Embed Size (px)
Citation preview

CLINICAL REPORT
Catel–Manzke Syndrome: A Clinical ReportSuggesting Autosomal Recessive InheritancePelin Ozlem Simsek Kiper,* G€ulen Eda Utine, Koray Boduro�glu, and Yasemin AlanayPediatric Genetics Unit, Department of Pediatrics, Hacettepe University Faculty of Medicine, Ankara, Turkey
Received 22 March 2011; Accepted 1 June 2011
We describe a 3-month-old male infant with cleft palate, glos-
soptosis,micrognathia, andbilateral clinodactyly, an association
which is characteristic of Catel–Manzke syndrome. In addition,
the patient had ligamentous laxity in the knee which is a rare
finding of this syndrome. The mode of inheritance of Catel–Manzke syndrome is unknown. Most cases are thought to be
sporadic but the present patient with consanguinity between
the parents and a possibly affected sib provide support for
autosomal recessive inheritance. � 2011 Wiley-Liss, Inc.
Key words: Catel–Manzke syndrome; cleft palate; autosomal
recessive inheritance; Pierre Robin sequence
INTRODUCTION
Catel–Manzke syndrome is a rare genetic disorder characterized by
the association of Pierre Robin sequencewith digital abnormalities.
It was first described in 1961 byCatel who reported on a 6-week-old
male infant with cleft palate, glossoptosis, micrognathia, and an
accessory ossification center at the base of the proximal phalanx of
the index finger leading to bilateral clinodactyly. In 1966, Manzke
reported additional details on the same patient, noting that the
supernumerary ossification center was a distinct form of hyper-
phalangism. Further descriptions of cases involving finger anoma-
lies in associationwithPierreRobin sequence led to thedefinitionof
this condition,whichwas given thenameCatel–Manzke syndrome.
The mode of inheritance of this rare syndrome is still unknown.
Themajority of reported patients have beenmale but an increasing
number of female patients are being reported, making an X-linked
inheritance unlikely. The present patient has classical features of the
Catel–Manzke syndrome. In addition, he has ligamentous laxity in
the knee. The consanguinity between the parents and the presence
of another affected sib in the family suggest autosomal recessive
inheritance.
CLINICAL REPORT
A 3-month-old male infant was referred to the clinic with atypical
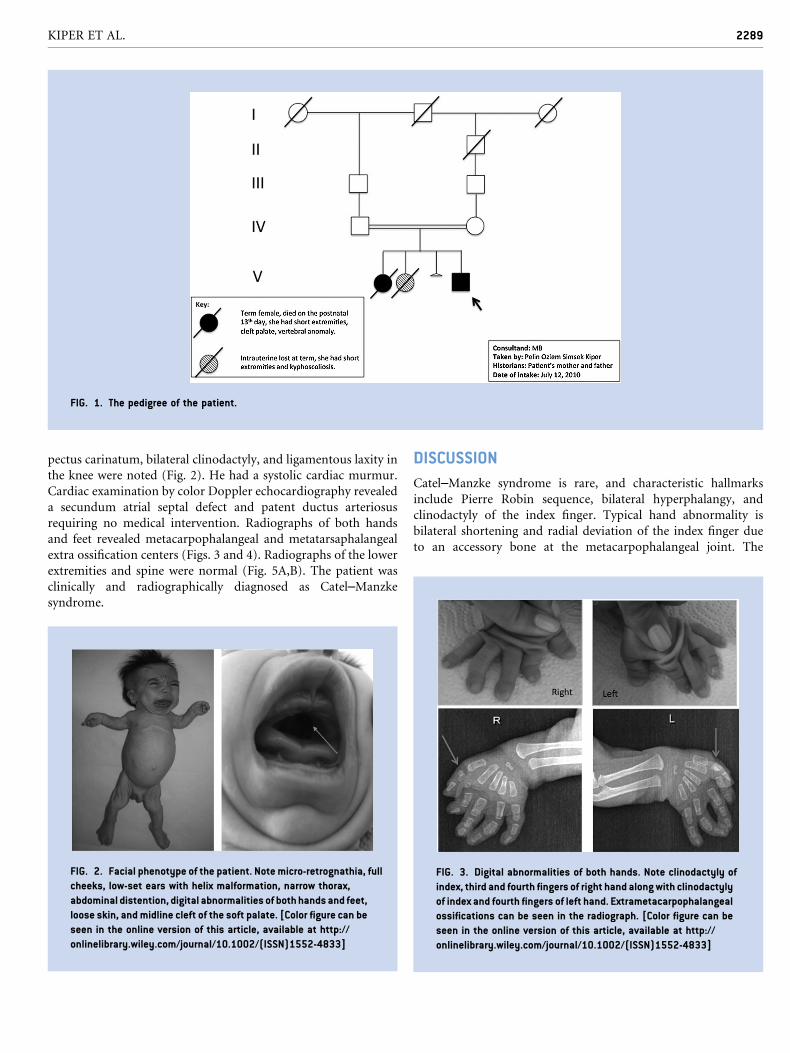
facial features and cleft palate. The patient was born to healthy
consanguineous parents (Fig. 1). Themother was 23 and father was
28 years old. Mother’s height and body weight were 163 cm (75th
centile) and 60 kg (75th centile), respectively and her head circum-
ference was 55 cm (mean); father’s height and body weight were
170 cm (25th centile) and 80 kg (90–97th centile), respectively. His
head circumference was 56 cm (mean). Both the mother and the
father did not have any systemic diseases. Their clinical examina-
