Embed Size (px)
Citation preview

Bleeding & Clotting
Disorders IRichard E. Freeman
MD2013


Overview
• The Players
• Factor Deficiencies causing bleeding
• Dissolving the Clot
• The Clinical Presentation
• Other Causes of Bleeding
• Hypercoagulability
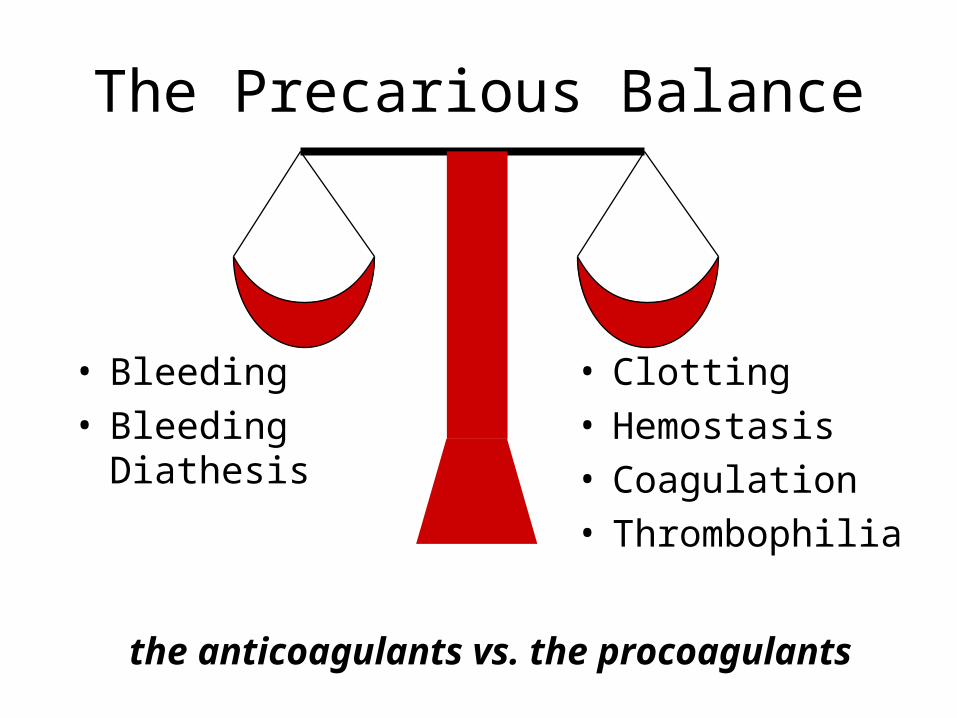
The Precarious Balance
• Bleeding• Bleeding Diathesis
• Clotting• Hemostasis• Coagulation• Thrombophilia
the anticoagulants vs. the procoagulants

• Hemostasis:– cessation of blood loss from a damaged
vessel
• Other Terms– bleeding disorders = bleeding diathesis =
coagulopathies– diathesis [Gr. Diathenai – to predispose] : A
constitutional predisposition to certain diseases or conditions
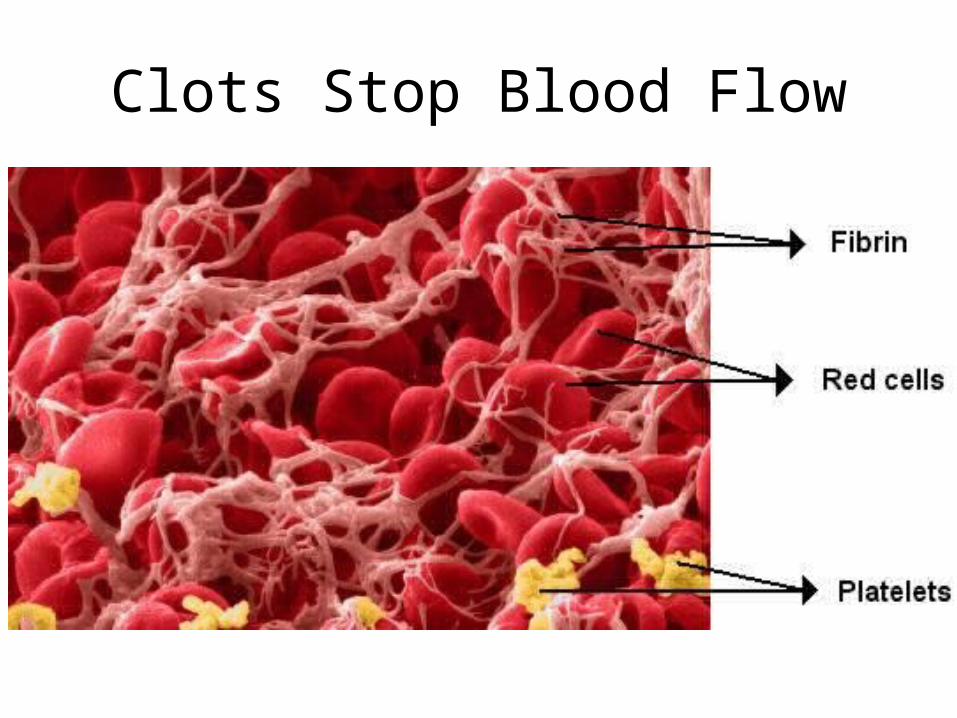
Clots Stop Blood Flow

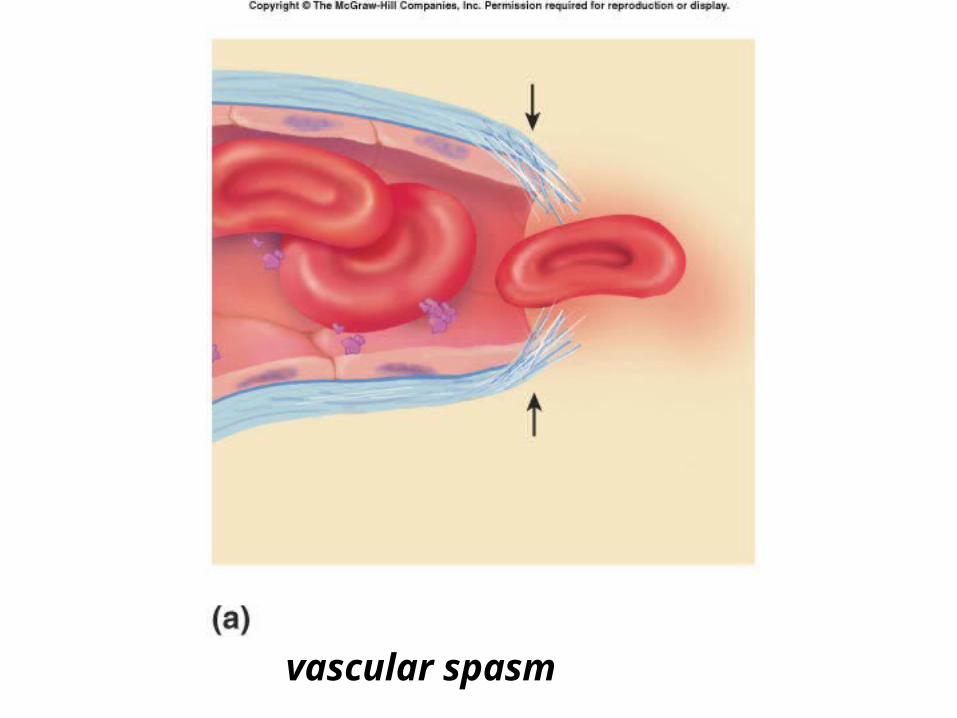
Phases of Clot FormationI. VASCULAR INJURY & SPASM
constriction of injured blood vessel– injury releases tissue factors, platelets
release serotonin (vasoconstrictor), and activates extrinsic pathway
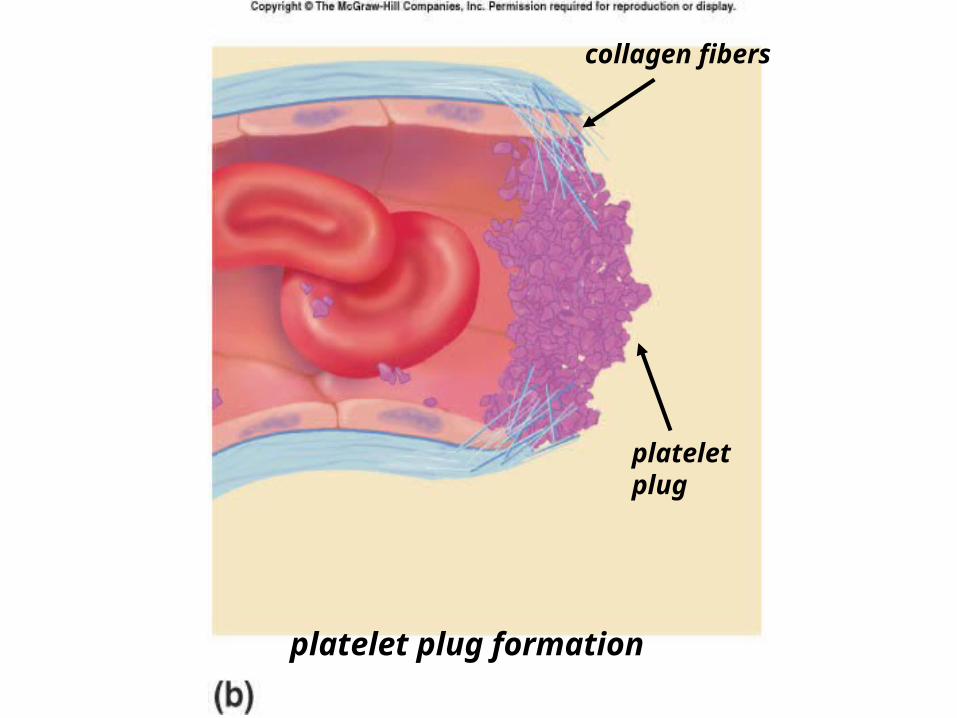
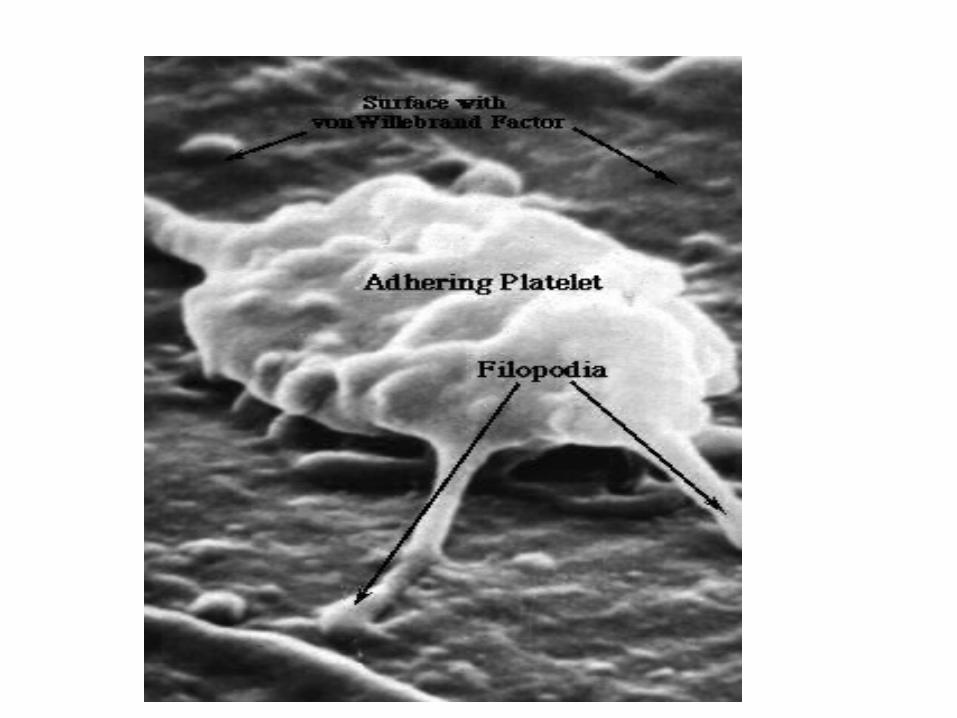
II. PLATELET PLUG FORMATIONcollagen in vessel exposed by injury.
von Willebrand factor (vWF) needed to bridge platelets and collagen.
– Calcium (Ca 2+) needed

