1 ml of 100% acetoni-trile. The eluate was dried using a Speed-Vac lyophilizer, and driedsamples were analysed by HPLC. Samples were first dissolved in10 μl of DMSO and then diluted in 150 μl of the HPLC mobilephase. Following brief centrifugation (10000 g for 1 min) andfiltration through a 0.45 μm poly(tetrafluoroethylene) syringefilter (Phenomax), tetrapyrroles were analysed with reverse-phase chromatography using an Agilent Technologies 1100 liquidchromatograph. The HPLC column used for all analyses was a4.6 mm × 250 mm Phenomenex Ultracarb 5 μm OD20 analyticalcolumn with a 4.6 mm × 30 mm guard column of the samematerial. The mobile phase consisted of acetone/20 mM formicacid (1:1; v/v), and the flow rate was 0.6 ml/min. Eluates weremonitored at 650 nm and 380 nm using an Agilent Technologies1100 series diode array detector. When required, complete spectrawere obtained for the peaks desired. Peak areas were quantifiedusing Agilent Technologies Chemstation software.
RESULTS
HO2, HO3 and HO4 are localized to the plastid
The HY1 protein contains a functional transit peptide forplastid localization and is processed to the mature plastidHO [21,31]. The N-termini of HO2, HO3 and HO4, deducedfrom cDNA sequences, also showed features of a transitpeptide when assessed using the ChloroP program ([36];http://www.cbs.dtu.dk/services/ChloroP/). The conservation ofprimary sequences in this region was much lower than in theHO region, also indicating that the stretches in the N-terminiare transit peptides. To demonstrate the function of the putativetransit peptides experimentally, we fused the potential transitpeptide regions of all four Arabidopsis HO genes to a geneencoding GFP under the control of the CaMV 35S promoter. Theconstructs were introduced to N. benthamiana by agro-infection[30]. GFP fluorescence was observed in oval structures in thesamples in which HY1 (HO1), HO2, HO3 and HO4 constructswere introduced (Figures 2a, 2d, 2g and 2j). These signals matchedthe chloroplast autofluorescence (Figures 2b, 2e, 2h and 2k; see themerges in Figures 2c, 2f, 2i and 2l), demonstrating that the fusionprotein was efficiently targeted to chloroplasts. A control constructwithout the putative transit peptide showed GFP fluorescencethroughout the cytoplasm (Figure 2m) that is clearly differentfrom the red autofluorescence from the chlorophyll in chloroplasts
Figure 2 Subcellular localization of HY1, HO2, HO3 and HO4 by transientexpression of the fused fluorescent proteins
Microscopic images of GFP or YFP, chloroplast autofluorescence (Chl) and the merged images(merge) from mesophyll cells of N. benthamiana infected with Agrobacteria harbouring theGFP/YFP constructs. All images were taken at the same magnification. GFP is fused to (a–c)the HY1 transit peptide, (d–f) the HO2 transit peptide, (g–i) the HO3 transit peptide and(j–l) the HO4 transit peptide. (m–o) Images of intact GFP. (p–r) YFP is fused to thetransmembrane stretch of the MIRO1 mitochondria marker. Scale bar, 20 μm. YFP fluorescencewas coloured green for a better quality merge figure.
(Figures 2n and 2o). Hence, as there are functional transit peptidesin the deduced HY1, HO2, HO3 and HO4 sequences, this impliesthat the proteins encoded by HY1, HO2, HO3 and HO4 are presentas a processed mature protein in the plastid compartment. As haemis also present in mitochondria, a mitochondrial marker (MIRO1)fused to YFP [29] was observed. The YFP signals for MIRO1 aredifferent from those of the HO proteins (Figures 2p, 2q and 2r)and therefore there is no indication of a mitochondrial localizationfor HO1–4 in this analysis.
Expression and purification of recombinant haem oxygenases
The Arabidopsis HO genes HY1, HO2, HO3 and HO4 wereexpressed without their predicted chloroplast transit peptideseither using a tac promoter-driven, N-terminal GST fusion,expression system (for HY1 and HO2) [21] or the pET vector
c© The Authors Journal compilation c© 2010 Biochemical Society

Arabidopsis haem oxygenases 429
Figure 3 Purification of recombinant proteins
(A) Scheme of the recombinant proteins used in the present study. GST, glutathionetransferase; m, mature protein (lacking the predicted transit peptide sequence); numbersdepict the amino acid residues in the full-length protein; syn, synthetic gene product;His6, His6 tag. (B) Coomassie-stained SDS/PAGE gel of recombinant purified HOs.Lane M, molecular-mass markers (values in kDa are on the right-hand side); lane1, purified HY1; lane 2, purified HO2 (native gene); lane 3, purified HO3; lane 4,purified HO4; lane 5, purified HO2 (synthetic gene adapted to E.coli codon usage) aftercleavage with PreScission® protease and purification with a second chromatographycolumn. HY1 and HO2 were purified by GST-affinity chromatography followed byprotease digestion and use of a second chromatography column. HO3 and HO4 were purifiedby cobalt-affinity chromatography.
system (for HO3 and HO4), which uses a T7-promoter andlabels proteins with a C-terminal His6 tag (Figure 3A) [8]. Afterinduction with IPTG and expression overnight at 17 C, the E. coliBL21 (DE3) cells had a brownish colour (except cells expressingHO2; results not shown). This is in contrast with the expressionof bacterial or mammalian HOs in E. coli, which most often showa green colour due to BV accumulation in the cells [23,37–39].Therefore the lack of green colour implies that the plant HOsmight not be fully active in E. coli.
HY1 was expressed and purified as described previously [21]and yielded a major band migrating at approx. 27 kDa onSDS/PAGE (Figure 3B). Recombinant HO3 and HO4 containeda C-terminal His6-tag and were purified by cobalt-affinitychromatography. This purification strategy led to over 90% purity(Figure 3B). The final yield of recombinant HY1, HO3, and HO4from 1 litre of bacterial culture was approx. 60 mg each. Theproteins were stored on ice and stayed fully active for up to 4 daysin 100 mM potassium phosphate buffer, pH 7.2. Unfortunately,the expression and purification of recombinant HO2 yielded onlylow amounts of soluble protein (Figure 3B, lane 2). Thereforea synthetic gene (without the transit peptide coding region)adapted to the codon usage of E. coli K12 was generated andcloned into pGEX-6-P1. The yield from this synthetic constructwas significantly higher than using the native gene (Figure 3B,
Table 1 Biochemical parameters of HY1, HO3 and HO4
Parameter HY1 HO3 HO4
ε (mM−1· cm−1) 203 271 175K d (μM) 1.6 2.3 2.8K m (μM) 1.3 2.7 5.7E a (J·mol−1) 36.6 14.78 10.76
lane 5) and resulted in approx. 24 mg recombinant HO2 from1 litre of bacterial culture. As the recombinant protein migratedslightly slower than predicted in SDS/PAGE, the integrity of therecombinant protein was verified by tryptic digest followed byMALDI (matrix-assisted laser-desorption ionization)–TOF (time-of-flight)/TOF MS analysis (results not shown).
Arabidopsis HOs are monomeric enzymes
Whereas the majority of HOs are monomeric enzymes, otherssuch as HO-2 from Synechocystis sp. PCC 6803 or HO-2 fromhuman are dimeric [40–42]. To probe the oligomerization stateof the Arabidopsis HOs, gel-permeation chromatography on aSuperdexTM 75 HR10/30 column was performed.
All four HO proteins eluted at approx. 11.5 ml as a majorsymmetric peak. However, whereas HY1 and HO3 showed noaggregation after purification, HO4 also displayed a minor peakat approx. 8.5 ml which was shown to be aggregated proteinby SDS/PAGE (results not shown). HO2, on the other hand,displayed two minor peaks at approx. 15 ml and 18 ml, whichare probable degradation products (results not shown). Under ourexperimental conditions and in comparison with the elution profileof standard proteins all four investigated recombinant HOs aremonomeric.
HO2 from Arabidopsis does not bind and convert haem
Arabidopsis possesses four annotated HOs with only hy1 and ho2mutants showing strong defects in photomorphogenesis, raisingthe question whether the HOs differ with regard to enzymeactivity. Thus we first attempted to analyse the haem-bindingproperties of the four HOs.
The formation of the haem–HO complex was determinedby absorption spectroscopy. Interestingly, only the three HOsbelonging to the HO1 subfamily displayed a strong Soret bandat 405 nm indicating enzyme-bound haem (Figures 4A, 4C and4D). This Soret band is identical to that reported for HOsfrom other sources [21,43–45]. Incubation of HO2 with haemdid not result in a significant absorbance change or in theformation of a distinct Soret band (Figure 4B). Even after extendedincubation time, no haem-binding was observed. The additionof reductant (Fd) also had no influence on the haem-bindingbehaviour (results not shown). As HO2 is not able to bindhaem, no haem-binding constant (Kd) and no activity could bedetermined. For the haem-binding HOs from the HO1 subfamily,the Kd values were calculated from titration data determinedby difference-absorption spectroscopy (results not shown). Allthree HO1 subfamily members have Kd values in the micromolarrange. Whereas HY1 displayed the highest affinity of approx.1.6 μM (in agreement with [21]), HO3 and HO4 had slightlyweaker affinities of 2.3 μM and 2.8 μM respectively (Table 1).All three HOs appeared to be saturated at a ratio of haem/proteinof 1:1.
Extinction coefficients of the recombinant HOs weredetermined using the method of Berry and Trumpower [33].
c© The Authors Journal compilation c© 2010 Biochemical Society
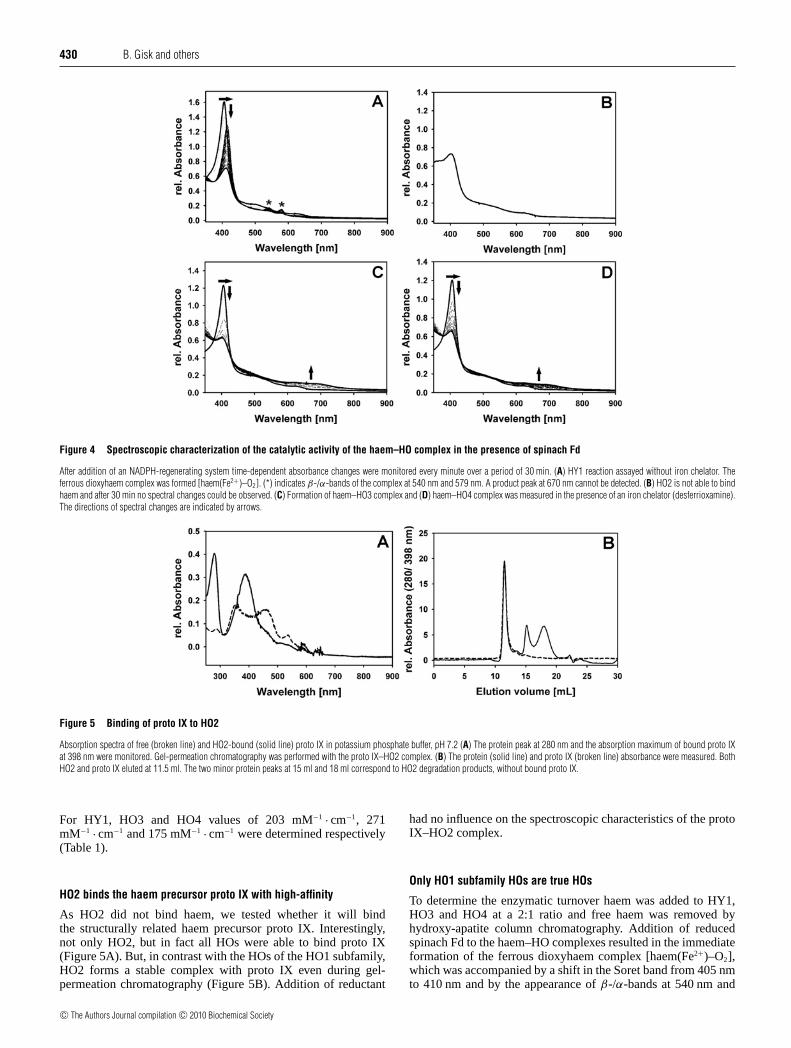
430 B. Gisk and others
Figure 4 Spectroscopic characterization of the catalytic activity of the haem–HO complex in the presence of spinach Fd
After addition of an NADPH-regenerating system time-dependent absorbance changes were monitored every minute over a period of 30 min. (A) HY1 reaction assayed without iron chelator. Theferrous dioxyhaem complex was formed [haem(Fe2+)–O2]. (*) indicates β-/α-bands of the complex at 540 nm and 579 nm. A product peak at 670 nm cannot be detected. (B) HO2 is not able to bindhaem and after 30 min no spectral changes could be observed. (C) Formation of haem–HO3 complex and (D) haem–HO4 complex was measured in the presence of an iron chelator (desferrioxamine).The directions of spectral changes are indicated by arrows.
Figure 5 Binding of proto IX to HO2
Absorption spectra of free (broken line) and HO2-bound (solid line) proto IX in potassium phosphate buffer, pH 7.2 (A) The protein peak at 280 nm and the absorption maximum of bound proto IXat 398 nm were monitored. Gel-permeation chromatography was performed with the proto IX–HO2 complex. (B) The protein (solid line) and proto IX (broken line) absorbance were measured. BothHO2 and proto IX eluted at 11.5 ml. The two minor protein peaks at 15 ml and 18 ml correspond to HO2 degradation products, without bound proto IX.
For HY1, HO3 and HO4 values of 203 mM−1 · cm−1, 271mM−1 · cm−1 and 175 mM−1 · cm−1 were determined respectively(Table 1).
HO2 binds the haem precursor proto IX with high-affinity
As HO2 did not bind haem, we tested whether it will bindthe structurally related haem precursor proto IX. Interestingly,not only HO2, but in fact all HOs were able to bind proto IX(Figure 5A). But, in contrast with the HOs of the HO1 subfamily,HO2 forms a stable complex with proto IX even during gel-permeation chromatography (Figure 5B). Addition of reductant
had no influence on the spectroscopic characteristics of the protoIX–HO2 complex.
Only HO1 subfamily HOs are true HOs
To determine the enzymatic turnover haem was added to HY1,HO3 and HO4 at a 2:1 ratio and free haem was removed byhydroxy-apatite column chromatography. Addition of reducedspinach Fd to the haem–HO complexes resulted in the immediateformation of the ferrous dioxyhaem complex [haem(Fe2+)–O2],which was accompanied by a shift in the Soret band from 405 nmto 410 nm and by the appearance of β-/α-bands at 540 nm and
c© The Authors Journal compilation c© 2010 Biochemical Society
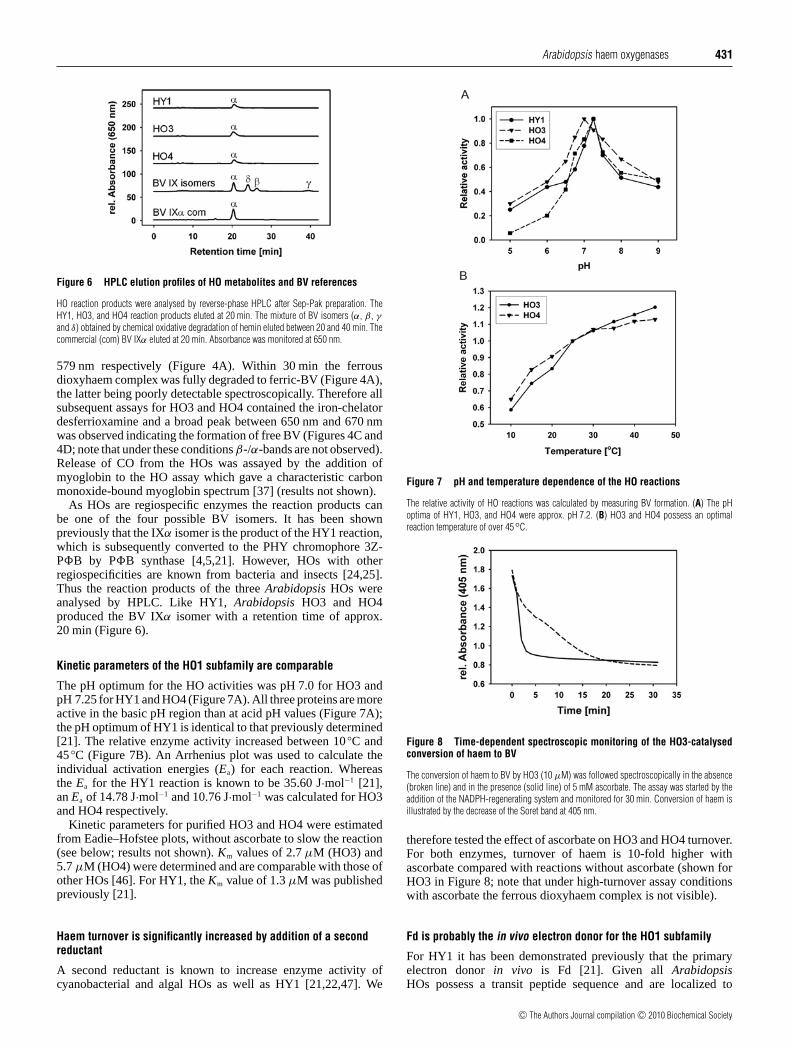
Arabidopsis haem oxygenases 431
Figure 6 HPLC elution profiles of HO metabolites and BV references
HO reaction products were analysed by reverse-phase HPLC after Sep-Pak preparation. TheHY1, HO3, and HO4 reaction products eluted at 20 min. The mixture of BV isomers (α, β , γ
and δ) obtained by chemical oxidative degradation of hemin eluted between 20 and 40 min. Thecommercial (com) BV IXα eluted at 20 min. Absorbance was monitored at 650 nm.
579 nm respectively (Figure 4A). Within 30 min the ferrousdioxyhaem complex was fully degraded to ferric-BV (Figure 4A),the latter being poorly detectable spectroscopically. Therefore allsubsequent assays for HO3 and HO4 contained the iron-chelatordesferrioxamine and a broad peak between 650 nm and 670 nmwas observed indicating the formation of free BV (Figures 4C and4D; note that under these conditions β-/α-bands are not observed).Release of CO from the HOs was assayed by the addition ofmyoglobin to the HO assay which gave a characteristic carbonmonoxide-bound myoglobin spectrum [37] (results not shown).
As HOs are regiospecific enzymes the reaction products canbe one of the four possible BV isomers. It has been shownpreviously that the IXα isomer is the product of the HY1 reaction,which is subsequently converted to the PHY chromophore 3Z-PB by PB synthase [4,5,21]. However, HOs with otherregiospecificities are known from bacteria and insects [24,25].Thus the reaction products of the three Arabidopsis HOs wereanalysed by HPLC. Like HY1, Arabidopsis HO3 and HO4produced the BV IXα isomer with a retention time of approx.20 min (Figure 6).
Kinetic parameters of the HO1 subfamily are comparable
The pH optimum for the HO activities was pH 7.0 for HO3 andpH 7.25 for HY1 and HO4 (Figure 7A). All three proteins are moreactive in the basic pH region than at acid pH values (Figure 7A);the pH optimum of HY1 is identical to that previously determined[21]. The relative enzyme activity increased between 10 C and45 C (Figure 7B). An Arrhenius plot was used to calculate theindividual activation energies (Ea) for each reaction. Whereasthe Ea for the HY1 reaction is known to be 35.60 J·mol−1 [21],an Ea of 14.78 J·mol−1 and 10.76 J·mol−1 was calculated for HO3and HO4 respectively.
Kinetic parameters for purified HO3 and HO4 were estimatedfrom Eadie–Hofstee plots, without ascorbate to slow the reaction(see below; results not shown). Km values of 2.7 μM (HO3) and5.7 μM (HO4) were determined and are comparable with those ofother HOs [46]. For HY1, the Km value of 1.3 μM was publishedpreviously [21].
Haem turnover is significantly increased by addition of a secondreductant
A second reductant is known to increase enzyme activity ofcyanobacterial and algal HOs as well as HY1 [21,22,47]. We
Figure 7 pH and temperature dependence of the HO reactions
The relative activity of HO reactions was calculated by measuring BV formation. (A) The pHoptima of HY1, HO3, and HO4 were approx. pH 7.2. (B) HO3 and HO4 possess an optimalreaction temperature of over 45C.
Figure 8 Time-dependent spectroscopic monitoring of the HO3-catalysedconversion of haem to BV
The conversion of haem to BV by HO3 (10 μM) was followed spectroscopically in the absence(broken line) and in the presence (solid line) of 5 mM ascorbate. The assay was started by theaddition of the NADPH-regenerating system and monitored for 30 min. Conversion of haem isillustrated by the decrease of the Soret band at 405 nm.
therefore tested the effect of ascorbate on HO3 and HO4 turnover.For both enzymes, turnover of haem is 10-fold higher withascorbate compared with reactions without ascorbate (shown forHO3 in Figure 8; note that under high-turnover assay conditionswith ascorbate the ferrous dioxyhaem complex is not visible).
Fd is probably the in vivo electron donor for the HO1 subfamily
For HY1 it has been demonstrated previously that the primaryelectron donor in vivo is Fd [21]. Given all ArabidopsisHOs possess a transit peptide sequence and are localized to
c© The Authors Journal compilation c© 2010 Biochemical Society
432 B. Gisk and others
Figure 9 Light dependence of the HO reaction
Light dependence of the HO reaction was observed using isolated thylakoid membranes as anelectron donor. (A) The reaction was incubated in the dark (broken line) or irradiated with lightafter 100 s (solid line, indicated by arrow). The BV formation was measured at 680 nm. Thereaction is shown for HO3. (B) The relative HO activity, determined as the rate of BV formation atA 680, was measured for the complete reaction in the light with all assay components (complet),in darkness (drak) and in light in the presence of 50 μM DCMU (DCMU), 50 μM DCMU andreduced 200 μM DCIP (DCIP) and in the absence of HO (HO) or Fd (Fd). Error bars indicatemeans +− S.E.M.
the chloroplast (Figure 2), Fd, receiving electrons from PSI(photosystem I), is probably the natural electron donor. Additionof isolated thylakoid membranes to the HO assay significantlyincreased the cleavage of haem to BV after illumination(Figure 9A). In the presence of the electron-transport inhibitorDCMU, BV formation decreases to a relative activity of about20% (Figure 9B). In this scenario cyclic electron flow does notresult in net production of reduced Fd, and subsequent haemturnover. Addition of reduced DCIP, an artificial electron donor, tothe DCMU-blocked assay resulted in an increased BV formation,as reduced DCIP is able to reduce PSI (Figure 9B).
DISCUSSION
Although the HO1 and HO2 subfamilies in Arabidopsis havebeen previously identified, direct evidence for the functionalityof the individual members was still lacking [6,8,31,48]. In thepresent study we provide the first biochemical data for all four
annotated Arabidopsis HOs using recombinant enzymes. WhereasHY1 appears to be the key HO responsible for PHY chromophorebiosynthesis, HO3 and HO4 are also classical α-hydroxylatingHOs. Analysis of the major biochemical parameters establishedthat their activity does not significantly differ from that of HY1.
The mechanism of haem cleavage is conserved between HOsof the HO1 subfamily from Arabidopsis, mammals and bacteria.Haem degradation results in production of BV IXα, carbonmonoxide and iron. Interestingly, higher plant HOs form a strongferric-BV complex and an iron-chelator (e.g. desferrioxamine)is essential to release BV in vitro (Figures 4C and 4D). Thisphenomenon has also been observed previously for HY1 fromArabidopsis [21] and HO from Cyandium caladanium [49].As approx. 90% of cellular iron is located in the chloroplast[50], it seems likely that the ferric–BV complex is not releasedspontaneously by HOs. One might hypothesize that only theinteraction between HO and PB synthase (HY2) releases BV,which is in turn converted by HY2 to PB.
Our experiments using isolated thylakoid membranes stronglysupport the role of Fd as the electron donor in vivo [21]. Thegenome of Arabidopsis possesses a total of four Fd genes (AtFd1,AtFd2, AtFd3 and AtFd4) coding for three different types of[2Fe-2S] Fd [51], the classic leaf-type Fds (AtFd1 and AtFd2),a root-type Fd (AtFd3) and a high-redox-potential Fd (AtFd4).All Fds contain chloroplast transit peptides and are transported tothe plastid. As predicted, Arabidopsis HOs were also shown tolocalize to the chloroplast. It is probable that one of the leaf-typeFds is the natural redox partner for the HOs in photosynthetic cells[51]. However, in non-photosynthetic cells one of the other Fd ismore likely to be the natural electron donor. In this case, the Fdis reduced by NADPH generated by the oxidative pentosephosphate pathway.
The localization of all HOs solely to the plastid, however,raises the question as to how haem turnover outside thechloroplast is achieved. As mitochondria contain a large numberof haemoproteins, one would also postulate the presence of anactive HO in these organelles. Although our experimental resultsdid not show mitochondrial localization in N. benthamiana, wecannot exclude mitochondrial localization in Arabidopsis. In thisregard, dual targeting of plant proteins to both the chloroplast andthe mitochondria has been described [52]. Furthermore, anotherenzyme of the tetrapyrrole pathway, protoporphyrinogen oxidaseof spinach, was shown to be localized to both compartments dueto the use of two in-frame initiation codons [53].
A general phenomenon of higher plant HOs is that additionof ascorbate as a second reductant significantly influences haemturnover. Endogenously varying ascorbate levels might be a toolfor modulating HO activity in planta. In this regard, it has beenreported that the chloroplastidic ascorbate concentration can varyfrom 15–46 mM, depending on light intensity, with high levelsof light leading to increased ascorbate levels [54]. ThereforeHO activity is indirectly regulated by light through the levelsof ascorbate, ensuring sufficient PHY chromophore biosynthesisduring the day. Furthermore, our biochemical results suggest apH optimum for HO activities which conform to the pH valueof the stroma at night (pH 7.2) [55]. During the day the pH ofthe chloroplast increases to pH 8.0, which might be expected todecrease the activity of all three HOs. In contrast, HO enzymeactivity increases with rising temperature. So the antagonisticdependency of HO activity on pH and temperature could be anadditional tool to fine-tune HO activity.
Although it has been proposed previously that HY1 is unableto use NADPH-cytochrome P450 reductase as a reductant, slowturnover of haem to the oxycomplex by HY1, HO3 and HO4could be detected. However, only further addition of ascorbate
c© The Authors Journal compilation c© 2010 Biochemical Society

Arabidopsis haem oxygenases 433
led to a complete turnover (results not shown) and therefore itis rather unlikely that NADPH-cytochrome P450 reductase is anatural electron donor in planta.
Considering the weak mutant phenotype of ho3 and ho4, itappears that the enzymes do not have a large impact on PHYchromophore biosynthesis due to their low expression levels[8]. However, the biochemical data suggests that they couldcompletely substitute for HY1. Nevertheless, there is still thepossibility that they might also function in the oxidative stressresponse as the product of the reaction, BV, has reasonableantioxidant properties, as shown for soybean nodules [15,16].However, there is currently no evidence that any of the HO genesin Arabidopsis are inducible by oxidative and/or salt stress.
The most interesting result of the present study is the productionand purification of soluble HO2 from a synthetic gene. Thisenabled us for the first time to address the question of whetherHO2 is indeed a HO. Our results clearly demonstrate that HO2is unable to bind and degrade haem and raises the question ofthe function of this protein. Phenotypic analysis of the ho2-1mutant plants suggested that HO2 does contribute to properphotomorphogenesis. This, however, does not necessarily demanda function in haem turnover and could suggest that HO2 has adifferent function in tetrapyrrole metabolism. One indication thatHO2 is not a true HO is the absence of the conserved histidineresidue in the active site, which is usually involved in haem–ironco-ordination. This residue is replaced by an arginine residue inHO2. However, the sequence similarity (approx. 38%) suggeststhe same overall fold is present in all HOs, including HO2 (exceptfor the inserted spacer sequence). It was therefore interesting forus to test whether HO2 might bind haem precursors like proto IX.HO2 is able to bind proto IX with high affinity (as the complexis stable during gel-permeation chromatography, see Figure 5B).Although the three other Arabidopsis HOs displayed the samebinding behaviour, proto IX in these enzymes can easily bereplaced by haem indicating a high affinity to the natural substratehaem (results not shown). It remains to be determined whetherthe HO2 binding to proto IX is of any biological significance butwe speculate that HO2 could either have a function as a protoIX storage protein, and hence it might regulate tetrapyrrole fluxthrough the pathway, or that it might function as a carrier proteinfor magnesium and/or ferrochelatase. In the future it will benecessary to analyse tetrapyrrole concentration in the chloroplastsin more detail to understand the role of the HOs from both theArabidopsis HO1 and HO2 subfamilies. The latter hypothesisshould also be addressed by protein–protein interaction studies,which are currently underway.
However, for the first time we have been able to show that allthree HOs of the HO1 subfamily are active in vitro, with nearlyidentical biochemical parameters, and they are able to converthaem to BV IXα. We have determined that the member of theHO2 subfamily (HO2) is not a true HO, but it might be involvedin the regulation of the two tetrapyrrole biosynthetic pathways.
AUTHOR CONTRIBUTION
Bjoern Gisk and Nicole Frankenberg-Dinkel designed the study. Bjoern Gisk performedall the experiments except the plastid localization experiment. Yukiko Yasui performed theplastid localization experiment. Yukiko Yasui and Takayuki Kohchi analysed the plastidlocalization experiment. Bjoern Gisk and Nicole Frankenberg-Dinkel analysed all the otherresults and wrote the paper.
ACKNOWLEDGEMENTS
We thank Dr Yuuki Sakai and Dt Takashi Araki for their support with the confocal microscopeexperiments, Dr Shohei Yamaoka (University of Tokyo, Japan) for the MIRO1 plasmid and
Dr Richard D. Vierstra (University of Wisconsin, Madison, U.S.A.) for the HO2, HO3and HO4 expression clones. We also thank Dr Lars I. Leichert (Ruhr-University Bochum,Germany) for help with the MALDI–TOF/TOF analysis and Dr Jessica Wiethaus and AndreaBusch (Ruhr-University Bochum, Germany) for critical reading of the manuscript prior tosubmission.
FUNDING
This work was supported by the Deutsche Forschungsgemeinschaft [project TeilprojektC8 of the Sonderforschungsbereich 480 (to N. F. D.)]; and by a Grant-in-Aid for ScientificResearch from the Japan Society for Promotion of Science [grant number 21027021 (toT. K.)].
REFERENCES
1 Tu, S.-L. and Lagarias, J. C. (2005) The phytochromes. In Handbook of PhotosensoryReceptors (Briggs, W. and Spudich, J., eds), pp. 121–149, Wiley-VCH, Weinheim
2 Frankenberg, N. and Lagarias, J. C. (2003) Biosynthesis and biological functions ofbilins. In The Porphyrin Handbook (Kadish, K. M., Smith, K. M. and Guilard, R., eds),pp. 211–213, Elsevier Science
3 Ortiz de Montellano, P. R. (2000) The mechanism of heme oxygenase. Curr. Opin. Chem.Biol. 4, 221–227
4 Kohchi, T., Mukougawa, K., Frankenberg, N., Masuda, M., Yokota, A. and Lagarias, J. C.(2001) The Arabidopsis HY2 gene encodes phytochromobilin synthase, aferredoxin-dependent biliverdin reductase. Plant Cell 13, 425–436
5 Frankenberg, N., Mukougawa, K., Kohchi, T. and Lagarias, J. C. (2001) Functionalgenomic analysis of the HY2 family of ferredoxin-dependent bilin reductases fromoxygenic photosynthetic organisms. Plant Cell 13, 965–978
6 Davis, S. J., Bhoo, S. H., Durski, A. M., Walker, J. M. and Vierstra, R. D. (2001) Theheme-oxygenase family required for phytochrome chromophore biosynthesis isnecessary for proper photomorphogenesis in higher plants. Plant Physiol. 126, 656–669
7 Terry, M. J., Linley, P. J. and Kohchi, T. (2002) Making light of it: the role of plant haemoxygenases in phytochrome chromophore synthesis. Biochem. Soc. Trans. 30, 604–609
8 Emborg, T. J., Walker, J. M., Noh, B. and Vierstra, R. D. (2006) Multiple heme oxygenasefamily members contribute to the biosynthesis of the phytochrome chromophore inArabidopsis. Plant Physiol. 140, 856–868
9 Matsumoto, F., Obayashi, T., Sasaki-Sekimoto, Y., Ohta, H., Takamiya, K.-I. and Masuda,T. (2004) Gene expression profiling of the tetrapyrrole metabolic pathway in Arabidopsiswith a mini-array system. Plant Physiol. 135, 2379–2391
10 Poss, K. D. and Tonegawa, S. (1997) Heme oxygenase 1 is required for mammalian ironreutilization. Proc. Natl. Acad. Sci. U.S.A. 94, 10919–10924
11 Poss, K. D. and Tonegawa, S. (1997) Reduced stress defense in heme oxygenase1-deficient cells. Proc. Natl. Acad. Sci. U.S.A. 94, 10925–10930
12 Otterbein, L. E., Bach, F. H., Alam, J., Soares, M., Tao Lu, H., Wysk, M., Davis, R. J.,Flavell, R. A. and Choi, A. M. (2000) Carbon monoxide has anti-inflammatory effectsinvolving the mitogen-activated protein kinase pathway. Nat. Med. 6, 422–428
13 McCoubrey, Jr, W. K., Huang, T. J. and Maines, M. D. (1997) Isolation andcharacterization of a cDNA from the rat brain that encodes hemoprotein hemeoxygenase-3. Eur. J. Biochem. 247, 725–732
14 Maines, M. D., Trakshel, G. M. and Kutty, R. K. (1986) Characterization of two constitutiveforms of rat liver microsomal heme oxygenase: only one molecular species of the enzymeis inducible. J. Biol. Chem. 261, 411–419
15 Balestrasse, K. B., Noriega, G. O., Batlle, A. and Tomaro, M. A. L. (2005) Involvement ofheme oxygenase as antioxidant defense in soybean nodules. Free Radic. Res. 39,145–151
16 Noriega, G. O., Balestrasse, K. B., Batlle, A. and Tomaro, M. L. (2004) Heme oxygenaseexerts a protective role against oxidative stress in soybean leaves. Biochem. Biophys.Res. Commun. 323, 1003–1008
17 Tenhunen, R., Marver, H. S. and Schmid, R. (1969) Microsomal heme oxygenase:characterization of the enzyme. J. Biol. Chem. 244, 6388–6394
18 Yoshinaga, T., Sassa, S. and Kappas, A. (1982) The oxidative degradation of heme c bythe microsomal heme oxygenase system. J. Biol. Chem. 257, 7803–7807
19 Yoshinaga, T., Sassa, S. and Kappas, A. (1982) The occurrence of molecular interactionsamong NADPH-cytochrome c reductase, heme oxygenase, and biliverdin reductase inheme degradation. J. Biol. Chem. 257, 7786–7793
20 Schacter, B. A., Nelson, E. B., Marver, H. S. and Masters, B. S. (1972) Immunochemicalevidence for an association of heme oxygenase with the microsomal electron transportsystem. J. Biol. Chem. 247, 3601–3607
21 Muramoto, T., Tsurui, N., Terry, M. J., Yokota, A. and Kohchi, T. (2002) Expression andbiochemical properties of a ferredoxin-dependent heme oxygenase required forphytochrome chromophore synthesis. Plant Physiol. 130, 1958–1966
c© The Authors Journal compilation c© 2010 Biochemical Society

