Embed Size (px)
Citation preview

Clinical Endocrinology (2000) 52, 241–250
Letters to the Editors
Complete resolution of protease inhibitor induceddiabetes mellitus
Sirs: Protease inhibitors (PI) are currently used to treat HIVinfection. They inhibit HIV protease which is necessary for theprocessing of viral proteins in the last stage of the viral cycle,and can achieve an undetectable viral load when used in com-bination with nucleoside analogues in triple therapy. However,PI are not free of adverse effects (Flexner, 1998). Hyper-lipidaemia and abnormal fat distribution were soon related tothe use of these drugs (Massipet al., 1997). The latter occurredbetween two and 12 months after beginning treatment and wasnot preceded by weight loss (Viraben & Aquilina, 1998). Thelipoatrophy was noted mainly in the legs, arms and face,sparing the central portion of the body (Carret al., 1998a).Hyperglycaemia was also noted with the use of PI (FDA, 1997)and new onset diabetes mellitus, easily controlled with the useof hypoglycaemic agents or insulin (Eastone & Decker, 1997),or even severe diabetes (Visnegarwalaet al., 1997) have beenreported. However, treatment with PI could not to be discon-tinued in the majority of patients (Ault, 1997). In 1998, anassociation between the syndrome of lipodystrophy, hyperlipi-daemia and insulin resistance, with the appearance of overtdiabetes mellitus in less than 2% of the cases was demonstrated(Carret al., 1998a). A recent publication indicated a prevalenceof 83% for lipodystrophy which can be severe in 11% of cases,and abnormalities in glucose homeostasis in 23% of patients,with less than 1% of cases with diabetes mellitus and less than3% with glucose intolerance, after 21 months of treatment withPI (Carret al., 1999). There is no information on the long-termnatural history or complications of the diabetes associated to theuse of PI nor about its reversibility after cessation of therapy.We report a patient with complete resolution of PI induceddiabetes mellitus. In all of these communications, nothing wassaid about the natural history of the diabetes associated to theuse of protease inhibitors. Nevertheless, it was proposed thatcessation of therapy might be considered for patients withdifficult control of severe diabetes mellitus.
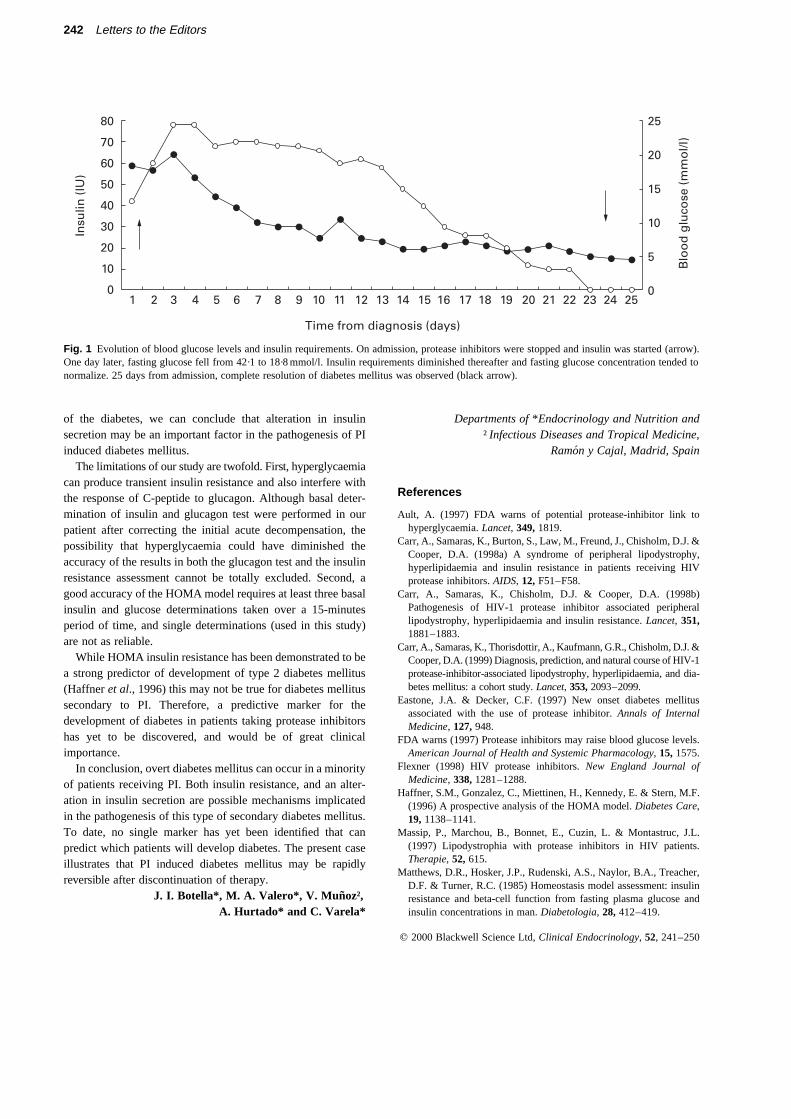
A 47-year-old man presented with polyuria, polydipsia,polyphagia, blurred vision and weight loss of four kilograms forone month. He had HIV infection in A2 stage and had receivedtreatment with indinavir and two nucleoside analogues(stavudine and lamivudine) for 10 months. He had no personalnor familial history of diabetes mellitus or hyperglycaemia. Hehad not suffered acute nor chronic pancreatitis and neverreceived treatment with anabolic steroids, pentamidine orother hyperglycaemia associated drugs apart from protease
inhibitors. Physical examination revealed fat wasting of face,legs and arms, sparing the abdomen and central part of thebody. Height was 1·79 m, weight 79 kg and BMI 24·6. Noacanthosis nigricans was noted. He had a CD4 level of 515 permm3 and a viral load of less than 1·7 logs (less than 50 virionsper mm3), showing a good response to treatment. He presentedwith a fasting glucose level of 43·6 mmol/l, venous pH 7·37,ketonuria almost undetectable, triglycerides 6·54 mmol/l,cholesterol 7·68 mmol/l, HDL 0·80 mmol/l, LDL 5·22 mmol/l,HbA1c 12·9%. Levels of CH-50 and C3 were 0·89 mmol/l and1·19 mmol/l, respectively. Islet cell autoantibody were absent.Treatment with the PI was stopped and insulin was started(Fig. 1). C-peptide rose after injection of glucagon was from0·72 nmol/l (00) to 1·39 nmol/l (60) with glucose levels of8·27 mmol/l and 9·27 mmol/l, respectively. Basal insulin was153 pmol/l for a fasting glucose level of 8·27 mmol/l. Insulinresistance calculated by HOMA was 7·86. The patient wasdischarged with insulin therapy, lipid lowering treatment withatorvastatin and treatment for HIV infection with nevirapine(a non-nucleoside antiretroviral), lamivudine and stavudine(both nucleoside analogues).