Phases of Clot Formation
III. COAGULATION CASCADE
INTRINSIC AND EXTRINSIC PATHWAYS
formation of cross-linked fibrin polymer clot “fibrin clot”
IV. DISSOLUTION OF CLOT
vascular spasm
platelet plug formation
collagen fibers
platelet plug
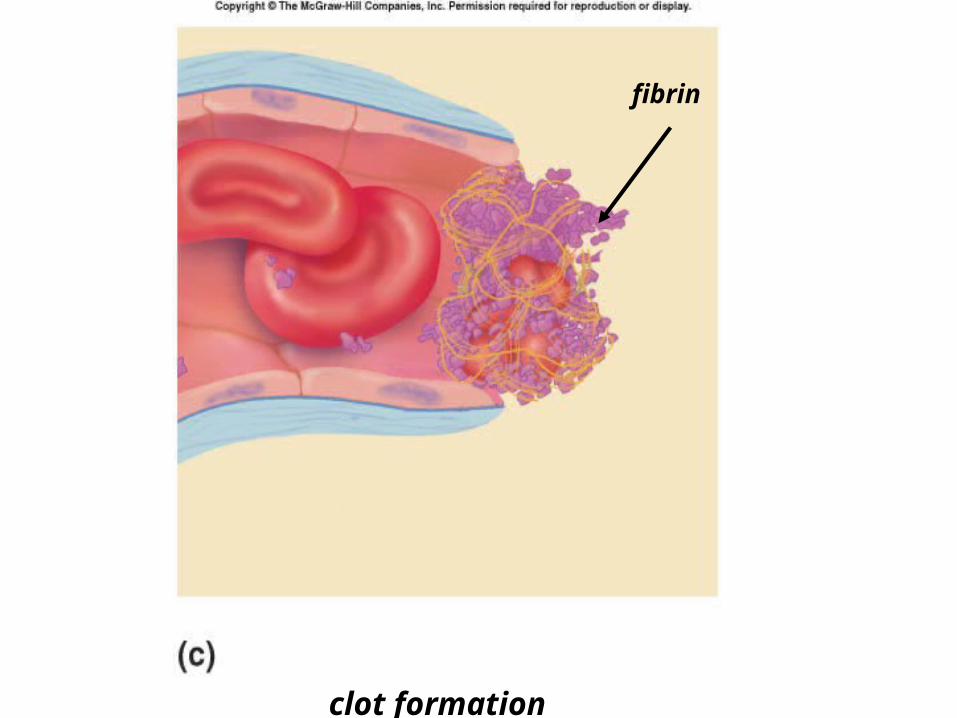
clot formation
fibrin

The Players

Complex Interactions
• ALL OF THESE MUST FUNCTION NORMALLY for EFFECTIVE CLOT FORMATION and HEMOSTASIS– Vascular endothelial cells– Platelets– Clotting factor cascade– Blood flow & shear– Antifibrinolysis

Blood Content & Hemostasis• Plasma – Unclotted liquid part of blood
– 90% Water
– 10% Dissolved and suspending particles• Organics: Proteins
– albumin 53%-LIVER– globulins 43%
» Antibodies-RE SYSTEM» Compliment & Kinin System (Inflamation)» Clotting Factors-LIVER AND ELSEWHERE
– Fibrinogen 4%
• Inorganics: salts
• Blood Cells– RBCs – Erythrocytes– WBCs – Leukocytes– Platelets – Thrombocytes

•ENDOTHELIAL CELLS
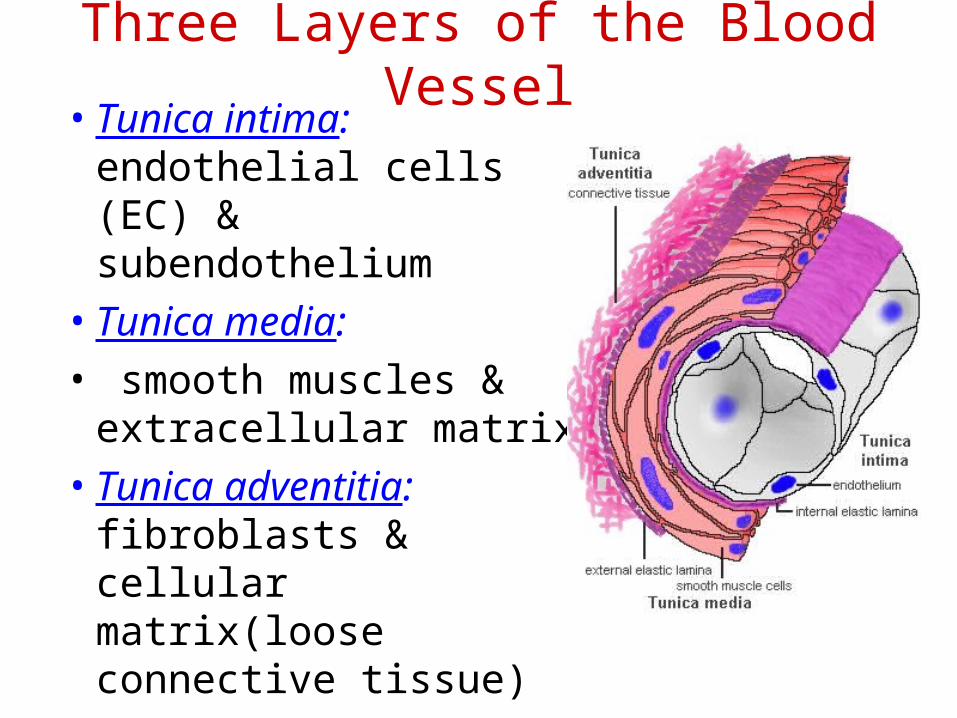
Three Layers of the Blood Vessel• Tunica intima:
endothelial cells (EC) & subendothelium
• Tunica media:
• smooth muscles & extracellular matrix
• Tunica adventitia: fibroblasts & cellular matrix(loose connective tissue)

The Endothelial Cell (EC)
• 1. Intact, healthy EC produces and is “teflon” coated with prostacyclin (PGI2)
• – RESTING STATE– A vasodilator– Inhibits platelet adhesion– Acts in opposition to the platelet thromboxane A2
• 2. EC coated w/ heparin sulfate which activates anti-thrombin III in plasma & stops thrombosis
• 3. Synthesis of Factor VIII – vonWillebrand factor– vWF synthesized in platelets & EC

3
•PLATELETS


The Amazing Platelet• 1. Secrete procoagulants which promote
clotting• 2. Secrete vasoconstrictors causing vascular
spasm in injured blood vessels• 3. as they aggregate they undergo
degranulation releasing:– serotonin (vasoconstrictor),– ADP (attractant)-calls for more platelets to help, – thromboxane A2 –(clot promoter)
• 4. form temporary platelet plugs to stop bleeding
• 5. dissolves blood clots that have outlasted their usefulness
• 6. inflammation and remodeling
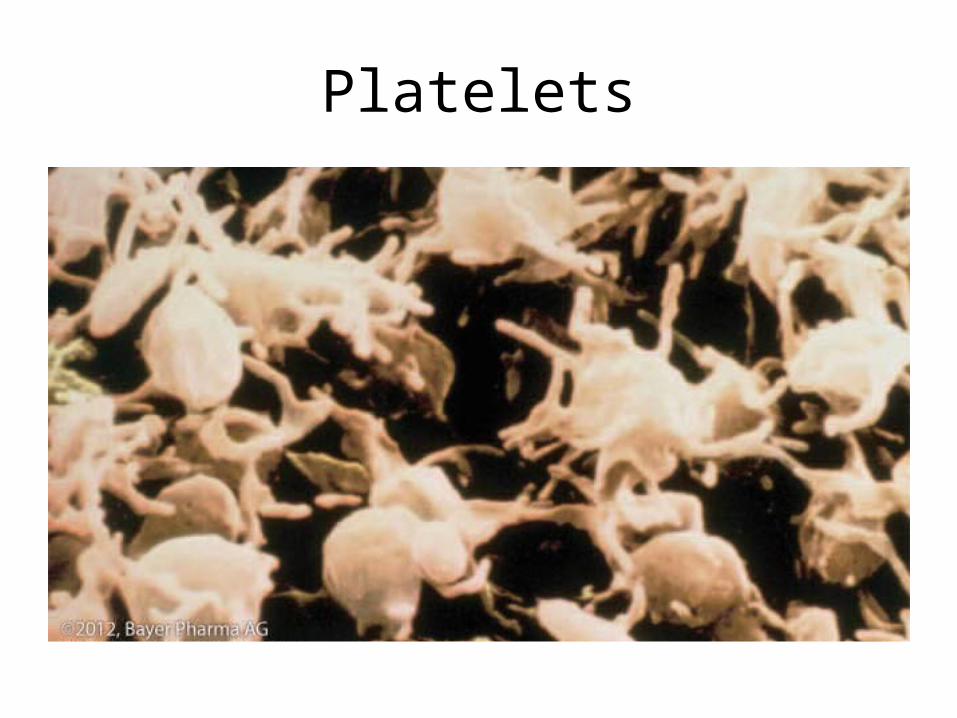
Platelets