434 B. Gisk and others
22 Cornejo, J., Willows, R. D. and Beale, S. I. (1998) Phytobilin biosynthesis: cloning andexpression of a gene encoding soluble ferredoxin-dependent heme oxygenase fromSynechocystis sp. PCC 6803. Plant J. 15, 99–107
23 Wegele, R., Tasler, R., Zeng, Y., Rivera, M. and Frankenberg-Dinkel, N. (2004) The hemeoxygenase(s)-phytochrome system of Pseudomonas aeruginosa. J. Biol. Chem. 279,45791–45802
24 Paiva-Silva, G. O., Cruz-Oliveira, C., Nakayasu, E. S., Maya-Monteiro, C. M., Dunkov,B. C., Masuda, H., Almeida, I. C. and Oliveira, P. L. (2006) A heme-degradation pathwayin a blood-sucking insect. Proc. Natl. Acad. Sci. U.S.A. 103, 8030–8035
25 Ratliff, M., Zhu, W., Deshmukh, R., Wilks, A. and Stojiljkovic, I. (2001) Homologuesof neisserial heme oxygenase in gram-negative bacteria: degradation of heme bythe product of the pigA gene of Pseudomonas aeruginosa. J. Bacteriol. 183,6394–6403
26 Caignan, G. A., Deshmukh, R., Wilks, A., Zeng, Y., Huang, H. W., Moenne-Loccoz, P.,Bunce, R. A., Eastman, M. A. and Rivera, M. (2002) Oxidation of heme to β- andδ-biliverdin by Pseudomonas aeruginosa heme oxygenase as a consequence of anunusual seating of the heme. J. Am. Chem. Soc. 124, 14879–14892
27 Friedman, J., Lad, L., Li, H., Wilks, A. and Poulos, T. L. (2004) Structural basis for novelδ-regioselective heme oxygenation in the opportunistic pathogen Pseudomonasaeruginosa. Biochemistry 43, 5239–5245
28 Nakagawa, T., Kurose, T., Hino, T., Tanaka, K., Kawamukai, M., Niwa, Y., Toyooka, K.,Matsuoka, K., Jinbo, T. and Kimura, T. (2007) Development of series of gateway binaryvectors, pGWBs, for realizing efficient construction of fusion genes for planttransformation. J. Biosci. Bioeng. 104, 34–41
29 Yamaoka, S. and Leaver, C. J. (2008) EMB2473/MIRO1, an Arabidopsis Miro GTPase, isrequired for embryogenesis and influences mitochondrial morphology in pollen. PlantCell 20, 589–601
30 Voinnet, O., Rivas, S., Mestre, P. and Baulcombe, D. (2003) An enhanced transientexpression system in plants based on suppression of gene silencing by the p19 protein oftomato bushy stunt virus. Plant J. 33, 949–956
31 Muramoto, T., Kohchi, T., Yokota, A., Hwang, I. and Goodman, H. M. (1999) TheArabidopsis photomorphogenic mutant hy1 is deficient in phytochrome chromophorebiosynthesis as a result of a mutation in a plastid heme oxygenase. Plant Cell 11,335–347
32 Gill, S. C. and von Hippel, P. H. (1989) Calculation of protein extinction coefficients fromamino acid sequence data. Anal. Biochem. 182, 319–326
33 Berry, E. A. and Trumpower, B. L. (1987) Simultaneous determination of hemes a, b, and cfrom pyridine hemochrome spectra. Anal. Biochem. 161, 1–15
34 Brindle, N. J., North, A. C. and Brown, S. B. (1986) Theoretical prediction andexperimental measurement of the bile-pigment isomer pattern obtained from degradationof catalase haem. Biochem. J. 236, 303–306
35 Arnon, D. I. and Whatley, F. R. (1949) Is chloride a coenzyme of photosynthesis? Science110, 554–556
36 Emanuelsson, O., Nielsen, H. and von Heijne, G. (1999) ChloroP, a neural network-basedmethod for predicting chloroplast transit peptides and their cleavage sites. Protein Sci. 8,978–984
37 Wilks, A. and Schmitt, M. P. (1998) Expression and characterization of a heme oxygenase(Hmu O) from Corynebacterium diphtheriae. Iron acquisition requires oxidative cleavageof the heme macrocycle. J. Biol. Chem. 273, 837–841
38 Suzuki, T., Sato, M., Ishikawa, K. and Yoshida, T. (1992) Nucleotide sequence of cDNA forporcine heme oxygenase and its expression in Escherichia coli. Biochem. Int. 28,887–893
39 Wilks, A. and Ortiz de Montellano, P. R. (1993) Rat liver heme oxygenase. High levelexpression of a truncated soluble form and nature of the meso-hydroxylating species.J. Biol. Chem. 268, 22357–22362
40 Sato, H., Sugishima, M., Sakamoto, H., Higashimoto, Y., Shimokawa, C., Fukuyama, K.,Palmer, G. and Noguchi, M. (2009) Crystal structure of rat haem oxygenase-1 in complexwith ferrous verdohaem: presence of a hydrogen-bond network on the distal side.Biochem. J. 419, 339–345
41 Bianchetti, C. M., Yi, L., Ragsdale, S. W. and Phillips, Jr, G. N. (2007) Comparison ofapo- and heme-bound crystal structures of a truncated human heme oxygenase-2. J. Biol.Chem. 282, 37624–37631
42 Sugishima, M., Hagiwara, Y., Zhang, X., Yoshida, T., Migita, C. T. and Fukuyama, K.(2005) Crystal structure of dimeric heme oxygenase-2 from Synechocystis sp. PCC 6803in complex with heme. Biochemistry 44, 4257–4266
43 Gohya, T., Zhang, X., Yoshida, T. and Migita, C. T. (2006) Spectroscopic characterizationof a higher plant heme oxygenase isoform-1 from Glycine max (soybean)–coordinationstructure of the heme complex and catabolism of heme. FEBS J. 273, 5384–5399
44 Frankenberg-Dinkel, N. (2004) Bacterial heme oxygenases. Antioxid. Redox Signal. 6,825–834
45 Wilks, A. (2002) Heme oxygenase: evolution, structure and mechanism. Antiox. RedoxSignal. 4, 603–614
46 Wilks, A., Black, S. M., Miller, W. L. and Ortiz de Montellano, P. R. (1995) Expression andcharacterization of truncated human heme oxygenase (hHO-1) and a fusion protein ofhHO-1 with human cytochrome P450 reductase. Biochemistry 34, 4421–4427
47 Rhie, G. and Beale, S. I. (1992) Biosynthesis of phycobilins. Ferredoxin-supportedNADPH-independent heme oxygenase and phycobilin-forming activities from Cyanidiumcaldarium. J. Biol. Chem. 267, 16088–16093
48 Davis, S. J., Kurepa, J. and Vierstra, R. (1999) The Arabidopsis thaliana HY1 locus,required for phytochrome-chromophore biosynthesis, encodes a protein related to hemeoxygenases. Proc. Natl. Acad. Sci. U.S.A. 96, 6541–6546
49 Rhie, G. and Beale, S. I. (1995) Phycobilin biosynthesis: reductant requirements andproduct identification for heme oxygenase from Cyanidium caldarium. Arch. Biochem.Biophys. 320, 182–194
50 Terry, N. and Abadia, J. (1986) Function of iron in chloroplasts. J. Plant Nutr. 9, 609–64651 Hanke, G. T., Kimata-Ariga, Y., Taniguchi, I. and Hase, T. (2004) A post genomic
characterization of Arabidopsis ferredoxins. Plant Physiol. 134, 255–26452 Peeters, N. and Small, I. (2001) Dual targeting to mitochondria and chloroplasts.
Biochim. Biophys. Acta 1541, 54–6353 Watanabe, N., Che, F. S., Iwano, M., Takayama, S., Yoshida, S. and Isogai, A. (2001) Dual
targeting of spinach protoporphyrinogen oxidase II to mitochondria and chloroplasts byalternative use of two in-frame initiation codons. J. Biol. Chem. 276, 20474–20481
54 Rautenkranz, A., Li, L., Machler, F., Martinoia, E. and Oertli, J. J. (1994) Transport ofascorbic and dehydroascorbic acids across protoplast and vacuole membranes isolatedfrom barley (Hordeum vulgare L. cv Gerbel) leaves. Plant Physiol. 106, 187–193
55 Enser, U. and Heber, U. (1980) Metabolic regulation by pH gradients. Inhibition ofphotosynthesis by indirect proton transfer across the chloroplast envelope. Biochim.Biophys. Acta 592, 577–591
Received 21 May 2009/26 October 2009; accepted 27 October 2009Published as BJ Immediate Publication 27 October 2009, doi:10.1042/BJ20090775
c© The Authors Journal compilation c© 2010 Biochemical Society

Publikationen/Manuskripte
38
2. Heme oxygenases from Arabidopsis thaliana reveal
different mechanisms of carbon monoxide binding
Gisk, B., Frankenberg-Dinkel, N. & Koetting, C.
(Manuskript bei
“Biochemical and Biophysical Research Communications”
eingereicht)

Heme oxygenases from Arabidopsis thaliana reveal different mechanisms
of carbon monoxide binding
Björn Gisk1, Nicole Frankenberg-Dinkel
1, and Carsten Kötting
2; *
1Physiology of Microorganisms, 2 Biophysics; Ruhr-University Bochum, 44780 Bochum,
Germany
*address correspondence to: [email protected]
Abstract
Heme oxygenases (HO) are widely distributed enzymes involved in the degradation of heme
to biliverdin, carbon monoxide and Fe2+. The model plant Arabidopsis thaliana possesses
three functional HOs (HY1, HO3 and HO4) which are thus far biochemically
indistinguishable. Here we investigate binding of the reaction product and putative inhibitor
CO to these three HOs with various spectroscopic techniques. Kinetics of CO rebinding were
found to differ substantially among the HOs. At low CO concentrations a novel intermediate
was identified for HO3 and HO4, substantially slowing down rebinding. All HOs show
relatively slow geminate rebinding of CO indicating the existence of an additional transient
binding niche for CO. The positions found for the IR absorptions of CO and FeC suggest a
nonpolar distal binding site for all three HOs. The frequency of the CO vibration was
calculated by a combination band on which we report here for the first time. Another band in
the FTIR difference spectrum could be assigned to a histidine residue, probably the proximal
ligand of the heme-iron. The observed different rebinding kinetics among the HOs could
indicate adaptation of the HOs to different environments.
Keywords: heme proteins, CO rebinding, flash photolysis, FTIR (Fourier-Transform
Infrared), heme oxygenase (HO), Arabidopsis thaliana.
39

Introduction
Heme oxygenases (HO; E.C.1.14.99.3) are regiospecific enzymes catalyzing the oxygen
dependent cleavage of heme to the open-chain tetrapyrrole biliverdin, carbon monoxide (CO),
and iron (Fe2+) via the intermediates a-meso-hydroxyheme, a-verdoheme, and iron(III)-BV
IXa [1]. HOs have been identified in a wide array of organisms, including mammals [2; 3],
higher plants [4], bacteria [5; 6; 7], insects [8], and photosynthetic organisms [9; 10]. In the
crystal structures of the HOs HmuO from Corynebacterium diphtheriae and human HO-1 the
heme is coordinated via a neutral form of a histidine residue of the proximal helix [11; 12; 13;
14; 15].
The higher plant Arabidopsis thaliana possesses four HOs whereof only three are active
(HY1, HO3, and HO4) [16]. These three HOs show different levels of gene expression [17],
but have nearly indistinguishable catalytic activities and biochemical parameters [16; 18]. All
HOs are postulated to play an important role in Arabidopsis as shown by the respective
mutants [17]. The question arises, whether differential gene expression is the only regulatory
step or enzymes activity is also regulated. A key step of the reaction mechanism of HOs is the
initial binding of oxygen, which becomes the sixth coordination partner of the central iron of
the heme. This binding competes with the binding of other diatomic gases like CO [1; 19].
The latter is a product of heme degradation [1] and could inhibit HO activity [20]. Thus
discrimination of CO and O2 binding is important for efficient functioning of HOs. This is
also crucial for other heme proteins and was investigated in many studies. The diatomic gases
have different binding angles relative to the heme plane. While O2 has a bending angle of
about 110o in order to maximize the overlap of the occupied π* electrons of the ligand with
the iron d orbitals, CO binds roughly perpendicular to the heme plane [14; 21; 22] because
backbonding is optimized by the overlap of occupied Fe(II) dπ orbitals with empty π* orbitals
of CO [23; 24; 25; 26]. The different geometries can be utilized by the HOs to increase ligand
discrimination [27; 28] e.g. by steric hindrance for one geometry and efficient hydrogen
bonding for the other geometry. Beside these thermodynamic factors, further discrimination is
possible by different binding kinetics.
The rebinding of CO can be studied spectroscopically after photolysis of the CO bound
protein complex. In the past investigation of gas binding to hemoproteins have focused on
hemoglobin and myoglobin, but also neuroglobin and HOs were investigated [19; 29].
In this study we analyzed the association of CO for all three active HOs of Arabidopsis using
laser flash photolysis. In addition to the conventional absorption spectroscopy in the visible,
40

we probed the structural environments of CO bound heme in HOs with Fourier transform
infrared (FTIR) spectroscopy. FTIR is a powerful tool for studying the details of CO-binding
to heme proteins because the CO vibration is very sensitive to its environment. FeCO
backbonding is modulated by polar interactions with protein residues, and by variations in the
donor strength of the trans ligand. This modulation can be monitored by the CO stretching
frequency [30]. The aim of this study was to investigate whether the three active HOs of
Arabidopsis thaliana differ in CO binding and thereby regulating their individual activities.
Material and Methods
Expression and purification of recombinant heme oxygenases
HY1, HO3, and HO4 from Arabidopsis thaliana have been expressed, purified and
characterized as described earlier [16]. Prior to analysis the proteins were dialyzed against
reaction buffer (100 mM potassium phosphate, pH 7.2).
Sample Preparation
15 mg of crystalline hemin was dissolved in 5 mL dimethylsulfoxid (DMSO) to generate 4.6
mM hemin solution. Hemin was added to the purified HOs to result a final 1:1 heme:HO
ratio. Then the ferric form of the heme:HO complex was diluted to a final protein
concentration of 10 µM within an anaerobic cuvette by anaerobic buffer solution containing
100 mM potassium phosphate, pH 7.2 and was aerated for 20 min with argon. To generate the
ferrous heme:HO form of the HOs an excess of sodium dithionite (50 mM) was added to the
ferric complex via a gas-tight Sample Lock Hamilton syringe. For the association rate
constants defined volumes of saturated stock solution of CO (1 mM) were added to the
ferrous complex. The formation of the complexes were followed by optical absorption spectra
using an Agilent Technologies 8453 UV-visible spectroscopy system. The obtained
absorption maxima are given in Table 1.
Sample preparation for FTIR measurement also started with the generation of the ferric
heme:HO complex using an excess of sodium dithionite and aeration with carbon monoxide
for 15 min in an anaerobic cuvette. The sample was concentrated to a final concentration of
about 4 mM by evaporation of the solvent on a CaF2 window in a stream of CO.
Photolysis experiments
Single wavelength kinetics were recorded by a home built setup: Photolysis was realized by a
frequency doubled Continuum Minilite Nd:YAG laser (532 nm). Analysis was done by a
mercury lamp equipped with a 440 nm interference filter. Another 440 nm interference filter
was placed in front of a photomultiplier. The output of the photomultiplier was recorded by a
41

LeCroy 9304M oscillosope (fast times) and an Adwin 8F A/D card (slow times). The sample
was thermostated at 280 K.
Multi-wavelength experiments were carried out with another home built setup. Photolysis was
achieved by a xenon flash-lamp and analysis by an Avalight deuterium-halogen lamp and a
diode array module (Zeiss MMS UV-VIS1). Experiments were done at room temperature
(293 K).
Steady state photolysis within the FTIR experiments was realized by a green light emitting
diode (520 nm). Detection was done by a Bruker Vertex 80v spectrometer equipped with a
KBr beamsplitter and a photovoltaic MCT detector. Spectra were recorded with 4
wavenumber resolution, zero-filling of 2 and Norton-beer weak apodization. After taking
spectra for 10 s in the dark, the same was done with 520 nm irradiation. This scheme was
repeated for 1-10 h. Absorbance difference spectra of the averaged measurements with and
without irradiation were calculated.
Results
The rebinding of CO to heme proteins can be studied by photolysis of the CO bound protein
complex. Here we performed flash photolysis experiment with a time resolution of 100 ns
with the three HO:heme:CO complexes. Figure 1A shows the kinetics observed at 440 nm
after flash photolysis of the HY1:heme:CO complex with different CO concentrations. The
data for each CO concentration can be described by a biexponential function. The derived
time constants are given in Table 1. Only the slow phase is dependent on the CO
concentration. Thus the fast rate describes a geminate rebinding of CO transiently bound in an
alternative cavity [31], whereas the slow rate describes an exogenous binding of CO. Figure
1B shows that the slow rate is linearly dependent on the CO concentration. From this linear fit
we obtained a kon for CO of 1.0 µM 1s 1. This value is very similar compared to other HOs
[19].
The kinetics of CO rebinding for HO3 and HO4 (Figure 1C&D, Table 1) are more complex.
We used a set of two CO-independent and two CO dependent rates and obtained a reasonable
description of the kinetics. However, there are still some residuals left and especially the
kinetics in the µs range appear to deviate from exponential behaviour. This was also found for
other heme proteins [32] and indicates the dependence of CO movements on fluctuation of the
protein between substates [33].
The amplitude of the geminate phase for HY1 is about 35% of the amplitude for exogenous
binding (Table 1 and Figure 1). For HO4 the amplitude for the geminate phase is larger and
42

the corresponding rate constant is slower, indicating a higher barrier for the transition of CO
from the alternative cavity to the heme binding site. The opposite is observed for HO3. At
very short times the absorbance at 440 nm even increases slightly, indicating less geminate
binding or an efficient escape of the CO from the alternative binding site into the bulk.
We further investigated CO rebinding of the three HOs using a photodiode array
spectrometer. In this case the time resolution was one millisecond, but we obtained complete
UV/vis spectra for each time point. Figure 2A shows the difference spectra obtained 1 ms
after the flash. All three spectra are governed by the Soret band. The negative band due to the
depleted CO bound state is found at 421 nm, in accordance with the absorbance spectrum of
this form. The positive band due to the population of the deoxy form is found at 437 nm. The
Q-bands for the CO state are found at 539 nm and 572 nm and for the deoxy state at 559 nm
again in accordance with the absorption spectra.
The obtained kinetic data was fitted by a global exponential fit [34]. Whereas for HY1 no
further kinetic compounds were revealed by this method, for HO3 and HO4 an additional
intermediate was found. This intermediate is populated more in HO4 and with low CO
concentrations. Thus we can assign this intermediate to a product of an endogenous reaction
competing with the exogenous CO binding. While this intermediate can not be seen at 440
nm, the absorbance change at 406 nm shows (Figure 2B) its build-up and decay. A global
exponential fit of the millisecond spectral data of HO4 needs as many as five exponential
functions and the rates are close to each other. Thus we can not divide them into simple
consecutive reactions steps and the available data does not allow elucidating a reaction
mechanism. Notably, the slowest rate is about 3.5 s 1, about 40 times slower than the slowest
rate observed upon detection at 440 nm.
By means of a multivariate curve resolution (MCR) [35], we were able to obtain
concentration profiles for two components (Figure 2C&D). Note that in the spectra obtained
by a MCR of difference spectra are not the spectra of the pure compounds but spectra with
positive peaks corresponding to formed products and negative peaks linked to the
disappearance of the related reactants. Component one (red) is nearly identical to the
difference spectrum shown in Figure 2A with the negative peaks from the CO bound state and
the positive peaks of the deoxy state. The absorbance peaks of the intermediate are found as
positive peaks in component two (black) at 408 nm and 560 nm. Thus the Q-band of the
intermediate is in the same position as for the deoxy state, while the Soret band is strongly
blue shifted. The concentration of the intermediate is still low at c(CO)=125 µM, however at
physiological CO concentrations (3-30µM) [36] it will be higher.
43

In order to obtain first structural information of the HOs we applied FTIR difference
spectroscopy [37]. Figure 3A-C shows the steady state difference spectrum of the CO bound
and photolyzed state. In this difference spectrum only the bands changing upon CO
association are seen. Figure 3A shows the complete MIR difference spectrum for HO3. As
expected this spectrum is governed by the strong CO absorption of iron bound CO (Figure
3B). The position of this absorption is identical for HO3 and HO4, and it is redshifted by three
wavenumbers for HY1. The free CO is always found at 2132 wavenumbers.
Further assignments are possible by isotopic labelling or mutagenesis. At this stage we used 15N labelled HO3. Bands at 1547 cm 1 and 1576 cm 1 shift and can be assigned to amide II
modes, indicating conformational changes of the protein. Further the band at 1108 cm-1 can be
assigned to a histidine residue (Figure 3C), due to its position and shift upon 15N labelling
[38; 39]. Probably the signal originates from a histidine in the proximal position as found for
other HOs [19]. A further interesting band is found at 2456 cm 1. No functional group that is
present in the system absorbs in this region. Thus we can assign this band to a combination
band of the strong CO and the FeC vibration. By subtracting CO=1954 cm 1 we can calculate
the position of the FeC vibration to 502 cm 1. Another result from the FTIR difference
spectrum is the absence of any band between 1700 cm-1 and 1800 cm-1. This indicates that no
protonated Glu or Asp are involved in the reaction. In order to obtain further information,
experiments with site specific mutants or group specific isotopic labelling are necessary.
Discussion
Indeed we found substantial differences in the mechanism of CO rebinding among the three
HOs. Besides the different rates and amplitudes of the geminate rate, the additional
intermediate for the reactions of HO3 and HO4 is notable. For some other heme-proteins an
intermediate with a hexacoordinated iron was found. The sixth coordination ligand is
subsequentially substituted by the CO [40]. However, in that case we should find for the
intermediate the same spectral features as for the deoxy-state, but the Soret band of the
intermediate at 408 nm is clearly different. On the other hand the same position is found for
the ferric form, thus one could speculate about a change in the electronic configuration that
could play a role as an additional factor in ligand discrimination. To our knowledge no similar
reaction intermediate is reported thus far. However, often ligand binding is solely monitored
at the absorption maximum of the pentacoordinated state (440 nm) and the intermediate can
not be observed at all in that case.
44

One feature found for all three HOs is a substantial CO concentration independent rebinding
step. This step is remarkably slow. For HY1 it is 8·104 s 1. In similar measurements with
neuroglobin (Ng) [32] or Arabidopsis hemoglobins (AtHb) [41] all CO concentration
independent steps are much faster. Thus in the Arabidopsis HOs the CO can be stored more
stable in a second cavity compared to Ng or AtHb with a higher barrier for rebinding at the
heme. This possibly helps to keep CO away from the active site, where it could inhibit HO
activity.
All three HOs show only one absorption of iron bound CO within the FTIR difference
spectrum, indicating a single conformer. Further the position of the absorption is very similar
among the HOs. The position of CO and FeC are negatively correlated [42; 43; 44].
Heme proteins with the same coordination number and proximal ligand are found on
displaced lines in a plot CO versus FeC, respectively. Our result of 502 cm 1 for the FeC and
1954 cm 1 for the CO coincides with the line for six-coordinated heme with neutral histidine
as the proximal ligand.
The shift along the line reflects the amount of -backbonding from the iron to CO. A positive
polar environment facilitates the backbonding and decreases the position of CO. For
comparison, horse radish peroxidise (HRP) with an arginine hydrogen bonded to the CO and
another positive charge (protonated His) in the distal pocket is found at 1904 cm-1.
Mutagenesis of the positively charged groups in HRP leads to a blueshift of the vibration. A
similar position as for the HOs is found for myoglobin, which has a more hydrophobic pocket.
Two measurements of the CO for other HOs are reported, 1964 cm 1 for Neisseriae
meningitidis (nm-HO) and 1954 cm-1 for Pseudomonas aeruginosa (pa-HO) [45].
Interestingly a chimeric protein (nm-HO with the distal helix of pa-HO) shifts the position
close to the position of pa-HO, indicating that CO is indeed a good probe for the distal
binding site. Thus we can propose that the distal pocket in our HOs should be similar to the
one in pa-HO [46]. Here water and serine are closest to the iron.
Conclusion
Using the position of CO as a probe of the environment enables us to get first structural
insights into the binding pocket. The slow geminate rates indicate a relatively stable
additional transient binding site for CO. Further we found for HO3 and HO4 an endogenous
competitive reaction resulting in an unknown intermediate, possibly with a modified
electronic configuration. The substantial differences found might reveal various attempts to
keep CO away from the binding site in order to keep the HOs functional under varying
environmental conditions.
45

Acknowledgements
Financial support from the Deutsche Forschungsgemeinschaft (SFB 480, Teilprojekt C8) to
NFD is greatly acknowledged. CK thanks Samir El-Mashtoly for helpful discussions.
Figure Captions:
Figure 1: CO rebinding behaviour of HY1 (A), HO3 (C) and HO4 (D) from Arabidopsis
thaliana with different CO concentrations (1000 µM (black), 500 µM (red), 250 µM (green),
125 µM (blue) and 62.5 µM (cyan)) monitored by the absorption change at 440 nm. All HOs
show a fast CO concentration independent rebinding and a slow CO concentration dependent
rebinding. The lines are exponential fits with the parameters given in Table 1. (B) A plot of
the single CO-concentration dependent rate against the CO concentration reveals a linear
relationship. From the slope kon was calculated to be 1.0 µm 1s 1.
Figure 2: (A) Difference spectra of the three Arabidopsis HOs (HY1 (red), HO3 (green), and
HO4 (blue)) obtained 1 ms after flash photolysis (c(CO)=62.5 µM)). All three spectra are
governed by the Soret band. The negatively band is due to the CO bound state, the positive
band due to the deoxy form. The Q-bands are found between 539 nm - 572 nm. (B) Time
resolved absorbance changes at 406 nm after flash photolysis for the three different HOs at a
CO concentration of 125 µM. (C) MCR-analysis of the time resolved spectra of HO4 after
flash photolysis (c(CO)=125µM) with two components. Component one (red) is almost
identical to the difference spectrum after 1 ms, while component 2 (black) reveals an
additional intermediate with a Soret band at 408 nm. (D) Concentration profiles for the two
components are shown in C. Note that detection at 440 nm monitors only component 1.
Figure 3: (A) FTIR difference spectrum of HO3 (black) and 15N-labeled HO3. Bands facing
downwards belong to the CO bound state, and bands facing upwards belong to the CO free
form. The changes in the amide II (1567 cm 1 and 1547 cm 1) reveal conformational changes,
while a band 1108 cm 1 can be assigned to a histidine residue (C). Shifts due to the 15N labels
are indicated by arrows. (B) Comparison of the CO vibration (Table 1) for all three analyzed
HOs. This absorption probes the binding pocket (see text).
46
Table 1: Spectroscopic data and apparent rate constants of CO rebinding for
the heme-HO complexes.
HY1 HO3 HO4
Ferric form
max (Soret) [nm]
max (vis) [nm]
406
631
406
633
407
633
Ferrous
deoxy form
max (Soret) [nm]
max (vis) [nm]
431
561
431
558
431
561
CO form max (Soret) [nm]
max ( , ) [nm]
419
567, 538
421
569, 540
421
569, 540
kapp (endogenous rebinding)
[s1]
8.1*104
9.8*105
5.0*104
5.4*106
1.2*105
kapp (exogenous rebinding)
1 mM CO [s1]
1.0*103 1.0*104
810
8.3*103
610
kapp (exogenous rebinding)
500 µM CO [s1]
490 2.6*103
280
4.8*103
370
kapp (exogenous rebinding)
250 µM CO [s1]
260 1.7*103
190
3.4*103
220
kapp (exogenous rebinding)
125 µM CO [s1]
140 1.8*103
110
2.7*103
140
kapp (exogenous rebinding)
67.5 µM CO [s1]
61 1.6*103
55
2.6*103
88
Ratio exogenous : endogenous
CO rebinding
1 : 0.35 1 : 0.50 1 : 0.29
CO form CO [cm1] 1951.1 1954.4
15N labeled
1953.7
1954.4
47

References
[1] P.R. Ortiz de Montellano, Curr. Opin. Chem. Biol. 4 (2000) 221-227. [2] A. Wilks, S.M. Black, W.L. Miller, and P.R. Ortiz de Montellano, Biochemistry 34
(1995) 4421-7. [3] R. Tenhunen, H.S. Marver, and R. Schmid, J. Biol. Chem. 244 (1969) 6388-94. [4] S.J. Davis, S.H. Bhoo, A.M. Durski, J.M. Walker, and R.D. Vierstra, Plant. Physiol. 126
(2001) 656-69. [5] M. Schmitt, Infect. Immun. 65 (1997) 4634-4641. [6] M. Ratliff, W. Zhu, R. Deshmukh, A. Wilks, and I. Stojiljkovic, J. Bacteriol. 183 (2001)
6394-6403. [7] U.A. Ochsner, and M.L. Vasil, Proc. Natl. Acad. Sci. U. S. A. 93 (1996) 4409-4414. [8] G.O. Paiva-Silva, C. Cruz-Oliveira, E.S. Nakayasu, C.M. Maya-Monteiro, B.C. Dunkov,
H. Masuda, I.C. Almeida, and P.L. Oliveira, Proc. Natl. Acad. Sci. U. S. A. 103 (2006) 8030-5.
[9] J. Cornejo, R.D. Willows, and S.I. Beale, Plant J. 15 (1998) 99-107. [10] X. Zhang, C.T. Migita, M. Sato, M. Sasahara, and T. Yoshida, FEBS J. 272 (2005)
1012-22. [11] J. Sun, T.M. Loehr, A. Wilks, and P.R. Ortiz de Montellano, Biochemistry 33 (1994)
13734-40. [12] A. Wilks, P.R. Ortiz de Montellano, J. Sun, and T.M. Loehr, Biochemistry 35 (1996)
930-6. [13] D.J. Schuller, A. Wilks, P.R. Ortiz de Montellano, and T.L. Poulos, Nat. Struct. Biol. 6
(1999) 860-867. [14] M. Unno, T. Matsui, G.C. Chu, M. Couture, T. Yoshida, D.L. Rousseau, J.S. Olson, and
M. Ikeda-Saito, J. Biol. Chem. 279 (2004) 21055-61. [15] M. Ito-Maki, K. Ishikawa, K.M. Matera, M. Sato, M. Ikeda-Saito, and T. Yoshida, Arch.
Biochem. Biophys. 317 (1995) 253-8. [16] B. Gisk, Y. Yasui, T. Kohchi, and N. Frankenberg-Dinkel, Biochem. J. 425 (2010) 425-
34. [17] T.J. Emborg, J.M. Walker, B. Noh, and R.D. Vierstra, Plant. Physiol. 140 (2006) 856-
68. [18] T. Muramoto, N. Tsurui, M.J. Terry, A. Yokota, and T. Kohchi, Plant. Physiol. 130
(2002) 1958-66. [19] J. Friedman, Y.T. Meharenna, A. Wilks, and T.L. Poulos, J. Biol. Chem. 282 (2007)
1066-71. [20] M. Noguchi, T. Yoshida, and G. Kikuchi, J. Biochem. 90 (1981) 1671-5. [21] M. Sugishima, H. Sakamoto, Y. Higashimoto, M. Noguchi, and K. Fukuyama, J. Biol.
Chem. 278 (2003) 32352-8. [22] M. Unno, T. Matsui, and M. Ikeda-Saito, Nat. Prod. Rep. 24 (2007) 553-70. [23] T.G. Spiro, and A.A. Jarzecki, Curr. Opin. Chem. Biol. 5 (2001) 715-23. [24] T.G. Spiro, and P.M. Kozlowski, Acc. Chem. Res. 34 (2001) 137-44. [25] M. Lim, T.A. Jackson, and P.A. Anfinrud, Science 269 (1995) 962-966. [26] F. De Angelis, A.A. Jarzecki, R. Car, and T.G. Spiro, J. Phys. Chem. B 109 (2005)
3065-3070. [27] G.S. Kachalova, A.N. Popov, and H.D. Bartunik, Science 284 (1999) 473-6. [28] R. Jain, and M.K. Chan, J. Biol. Inorg. Chem. 8 (2003) 1-11. [29] J.L. Wang, S. Lu, P. Moenne-Loccoz, and P.R.O. de Montellano, J. Biol. Chem. 278
(2003) 2341-2347. [30] T.G. Spiro, and I.H. Wasbotten, J. Inorg. Biochem. 99 (2005) 34-44.
48

[31] M. Sugishima, H. Sakamoto, M. Noguchi, and K. Fukuyama, J. Mol. Biol. 341 (2004) 7-13.
[32] J.M. Kriegl, A.J. Bhattacharyya, K. Nienhaus, P.C. Deng, O. Minkow, and G.U. Nienhaus, Proc. Natl. Acad. Sci. U. S. A. 99 (2002) 7992-7997.
[33] H. Frauenfelder, P.G. Wolynes, and R.H. Austin, Rev. Mod. Phys. 71 (1999) S419-S430.
[34] B. Hessling, G. Souvignier, and K. Gerwert, Biophys. J. 65 (1993) 1929-1941. [35] L. Blanchet, C. Ruckebusch, A. Mezzetti, J.P. Huvenne, and A. de Juan, J. Phys. Chem.
B 113 (2009) 6031-6040. [36] C.L. Weeks, S. Singh, P. Madzelan, R. Banerjee, and T.G. Spiro, J. Am. Chem. Soc. 131
(2009) 12809-12816. [37] C. Kötting, and K. Gerwert, ChemPhysChem 6 (2005) 881-888. [38] A. Barth, and C. Zscherp, Q. Rev. Biophys. 35 (2002) 369-430. [39] W.J. Ingledew, and P.R. Rich, Biochem. Soc. Trans. 33 (2005) 886-889. [40] J.T. Trent, R.A. Watts, and M.S. Hargrove, J. Biol. Chem. 276 (2001) 30106-30110. [41] S. Bruno, S. Faggiano, F. Spyrakis, A. Mozzarelli, S. Abbruzzetti, E. Grandi, C.
Viappiani, A. Feis, S. Mackowiak, G. Smulevich, E. Cacciatori, and P. Dominici, J. Am. Chem. Soc. 129 (2007) 2880-2889.
[42] T. Egawa, and S.R. Yeh, Journal of Inorganic Biochemistry 99 (2005) 72-96. [43] M. Ibrahim, C.L. Xu, and T.G. Spiro, J. Am. Chem. Soc. 128 (2006) 16834-16845. [44] J. Igarashi, A. Sato, T. Kitagawa, T. Yoshimura, S. Yamauchi, I. Sagami, and T.
Shimizu, J. Biol. Chem. 279 (2004) 15752-15762. [45] R. Deshmukh, Y.H. Zeng, L.M. Furci, H.W. Huang, B.N. Morgan, S. Sander, A.Y.
Alontaga, R.A. Bunce, P. Moenne-Loccoz, M. Rivera, and A. Wilks, Biochemistry 44 (2005) 13713-13723.
[46] J. Friedman, L. Lad, H. Li, A. Wilks, and T.L. Poulos, Biochemistry 43 (2004) 5239-45.
49
Figure 1
50
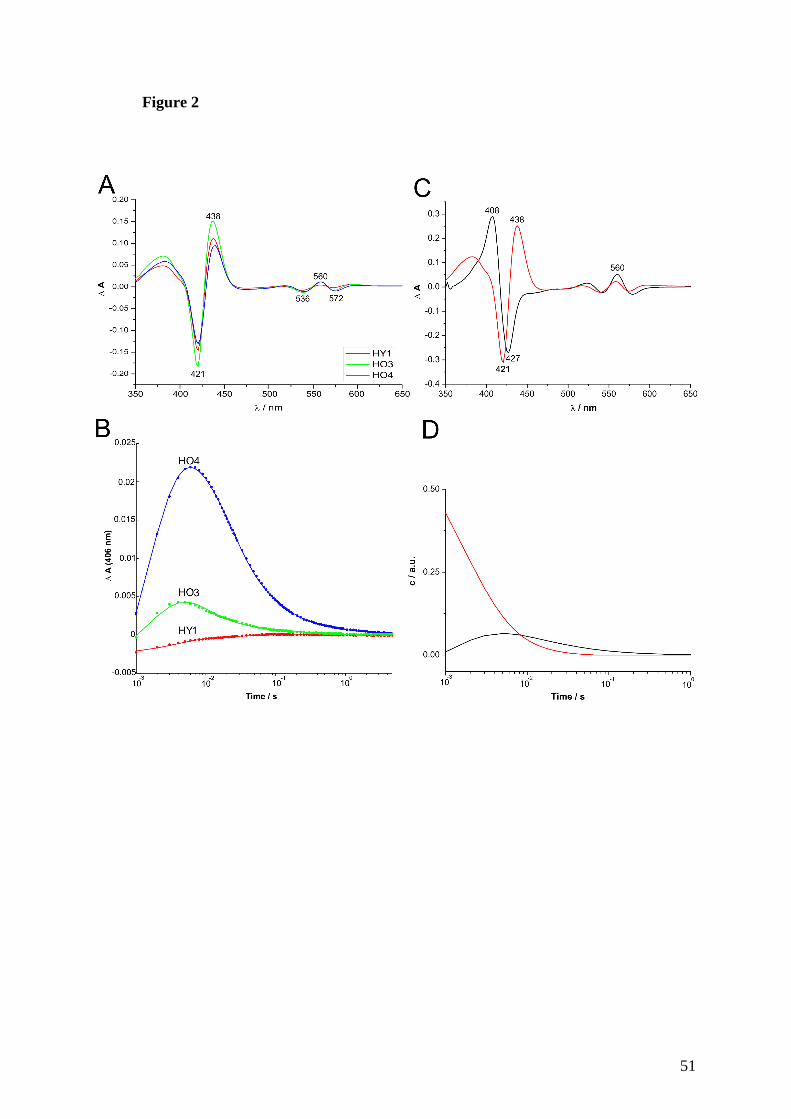
Figure 2
51
Figure 3
52