One month later, insulin was withdrawn, and basal andpostprandial glucose levels were normal (Fig. 1). The patientwas admitted into our Metabolic Unit in order to perform aglucagon test and basal measurements of insulin. C-peptidelevels were of 1·25 nmol/l (00) to 2·44 nmol/l (60), and glucoselevels of 4·7 mmol/l and 5·38 mmol/l, respectively. Basalinsulin determination was of 406 pmol/l for a glucose level of4·7 mmol/l. Insulin resistance was 11·83 determined by HOMAmodel. Triglycerides were 2·23 mmol/l and cholesterol6·1 mmol/l on atorvastatin treatment, and lipodystrophy wasstill clinically evident.
The present case shows that PI induced diabetes mellitus canbe a rapidly reversible phenomenon after cessation of the drug.The inhibition of proteins involved in regulation of blood lipidlevels and in adipocyte proliferation, has been proposed as acause of lipodystrophy and insulin resistance (Carret al., 1998b).However, decreased insulin secretion may also play a role in thepathogenesis of associated diabetes mellitus (Visnegarwalaet al., 1997). In our patient, both insulin resistance and defec-tive insulin secretion were shown. HOMA model is accurate forthe estimation of the degree of insulin resistance (Matthewset al., 1985) and the response of C-peptide to glucagon wasused as an estimation ofb-cell function. Since insulin resistancewas present even after the complete resolution of clinical andbiochemical diabetes mellitus in our patient, and the responseof C-peptide to a glucagon stimulus improved after resolution
241q 2000 Blackwell Science Ltd
of the diabetes, we can conclude that alteration in insulinsecretion may be an important factor in the pathogenesis of PIinduced diabetes mellitus.
The limitations of our study are twofold. First, hyperglycaemiacan produce transient insulin resistance and also interfere withthe response of C-peptide to glucagon. Although basal deter-mination of insulin and glucagon test were performed in ourpatient after correcting the initial acute decompensation, thepossibility that hyperglycaemia could have diminished theaccuracy of the results in both the glucagon test and the insulinresistance assessment cannot be totally excluded. Second, agood accuracy of the HOMA model requires at least three basalinsulin and glucose determinations taken over a 15-minutesperiod of time, and single determinations (used in this study)are not as reliable.
While HOMA insulin resistance has been demonstrated to bea strong predictor of development of type 2 diabetes mellitus(Haffneret al., 1996) this may not be true for diabetes mellitussecondary to PI. Therefore, a predictive marker for thedevelopment of diabetes in patients taking protease inhibitorshas yet to be discovered, and would be of great clinicalimportance.
In conclusion, overt diabetes mellitus can occur in a minorityof patients receiving PI. Both insulin resistance, and an alter-ation in insulin secretion are possible mechanisms implicatedin the pathogenesis of this type of secondary diabetes mellitus.To date, no single marker has yet been identified that canpredict which patients will develop diabetes. The present caseillustrates that PI induced diabetes mellitus may be rapidlyreversible after discontinuation of therapy.
J. I. Botella*, M. A. Valero*, V. Mun˜oz†,A. Hurtado* and C. Varela*
Departments of*Endocrinology and Nutrition and†Infectious Diseases and Tropical Medicine,
Ramon y Cajal, Madrid, Spain
References
Ault, A. (1997) FDA warns of potential protease-inhibitor link tohyperglycaemia.Lancet, 349,1819.
Carr, A., Samaras, K., Burton, S., Law, M., Freund, J., Chisholm, D.J. &Cooper, D.A. (1998a) A syndrome of peripheral lipodystrophy,hyperlipidaemia and insulin resistance in patients receiving HIVprotease inhibitors.AIDS, 12, F51–F58.
Carr, A., Samaras, K., Chisholm, D.J. & Cooper, D.A. (1998b)Pathogenesis of HIV-1 protease inhibitor associated peripherallipodystrophy, hyperlipidaemia and insulin resistance.Lancet, 351,1881–1883.
Carr, A., Samaras, K., Thorisdottir, A., Kaufmann, G.R., Chisholm, D.J. &Cooper, D.A. (1999) Diagnosis, prediction, and natural course of HIV-1protease-inhibitor-associated lipodystrophy, hyperlipidaemia, and dia-betes mellitus: a cohort study.Lancet, 353,2093–2099.
Eastone, J.A. & Decker, C.F. (1997) New onset diabetes mellitusassociated with the use of protease inhibitor.Annals of InternalMedicine, 127,948.
FDA warns (1997) Protease inhibitors may raise blood glucose levels.American Journal of Health and Systemic Pharmacology, 15, 1575.
Flexner (1998) HIV protease inhibitors.New England Journal ofMedicine, 338,1281–1288.
Haffner, S.M., Gonzalez, C., Miettinen, H., Kennedy, E. & Stern, M.F.(1996) A prospective analysis of the HOMA model.Diabetes Care,19, 1138–1141.
Massip, P., Marchou, B., Bonnet, E., Cuzin, L. & Montastruc, J.L.(1997) Lipodystrophia with protease inhibitors in HIV patients.Therapie, 52, 615.
Matthews, D.R., Hosker, J.P., Rudenski, A.S., Naylor, B.A., Treacher,D.F. & Turner, R.C. (1985) Homeostasis model assessment: insulinresistance and beta-cell function from fasting plasma glucose andinsulin concentrations in man.Diabetologia, 28, 412–419.
242 Letters to the Editors
q 2000 Blackwell Science Ltd,Clinical Endocrinology, 52, 241–250
Time from diagnosis (days)
Insu
lin (
IU)
Blo
od
glu
cose
(m
mo
l/l)
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25
80
70
60
50
40
30
20
10
0
25
20
15
10
5
0
Fig. 1 Evolution of blood glucose levels and insulin requirements. On admission, protease inhibitors were stopped and insulin was started (arrow).One day later, fasting glucose fell from 42·1 to 18·8 mmol/l. Insulin requirements diminished thereafter and fasting glucose concentration tended tonormalize. 25 days from admission, complete resolution of diabetes mellitus was observed (black arrow).

Viraben, R. & Aquilina, C. (1998) Indinavir-associated lipodystrophy.AIDS, 12, F37–F39.
Visnegarwala, F., Krause, K.L. & Musher, D.M. (1997) Severe diabetesassociated with protease inhibitor therapy.Annals of InternalMedicine, 127,947.
Metformin and ovarian steroidogenesis in PCOSwomen
Sirs, We read with interest the recent paper by Unluhizarciet al.(1999) about the effects of the reduction of hyperinsulinaemiainduced by metformin on steroidogenic activity in women withPCOS. The results indicate that although metformin reducedinsulin resistance and plasma levels of free testosterone andincreased those of SHBG, it did not alter the pituitary-ovarianresponse to GnRH.