Life of a Platelet
• MEGAKARYOCYTE (“mama” cell) fragmentation in marrow
• thrombopoietin (TPO) stimulates production produced in liver, bone marrow & kidney
• 1/3 sequestered to the spleen
• 2/3 into circulation (150-450,000 per microliter of blood).
• LIFE SPAN 7 TO 10 DAYS– (RBCs 90-120 days, WBCs 1 day)
– ASA (aspirin) thus effect lasts this long • irreversible acetylation of cyclooxygenase 1
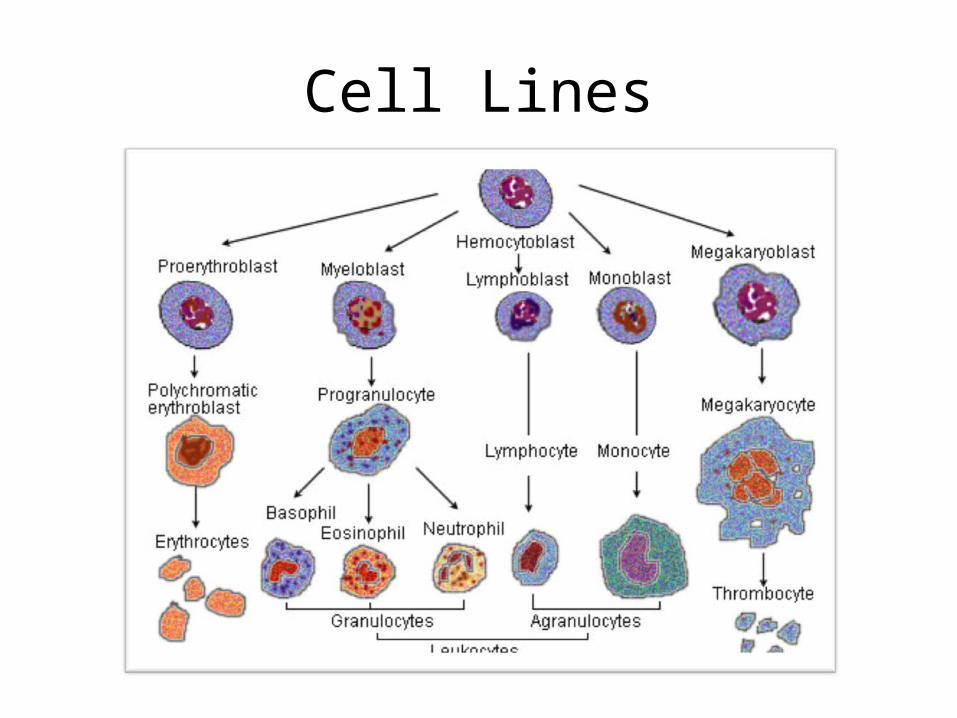
Cell Lines

Platelet-Chemistry• ACTIN-MYOSIN
– Contraction – pulls clot together
• SURFACE: GP IIb/IIIa glycoprotein– Important in adhesion and aggregation
• DENSE GRANULE: – Calcium, ADP, Serotonin
• ALPHA-GRANULE: – Growth factor, fibrinogen, Factor V, von
Willebrand factor (vWF), fibronectin, beta-thromboglobulin, heparin antagonist (PF4), thrombospondin
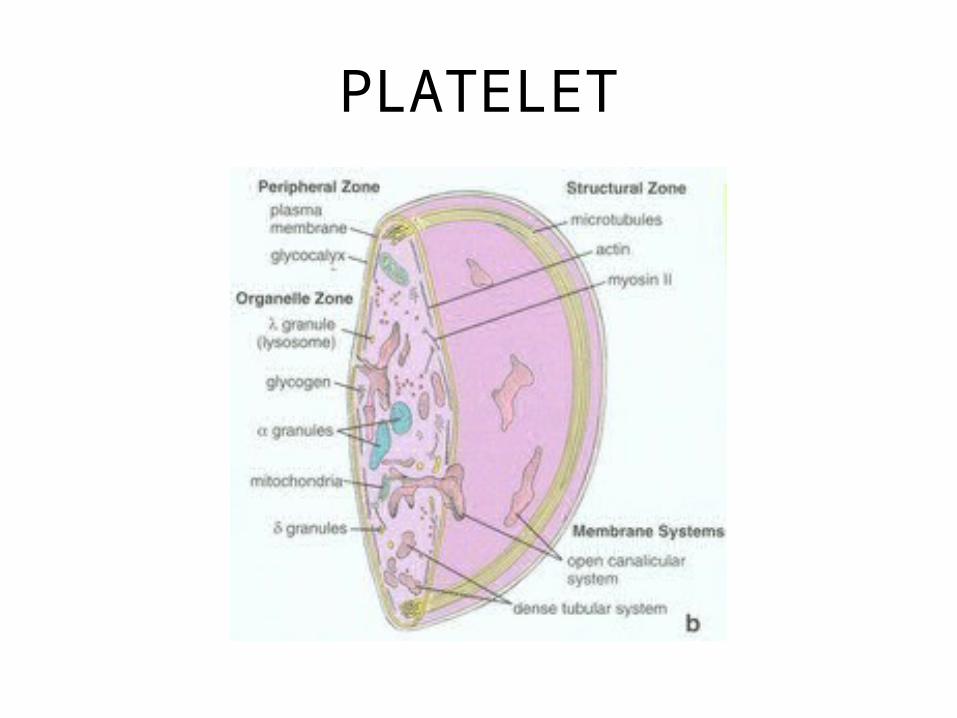
PLATELET

PLATELETS-FOUR MAJOR FUNCTIONS
• 1. ADHESION ACTIVATION
• 2. AGGREGATION
• 3. SECRETION
• 4. PROCOAGULANT ACTIVATION

1. ADHESION ACTIVATION:
– platelet on subendothelial matrix (surface)
– glycoprotein IIb/IIIa surface receptor binds vWF in subendo matrix

2. AGGREGATION:
– cohesion of platelets
– fibrinogen binds activated glycoprotein IIb/IIIa receptor
• Inhibited by – abciximab IV (ReoproR), tirofiban (AggrastatR),
eptifibatide (IntegrelinR) all IV; used in angioplasty; often used along with aspirin and heparin*
– ADP-receptor involved in GP IIb/IIIa-fibrinogen interaction and possibly also the vWF site • Inhibited by
– clopidogrel (PlavixR), ticlopidine (TicilidR) inhibit; both given PO*
*all treat and prevent arterial thrombosis

3. SECRETION:
• release of plt. granule proteins– ADP, serotonin, adhesive protein (fibronectin,
others), Factor V, thromboxane A2
– many growth factors (smooth muscle etc)• may be involved in restenosis post PTCA

4. PROCOAGULANT ACTIVITY:
• enhancement of thrombin generation– assembly of the clotting cascade on the
platelet surface
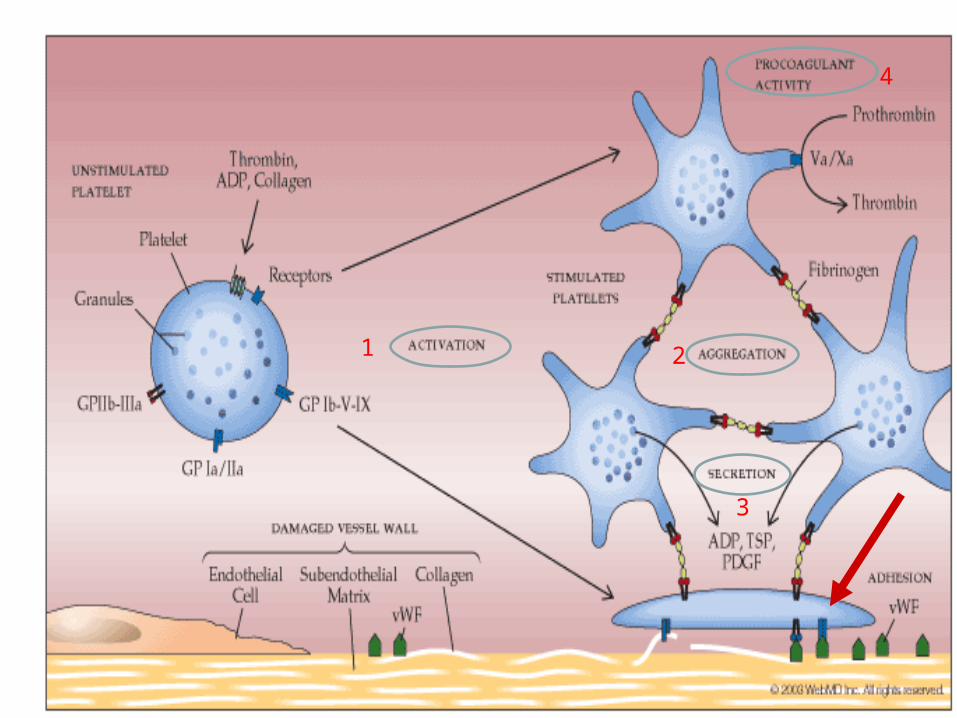
1 2
3
4
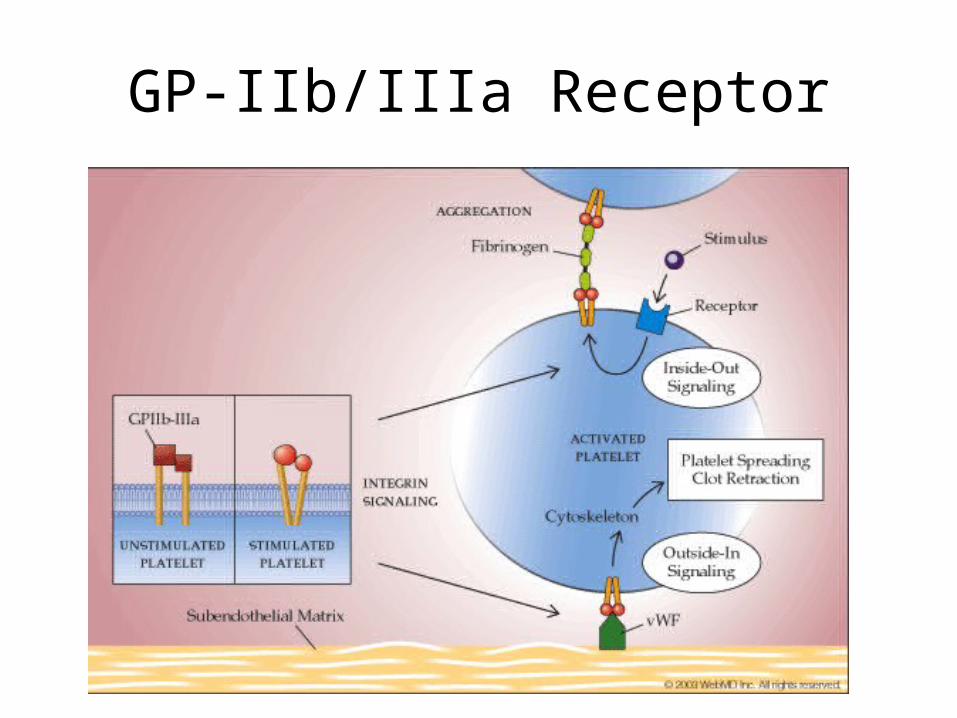
GP-IIb/IIIa Receptor