Publikationen/Manuskripte
53
3. Enzymatic ring opening of an iron corrole by plant-type
heme oxygenases:
Unexpected substrate and protein selectivities
Gisk, B., Brégier, F., Krueger, R. A., Broering, M. &
Frankenberg-Dinkel, N.
(Manuskript bei “Biochemistry” in Revision)

Enzymatic ring opening of an iron corrole by plant-type
heme oxygenases: Unexpected substrate and protein selectivities
Journal: Biochemistry
Manuscript ID: bi-2010-014369.R1
Manuscript Type: Rapid Report
Date Submitted by the Author:
n/a
Complete List of Authors: Gisk, Björn; Ruhr-University, Physiology of Microorganisms Brégier, Frédérique; Philipps-Universität Marburg, Fachbereich Chemie Krueger, Robin; Philipps-Universität Marburg, Fachbereich Chemie Bröring, Martin; TU Braunschweig, Institut für Anorganische und Analytische Chemie; Philipps-Universität Marburg, Fachbereich Chemie Frankenberg-Dinkel, Nicole; Ruhr-University, Physiology of Microorganisms
ACS Paragon Plus Environment
Biochemistry

1
Enzymatic ring opening of an iron corrole by plant-type heme oxygenases: Unexpected substrate and protein selectivities†
Björn Gisk, Frédérique Brégier
‡,∫, Robin A. Krüger‡,†, Martin Bröring
‡,§,*, and Nicole Frankenberg-
Dinkel,*
Physiology of Microorganisms, Ruhr-University Bochum, 44780 Bochum, Germany, and ‡Fachbereich Chemie, Philipps-University Marburg, 35032 Marburg, Germany
RECEIVED DATE (automatically inserted by publisher);
† This work was supported by grants from the Volkswagen foundation and the Deutsche Forschungsgemienschaft to MB and NFD. ∫ current address: ICMUB, Dijon, France † current address: University of Calgary, Chemistry, Calgary, AB, CAN, T2N, 1N4 § current address: Institute for Inorganic and Analytical chemistry, Technical University Braunschweig, 38106 Braunschweig, Germany * To whom the correspondence should be addressed: Email: [email protected]. Telephone +49 234 32 23101, FAX +49 234 32 14620 or m.broering@tu-
bs.de, Telefone +49 531 391 5302
ABSTRACT Heme oxygenases are widely distributed enzymes involved in the oxidative cleavage of the heme macrocycle to yield the open-chain tetrapyrrole biliverdin IX, CO and iron. For the first time two regioisomeric iron corroles (α-CH- and γ-CH-Fe(cor)) have been employed as artificial substrate and cofactor analogs to mammalian, plant, cyanobacterial and bacterial heme oxygenases. The non-natural enzymatic cleavage of γ-CH-Fe(cor), catalyzed by plant-type heme oxygenases from Arabidopsis thaliana and Synechocystis sp. happens selectively at the unexpected bipyrrolic position, and yields a biomimetic biliverdin-like product. The reaction is selective for this corrole regioisomer AND for plant-type heme oxygenases, and is the first report of an enzymatic corrole ring opening.
Metal corroles are artificial one-carbon short relatives of
the ubiquitous metal porphyrins that have attracted considerable attention over the last decade (1, 2). These metal complexes are special with respect to their electronic structures and – often – the non-innocent behavior of the organic ligands which tend to form magnetically stabilized radicals (3-8). The unique properties of metal corroles have more recently advanced into topical fields like catalysis, photophysics, medicinal chemistry, and others (9-11). In many of these applications, the performance of metal corroles was found clearly superior over related metal porphyrins.
Besides the electronic peculiarities, metal corroles differ from the parent porphyrin complexes mainly by their reduced symmetry (12). We have attempted to make use of this property variation by introducing regioisomeric iron corroles 1 and 2 as heme cofactor and substrate analogs for heme oxygenases (HOs, Scheme 1). HOs are a family of enzymes which catalyze the oxygen-dependent degradation of heme to biliverdin IX, CO, and iron (13). Three molecules of O2 and seven reducing equivalents are required for this conversion. In mammals, HOs have a key function in physiological heme metabolism, whereas in certain pathogenic bacteria HOs are used for iron utilization from heme obtained from the host (14-16). An additional role of mammalian HOs in the production of CO as a
neurotransmitter has been demonstrated, and recently, bacterial HO has become a target for drug research (17-20). In contrast, higher plant, cyanobacterial, and algal HOs are involved in the biosynthesis of open-chain tetrapyrroles which are used as the chromophores in light-sensing phytochromes or in the light harvesting phycobiliproteins (21-23). The majority of HOs function on heme upon the regioselective extrusion of the α situated methine bridge (Scheme 1). Propionate substituted metal corroles (24, 25) are thought to be useful probes for this reaction. Employing α-CH- and γ-CH-Fe(cor) 1 and 2 as substrate analogs, it could be speculated, that the α-CH isomer 1 is transformed to ring oxygenated or ring opened products while the γ-CH isomer 2 should not be converted by the enzymes. Our results, however, show that in fact the opposite scenario occurs, and that a remarkable reactivity difference exists between HOs from different organisms.
SCHEME 1. Heme and regioisomeric iron corroles 1 and 2 as substrate analogs. For illustration purposes the nomenclature for the heme methine bridges has been adapted for the employed corroles as well.
First we on the HOs from Pseudomonas aeruginosa (15, 26, 27). This bacterium is the first biological example
NN
NN
HO2CHO2C
Fe
α
β
γ
δ
N N
NNFe
Cl
NN
N NFe
Cl
HO2CHO2C
α
γ
HO2CHO2C
α
γ
heme
α-CH-Fe(cor)
1
γ-CH-Fe(cor)
2
ACS Paragon Plus Environment
Biochemistry
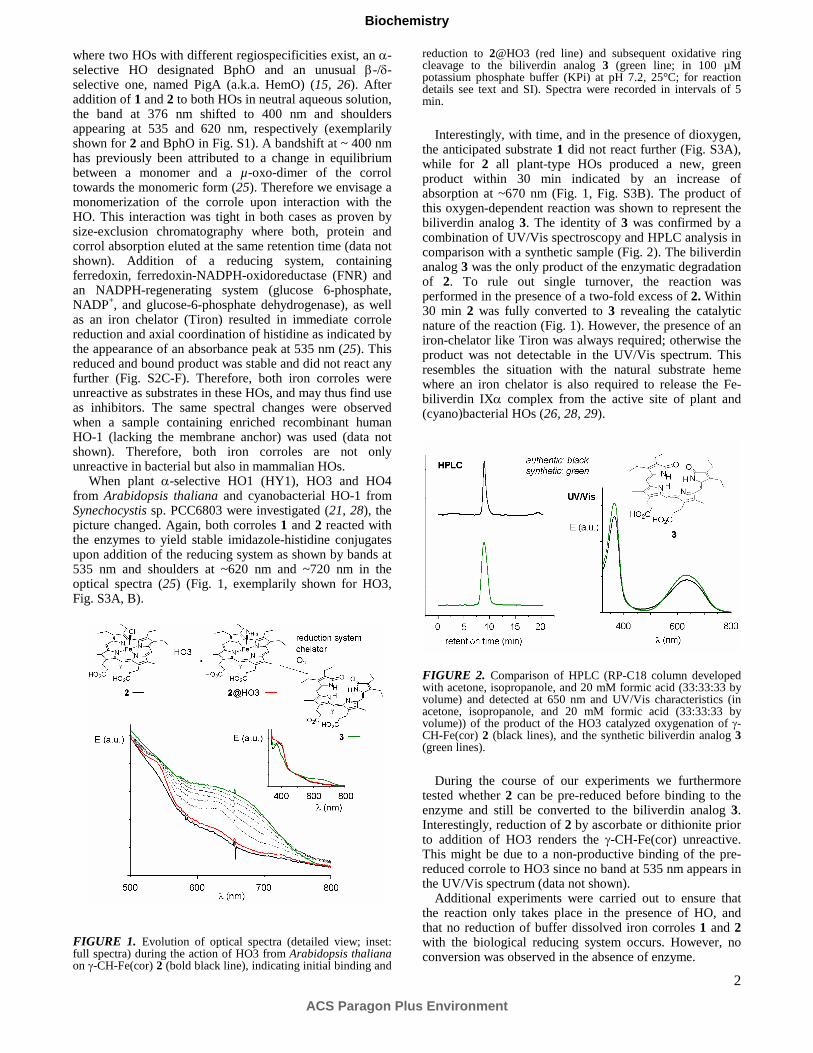
2
where two HOs with different regiospecificities exist, an α-selective HO designated BphO and an unusual β-/δ-selective one, named PigA (a.k.a. HemO) (15, 26). After addition of 1 and 2 to both HOs in neutral aqueous solution, the band at 376 nm shifted to 400 nm and shoulders appearing at 535 and 620 nm, respectively (exemplarily shown for 2 and BphO in Fig. S1). A bandshift at ~ 400 nm has previously been attributed to a change in equilibrium between a monomer and a µ-oxo-dimer of the corrol towards the monomeric form (25). Therefore we envisage a monomerization of the corrole upon interaction with the HO. This interaction was tight in both cases as proven by size-exclusion chromatography where both, protein and corrol absorption eluted at the same retention time (data not shown). Addition of a reducing system, containing ferredoxin, ferredoxin-NADPH-oxidoreductase (FNR) and an NADPH-regenerating system (glucose 6-phosphate, NADP+, and glucose-6-phosphate dehydrogenase), as well as an iron chelator (Tiron) resulted in immediate corrole reduction and axial coordination of histidine as indicated by the appearance of an absorbance peak at 535 nm (25). This reduced and bound product was stable and did not react any further (Fig. S2C-F). Therefore, both iron corroles were unreactive as substrates in these HOs, and may thus find use as inhibitors. The same spectral changes were observed when a sample containing enriched recombinant human HO-1 (lacking the membrane anchor) was used (data not shown). Therefore, both iron corroles are not only unreactive in bacterial but also in mammalian HOs.
When plant α-selective HO1 (HY1), HO3 and HO4 from Arabidopsis thaliana and cyanobacterial HO-1 from Synechocystis sp. PCC6803 were investigated (21, 28), the picture changed. Again, both corroles 1 and 2 reacted with the enzymes to yield stable imidazole-histidine conjugates upon addition of the reducing system as shown by bands at 535 nm and shoulders at ~620 nm and ~720 nm in the optical spectra (25) (Fig. 1, exemplarily shown for HO3, Fig. S3A, B).
FIGURE 1. Evolution of optical spectra (detailed view; inset: full spectra) during the action of HO3 from Arabidopsis thaliana on γ-CH-Fe(cor) 2 (bold black line), indicating initial binding and
reduction to 2@HO3 (red line) and subsequent oxidative ring cleavage to the biliverdin analog 3 (green line; in 100 µM potassium phosphate buffer (KPi) at pH 7.2, 25°C; for reaction details see text and SI). Spectra were recorded in intervals of 5 min.
Interestingly, with time, and in the presence of dioxygen, the anticipated substrate 1 did not react further (Fig. S3A), while for 2 all plant-type HOs produced a new, green product within 30 min indicated by an increase of absorption at ~670 nm (Fig. 1, Fig. S3B). The product of this oxygen-dependent reaction was shown to represent the biliverdin analog 3. The identity of 3 was confirmed by a combination of UV/Vis spectroscopy and HPLC analysis in comparison with a synthetic sample (Fig. 2). The biliverdin analog 3 was the only product of the enzymatic degradation of 2. To rule out single turnover, the reaction was performed in the presence of a two-fold excess of 2. Within 30 min 2 was fully converted to 3 revealing the catalytic nature of the reaction (Fig. 1). However, the presence of an iron-chelator like Tiron was always required; otherwise the product was not detectable in the UV/Vis spectrum. This resembles the situation with the natural substrate heme where an iron chelator is also required to release the Fe-biliverdin IXα complex from the active site of plant and (cyano)bacterial HOs (26, 28, 29).
FIGURE 2. Comparison of HPLC (RP-C18 column developed with acetone, isopropanole, and 20 mM formic acid (33:33:33 by volume) and detected at 650 nm and UV/Vis characteristics (in acetone, isopropanole, and 20 mM formic acid (33:33:33 by volume)) of the product of the HO3 catalyzed oxygenation of γ-CH-Fe(cor) 2 (black lines), and the synthetic biliverdin analog 3 (green lines).
During the course of our experiments we furthermore tested whether 2 can be pre-reduced before binding to the enzyme and still be converted to the biliverdin analog 3. Interestingly, reduction of 2 by ascorbate or dithionite prior to addition of HO3 renders the γ-CH-Fe(cor) unreactive. This might be due to a non-productive binding of the pre-reduced corrole to HO3 since no band at 535 nm appears in the UV/Vis spectrum (data not shown).
Additional experiments were carried out to ensure that the reaction only takes place in the presence of HO, and that no reduction of buffer dissolved iron corroles 1 and 2 with the biological reducing system occurs. However, no conversion was observed in the absence of enzyme.
ACS Paragon Plus Environment
Biochemistry

3
As γ-CH-Fe(cor) 2 is regioselectively cleaved at the bipyrrolic position, no CO would be expected to develop. This additional peculiarity has been independently confirmed by a myoglobin addition assay (16). No specific myoglobin-CO complex absorption in the UV/Vis spectrum could be detected indicating that indeed no CO is produced during the course of the reaction (data not shown). Furthermore, assays performed under anaerobic conditions stopped at the reduction step as indicated by absorption at ~535 nm. Therefore, at least one oxygen atom of the linear tetrapyrrole 3 might originate from dioxygen, rather than from water. From a balanced chemical equation this reaction is categorized as an addition of one equivalent of O2 and two equivalents of H2O to a molecule of γ-CH-Fe(cor) 2, with elimination of one equivalent of FeO(OH).
Taking the currently postulated mechanism of HO into consideration one would speculate that the employed γ-CH-Fe(cor) 2 enters the active site pocket of plant-type HOs with the propionate side chains facing the solvent. Stable coordination via the axial histidine ligand requires reduction of γ-CH-Fe(cor) 2 within the enzyme. In analogy to the degradation of heme oxygen will be required and could likely generate a short-lived peroxo-species which specifically targets the bipyrrolic position which in case of the γ-CH-Fe(cor) 2 will be deeply buried in the HO active site pocket. The reason why only γ-CH-Fe(cor) 2 is converted is currently unknown. Our results appear peculiar and not understood at the first glance, but should be explainable on the basis of a thorough analysis of the different ligand binding and electron transfer kinetics in each case. We are currently performing detailed investigations towards this end. Another important issue of this result, however, is the biological aspect that heme oxygenases of different origin employ different substrate specificity towards the iron corroles. This would point towards a slightly different architecture of the active site of plant-type HOs compared to mammalian and bacterial HOs. Based on a structural model of pea HO it has been postulated that the active site of plant HOs is more spacious than the respective enzymes from other sources due to the requirement of a second reductant (ascorbate) for full catalytic activity (29, 30). If space is the only feature that allows iron corrole degradation and what is the chemical nature of this reaction will be addressed in the future and will be highly dependent on the crystal structure of a plant HO.
SUPPORTING INFORMATION AVAILABLE: Detailed procedures for the preparation of 1 and 3, protein purification, data of bacterial HO (BphO, PigA) and cyanobacterial HO-1 action on 1 and 2, and data of plant HO3 action on 1. This material is available free of charge via the Internet at http://pubs.acs.org.
References 1. Erben, C., Will, S., and Kadish, K. M. (2000)
Metallocorroles: Molecular Structure, Spectroscopy and Electronic States, in The Porphyrin Handbook (Kadish, K. M., Smith, K. M., and Guilard, R., Eds.), pp 233-300, Academic Press, San Diego.
2. Paolesse, R. (2000) Syntheses of Corroles, in The
Porphyrin Handbook (Kadish, K. M., Smith, K. M., and
Guilard, R., Eds.), pp 201-233, Academic Press, San Diego.
3. Broering, M., Bregier, F., Consul Tejero, E., Hell, C., and Holthausen, M. C. (2007) Revisiting the electronic ground state of copper corroles, Angew Chem Int Ed
Engl 46, 445-448. 4. Hocking, R. K., DeBeer George, S., Gross, Z., Walker,
F. A., Hodgson, K. O., Hedman, B., and Solomon, E. I. (2009) “Fe L- and K-edge XAS of Low-Spin Ferric Corrole: Bonding and Reactivity Relative to Low-Spin Ferric Porphryin, Inorg Chem 48, 1678-1688.
5. Nardis, S., Paolesse, R., Licoccia, S., Fronczek, F. R., Vicente, M. G., Shokhireva, T. K., Cai, S., and Walker, F. A. (2005) NMR and structural investigations of a nonplanar iron corrolate: modified patterns of spin delocalization and coupling in a slightly saddled chloroiron(III) corrolate radical, Inorg Chem 44, 7030-7046.
6. Roos, B. O., Veryazov, V., Conradie, J., Taylor, P. R., and Ghosh, A. (2008) Not innocent: verdict from ab initio multiconfigurational second-order perturbation theory on the electronic structure of chloroiron corrole, J Phys Chem B 112, 14099-14102.
7. Wasbotten, I., and Ghosh, A. (2006) Biliverdine-based metalloradicals: sterically enhanced noninnocence, Inorg Chem 45, 4914-4921.
8. Ye, S., Tuttle, T., Bill, E., Simkhovich, L., Gross, Z., Thiel, W., and Neese, F. (2008) The electronic structure of iron corroles: a combined experimental and quantum chemical study, Chemistry 14, 10839-10851.
9. Aviv, I., and Gross, Z. (2007) Corrole-based Applications, Chem. Commun 1987-1999.
10. Aviv-Harel, I., and Gross, Z. (2009) Aura of corroles, Chemistry 15, 8382-8394.
11. Flamigni, L., and Gryko, D. T. (2009) Photoactive corrole-based arrays, Chem Soc Rev 38, 1635-1646.
12. Kuck, S., Hoffmann, G., Broring, M., Fechtel, M., Funk, M., and Wiesendanger, R. (2008) "Naked" iron-5,10,15-triphenylcorrole on Cu(111): observation of chirality on a surface and manipulation of multiple conformational states by STM, J Am Chem Soc 130, 14072-14073.
13. Matsui, T., Unno, M., and Ikeda-Saito, M. (2010) Heme oxygenase reveals its strategy for catalyzing three successive oxygenation reactions, Acc Chem Res 43, 240-247.
14. Abraham, N. G., Drummond, G. S., Lutton, J. D., and Kappas, A. (1996) The Biological Significance and Physiological Role of Heme Oxygenase, Cell Physiol.
Biochem. 6, 129-168. 15. Ratliff, M., Zhu, W., Deshmukh, R., Wilks, A., and
Stojiljkovic, I. (2001) Homologues of neisserial heme oxygenase in gram-negative bacteria: degradation of heme by the product of the pigA gene of Pseudomonas
aeruginosa, J Bacteriol 183, 6394-6403. 16. Wilks, A., and Schmitt, M. P. (1998) Expression and
characterization of a heme oxygenase (Hmu O) from Corynebacterium diphtheriae. Iron acquisition requires oxidative cleavage of the heme macrocycle, J Biol
Chem 273, 837-841. 17. Boehning, D., Moon, C., Sharma, S., Hurt, K. J.,
Hester, L. D., Ronnett, G. V., Shugar, D., and Snyder, S. H. (2003) Carbon Monoxide Neurotransmission Activated by CK2 Phosphorylation of Heme Oxygenase-2, Neuron 40, 129-137.
18. Furci, L. M., Lopes, P., Eakanunkul, S., Zhong, S., MacKerell, A. D., Jr., and Wilks, A. (2007) Inhibition of the bacterial heme oxygenases from Pseudomonas
ACS Paragon Plus Environment
Biochemistry

4
aeruginosa and Neisseria meningitidis: novel antimicrobial targets, J Med Chem 50, 3804-3813.
19. Otterbein, L. E., Bach, F. H., Alam, J., Soares, M., Tao Lu, H., Wysk, M., Davis, R. J., Flavell, R. A., and Choi, A. M. (2000) Carbon monoxide has anti-inflammatory effects involving the mitogen-activated protein kinase pathway, Nat Med 6, 422-428.
20. Piepoli, A. L., de Salvatore, G., Lemoli, M., de Benedictis, L., Mitolo-Chieppa, D., and de Salvia, M. A. (2008) Modulation of heme oxygenase/carbon monoxide system affects the inhibitory neurotransmission involved in gastrointestinal motility of streptozotocin-treated diabetic rats, Neurogastroenterol Motil 20, 1251-1262.
21. Cornejo, J., Willows, R. D., and Beale, S. I. (1998) Phytobilin biosynthesis: cloning and expression of a gene encoding soluble ferredoxin-dependent heme oxygenase from Synechocystis sp. PCC 6803, Plant J 15, 99-107.
22. Muramoto, T., Kohchi, T., Yokota, A., Hwang, I., and Goodman, H. M. (1999) The Arabidopsis photomorphogenic mutant hy1 is deficient in phytochrome chromophore biosynthesis as a result of a mutation in a plastid heme oxygenase, Plant Cell 11, 335-348.
23. Rhie, G., and Beale, S. I. (1992) Biosynthesis of phycobilins. Ferredoxin-supported NADPH-independent heme oxygenase and phycobilin-forming activities from Cyanidium caldarium, J Biol Chem 267, 16088-16093.
24. Broering, M., Bregier, F., Krueger, R., and Kleeberg, C. (2008) Functional Porphyrinoids from a Biomimetically Decorated Bipyrrole, Eur. J. Inorg. Chem. 35, 5505-5512.
25. Matsuo, T., Hayashi, A., Abe, M., Matsuda, T., Hisaeda, Y., and Hayashi, T. (2009) Meso-unsubstituted iron corrole in hemoproteins: remarkable differences in effects on peroxidase activities between myoglobin and horseradish peroxidase, J Am Chem Soc 131, 15124-15125.
26. Wegele, R., Tasler, R., Zeng, Y., Rivera, M., and Frankenberg-Dinkel, N. (2004) The heme oxygenase(s)-phytochrome system of Pseudomonas aeruginosa, J
Biol Chem 279, 45791-45802. 27. Wilks, A., Black, S. M., Miller, W. L., and Ortiz de
Montellano, P. R. (1995) Expression and characterization of truncated human heme oxygenase (hHO-1) and a fusion protein of hHO-1 with human cytochrome P450 reductase, Biochemistry 34, 4421-4427.
28. Gisk, B., Yasui, Y., Kohchi, T., and Frankenberg-Dinkel, N. (2010) Characterization of the haem oxygenase protein family in Arabidopsis thaliana reveals a diversity of functions, Biochem J 425, 425-434.
29. Muramoto, T., Tsurui, N., Terry, M. J., Yokota, A., and Kohchi, T. (2002) Expression and biochemical properties of a ferredoxin-dependent heme oxygenase required for phytochrome chromophore synthesis, Plant
Physiol 130, 1958-1966. 30. Linley, P. J., Landsberger, M., Kohchi, T., Cooper, J.
B., and Terry, M. J. (2006) The molecular basis of heme oxygenase deficiency in the pcd1 mutant of pea, FEBS J 273, 2594-2606.
ACS Paragon Plus Environment
Biochemistry

5
TABLE OF CONTENT ARTWORK
ACS Paragon Plus Environment
Biochemistry

Supporting Information:
Enzymatic ring opening of an iron corrole by plant-type heme oxygenases:
Unexpected substrate and protein selectivities
Björn Gisk, Frédérique Brégier, Robin A. Krüger, Martin Bröring, and Nicole Frankenberg-Dinkel
Material and Methods
Expression and purification of recombinant heme oxygenases Details on expression and purification of the various heme oxygenases have been described earlier (1, 2). In brief, Arabidopsis thaliana HO1 (HY1), Synechocystis sp. PCC6803 HO-1, BphO and PigA of Pseudomonas aeruginosa were recombinantely produced as N-terminal glutathione-S-transferase (GST)-fusions in Escherichia coli while Arabidopsis thaliana HO3 and HO4 both contained a C-terminal hexa-histidine tag. All plant HOs lacked the predicted chloroplast transit peptide sequence. Truncated human HO1 (3) was expressed in E. coli and subjected to a 60 % (NH4)2SO4 cut, dialyzed against reaction buffer and used as an enriched protein fraction similar to a method described in (4). All proteins were dialyzed against reaction buffer (100 mM potassium phosphate, pH 7.2) prior touse. Analytical size-exclusionchromatography Analytical size-exclusion chromatography was employed to investigate the complex formation between the iron corroles of all five heme oxygenases. A SuperdexTM 75 10/300 GL column (GE Healthcare) equilibrated with 100 mM potassium phosphate, pH 7.2, at a flow rate of 0.5 mL/min, was used to analyze iron corrole:heme oxygenase complexes (1:1). During the run the absorbance at 280 nm and 398 nm / 535 nm using a UV-900 detector of an AEKTA-Explorer system (GE Healthcare) was detected. Heme oxygenase assay Iron corrole isomers were dissolved in DMSO and incubated with equimolar amounts of each HO for 10 minutes in reaction buffer (100 mM potassium phosphate, pH 7.2). Spectral changes were monitored using an Agilent Technologies 8453 UV-visible Spectroscopy system. For catalytic turnover, the assay contained 10 μM HO, 20 μM iron corrole, 0.15 mg/mL bovine serum albumin, 4.6 μM ferredoxin (spinach), 0.025 unit/mL spinach ferredoxin-NADP+ oxidoreductase and 10 μM catalase from Aspergillus niger in 100 mM potassium phosphate buffer (pH 7.2) in a total volume of 500 µl. The reaction was started by addition of an NADPH-regenerating system containing 6.5 mM glucose 6-phosphate, 0.82 mM NADP+, and 1.1 unit/mL glucose-6-phosphate dehydrogenase. Spectral changes between 300 and 900 nm were monitored for 30 min at 25 oC using an Agilent Technologies 8453 UV-visible Spectroscopy system. To detect the reaction product 5 mM of an iron chelator (e.g. Tiron) was added to the assay. HPLC analyses HO reaction products were acidified with tenfold 0.1% trifluoroacetic acid (TFA) and loaded onto a Waters (Milford, MA, USA) C18- Sep-Pak Light cartridge preconditioned as follows: 3 mL acetonitrile to wet the Sep-Pak, 3 mL MilliQ water, 3 mL 0.1% TFA, 3 ml 10% methanol in 0.1% TFA, 3 mL acetonitrile, 3 mL MilliQ water, 3 mL 10% methanol in TFA. After the sample was loaded onto the Sep-Pak, it was washed with 6 mL 0.1% TFA, and 6 mL 20% methanol / 80 % TFA (0.1%). The pyrroles were then eluted from the Sep-Pak using 1 mL of 100% acetonitrile. The eluate was dried using a Speed-Vac lyophilizer and
60

subsequently analyzed by HPLC. The dried sample was first dissolved in 10 μL DMSO and then diluted with 150 μL of the HPLC mobile phase. Following brief centrifugation (10.000 × g for 1 min) and filtration through a 0.45 μm polytetrafluoroethylene syringe filter, the pyrroles were analyzed by reversed phase chromatography using an Agilent Technologies 1100 liquid chromatograph. The HPLC column used for all analyses was a 4.6 x 250 mm Phenomenex Ultracarb 5 μm ODS(20) analytical column with a 4.6 x 30 mm guard column of the same material. The mobile phase consisted of acetone, isopropanole, and 20 mM formic acid (33:33:33 by volume), and the flow rate was 0.6 mL/min. The eluate was monitored at 650 nm and 380 nm using an Agilent Technologies 1100 series diode array detector. A standard (synthetic bilindione 3) was analyzed the same way. Material and instruments for the chemical compounds Starting materials and reagents were obtained from commercial sources and used as received. All solvents were dried by standard procedures and stored under a blanket of nitrogen. 1H NMR spectra were obtained with a Bruker Avance 300 or a Bruker DRX-400 spectrometer. Chemical shifts (δ) are given in ppm relative to residual protio solvent resonances. High resolution mass spectra (APCI, ESI) were recorded with an IonSpec Ultima, a Finnigan LTQ FT, or a QStarPulsar i. UV-Vis spectra were measured on a Shimadzu UV-1601 PC in concentrations of about 10-5 mol L-1. CHN analyses were recorded on a Elementar Vario EL. Synthesis of tetrapyrroles [α-CH-Fe(cor)] 1:
S1HBr
N
HO2C
N
CO2HH H
NOHC I
CO2Me
H
S2
N NH H
N N
I I
CO2MeMeO2C
H H2 Br-
S3
N N
N N
MeO2C CO2Me
Fe
Cl
S4
N N
N N
HO2C CO2H
Fe
Cl
1
FeCl2 x 4 H2ONaOAc
LiOH x H2O
Dipyrrylmethane S1 (5) (319.5 mg, 0.92 mmol) and iodopyrrole aldehyde S2 (6) (591 mg, 1.84 mmol) were dissolved in hot ethanol (15 mL) and treated with hydrobromic acid (48% in water, 4 mL) at once. The solution was refluxed for three minutes and then cooled to 4°C to allow the product to crystallize over night as the bis(hydrobromide) S3 (646.8 mg, 68%). This
61

material was dissolved in DMF (100 mL) under an atmosphere of nitrogen. FeCl2 × 4 H2O (635 mg, 3.19 mmol) and NaOAc (262 mg, 3.19 mmol) were added, and the mixture was heated to 80°C for 7 hours. After cooling the solvent was removed in vacuo, the residue was redissolved in dichloromethane (50 mL) and washed twice with each, hydrochloric acid (1 mol/L, 100 mL) and water (100 mL). After drying with sodium sulfate all volatiles were removed in vacuo, and the residue was subjected to column chromatography on silica with dichloromethane/methanol (99:1). Recrystallization from dichloromethane/n-hexane yielded the iron corrole S4 as a brown solid (207.1 mg, 47%). Analytical data for S4: C37H43ClFeN4O4 (699.06), calc. C: 63.57; H: 6.26; N: 7.91. Found C: 63.28; H: 6.27; N: 7.96. HRMS (ESI): calc. for [C37H44FeN4O4]+ 664.2706; found: 664.2717; Δ = -1.1 mmu. 1H NMR (400 MHz, CD2Cl2): δ = 192.08 (br. s, 1H), 178.98 (br. s, 2H), 32.14 (br. s, 2H), 27.62 (br. s, 2H), 19.13 (br. s, 2H), 16.71 (br. s, 2H), 15.82 (br. s, 6H), 3.93 (br. s, 2H), 3.66 (br. s, 6H), 1.67 (br. s, 2H), 1.35 (br. s, 6H), 0.08 (br. s, 4H), -0.14 (br. s, 6H). UV-Vis (CH2Cl2): λmax (ε) = 295 (20600), 330 (30900), 371 (50100), 480 (8900), 524 (6600), 935 nm (530 M-1
λmax ) = 358 (35100), 520 sh (4400), 997 nm (2500 M cm ).
cm-1). Saponification: Iron corrole S4 (128.5 mg, 0.18 mmol) and LiOH × H2O (101.3 mg, 2.43 mmol) were dissolved in THF (20 mL) and water (2 mL) and stirred at ambient temperature over night. Dichloromethane (20 mL) and hydrochloric acid (1 mol/L, 20 mL) were added, the layers were separated and the organic layer containing the title compound α-CH-Fe(cor) 1 was washed with water (50 mL). Drying with sodium sulfate and recrystallization from dichloromethane/n-hexane yielded a brown powder (86.4 mg, 70%). Analytical data for 1: HRMS (APCI): calc. for [C35H39FeN4O4]+ 635.2315; found: 635.2336; Δ = -2.1 mmu. 1H NMR (400 MHz, DMSO-d6): δ = 194.2 (br. s, 1H), 179.3 (br. s, 2H), 30.9 (br. s, 2H), 28.0 (br. s, 2H), 21.0 (br. s, 2H), 19.1 (br. s, 6H), 18.2 (br. s, 2H), 4.1 (br. s, 2H), 2.5 (br. s, 6H), 1.5 (br. s, 2H), 1.0 (br. s, 2H), 0.7 (br. s, 6H), -6.2 (br. s, 2H). UV-Vis (KPi 50 mmol, pH 7.0):
-1 -1(ε [γ-CH-Fe(cor)] 2: This compound was prepared as reported in Ref. (7).
Bilindione 3:
p-chloranilN NH H
N NH H2 Br-
S5
MeO2C CO2Me
HClN N
N NH
S6
MeO2C CO2Me
HH
O O
N N
N NH
3
HO2C CO2H
HH
O O
a,c-Biladiene as bis(hydrobromide) S5 (8) (775 mg, 1.00 mmol) and p-chloranil (740 mg, 3 mmol) were dissolved in water-saturated chloroform and refluxed for 2 hours. After cooling the mixture was washed with water (3 × 20 mL), dried with sodium sulfate, and all volatiles were evaporated at reduced pressure. Chromatographic separation of the residue with dichloromethane on alumina and collection of the blue band yielded the bilindione dimethylester S6 as a dark blue solid (290 mg, 45%). Analytical data for S6: HRMS (ESI): calc. for [C37H46N4O6Na]+ 665.3310; found: 665.3318; Δ = 0.8 mmu. 1H NMR (400 MHz, CDCl3): δ = 8.22 (br. s, 3H), 6.73 (s, 1H), 5.90 (s, 2H), 3.67 (s, 6H), 2.92 (t, J = 7.6 Hz, 4H), 2.55 (t, J = 7.6 Hz, 4H), 2.51 (q, J = 7.6 Hz, 4H), 2.28 (q, J = 7.6 Hz, 4H), 2.08 (s, 6H), 1.24
62