In our opinion the experimental protocol has some defects. Inorder to eliminate the contribution of adrenal androgens, 2 mg/day dexamethasone was administered for 4 days before theGnRH test. To standardize the experimental protocol, GnRHwas administered after as much as a month of metformintherapy. However, the possible effects of dexamethasone onsugar metabolism were not considered. This drug is known topromote diabetes. Indeed, glucocorticoids have this propertybecause they decrease glucose uptake and increase hepaticglucose production (Delaunayet al., 1997). Administration ofdexamethasone (2 mg/day for 3 days) reduces sensitivity toinsulin by 46% (Matsumotoet al., 1996). Hence glucocorti-coids induce an increase in hepatic glucose production andperipheral resistance to insulin.
A significant reduction in insulin resistance is reported in thepaper after metformin therapy. This is probably due to thestandardization of the experimental protocol (administration ofdexamethasone before both GnRH tests). However, the areaunder the insulin curve after metformin was much greater thanthat observed in normal women (430666 SD vs. 130756 SDpmol/time).
Little is yet known about the interaction between insulin andovarian steroidogenesis. We therefore do not think that theresults reported in the paper can be compared with those ofother authors (Nestler & Jakubowicz, 1996).
Unluhizarci et al. (1999) also report a response of LH toGnRH after metformin therapy, similar to the basal test (beforetherapy), despite the reduction in insulin resistance. In a recentstudy (la Marcaet al., 1999) we demonstrated that oraladministration of 2 mg dexamethasone in healthy women wasfollowed by a reduction in the area under the curve of the LHresponse to GnRH. Glucocorticoids inhibit the GnRH-LH axis,either directly, or indirectly through an increase in opioid tonus.It is therefore possible that the dexamethasone given in thebasal test induced a reduction in the response of LH to GnRH
which masked the changes obtained with metformin. It is alsonot possible to exclude little known effects of dexamethasoneon ovarian steroidogenesis. Dexamethasone has a synergisticeffect with FSH in the accumulation of progesterone, and thismay be due to stimulation of 3b-hydroxysteroid dehydrogenaseand inhibition of 20a hydroxysteroid dehydrogenase (Adashiet al., 1981). Other authors have reported that glucocorticoidsinhibit FSH-induced stimulation of hCG binding to granulosacells (Danisovaet al., 1978).
Since glucocorticoids can affect insulin sensitivity andhypothalamic-pituitary and ovarian activity, we consider thatstudies to explore the role of insulin resistance in PCOS shouldbe carried out without administration of these drugs.
Vincenzo De Leo, Antonio la Marca andGiuseppe Morgante
Department of Obstetrics and Gynaecology,University of Siena,
Policlinico Le Scotte,53100 Siena (SI), Italy
References
Adashi, E.Y., Jones, P.B. & Hsueh, A.J. (1981) Synergistic effects ofglucocorticoids on the stimulation of progesterone production byfollicle-stimulating hormone in cultured rat granulosa cells.Endocrinology, 109, 1888–1894.
Danisova, A., Sebokova, E. & Kolena, J. (1978) Glucocorticoidinhibition of FSH induced estrogen production in cultured ratgranulosa cells.Steroids, 21, 639–648.
Delaunay, F., Khan, A., Cintra, A., Davani, B., Ling, Z.C., Andersson,A., Ostenson, C.G., Gustafsson, J., Efendic, S. & Okret, S. (1997)Pancreatic beta cells are important targets for the diabetogenic effectsof glucocorticoids.Journal Clinical Investigation, 100, 2094–2098.
la Marca, A., Torricelli, M., Morgante, G., Lanzetta, D. & De Leo, V.(1999) Effects of dexamethasone and dexamethasone plus naltrexoneon pituitary responses to GnRH and TRH in normal women.Hormone Research, 51, 85–90.
Matsumoto, K., Yamasaki, H., Akazawa, S., Sakamaki, H., Ishibashi,M., Abiru, N., Uotani, S., Matsuo, H., Yamaguchi, Y., Tokuyama, K.& Nagataki, S. (1996) High-dose but not low-dose dexamethasoneimpairs glucose tolerance by inducing compensatory failure ofpancreatic beta-cells in normal men.Journal of Clinical Endocrinologyand Metabolism, 81, 2621–2626.
Nestler, J.E. & Jakubowicz, D.J. (1996) Decreases in ovariancytochrome P450c17a activity and serum free testosterone afterreduction of insulin secretion in polycystic ovary syndrome.NewEngland Journal of Medicine, 335, 617–623.
Unluhizarci, K., Kelestimur, F., Bayram, F., Sahin, Y. & Tutus, A.(1999) The effects of metformin on insulin resistance and ovariansteroidogenesis in women with polycystic ovary syndrome.ClinicalEndocrinology, 51, 231–236.
Metformin and polycystic ovary syndrome
Sirs, We write regarding the article by U¨ nluhizarciet al. (1999)entitled ‘The effects of metformin on insulin resistance and
Letters to the Editors 243
q 2000 Blackwell Science Ltd,Clinical Endocrinology, 52, 241–250

ovarian steroidogenesis in women with polycystic ovarysyndrome.
We have evaluated the effects of metformin on insulinsensitivity and androgenic profile in women with polycysticovary syndrome (PCOS) over a total of six months, which is thelongest investigation to date (Diamanti-Kandarakiset al.,1998). Sixteen obese women with PCOS on a weightmaintaining diet were treated with 1700 mg of metformindaily. Insulin sensitivity was assessed by the estimation ofglucose utilization during the euglycaemic hyperinsulinaemicclamp technique before and after therapy. Glucose utilizationwas markedly enhanced at six months and this improvementwas accompanied by increase of SHBG levels and reduction offree testosterone levels. These findings are in agreement withthe ones reported by U¨ nluhizarci et al. (1999). However, wealso observed a significant decrease of androstenedione levels,a finding which could be mostly explained by the ameliorationof ovarian androgen production after metformin therapy(Velazquez et al., 1994; Nestler & Jacobowicz, 1997).Additionally, the above biochemical changes were associatedwith clinical improvements such as, the resumption of regularmenstruation in about 50% of the studied women and two casesof spontaneous pregnancy.
Although, Unluhizarci et al. (1999), reported the beneficialeffects of metformin (given at a dose of 1000 mg daily) oninsulin sensitivity in women with PCOS, they did not observeany change in the ovarian cytochrome P450c17a activityassessed by the buserelin test. Androstenedione levels wereunchanged and the reduction of free testosterone levels wasconsidered to be a consequence of the increase of SHBG levels.However, a higher dose of metformin and/or longer duration ofadministration might had resulted in significant reductions inother adrogens too, (Diamanti-Kandarakiset al., 1998).
Regarding the buserelin test after metformin treatment, wesuggest that the administration of dexamethasone (0·5 mg fourtimes daily) for four days before the test, could have aggravatedinsulin resistance and ameliorated the improvement ofmetformin on hyperinsulinaemia at that particular time.Subsequently, this event might have been responsible for theunchanged P450c17a response. Thus, it would have been ofinterest to measure insulin levels just before buserelin wasadministered.