tions including cardiovascular, respiratory, genitourinary, gastro-
intestinal, and nervous systemswere normal. The first pregnancy of
this couple was a term female who died on the postnatal 13th day.
She had short extremities, cleft palate, and vertebral anomalies. The
second pregnancy was an intrauterine demise lost at term. This
female fetus also had short extremities and kyphoscoliosis. The
third pregnancywas a spontaneous abortion. The fourth pregnancy
was the present patient. Prenatally, nuchal edema and polyhy-
dramnios were noted in the second trimester of the present
patient. Detailed fetal ultrasonography showed bilateral clinodac-
tyly, shortness in the extremities, and scoliosis. Amniocentesis
revealed 46, XY. Medical termination was suggested but the family
declined.
The patient was born at term, via normal delivery, with a birth
weight of 3,000 g (25th centile). No resuscitation was required
initially but later on he had cyanosis and difficulty in breathing. He
was then noted to have a cleft palate. After the improvement of
respiratory distress and stabilization of the patient, he was referred
to our hospital for further evaluation. At admission, body weight
was 2,800 g (<3rd centile), length was 44.5 cm (<3rd centile), and
head circumference was 37 cm (25th centile). He had eye contact
but no head control yet. On physical examination broad base to
nose, cleft palate, glossoptosis, micro-retrognathia, narrow thorax,
*Correspondence to:
Pelin Ozlem Simsek Kiper, M.D., Pediatric Genetics Unit, Hacettepe
University, _Ihsan Do�gramacı Children’s Hospital, Sihhiye 06100,
Ankara, Turkey. E-mail: [email protected]
Published online 10 August 2011 in Wiley Online Library
(wileyonlinelibrary.com).
DOI 10.1002/ajmg.a.34163
How to Cite this Article:Kiper POS, UtineGE, Boduro�gluK, Alanay Y.2011. Catel–Manzke syndrome: A clinical
report suggesting autosomal recessive
inheritance.
Am J Med Genet Part A 155:2288–2292.
� 2011 Wiley-Liss, Inc. 2288
pectus carinatum, bilateral clinodactyly, and ligamentous laxity in
the knee were noted (Fig. 2). He had a systolic cardiac murmur.
Cardiac examination by color Doppler echocardiography revealed
a secundum atrial septal defect and patent ductus arteriosus
requiring no medical intervention. Radiographs of both hands
and feet revealed metacarpophalangeal and metatarsaphalangeal
extra ossification centers (Figs. 3 and 4). Radiographs of the lower
extremities and spine were normal (Fig. 5A,B). The patient was
clinically and radiographically diagnosed as Catel–Manzke
syndrome.
DISCUSSION
Catel–Manzke syndrome is rare, and characteristic hallmarks
include Pierre Robin sequence, bilateral hyperphalangy, and
clinodactyly of the index finger. Typical hand abnormality is
bilateral shortening and radial deviation of the index finger due
to an accessory bone at the metacarpophalangeal joint. The
FIG. 1. The pedigree of the patient.