•CLOTTING FACTOR CASCADE

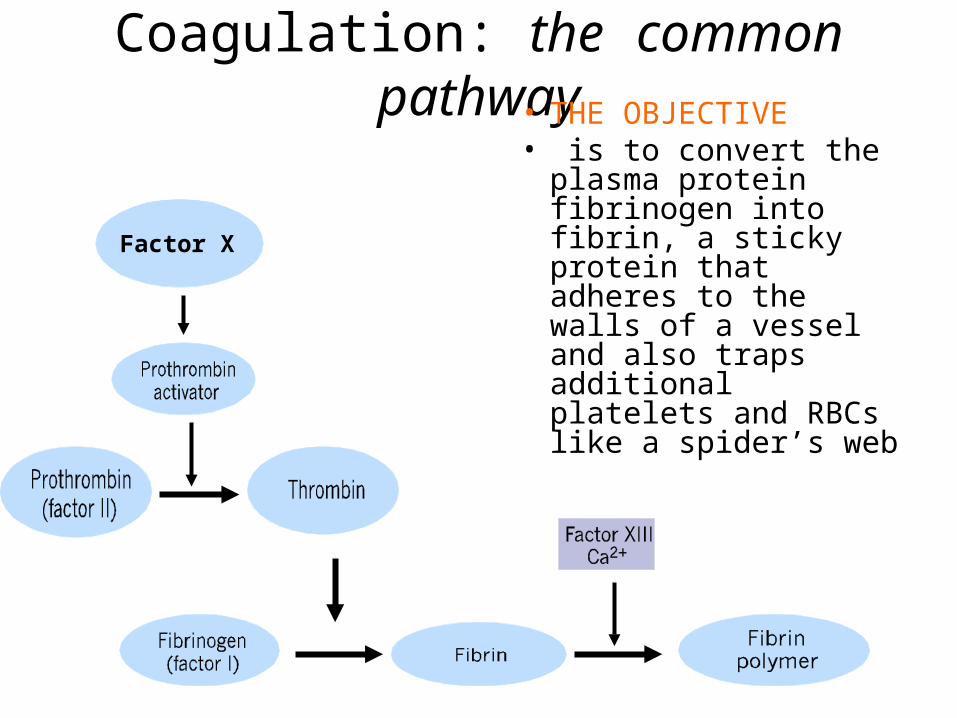
Coagulation: the common pathway• THE OBJECTIVE• is to convert the
plasma protein fibrinogen into fibrin, a sticky protein that adheres to the walls of a vessel and also traps additional platelets and RBCs like a spider’s web
Factor X
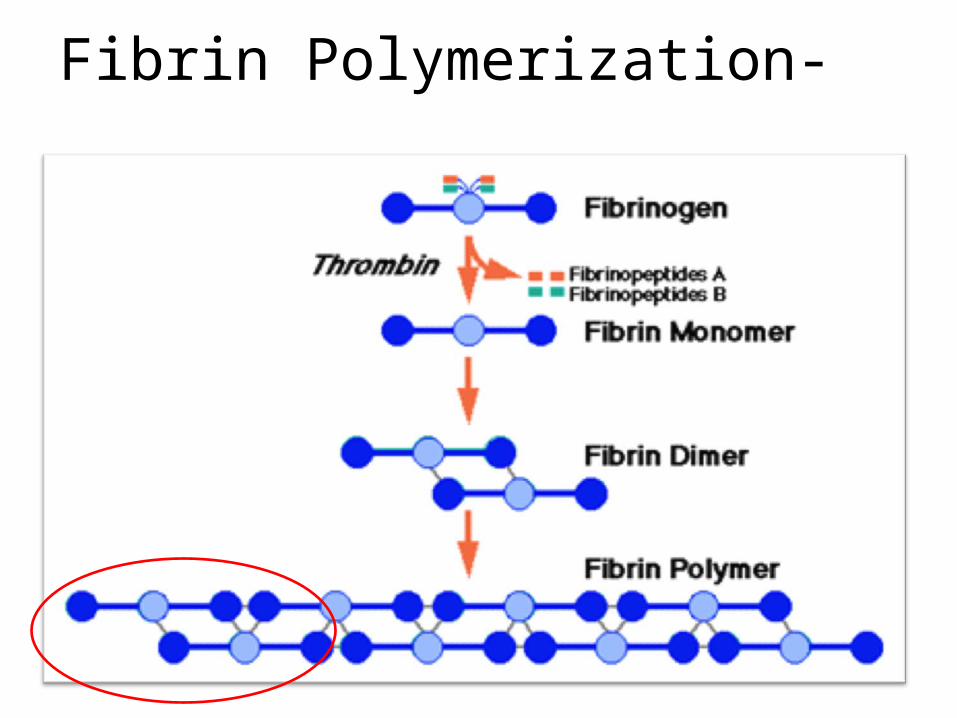
Fibrin Polymerization-