Lastly, from our clinical experience metformin has proved tobe sufficient, even as monotherapy, in some lean and obesewomen with PCOS, in terms of improvement metabolic profile,menstruation and fertility.
E. Diamanti-Kandarakis, C. Kouli,T. Tsianateli and A. Bergiele
University of Athens Medical School, 1st Department ofMedicine, Endocrine Section, Laiko General Hospital, Greece
References
Diamanti-Kandarakis, E., Kouli, C., Tsianateli, T. & Bergiele, A.(1998) Therapeutics effects of metformin on insulin resistance andhyperandrogenism in polycystic ovary syndrome.European Journalof Endocrinology, 138, 269–274.
Nestler, J., E. & Jacubowicz, D.J. (1997) Lean women with polycysticovary syndrome respond to insulin reduction with decreases inovarian P450c17a activity and serum androgens.Journal of ClinicalEndocrinology and Metabolism, 82, 4075–4079.
Unluhizarci, K., Kelestimur, F., Bayram, F., Sahin, Y. & Tutus, A.(1999) The effects of metformin on insulin resistance and ovariansteroidogenesis in women with polycystic ovary syndrome.ClinicalEndocrinology, 51, 231–236.
Velazquez, E.M., Mendoza, S., Hamer, T., Sosa, F. & Glueck, C.J.(1994) Metformin therapy in polycystic ovary syndrome reduceshyperinsulinemia, insulin resistance, hyperandrogenemia and systo-lic blood pressure, while facilitating normal menses and pregnancy.Metabolism, 43, 647–654.
Sirs, We would like to thank Dr. Vincenzoet al. and Diamanti-Kandarakis et al. for their comments. Polycystic ovarysyndrome (PCOS) reflects multiple potential aetiologies andvariable clinical presentations. As we have previously shown,PCOS is characterized by increased ovarian volume andabnormality in ovarian and adrenal androgen biosynthesislocalized to the 17a-hydroxylase/17-20 lyase reaction (Sahin &Kelestimur, 1993; U¨ nluhizarci et al., 1999a). In addition tochronic anovulation and hyperandrogenism, women with PCOSalso have insulin resistance with compensatory hyperinsuli-naemia (Sahinet al., 1997). Insulin resistance appears to have akey pathogenetic role in the hyperandrogenism and glucoseintolerance of PCOS (Barbieriet al., 1986; Legroet al., 1999).Thus, a number of studies were performed to test the effect ofamelioration of hyperinsulinaemia on the androgen excess ofPCOS. In our study, our aim was to determine whetherreduction of insulin levels by metformin would attenuate FSH,LH, 17-hydroxyprogesterone (17-OHP) and androstenedionehyperresponsiveness to buserelin testing in PCOS women(Unluhizarci et al., 1999b). We have performed buserelintesting after administration of dexamethasone. This issue iscritical since the extent to which adrenal steroid production iscontributing to the apparent ovarian 17-OHP levels wouldconfound the interpretation. The only way to be certain that the17-OHP level after buserelin testing is of ovarian origin wouldbe to pre-treat subjects with dexamethasone. We gavedexamethasone (0·5 mg four times a day, beginning 4 daysbefore the buserelin test and continuing through the day of thestudy) before and after the metformin treatment. If dexametha-sone is given only before but not after the metformin treatmentto eliminate the adrenal contribution of 17-OHP, as it waspreviously done in some studies, the interpretation of the resultsmay not be correct. Because adrenal contribution might havebeen eliminated before but not after the metformin therapy. For
244 Letters to the Editors
q 2000 Blackwell Science Ltd,Clinical Endocrinology, 52, 241–250

this reason, we have given dexamethasone before and after themetformin therapy.
We are completely agree with Vincenzoet al. thatdexamethasone may have some possible effects on insulinsensitivity. But, in our study we carried out the ITT and OGTTfor the evaluation of insulin resistance before the dexametha-sone treatment (unfortunately, this is not very clear in theexperimental protocol). For this reason, dexamethasonepretreatment did not affect insulin sensitivity in our patients.We have found that metformin therapy resulted in someimprovement in insulin sensitivity and reduced basal and postglucose load insulin levels. But, the area under the insulin curveafter metformin therapy was still greater than that observed innormal women. It means that metformin could not improve theinsulin sensitivity to the levels of healthy women. This partialeffect of metformin may be due to the smaller dose (1 g/day) weused or it may not be very potent drug for the treatment ofinsulin resistance of PCOS women. Ehrmannet al. (1997)sought to determine whether metformin, when given to obesenondiabetic women with PCOS, results in a reduction ofhyperinsulinemia while the body weight is maintained andwhether the reduction in insulin levels would attenuate the 17-OHP hyperresponsiveness to a GnRH agonist challenge. Theyfound that hyperinsulinemia and androgen excess in obesenondiabetic women with PCOS are not improved by theadministration of high dose metformin (2250 mg/day) and bothbasal and stimulated LH and FSH levels were unaffected bymetformin. In that study, dexamethasone therapy was notadministered to eliminate the adrenal contribution of 17-OHP(Ehrmannet al., 1997). It seems that the dose of metformin isnot responsible for the incomplete improvement in insulinresistance.
We are agree with Vincenzoet al. that glucocorticoids mayhave some effects on GnRH-LH axis. We did not compare theLH response to buserelin in PCOS women with the levelsobtained in healthy women. So, we are not able to make anycomment on the possible effects of dexamethasone onGnRH-LH axis.
Diamanti-Kandarakis et al. cite their recent report(Diamanti-Kandarakiset al., 1998) in which they treated 16obese women with PCOS with 1700 mg metformin on weightmaintaining diet for six months and found that metformintreatment increased glucose utilization and decreased freetestosterone and androstenedione levels. The biochemicalchanges were associated with the resumption of regularmenstruation in about 50% of the women. The reduction infree testosterone levels was clearly due to increased SHBGlevels after metformin treatment. In that study they did notevaluate the effects of metformin on androstenedione responsesto GnRH analogue testing. We have evaluated not only thebasal androstenedione levels but also peak and AUC andros-
tenedione levels after buserelin testing. On the other hand,Morin-Papunenet al. (1998) treated 20 obese patients withPCOS with 1500 mg metformin daily for 4–6 months. 68·8% ofthe women with menstrual disturbances experienced moreregular cycles during therapy. Free testosterone levelsdecreased significantly during the treatment but, there was nosignificant change in the levels of other sex steroids includingandrostenedione.