(t, J = 7.6 Hz, 6H), 1.07 (t, J = 7.6 Hz, 6H). UV-Vis (MeOH): λmax (ε) = 364 (50100), 640 nm (14600 M-1 cm-1). Saponification: Bilindione S6 (12,8 mg, 20 μmol) and hydrochloric acid (conc., 2.5 mL) were mixed with glacial acetic acid (2.5 mL) and stirred at ambient temperature in the dark for 24 hours. Chloroform (20 mL) was added, the layers were separated and the organic layer containing the title bilindione 3 was carefully washed with aqueous sodium acetate (conc., 5 mL) and water (5 mL). Drying with sodium sulfate and recrystallization from dichloromethane/n-hexane yielded a blue powder (7.0 mg, 55%). Analytical data for 3: HRMS (ESI): calc. for [C35H42N4O6Na]+ 637.2997; found: 637.2984; Δ = -1.3 mmu. 1H NMR (300 MHz, D2O): δ = 6.76 (s, 1H), 5.93 (s, 2H), 2.71 (t, J = 7 Hz, 4H), 2.43 (q, J = 7 Hz, 4H), 2.27 (t, J = 7 Hz, 4H), 2.11 (q, J = 7 Hz, 4H), 1.93 (s, 6H), 1.12 (t, J = 7 Hz, 6H), 0.94 (t, J = 7 Hz, 6H). UV-Vis (DMSO): λmax (ε) = 374 (43300), 635 nm (14400 -1 -1 M cm ).
References
1. Gisk, B., Yasui, Y., Kohchi, T., and Frankenberg-Dinkel, N. (2010) Characterization
of the haem oxygenase protein family in Arabidopsis thaliana reveals a diversity of functions, Biochem J 425, 425-434.
2. Wegele, R., Tasler, R., Zeng, Y., Rivera, M., and Frankenberg-Dinkel, N. (2004) The heme oxygenase(s)-phytochrome system of Pseudomonas aeruginosa, J Biol Chem 279, 45791-45802.
3. Wilks, A., Black, S. M., Miller, W. L., and Ortiz de Montellano, P. R. (1995) Expression and characterization of truncated human heme oxygenase (hHO-1) and a fusion protein of hHO-1 with human cytochrome P450 reductase, Biochemistry 34, 4421-4427.
4. Wilks, A., and Ortiz de Montellano, P. R. (1993) Rat liver heme oxygenase. High level expression of a truncated soluble form and nature of the meso-hydroxylating species, J Biol Chem 268, 22357-22362.
5. Murakami, Y., Yamada, S., Matsuda, Y., and Sakata, K. (1978) Transition-metal Complexes of Pyrrole Pigments. XV. Coordination of Pyridine Bases to the Axial Sites of Cobalt Corroles, Bull Chem Soc Jap 51, 123-129.
6. Tu, B., Ghosh, B., and Lightner, D. A. (2003) A new class of linear tetrapyrroles: acetylenic 10,10a-didehydro-10a-homobilirubins, J Org Chem 68, 8950-8963.
7. Matsuo, T., Hayashi, A., Abe, M., Matsuda, T., Hisaeda, Y., and Hayashi, T. (2009) Meso-unsubstituted iron corrole in hemoproteins: remarkable differences in effects on peroxidase activities between myoglobin and horseradish peroxidase, J Am Chem Soc 131, 15124-15125.
8. Sengupta, D., and Robinson, B. C. (2002) Synthesis of chlorins possessing a fused naphthalene ring, Tetrahedron 58, 5497-5502.
63
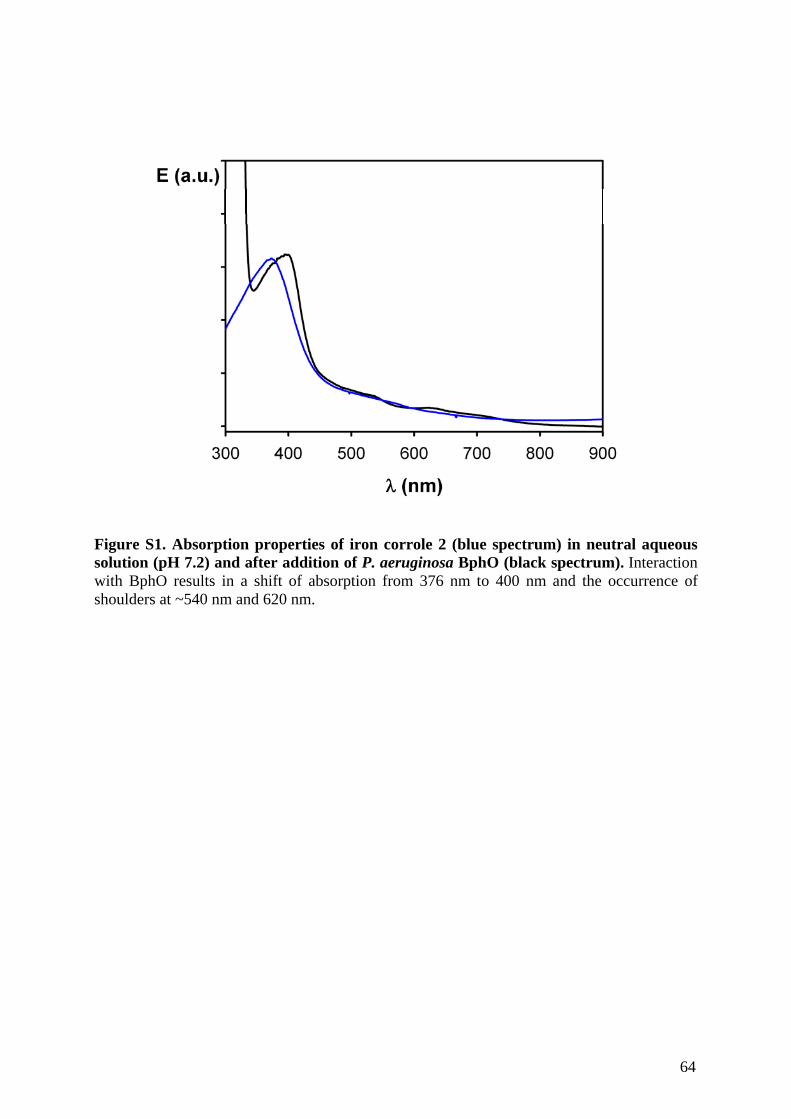
Figure S1. Absorption properties of iron corrole 2 (blue spectrum) in neutral aqueous solution (pH 7.2) and after addition of P. aeruginosa BphO (black spectrum). Interaction with BphO results in a shift of absorption from 376 nm to 400 nm and the occurrence of shoulders at ~540 nm and 620 nm.
64
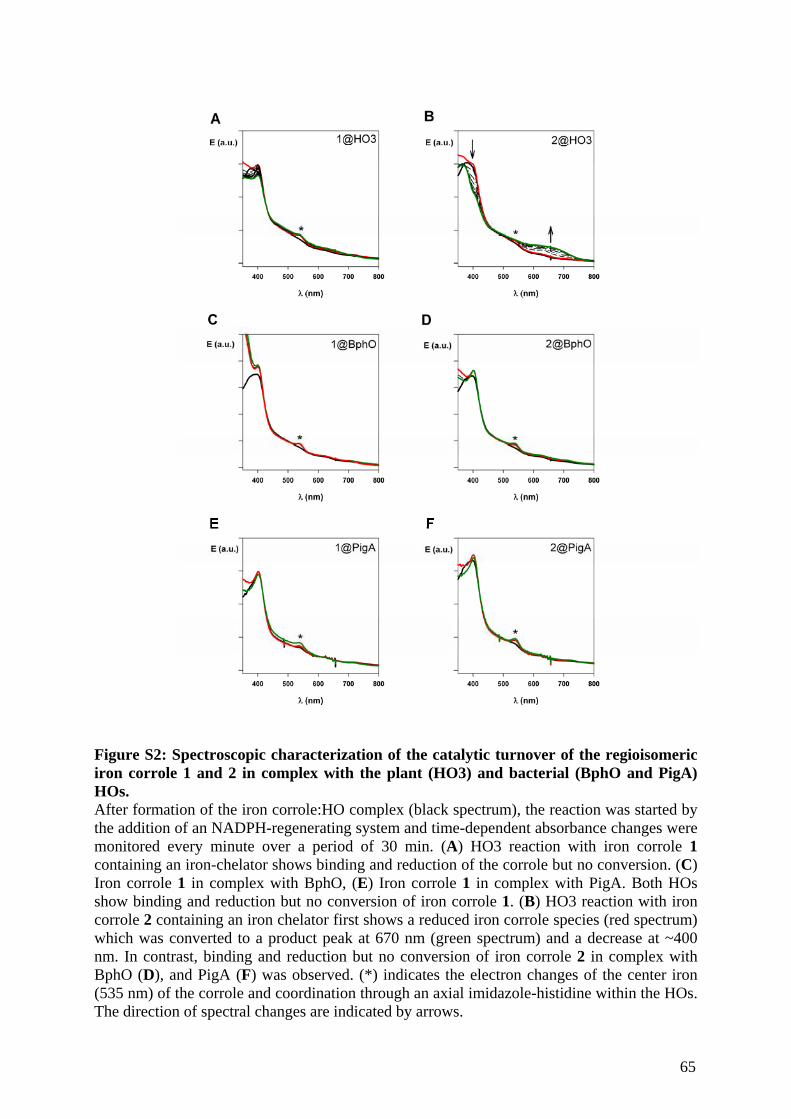
Figure S2: Spectroscopic characterization of the catalytic turnover of the regioisomeric iron corrole 1 and 2 in complex with the plant (HO3) and bacterial (BphO and PigA) HOs. After formation of the iron corrole:HO complex (black spectrum), the reaction was started by the addition of an NADPH-regenerating system and time-dependent absorbance changes were monitored every minute over a period of 30 min. (A) HO3 reaction with iron corrole 1 containing an iron-chelator shows binding and reduction of the corrole but no conversion. (C) Iron corrole 1 in complex with BphO, (E) Iron corrole 1 in complex with PigA. Both HOs show binding and reduction but no conversion of iron corrole 1. (B) HO3 reaction with iron corrole 2 containing an iron chelator first shows a reduced iron corrole species (red spectrum) which was converted to a product peak at 670 nm (green spectrum) and a decrease at ~400 nm. In contrast, binding and reduction but no conversion of iron corrole 2 in complex with BphO (D), and PigA (F) was observed. (*) indicates the electron changes of the center iron (535 nm) of the corrole and coordination through an axial imidazole-histidine within the HOs. The direction of spectral changes are indicated by arrows.
65
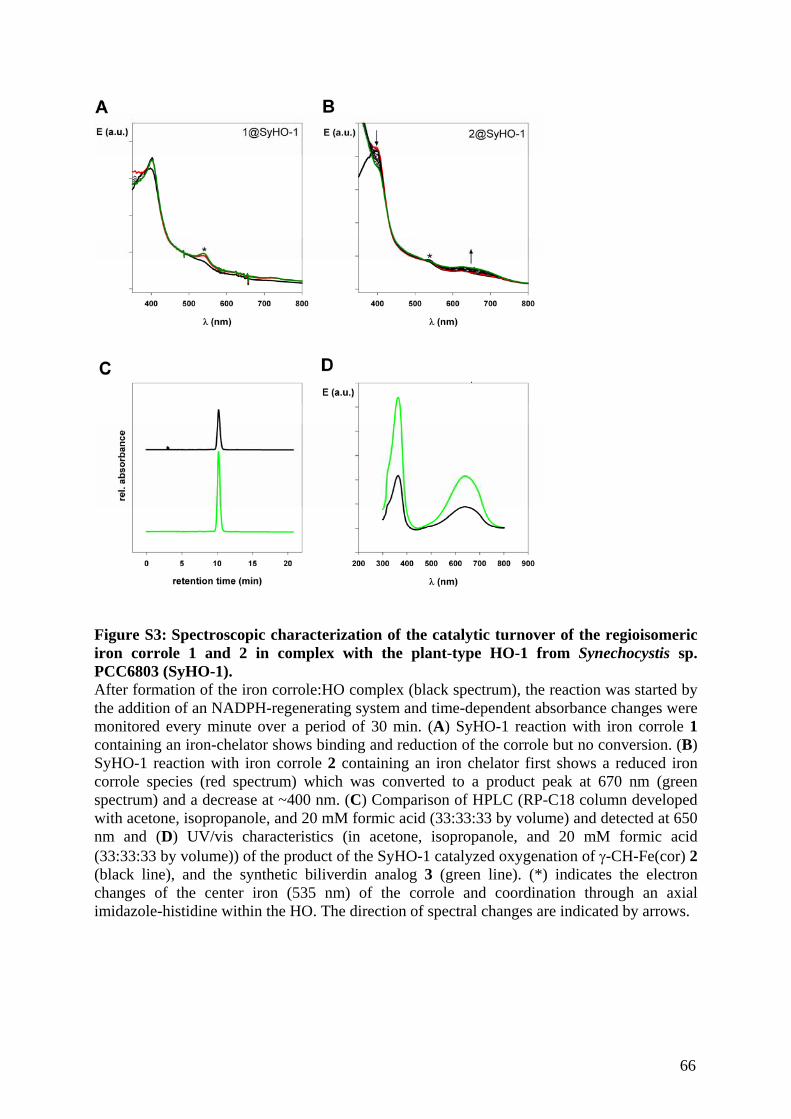
Figure S3: Spectroscopic characterization of the catalytic turnover of the regioisomeric iron corrole 1 and 2 in complex with the plant-type HO-1 from Synechocystis sp. PCC6803 (SyHO-1). After formation of the iron corrole:HO complex (black spectrum), the reaction was started by the addition of an NADPH-regenerating system and time-dependent absorbance changes were monitored every minute over a period of 30 min. (A) SyHO-1 reaction with iron corrole 1 containing an iron-chelator shows binding and reduction of the corrole but no conversion. (B) SyHO-1 reaction with iron corrole 2 containing an iron chelator first shows a reduced iron corrole species (red spectrum) which was converted to a product peak at 670 nm (green spectrum) and a decrease at ~400 nm. (C) Comparison of HPLC (RP-C18 column developed with acetone, isopropanole, and 20 mM formic acid (33:33:33 by volume) and detected at 650 nm and (D) UV/vis characteristics (in acetone, isopropanole, and 20 mM formic acid (33:33:33 by volume)) of the product of the SyHO-1 catalyzed oxygenation of γ-CH-Fe(cor) 2 (black line), and the synthetic biliverdin analog 3 (green line). (*) indicates the electron changes of the center iron (535 nm) of the corrole and coordination through an axial imidazole-histidine within the HO. The direction of spectral changes are indicated by arrows.
66

Publikationen/Manuskripte
67
4. The existence of two heme oxygenases with different
regiospecificities is a characteristic trait of pathogenic and
non-pathogenic Pseudomonas species
Gisk, B., Aras, M., Wiethaus, J., & Frankenberg-Dinkel, N.
(Manuskript in Vorbereitung)

68
The existence of two heme oxygenases with different regiospecificities is a characteristic
trait of pathogenic and non-pathogenic Pseudomonas species
Bjoern Gisk, Marco Aras, Jessica Wiethaus and Nicole Frankenberg-Dinkel*
Physiology of Microorganisms, Ruhr-University Bochum, 44780 Bochum, Germany
Corresponding author*: [email protected]
Abstract:
Heme oxygenases (HO) degrade heme yielding iron, carbon monoxide and biliverdin (BV).
Depending on the HO cleavage site the four BV isomers IXα, IX, IXγ and IX are produced.
Bioinformatic studies revealed that all Pseudomonas species encode two phylogenetic
different HOs, BphO and PigA. In Pseudomonas aeroginosa PAO1 products of heme
degradation by PigA are BV IX and IX while degradation by BphO yields BV IXα.
To verify whether this is a common feature of Pseudomonas we tested HOs of diverse
Pseudomonas strains (P. aeruginosa PA14, P. fluorescence, P. putida, and P. syringae) with
regard to their heme regiospecificity. Tested PigAs cleave heme under production of BV
IXand IX. The additional release of carbon monoxide indicates a typical HO reaction
mechanism for heme degradation. All PigAs are monomeric and have nearly identical
biochemical parameters. In contrast, BphOs are specific for cleavage of the heme α-meso
carbon bridge. Therefore, it seems to be a general and unique characteristic of pathogenic and
non-pathogenic Pseudomonas species to possess two HOs with different regiospecificities.

69
1. Introduction
Heme oxygenases (HOs [EC 1.14.99.3]) degrade heme to biliverdin (BV), iron and carbon
monoxide. The oxygen dependent HO reaction needs three molecules of molecular oxygen
and seven electrons (Ortiz de Montellano 2000). HOs are regiospecific with regard to the
cleaved meso carbon bridge of heme. Depending on the cleavage position HOs produce the
BV isomers IXα, IX, IX, or IX. HOs are widely distributed enzymes which were found in
mammals (Tenhunen, Marver et al. 1969; Wilks, Black et al. 1995), higher plants (Davis,
Bhoo et al. 2001), insects (Paiva-Silva, Cruz-Oliveira et al. 2006), pathogenic (Ochsner and
Vasil 1996; Ratliff, Zhu et al. 2001), and non-pathogenic bacteria (Wilks and Schmitt 1998),
and in photosynthetic organisms (Cornejo and Beale 1997; Richaud and Zabulon 1997;
Zhang, Migita et al. 2005). The opportunistic pathogenic non-photosynthetic bacterium
Pseudomonas aeruginosa PAO1 is the only described organism which encodes two functional
HOs, BphO and PigA, with different regiospecificities. BphO is specific for cleavage of the α-
meso carbon bridge and PigA for cleavage of the - and -meso carbon bridge of heme
(Fig. 1) (Ratliff, Zhu et al. 2001; Wegele, Tasler et al. 2004). The bphO gene is located
upstream of a gene, coding for the bacterial phytochrome BphP, and transcribed cell density
dependent under control of RpoS (Barkovits, Harms et al. 2008). In contrast to plant and
cyanobacterial phytochromes, which carry phytochromobilin (Kohchi, Mukougawa et al.
2001) or phycocyanobilin as chromophore, the chromophore of BphP is BV (Bhoo, Davis et
al. 2001). It was demonstrated that both BV IX and BV IX can bind to BphP (Wegele,
Tasler et al. 2004; Tasler, Moises et al. 2005). PigA is located in the polycistroninc pig
operon, which gene products are partially involved in iron utilization (Ochsner and Vasil
1996). The pig gene cluster is regulated by Fur and expressed under iron limiting conditions.
(Ochsner and Vasil 1996).
In this study we biochemically characterized potential PigAs and BphOs from diverse
Pseudomonads sp. (P. fluorescence (P.f.) 5, P. putida (P.p.) KT2440, P. syringae (P.s.)

70
DC3000 pv tomato and the P. aeruginosa strain PA14 (PA)). In addition, for PigA catalyzed
reactions an artificial electron donor system was compared with the hypothesized natural
donor pa-FPR (Ferredoxin NADP+ reductase). We demonstrate that heme degradation by
different PigAs produces BV IXund IX while BphO cleavage of heme exclusively yields
BV IXα. We conclude that it is a specific triad of pathogenic and non-pathogenic
Pseudomonas species to possess two HOs with different regiospecificities.
2. Material and Methods
2.1 Construction of expression plasmids
Pseudomonas pigAs and bphO were PCR amplified with High-Fidelity© Phusion Polymerase
(New England Biolabs) from genomic Pseudomonas DNA using oligos introducing
restriction site for BamHI and XhoI:
5'-GCGGATCCATGTGGTGTGGG-3', 5'-CGCTCGAGTCATGCCTGTACC-3' (P.s. DC3000 pigA);
5'-GCGGATCCATGACCGACCGC-3', 5'-CGCTCGAGTCAGACCAGCGC-3' (P.p. KT2440 pigA);
5'-GCGGATCCATGACCTCCCAGG-3', 5'-CGCTCGAGTCAGGCCGGTTGTG-3' (P.f. 5 pigA);
5'-GCGGATCCATGGATACCCTGG-3', 5'-CGCTCGAGTCAGGCGAAGGTAC-3' (PA14 pigA);
5' CGGGATCCATGCCCAAACCGCACCAAG-3',
5'-CGGCTCGAGTCATAGCAGCACCTCGCGG-3' (P.f. 5 bphO).
Amplified DNA fragments were cloned into pGEX-6P-1 (GE healthcare) generating pGEX-
6P-1-PigA and pGEX-6P-1-BphO plasmids which were verified by sequencing. Plasmid
pET14-BphO for expression of P.s. DC3000 BphO was a gift from Dr. W. Gaertner (Max-
Planck Institute, Muehlheim).
2.2 Expression and purification of recombinant proteins
Expression and purification of PigAs and BphOs was performed according to manufacturers
instructions (GE healthcare) and to (Frankenberg, Mukougawa et al. 2001; Muramoto, Tsurui

71
et al. 2002) with following modifications. A culture of BL21(DE3) carrying the respective
overexpression plasmid was grown at 37oC to an OD578 of 0.6 - 0.8, shifted to 30oC, and
induced by addition of 0.5 mM isopropyl -D-thiogalactoside. Cells were cultured for
additional 3h. After purification proteins were dialysed against reaction buffer (100mM
potassium phosphate buffer, pH 7.0). BphO from P.s. DC3000 and Ferredoxin NADP+
reductase from P. aeruginosa (pa-FPR) were expressed and purified as described before
(Wang, Zeng et al. 2007; Gisk, Yasui et al. 2010).
2.3 Size-exclusion chromatography
Oligomerization states of the soluble PigA proteins were analysis was described before (Gisk,
Yasui et al. 2010) with an SuperdexTM 75 10/300 GL colum (GE healthcare).
2.4 Reconstitution and titration of HOs with heme
The reconstitution and titration of HOs with heme was described before (Gisk, Yasui et al.
2010).
2.5 Determination of extinction coefficients of PigA-heme complexes
The determination of extinction coefficients of PigA-heme complexes was done according to
(Gisk, Yasui et al. 2010).
2.6 Heme cleavage assays with PigAs
Reactions were performed in reaction buffer (100 mM potassium phosphate, pH 7.0) at room
temperature with 10 µM PigA, 10 µM heme, 0.15 mg/ml BSA, 20µM pa-FPR, 5 mM catalase
from Aspergillus niger. Unless otherwise noted 5 mM iron chelator (Tiron) was added. In
case of P.s. DC3000 PigA lysate of the respective overexpression culture and 50 µM hemin
were used. Reactions were started by addition of an NADPH-regeneration system containing

72
6.5 mM glucose 6-phosphate, 0.82 mM NADP+, and 1.1 units/ml glucose 6-phosphate
dehydrogenase. Spectral changes between 350 and 900 nm were monitored for 30 min at 25oC
using an Agilent Technologies 8453 UV-visible spectroscopy system.
BphOs expressed in E. coli were purified with bound BV due to cleavage of E. coli
endogenous heme. BV was extracted from these BphOs and HPLC analyzed (2.9).
2.7 Determination of pH and temperature dependency and kinetic parameters of PigA
reactions
10 µM PigA was mixed with 10 µM heme, 0.15 mg/ml BSA, 4.6 µM Fd (spinach),
0.025unit/ml spinach Fd-NADP+-oxidoreductase, 5 mM catalase from Aspergillus niger,
5 mM sodium ascorbate, and 5 mM iron chelator. Reactions were started by addition of an
NADPH-regeneration system. The rate of BV formation was calculated using the absorbance
changes at 670 nm and the extinction coefficient of BV in 100 mM potassium phosphate
buffer at pH 7.0 (ε670 6.25 mM-1cm-1) (Brindle, North et al. 1986). The pH dependency was
measured between pH 6.0 and 9.0. The temperature dependency was measured between 10°C
and 45°C. Km values were calculated using Eadie-Hofstee and Lineweaver Burk plots
(standard assay with 10 µM PigA, and heme concentrations between 0.58 µM and 100 µM
heme). The activation energy (Ea) was calculated based on an Arrhenius plot.
2.8. Detection of carbon monoxide as reaction product
10µM PigA, 10µM heme, 50µM NADPH and 4.6 µM Fd (spinach), 0.025unit/ml spinach Fd-
NADP+-oxidoreductase, 5 mM catalase from Aspergillus niger, and 5 mM sodium ascorbate
in a final volume of 500 µl were filled in the reference and the reaction cuvette and blanked
immediately. To the reaction solution 100µM myoglobin and to the reference an identical

73
volume of buffer was added. The spectra were taken between 350 and 900 nm every minute,
and the maximum shift from 405 nm to 420 nm and the decrease of Q-bands was monitored.
2.9 HPLC analysis of HO reaction products
The HPLC analysis were performed as described before (Gisk, Yasui et al. 2010).
3. Results
3.1 Expression and purification of recombinant Pseudomonas sp. PigAs
Four different Pseudomonas sp. PigAs and the P.f. 5 BphO were expressed in E. coli as
N-terminal GST fusion proteins. BphO from P.s. DC3000 was expressed as N-terminal His6-
tag fusion protein. HOs overexpressing cells are often of green color due to BV accumulation
after degradation of E. coli endogenous heme (Suzuki, Sato et al. 1992; Wilks and Ortiz de
Montellano 1993; Wilks and Schmitt 1998; Wegele, Tasler et al. 2004). This was observed for
Pseudomonas BphO but not for PigA overexpressing cells (data not shown). Migration of
purified PigAs from PA14, P.f 5, and P.p. KT2440 during SDS-PAGE was in agreement with
their calculated molecular masses of about 22 kDa (Fig. 2). Proteins were obtained to almost
100 % purity and in high amounts (40-50 mg/l culture). However, for PA14 PigA an
additional band corresponding to the PigA-GST fusion protein was detected. P.s. DC3000
PigA was only soluble in lysate (data not shown) and thus just used for analysis of heme
cleavage products. BphOs from P.s. DC3000, and P.f 5 were obtained to almost 100 % purity
and with bound BV, which was the reaction product of heme degradation in E. coli (data not
shown).
3.2 Pseudomonas PigAs are monomers
With the exception of dimeric HO-2 from Synechocystis sp. PCC 6803 and human all
characterized HOs are monomeric enzymes (Sugishima, Hagiwara et al. 2005; Bianchetti, Yi

74
et al. 2007; Sato, Sugishima et al. 2009). Purified PigAs were subjected to size exclusion
chromatography and eluted mainly at approx. 11.5 ml (data not shown). This corresponds
well with the calculated molecular weights of PigA monomers. A minor peak at approx. 8.5
ml represented aggregated PigAs as verified by SDS-PAGE (data not shown). As jugged from
the absorbance at 406 nm purified proteins had no endogenous heme from E. coli bound,
which is in line with the colorless PigA overexpressing E. coli cultures.
3.3 Pseudomonas PigAs bind heme
Pseudomonas PigAs bind heme with a distinct Soret peak of the heme-PigA complexes at 406
nm (Fig. 3). The heme binding constants (Kd) of PigAs were calculated to be between 1.5 µM
and 2.7 µM (Tab.1) and are comparable to Kd values of other HOs (Wilks and Schmitt 1998).
The extinction coefficients at 406 nm of the different heme-PigA complexes were determined
via the pyridine hemochrome method to be nearly identical (151-166 mM-1cm-1) (Tab. 1) and
to fit the value of heme-PigA from P. aeruginosa PAO1 (ε406 129 mM-1cm -1) (Ratliff, Zhu et
al. 2001).
3.4 BV and CO are products of heme degradation by PigAs
Heme turnover by PigAs was performed in the presence of ascorbate, FNR and Fd as
exogenous reductant system and followed spectroscopically. Independent of the tested PigA
heme was converted during the catalytic turnover to ferric-biliverdin (Fig. 3). After addition
of electrons (NAPH-regenerating system) the Soret peak shifted from 406 nm to 411 nm and
the-/bands increased at 536 nm and 576 nm indicating formation of the oxyferrous
complex. During the reaction Soret peak and Q-bands decreased. In presence of an iron
chelator BV was released from the protein and a broad product peak (BV) increased between
650 nm and 700 nm. Without an iron chelator no BV peak was detectable (data not shown).

75
Detected absorption spectra are typical for heme catalysis by HOs (Wilks and Schmitt 1998;
Ratliff, Zhu et al. 2001; Wegele, Tasler et al. 2004; Gisk, Yasui et al. 2010).
The natural electron donor for PigA form P. aeruginosa PAO1 is pa-FPR. Therefore we
repeated our experiments with pa-FPR as electron donor instead of the artificial reductant
system. The course of reaction was identical for all tested PigAs with a representative
example shown in Fig. 1A. Results were indistinguishable from those obtained with Fd, FNR
and Asc as reductant system. However, reactions proceeded significantly slower with pa-FPR.
In case of PigA from P. syringae in lyaste a small peak at 406 nm representing the ferric
heme-PigA complex was monitored and after addition of the regenerating system a small
increase of BV absorbance was detectable (Fig. 3D). During the reaction no oxyferrous
complex was observed.
The release of carbon monoxide during cleavage of heme by PigA was verified by addition of
myoglobin to the assay. Independent of the used PigA ferric myoglobin was completely
converted to the ferrous carbon monoxide complex as jugged from the typical Soret shift from
405 nm to 420 nm and an increase of the Q-bands (data not shown). This indicates that
besides BV and CO is produced during heme cleavage by PigAs.
3.5 Kinetic parameters of heme cleavage by tested PigAs are nearly identical
PigAs had high activities over the complete analyzed pH range from pH 6.0 to pH 9.0 (Fig.
5A). The pH optima were between pH 6.0 and 6.75. Under acid conditions no authentic
values could be determined as assay components were no longer stable. PigA activity
increased with temperature and the optimum was beyond 45oC (Fig. 5B). At higher
temperatures the assay components were not stable. With the Arrhenius plot the individual
activation energies (Ea) for the different PigAs were calculated as followed: Ea = 9.8 Jmol-1
(PA14); Ea = 4.4 Jmol-1 (P.f. 5); Ea = 8.8 Jmol-1 (P.p. KT2400). The reaction kinetic

76
parameters were estimated from Eadie Hofstee plots and the Michaelis-Menten constants
(Km) for the different PigAs were determined to be about 24 µM (Tab. 1).
3.6 Heme is cleaved by HOs from Pseudomonas sp. with different regiospecificities
After the catalytic turnover of heme by PigAs the reaction products were extracted and
subsequently subjected to reversed phase HPLC. The four BV IX isomers ( , , and ),
were used as standards (Fig. 4A/B). All four PigAs converted heme to the BV isomers IX
and IX (Fig. 4B). Note that a very small BV IX peak was detected in each case, too. The
BV IX peak was slightly higher then the peak of BV IX. PigA from P. aeruginosa PAO1
was reported to produce more BV IX then BV IX(Ratliff, Zhu et al. 2001). However,
under our test conditions PAO1 PigA produced mainly BV IX (data not shown) (Wegele,
Tasler et al. 2004). There was no difference in product formation whether pa-FPR or Fd,
FNR, and Asc were used as electron donor for heme cleavage (data not shown). Isolated BV
from BphOs was identified as BV IX (Fig. 4A).
4. Discussion
Pseudomonas species encode two different types of HOs, PigA and BphO. This is unique as
all other organisms possess either a PigA or a BphO homolog. We biochemically
characterized different PigAs and BphOs from pathogenic- and non-pathogenic Pseudomonas
species (PA14, P.f. 5, P.p. KT2440, and P.s. DC3000) proving the functionality of both HOs.
PigAs and BphOs exhibit different regiospecificities for the meso carbon bridges of heme
(Fig. 1). While all tested PigAs cleave heme at the - and β-position, the BphOs produce
solely BV IXα. As various “classical” HOs like BphO from diverse species are already well
characterized (Bhoo, Davis et al. 2001; Wegele, Tasler et al. 2004), we focused on the
biochemical characterization of PigAs. Like all other HOs except of HO-2 from Synechocystis

77
sp. PCC 6803 and human (Sugishima, Hagiwara et al. 2005; Bianchetti, Yi et al. 2007; Sato,
Sugishima et al. 2009) Pseudomonas PigAs are monomeric enzymes. PigAs possess
extinction coefficients (406) and heme binding constants (Kd) which are in range with values
obtained for other bacterial HOs (Wilks and Schmitt 1998; Gisk, Yasui et al. 2010). The
activity of PigAs is very high in a broad pH and temperature range. For plant HOs the activity
rises with the temperature, too, but those enzymes have a more defined pH optimum (Gisk,
Yasui et al. 2010). All PigAs are able to convert heme with high efficiency and low activation
energy (Ea) (Tab. 1). Spectra obtained during heme cleavage by PigAs are typical HO
dependent heme degradation spectra (Wilks and Schmitt 1998; Gohya, Zhang et al. 2006).
Carbon monoxide is released during PigA catalyzed heme cleavage. These data indicate a
typical HO mechanism for heme degradation by PigA with BV, CO and iron as reaction
products (Ortiz de Montellano 2000).
PigA mediated heme cleavage results in formation of BV IX and -IX as previously shown
for PigA from P. aeruginosa PAO1 (Ratliff, Zhu et al. 2001). Product formation is
independent of the used electron sources pa-FPR or Fd, FNR, and Asc. This indicates that the
artificial electron donor system is as efficient as pa-FPR, which is the PigA electron donor in
P. aeruginosa PAO1 (Wang, Zeng et al. 2007). Most likely pa-FPR is the natural electron
donor in all tested Pseudomonas sp.. The BV IX is produced in higher amounts than the
-isomer by PigA. In addition, minor amounts of BV IX are produced. However, in the
crystal structure of PigA from P. aeruginosa PAO1 heme is orientated with the -meso carbon
bridge facing the catalytic active site of the enzyme (Friedman, Lad et al. 2004). The
appearance of the β-isomer in in vitro assays most likely arises from a 180° rotation around
the /-axis of heme, that might not occur in vivo (Friedman, Lad et al. 2004). Interestingly,
in P. aeruginosa the heme binding and transfer protein PhuS interacts with PigA but not with
BphO (Bhakta and Wilks 2006). PhuS is part of the phu (Pseudomonas heme utilization)

78
system for uptake of external heme. One might speculate, that PhuS shuttles external heme
directly to PigA. Stereospecific transfer of heme from PhuS to PigA might ensure the correct
heme orientation in the catalytic cave of PigA so that BV IX is the only degradation product.
Detected - and α-isomers would therefore be site products in in vitro assays due to incorrect
“loading” of PigA with heme. However, the phu system is characteristic for opportunistic
pathogenic Pseudomonas species and PhuS homologs are not encoded by the other tested
Pseudomonas sp. in which the heme supply of PigAs is still unknown.
In contrast to PigAs, different BphOs from Pseudomonas sp. cleave heme at the α-meso
carbon bridge resulting in formation of BV IXα. Interestingly, both the BphO produced BV
IX as well as PigA released BV IX can interact with the bacterial phytochrome BphP of P.
aeruginosa PAO1 (Wegele, Tasler et al. 2004; Tasler, Moises et al. 2005). Difference spectra
of BphP with either BV IX or BV IX bound differ indicating divergent signal outputs to
possible response regulators. Due to the Fur dependent transcription of pigA simultaneously
with the phu genes and due to the interaction of PigA with PhuS, PigA may play a role in iron
acquisition from external heme (Bhakta and Wilks 2006; Lansky, Lukat-Rodgers et al. 2006).
Under iron limiting conditions the IX isomer of BV is produced by PigA and might
outcompete the IX isomer for binding to the BphP phytochrome. Thereby informations
about the iron status of the cell might be integrated into the phytochrome system. However,
function or corresponding response regulators of BphO are unknown.
5. Acknowledgements
We thank Dr. M. Rivera (Ralph N. Adams Institute for Bioanalytical Chemistry &
Department of Chemistry, University of Kansas) for sending us the pa-FPR expression
plasmid. We thank Dr. W. Gaertner (Max Planck Institute for Bioinorganic Chemistry,

79
Muehlheim) for sending us the pET14-BphO P.s DC3000 expression plasmid. This study was
supported by the Deutsche Forschungsgemeinschaft.
6. References
Barkovits, K., A. Harms, et al. (2008). "Expression of the phytochrome operon in
Pseudomonas aeruginosa is dependent on the alternative sigma factor RpoS." FEMS Microbiol Lett 280(2): 160-8.
Bhakta, M. N. and A. Wilks (2006). "The mechanism of heme transfer from the cytoplasmic heme binding protein PhuS to the delta-regioselective heme oxygenase of Pseudomonas aeruginosa." Biochemistry 45(38): 11642-9.
Bhoo, S. H., S. J. Davis, et al. (2001). "Bacteriophytochromes are photochromic histidine kinases using a biliverdin chromophore." Nature 414(6865): 776-9.
Bianchetti, C. M., L. Yi, et al. (2007). "Comparison of apo- and heme-bound crystal structures of a truncated human heme oxygenase-2." J Biol Chem 282(52): 37624-31.
Brindle, N. J., A. C. North, et al. (1986). "Theoretical prediction and experimental measurement of the bile-pigment isomer pattern obtained from degradation of catalase haem." Biochem J 236(1): 303-6.
Cornejo, J. and S. I. Beale (1997). "Phycobilin biosynthetic reactions in extracts of cyanobacteria." Photosynth Research 51(3): 223-230.
Davis, S. J., S. H. Bhoo, et al. (2001). "The heme-oxygenase family required for phytochrome chromophore biosynthesis is necessary for proper photomorphogenesis in higher plants." Plant Physiol 126(2): 656-69.
Frankenberg, N., K. Mukougawa, et al. (2001). "Functional genomic analysis of the HY2 family of ferredoxin-dependent bilin reductases from oxygenic photosynthetic organisms." Plant Cell 13(4): 965-78.
Friedman, J., L. Lad, et al. (2004). "Structural basis for novel delta-regioselective heme oxygenation in the opportunistic pathogen Pseudomonas aeruginosa." Biochemistry 43(18): 5239-45.
Gisk, B., Y. Yasui, et al. (2010). "Characterization of the haem oxygenase protein family in Arabidopsis thaliana reveals a diversity of functions." Biochem J 425(2): 425-34.
Gohya, T., X. Zhang, et al. (2006). "Spectroscopic characterization of a higher plant heme oxygenase isoform-1 from Glycine max (soybean)--coordination structure of the heme complex and catabolism of heme." FEBS J 273(23): 5384-99.
Kohchi, T., K. Mukougawa, et al. (2001). "The Arabidopsis HY2 gene encodes phytochromobilin synthase, a ferredoxin-dependent biliverdin reductase." Plant Cell 13(2): 425-36.
Lansky, I. B., G. S. Lukat-Rodgers, et al. (2006). "The cytoplasmic heme-binding protein (PhuS) from the heme uptake system of Pseudomonas aeruginosa is an intracellular heme-trafficking protein to the delta-regioselective heme oxygenase." J Biol Chem 281(19): 13652-62.
Muramoto, T., N. Tsurui, et al. (2002). "Expression and biochemical properties of a ferredoxin-dependent heme oxygenase required for phytochrome chromophore synthesis." Plant Physiol 130(4): 1958-66.
Ochsner, U. A. and M. L. Vasil (1996). "Gene repression by the ferric uptake regulator in Pseudomonas aeruginosa: Cycle selection of iron-regulated genes." Proc Natl Acad Sci U S A 93(9): 4409-4414.
Ortiz de Montellano, P. R. (2000). "The mechanism of heme oxygenase." Curr Opin in Chem Biol 4(2): 221-227.