Coagulation Cascade
• TWO reaction pathways (roads)
– INTRINSIC PATHWAY uses only clotting factors found inside the blood itself (plasma, platelets) 3- 6 seconds
– EXTRINSIC PATHWAY initiated by clotting factors released in damaged vessels (tissue factor or thromboplastin) and perivascular tissues -15 seconds
• Roman numerals indicate the order discovered, however some numbers are not used: – Factor IV is Ca++; Factor VI is activated Factor V
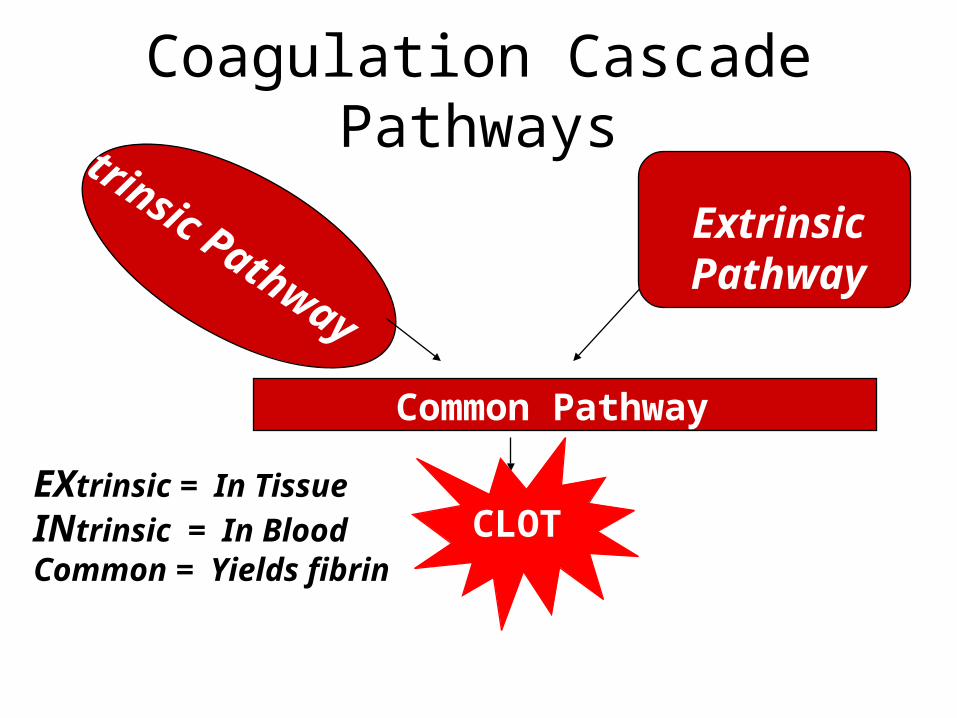
Coagulation Cascade PathwaysIntrinsic Pathway
Extrinsic Pathway
Common Pathway
CLOTEXtrinsic = In Tissue
INtrinsic = In BloodCommon = Yields fibrin
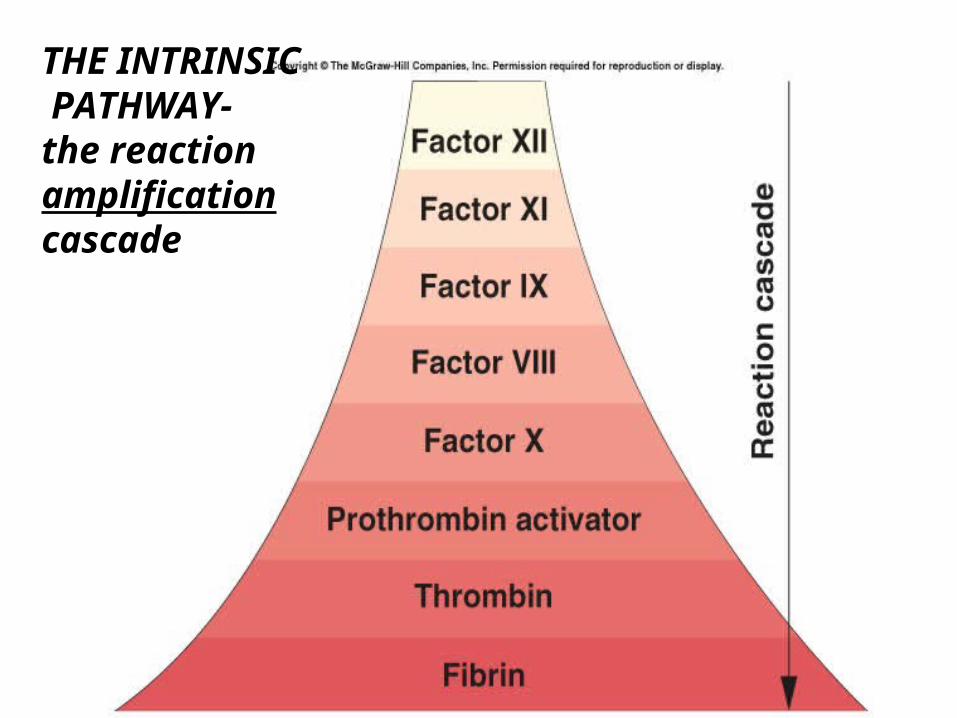
THE INTRINSIC PATHWAY-the reaction amplification cascade
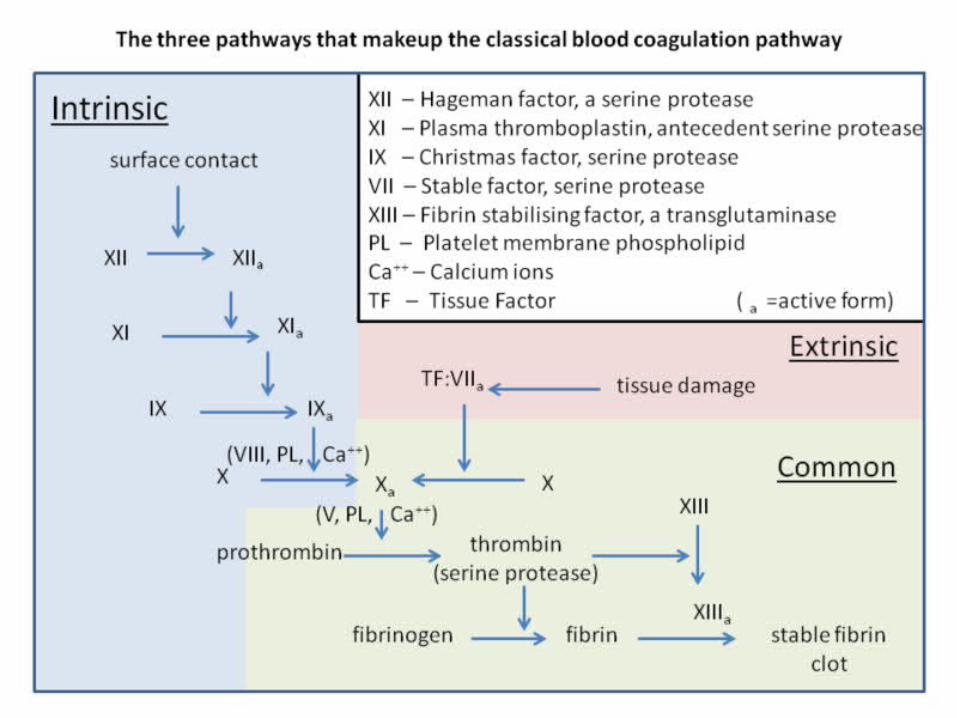
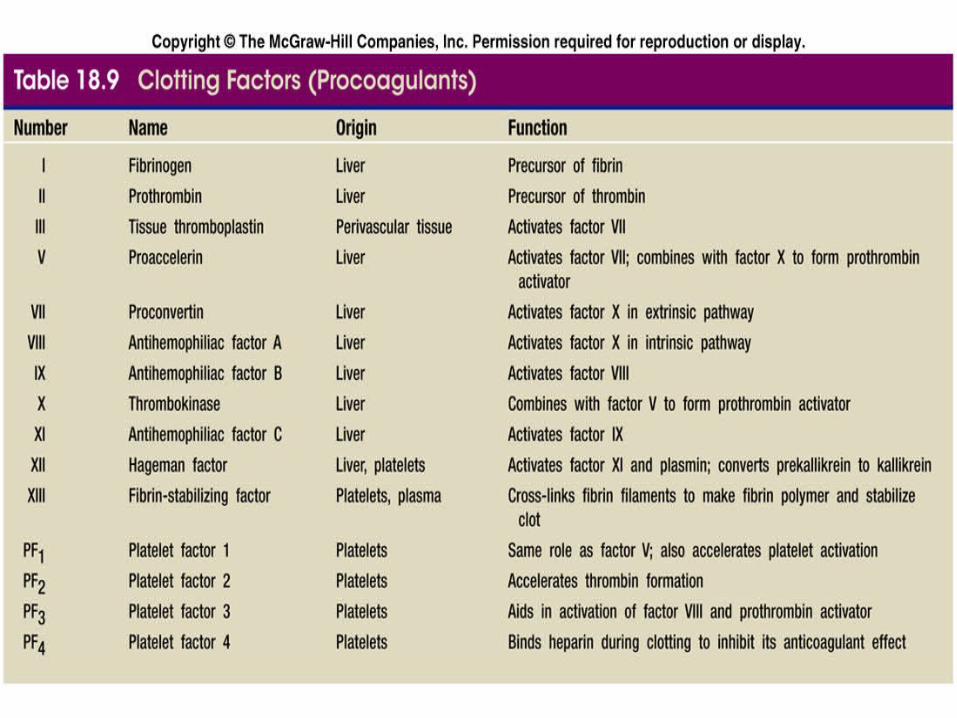
Table 18.9

PROCOAGULANT “CLOTTING FACTORS”
Synthesis
• ALL synthesized in the LIVER except– von Willebrand Factor in megakaryocytes and
endothelial cells• vWF is involved in
– PLATELET ACTIVATION– Maintaining normal factor VIII levels

Vitamin K
• Vitamin K-dependent procoagulants– II (Prothrombin), VII, IX, X
• Vitamin K-dependent anticoagulants– Warfarin-dicoumarol (COUMADIN)
– Vit K antagonist– Inhibits Vit K epoxide reductase
• (recycles oxidized Vit K – after K has been used in carboxylation of clotting factor production)
•

“THE REGULATORS”
• PROTEIN C & S
• ANTITHROMBIN
• TISSUE FACTOR PATHWAY INHIBITOR
• PLASMINOGEN->PLASMA
• PROSTACYCLIN ( PGI2)

PROTEIN C & PROTEIN S
Protein C & Protein S• ACTION: inactivates Factor Va and VIIIa• “Natural anticoagulant” – keeps system in balance• Protein C:
– vitamin K-dependent serine protease enzyme
• Thrombomodulin (on endothelial cell surfaces) and Thrombin stimulate Protein C and when activated by Protein S- inactivates Factor V
• .see flow diagram
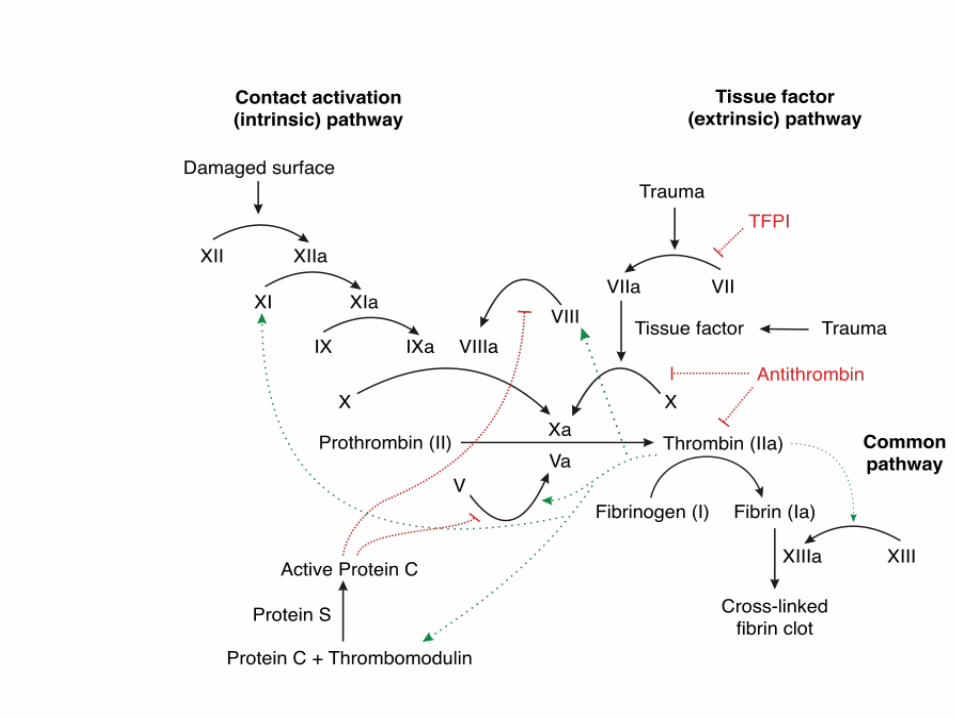
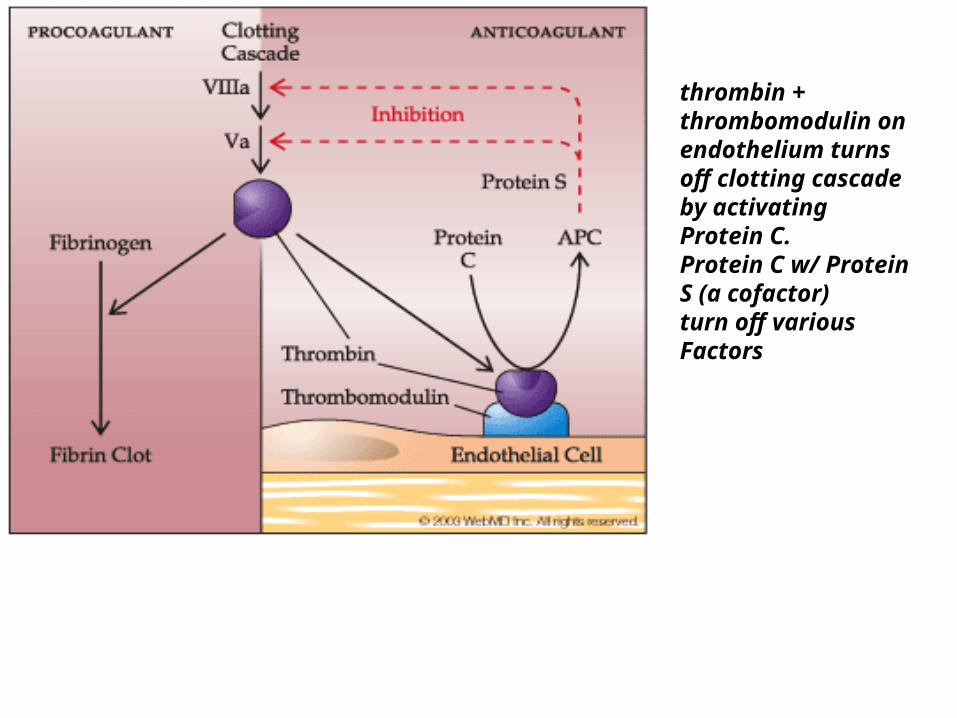
thrombin + thrombomodulin on endothelium turns off clotting cascade by activating Protein C.Protein C w/ Protein S (a cofactor) turn off various Factors