In conclusion, our study has shown that metformin therapyimproved menstrual disturbances in 25% of the women withPCOS and also resulted in some improvement in insulinsensitivity. Amelioration of hyperinsulinemia has no significanteffect on ovarian cytochrome P450c17a enzyme activity. But,we could not achieve a normal insulin sensitivity aftermetformin therapy. So, it is not possible to completely excludethe effects of hyperinsulinemia on P450c17a enzyme activity.We agree with Vincenzoet al. that glucocorticoid administra-tion before the tests performed to assess insulin resistance mayaffect insulin sensitivity. But the effects of glucocorticoidpretreatment for a short time on the GnRH-LH axis are not soclear and further studies are needed to clarify this issue.
Fahrettin Kelestimur*, Ku¨rsad Unluhizarci*,Fahri Bayram,* Yilmaz Sahin† and Ahmet Tutus‡
Departments of *Endocrinology, †Obstetrics and Gynecologyand‡Nuclear Medicine, Erciyes University School of
Medicine, Kayseri, Turkey
References
Barbieri, R.L., Makris, A., Randall, R.W., Daniels, G., Kistner, R.W. &Ryan, K.J. (1986) Insulin stimulates androgen accumulation inincubations of ovarian stroma obtained from women with hyperan-drogenism.Journal of Clinical Endocrinology and Metabolism, 62,904–910.
Diamanti-Kandarakis, E., Kouli, C., Tsianateli, T. & Bergiele, A.(1998) Therapeutic effects of metformin on insulin resistance andhyperandrogenism in polycystic ovary syndrome.European Journalof Endocrinology, 138, 269–274.
Ehrmann, D.A., Cavaghan, M.K., Imperial, J., Sturis, J., Rosenfield,R.L. & Polonsky, K.S. (1997) Effect of metformin on insulinsecretion, insulin action and ovarian steroidogenesis in women withpolycystic ovary syndrome.Journal of Clinical Endocrinology andMetabolism, 82, 524–530.
Legro, R.S., Kunselman, A.R., Dodson, W.C. & Dunaif, A. (1999)Prevalence and predictors of risk for type 2 diabetes mellitus andimpaired glucose tolerance in polycystic ovary syndrome: aprospective, controlled study in 254 affected women.Journal ofClinical Endocrinology and Metabolism, 84, 164–169.
Morin-Papunen, L.C., Koivunen, R.M., Ruokonen, A. & Martikainen,H.K. (1998) Metformin therapy improves the menstrual pattern withminimal endocrine and metabolic effects in women with polycysticovary syndrome.Fertility and Sterility, 69, 691–696.
Sahin, Y. & Kelestimur, F. (1993) 17-Hydroxyprogesterone response tobuserelin testing in the polycystic ovary syndrome.ClinicalEndocrinology, 39, 151–155.
Letters to the Editors 245
q 2000 Blackwell Science Ltd,Clinical Endocrinology, 52, 241–250

Sahin, Y., Ayata, D. & Kelestimur, F. (1997) Lack of relationshipbetween 17-hydroxyprogesterone response to buserelin testing andhyperinsulinemia in polycystic ovary syndrome.European Journalof Endocrinology, 136, 410–415.
Unluhizarci, K., Kelestimur, F., Sahin, Y. & Bayram, F. (1999a) Thetreatment of insulin resistance does not improve adrenal cytochromeP450c17a enzyme dysregulation in polycystic ovary syndrome.European Journal of Endocrinology, 140, 56–61.
Unluhizarci, K., Kelestimur, F., Bayram, F., Sahin, Y. & Tutus, A.(1999b) The effects of metformin on insulin resistance and ovariansteroidogenesis in women with polycystic ovary syndrome.ClinicalEndocrinology, 51, 231–236.
Carbenoxolone effects in congenital adrenalhyperplasia
Sirs, A recent paper by Irony & Cutler (1999) demonstrated thatthe addition of carbenoxolone to hydrocortisone therapy inpatients with 21-hydroxylase deficiency (congenital adrenalhyperplasia; CAH) potentiated the suppression of ACTH-dependent adrenal androgen excess and allowed withdrawal offludrocortisone. This was greeted enthusiastically by Speiser(1999) in her commentary. While the approach has some merit,we are concerned by several misconceptions which readers mayinfer from these data.
The principal difficulty in adjusting glucocorticoid replace-ment therapy in CAH is matching the timing of the highestglucocorticoid levels to suppress the highest ACTH levels inearly morning. To achieve this, many clinicians use syntheticglucocorticoids with a longer half-life than hydrocortisone(dexamethasone, prednisolone) given as so-called ‘reverse-phase’ therapy, with the highest dose at bedtime. Carbenox-olone inhibits conversion of cortisol to cortisone by 11b-hydroxysteroid dehydrogenase type 2 (11-HSD2) in the kidney,thereby impairing one of the major routes of metabolism ofcortisol and extending the half life of hydrocortisone (Stewartet al., 1990). This alone could account for enhanced suppres-sion of adrenal androgens by hydrocortisone when carbenox-olone is added.
The authors, however, refer to the expression of anotherenzyme, 11-HSD type 1, in the brain (specifically, in pituitary,hypothalamus and hippocampus). They suggest that inhibitionof this isozyme by carbenoxolone will prevent inactivation ofcortisol in these sites, thereby enhancing negative feedbacksuppression of ACTH. This interpretation would have beenwidely accepted in the early 1990s, but not today. Severalrecent studies confirm that—by contrast with the dehydrogen-ase activity of 11-HSD2—the 11-HSD type 1 isozyme is areductase, converting cortisone to cortisol in whole organs andintact cells (Jamiesonet al., 1995; Bujalskaet al., 1997),including neurones (Rajanet al., 1996). Inhibition of thisisozyme therefore results in lower local cortisol levels andpredicts impaired suppression of ACTH. Indeed, 11-HSD1
knockout mice have higher, not lower, circulating glucocorticoidconcentrations (Kotelevtsevet al., 1997).
Similar concerns apply to peripheral targets for glucocorti-coid action. The authors imply that carbenoxolone mayexacerbate glucocorticoid-induced insulin resistance by inhi-biting 11-HSDs in liver, fat and skeletal muscle; in fact, insulinsensitivity isenhancedby carbenoxolone in man (Walkeret al.,1995) and in 11-HSD1 knockout mice (Kotelevtsevet al.,1997). Indeed, this insulin-sensitising effect may contribute toreducing the androgen excess (Dunaifet al., 1996). In othertissues, however, carbenoxolone may not be beneficial. 11-HSD2 is expressed in lung, colon and vascular endothelium,sites where enhanced cortisol action is undesirable. Preliminarydata suggests that both 11-HSD isozymes are expressed inhuman bone where they may regulate both glucocorticoid-dependant differentiation and bone resorption. Again, enhancedcortisol action within bone could have detrimental effects in thelong-term. Similarly it would be unwise to use carbenoxolonein patients seeking fertility, since inhibition of 11-HSD2 in fetaltissues including the placenta, may result in enhanced exposureof the fetus to maternal cortisol. This may impair fetal growthand ‘programme’ future diseases such as diabetes andcardiovascular disease (Lindsayet al., 1996a, 1996b; Shamset al., 1998).