80
Paiva-Silva, G. O., C. Cruz-Oliveira, et al. (2006). "A heme-degradation pathway in a blood-sucking insect." Proc Natl Acad Sci U S A 103(21): 8030-5.
Ratliff, M., W. Zhu, et al. (2001). "Homologues of Neisserial Heme Oxygenase in Gram-Negative Bacteria: Degradation of Heme by the Product of the pigA Gene of Pseudomonas aeruginosa." J Bacteriol 183(21): 6394-6403.
Richaud, C. and G. Zabulon (1997). "The heme oxygenase gene (pbsA) in the red alga Rhodella violacea is discontinuous and transcriptionally activated during iron limitation." Proc Natl Acad Sci U S A 94(21): 11736-11741.
Sato, H., M. Sugishima, et al. (2009). "Crystal structure of rat haem oxygenase-1 in complex with ferrous verdohaem: presence of a hydrogen-bond network on the distal side." Biochem J 419(2): 339-45.
Sugishima, M., Y. Hagiwara, et al. (2005). "Crystal structure of dimeric heme oxygenase-2 from Synechocystis sp. PCC 6803 in complex with heme." Biochemistry 44(11): 4257-66.
Suzuki, T., M. Sato, et al. (1992). "Nucleotide sequence of cDNA for porcine heme oxygenase and its expression in Escherichia coli." Biochem Int 28(5): 887-93.
Tasler, R., T. Moises, et al. (2005). "Biochemical and spectroscopic characterization of the bacterial phytochrome of Pseudomonas aeruginosa." Febs J 272(8): 1927-36.
Tenhunen, R., H. S. Marver, et al. (1969). "Microsomal heme oxygenase. Characterization of the enzyme." J Biol Chem 244(23): 6388-94.
Wang, A., Y. Zeng, et al. (2007). "Biochemical and structural characterization of Pseudomonas aeruginosa Bfd and FPR: ferredoxin NADP+ reductase and not ferredoxin is the redox partner of heme oxygenase under iron-starvation conditions." Biochemistry 46(43): 12198-211.
Wegele, R., R. Tasler, et al. (2004). "The heme oxygenase(s)-phytochrome system of Pseudomonas aeruginosa." J Biol Chem 279(44): 45791-802.
Wilks, A., S. M. Black, et al. (1995). "Expression and characterization of truncated human heme oxygenase (hHO-1) and a fusion protein of hHO-1 with human cytochrome P450 reductase." Biochemistry 34(13): 4421-7.
Wilks, A. and P. R. Ortiz de Montellano (1993). "Rat liver heme oxygenase. High level expression of a truncated soluble form and nature of the meso-hydroxylating species." J Biol Chem 268(30): 22357-62.
Wilks, A. and M. P. Schmitt (1998). "Expression and characterization of a heme oxygenase (HmuO) from Corynebacterium diphtheriae. Iron acquisition requires oxidative cleavage of the heme macrocycle." J Biol Chem 273(2): 837-41.
Zhang, X., C. T. Migita, et al. (2005). "Protein expressed by the ho2 gene of the cyanobacterium Synechocystis sp. PCC 6803 is a true heme oxygenase. Properties of the heme and enzyme complex." Febs J 272(4): 1012-22.
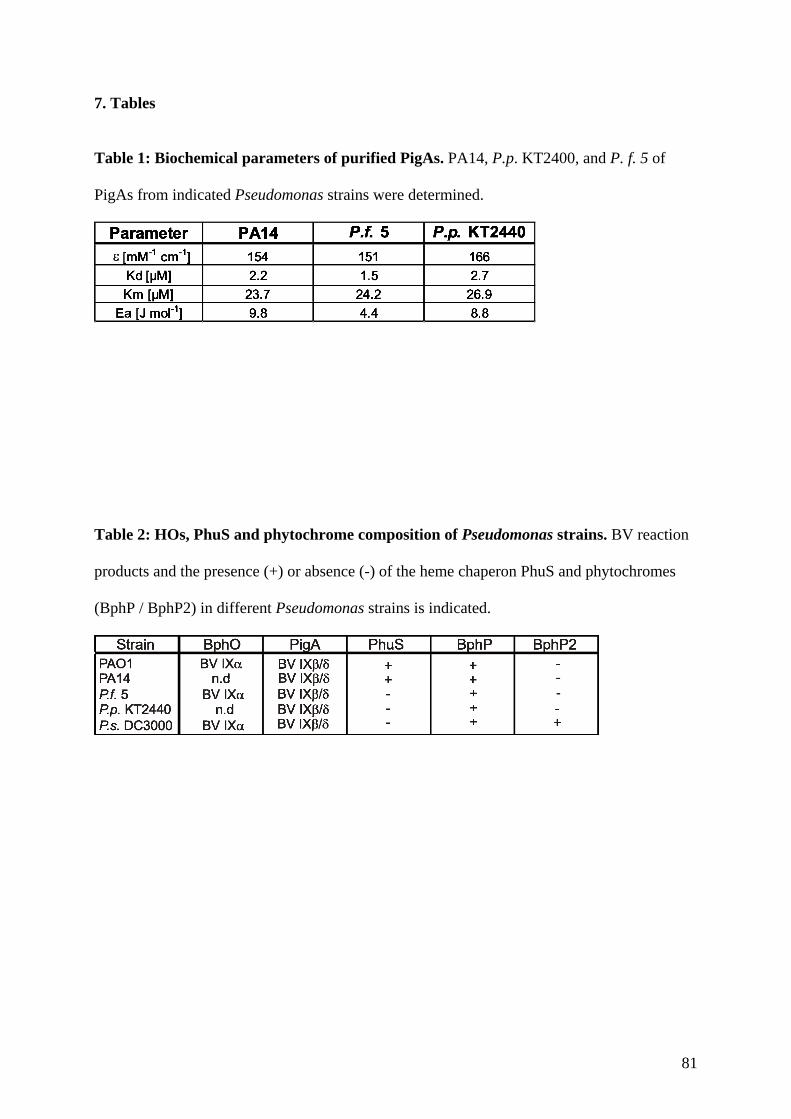
81
7. Tables
Table 1: Biochemical parameters of purified PigAs. PA14, P.p. KT2400, and P. f. 5 of
PigAs from indicated Pseudomonas strains were determined.
Table 2: HOs, PhuS and phytochrome composition of Pseudomonas strains. BV reaction
products and the presence (+) or absence (-) of the heme chaperon PhuS and phytochromes
(BphP / BphP2) in different Pseudomonas strains is indicated.
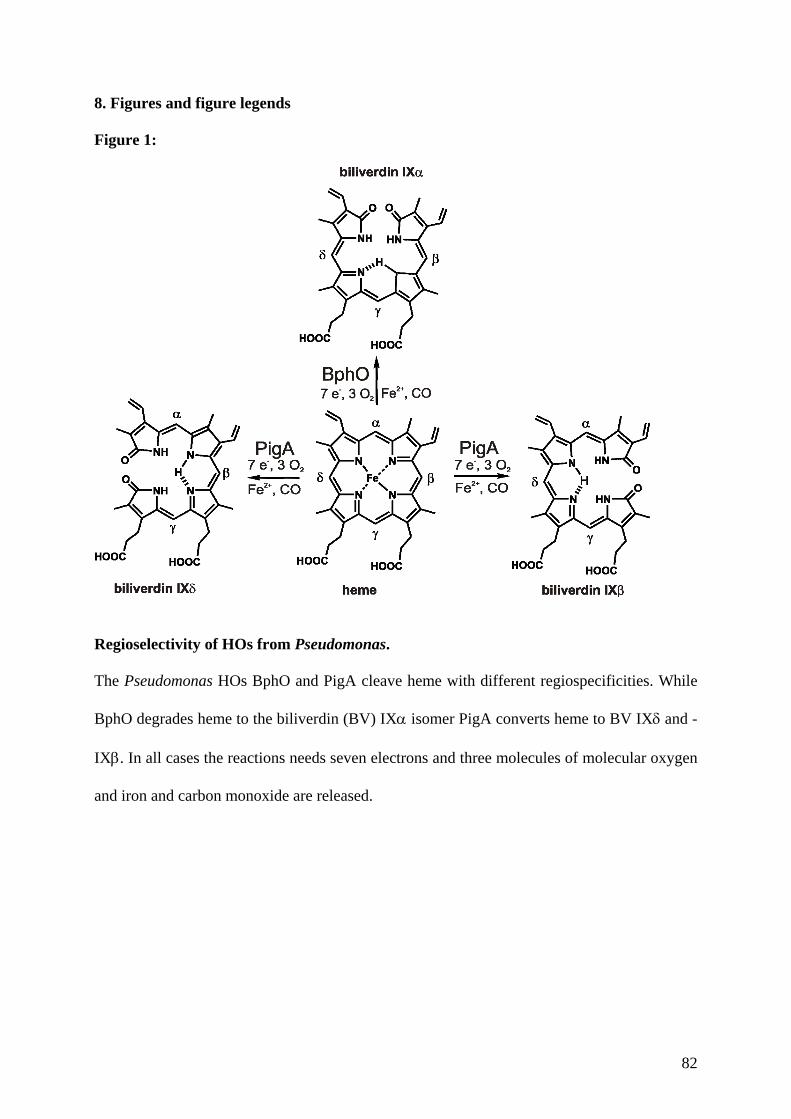
82
8. Figures and figure legends Figure 1:
Regioselectivity of HOs from Pseudomonas.
The Pseudomonas HOs BphO and PigA cleave heme with different regiospecificities. While
BphO degrades heme to the biliverdin (BV) IX isomer PigA converts heme to BV IX and -
IX. In all cases the reactions needs seven electrons and three molecules of molecular oxygen
and iron and carbon monoxide are released.

83
Figure 2:
SDS-PAGE analysis of recombinantly purified Pseudomonas PigAs.
Lane M: molecular mass marker (values are given in kDa on the left); lane 1: PA14 PigA;
lane 2: P.f. 5 PigA; lane 3: P. p. KT2440 PigA.
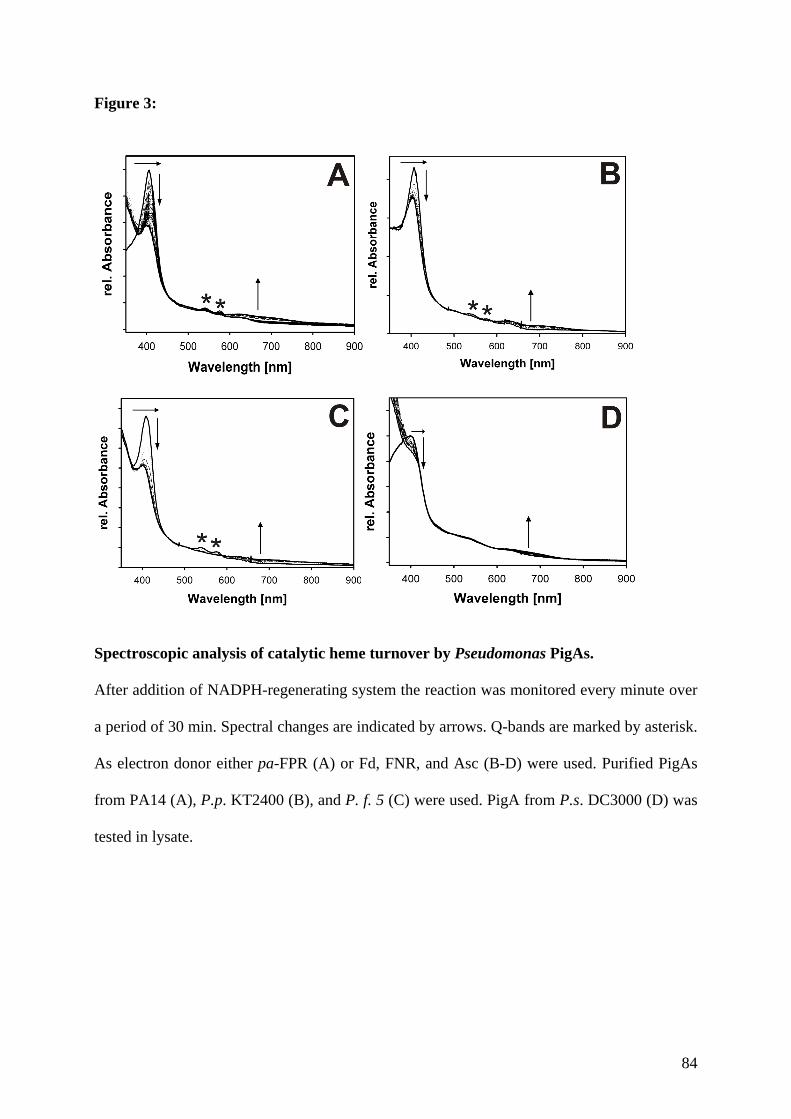
84
Figure 3:
Spectroscopic analysis of catalytic heme turnover by Pseudomonas PigAs.
After addition of NADPH-regenerating system the reaction was monitored every minute over
a period of 30 min. Spectral changes are indicated by arrows. Q-bands are marked by asterisk.
As electron donor either pa-FPR (A) or Fd, FNR, and Asc (B-D) were used. Purified PigAs
from PA14 (A), P.p. KT2400 (B), and P. f. 5 (C) were used. PigA from P.s. DC3000 (D) was
tested in lysate.

85
Figure 4:
HPLC analysis of BV isomers products during heme turnover by Pseudomonas sp. HOs.
After cleavage of heme by BphO (A) or PigA (B) BV was isolated and analyzed by reversed
phase HPLC. BphOs and PigAs from different Pseudomonas strains were tested as indicated
above each elution profile. A mixture of BV isomers was used as standard (BV isomers).
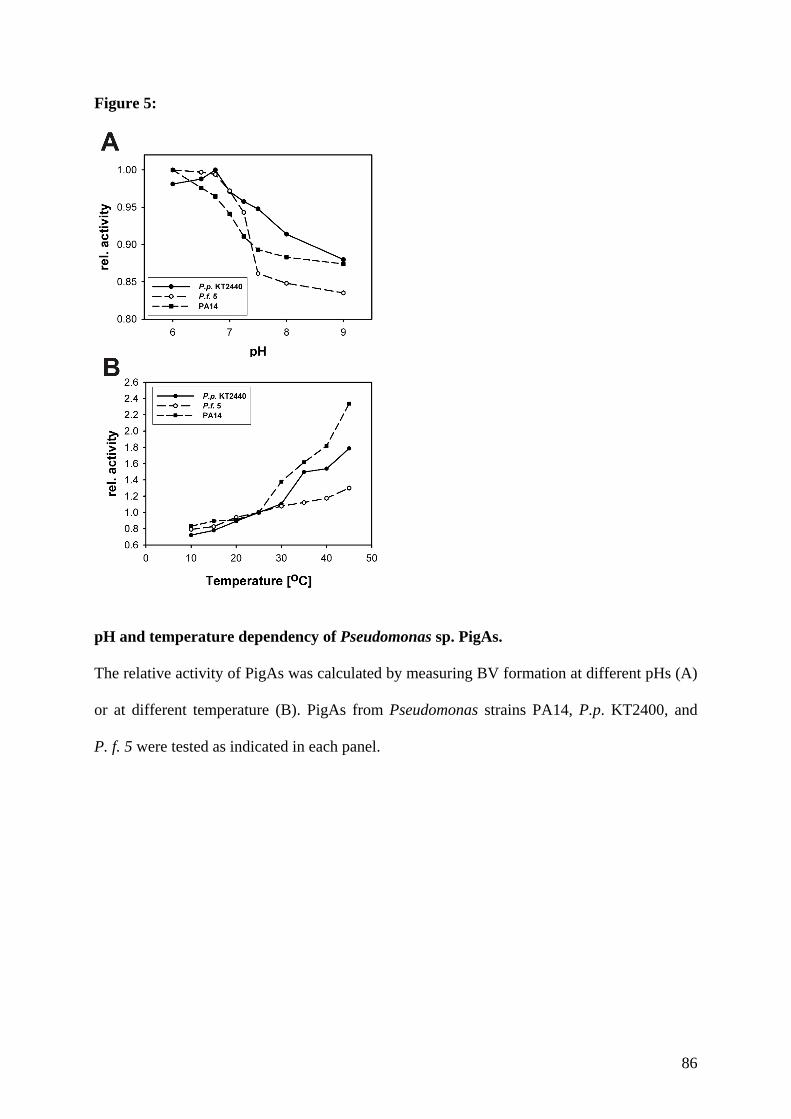
86
Figure 5:
pH and temperature dependency of Pseudomonas sp. PigAs.
The relative activity of PigAs was calculated by measuring BV formation at different pHs (A)
or at different temperature (B). PigAs from Pseudomonas strains PA14, P.p. KT2400, and
P. f. 5 were tested as indicated in each panel.

Diskussion
87
D Diskussion
1. Hämoxygenasen aus Arabidopsis thaliana
Das Arabidopsis thaliana Genom besitzt vier Gene, die für putative HOs (HY1, HO2, HO3
und HO4) kodieren. Für alle HOs wird eine Beteiligung an der Synthese des Phytochrom-
Chromophors PΦB postuliert, wobei die HY1 das Hauptenzym darstellt. Da alle HOs unter
gleichen Bedingungen in identischen Geweben exprimiert werden, stellt sich die Frage,
warum A. thaliana über vier HOs verfügt bzw. was deren spezifische Funktionen sind. Ob die
HOs in ihrer Enzymaktivität differieren, sollte in Experimenten dieser Arbeit geklärt werden.
Lediglich die HY1 wurde bis dato biochemisch näher untersucht und ihre HO-Aktivität
in vitro nachgewiesen. Die HO2, HO3 und HO4 sollten daher erstmalig biochemisch
charakterisiert und mit der HY1 verglichen werden um letztlich Rückschlüsse auf mögliche
Funktionsunterschiede der Enzyme zuzulassen.
Die biochemische Charakterisierung der vier HOs aus A. thalina zeigte, dass alle Mitglieder
der HO1-Unterfamilie (HY1, HO3 und HO4) in vitro aktiv sind und BV IXα produzierten
(Muramoto et al., 2002; Gisk et al., 2010). Die HO2 konnte weder Häm binden noch
umsetzen. Entsprechend stellt die HO2 keine aktive HO dar. Eine Gemeinsamkeit aller vier
Arabidopsis HOs besteht darin, dass sie eine N-terminale Transit-Peptid-Lokalisationssequenz
für den Chloroplast oder das Mitochondrium aufweisen. In beiden Kompartimenten ist die
Anwesenheit von HOs vorstellbar. Im Chloroplast findet die Phytochrom-
Chromophorbiosynthese statt, an der die HOs maßgeblich beteiligt sind. Im Mitochondrium
könnten die HOs zum Hämabbau benötigt werden, auf Grund der dort vorliegenden hohen
Hämkonzentrationen.
1.1 Hämoxygenasen aus A. thaliana sind nicht im Mitochondrium, sondern im Chloroplast lokalisiert
Im Mitochondrium sind große Mengen an Häm bzw. Hämproteinen vorhanden um z. B. die
Zelle mit Energie (ATP) durch oxidative Phosphorylierung zu versorgen (Bonner&Millerd,
1953). Durch den Abbau der im Mitochondrium vorhandenen Hämproteine wird das in den
Proteinen als Cofaktor eingebaute Häm freigesetzt und muss degradiert werden, bevor es
zytotoxisch wirkt. Von einigen Proteinen mit N-terminaler Transit-Peptid-
Lokalisationssequenz, wie sie alle vier HOs aufweisen, ist bekannt, dass sie sowohl in den
Chloroplast, als auch in das Mitochondrium transportiert werden (Chabregas et al., 2001;
Bhushan et al., 2006). Es ist somit möglich, dass die HOs ebenfalls im Mitochondrium

Diskussion
88
lokalisiert und dort am Hämabbau beteiligt sind. Da gezeigt werden konnte, dass die HY1
maßgeblich in die Phytochrom-Chromophorbildung involviert ist (Emborg et al., 2006), wäre
als mögliche Funktion der anderen beiden aktiven HOs (HO3 und HO4) der Hämabbau im
Mitochondrium denkbar. Unsere Untersuchungen ergaben jedoch, dass alle vier HOs im
Chloroplast der Pflanzenzelle lokalisiert sind (Gisk et al., 2010). Sind die HOs nicht die Häm
abbauenden Proteine im Mitochondrium, so muss dort ein alternatives Häm-abbauendes
Protein, wie eine Monooxygenase, vorliegen.
Da sich die HOs nicht durch die Lokalisation unterschieden haben, müssen die biochemischen
Reaktionen und Kinetiken der HOs näher betrachtet werden, um eventuelle Unterschiede zu
finden.
1.2 HY1, HO3 und HO4 aus A. thaliana sind biochemisch nahezu identisch
Um die unterschiedlichen Funktionen der drei Mitglieder der HO1-Unterfamile (HY1, HO3
und HO4) aus A. thaliana zu identifizieren, wurden ihre biochemischen Parameter analysiert.
Die drei HOs zeigten nahezu identische biochemische Parameter, wie Kd, Km, Ea und die
gleiche Regiospezifität für den α-meso-Kohlenstoff des Häms. Im Gegensatz zu bakteriellen
und Säuger HOs, die während der heterologen Expression in E. coli eine grünliche Färbung
aufweisen, ist bei der Expression der pflanzlichen HOs diese Färbung nicht zu beobachten.
Die grüne Färbung der Zellen ist u. a. für die BphO aus P. aeruginosa (Wegele et al., 2004)
und die hHO-1 aus dem Säuger (Wilks et al., 1995) beschrieben worden. Sie ist auf die
Umsetzung von endogenem Häm in E. coli zurückzuführen. Als Elektronendonator dient
vermutlich das E. coli eigene Ferredoxin (Fd) (Knoell&Knappe, 1974). Die Arabidopsis HOs
hingegen konnten das E. coli eigene Häm nicht umsetzen. Es lässt sich vermuten, dass das
E. coli Fd kein funktioneller Redoxpartner für diese HOs darstellt und die pflanzlichen HOs
ein anderes Fd als Redoxparter benötigen um aktiv zu sein.
In den Experimenten dieser Arbeit konnte gezeigt werden, dass alle drei aktiven HOs ein
pflanzliches Fd als Elektronendonator verwendeten und in Anwesenheit eines zweiten
Elektrondonators, wie Ascorbat, einen schnelleren Hämumsatz zeigten. Welches Fd primär
von den HOs der HO1-Unterfamilie in Arabidopsis verwendet wird, ist unklar. Arabidopsis
besitzt vier Fds (Fd1-4) und zwei Fd-ähnliche Proteine (Fd-like1- und Fd-like2-Protein). Alle
Fds besitzen ein [2Fe-2S]-Zentrum, welches durch vier Cystein-Reste koordiniert wird. Sie
weisen ein sehr negatives Redoxpotential von -350 mV zu -450 mV auf, was sie zu guten
Eletronendonatoren macht (Hanke et al., 2004). Unterschieden werden die vier Fds durch ihr
gewebespezifisches Vorkommen. Fd1 und Fd2 sind Blatttyp-Fd, Fd3 ist ein Wurzeltyp-Fd

Diskussion
89
und Fd4 ein dem Wurzeltyp ähnliches Fd. Es konnte außerdem gezeigt werden, dass Fd3 und
Fd4 eine geringere Affinität zum Photosystem I (PSI) aufweisen, als Fd1 und Fd2 (Hanke et
al., 2004). Da durch in vitro Experimente mit isolierten Thylakoidmembranen aus Spinat die
HO-Aktivität aller drei Arabidopsis HOs nachgewiesen werden konnte ist anzunehmen, dass
diese entweder mit dem Fd1 oder Fd2 interagieren, da Fd3 und Fd4 nicht in diesem
Kompartiment vorkommen.
Das PSI überträgt die Elektronen auf die Blatttyp-Fds an der äußeren Thylakoidmembran
(Abb. 9). Die Elektronen werden anschließend vermutlich von Fd1 und/oder Fd2 direkt weiter
auf die HOs geleitet.
Abbildung 9: Vereinfachte schematische Darstellung der Elektronenübertragung auf eine pflanzliche HO. Dargestellt sind das Photosystem II (PSII), der Plastochinonpool (PQ), der Cytochrom b6f-Komplex, das Plastocyanin, das Photosystem I (PSI) sowie das Ferredoxin (Fd) und die Hämoxygenase (HO). Die Elektronentransportkette zum Ferredoxin ist mit schwarzen Pfeilen und der Protonentransport ist mit grauen Pfeilen gekennzeichnet. Der Das Fd überträgt die Elektronen auf die HO, die dann das Häm zum BV spaltet.
Obwohl alle drei aktiven HOs der HO1-Unterfamilie (HY1, HO3 und HO4) in vitro
annähernd identische biochemische Parameter, wie Kd, Km, Ea, zeigten, können HO3 und
HO4 die HY1 in ihrer Funktion in vivo nicht ersetzen. Dies wird durch den ausgeprägten
Phänotyp der HY1 T-DNA Insertionsmutante (hy1) in Arabidopsis deutlich. Diese Pflanze ist
in ihrem Wachstum stark limitiert, weist ein langes Hypocotyl auf und besitzt nur eine geringe
Chlorophyllkonzentration (Emborg et al., 2006).
Über DNA-Microarrays konnte gezeigt werden, dass alle vier Arabidopsis HO Gene
exprimiert werden, wobei HO2, HO3 und HO4 ein deutlich niedrigeres Expressionsniveau
aufweisen. Die Expressionslevel nehmen von HY1 (~ drei-fach höher als HO2) über HO2
(~ zehn-fach höher als HO3 und HO4) zu HO3 und HO4 deutlich ab. Die höchste Expression
aller vier HO Gene ist in den Blättern der Pflanze durch β-Glucuronidase-Fusionen (GUS)
detektiert worden (Emborg et al., 2006). Die niedrigen Expressionslevel von HO3 und HO4
könnten erklären, warum beide aktiven HOs (HO3 und HO4) die HY1 nicht ersetzen können.
PSII
PSI
Plasto-cyanin
HO
Cytb6f- Komplex Thylakoidmembran
Stroma
Lumen
PQ
PQH2
2H2O 4H+ + O2
Licht Licht
2H+
2H+
Fd

Diskussion
90
Bislang konnten keine Bedingungen bestimmt werden, unter denen HO3 und HO4 stärker
induziert werden. Durch Studien bei anderen Pflanzen, wie der Sojabohne, konnte bereits
gezeigt werden, dass unter extremen Umweltbedingungen die Expression der HO1 gesteigert
wird. Das von der HO1 gebildete BV beugt dann als Antioxidant Zellschädigungen vor
(Balestrasse et al., 2005; Yannarelli et al., 2006). Durch zukünftige weitere Untersuchungen
auf DNA und Proteinebene könnten zusätzliche Informationen über die Funktionen der HOs
aus der HO1-Unterfamilie gewonnen werden.
Einen deutlicheren Unterschied in seiner Biochemie zeigte die HO2 im Vergleich zu den
anderen drei A. thaliana HOs.
1.3 HO2 aus A. thaliana und ihre mögliche Funktion
Nachdem die aktiven HOs aus A. thaliana scheinbar nur in ihrer Expression Unterschiede
zeigten, wurde die nicht aktive HO2 der A. thaliana untersucht. Sie konnte weder Häm binden
noch umsetzen. Interessanterweise weist die HO2 an der Position des für die Hämbindung
notwendigen Histidin-Restes einen Arginin-Rest auf (Abb. 10).
Abbildung 10: Aminosäuresequenzvergleich der vier HOs aus A. thaliana. In dem senkrecht markierten Bereich befindet sich bei der HY1, HO3 und HO4 ein Histidin-Rest, welchem für die Koordination des Eisenzentralatoms des Häms eine wichtige Bedeutung zugemessen wird. Im waagerecht markierten Bereich bei der HO2 befinden sich die Spacer-Region der HO2 und eine Abfolge von Glutamat- und Aspartat-Resten. Identische, konservierte, Aminosäure-Reste (AS) sind dunkelgrau (*), konservierte Aminosäureaustausche hell grau (:) hinterlegt. Semi- konservierte, ausgetauschte AS sind mit einem Punkt markiert (.). Hellgrau hinterlegte AS ohne zusäzliches Symbol sind AS, deren Mehrheit im Alignment vorkommt.

Diskussion
91
Durch einen indirekten in vivo Nachweis des Produktes BV IX konnte gezeigt werden, dass
weder die HO2, noch eine HO2 Variante mit einem Austausch des Arginin-Restes zu einem
Histidin-Rest (HO2_R88H) eine funktionelle HO ist (Emborg et al., 2006). Allerdings stellte
sich in dieser Studie die schlechte Lößlichkeit der Proteine als großes Problem dar (Emborg et
al., 2006). In den Experimenten dieser Arbeit konnte somit erstmalig die HO2 in größeren
Mengen gereinigt und die Inaktivität eindeutig demonstriert werden.
Die HO2 konnte allerdings Protoporphyrin IX (Proto IX), die Vorstufe des Häms, mit hoher
Affinität binden. Diese Bindung des Proto IX in der HO2 wurde durch
Gelfiltrationsexperimente bestätigt (Gisk et al., 2010). Die hohe Affinität der HO2 zum Proto
IX könnte durch die zahlreich vorhandenen Aspartat- und Glutamat-Reste, die sich hinter der
Spacer-Region des Proteins befinden (Abb. 10), zu erklären sein. Die Sauerstoff-Atome der
Aspartat- und Glutamat-Reste könnten die Stickstoffe der vier Pyrrolringe durch ionische
Wechselwirkungen koordinieren und somit das Proto IX in der Substrattasche binden. Eine
ähnliche Bindung wurde bereits für die Porphobilinogen-Deaminase nachgewiesen (Louie et
al., 1992). Auch bei diesem Protein sind Aspartat-Reste in unmittelbarer Umgebung des
Porphobilinogens, einer frühen Vorstufe des Proto IX, an der Bindung des Tetrapyrrol-
Derivates im Protein maßgeblich beteiligt. Da jedoch die Tertiärstruktur einer pflanzlichen
HO noch nicht aufgeklärt werden konnte, bleibt offen, ob tatsächlich die zahlreichen
Aspartat- und Glutamat-Reste hinter der Spacer-Region der HO2 in der Substrattasche
lokalisiert sind und die Koordination des Proto IX über diese bestimmten Aminosäure-Reste
geschieht.
Durch die hohe Affinität der HO2 zum Proto IX könnte dieses Protein allerdings in der
Pflanzenzelle als Transport- und/oder Speicherprotein für Proto IX fungieren. Proto IX ist
nicht nur die Vorstufe des Häms, sondern in photosynthetischen Organismen auch die
Vorstufe der Chlorophylle. Die Insertion eines Magnesiumions in den Proto IX-Ring durch
die Magnesiumchelatase leitet die Chlorophyllbiosynthese ein (Walker&Willows, 1997),
durch den Einbau eines Eisenions hingegen entsteht Häm (Dailey et al., 2000). Die HO2 als
Proto IX Transport- und/oder Speicherprotein könnte regulieren, welcher Biosyntheseweg
bedient wird.
Dass HO2, HO3 und HO4 eine Funktion in der Photomorphogenese, besonders in
Abwesenheit der HY1, haben, wurde bereits durch phänotypische Analysen von T-DNA
Insertionsmutanten gezeigt (Emborg et al., 2006). Die HO2 T-DNA Insertionsmutante (ho2)
in Arabidopsis zeigt einen leichten Phänotyp, welcher mit einer Rolle der HO2 als Proto IX
Transport- und/oder Speicherprotein zu erklären wäre. Die ho2 Pflanze ist trotz andauernder
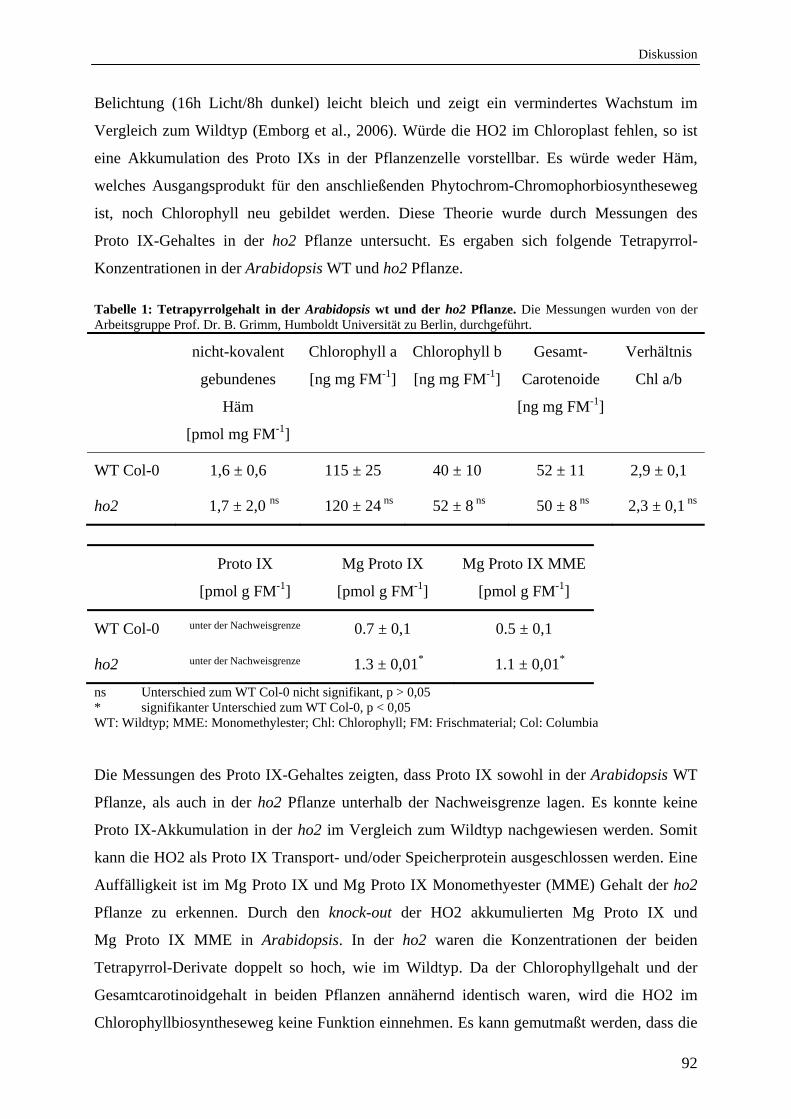
Diskussion
92
Belichtung (16h Licht/8h dunkel) leicht bleich und zeigt ein vermindertes Wachstum im
Vergleich zum Wildtyp (Emborg et al., 2006). Würde die HO2 im Chloroplast fehlen, so ist
eine Akkumulation des Proto IXs in der Pflanzenzelle vorstellbar. Es würde weder Häm,
welches Ausgangsprodukt für den anschließenden Phytochrom-Chromophorbiosyntheseweg
ist, noch Chlorophyll neu gebildet werden. Diese Theorie wurde durch Messungen des
Proto IX-Gehaltes in der ho2 Pflanze untersucht. Es ergaben sich folgende Tetrapyrrol-
Konzentrationen in der Arabidopsis WT und ho2 Pflanze.
Tabelle 1: Tetrapyrrolgehalt in der Arabidopsis wt und der ho2 Pflanze. Die Messungen wurden von der Arbeitsgruppe Prof. Dr. B. Grimm, Humboldt Universität zu Berlin, durchgeführt.
nicht-kovalent
gebundenes
Häm
[pmol mg FM-1]
Chlorophyll a
[ng mg FM-1]
Chlorophyll b
[ng mg FM-1]
Gesamt-
Carotenoide
[ng mg FM-1]
Verhältnis
Chl a/b
WT Col-0 1,6 ± 0,6 115 ± 25 40 ± 10 52 ± 11 2,9 ± 0,1
ho2 1,7 ± 2,0 ns 120 ± 24 ns 52 ± 8 ns 50 ± 8 ns 2,3 ± 0,1 ns
Proto IX
[pmol g FM-1]
Mg Proto IX
[pmol g FM-1]
Mg Proto IX MME
[pmol g FM-1]
WT Col-0 unter der Nachweisgrenze 0.7 ± 0,1 0.5 ± 0,1
ho2 unter der Nachweisgrenze 1.3 ± 0,01* 1.1 ± 0,01*
ns Unterschied zum WT Col-0 nicht signifikant, p > 0,05 * signifikanter Unterschied zum WT Col-0, p < 0,05 WT: Wildtyp; MME: Monomethylester; Chl: Chlorophyll; FM: Frischmaterial; Col: Columbia
Die Messungen des Proto IX-Gehaltes zeigten, dass Proto IX sowohl in der Arabidopsis WT
Pflanze, als auch in der ho2 Pflanze unterhalb der Nachweisgrenze lagen. Es konnte keine
Proto IX-Akkumulation in der ho2 im Vergleich zum Wildtyp nachgewiesen werden. Somit
kann die HO2 als Proto IX Transport- und/oder Speicherprotein ausgeschlossen werden. Eine
Auffälligkeit ist im Mg Proto IX und Mg Proto IX Monomethyester (MME) Gehalt der ho2
Pflanze zu erkennen. Durch den knock-out der HO2 akkumulierten Mg Proto IX und
Mg Proto IX MME in Arabidopsis. In der ho2 waren die Konzentrationen der beiden
Tetrapyrrol-Derivate doppelt so hoch, wie im Wildtyp. Da der Chlorophyllgehalt und der
Gesamtcarotinoidgehalt in beiden Pflanzen annähernd identisch waren, wird die HO2 im
Chlorophyllbiosyntheseweg keine Funktion einnehmen. Es kann gemutmaßt werden, dass die