Protein C & S deficiency
• Spontaneous Thrombosis/thromboemboli
• Suspect in young person with DVT/Stroke with significant risk factors
• Most will be need Lifelong Anticoagulant therapy

FACTOR V - LEIDEN
• MUTATIONS OF FACTOR VA –
–resistant to APC (activated protein C)–Factor V Leiden mutation (present in 20
– 60% of pts with spontaneous venous thromboembolism
–5% of European decent, rare in Asian and African decent

ANTITHROMBIN III
• Serine protease inhibitor
• Active at all times
• Degrades:– Thrombin, Factors IXa, Xa, XIa, XIIa,
• Enhanced by Heparin sulfate
• Deficiency- leads to thrombophilia
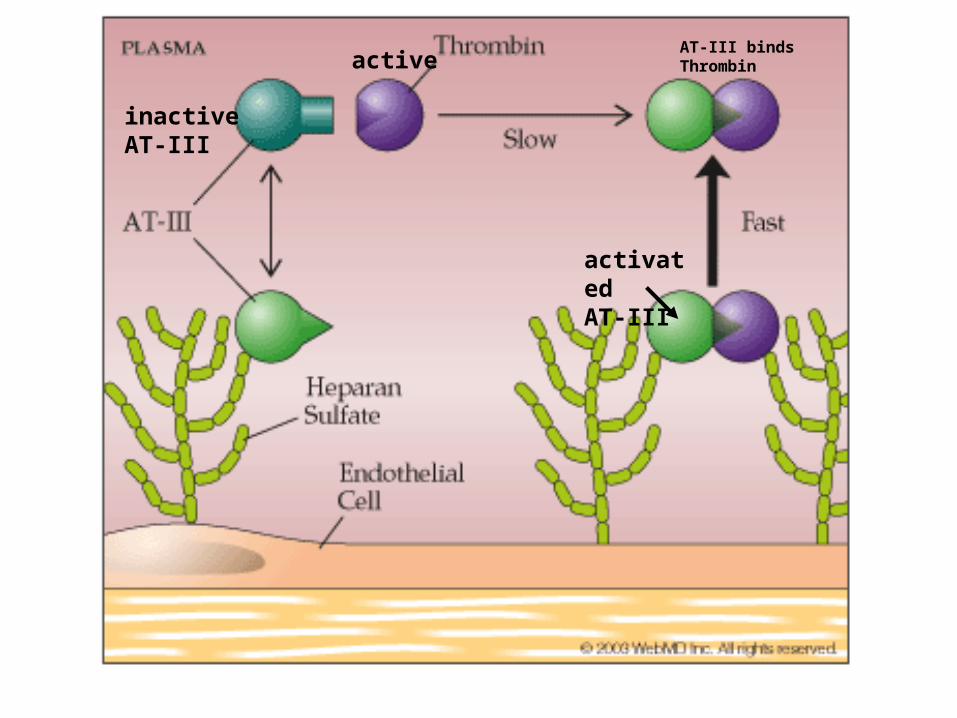
activatedAT-III
activeAT-III binds Thrombin
inactiveAT-III

TISSUE FACTOR PATHWAY INHIBITOR (TFPI)
• limits the action of tissue factor (TF)
• inhibits excessive TF-mediated activation of FVII and FX
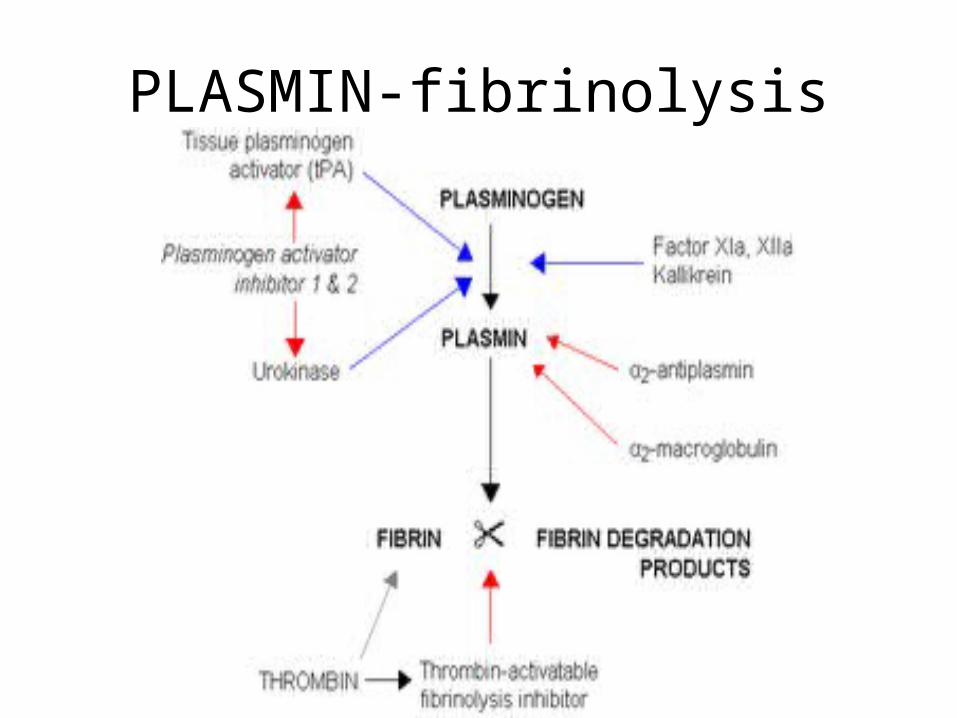
PLASMIN-fibrinolysis

Fibrinolysis: Dissolution of Clot
• Plasminogen Plasminogen a– t-Plasminogen activator (t-PA)
• cleaves plasminogen into the activated form: a two chain disulfide
• Plasminogen ----kallikrein----Plasmin• PLASMIN DISSOLVES THE CLOT
– limits fibrin clot to avoid ischemia in tissue – localizes clot to prevent widespread thrombosis
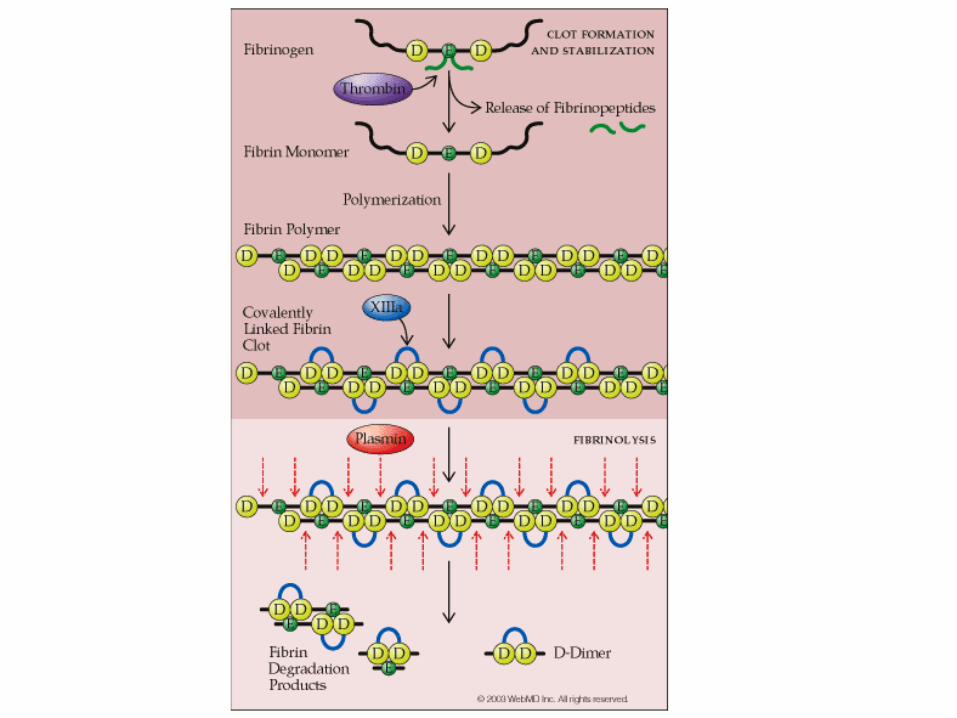
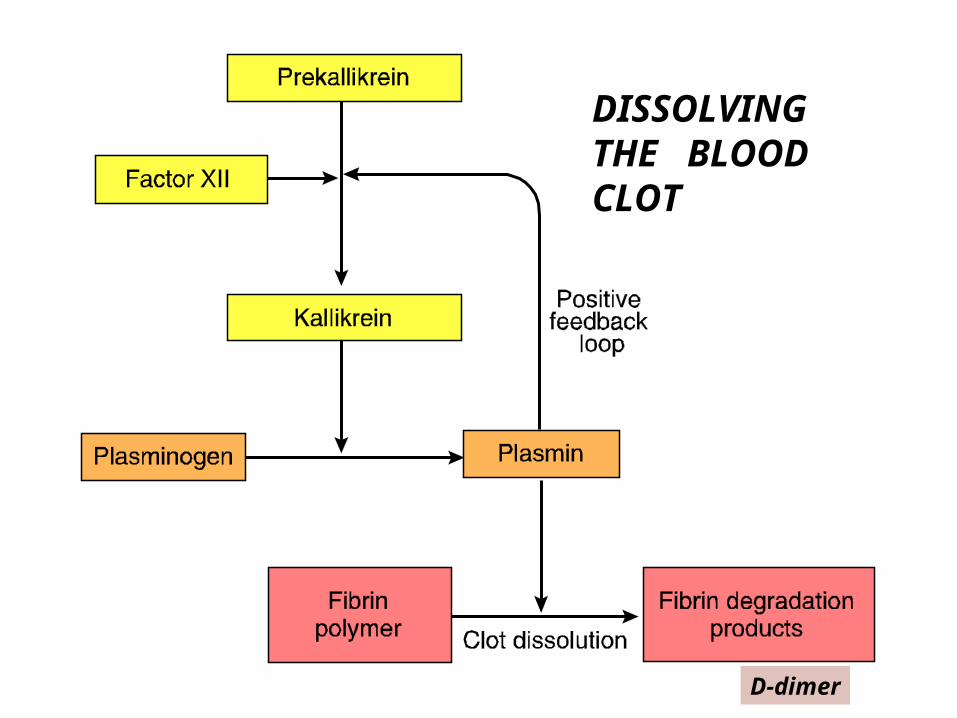
DISSOLVING THE BLOOD CLOT
D-dimer