Finally, a key premise of this novel approach in CAH is thatcarbenoxolone could substitute for fludrocortisone as miner-alocorticoid replacement. We believe this is a potentiallydangerous strategy. Because of the 100–1000 fold higherconcentrations of cortisol than aldosterone, patients withimpaired 11-HSD2 have dramatic mineralocorticoid excesswhich, in its congenital form, is often fatal in childhood(Stewart & Krozowski, 1999). Thus the risks of mineralo-corticoid excess with chronic carbenoxolone/hydrocortisonetherapy in the long-term is high, and titration of carbenoxoloneto a dose which only permits 1/100th to 1/1000th of cortisol toreach mineralocorticoid receptors is unlikely to be feasible.
The concept that the modulation of the tissue metabolism ofcortisol can be used therapeutically to alter glucocorticoidhormone action is both exciting and achievable. Other enzymesin addition to the 11-HSDs such as 5a-reductase and 20-HSDmay be other worthwhile therapeutic targets. At present,however, there are no specific compounds which are selectiveinhibitors of 11-HSD1 or 11-HSD2. Carbenoxolone is certainlyan inhibitor of both 11-HSD isozymes and a critical appraisal ofthe functional significance of 11-HSD isozyme expression inmany human tissues is required before sanctioning the use ofcarbenoxolone instead of fludrocortisone in patients with CAH.
Brian R. Walker* and Paul M. Stewart†*Department of Medical Sciences, University of Edinburgh,Western General Hospital, Edinburgh and†Department of
Medicine, University of Birmingham, Birmingham, UK
246 Letters to the Editors
q 2000 Blackwell Science Ltd,Clinical Endocrinology, 52, 241–250

References
Bujalska, I.J., Kumar, S. & Stewart, P.M. (1997) Does central obesityreflect ‘Cushing’s disease of the omentum’?Lancet, 349, 1210–1213.
Dunaif, A., Scott, D., Finegood, D., Quintana, B. & Whitcomb, R.(1996) The insulin-sensitising agent troglitazone improves metabolicand reproductive abnormalities in the polycystic ovary syndrome.Journal of Clinical Endocrinology and Metabolism, 81, 3299–3306.
Irony, I. & Cutler, G.B. (1999) Effect of carbenoxolene on the plasmarenin activity and hypothalamic-pituitary-adrenal axis in congenitaladrenal hyperplasia due to 21-hydroxylase deficiency.ClinicalEndocrinology, 51, 285–291.
Jamieson, P.M., Chapman, K.E., Edwards, C.R.W. & Seckl, J.R. (1995)11b-Hydroxysteroid dehydrogenase is an exclusive 11b-reductase inprimary cultures of rat hepatocytes: effect of physicochemical andhormonal manipulations.Endocrinology, 136, 4754–4761.
Kotelevtsev, Y.V., Holmes, M.C., Burchell, A., Houston, P.M., Scholl,D., Jamieson, P.M., Best, R., Brown, R.W., Edwards, C.R.W., Seckl,J.R. & Mullins, J.J. (1997) 11b-Hydroxysteroid dehydrogenase type1 knockout mice show attenuated glucocorticoid inducible responsesand resist hyperglycaemia on obesity and stress.Proceedings of theNational Academy of Sciences USA, 94, 14924–14929.
Lindsay, R.S., Lindsay, R.M., Edwards, C.R.W. & Seckl, J.R. (1996a)Inhibition of 11b-hydroxysteroid dehydrogenase in pregnant rats andthe programming of blood pressure in the offspring.Hypertension,27, 1200–1204.
Lindsay, R.S., Lindsay, R.M., Waddell, B.J. & Seckl, J.R. (1996b)Prenatal glucocorticoid exposure leads to offspring hyperglycaemiain the rat: studies with the 11b-hydroxysteroid dehydrogenaseinhibitor carbenoxolone.Diabetologia, 39, 1299–1305.
Rajan, V., Edwards, C.R.W. & Seckl, J.R. (1996) 11b-Hydroxysteroiddehydrogenase in cultured hippocampal cells reactivates inert 11-dehydrocorticosterone, potentiating neurotoxicity.Journal ofNeuroscience, 16, 65–70.
Shams, M., Kilby, M.D., Somerset, D.A., Howie, A.J., Gupta, A.,Wood, P.J., Afnan, M. & Stewart, P.M. (1998) 11b-hydroxysteroiddehydrogenase type 2 in human pregnancy and reduced expression inintrauterine growth restriction.Human Reproduction, 13, 799–804.
Speiser, P.W. (1999) Toward better treatment of congenital adrenalhyperplasia.Clinical Endocrinology, 51, 273–274.
Stewart, P.M., Wallace, A.M., Atherden, S.M., Shearing, C.H. &Edwards, C.R.W. (1990) Mineralocorticoid activity of carben-oxolone: contrasting effects of carbenoxolone and liquorice on11b-hydroxysteroid dehydrogenase activity in man.Clinical Science,78, 49–54.
Stewart, P.M., Krozowski, Z.S. (1999) 11b-hydroxysteroid dehydro-genase.Vitamins and Hormones, 57, 249–324.
Walker, B.R., Connacher, A.A., Lindsay, R.M., Webb, D.J. & Edwards,C.R.W. (1995) Carbenoxolone increases hepatic insulin sensitivity inman: a novel role for 11-oxosteroid reductase in enhancingglucocorticoid receptor activation.Journal of Clinical Endocrinologyand Metabolism, 80, 3155–3159.
Sirs, We want to thank Dr Walker and Prof. Stewart for theinsightful and up-to-date comments on our paper (Irony &Cutler, 1999). We would like to use this opportunity to addresssome of their concerns.
Regarding the first paragraph: the main idea, taken from thefindings in Stewartet al. (1990), that the rise in plasma half-life
of 11alpha 3Hcortisol in 6 volunteers given carbenoxolone is aresult of decrease in the kidney dehydrogenase activity, doesnot automatically imply an increased systemic glucocorticoidactivity. One of our stated objectives was to determine if thereduced clearance of cortisol (and the increase in urine freecortisol noted) is a local phenomenon in the kidney or if there isa ‘spillover’ of cortisol into the systemic circulation. We couldnot detect an elevation in serum cortisol levels, and our attemptto detect biological effects of increased circulating cortisolwere unsuccessful except for the slight decrease in TRH-induced TSH secretion. On the other hand, our preliminaryobservations cannot answer with certainty that suppression ofadrenal androgens did not occur simply because oral hydro-cortisone doses had prolonged (and potentiated) effects on theseas well as in other cortisol targets. Long-term, well-designedstudies are needed in order to address this concern.