Diskussion
93
HO2 Mg Proto IX und Mg Proto IX MME bindet und zu einem anderen, bis jetzt
unbekannten Protein oder Ort der Zelle transportiert. Dieser Ort könnte der Zellkern sein. Es
wird vermutet, dass Mg Proto IX und Mg Proto IX MME retrograde Signale in der
Pflanzenzelle sind, durch die die Chlorophyllbiosynthesegene feedback reguliert werden. Bei
dieser Theorie wird davon ausgegangen, dass Mg Proto IX und Mg Proto IX MME an ein
noch unbekanntes Signalprotein binden und mittels dieses Proteins in den Zellkern gelangen
(Jarvis, 2003; Shepherd et al., 2003; Strand et al., 2003; Ankele et al., 2007). Da in der ho2
Pflanze genau diese beiden Tetrapyrrol-Derivate akkumulierten, könnte die HO2 das gesuchte
Signalprotein sein. Sie könnte Mg Proto IX und Mg Proto IX MME binden und in den
Zellkern transportieren (Abb. 11).
Abbildung 11: Potentielle Funktion der HO2 als Signalprotein. Die HO2 bindet die in der ho2 Pflanze akkumulierten Mg Proto IX und Mg Proto IX MME (Monomethylester) im Cytosol und transferiert diese Moleküle in den Zellkern, wo die Genregulation retrograd reguliert wird.
Bislang ist jedoch nicht bekannt, dass gefaltete Proteine aus dem Chloroplasten in das Cytosol
gelangen können. Es könnte daher sein, dass HO2 nicht im Chloroplast translokalisiert wird,
sondern im Cytosol und die durch die Chloroplastenhülle ausgeschleusten akkumulierten
Tetrapyrrol-Derivate bindet. Im Cytosol ist die HO2 dann das Trägerprotein, was den
Transport von Mg Proto IX und Mg Proto IX MME zum Zellkern vermittelt.
Gelangt die HO2 mit den gebundenen Tetrapyrrol-Derivaten nicht in den Zellkern, so könnte
die HO2 dennoch Mg Proto IX und Mg Proto IX MME binden und an ein weiteres, bis jetzt
noch unbekanntes Protein übergeben, welches stattdessen in den Zellkern gelangt.
ALA Proto IX
Vakuole
Fe Proto IX Mg Proto IX
Mg Proto IX MME
Chlorophyll
Mg Proto IX
HO2
HO2

Diskussion
94
Da in den Experimenten der Arbeit jedoch gezeigt werden konnte, dass auch die HO2 im
Chloroplast lokalisiert ist, könnte sie bereits die beiden akkumulierten Tetrapyrrol-Derivate
im Chloroplast binden. Nach dortiger Bindung transportiert sie diese dann zur
Chloroplastenhülle, wo Mg Proto IX und Mg Proto IX MME anschließend ausgeschleust
werden um an ein noch unbekanntes Protein im Cytosol zu binden und in den Zellkern zu
gelangen. Um diese Hypothese zu verifizieren oder zu falsifizieren sind weitere
Untersuchungen notwendig.
1.4 Arabidopsis Hämoxygenasen unterliegen der Produktinhibierung durch Eisen und BV, jedoch der durch CO nur bedingt
Enzyme können durch eigene Reaktionsprodukte in ihrer katalytischen Aktivität inhibiert
werden. Dieses ist auch bei der HO-Reaktion zu beobachten. Das bei der Hämumsetzung
gebildete BV liegt nach Beendigung der katalytischen Reaktion noch chelatisiert als Fe3+-BV
vor. Zur Freisetzung des BVs muss dieser Komplex mit einem weiteren Elektron zum Fe2+-
BV reduziert werden, bevor das BV frei ohne Eisen vorliegen kann (Yoshida et al., 1980;
Liu&Ortiz de Montellano, 2000). Für einige pflanzliche HOs wurde bereits gezeigt, dass ohne
Anwesenheit eines starken Eisenchelators das Eisen-BV in der HO gebunden bleibt und eine
weitere Umsetzung eines neuen Hämmoleküls ausgeschlossen ist (Muramoto et al., 2002;
Linley et al., 2006). Dies traf auch auf die drei aktiven HOs aus Arabidopsis zu (Gisk et al.,
2010). Die hohe Affinität des Eisen-BV zur HO kann dadurch erklärt werden, dass 90% des
zellulären Eisens im Chloroplasten vorliegen und folglich die Freisetzung des Eisens mit einer
hohen Barriere verbunden ist (Terry&Abadia, 1986). Somit unterliegt der Hämabbau der
Produktinhibierung durch Eisen-BV. Diese Produktinhibierung wird auch als negative
feedback Inhibierung bezeichnet und wurde in vitro und in vivo ebenfalls für das
Reaktionsprodukt BV nachgewiesen. Es konnte gezeigt werden, dass die Zugabe von freiem
BV in vitro bzw. das in vivo beim Hämabbau akkumulierte BV die katalytische Aktivität der
HO aus Ratte stark reduzierte (Kutty&Maines, 1984; Maines, 1992). Die Weitergabe des BV
aus der HO zum nachfolgenden Enzym ist daher unabdingbar für die Aktivität der HOs
(Gohya et al., 2006).
Im Gegensatz zur Produktinhibierung durch BV ist der Einfluss des bei der HO-Reaktion
entstandenen COs gering. Die Schritte der Hämspaltung, bei denen CO an die Intermediate
des Hämabbaus binden kann, zeigen bei näherer Betrachtung im ersten Schritt der Reaktion
(Abb. 8), dass die Affinität des Eisenzentralatoms des Häms für molekularen O2 sehr hoch ist.
Im Vergleich zum Myoglobin ist die Affinität der HO zum O2 sogar 30-fach höher. Im

Diskussion
95
Gegensatz dazu sind die CO-Affinitäten beider Proteine dieselben, so dass in diesem Schritt
eine Produktinhibierung von CO ausgeschlossen werden kann (Migita et al., 1998). Im letzten
Schritt ist die Bindung des CO an den HO-Verdohäm Komplex sehr schwach und die
Dissoziationskonstante ist sehr groß (koff(CO) ≈ 3 s-1), so dass CO auch diesen Schritt von
Verdohäm zu BV nicht hemmen kann (Abb. 8, Stufe 5) (Migita et al., 1998). Es wurde zudem
anhand der Kristallstruktur der HO1 aus Rattus norvegicus eine „CO-trapping site“ gefunden,
in der das bei der Freisetzung des Hämabbaus entstandene eigene CO eingelagert wird. Diese
„CO-trapping site“ wird auch als hydrophobe Tasche bezeichnet und befindet sich nahe der
Hämtasche über dem α-meso-Kohlenstoff des Häms. Durch die sofortige Speicherung des
beim Hämabbau freigesetzten COs kann dieses nicht weiter mit dem Eisenzentralatom des
Häms reagieren und die Reaktion inhibieren (Sugishima et al., 2004).
Liegt CO allerdings in höheren Konzentrationen vor, ist die Bindung von CO an das Fe2+ des
Häms möglich, und es kann die Reaktivität der HO beeinflussen. Aber nicht nur CO, sondern
auch andere diatomische Gase, wie Stickstoffmonoxid (NO) und O2 können sehr effizient an
das Fe2+-Atom der prosthetischen Hämgruppe der HOs binden. Dabei erfüllen die Gase als
sechster Koordinationspartner des Eisenzentralatoms des Häms neben dem Stickstoffatom des
neutralen Imidazol vom Histidin-Rest sowie der vier Stickstoffatome des Pyrrolrings eine
wichtige Funktion bei der Stabilisierung des Häms in der Substrattasche (Sugishima et al.,
2002). Im Falle des O2 ist diese Stabilisierung nur transient, da O2 durch die Bindung an Häm
aktiviert wird und dieses letztendlich umsetzt (Ortiz de Montellano&Wilks, 2001; Matsui et
al., 2010).
Diatomische Gase spielen auch eine kritische Rolle in physiologischen Prozessen vieler
Organismen, wie in der Atmung, der Erweiterung der Blutgefäße und als Neurotransmitter
(Maines, 1997; Zhao et al., 1999; Shikama, 2006). In Pflanzen ist CO an der Auxin-
induzierten Adventivwurzelbildung beteiligt (Guo et al., 2008; Xuan et al., 2008). Eine
weitere wichtige Funktion von CO in Pflanzen ist der Einfluss auf die Eisen-Homöostase und
die daraus resultierende verbesserte Anpassung an Eisenmangelbedingungen. So reguliert CO
in Arabidopsis die Expression von Genen, die für die Eisenaufnahme notwendig sind (Kong
et al., 2010). Es konnte gezeigt werden, dass der interne CO-Gehalt im Gewebe ansteigt,
wenn der Eisengehalt in Arabidopsis limitiert ist. Dieser Anstieg von internem CO ist auf eine
verstärkte Expression der HY1 zurückzuführen (Kong et al., 2010). Eine
Endprodukthemmung der HY1 würde somit den CO-Gehalt im Gewebe senken und zu einer
verminderten Eisenaufnahme führen. Daher ist in dieser Arbeit die Bindung von CO an alle
drei aktiven Arabidopsis HOs untersucht worden. Hämproteine können nicht nur ihr

Diskussion
96
eigentliches „Zielgas“ binden, sondern auch ihm ähnliche Gase gleicher Größe und Form. Die
Art der Bindung der verschiedenen Gase an die Hämproteine kann entscheidend sein, welches
Gas an die Proteine binden kann (Abb. 13).
R
O
NH
N
Fe
NH2
C
O
R
O
NH
N
Fe
NH2
OO
Abbildung 13: Schematische Darstellung der Bindung von CO und O2 an Häm. Das Häm ist in der horizontalen Ebene gezeigt mit dem Eisen als Zentralatom. Die fünfte Koordination des Eisens findet über einen Histidin-Rest des Proteins statt. A: CO bindet mit einem Winkel von ~ 158° an des Eisen des Häms. B: O2 bindet in einem Winkel von ~ 110° an das Eisen des Hämmoleküls.
Das CO bindet fast linear (axial) an das Fe2+ des Häms. Der Bindungswinkel zwischen dem
Eisen und CO beträgt ~ 158°. Dies ist der Fall, weil die dπ-Orbitale des Fe2+ mit den leeren
π*-Orbitalen des CO überlappen (Spiro&Jarzecki, 2001; Spiro&Kozlowski, 2001). In
Gegensatz zu CO binden O2 und NO gewinkelt an das Fe2+. Die π*-Elektronen des O2
überlappen maximal mit den d-Orbitalen des Eisens. Ihr Bindungswinkel zum Eisen ist mit
~ 110° deutlich kleiner, als bei der Bindung von CO (Kachalova et al., 1999; Jain&Chan,
2003).
Diese verschiedenen intrinsischen Bindungsgeometrien der Liganden bzw. der jeweiligen
„Zielgase“ entscheiden u. a. darüber, welches diatomische Gas in den Hämproteinen
gebunden werden kann.
Hämproteinen, wie Myoglobin und HOs, weisen allerdings eine erhöhte Affinität zu O2
relativ zu CO auf, welche durch die höhere Polarität des gebundenen O2-Ligand begründet ist.
Daran sind elektrostatische Interaktionen der distalen Tasche zur Stabilisierung des stark
polaren Fe-O2-Komplexes beteiligt. Ebenfalls wird gebundener Sauerstoff durch zahlreiche
Wasserstoffbrückenbindungen in der Substrattasche der HOs stärker stabilisiert, als CO
(Ohlson&Phillips, 1997). Für Säuger HOs (Migita et al., 1998; Unno et al., 2004) und
bakterielle HOs (Unno et al., 2004; Friedman et al., 2007) sind die Affinitäten und
Bindungsparameter für diatomische Gase bereits beschrieben worden. Die PigA aus
P. aeruginosa weist eine Assoziationskonstante für die CO-Bindung von 0,90 µM-1 s-1 auf.
Dieser Wert ist in gleicher Größenordnung mit dem für Myoglobin (0,76 µM-1 s-1) und für die
menschliche hHO-1 (0,90 µM-1 s-1) (Friedman et al., 2007). Die untersuchten pflanzlichen
158° 110°
A B

Diskussion
97
HOs haben mit einem kon von 1,0 µM-1 s-1 (für HY1) ebenfalls in der gleichen Größenordnung
gelegen und unterscheiden sich nicht zu den bereits untersuchten HOs.
Ein wesentlicher Unterschied der Pflanzen HOs untereinander und zu den bereits untersuchten
HOs bestand in der Tatsache, dass die HO3 und HO4 ein nicht eindeutig charakterisierbares
Intermediat während der CO-Bindung aufwiesen. Dieses Intermediat wurde spektroskopisch
bei 408 nm detektiert. Für die HO4 war es deutlicher nachweisbar, als für die HO3. Eine
mögliche Erklärung für dieses Intermediat in beiden HOs könnte in einer veränderten
elektronischen Umgebung des Eisenzentralatoms des Häms in der HO3 und HO4 während der
CO-Bindung sein. Es ist vorstellbar, dass das Eisenzentralatom des Häms in beiden HOs, bei
denen das Intermediat nachgewiesen werden konnte, während der CO-Bindung in einem
anderen spin Zustand (z. B. high-spin oder low-spin) vorliegt. Die daraus resultierende
veränderte Überlappung der d-Orbitale des Fe2+ mit den leeren π*-Orbitalen des CO könnte
die spektralen Veränderung bei 408 nm erklären.
Bei der CO-Bindung an das Häm der drei Arabidopsis HOs konnte ein CO-Konzentrations-
unabhängiger-Bereich gefolgt von einem CO-Konzentrationsabhängigen-Bereich ausfindig
gemacht werden (Manuskript 2, Fig. 1). Daher kann postuliert werden, dass die oben
beschriebene „CO-trapping site“ oder hydrophobe Tasche CO-Gas speichern kann. Da die
hydrophobe Tasche sehr nahe am Eisenzentralatom des Häms liegt, ist zu vermuten, dass das
an das Eisen des Häms gebundene CO zunächst aus der hydrophoben Tasche (CO-Speicher)
freigesetzt wird und anschließend aus dem CO angereicherten Solvent stammt. Somit bindet
erst das CO-Gas aus der Tasche konzentrationsunabhängig an das Häm der HOs. Nach der
Freisetzung des COs aus der Tasche ist es von Bedeutung wie hoch die CO-Konzentration im
Solvent ist um an das Eisen des Häm zu binden. Dadurch gibt es einen zweiten
konzentrationsabhängigen Bereich in der CO-Assoziation. Die CO-Assoziationskonstante der
Geminate war im Vergleich zu anderen Hämproteinen mit 8 * 104 s1 (für HY1) sehr klein
(Kriegl et al., 2002; Bruno et al., 2007). Das deutet darauf hin, dass diese hydrophobe CO-
Speichertasche in den Arabidopsis HOs sehr groß ist und somit der Reaktionsinhibierung
vorbeugt.
Ebenfalls können erste Aussagen über die an der CO-Bindung beteiligten Aminosäure-Reste
getroffen werden. Da im FTIR-Differenzspektrum keine Banden zwischen 1700 cm-1 und
1800 cm-1 detektiert wurden, kann davon ausgegangen werden, dass keine Aspartat- und
Glutamat-Reste an der CO-Bindung beteiligt sind.

Diskussion
98
1.5 Arabidopsis Hämoxygenasen sind strukturell ähnlich zu anderen Hämoxygenasen
Da allein auf Grund der biochemischen Charakterisierung die Funktionen der HOs aus
A. thaliana nicht eindeutig geklärt werden konnten, ist ein Vergleich der Strukturen dieser
HOs mit schon bekannten HOs notwendig um zusätzliche Informationen zu erhalten.
Auf Grund fehlender Strukturdaten einer pflanzlichen HO ist, basierend auf den vorliegenden
Kristallstrukturen der bekannten HOs, 2006 von Linley et al. ein Strukturmodell der HO1 aus
der Erbse mit Hilfe eines Strukturvorhersage-Programms erstellt worden (Linley et al., 2006).
Demnach ist auch die pflanzliche HO1 aus der Erbse ein α-helikales monomeres Protein. Das
Hämmolekül wird ebenfalls, wie bei anderen HOs, deren Struktur bereits gelöst wurde, über
eine distale und eine proximale Helix sowie über ein neutrales Imidazol eines Histidin-Restes
koordiniert. Auffallend ist die wesentlich größere Substrattasche oberhalb des gebundenen
Häms im Vergleich zu anderen bekannten HOs. Es wird postuliert, dass dort ein Ascorbat
(Asc) koordiniert werden kann, welches als ergänzender Cofaktor zum Ferredoxin der HO die
Elektronen zur Hämspaltung liefert (Muramoto et al., 2002). Der vorhergesagte Abstand
zwischen Häm und Asc beträgt 3,3 Å. Sechs Aminosäure-Reste (Glu 96, Phe 120, His 207, Ile
214, Tyr 231 and Ser 274), die mit dem potentiellen Asc interagieren könnten, wären mit 2,4-
3,8 Å in adequater Nähe (Linley et al., 2006). Diese größere Substrattasche wird auch für die
cyanobakteriellen HOs postuliert, da deren Reaktivität unter Anwesenheit von Asc deutlich
gesteigert werden konnte (Cornejo et al., 1998).
In der vorliegenden Arbeit ist der Versuch unternommen worden, die erste Tertiärstruktur
einer pflanzlichen HO zu lösen. Da jedoch die Versuche, Kristalle der Arabiopsis HOs zu
erlangen, wiederholt scheiterten, wurde für die HO3 aus Arabidopsis mit Hilfe des
Programms ExPASy3D eine Tertiärstruktur vorhergesagt (Abb. 12).
Ähnlich der bekannten Kristallstrukturen anderer HOs zeigt das Modell der HO3 ein
monomeres Protein mit acht rechtsgängigen α-Helices. Das Hämmolekül könnte auch in
diesem vorhergesagten Strukturmodell durch eine proximale und eine distale Helix
koordiniert werden (Abb. 12). Die Substrattasche weist eine diagonale Länge von 24,16 Å
und eine vertikale Länge von 13,51 Å auf. Somit kann die Substrattasche genügend Raum
bieten um ein Hämmolekül, das einen Durchmesser von ~ 15 Å hat, zu binden und zugleich
noch ein Asc in der Substrattasche als zweiten Elektronendonator zu koordinieren. Dieses
Modell ist im Einklang mit dem biochemischen Befund, dass in Kombination von Fd und Asc
die katalytische Aktivität der pflanzlichen HOs, im Vergleich zu der alleinigen Verwendung
von Fd, deutlich gesteigert werden konnte (Muramoto et al., 2002; Gisk et al., 2010).

Diskussion
99
Abbildung 12: Modell der Arabidopsis HO3 als Bänder-Modell. Die postulierte Tertiärstruktur der Arabidopsis HO3, die mit dem Programm ExPASy3D auf Grund vorhandener Kristallstrukturen ermittelt wurde, zeigt ein α-helikales Protein. Die distale und proximale Helix, zwischen denen das Hämmolekül koordiniert sein wird, sind beschriftet. Die Taschengröße wurde mit Hilfe des Programms PyMOL vermessen und hat eine diagonale Länge von ~ 24,16 Å und eine vertikale Länge von ~ 13,51 Å.
Über strukturelle Ähnlichkeiten der Mitglieder der HO1-Unterfamilie kann nur spekuliert
werden. Es konnte der Oligomerisierungsgrad aller vier Arabidopsis HOs mit Hilfe der
Gelfiltrationschromatographie bestimmt werden. Alle vier HOs sind monomere Proteine, die
eine kompakt gefaltete globuläre Domänenstruktur aufweisen. Diese strukturelle Annahme
kann auf Grund der Übereinstimmung der theoretisch berechneten und experimentell
bestimmten relativen molekularen Massen getroffen werden.
Durch FTIR-Messungen konnten erste Aminosäure-Reste, die an der Koordinierung des
Eisenzentralatoms des Häms beteiligt sind, bei allen drei aktiven Arabidopsis HOs bestimmt
werden. Die Koordination des Eisenzentralatoms des Häms durch den konservierten Histidin-
Rest in der proximalen Helix konnte exemplarisch für die HO3 gezeigt werden. Für HY1 und
HO4 ist dieses ebenfalls wahrscheinlich, da auch sie den konservierten Histidin-Rest in ihrer
Aminosäuresequenz an der gleichen Position, wie die HO3, aufweisen. In der distalen Helix
koordinieren ein Wasser-Molekül und ein Serin-Rest das Eisenzentralatom des Häms. Diese
Aussage kann auf Grund der Schwingungsbande des COs im FTIR (CO ~1954 cm1)
getroffen werden. Diese CO-Bande wurde bereits für PigA aus P. aeruginosa beschrieben
(Friedman et al., 2004). Mit Hilfe der vorhandenen PigA Kristallstruktur und in Kombination
mit FTIR-Messungen konnten in diesem Bakterium in der distalen Helix sowohl ein Wasser-
Molekül als auch ein Serin-Rest nahe dem Eisenzentralatom des Häms ausfindigt gemacht
werden (Friedman et al., 2004). Auf Grund der gleichen Bandenlage der CO-Schwingung ist
dieses Ergebnis auch auf die Arabidopsis HOs abzuleiten.
proximale Helix
distale Helix

Diskussion
100
Um eindeutige Aussagen über die Tertiärstruktur der pflanzlichen HOs zu treffen, ist es für
zukünftige Untersuchungen essentiell die Struktur der Proteine zu lösen oder alternativ
weitere, für die Hämbindung wichtige, Aminosäure-Reste in der Substrattasche zu
bestimmen.
2. Hämoxygenasen aus Pseudomonaden
2.1 Verschiedene Pseudomonas Spezies besitzen alle zwei aktive und regiospezifisch unterschiedliche Hämoxygenasen
Bakterielle HOs können auf Grund ihrer Funktion in zwei Gruppen eingeteilt werden. Die
eine Gruppe der HOs (PigAs) kann als Eisenaufnahme-Proteine bezeichnet werden, da sie an
der Gewinnung von Eisen aus Häm beteiligt sind. Die HOs der zweiten Gruppe sind
funktionell mit einem Phytochrom verbunden, wobei die Hauptaufgabe dieser Enzyme die
Produktion von BV als Chromophor für das bakterielle Phytochrome ist. Daher werden sie
auch als bakterielle Phytochrom-HOs bezeichnet (BphOs). Interessanterweise wurde bis jetzt
immer nur eine der beiden Gruppen in einem Bakterium beschrieben, mit Ausnahme des
opportunistisch pathogenen Bakterium P. aeruginosa. Dieser Organismus unterscheidet sich
nicht nur durch den Besitz der beiden funktionellen HOs von anderen Bakterien, sondern war
bis jetzt das einzige beschriebene Bakterium, das zwei HOs mit unterschiedlicher
Regiospezifität besitzt (Ratliff et al., 2001; Wegele et al., 2004). Die BphO katalysiert die
Spaltung des Häms zu BV IXα, Fe2+ und CO (Wegele et al., 2004), PigA hingegen setzt das
Häm zu Fe2+, CO, und den beiden BV Isomeren IXδ und IXβ im Verhältnis 70:30 um
(Caignan et al., 2002).
Im Rahmen dieser Arbeit wurden die bis dato sequenzierten und zugänglichen Genome der
Gattung Pseudomonas bioinformatisch auf die Existenz der beiden regiospezifisch
unterschiedlichen HOs untersucht. Alle Pseudomonaden verfügen über eine putative PigA
und eine putative BphO. Die beiden putativen HOs (PigAs und BphOs) aus beispielhaft
ausgewählten Pseudomonas sp. (P. aeruginosa PA14, P. fluorescence 5,
P. putida KT2440, und P. syringae DC3000) wurden biochemisch charakterisiert.
Die untersuchten PigAs und BphOs setzten Häm zu BV um und sind somit aktive HOs. Alle
PigAs zeigten eine Regiospezifität für den δ- und β-meso-Kohlenstoff des Häms auf, so dass
die beiden BV-Isomere IXδ und IXβ entstanden. Alle PigAs wiesen untereinander annähernd
gleiche biochemischen Parameter, wie, Kd, Km und Ea, auf. Die BphOs der untersuchten
Pseudomonas sp. hingegen haben das Häm ausschließlich in das BV IXα Isomer gespalten.

Diskussion
101
Dieses Phänomen wurde bis dato nur für P. aeruginosa gezeigt (Ratliff et al., 2001; Wegele et
al., 2004). Eine weitere Gemeinsamkeit aller untersuchen Pseudomonas sp. ist das
Vorkommen eines, bzw. im Falle von P. syringae zweier Phytochrome (Bhoo et al., 2001).
In vitro konnte gezeigt werden, dass das bakterielle Phytochrom BphP die beiden BV Isomere
IXα und BV IXδ kovalent binden kann. Eine Bindung des BV IXβ ist ausgeschlossen, weil
die Bindung eines BVs an das apo-Phytochrom ausschließlich über die Endovinylgruppe am
A-Ring des BV geschieht. BV IX besitzt diese Gruppe nicht (Wegele et al., 2004; Tasler et
al., 2005) (Abb. 3). Es ist jedoch zu vermuten, dass der natürliche Phytochrom-Chromophor
in vivo BV IXα ist, da bphO und bphP in einem Operon organisiert sind und gemeinsam
reguliert werden (Barkovits et al., 2008). Ob BV IXδ in vivo an BphP bindet ist unklar.
Ebenfalls unklar ist auch, ob beide BV Isomer in vivo von PigA gebildet werden. In der
Kristallstruktur der PigA aus P. aeruginosa ist das Häm mit dem -meso-Kohlenstoff zur
aktiven Seite der Tasche hin positioniert, so dass bei der Hämspaltung nur BV IXδ entstehen
kann (Abb. 7) (Friedman et al., 2004). Die Entstehung des BV IXβ in vitro ist dadurch
bedingt, dass durch eine Rotation entlang der /-Achse des Häms der β-meso-Kohlenstoff in
den reaktiven Bereich der Substrattasche kommt, so dass BV IXβ entsteht (Friedman et al.,
2004). Durch in vivo Experimente kann aber angenommen werden, dass das natürliche
Hämabbauprodukt der PigA das BV IXδ ist, da in P. aeruginosa ein Hämtransport-Protein
bzw. Hämchaperon gefunden wurde (Lansky et al., 2006). Es wird spekuliert, dass das
Hämchaperon PhuS (Pseudomonas heme utilization system), das Teil eines
Hämaufnahmesystems ist, externes Häm sterisch gerichtet zu PigA transferiert, wodurch das
Häm nur in einer Orientierung in PigA vorliegt und somit einzig zu BV IXδ gespalten werden
kann (Lansky et al., 2006). Da PhuS nicht mit BphO interagiert (Bhakta&Wilks, 2006), kann
angenommen werden, dass PigA externes Häm unter Eisenmangelbedingungen spaltet, um
den Organismus mit Eisen zu versorgen. BphO hingegen nutzt vermutlich internes Häm, um
das bakterielle Phytochrom BphP mit dem Chromophor für den putativen Rotlichtsensor zu
versorgen. Falls BV IXδ tatsächlich ebenfalls an das Phytochrom bindet, könnte auf diese
Weise ein Signal über den Eisenstatus der Zelle über das Phytochromsystem integriert
werden. Die anderen drei untersuchten Pseudomonas sp. (P.f. 5, P.p. KT2440, und P.s.
DC3000) besitzen kein Phu-ähnliches Hämaufnahmesystem, welches das Häm gerichtet zu
PigA transferieren könnte. Daher ist bei diesen Pseudomonas sp. zu vermuten, dass beide BV-
Isomere durch PigA gebildet werden und in der Zelle vorliegen. Alternativ könnte ein bis jetzt
noch unbekanntes Hämchaperon, ähnliche dem PhuS, das Häm gerichtet zu PigA
transferieren und dadurch nur ein BV-Isomer gebildet werden.

Diskussion
102
Zum jetzigen Wissensstand ist es wahrscheinlich, dass nur potentiell Säuger pathogene
Pseudomonaden (P. aeruginosa PAO1 und PA14) externes Häm aus dem Wirt aufnehmen
können um sich mit Eisen zu versorgen. Da der Säugerorganismus große Mengen an Häm
besitzt (u. a. Hämoglobin, Myoglobin) (Hayashi et al., 1973) ist durch ein externes
Hämaufnahmesystem und ein Häm abbauendes Protein (PigA) die Versorgung von
P. aeruginosa mit Eisen gewährleistet. Die Funktion von PigA in den anderen
Pseudomonaden bleibt allerdings offen. Interessanterweise besitzen alle untersuchten
Pseudomonas sp., auch diese die kein PhuS besitzen, Fur-Boxen (ferric-uptake-regulater).
Durch diesen Eisenaufnahme-Regulator wird die Expression der Eisen-Homöostasegene
durch Integration des intrazellulären Eisengehaltes reguliert (Ochsner&Vasil, 1996). Welche
Funktion die HOs innerhalb dieses Systems einnehmen ist jedoch noch unklar.
Die Funktion des BphO hingegen besteht vermutlich in allen Pseudomonas sp. in der
Bereitstellung des Phytochrom-Chromophors BV IXα. Das bakterielle Phytochrom BphP ist
ein putativer Rotlichtsensor, welcher externe Signale in interne Signale der Zelle verarbeitet.
Strukturell haben alle untersuchten PigAs die Gemeinsamkeit, dass sie einen konservierten
Histidin-Rest in der proximalen Helix und in der distalen Helix das G-X-X-X-G-Motiv zur
Hämkoordination aufweisen (Abb. 14). Durch die vorhandene Kristallstruktur der PigA aus
P. aeruginosa und mit Hilfe von Punktmutationen konnten drei für die δ-Regiospezifität
essentielle Aminosäure-Reste bestimmt werden. Diese sind Lysin 34 und 132 und ein
Phenylalanin an Position 189 (Fujii et al., 2004). Alle vier untersuchten Pseudomonas sp.
haben an der entsprechenden Position ebenfalls ein Lysin- bzw. ein Phenylalanin-Rest. An
Stelle des Lysins 132 weisen die PigAs aus P.f. 5, P.p. KT2440 und P.s. DC3000 einen
Arginin-Rest auf (Abb. 14). Dieser Aminosäureaustausch in den PigAs kann als konservativer
Ausstausch bezeichntet werden. Der Arginin-Rest könnte dieselbe Funktion wie der Lysins-
Rest erfüllen. Alle untersuchten Proteine, die diese drei Aminosäure-Reste besitzen, zeigten in
vitro die Regiospezifität für den δ-meso-Kohlenstoff. Somit könnte in einem Aminosäure-
sequenzalignment von bakteriellen HOs anhand dieser drei Aminosäure-Reste die
Regiospezifität für den δ-meso-Kohlenstoff zu erkennen sein. Interessanterweise haben
BLAST Analysen ergeben, dass die am nächsten verwandten „PigAs“ in den Datenbanken,
nach den aufgeführten Pseudomonas sp., die PigAs aus z. B. Agrobacterium tumefaciens und
Neisseria meningitids sind. Diese beiden PigA Hämoxygenasen sind ausschließlich für den -
meso-Kohlenstoff spezifisch und spalten das Häm nur zu BV IX (Zhu et al., 2000;
Lamparter et al., 2002). Somit scheint die Regiospezifität für den - bzw. -meso Kohlenstoff
exklusiv in Pseudomonaden vorzuliegen.

Diskussion
103
Abbildung 14: Aminosäuresequenzvergleich verschiedener bakterieller PigAs und BphOs. Die PigAs und BphOs aus P. aeruginosa PAO1 und PA14, P. fluorescence (Pf), P. putida (Pp), P. syringae (Ps) und die PigAs aus Neisseria meningitidis (Nm), Agrobacterium tumefaciens (Atu) und die BphO aus Deinococcus radiodurans (Dr) sind gezeigt. Der konservierte Histidin-Rest in der proximalen Helix und das in Bakterien konservierte Glycin-Motiv in der distalen Helix zur Hämkoordination sind mit ( ) markiert. Potentielle Aminosäure-Reste (AS) für die -Regiospezifität (K und F) sind mit ( ) gekennzeichnet. (*) Konservierte AS-Reste; (:) Konservierte AS-Austausche; (.) Semi-konservierte AS-Austausche; (hell-grau) Mehrheitlich vorkommende AS-Reste; (dunkel-grau) konservierte AS-Reste

Diskussion
104
3. Corrole als alternative Hämoxygenase-Substrate
Hämproteine können nicht nur Häm als prosthetische Gruppe binden, sondern auch dem Häm
ähnliche Moleküle, die modifizierten Metalloporphyrine. Da die modifizierten
Metalloporphyrine eine ähnliche Struktur aufweisen, wie die natürlichen Hämgruppen,
können dem Protein durch eine Substitution neue Reaktivitäten verliehen werden. Dabei sind
keine entscheidenden strukturellen Änderungen an der Metallbindungsstelle erforderlich. In
dieser Arbeit sollten erstmals zwei regioisomere Corrole (Tetradehydrocorrine: α-CH-Fe(cor)
und γ-CH-Fe(cor)) in Pflanzentyp-HOs (Arabidopsis HY1, HO3 und HO4 sowie
Synechocystis sp. PCC 6803 HO-1), in die Säuger hHO-1 und in die beiden regiospezifisch
unterschiedlichen Pseudomonas HOs (BphO und PigA) eingebaut und auf ihre Aktivität hin
untersucht werden. Es konnte gezeigt werden, dass alle Pflanzentyp-HOs das γ-CH-Fe(cor)
gebunden und zu einem BV-Analog umgesetzt haben. Das regioisomere α-CH-Fe(cor) wurde
zwar von allen HOs gebunden, allerdings nicht umgesetzt (Abb. 15).
N
N
N
N
COOHHOOC
Fe
N
N
N
N
Fe
Cl
HOOC COOH
Cl
α-CH-Fe(cor) γ-CH-Fe(cor)
Abbildung 15: Die regioisomeren Eisencorrole α-CH-Fe(cor) und γ-CH-Fe(cor). Markiert sind die jeweilig reaktiven Kohlenstoffe in beiden Molekülen.
Dieses Ergebnis war nicht zu erwartet, da beim γ-CH-Fe(cor) kein meso-Kohlenstoff, ähnlich
wie beim Häm und beim α-CH-Fe(cor), vorliegt. Dieser meso-Kohlenstoff ist für die
katalytische Spaltung des Häms essentiell (Ortiz de Montellano&Wilks, 2001; Matsui et al.,
2010). Es wäre also ein genau entgegengesetztes Ergebnis (keine Umsetzung von γ-CH-
Fe(cor) und Umsetzung von α-CH-Fe(cor) zu erwarten gewesen.
Die Säuger hHO-1 und die regiospezifisch unterschiedlichen HOs, BphO und PigA, aus
P. aeruginosa konnten ebenfalls beide regioisomere Eisencorrole binden, jedoch keines der
beiden Moleküle umsetzen. Dieses unterschiedliche Reaktionsverhalten der HOs könnte auf
Unterschiede in der Substrattasche zwischen den Pflanzentyp-HOs und den bakteriellen bzw.