PROSTACYCLIN (PGI2)
• Produced by endothelial cell via PGH1
• chiefly prevents formation of the PLATELET plug
• inhibiting platelet activation
• Antagonist to Thromboxane (TXA2)
• Vasodilator– epoprostenol –FLOLAN
• primary pulmonary hypertension

Control Mechanisms-BLOOD FLOW AND SHEAR
• DILUTION of procoagulants by flowing blood
• Removal of activated Factors via RE cells
• Stasis promotes clotting

Antithrombotic Mechanisms-summary• INTRINSIC REGULATION OF CLOTTING CASCADE
– ANTITHROMBIN III – PROTEIN C/PROTEIN S– TF PATHWAY INHIBITOR
• TISSUE FACTOR (thromboplastin or factor III)• MODULATION OF VESSEL AND PLATELET REACTIVITY
– PROSTACYCLIN (PGI2) – repulses platelets – nitric oxide- Vasodilation
• INHIBITION OF PLATELET RECRUITMENT
– Ecto-ADPase (CD39) – inactivated ADP • REMOVAL OF FIBRIN CLOT (CLOT BUSTERS )
– Fibrinolysis– tPA and plasminogen

Prevention of Inappropriate Coagulation• Platelet repulsion (PGI2; nitric oxide-- vasodilation)• Dilution of activated factor
– Nitric oxide-vasodilator more blood flow– STASIS IS THE ENEMY
• in low flow states may see thrombosis
• Circulating anticoagulants– antithrombin-III (circulating protease inhibitor)
• inhibits thrombin & Factor X – two big players in common pathway !!!!
– heparin-blocks thrombin quickly, • prothrombin activator, and promotes antithrombin III activity
(supercharges it)• Source: Mast cells
– Protein C & Protein S – negative feedback of pathways; important due to
common deficiencies found in pts• Fibrinolysis removes clot
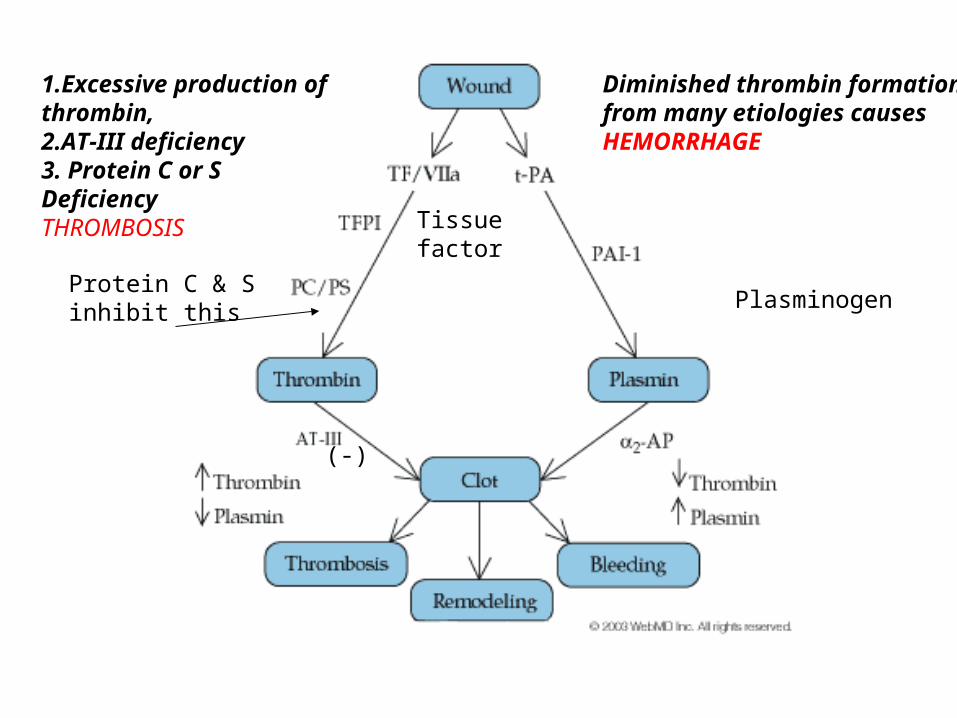
Diminished thrombin formationfrom many etiologies causesHEMORRHAGE
1.Excessive production of thrombin, 2.AT-III deficiency3. Protein C or SDeficiencyTHROMBOSIS
Protein C & Sinhibit this
(-)
Plasminogen
Tissue factor

Thrombotic Disorders
• Genetic– Factor V Leiden
mutation– Protein C deficiency– Protein S deficiency– Antithrombin III
deficiency– prothrombin 20210
mutation
• Acquired– Antiphospholipid
antibodies• (Anti-Cardiolipin
antibodies)
– Cancer– Atherosclerosis– Hyper-homocysteinemia– Infection– Stasis– Injury

Hypercoagulability
-more deaths from clotting than
bleeding

“KILLER CLOTS”
• Myocardial infarction
• Deep venous ThrombosisPulmonary Emboli
• Cerebral vascular accidents-
• Thrombotic Emboli-sources– Carotids– Heart – Atrial fibrillation, CHF

Deep Venous Thrombosis/Venous Thrombo Embolism
• VenousThromboEmbolism: DVT and/orPE– 1:1000 Americans, yearly– high death rate
• 250,000 Americans/year
– objective of DVT treatment to prevent PE– 59 % of VTE from current or recent
hospitalization or nursing home• 24 % hospital-surgery• 22 % hospital-medical• 13 % nursing home

VTE risk
• Malignancy• Trauma• Surgery- Extremity• Smoking• Drugs- Birth Control – high estrogen• Congestive Heart failure• Central Venous Pressure (CVP) catheter• Pacemaker/Defibrillator placement• Neurological deficits- CVA, MS, Spinal cord• Superficial vein thrombosis• Hereditary deficiencies- ProteinC/S, antithrombin III
• STASIS, STASIS, STASIS
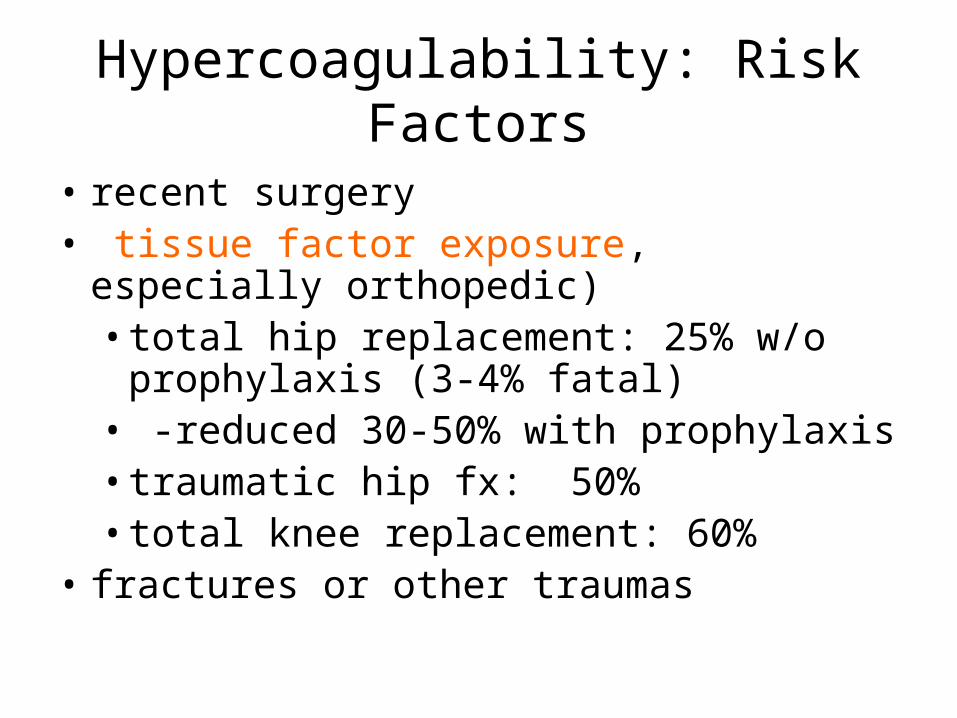
Hypercoagulability: Risk Factors
• recent surgery • tissue factor exposure,
especially orthopedic)• total hip replacement: 25% w/o
prophylaxis (3-4% fatal)• -reduced 30-50% with prophylaxis• traumatic hip fx: 50%• total knee replacement: 60%
• fractures or other traumas

Virchow’s Classic Triad
• THREE MAJOR ELEMENTS – that promote thrombosis
– Endothelial injury
– Decrease in blood flow
– Imbalance between procoagulants and anticoagulants
• (Hypercoagulable state)

Endothelial Injury
• Mechanical trauma
• Atherosclerosis- (intrinsic pathway)
• Endotoxins from bacteria
• Proteases & cytokines of inflammation
• Immune-autoimmune
• Hypoxia

Decrease in Blood Flow
• Heart failure (HF)-causes stasis
• Atrial fib/flutter,
• Myocardial infarction
• Immobilization
• Long travel & long flight – • “economy class syndrome”
• Casting
• Bedridden (stroke)

Thrombophilia (hypercoagulable)
• GENETIC:– 5 – 8 % of population has one genetic clotting disorder;
25-50% will have F. V Leiden
• AGE: at onset < 50 yrs.• IDENTIFIABLE RISK FACTORS: may be none
– Frequently triggered by 2nd risk factor
• FAMILY HISTORY: frequently positive• PAST HISTORY: recurrent events• get into trouble when 2nd risk factor is present

INHERITED Hypercoagulable Disorders
• activated Protein C resistance– (Factor V Leiden genetic mutation)– 25% in pt w/ family hx of thrombosis– 5% of white population;– #1 cause of inherited thrombophilia– named for city in Netherlands
• Hyper-homocysteinemia - (under debate)• Antithrombin III deficiency• Protein C deficiency (1:100)• Protein S deficiency• prothrombin 20210 mutation -altered thrombin
production

ACQUIRED Hypercoagulable Disorders
• ANTIPHOSPHOLIPID ANTIBODY SYNDROME:– found in some cases of SLE, syphilis, rheumatoid arthritis
• ACQUIRED HYPERCOAGULABLE STATE – physiologic or thrombogenic stimulus
• acute-phase reactants from inflammation• advanced age (fibrinogen levels increase)• OCP, pregnancy• surgery, trauma
– hypercoagulable state from other disease• Malignancy,• renal- nephrotic syndrome, • “thick blood”-polycythemia, Sickle Cell

Treatment of Venous Thromboembolism
• RISK STRATIFY
• LOW risk & clearly identifiable cause– 3 months oral anticoagulation
• MEDIUM risk– 6 months oral anticoagulation
• HIGH risk– life long anticoagulation w/ INR - 2-3