In the second paragraph, Dr Walker and Prof. Stewartmention the recent studies by Jamiesonet al. (1995) (using rathepatocytes) and Bujalskaet al. (1997) (using omental fat),which do not describe or predict the 11b hydroxysteroiddehydrogenase-1 activity in the central nervous system. Thepaper published by Rajanet al. (1996), however, deservescloser attention: in thisin vitro study, utilizing rodenthippocampal cell cultures, the observed effect of carbenox-olone in intact cells was 11b reduction, but when the cells werehomogenized, they exhibited both dehydrogenase and reduc-tase activities. One should be careful about extrapolating thesefindings to the clinical trials in humans; the predominantenzymatic direction is not known in the human anteriorpituitary gland, or whether the enhanced cortisol activitywould modulate local hypothalamic CRH and/or vasopressinsecretion, with suppression of ACTH. The fact that the naturalhuman model of our experimental condition, i.e. the patientswith apparent mineralocorticoid excess, exhibit suppressedplasma levels of ACTH (DiMartino-Nardiet al., 1987) can beinterpreted as a genetic deficiency of pituitary 11b dehydro-genase activity. Also, as an important consideration, thepatients with apparent mineralocorticoid excess syndrome donot manifest any glucocorticoid excess features, only thoserelated to mineralocorticoid excess.
In the third paragraph, Dr Walker and Prof. Stewart focus onperipheral targets of glucocorticoid action. We did not implythat carbenoxolone exacerbates glucocorticoid-induced insulinresistance by inhibiting the 11b hydroxysteroid dehydrogenase-1 in the liver, fat and skeletal muscle, as their statementsuggests. Our data (in Table 2) indicate the opposite: a slightdecrease in fasting insulin levels, while maintaining the samefasting glucose levels during phase 2 (when carbenoxolone wasadded), suggesting enhanced insulin sensitivity. Therefore ourdata agree with previously publishedin vivo (Walker et al.,1995) andin vitro (Kotelevtsevet al., 1997) studies. We did not
Letters to the Editors 247
q 2000 Blackwell Science Ltd,Clinical Endocrinology, 52, 241–250

mention in our conclusions that the insulin-sensitizing effect ofcarbenoxolone is another potential reason for the observedreduction in adrenal androgen production. In our view thiswould be a ‘desirable’ side effect, in addition to the ACTH-suppressing decrease in adrenal androgens.
Dr Walker and Prof. Stewart are also concerned about 11b
hydroxysteroid dehydrogenase-2 inhibition in (a) lung, (b)colon, (c) vascular endothelium, (d) bone and (e) placenta bycarbenoxolone. We would like to address each specific targetorgan concern separately:
a) Local glucocorticoid excess in the lung, by use ofglucocorticoid inhalers in asthma, has not been shown, forthe most part, to suppress the hypothalamic-pituitary-adrenal axis, exert other systemic glucocorticoid activity,or have harmful effects on lung function;
b) In the colonic mucosa, it is desirable to obtain enhancedmineralocorticoid activity, similar to what we strive to achieveat the kidney level (and colon) by using fludrocortisone, inboth patients with the salt-losing and the non salt-losing forms.
c) We could not demonstrate any negative effects uponvascular endothelium (no increase in mean blood pressure,no increase in serum calcium levels), and we infer that anincreased insulin sensitivity (as we discussed previously)might be beneficial to the vascular endothelium. Althoughthere were no changes in blood pressure or other risk factorsfor vascular damage, these short-term studies cannot addressthe possibility of adverse vascular effects of carbenoxolone.
d) We do not have information about the preliminary data thatboth 11b hydroxysteroid dehydrogenase isozymes arepresent in bone tissue. We agree that ‘enhanced glucocorti-coid action within the bone could have detrimental effects inthe long-term’. However, our data reveal unchangedmarkers of bone turnover and positive nitrogen balance.Additionally, calcium excretion and serum calcium levelsdid not change with the addition of carbenoxolone.
e) We agree that carbenoxolone should pose risks if used infemale patients during their reproductive years. If carbe-noxolone can cross the placenta, it is reasonable to predictthat fetal growth retardation and future metabolic anddevelopmental problems may occur. The British NationalFormulary (1991), in its chapter on ulcer-healing drugs,does not mention pregnancy as a contra-indication forcarbenoxolone, as it is very explicit about contra-indicateduse of the synthetic prostaglandin misoprostol duringpregnancy, in the same chapter. However, although ourpilot study was conducted in adult patients with CAH, amajor potential therapeutic use of carbenoxolone lies in thegrowing child and adolescent.
In the fourth paragraph, Dr Walker and Prof. Stewartexpress caution about the difficulty in titrating adequately
carbenoxolone doses, in order to avoid the catastrophicmineralocorticoid effects seen in children with apparentmineralocorticoid excess syndrome. Although we share thisconcern, our data, with the exception of mild hypokalemiashowed mineralocorticoid activity that is similar to thatachieved with current doses of fludrocortisone. Finally, wenote that carbenoxolone, at average doses higher than the onesused in our study, has been in use for decades throughoutEurope and Canada, by thousands of patients, for the treatmentof peptic ulcer disease.
Ilan Irony* and Gordon B. Cutler, Jr.†*Suburban Primary Care Physicians,
Bethesda MD and†Lilly Research Laboratories,
Indianapolis IN, USA
References
Bujalska, I.J., Kumar, S. & Stewart, P.M. (1997) Does central obesityreflect ‘Cushing’s disease of the omentum’?Lancet, 349, 1210–1213.
DiMartino-Nardi, J., Stoner, E., Martin, K., Balfe, J.W., Jose, P.A. &New, M.I. (1987) New findings in apparent mineralocorticoid excess.Clinical Endocrinology, 27, 49–62.
Irony, I. & Cutler, G.B. (1999) Effect of carbenoxolone on the plasmarenin activity and hypothalamic-pituitary-adrenal axis in congenitaladrenal hyperplasia due to 21-hydroxylase deficiency.ClinicalEndocrinology, 51, 285–291.
Jamieson, P.M., Chapman, K.E., Edwards, C.R.W. & Seckl, J.R. (1995)11b-hydroxysteroid dehydrogenase is an exclusive 11b reductase inprimary cultures of rat hepatocytes: effect of physicochemical andhormonal manipulations.Endocrinology, 136, 4754–4761.
Kotelevtsev, Y.V., Holmes, M.C., Burchell, A., Houston, P.M., Scholl,D., Jamieson, P.M., Best, R., Brown, R.W., Edwards, C.R.W., Seckl,J.R. & Mullins, J.J. (1997) 11b hydroxysteroid dehydrogenase type 1knockout mice show attenuated glucocorticoid inducible responsesand resist hyperglycaemia on obesity and stress.Proceedings of theNational Academy of Sciences USA, 94, 14924–14929.
Rajan, V., Edwards, C.R.W. & Seckl, J.R. (1996) 11b hydroxysteroiddehydrogenase in cultured hyppocampal cells reactivate inert 11-dehydrocorticosterone, potentiating neurotoxicity.Journal ofNeuroscience, 16, 65–70.