Diskussion
105
Säuger HOs zurückzuführen sein. Wie oben bereits beschrieben besitzen die Arabidopsis
HOs, wie auch die cyanobakterielle HO-1, wahrscheinlich eine größere aktive Substrattasche
(Kap. 1.4), in der ein Asc zur effizienteren Katalyse Raum findet. Die Corrole sind im
Gegensatz zum Häm nicht planar, da durch die Abstoßung der inneren Wasserstoffatome eine
leichte Verzerrung des Corrolring-Rückgrates aus der Ebene der vier zentralen
Stickstoffatome heraus vorliegt (Harrison et al., 1971). Daher können die Eisencorrole
vermutlich nur durch HOs mit einer größeren Substrattasche umgesetzt werden. Über den
Reaktionsmechanismus der Corrolöffnung kann nur spekuliert werden (Abb. 16).
Fe
N His
Fe
N His
OO
Superoxo-Komplex unreaktiv bei Eisenporphyrin
e-, H+
Häm
O2
Fe
N His
Superoxo-Komplex reaktiv bei Eisencorrol
Eisencorrol
O2
X e-, H+
Hydroperoxo-Komplex reaktiv bei Eisenporphyrin
Fe
N His
OO
Fe
N His
OO
Abbildung 16: Gegenüberstellung der O2-Aktivierungsschritte bei der natürlichen Hämspaltung (links) mit dem postulierter Verlauf der Eisencorrol-Öffnung durch die Pflanzentyp- HOs (rechts). Schematisch dargestellt sind das Hämmolekül, das über den neutralen Histidin-Rest des Proteins koordiniert wird sowie die Aktivierung durch den Sauerstoff.
Die Ringöffnung des Eisencorrols muss sich von der des Häms allerdings deutlich
unterscheiden, da beim geöffneten Eisencorrol kein meso-Kohlenstoff zwischen den beiden
Pyrrolringen vorhanden ist. Dieser meso-Kohlenstoff ist für die Hämspaltung entscheidend
(Teil A Kap. 3.5). Entsprechend wird bei der Corrolöffnung auch kein CO freigesetzt. Zieht
man eine ausgeglichene chemische Gleichung als Grundlage zur Beschreibung dieser
Umsetzung heran, so lässt sich die Reaktion als einfache Addition von O2 und zwei
Äquivalenten H2O an ein Molekül -CH-Fe(cor), gefolgt von der Eliminierung eines
Äquivalents Fe-O-OH formulieren. Der Disauerstoff wird in diesem Prozess offenbar durch

Diskussion
106
Eisencorrole reversibel gebunden und bei passender Orientierung im Protein auch auf eine
Bipyrroleinheit übertragen. Für die CH-Brücke wird hingegen keine Reaktion beobachtet. Die
natürliche HO-Reaktion, bei der eine solche Methingruppe der Hämeinheit angegriffen wird,
durchläuft zuvor einen weiteren, aktivierenden Reduktionsschritt unter Ausbildung einer
Eisenperoxo-Spezies (Abb. 16). Ein solcher vorgelagerte Elektronenübertrag kann beim
Eisencorrol jedoch auf Grund der ungünstigen Lage des O2-Bindungsgleichgewichts nicht mit
der Dissoziation der Fe-O2-Einheit konkurrieren (Collman et al., 1976; Vogel et al., 1994).
Die regioselektive Öffnung von -CH-Fe(cor) an der Bipyrrolseite deutet somit auf einen
nicht natürlichen Mechanismus hin, in dem schon die durch Assoziation gebildete Eisen-O2-
Einheit als aktive Spezies auftritt.
Die einzelnen Reaktionschritte bei der Eisencorrolöffnung und die dabei evtl. auftretenden
Intermediate sind noch unbekannt und müssen durch weitere Experimente überprüft werden.
Es ist jedoch bereits jetzt schon möglich, durch die unterschiedliche Regiochemie der beiden
untersuchten Eisencorrole (α-CH-Fe(cor) und γ-CH-Fe(cor)), die Reaktionsfähigkeit eines
Häm-Cofaktor-Analogons zu steuern. Dadurch ergeben sich viele wertvolle Möglichkeiten für
die Inaktivierung bakterieller HOs, für ein partielles knock-out der biologischen CO-
Produktion und für die Erforschung strukturell noch unbekannter aktiver Taschen, speziell die
pflanzlicher HOs.

Zusammenfassung
107
E Zusammenfassung
In dieser Arbeit sollten die Hämoxygenasen (HOs) aus Arabidopsis thaliana und aus
verschiedenen Pseudomonas Spezies biochemisch und biophysikalisch untersucht werden.
Alle vier HOs aus A. thaliana besitzen eine N-terminale Transit-Peptid-Lokalisationssequenz
und sind im Chloroplast lokalisiert. Durch Kombination von UV/vis-Spektroskopie und
HPLC-Untersuchungen konnte die Aktivität der Arabidopsis HOs aus der HO1-Unterfamilie
(HY1, HO3 und HO4) gezeigt werden. Alle drei Proteine spalten das Häm zum offenkettigen
Tetrapyrrol Biliverdin IXα (BV IX), der Vorstufe des Phytochrom-Chromophors. Da
bestimmte biochemische Parameter, wie Kd, Km und Ea, der HOs der HO1-Unterfamilie
annähernd identisch sind, kann davon ausgegangen werden, dass jede einzelne dieser HOs in
der Lage ist, BV IXα mit gleicher Effizienz zu bilden. Ebenfalls verwenden alle drei HOs
Ferredoxin als natürlichen Elektronendonator. Dies konnte mit Hilfe von isolierten
Thylakoidmembranen in vitro gezeigt werden. Die Verwendung eines zweiten
Elektronendonators (Ascorbat) steigert die Aktivität aller drei aktiven HOs um das zehn-
fache. Die HO2, die das einzige Protein der HO2-Unterfamilie in A. thaliana darstellt, ist
keine aktive HO. Die HO2 bindet weder Häm, noch ist das Protein in der Lage Häm zu
spalten. Allerdings kann eine Bindung des Häm- und Chlorophyll-Vorläufermolekül
Protoporphyrin IX (Proto IX) nachgewiesen werden. Ob es sich hierbei um eine physiologisch
bedeutende Interaktion handelt ist derzeit unklar.
Alle untersuchten pflanzlichen und bakteriellen HOs sind monomere Proteine und besitzen
einen konservierten Histidin-Rest, welcher zur Koordinierung des Eisenzentralatoms des
Häms erforderlich ist. Das Vorhandensein des proximalen Histidin-Restes wurde durch FTIR-
Untersuchungen der HO3 und der 15N angereicherten HO3 aus Arabidopsis bestätigt. Die
A. thalinana HOs der HO1-Unterfamilie unterscheiden sich lediglich in der Bindung zu
Kohlenmonoxid (CO), was durch Flash-Photolyse und FTIR-Messungen gezeigt werden
konnte. HO3 und HO4 weisen während der CO-Bindung ein detektierbares Intermediat auf,
welches in der HY1 nicht auftritt. Die eindeutige Charakterisierung dieses Intermediates ist
jedoch auf Grund der verwendeten Methoden nicht möglich gewesen. Die
Assoziationskonstante für CO für die pflanzlichen HOs ist, im Vergleich zu anderen bereits
untersuchten HOs, mit 8 * 104 s1 (für HY1) sehr klein, was auf eine veränderte Substrattasche
des Proteins hindeutet.
Der Einbau von zwei regioisomeren, artifiziellen, chemisch synthetisierten
Metalloporphyrinen (Eisencorrolen) konnte in verschiedenen HOs erfolgreich durchgeführt

Zusammenfassung
108
werden. Dabei ist ein Unterschied in der Reaktivität zwischen den zwei verwendeten
regioisomeren Eisencorrolen (α-CH-Fe(cor) und γ-CH-Fe(cor)) in Pflanzentyp-HOs (HY1,
HO3 und HO4 aus A. thaliana und HO-1 aus Synechocystis sp. PCC 6803) zu beobachten.
Alle getesteten Pflanzentyp-HOs können das γ-CH-Fe(cor) zu einem Biliverdin-Analog
umsetzen, hingegen das α-CH-Fe(cor) nur binden. Die Säuger hHO-1 und die untersuchten
regiospezifisch unterschiedlichen bakteriellen HOs (BphO und PigA) aus Pseudomonas
aeruginosa können beide Eisencorrole binden, aber nicht umsetzen.
Diese beiden regiospezifisch unterschiedlichen HOs findet man nicht nur in P. aeruginosa,
sondern sie scheinen innerhalb der Pseudomonaden weit verbreitet zu sein. Die untersuchten
rekombinanten PigAs aus P. aeruginosa (PA14), P. fluorescence 5, P. putida KT2440 und
P. syringae DC3000 zeigen eindeutig eine Regiospezifität für den - und -meso-Kohenstoff
des Häms, so dass die Produkte BV IX und BV IX gebildet werden. Zusätzliche
Untersuchungen an BphOs aus P.s. DC3000 und P.f. 5 haben gezeigt, dass es sich bei diesen
HOs um „klassische“, für den -meso-Kohlenstoff spezifische HOs handelt. Somit scheint der
Besitz zweier HOs mit unterschiedlicher Regiospezifität innerhalb eines Organismus eine
Besonderheit unter den Pseudomonaden zu sein.

Summary
109
F Summary
The aim of this study was the biochemical und biophysical characterization of heme
oxygenases (HOs) from Arabidopsis thaliana and from different selected Pseudomonas
species. All Arabidopsis HOs possess an N-terminal transit-peptide-localization sequence and
are localized in the chloroplast. The enzymatic activity was demonstrated using a combination
of UV/vis spectroscopy and HPLC analyses of all three members (HY1, HO3 und HO4) of
the Arabidopsis HO1-subfamily. All three proteins cleave the heme molecule to the open-
chain tetrapyrrole biliverdin IX (BV IX), the precursor of the phytochrome chromophore.
The investigated biochemical parameters (Kd, Km, Ea) of HOs from the HO1-subfamily are
nearly identical suggesting that all three HOs are able to synthesize BV IX with the same
efficiency. Also, all three HOs utilize ferredoxin as the natural electron donor. This was
demonstrated using isolated thylakoid membranes in vitro. All three HOs require a second
electron donor which leads to a ten-fold increase of heme turnover. The only protein of the
HO2-subfamily, HO2, is not able to bind or degrade heme. It is able to bind the precursor of
heme and chlorophyll, protoporphyrin IX (proto IX). If this binding of proto IX to HO2 is
physiological relevant is currently unknown.
All analyzed plant and bacterial HOs are monomeric proteins and possess a conserved
histidine residue in the proximal helix to coordinate the central iron of heme. These results
were confirmed with FTIR measurements of the Arabidopsis HO3 and the 15N labeled
version. The HOs of the HO1-subfamily differ in their carbon monoxide (CO) binding. This
was detected via Flash-Photolysis and FTIR measurements. The HO3 and HO4 show an
intermediate during CO binding in comparison to HY1. A distinct characterization of the
intermediate was impossible with the employed methods. The association constant of CO
rebinding to HY1 is very low (8 104 s1) in comparison to other HOs. These results point
towards an additional substrate pocket in plant HOs.
Two regioisomeric, artificial, chemical synthesized metalloporphyrins (iron corroles) could
successfully be assembled into various HOs. Thereby a difference in the reactivity between
the two tested regioisomeric iron corroles (α-CH-Fe(cor) and γ-CH-Fe(cor)) of plant-type
HOs (HY1, HO3 and HO4 from Arabidopsis and HO-1 from Synechocystis sp. PCC 6803)
was detected. All tested plant-type HOs were able to convert the γ-CH-Fe(cor) to a BV
analog. In contrast, α-CH-Fe(cor) was only bound but not converted. On the other hand, the
investigated human hHO-1 and the regiospecific bacterial HOs of Pseudomonas aeruginosa
(BphO and PigA) are only able to bind both molecules.

Summary
110
Two regiospecific different HOs are not only a specific trait for P. aeruginosa but can also be
found in other Pseudomonads. The investigated recombinant PigAs from P. aeruginosa
(PA14), P. fluorescence 5, P. putida KT2440 and P. syringae DC3000 show distinct
regiospecificities for the δ- and β-meso-carbon of the heme. All four PigAs produce both BV
isomers, BV IXδ and BV IXβ. The characterized BphOs from P.f. 5, and P.p. KT2440 are
“classical” HOs and cleave the heme at the -meso-carbon bridge. It could be demonstrated
that it is a specific trait of all Pseudomonads to possess two regiospecifically different and
active HOs.

Anhang
111
G Anhang
1. Strukturaufklärung der HO3 aus A. thaliana
Auf Grund fehlender Strukturdaten wurde im Rahmen dieser Arbeit der Versuch
unternommen, die Struktur einer Arabidopsis HO aus der HO1-Unterfamilie ohne die
N-terminale Transit-Peptid-Lokalisationssequenz zu ermitteln. Zuerst wurde dieser Verusch
mit Hilfe der Proteinkristallisation unternommen. Dazu wurde versucht alle drei aktiven HOs
in zahlreichen Variationen mit und ohne Häm, bzw. Inhibitor im Protein, in variierenden
Proteinkonzentrationen von 5 mg/mL bis zu 20 mg/mL und in der Variation von acht
screening-Bedingungen mit je 96 unterschiedlichen Reagenzien zu kristallisieren. Da sich
aber alle drei HOs nicht unter diversen getesteten Bedingungen kristallisieren ließen, wurde
die Methode der Kernspinresonanzspektroskopie (Nuclear Magnetic Resonance (NMR))
verwendet. Es wurden dazu zwei verschiedene Messungen durchgeführt. Die ein-
dimensionale (1D) 1H-NMR Messung sowie die zwei-dimensionale (2D) 1H-15N HSQC-
NMR Messung (Heteronuclear Single Quantum Coherence). Das Ziel der Messungen war die
Aufklärung der 3D-Struktur bzw. der Zuordnung des Proteinrückgrates. Für diese
Untersuchungen wurde die HO3 auf Grund hoher produzierbarer Proteinmengen sowie hoher
Reinheit und Löslichkeit exemplarisch für eine Arabidopsis HO ausgesucht.
1.1 D 1H-NMR an Arabidopsis HO3
Die einfachste und empfindlichste NMR Messung ist die 1D 1H-NMR. Mit ihr kann man
qualitativ zwischen ungefalteten und gefalteten Proteinen unterscheiden. Signale ungefalteter
Proteine zeigen eine geringe Dispersion. Damit ist gemeint, dass die 1H-Frequenzen der
verschiedenen Aminosäuren sehr ähnlich sind und sich die Signale stark überlagern. Gefaltete
Proteine hingegen zeigen eine, in Abhängigkeit von der Sekundärstruktur, mehr oder weniger
große Dispersion, wodurch einzelne Signale oberhalb von etwa 8,5 ppm und unterhalb von
etwa 0,5 ppm auftreten. Dabei kann man vereinfacht sagen, dass der Grad der
Signaldispersion mit dem Tertiärstrukturanteil einhergeht. Vorwiegend helikale Proteine
zeigen hingegen eher eine geringe Dispersion. Mit Hilfe der 1D 1H-Messung kann man,
indem man die äußeren Parameter, pH, Salzkonzentration und Temperatur variiert, die
optimale Pufferzusammensetzung bezüglich höchster Löslichkeit und optimaler Faltung
bestimmen. Dabei ist darauf zu achten, möglichst nahe am physiologischen pH-Wert zu
bleiben, um biologisch relevante Ergebnisse zu erhalten.

Anhang
112
1.2 2D 1H-15N HSQC-NMR an Arabidopsis HO3
Die HSQC-NMR Spektroskopie gehört zur Klasse der heteronuklearen 2D Messungen. Sie
beruht auf der Übertragung von Magnetisierung vom 1H-Kern auf einen direkt daran
gebundenen Heterokern und wieder zurück. Im Rahmen dieser Arbeit wurden 1H-15N HSQC
Messungen durchgeführt (Kay et al., 1992; Schleucher et al., 1994). Im Falle der 1H-15N
HSQC Messungen liefern alle Aminosäure-Reste ein Signal, welches der Amidgruppe NH
entspricht. Da die Iminosäure Prolin kein stickstoffgebundenes Wasserstoffatom besitzt,
bilden Proline eine Ausnahme von dieser Regel und sind in einem 1H-15N HSQC Spektrum
nicht zu detektieren. Zusätzlich können in Abhängigkeit vom pH-Wert und der Exponiertheit
gegenüber dem Lösungsmittel auch Seitenketten 1HN Signale sichtbar sein. Das 1H-15N HSQC
Spektrum stellt die Grundlage für viele mehrdimensionale Experimente dar. Bei Variation des
pH-Werts, Änderung der Temperatur oder Zugabe eines Liganden kann man im 1H-15N
HSQC Spektrum unmittelbar erkennen, welche Aminosäure eine Änderung in ihrer
chemischen Umgebung erfährt. Es eignet sich somit hervorragend für Bindungsstudien.
Um die Informationen sequenzspezifisch den Aminosäuren zuordnen zu können, müssen alle 1HN und 15N Frequenzen einer bestimmten Aminosäure zugeordnet werden. In diesem
Zusammenhang spricht man von einer Zuordnung (assignment). Das Proteinrückgrat wäre
dementsprechend vollständig zugeordnet (assigned).
1.3 Material und Methoden
Für die 1D 1H- und die 2D 1H-15N HSQC Messungen wurde die HO3, wie in Gisk et al.
beschrieben, gereinigt (Gisk et al., 2010). Es wurden 10 mg/mL HO3 in 500 µL KPi, pH 7,2
mit 10 % dH2O (v/v) in ein NMR-Röhrchen gegeben und das Protein mit dem in der Tabelle 3
stehenden Pulsprogramm vermessen. Zusätzlich wurde in gleicher Konzentration das in
dDMSO (deuteriertem Dimethylsulfoxid) gelöste Häm im Verhältnis 1:1 bzw. in deutlichem
Überschuss zum Protein hinzugegeben.
Für die 1H-15N HSQC Messungen wurde die HO3 in Minimalmedium angezogen.

Anhang
113
Medienzusammensetzung für 1L:
M9-Medium (10-fach):
60 g Na2HPO4
30 g KH2PO4
5 g NaCl
Spurenelementelösung (100-fach):
5 g EDTA
0.83 g FeCl3 6H2O
84 mg ZnCl2
13 mg CuCl2 2H2O
10 mg CoCl 6H2O
1,35 mg MnCl2 4H2O
Minimalmedium:
0,3 mL 1 M CaCl2-Lösung
100 mL M9 Medium (10-fach)
10 mL Spurenelementelösung (100-fach)
20 mL 20 % (v/v) Glukose-Lösung
1 mL 1 M MgSO4-Lösung
1 mL Biotin-Lösung (1 mg/mL)
1 mL Thiamin-Lösung (1 mg/mL)
0,5 g 15N-Ammoniumchlorid
Die jeweiligen Messungen wurden mit nachstehendem Programm durchgeführt:
Tabelle 3: Auflistung der zu jeweiligen Messung gehörenden Parameter.
Messung Pulsprogramm (nach Bruker) Parameter 1H 1D Watergate NS 16;SW 9615.4 Hz 1H-15N HSQC 2D Heteronukleare Single-
Quanten-Koharenz NS 32;TD (1H/F1) 2048, (15N/F2) 128; SW (1H/F1) 12019.2 Hz, (15N/F2) 2736.7 Hz
NS: Number of Scans; SW: Sweep width; F: Frequenzdomäne
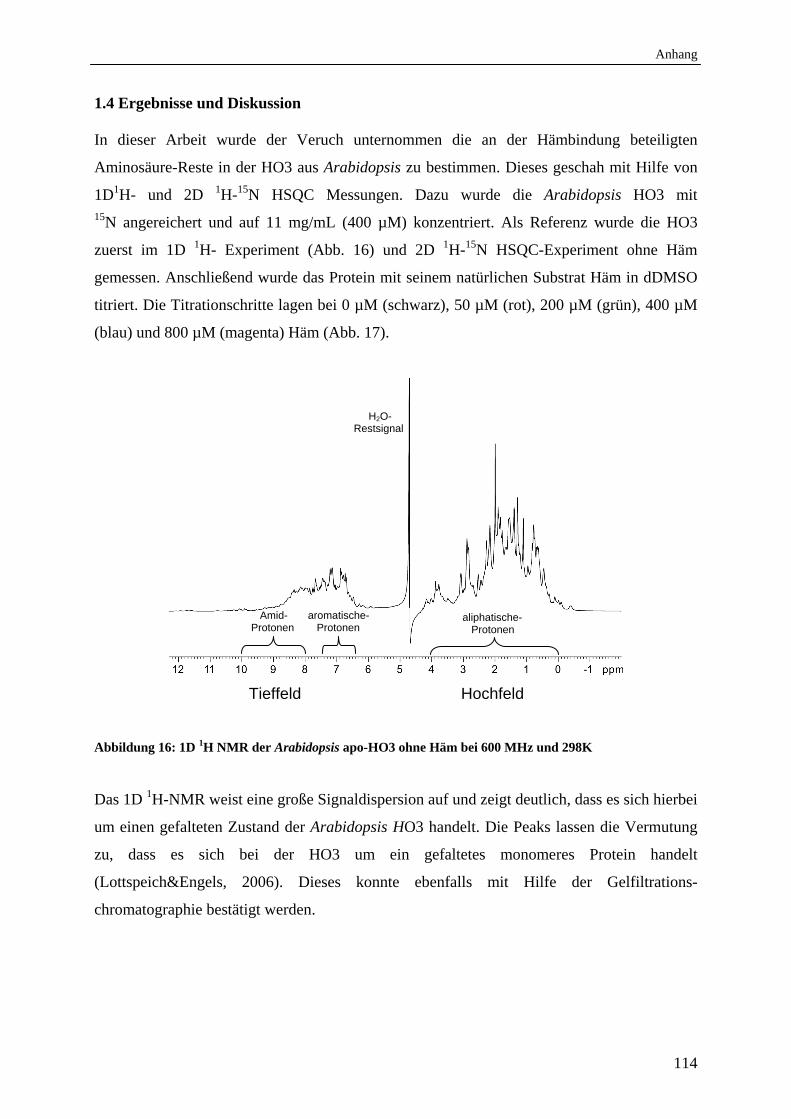
Anhang
114
1.4 Ergebnisse und Diskussion
In dieser Arbeit wurde der Veruch unternommen die an der Hämbindung beteiligten
Aminosäure-Reste in der HO3 aus Arabidopsis zu bestimmen. Dieses geschah mit Hilfe von
1D1H- und 2D 1H-15N HSQC Messungen. Dazu wurde die Arabidopsis HO3 mit 15N angereichert und auf 11 mg/mL (400 µM) konzentriert. Als Referenz wurde die HO3
zuerst im 1D 1H- Experiment (Abb. 16) und 2D 1H-15N HSQC-Experiment ohne Häm
gemessen. Anschließend wurde das Protein mit seinem natürlichen Substrat Häm in dDMSO
titriert. Die Titrationschritte lagen bei 0 µM (schwarz), 50 µM (rot), 200 µM (grün), 400 µM
(blau) und 800 µM (magenta) Häm (Abb. 17).
Tieffeld Hochfeld
Abbildung 16: 1D 1H NMR der Arabidopsis apo-HO3 ohne Häm bei 600 MHz und 298K
Das 1D 1H-NMR weist eine große Signaldispersion auf und zeigt deutlich, dass es sich hierbei
um einen gefalteten Zustand der Arabidopsis HO3 handelt. Die Peaks lassen die Vermutung
zu, dass es sich bei der HO3 um ein gefaltetes monomeres Protein handelt
(Lottspeich&Engels, 2006). Dieses konnte ebenfalls mit Hilfe der Gelfiltrations-
chromatographie bestätigt werden.
Amid- Protonen
aromatische- Protonen
aliphatische- Protonen
H2O- Restsignal
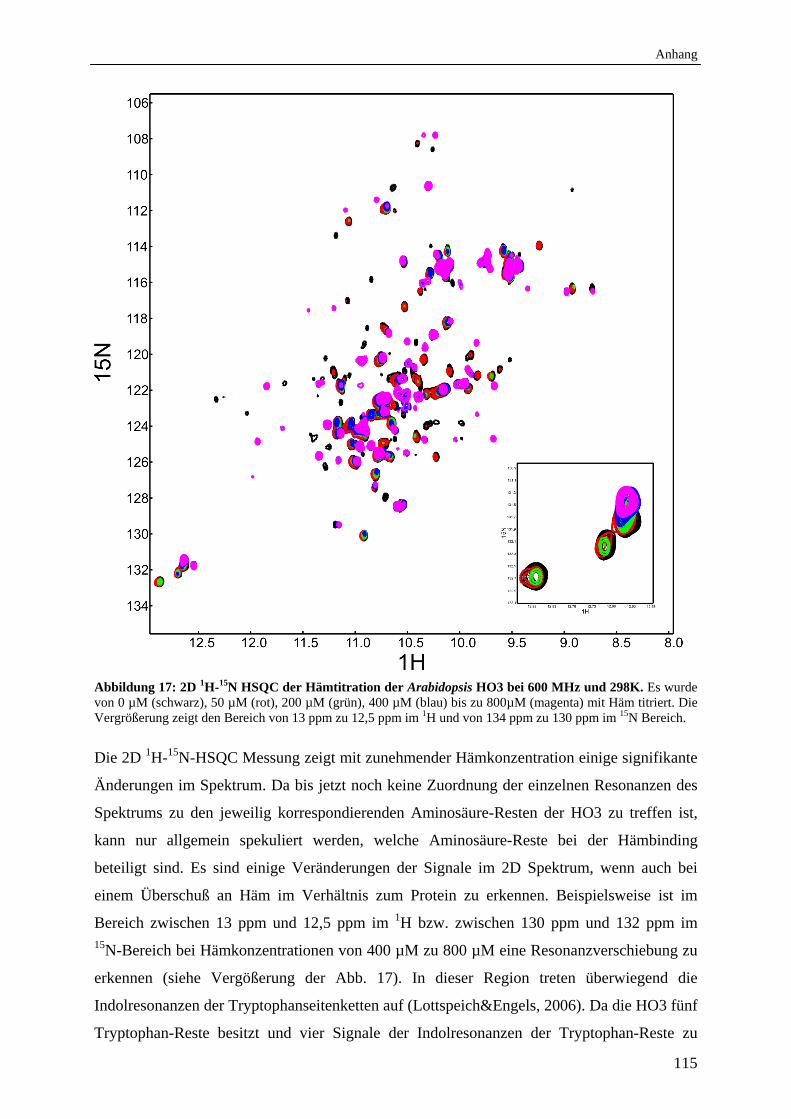
Anhang
115
Abbildung 17: 2D 1H-15N HSQC der Hämtitration der Arabidopsis HO3 bei 600 MHz und 298K. Es wurde von 0 µM (schwarz), 50 µM (rot), 200 µM (grün), 400 µM (blau) bis zu 800µM (magenta) mit Häm titriert. Die Vergrößerung zeigt den Bereich von 13 ppm zu 12,5 ppm im 1H und von 134 ppm zu 130 ppm im 15N Bereich.
Die 2D 1H-15N-HSQC Messung zeigt mit zunehmender Hämkonzentration einige signifikante
Änderungen im Spektrum. Da bis jetzt noch keine Zuordnung der einzelnen Resonanzen des
Spektrums zu den jeweilig korrespondierenden Aminosäure-Resten der HO3 zu treffen ist,
kann nur allgemein spekuliert werden, welche Aminosäure-Reste bei der Hämbinding
beteiligt sind. Es sind einige Veränderungen der Signale im 2D Spektrum, wenn auch bei
einem Überschuß an Häm im Verhältnis zum Protein zu erkennen. Beispielsweise ist im
Bereich zwischen 13 ppm und 12,5 ppm im 1H bzw. zwischen 130 ppm und 132 ppm im 15N-Bereich bei Hämkonzentrationen von 400 µM zu 800 µM eine Resonanzverschiebung zu
erkennen (siehe Vergößerung der Abb. 17). In dieser Region treten überwiegend die
Indolresonanzen der Tryptophanseitenketten auf (Lottspeich&Engels, 2006). Da die HO3 fünf
Tryptophan-Reste besitzt und vier Signale der Indolresonanzen der Tryptophan-Reste zu

Anhang
116
erkennen sind, ist ein Signal vermutlich überlagert. Die neu hinzugekommenden 17 Signale
(Abb. 17 (magenta)) können wahrscheinlich durch Veränderungen der an der Hämbindung
beteiligten Aminosäure-Reste erklärt werden, dessen Signale vor der Hämbindung von den
Signalen der apo-Form überlagert oder durch einen chemischen Austausch bedingt nicht
detektierbar gewesen sind.
In Zukunft sollten die HO3 mit 15N- und 13C Isotopen angereichert werden, um durch die
dritte Ebene eine einfachere und genauere Zuordnung der jeweiligen Resonanzen zur
Aminosäuresequenz des Proteins mittels Tripelresonanzspektren zu bekommen und die
Konformation der HO3 zu bestimmen.

Anhang
117
2. Prozentuale Verteilung der Eigenleistungen an den Publikationen/ Manuskripte
Characterization of the haem oxygenase protein family in Arabidopsis thaliana reveals a diversity of functions Gisk, B., Yasui, Y., Kohchi, T. & Frankenberg-Dinkel, N. (2010) Biochemical Journal 425, 425-434 Planung (P): 90%
Experimentelle Durchführung (E): 90 % Verfassen des Manuskripts (M): 70 %
Heme oxygenases from Arabidopsis thaliana reveal different mechanisms of carbon monoxide binding Gisk, B., Frankenberg-Dinkel, N. & Koetting, C. (Manuskript bei “Biochemical and Biophysical Research Communications“ eingereicht) Planung (P): 80%
Experimentelle Durchführung (E): 90 % Verfassen des Manuskripts (M): 60 %
Enzymatic ring opening of an iron corrole by plant-type heme oxygenases: Unexpected substrate and protein selectivities Gisk, B., Brégier, F., Krueger, R. A., Broering, M. & Frankenberg-Dinkel, N. (Manuskript bei “Biochemistry“ in Revision) Planung (P): 90%
Experimentelle Durchführung (E): 95 % Verfassen des Manuskripts (M): 70 %
The existence of two heme oxygenases with different regiospecificities is a characteristic trait of pathogenic and non-pathogenic Pseudomonas species Gisk, B., Aras, M., Wiethaus, J., & Frankenberg-Dinkel, N. (Manuskript in Vorbereitung) Planung (P): 90%
Experimentelle Durchführung (E): 70 % Verfassen des Manuskripts (M): 80 %

Anhang
118
3. Konferenzbeiträge
Gisk, B. & Frankenberg-Dinkel, N. (2007) Biochemical characterization of heme oxygenases from Arabidopsis thaliana (Poster)
SFB480 Symposium in Vehlen/Deutschland Gisk, B. (2008) Initial characterization of heme oxygenases from Arabidopsis thaliana (Organisator und Redner)
1st International PhD Student Symposium of the SFB 480 in Bochum/Deutschland Gisk, B. & Frankenberg-Dinkel, N. (2008) Biochemical characterization of heme oxygenases from Arabidopsis thaliana (Poster)
1st International PhD Student Symposium of the SFB 480 in Bochum/Deutschland Gisk, B. & Frankenberg-Dinkel, N. (2008) Biochemical characterization of heme oxygenases from Arabidopsis thaliana (Poster)
17. Photosyntheseworkshop Nord-West 2008 in Bochum/Deutschland Gisk, B. & Frankenberg-Dinkel, N. (2008) Biochemical characterization of heme oxygenases from Arabidopsis thaliana (Poster)
Tetrapyrrole Discussion Group Meeting; Trinity College in Dublin/Irland Gisk, B., Kohchi, T. & Frankenberg-Dinkel, N. (2009) Heme Oxygenases of Arabidopsis thaliana - Two families - two functions? (Poster)
Tetrapyrrole Photoreceptors of Photosynthetic Organisms ICTPPO 2009 in Asilomar/ USA
Gisk, B., Kohchi, T. & Frankenberg-Dinkel, N. (2009) Heme Oxygenases of Arabidopsis thaliana - Two families - two functions? (Poster)
Abschlusssymposium des SFB 480 in Bochum/Deutschland Gisk, B., Brégier,F., Krueger, R. A., Broering, M. & Frankenberg-Dinkel, N. (2010) Enzymatic ring opening of an iron corrole by Arabidopsis thaliana heme oxygenases (Poster)
Gordon Research Conference (GRC) “Chemistry and Biology of Tetrapyrroles” in Newport/USA
Gisk, B., Aras, M., & Frankenberg-Dinkel, N. (2010) The existence of two heme oxygenases with different regiospecificities is a characteristic trait of pathogenic and non-pathogenic Pseudomonas species (Poster)
Gordon Research Conference (GRC) “Chemistry and Biology of Tetrapyrroles” in Newport/USA

Anhang
119
4. Lebenslauf
PERSÖNLICHE DATEN
Björn Gisk, M.Sc. Biochemie
Geburtsdatum: 06.11.1980
Geburtsort: Herne
Nationalität: deutsch
HOCHSCHULAUSBILDUNG
11/2007 Beginn der Doktorarbeit an der Ruhr-Universität Bochum, Fakultät für
Biologie und Biotechnologie in der Arbeitsgruppe „Physiologie der
Mikroorganismen“ (Prof. Dr. N. Frankenberg-Dinkel)
Thema: Biochemische und biophysikalische Untersuchungen an
pflanzlichen und bakteriellen Hämoxygenasen
11/2007 Master of Science (M.Sc.) in Biochemie
Schwerpunkt: Proteine: Struktur und biologische Funktion
11/2006 - 10/2007 Masterarbeit
Ruhr-Universität Bochum an der Fakultät Biologie in der Arbeitsgruppe
„Physiologie der Mikroorganismen“ (Prof. Dr. N. Frankenberg-Dinkel)
Thema: Biosynthese offenkettiger Tetrapyrrole in Pflanzen und
Bakterien
10/2005 - 10/2006 Studium der Biochemie (M.Sc. Studiengang)
Ruhr-Universität Bochum
09/2005 Bachelor of Science (B.Sc.) in Biochemie
04/2005 - 09/2005 Bachelorarbeit
Ruhr-Universität Bochum an der Fakultät Biologie am Lehrstuhl
„Biochemie der Pflanzen“ (Prof. Dr. M. Rögner)
Thema: Optimierung des PS II -Photostroms im biomimetrischen
System zur Biowasserstofferzeugung
10/2001 - 03/2005 Studium der Biochemie (B.Sc. Studiengang)
Ruhr-Universität Bochum

Anhang
120
WEHRERSATZDIENST
09/2000 – 08/2001 Zivildienst im Alten- und Pflegeheim Jochen-Klepper-Haus der
Diakonie Ruhr
SCHULISCHER WERDEGANG
1997 – 2000 Gymnasiale Oberstufe des Heinrich von Kleist Gymnasiums
Abschluss: Allgemeine Hochschulreife
1987 - 1997 Gymnasium Heinrich von Kleist in Bochum Gerthe
1984 - 1987 Grundschule Maischützen in Bochum Harpen
QUALIFIKATIONEN/SEMINARE
Sachkenntnis - in Tierversuchen nach FELASA Kat.B bei Bayer Health Care
- in Strahlenschutz
- im Umgang mit giftigen und hoch giftigen Stoffen aktive Teilnahme an - Kreativ und effizient zum Professionellen Text
- Rund um den Vortrag – souverän und professionell präsentieren
- Präsentation techniques - zielorientiertes Training für
englischsprachige Präsentationen
- Zwischen Chaos und Perfektion- Projekt und Zeitmanagement für
Promovierende
- Kristallisationsworkshop in Zusammenarbeit mit der Firma Qiagen
KONGRESSE/TAGUNGEN
07/2010 Posterpräsentationen auf der Gordon Research Conference (GRC)
Chemistry and Biology of Tetrapyrroles; Newport/USA
02/2010 Posterpräsentation beim Abschlusssymposium des SFB 480;
Bochum/Deutschland
09/2009 - 10/2009 einmonatiger Forschungsaufenthalt an der Academica Sinica in der
Arbeitsgruppe von Prof. Dr. Shih-Long Tu am Institute of Plant and
Microbial Biology; Taipei/Taiwan

Anhang
121
07/2009 Posterpräsentation auf der Konferenz Tetrapyrrole Photoreceptors of
Photosynthetic Organisms ICTPPO 2009; Asilomar/USA
09/2008 Posterpräsentation beim Tetrapyrrole Discussion Group Meeting;
Trinity College, Dublin/Irland
09/2008 Posterpräsentation beim 17. Photosyntheseworkshop Nord-West 2008;
Bochum/Deutschland
03/2008 Organisator und Redner beim 1st International PhD Student
Symposium of the SFB 480; Bochum/Deutschland
11/2007 SFB480 Symposium; Vehlen/Deutschland
09/2007 5th International Heme Oxygenase Congress; Krakau/Polen
PUBLIKATIONEN
Gisk, B., Yasui, Y., Kohchi, T. & Frankenberg-Dinkel, N. (2010) Characterization of the haem oxygenase protein family in Arabidopsis thaliana reveals a diversity of functions. Biochemical Journal, 425, 425-434 Gisk, B., Bregier, F., Krueger, R. A., Broering, M. & Frankenberg-Dinkel, N. (2010) Enzymatic ring opening of an iron corrole by plant heme oxygenases: Unexpected substrate and protein selectivities (Manuskript in Revision) Gisk, B., Frankenberg-Dinkel, N. & Koetting, C. (2010) Heme oxygenases from Arabidopsis thaliana reveal different mechanisms of carbon monoxide binding (Manuskript eingereicht)
WEITERE KENNTNISSE
Fremdsprachen Englisch
Latein
EDV MS Windows
MS Word, MS Excel, MS Powerpoint
MS Origin
Sigma Plot
Adobe Photoshop
Corel DRAW
Gene Construction Kit, DNAStar
Bochum, den ___________________ Björn Gisk