Treatment for Hypercoagulation• ANTICOAGULANTS:
• heparin, low-molecular heparin, warfarin (Coumadin)
• PLATELET ANTAGONISTS• INACTIVATES COX – • necessary for production of Thromboxane A2
– Aspirin –irreversibly
– NSAIDS: reversible inactivation of platelets• ADP INHIBITIORS
– Ticlopidine-Ticlid– Clopidogrel-Plavix
• IIB/IIIA INHIBITORS (INHIBIT PLATELET AGGREGATION)– Abciximab(Reopro), Eptifibatide(Integrilin) – Tirofiban (Aggrastat)
• FIBRINOLYTIC AGENTS• Streptokinase, urokinase, tissue plasminogen activators (t-PA)

Heparin-Low molecular weight
• LMWH - depolymerized (fractionated)– more reliable, predictable– Monitoring PTT not required– SQ administration; outpatient therapy possible– Pregnancy-prophylaxis only- 2 & 3 trimester– lower risk of thrombocytopenia– Enoxaparin(Lovenox), Dalteparin (Fragmin)

Coumadin (warfarin)
• reduce clotting of the blood via blocking the production of Factors VII, IX, X, and II; competes w/ Vitamin K uptake and recycling by the liver
• PO, daily• Indications: DVT/PE, AFib, mechanical
valve replacement• - must follow PT/INR• Caution: when using platelet inhibitory Rx

Dabigatran-PRADAXA
• Direct Thrombin inhibitor– No Protime necessary
• Indications: – Stroke prevention, DVT Treatment
• Problems:– Bleeding– Renal adjustment necessay

INDICATIONS for THROMBOLYTICS –
”CLOT BUSTERS”
• Acute myocardial infarction (AMI)
• Deep vein thrombosis (DVT)
• Pulmonary embolism (PE)
• Acute ischemic stroke (AIS)
• Acute peripheral arterial occlusion
• Occlusion of indwelling catheters

Contraindications to Thrombolytics• history of hemorrhagic stroke < 2 months• CNS neoplasm, AV malformation, or aneurysm, or
CNS surgery < 2months• Severe uncontrolled hypertension (over 200/130 or
complicated by retinovascular disease or encephalopathy)
• ongoing (active) bleeding• s/p recent significant surgery• known bleeding disorder• MI due to aortic dissection• allergy to agent planned• many relative contraindications

Thrombolytics – clot busters
• FIBRIN-SPECIFIC AGENTS– Alteplase (t-PA) Activase, t-PA– Retaplase (recombinant-PA) Retavase– Tenecteplase TNKase
• NON–FIBRIN-SPECIFIC AGENTS-– Urokinase or Prourokinase (direct
plasminogen cleavage) - nonallergenic– Streptokinase- allergenic- inexpensive

DISSEMINATED INTRAVASCULAR
COAGULATION
DIC

DISSEMINATED INTRAVASCULAR COAGULATION
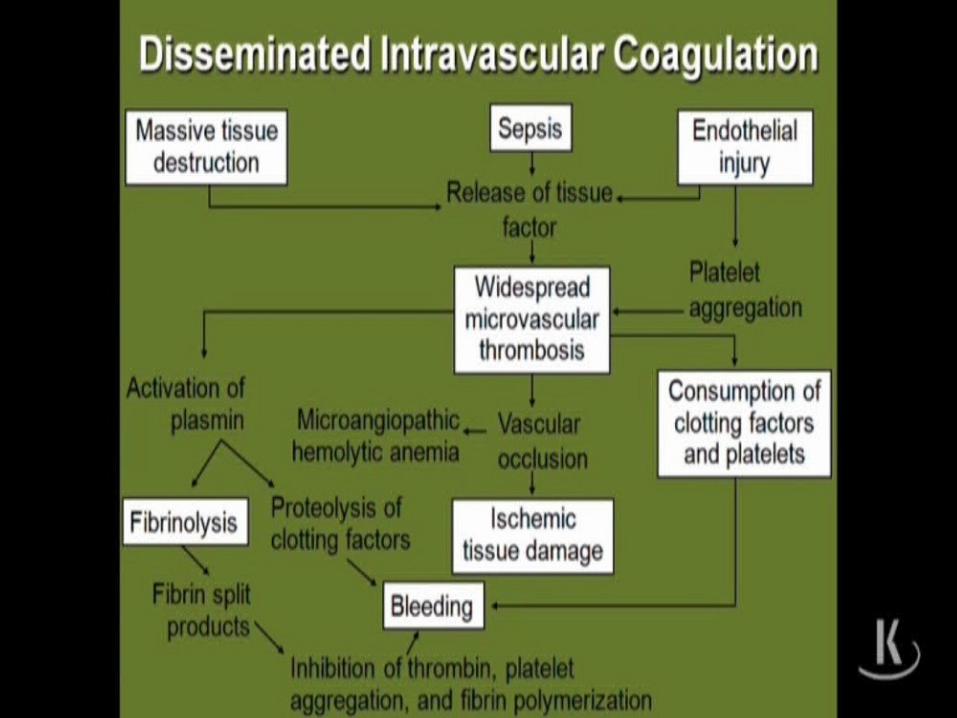

DICClinical: Hemorrhage - internal and external
Mucosal“shock”Petechia/purpura
Labs:• thrombocytopenia• normal or slightly prolonged aPT• prolonged aPTT• Low Fibrinogen levels• FIBRIN SPLIT PRODUCTS PRESENT• RBC’s – Helmet cells/schiztocytes(fragmented)
– Due to RBC’s being cut up by Fibrin strands
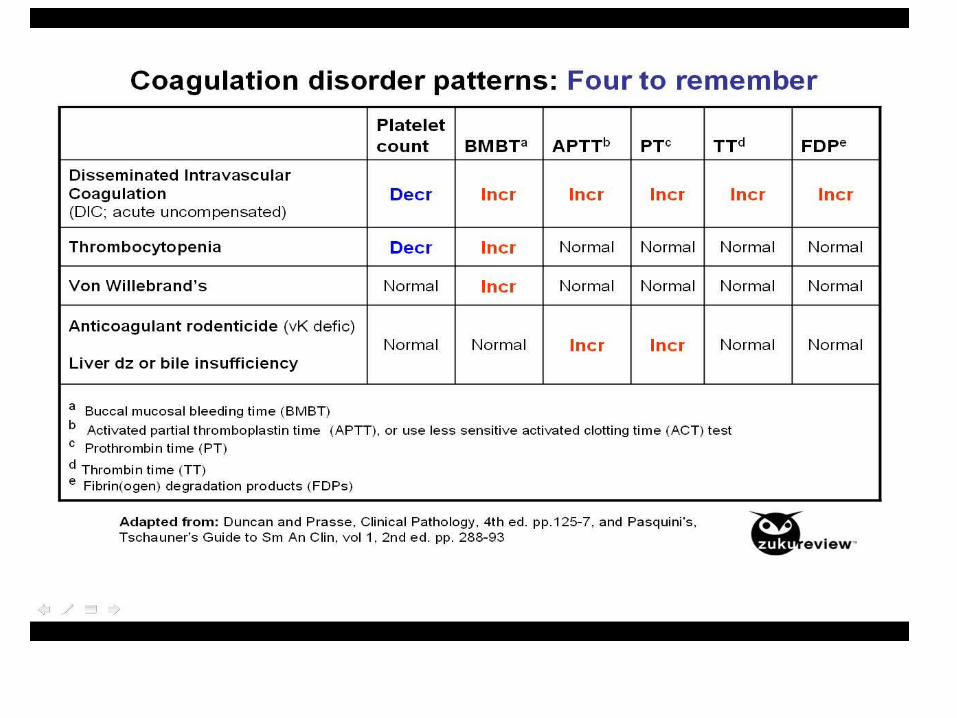
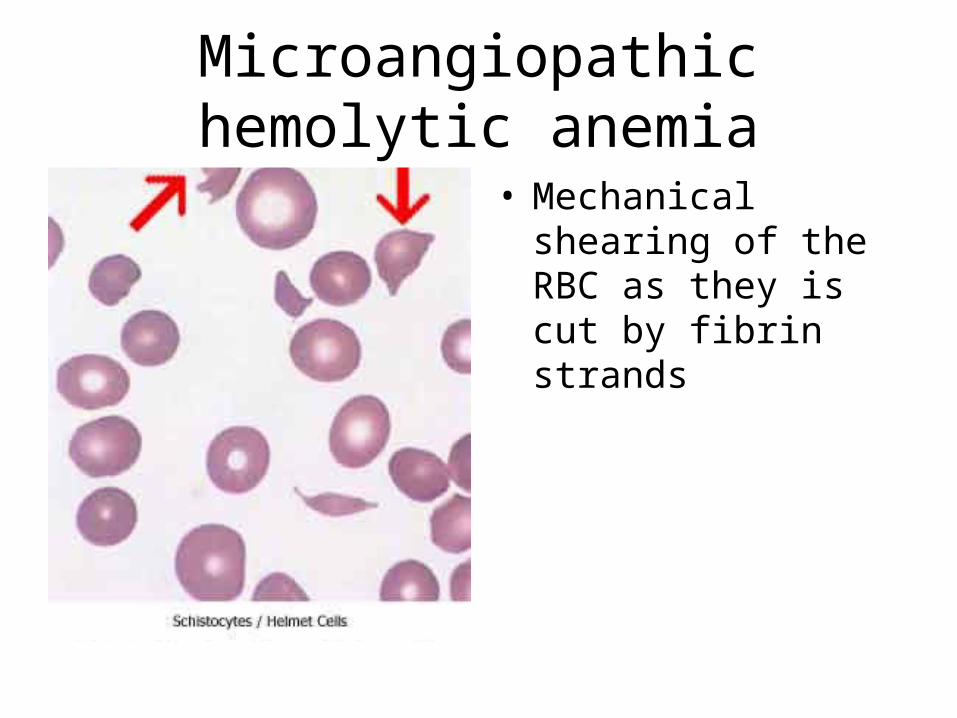
Microangiopathic hemolytic anemia
• Mechanical shearing of the RBC as they is cut by fibrin strands

DIC -TREATMENT• Treatment should primarily focus on
addressing the underlying disorder.• Monitor vital signs, assess and document
extent of hemorrhage and thrombosis, correct hypovolemia, and administer basic hemostatic procedures-ICU-Transfer
• Platelet and factor replacement.as indicated- Fresh frozen plasma
• HEPARIN should be provided to those patients who demonstrate extensive fibrin deposition without evidence of substantial hemorrhage





