Stewart, P.M., Wallace, A.M., Atherden, S.M., Shearing, C.H. &Edwards, C.R.W. (1990) Mineralocorticoid activity of carbenox-olone: contrasting effects of carbenoxolone and liquorice on 11b-hydroxysteroid dehydrogenase activity in man.Clinical Science, 78,49–54.
Walker, B.R., Connacher, A.A., Lindsay, R.M., Webb, D.J. & Edwards,C.R.W. (1995) Carbenoxolone increases hepatic insulin sensitivity inman: a novel role for 11-oxosteroid reductase in enhancingglucocorticoid receptor activation.Journal of Clinical Endocrinologyand Metabolism, 80, 3155–3159.
Screening for diabetes in obese women: comparisonbetween the New American Diabetes Associationand WHO criteria
Sirs, Recently the American Diabetes Association (ADA)
248 Letters to the Editors
q 2000 Blackwell Science Ltd,Clinical Endocrinology, 52, 241–250

adopted a new, lower, fasting plasma glucose level (>7·0 mmol/L) for the diagnosis of diabetes mellitus (ADA, 1997) andencourages the use of fasting glucose as the main diagnostic testrather than the 2 h OGTT glucose as currently recommended byWHO (WHO, 1985). The new ADA criteria also include a newcategory, impaired fasting glucose (IFG), which is described asbeing analogous to the category of impaired glucose tolerance(IGT) based on the 2 h OGTT result (ADA, 1997).
We compared the current WHO criteria with the new ADAfasting criteria in a consecutive series of 100 obese women(BMI >30 kg/m2, mean6SE¼ 38·66 0·7 kg/m2), aged 20–67years (mean6 SE¼ 41·86 1·5), without previous history ofdiabetes, who attended the Obesity Outpatient Clinic of 1stPropaedeutic Department of Internal Medicine of the Uni-versity of Athens. In all patients, fasting plasma glucose wasdetermined at 0800 h after an overnight fast, followed by astandard OGTT, measuring plasma glucose at 120 min after theadministration of a 75-g oral glucose load.
Using the new diagnostic criteria proposed by the ADA, 6cases of diabetes (6%) were identified, as compared with 10cases (10%) of diabetes when the WHO criteria were applied(Table 1). Of the latter cases, only 2 (20%) were also classifiedas having diabetes by the ADA criteria, 4 (40%) as having IFGand 4 (40%) as normal. Further, the prevalence of IGT by WHOcriteria was more than 2-fold higher than the prevalence of IFGbased on the ADA criteria (23%vs. 10%,P< 0·01 byx2 test).The overall prevalence of diabetes and related abnormalitieswas 16% using the new ADA criteria (diabetes plus IFG),which was less than half of that based on the WHO criteria(33%), a significant difference (P<0·01 by x2 test and byCramer’s V measure).
Thus, if the results of the OGTT had not been considered,4 out of 10 cases with diabetes (40%) and 17 out of 23 caseswith IGT (74%) would have been completely missed andcharacterised as having no abnormality in glucose tolerance(Table 1), a matter of concern because of the potential adverseeffects of hyperglycaemia on microvascular and macrovasculardisease. Further, 2 of the 6 cases with diabetes by ADA criteria(30%) had completely normal glucose tolerance, thus carrying a
diagnosis with important socioeconomic consequences, despitethe lack of longitudinal studies that examine whether theseindividuals would also be subject to increased risk for diabetes-associated chronic complications.
The discordance between the two sets of criteria inidentifying diabetes and IGT in obese individuals is indeedalarming (Table 1). Thek statistic to measure overall agreementbetween the two classification schemes was very low: 0·24(95% CI 0·16–0·32) (P<0·001). Thus, only 14·3% of thoseindividuals identified by either criterion as having diabetesmeet both. This is a much lower percentage from the 48%concordance reported in the US NHANES III population study(Harriset al., 1997), from the 45% concordance reported in theCardiovascular Health Study (Wahlet al., 1998), and from the28% concordance reported in the European DECODE study(DECODE Study Group, 1998). Interestingly, in the latterstudy, obesity was identified as the main confounder in theassociation between fasting and the 2 h glucose (DECODEStudy Group 1998, 1999).
In conclusion, although the use of fasting plasma glucose mayconsiderably simplify the screening procedures for diagnosis ofdiabetes in unaffected subjects, it failed to identify 80% ofdiabetes cases and 74% of impaired glucose tolerance in ourunselected population of obese women without known diabetes.
Constantine Tsigos*‡, Ioannis Ioannidis*,Sotirios A. Raptis†‡ and Nikolaos Katsilambros*‡*1st Propaedeutic Department of Internal Medicine,
University of Athens, Laiko Hospital, †2nd PropaedeuticDepartment of Internal Medicine, Research Institute and
Diabetes Centre, University of Athens, Evangelismos Hospitaland‡Hellenic National Centre for the Research, Prevention
and Treatment of Diabetes Mellitus and its Complications,Athens, Greece
References
American Diabetes Association (1997) Report of the Expert Committeeon the diagnosis and classification of diabetes mellitus.DiabetesCare, 7, 1183–1197.
DECODE Study Group on behalf of the European DiabetesEpidemiology Study Group (1998) Will new diagnostic criteria fordiabetes mellitus change phenotype of patients with diabetes?Reanalysis of European epidemiological data.British MedicalJournal, 317, 371–375.
DECODE Study Group on behalf of the European DiabetesEpidemiology Study Group (1999) Is fasting glucose sufficient todefine diabetes? Epidemiological data from 20 European countries.Diabetologia, 42, 647–654.
Harris, M.I., Eastman, R.C., Cowie, C.C., Flegal, K.M. & Eberhardt,M.S. (1997) Comparison of diabetes diagnostic categories in the USpopulation according to 1997 American Diabetes Association and1980–1985 World Health Organization diagnostic criteria.DiabetesCare, 7, 1859–1862.
Letters to the Editors 249
q 2000 Blackwell Science Ltd,Clinical Endocrinology, 52, 241–250
Table 1 Comparison of WHO and ADA diagnostic categories inobese women
1997 ADA criteria1985 WHO WHOcriteria Normal IFG Diabetes total
NGT 63 2 2 67IGT 17 4 2 23Diabetes 4 4 2 10ADA total 84 10 6 100

Wahl, P.W., Savage, P.J., Psaty, B.M., Orchard, T.I., Robbins, J.A. &Tracy, R.P. (1998) Diabetes in older adults: comparison of 1997American Diabetes Association classification of diabetes mellitus
with 1985 WHO classification.Lancet, 352, 1012–1015.WHO (1985)Diabetes Mellitus: Report of the a WHO Study Group.
Geneva, WHO, (Technical Report Service, no. 727).
250 Letters to the Editors
q 2000 Blackwell Science Ltd,Clinical Endocrinology, 52, 241–250





