Anhang
122
5. Erklärung
Hiermit erkläre ich, dass ich die Arbeit selbständig verfasst und bei keiner anderen Fakultät
eingereicht und dass ich keine anderen als die angegebenen Hilfsmittel verwendet habe. Es
handelt sich bei der heute von mir eingereichten Dissertation um sechs in Wort und Bild
völlig übereinstimmende Exemplare.
Weiterhin erkläre ich, dass digitale Abbildungen nur die originalen Daten enthalten und in
keinem Fall inhaltsverändernde Bildbearbeitung vorgenommen wurde.
Bochum, den
___________________ Björn Gisk

Literaturverzeichnis
123
H Literaturverzeichnis
Akashi T, Matsumura T, Ideguchi T, Iwakiri K, Kawakatsu T, Taniguchi I, Hase T (1999) Comparison of the electrostatic binding sites on the surface of ferredoxin for two ferredoxin-dependent enzymes, ferredoxin-NADP+ reductase and sulfite reductase. J Biol Chem 274: 29399-29405
Alfonso-Prieto M, Biarnes X, Vidossich P, Rovira C (2009) The molecular mechanism of the catalase reaction. J Am Chem Soc 131: 11751-11761
Ankele E, Kindgren P, Pesquet E, Strand A (2007) In vivo visualization of Mg-protoporphyrin IX, a coordinator of photosynthetic gene expression in the nucleus and the chloroplast. Plant Cell 19: 1964-1979
Balestrasse KB, Noriega GO, Batlle A, Tomaro MaL (2005) Involvement of heme oxygenase as antioxidant defense in soybean nodules. Free Radical Research 39: 145 - 151
Baranano DE, Rao M, Ferris CD, Snyder SH (2002) Biliverdin reductase: a major physiologic cytoprotectant. Proc Natl Acad Sci U S A 99: 16093-16098
Barkovits K, Harms A, Benkartek C, Smart JL, Frankenberg-Dinkel N (2008) Expression of the phytochrome operon in Pseudomonas aeruginosa is dependent on the alternative sigma factor RpoS. FEMS Microbiol Lett 280: 160-168
Beal SI (1993) Biosynthesis of Phycobilins. Chemical Reviews 93: 782-802 Beri R, Chandra R (1993) Chemistry and biology of heme. Effect of metal salts,
organometals, and metalloporphyrins on heme synthesis and catabolism, with special reference to clinical implications and interactions with cytochrome P-450. Drug Metab Rev 25: 49-152
Bhakta MN, Wilks A (2006) The mechanism of heme transfer from the cytoplasmic heme binding protein PhuS to the delta-regioselective heme oxygenase of Pseudomonas aeruginosa. Biochemistry 45: 11642-11649
Bhoo SH, Davis SJ, Walker J, Karniol B, Vierstra RD (2001) Bacteriophytochromes are photochromic histidine kinases using a biliverdin chromophore. Nature 414: 776-779
Bhushan S, Kuhn C, Berglund AK, Roth C, Glaser E (2006) The role of the N-terminal domain of chloroplast targeting peptides in organellar protein import and miss-sorting. FEBS Lett 580: 3966-3972
Bonner J, Millerd A (1953) Oxidative phosphorylation by plant mitochondria. Biochemistry and Biophysics 42: 135-148
Bonnett R, McDonagh AF (1973) The meso-reactivity of porphyrins and related compounds. VI. Oxidative cleavage of the haem system. The four isomeric biliverdins of the IX series. J Chem Soc Perkin 1 9: 881-888
Brandlmeier T, Scheer H, Rüdiger W (1981) Chomophore content and molar absorptivity of phytochrome in the Pr form. Z. Naturforsch. 36: 1057-1063
Bruno S, Faggiano S, Spyrakis F, Mozzarelli A, Abbruzzetti S, Grandi E, Viappiani C, Feis A, Mackowiak S, Smulevich G, Cacciatori E, Dominici P (2007) The reactivity with CO of AHb1 and AHb2 from Arabidopsis thaliana is controlled by the distal HisE7 and internal hydrophobic cavities. Proc Natl Acad Sci U S A 129: 2880-2889
Caignan GA, Deshmukh R, Wilks A, Zeng Y, Huang HW, Moenne-Loccoz P, Bunce RA, Eastman MA, Rivera M (2002) Oxidation of heme to beta- and delta-biliverdin by Pseudomonas aeruginosa heme oxygenase as a consequence of an unusual seating of the heme. J Am Chem Soc 124: 14879-14892
Chabregas SM, Luche DD, Farias LP, Ribeiro AF, van Sluys MA, Menck CF, Silva-Filho MC (2001) Dual targeting properties of the N-terminal signal sequence of

Literaturverzeichnis
124
Arabidopsis thaliana THI1 protein to mitochondria and chloroplasts. Plant Mol Biol 46: 639-650
Chu GC, Katakura K, Zhang X, Yoshida T, Ikeda-Saito M (1999) Heme degradation as catalyzed by a recombinant bacterial heme oxygenase (HmuO) from Corynebacterium diphtheriae. J Biol Chem 274: 21319-21325
Colas C, Ortiz de Montellano PR (2003) Autocatalytic radical reactions in physiological prosthetic heme modification. Chemical Reviews 103: 2305-2332
Collman JP, Brauman JI, Halbert TR, Suslick KS (1976) Nature of O2 and CO binding to metalloporphyrins and heme proteins. Proc Natl Acad Sci U S A 73: 3333-3337
Cornejo J, Beale SI (1988) Algal heme oxygenase from Cyanidium caldarium. Partial purification and fractionation into three required protein components. J Biol Chem 263: 11915-11921
Cornejo J, Beale SI (1997) Phycobilin biosynthetic reactions in extracts of cyanobacteria. Photosynth Research 51: 223-230.
Cornejo J, Willows RD, Beale SI (1998) Phytobilin biosynthesis: cloning and expression of a gene encoding soluble ferredoxin-dependent heme oxygenase from Synechocystis sp. PCC 6803. Plant J 15: 99-107
Dailey HA, Dailey TA, Wu CK, Medlock AE, Wang KF, Rose JP, Wang BC (2000) Ferrochelatase at the millennium: structures, mechanisms and [2Fe-2S] clusters. Cell Mol Life Sci 57: 1909-1926
Dammeyer T, Bagby SC, Sullivan MB, Chisholm SW, Frankenberg-Dinkel N (2008) Efficient phage-mediated pigment biosynthesis in oceanic cyanobacteria. Curr Biol 18: 442-448
Dammeyer T, Frankenberg-Dinkel N (2006) Insights into phycoerythrobilin biosynthesis point toward metabolic channeling. J Biol Chem 281: 27081-27089
Dammeyer T, Frankenberg-Dinkel N (2008) Function and distribution of bilin biosynthesis enzymes in photosynthetic organisms. Photochem Photobiol Sci 7: 1121-1130
Davis SJ, Bhoo SH, Durski AM, Walker JM, Vierstra RD (2001) The heme-oxygenase family required for phytochrome chromophore biosynthesis is necessary for proper photomorphogenesis in higher plants. Plant Physiol 126: 656-669
Davis SJ, Kurepa J, Vierstra RD (1999) The Arabidopsis thaliana HY1 locus, required for phytochrome-chromophore biosynthesis, encodes a protein related to heme oxygenases. Proc Natl Acad Sci U S A 96: 6541-6546
Davydov R, Kofman V, Fujii H, Yoshida T, Ikeda-Saito M, Hoffman BM (2002) Catalytic mechanism of heme oxygenase through EPR and ENDOR of cryoreduced oxy-heme oxygenase and its Asp 140 mutants. J Am Chem Soc 124: 1798-1808
de Vitry C, Desbois A, Redeker V, Zito F, Wollman FA (2004) Biochemical and spectroscopic characterization of the covalent binding of heme to cytochrome b6. Biochemistry 43: 3956-3968
Douglas SE, Penny SL (1999) The plastid genome of the cryptophyte alga, Guillardia theta: complete sequence and conserved synteny groups confirm its common ancestry with red algae. J Mol Evol 48: 236-244
Emborg TJ, Walker JM, Noh B, Vierstra RD (2006) Multiple heme oxygenase family members contribute to the biosynthesis of the phytochrome chromophore in Arabidopsis. Plant Physiol 140: 856-868
Everse J, Hsia N (1997) The toxicities of native and modified hemoglobins. Free Radic Biol Med 22: 1075-1099
Falk H (1989) The Chemistry of Linear Oligopyrroles and Bile Pigments. Springer-Verlag, Vienna

Literaturverzeichnis
125
Frankenberg N, Lagarias JC (2003) Phycocyanobilin: ferredoxin oxidoreductase of Anabaena sp. PCC 7120. Biochemical and spectroscopic. J Biol Chem 278: 9219-9226
Frankenberg N, Mukougawa K, Kohchi T, Lagarias JC (2001) Functional genomic analysis of the HY2 family of ferredoxin-dependent bilin reductases from oxygenic photosynthetic organisms. Plant Cell 13: 965-978
Friedman J, Lad L, Li H, Wilks A, Poulos TL (2004) Structural basis for novel delta-regioselective heme oxygenation in the opportunistic pathogen Pseudomonas aeruginosa. Biochemistry 43: 5239-5245
Friedman J, Meharenna YT, Wilks A, Poulos TL (2007) Diatomic ligand discrimination by the heme oxygenases from Neisseria meningitidis and Pseudomonas aeruginosa. J Biol Chem 282: 1066-1071
Fujii H, Zhang X, Yoshida T (2004) Essential amino acid residues controlling the unique regioselectivity of heme oxygenase in Pseudomonas aeruginosa. J Am Chem Soc 126: 4466-4467
Gisk B, Yasui Y, Kohchi T, Frankenberg-Dinkel N (2010) Characterization of the haem oxygenase protein family in Arabidopsis thaliana reveals a diversity of functions. Biochem J 425: 425-434
Gohya T, Zhang X, Yoshida T, Migita CT (2006) Spectroscopic characterization of a higher plant heme oxygenase isoform-1 from Glycine max (soybean)-coordination structure of the heme complex and catabolism of heme. FEBS J 273: 5384-5399
Gordon SM, Serkey JM, Keys TF, Ryan T, Fatica CA, Schmitt SK, Borsh JA, Cosgrove DM, 3rd, Yared JP (1998) Secular trends in nosocomial bloodstream infections in a 55-bed cardiothoracic intensive care unit. Ann Thorac Surg 65: 95-100
Gossauer A, Hirsch W (1974) Bile pigments. IV. Total synthesis of racemic phycocyanobilin (phycobiliverdin) and of homophycobiliverdin. Justus Liebigs Ann. Chem. 9: 1496-1513
Guilard R, Barbe JM, Stern C, Kadish KM (2003) The Porphyrin Handbook. Elservier Science 303: 18
Guo K, Xia K, Yang ZM (2008) Regulation of tomato lateral root development by carbon monoxide and involvement in auxin and nitric oxide. J Exp Bot 59: 3443-3452
Halliwell B, Gutteridge JM (1984) Oxygen toxicity, oxygen radicals, transition metals and disease. Biochem J 219: 1-14
Hanke GT, Kimata-Ariga Y, Taniguchi I, Hase T (2004) A post genomic characterization of Arabidopsis ferredoxins. Plant Physiol 134: 255-264
Harrison HR, Hodder OJR, Hodgkin DC (1971) Crystal and molecular structure of 8,12-diethyl-2,3,7,13,17,18-hexamethylcorrole. J Chem Soc B: 640-645
Hayashi A, Suzuki T, Shin M (1973) An enzymic reduction system for metmyoglobin and methemoglobine, and its application to functional studies of oxygen carriers. Biochimica Et Biophysica Acta 310: 309-316
Hayashi T, Dejima H, Matsuo T, Sato H, Murata D, Hisaeda Y (2002) Blue myoglobin reconstituted with an iron porphycene shows xtremely high oxygen affinity. J Am Chem Soc 124: 11226-11227
Heilesen AM, Permin H, Koch C, Hoiby N (1983) Treatment of chronic Pseudomonas aeruginosa infection in cystic fibrosis patients with ceftazidime and tobramycin. Scand J Infect Dis 15: 271-276
Hernandez G, Wilks A, Paolesse R, Smith KM, Ortiz de Montellano PR, La Mar GN (1994) Proton NMR investigation of substrate-bound heme oxygenase: evidence for electronic and steric contributions to stereoselective heme cleavage. Biochemistry 33: 6631-6641

Literaturverzeichnis
126
Hess WR, Rocap G, Ting CS, Larimer F, Stilwagen S, Lamerdin J, Chisholm SW (2001) The photosynthetic apparatus of Prochlorococcus: Insights through comparative genomics. Photosynth Research 70: 53-71
Hirotsu S, Chu GC, Unno M, Lee D-S, Yoshida T, Park S-Y, Shiro Y, Ikeda-Saito M (2004) The crystal structures of the ferric and ferrous forms of the heme complex of HmuO, a heme oxygenase of Corynebacterium diphtheriae. J Biol Chem 12:11937-11947
Jain R, Chan MK (2003) Mechanisms of ligand discrimination by heme proteins. J Biol Inorg Chem 8: 1-11
Jarvis P (2003) Intracellular signalling: the language of the chloroplast. Curr Biol 13: R314-316
Jeney V, Balla J, Yachie A, Varga Z, Vercellotti GM, Eaton JW, Balla G (2002) Pro-oxidant and cytotoxic effects of circulating heme. Blood 100: 879-887
Kachalova GS, Popov AN, Bartunik HD (1999) A steric mechanism for inhibition of CO binding to heme proteins. Science 284: 473-476
Kay LE, Keifer T, T. S (1992) Pure absorption gradient enhanced heteronuclear single quantum correlation spectroscopy with improved sensitivity. J Am Chem Soc 114: 10663-10665
Kharitonov VG, Sharma VS, Pilz RB, Magde D, Koesling D (1995) Basis of guanylate cyclase activation by carbon monoxide. Proc Natl Acad Sci U S A 92: 2568-2571
Kim D, Yukl ET, Moenne-Loccoz P, Montellano PR (2006) Fungal heme oxygenases: Functional expression and characterization of Hmx1 from Saccharomyces cerevisiae and CaHmx1 from Candida albicans. Biochemistry 45: 14772-14780
Knoell HE, Knappe J (1974) Escherichia coli ferredoxin, an iron-sulfur protein of the adrenodoxin type. Eur J Biochem 50: 245-252
Kohchi T, Mukougawa K, Frankenberg N, Masuda M, Yokota A, Lagarias JC (2001) The Arabidopsis HY2 gene encodes phytochromobilin synthase, a ferredoxin-dependent biliverdin reductase. Plant Cell 13: 425-436
Kong WW, Zhang LP, Guo K, Liu ZP, Yang ZM (2010) Carbon monoxide improves adaptation of Arabidopsis to iron deficiency. Plant Biotechnol J 8: 88-99
Koornneef M, Rolff E, Spurit C (1980) Genetic-control of light inhibit hypocotyl elongation in Arabidopsis thaliana (L.) Heynh. Z. Pflanzenphysiol. 100: 147-160
Kriegl JM, Bhattacharyya AJ, Nienhaus K, Deng P, Minkow O, Nienhaus GU (2002) Ligand binding and protein dynamics in neuroglobin. Proc Natl Acad Sci U S A 99: 7992-7997
Kutty RK, Maines MD (1984) Hepatic heme metabolism: possible role of biliverdin in the regulation of heme oxygenase activity. Biochem Biophys Res Commun 122: 40-46
Laibach F (1907) Zur Frage nach der Individualität der Chromosomen im Pflanzenreich. Botanisches Centralblatt Begleithefte 1 22: 191-219
Laibach F (1943) Arabidopsis thaliana (L) Heynh. als Objekt für genetische und entwicklungsphysiologische Untersuchungen. Botanisches Archiv 44: 439-455
Lamparter T, Michael N, Mittmann F, Esteban B (2002) Phytochrome from Agrobacterium tumefaciens has unusual spectral properties and reveals an N-terminal chromophore attachment site. Proc Natl Acad Sci U S A 99: 11628-11633
Lansky IB, Lukat-Rodgers GS, Block D, Rodgers KR, Ratliff M, Wilks A (2006) The cytoplasmic heme-binding protein (PhuS) from the heme uptake system of Pseudomonas aeruginosa is an intracellular heme-trafficking protein to the delta-regioselective heme oxygenase. J Biol Chem 281: 13652-13662
Lee WC, Reniere ML, Skaar EP, Murphy ME (2008) Ruffling of metalloporphyrins bound to IsdG and IsdI, two heme-degrading enzymes in Staphylococcus aureus. J Biol Chem 283: 30957-30963

Literaturverzeichnis
127
Linley PJ, Landsberger M, Kohchi T, Cooper JB, Terry MJ (2006) The molecular basis of heme oxygenase deficiency in the pcd1 mutant of pea. FEBS J 273: 2594-2606
Liu Y, Moenne-Loccoz P, Loehr TM, Ortiz de Montellano PR (1997) Heme oxygenase-1, intermediates in verdoheme formation and the requirement for reduction equivalents. J Biol Chem 272: 6909-6917
Liu Y, Ortiz de Montellano PR (2000) Reaction intermediates and single turnover rate constants for the oxidation of heme by human heme oxygenase-1. J Biol Chem 275: 5297-5307
Lottspeich F, Engels JW (2006) Bioanalytik. Spektrum. Akademischer Verlag 2: 377-396 Louie GV, Brownlie PD, Lambert R, Cooper JB, Blundell TL, Wood SP, Warren MJ,
Woodcock SC, P.M. J (1992) The three dimensional structure of porphobilinogen deaminase: a flexible multidomain polymerase with one catalytic site. Nature 359: 33-39
Maines DM (1988) Heme oxygenase, function, multiplicity, regulatory mechanisms, and clinical applications. FASEB J 2: 2557-2568.
Maines MD (1992) Heme Oxygenase: Clinical Applications and Functions. CRC Press, Inc. Boca Raton, FL: 1-276
Maines MD (1997) The heme oxygenase system: a regulator of second messenger gases. Annu Rev Pharmacol Toxicol 37: 517-554
Maines MD, Trakshel GM (1993) Purification and characterization of human biliverdin reductase. Archives of Biochemistry and Biophysics 300: 320-326
Matera KM, Takahashi S, Fujii H, Zhou H, Ishikawa K, Yoshimura T, Rousseau DL, Yoshida T, Ikeda-Saito M (1996) Oxygen and one reducing equivalent are both required for the conversion of alpha-hydroxyhemin to verdoheme in heme oxygenase. J Biol Chem 271: 6618-6624
Matsui T, Unno M, Ikeda-Saito M (2010) Heme oxygenase reveals its strategy for catalyzing three successive oxygenation reactions. Acc Chem Res 43: 240-247
Matsuo T, Hayashi A, Abe M, Matsuda T, Hisaeda Y, Hayashi T (2009) Meso-unsubstituted iron corrole in hemoproteins: Remarkable differences in effects on peroxidase activities between myoglobin und horseradish peroxidase. J Am Chem Soc 131: 15124-15124
McCoubrey WK, Jr., Huang TJ, Maines MD (1997) Isolation and characterization of a cDNA from the rat brain that encodes hemoprotein heme oxygenase-3. Eur J Biochem 247: 725-732
Migita CT, Matera KM, Ikeda-Saito M, Olson JS, Fujii H, Yoshimura T, Zhou H, Yoshida T (1998) The oxygen and carbon monoxide reactions of heme oxygenase. J Biol Chem 273: 945-949
Migita CT, Zhang X, Yoshida T (2003) Expression and characterization of cyanobacterium heme oxygenase, a key enzyme in the phycobilin synthesis. Properties of the heme complex of recombinant active enzyme. Eur J Biochem 270: 687-698
Muramoto T, Kohchi T, Yokota A, Hwang I, Goodman HM (1999) The Arabidopsis photomorphogenic mutant hy1 is deficient in phytochrome chromophore biosynthesis as a result of a mutation in a plastid heme oxygenase. Plant Cell 11: 335-347.
Muramoto T, Tsurui N, Terry MJ, Yokota A, Kohchi T (2002) Expression and biochemical properties of a ferredoxin-dependent heme oxygenase required for phytochrome chromophore synthesis. Plant Physiol 130: 1958-1966
Ochsner UA, Vasil ML (1996) Gene repression by the ferric uptake regulator in Pseudomonas aeruginosa: Cycle selection of iron-regulated genes. Proc Natl Acad Sci U S A 93: 4409-4414
Ohlson J, Phillips GNJ (1997) Myoglobin discriminates between O2, NO, and CO by electrostatic interactions with the bound ligand. J Biol Inorg Chem 2: 544-552

Literaturverzeichnis
128
Ortiz de Montellano PR (2000) The mechanism of heme oxygenase. Curr Opin Chem Biol 4: 221-227
Ortiz de Montellano PR, Wilks A (2001) Heme oxygenase structure and mechanism. Advan Inorg Chem 51: 359-407
Otterbein LE, Choi AM (2000) Heme oxygenase: colors of defense against cellular stress. Am J Physiol Lung Cell Mol Physiol 279: L1029-1037
Paiva-Silva GO, Cruz-Oliveira C, Nakayasu ES, Maya-Monteiro CM, Dunkov BC, Masuda H, Almeida IC, Oliveira PL (2006) A heme-degradation pathway in a blood-sucking insect. Proc Natl Acad Sci U S A 103: 8030-8035
Parks BM, Quail PH (1991) Phytochrome-deficient hy1 and hy2 long hypocotyl mutants of Arabidopsis are defective in phytochrome chromophore biosynthesis. Plant Cell 3: 1177-1186
Ponka P (1999) Cell biology of heme. Am J Med Sci 318: 241-256 Poss KD, Tonegawa S (1997a) Heme oxygenase 1 is required for mammalian iron
reutilization. Proc Natl Acad Sci U S A 94: 10919-10924 Poss KD, Tonegawa S (1997b) Reduced stress defense in heme oxygenase 1-deficient cells.
Proc Natl Acad Sci U S A 94: 10925-10930 Poulos TL (1988) Cytochrome P450: molecular architecture, mechanism, and prospects for
rational inhibitor design. Pharm Res 5: 67-75 Ratliff M, Zhu W, Deshmukh R, Wilks A, Stojiljkovic I (2001) Homologues of neisserial
heme oxygenase in Gram-negative bacteria: Degradation of heme by the product of the pigA gene of Pseudomonas aeruginosa. J Bacteriol 183: 6394-6403
Reniere ML, Ukpabi GN, Harry SR, Stec DF, Krull R, Wright DW, Bachmann BO, Murphy ME, Skaar EP (2010) The IsdG-family of haem oxygenases degrades haem to a novel chromophore. Mol Microbiol 75: 1529-1538
Richaud C, Zabulon G (1997) The heme oxygenase gene (pbsA) in the red alga Rhodella violacea is discontinuous and transcriptionally activated during iron limitation. Proc Natl Acad Sci U S A 94: 11736-11741
Sakamoto H, Omata Y, Palmer G, Noguchi M (1999) Ferric alpha-hydroxyheme bound to heme oxygenase can be converted to verdoheme by dioxygen in the absence of added reducing equivalents. J Biol Chem 274: 18196-18200
Schleucher J, Schwendinger M, Sattler M, Schmidt P, Schedletzky O, Glaser SJ, Sorensen OW, Griesinger C (1994) A general enhancement scheme in heteronuclear multidimensional NMR employing pulsed field gradients J Biomol NMR 4: 301-306
Schluchter WM, Shen G, Alvey RM, Biswas A, Saunee NA, Williams SR, Mille CA, Bryant DA (2010) Phycobiliprotein biosynthesis in cyanobacteria: structure and function of enzymes involved in post-translational modification. Adv Exp Med Biol 675: 211-228
Schmitt MP (1997a) Transcription of the Corynebacterium diphtheriae hmuO gene is regulated by iron and heme. Infect Immun 65: 4634-4641
Schmitt MP (1997b) Utilization of host iron sources by Corynebacterium diphtheriae: identification of a gene whose product is homologous to eukaryotic heme oxygenases and is required for acquisition of iron from heme and hemoglobin. J Bacteriol 179: 838-845
Schuller DJ, Wilks A, Ortiz de Montellano PR, Poulos TL (1999) Crystal structure of human heme oxygenase-l. Nature Structural Biology 6: 860-867
Schuller DJ, Zhu WM, Stojiljkovic I, Wilks A, Poulos TL (2001) Crystal structure of heme oxygenase from the gram-negative pathogen Neisseria meningitidis and a comparison with mammalian heme oxygenase-1. Biochemistry 40: 11552-11558

Literaturverzeichnis
129
Sciara G, Kendrew SG, Miele AE, Marsh NG, Federici L, Malatesta F, Schimperna G, Savino C, Vallone B (2003) The structure of ActVA-Orf6, a novel type of monooxygenase involved in actinorhodin biosynthesis. Embo J 22: 205-215
Sevrioukova IF, Peterson JA (1995) NADPH-P-450 reductase: structural and functional comparisons of the eukaryotic and prokaryotic isoforms. Biochemistry 77: 562-572
Shalloe F, Elliott G, Ennis O, Mantle TJ (1996) Evidence that biliverdin-IXb reductase and flavin reductase are identical. Biochem J 316: 385-387
Shepherd M, Reid JD, Hunter CN (2003) Purification and kinetic characterization of the magnesium protoporphyrin IX methyltransferase from Synechocystis PCC6803. Biochem J 371: 351-360
Shikama K (2006) Nature of the FeO2 bonding in myoglobin and hemoglobin: A new molecular paradigm. Prog. Biophys. Mol. Biol. 91: 83-162
Skaar EP, Gaspar AH, Schneewind O (2003) IsdG and IsdI, heme degrading enzymes in the cytoplasm of staphylococcus aureus. J. Biol. Chem.: M307952200
Skaar EP, Gaspar AH, Schneewind O (2006) Bacillus anthracis IsdG, a heme-degrading monooxygenase. J Bacteriol 188: 1071-1080
Sono M, Roach MP, Coulter ED, Dawson JH (1996) Heme-Containing Oxygenases. Chem Rev 96: 2841-2888
Sorenson JC, Scandalios JG (1980) Biochemical characterization of a catalase inhibitor from maize. Plant Physiol 66: 688-691
Spiro TG, Jarzecki AA (2001) Heme-based sensors: theoretical modeling of heme-ligand-protein interactions. Curr Opin Chem Biol 5: 715-723
Spiro TG, Kozlowski PM (2001) Is the CO adduct of myoglobin bent, and does it matter? Acc Chem Res 34: 137-144
Stocker R, Glazer AN, Ames BN (1987b) Antioxidant activity of albumin-bound bilirubin. Proc Natl Acad Sci U S A 84: 5918-5922
Stocker R, Yamamoto Y, McDonagh AF, Glazer AN, Ames BN (1987a) Bilirubin is an antioxidant of possible physiological importance. Science 235: 1043-1046
Strand A, Asami T, Alonso J, Ecker JR, Chory J (2003) Chloroplast to nucleus communication triggered by accumulation of Mg-protoporphyrinIX. Nature 421: 79-83
Sugishima M, Hagiwara Y, Zhang X, Yoshida T, Migita CT, Fukuyama K (2005) Crystal structure of dimeric heme oxygenase-2 from Synechocystis sp. PCC 6803 in complex with heme. Biochemistry 44: 4257-4266
Sugishima M, Migita CT, Zhang X, Yoshida T, Fukuyama K (2004) Crystal structure of heme oxygenase-1 from cyanobacterium Synechocystis sp. PCC 6803 in complex with heme. Eur J Biochem 271: 4517-4525
Sugishima M, Omata Y, Kakuta Y, Sakamoto H, Noguchi M, Fukuyama K (2000) Crystal structure of rat heme oxygenase-1 in complex with heme. FEBS Lett 471: 61-66
Sugishima M, Sakamoto H, Kakuta Y, Omata Y, Hayashi S, Noguchi M, Fukuyama K (2002) Crystal structure of rat apo-heme oxygenase-1 (HO-1): mechanism of heme binding in HO-1 inferred from structural comparison of the apo and heme complex forms. Biochemistry 41: 7293-7300
Sugishima M, Sakamoto H, Noguchi M, Fukuyama K (2004) CO-trapping site in heme oxygenase revealed by photolysis of its co-bound heme complex: mechanism of escaping from product inhibition. J Mol Biol 341: 7-13
Takahashi S, Matera KM, Fujii H, Zhou H, Ishikawa K, Yoshida T, Ikeda-Saito M, Rousseau DL (1997) Resonance Raman spectroscopic characterization of alpha-hydroxyheme and verdoheme complexes of heme oxygenase. Biochemistry 36: 1402-1410

Literaturverzeichnis
130
Tasler R, Moises T, Frankenberg-Dinkel N (2005) Biochemical and spectroscopic characterization of the bacterial phytochrome of Pseudomonas aeruginosa. FEBS J 272: 1927-1936
Tenhunen R, Marver HS, Schmid R (1969) Microsomal heme oxygenase. Characterization of the enzyme. J Biol Chem 244: 6388-6394
Terry MJ, Kendrick RE (1999) Feedback inhibition of chlorophyll synthesis in the phytochrome chromophore-deficient aurea and yellow-green-2 mutants of tomato. Plant Physiol 119: 143-152
Terry MJ, Linley PJ, Kohchi T (2002) Making light of it: the role of plant haem oxygenases in phytochrome chromophore synthesis. Biochem Soc Trans 30: 604-609
Terry MJ, McDowell MT, Lagarias JC (1995) (3Z)- and (3E)-phytochromobilin are intermediates in the biosynthesis of the phytochrome chromophore. J Biol Chem 270: 11111-11118
Terry MJ, Wahleithner JA, Lagarias JC (1993) Biosynthesis of the plant photoreceptor phytochrome. Arch Biochem Biophys 306: 1-15
Terry N, Abadia J (1986) Function of iron in chloroplasts. Journal of Plant Nutrition 9: 609 - 646
The-Arabidopsis-Genome-Initiative (2000) Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature 408: 796-815
Unno M, Matsui T, Chu GC, Couture M, Yoshida T, Rousseau DL, Olson JS, Ikeda-Saito M (2004) Crystal structure of the dioxygen-bound heme oxygenase from Corynebacterium diphtheriae: implications for heme oxygenase function. J Biol Chem 279: 21055-21061
Vogel E, Will S, Schulze Tilling A, Neumann L, Lex J, Bill E, Trautwein AX, Wieghardt K (1994) Metallocorroles with Formally Tetravalent Iron. Angew Chem Int Ed 33: 731-735
Wagener FA, Volk HD, Willis D, Abraham NG, Soares MP, Adema GJ, Figdor CG (2003) Different faces of the heme-heme oxygenase system in inflammation. Pharmacol Rev 55: 551-571
Wagner M, Cadetg P, Ruf R, Mazzucchelli L, Ferrari P, Redaelli CA (2003) Heme oxygenase-1 attenuates ischemia/reperfusion-induced apoptosis and improves survival in rat renal allografts. Kidney Int 63: 1564-1573
Walker CJ, Willows RD (1997) Mechanism and regulation of Mg-chelatase. Biochem J 327 ( Pt 2): 321-333
Wang A, Zeng Y, Han H, Weeratunga S, Morgan BN, Moenne-Loccoz P, Schonbrunn E, Rivera M (2007) Biochemical and structural characterization of Pseudomonas aeruginosa Bfd and FPR: ferredoxin NADP+ reductase and not ferredoxin is the redox partner of heme oxygenase under iron-starvation conditions. Biochemistry 46: 12198-12211
Wang M, Roberts DL, Paschke R, Shea TM, Masters BS, Kim JJ (1997) Three-dimensional structure of NADPH-cytochrome P450 reductase: prototype for FMN- and FAD-containing enzymes. Proc Natl Acad Sci U S A 94: 8411-8416
Wegele R, Tasler R, Zeng Y, Rivera M, Frankenberg-Dinkel N (2004) The heme oxygenase(s)-phytochrome system of Pseudomonas aeruginosa. J Biol Chem 279: 45791-45802
Wilks A (2002) Heme Oxygenase: Evolution, Structure and Mechanism. Antioxidants and Redox Signaling 4: 603-614
Wilks A, Black SM, Miller WL, Ortiz de Montellano PR (1995) Expression and characterization of truncated human heme oxygenase (hHO-1) and a fusion protein of hHO-1 with human cytochrome P450 reductase. Biochemistry 34: 4421-4427

Literaturverzeichnis
131
Wilks A, Ortiz de Montellano PR (1993) Rat liver heme oxygenase. High level expression of a truncated soluble form and nature of the meso-hydroxylating species. J Biol Chem 268: 22357-22362
Wilks A, Schmitt MP (1998) Expression and characterization of a heme oxygenase (HmuO) from Corynebacterium diphtheriae. Iron acquisition requires oxidative cleavage of the heme macrocycle. J Biol Chem 273: 837-841
Wu R, Skaar EP, Zhang R, Joachimiak G, Gornicki P, Schneewind O, Joachimiak A (2005) Staphylococcus aureus IsdG and IsdI, heme-degrading enzymes with structural similarity to monooxygenases. J Biol Chem 280: 2840-2846
Xuan W, Zhu FY, Xu S, Huang BK, Ling TF, Qi JY, Ye MB, Shen WB (2008) The Heme Oxygenase/Carbon Monoxide System Is Involved in the Auxin-Induced Cucumber Adventitious Rooting Process. Plant Physiol 148: 881-893
Yamaguchi T, Komoda Y, Nakajima H (1994) Biliverdin-IX alpha reductase and biliverdin-IX beta reductase from human liver. Purification and characterization. J Biol Chem 269: 24343-24348
Yannarelli GG, Noriega GO, Batlle A, Tomaro ML (2006) Heme oxygenase up-regulation in ultraviolet-B irradiated soybean plants involves reactive oxygen species. Planta 224: 1154-1162
Yi L, Jenkins PM, Leichert LI, Jakob U, Martens JR, Ragsdale SW (2009) Heme regulatory motifs in heme oxygenase-2 form a thiol/disulfide redox switch that responds to the cellular redox state. J Biol Chem 284: 20556-20561
Yi L, Ragsdale SW (2007) Evidence that the heme regulatory motifs in heme oxygenase-2 serve as a thiol/disulfide redox switch regulating heme binding. J Biol Chem 282: 21056-21067
Yoshida T, Kikuchi G (1978) Features of the reaction of heme degradation catalyzed by the reconstituted microsomal heme oxygenase system. J Biol Chem 253: 4230-4236
Yoshida T, Kikuchi G (1978) Purification and properties of heme oxygenase from pig spleen microsomes. J Biol Chem 253: 4224-4229
Yoshida T, Kikuchi G (1979) Purification and properties of heme oxygenase from rat liver microsomes. J Biol Chem 254: 4487-4491
Yoshida T, Noguchi M, Kikuchi G (1980) Oxygenated form of heme-heme oxygenase complex and requirement for second electron to initiate heme degradation from the oxygenated complex. J Biol Chem 255: 4418-4420
Zhang X, Fujii H, Matera KM, Migita CT, Sun D, Sato M, Ikeda-Saito M, Yoshida T (2003) Stereoselectivity of each of the three steps of the heme oxygenase reaction: hemin to meso-hydroxyhemin, meso-hydroxyhemin to verdoheme, and verdoheme to biliverdin. Biochemistry 42: 7418-7426
Zhang X, Migita CT, Sato M, Sasahara M, Yoshida T (2005) Protein expressed by the ho2 gene of the cyanobacterium Synechocystis sp. PCC 6803 is a true heme oxygenase. Properties of the heme and enzyme complex. FEBS J 272: 1012-1022
Zhang X, Sato M, Sasahara M, Migita CT, Yoshida T (2004) Unique features of recombinant heme oxygenase of Drosophila melanogaster compared with those of other heme oxygenases studied. Eur J Biochem 271: 1713-1724
Zhao Y, Brandish PE, Ballou DP, Marletta MA (1999) A molecular basis for nitric oxide sensing by soluble guanylate cyclase. Proc Natl Acad Sci U S A 96: 14753-14758
Zhou H, Migita CT, Sato M, Sun D, Zhang X, Ikeda-Saito M, Fujii H, Yoshida T (2000) Participation of carboxylate amino acid side chain in regiospecific oxidation of heme by heme oxygenase. J Am Chem Soc 122: 8311-8312
Zhu W, Hunt DJ, Richardson AR, Stojiljkovic I (2000) Use of heme compounds as iron sources by pathogenic Neisseriae requires the product of the hemO gene. J Bacteriol 182: 439-447

Literaturverzeichnis
132
Zhu W, Wilks A, Stojiljkovic I (2000) Degradation of heme in Gram-negative bacteria: the product of the hemO gene of Neisseriae is a heme oxygenase. J Bacteriol 182: 6783-6790





