FIG. 2. Facial phenotype of the patient. Note micro-retrognathia, full
cheeks, low-set ears with helix malformation, narrow thorax,
abdominal distention, digital abnormalities of both hands and feet,
loose skin, and midline cleft of the soft palate. [Color figure can be
seen in the online version of this article, available at http://
onlinelibrary.wiley.com/journal/10.1002/(ISSN)1552-4833]
FIG. 3. Digital abnormalities of both hands. Note clinodactyly of
index, third and fourth fingers of right hand along with clinodactyly
of index and fourth fingers of left hand. Extrametacarpophalangeal
ossifications can be seen in the radiograph. [Color figure can be
seen in the online version of this article, available at http://
onlinelibrary.wiley.com/journal/10.1002/(ISSN)1552-4833]
KIPER ET AL. 2289

hyperphalangism in Catel–Manzke syndrome is a distinct form of
hyperphalangism as Manzke first discussed in 1966. The present
patient withmicrognathia, glossoptosis, cleft palate, and associated
upper respiratory obstruction, together with digital defect readily
fulfills the diagnostic criteria for Catel–Manzke syndrome.
There are over 30 reported cases of Catel–Manzke syndrome,
some of which do not fulfill the criteria of this specific syndrome
[Manzke et al., 2008]. Pierre Robin sequence, which is an associated
finding in about 80% of cases, is normally a combination of
micrognathia and glossoptosis, with or without cleft palate
[Sheffield et al., 1987]. Not all patients affected with Catel–Manzke
Manzke syndrome show the full range of this sequence [Thompson
and Winter, 1986; Puri and Phadke, 2003; Clarkson et al., 2004;
Manzke et al., 2008]. Therefore, we agree withManzke et al. [2008]
that the term ‘‘palato-digital syndrome’’ is incorrect and should
be replaced by the term ‘‘micrognathia-digital syndrome’’ if an
alternative to Catel–Manzke is to be preferred. The present patient
had cleft palate, micrognathia, and glossoptosis typically showing
the full range of Pierre Robin sequence.
About 70% of the patients with Catel–Manzke syndrome are
known to have additional congenital abnormalities [Manzke et al.,
2008]. Congenital heart defects are reported in 40% of cases [Puri
and Phadke, 2003]. Our patient had secundum atrial septal defect
and patent ductus arteriosus requiring no medical intervention.
He also had pectus carinatum. Thoracic deformities, such as pectus
carinatum and pectus excavatum, have been reported along with
congenital heart defects in Catel–Manzke syndrome [Stevenson
et al., 1980].Kneedislocationand talipes equinovarus in association
with Pierre Robin sequence were described before [Thompson and
Winter, 1986].Thepresent patient alsohadprominent ligamentous
laxity specifically in the knee.
Skeletal abnormalities such as index delta phalanx, clinodactyly
of the fifth finger, and an extra delta phalanx at the base of the
third phalanx are very common and among the hallmarks of this
syndrome. Metatarsal and toe abnormalities, bifurcation of first
metatarsal, hypoplastic proximal phalanx hallus, or short toes can
also be seen. There is resemblance between the hyperphalangy
phenotype in Catel–Manzke syndrome and isolated brachydactyly
type C which is caused by GDF5 mutations in the majority
of patients. However sequencing of three genes involved in
brachydactyly including GDF5, BMPR1B, NOG revealed no caus-
ative mutations in three Catel–Manzke patients [Manzke et al.,
2008].
Desbuquois dysplasia (DBQD), which is characterized by severe
prenatal andpostnatal growth retardation (<�5 SD), short stature,
joint laxity, progressive scoliosis, and advanced carpal ossification
with a delta phalanx should be kept in mind in the differential
diagnosis. DBQD shares some of the clinical features, such as hand
and foot changes and cleft palate, with Catel–Manzke syndrome.
The main radiological features in DBQD are short long bones with
metaphyseal splay, a ‘‘swedish key’’ appearance of the proximal
femur (exaggerated trochanter), and advanced carpal and tarsal
bone age with a delta phalanx [Desbuquois et al., 1966; Faivre et al.,
2004] which were not present in this patient. Figure 5A,B demon-
strate normal proximal femora and absence of platyspondyly.
Several distinct mutations in the Calcium-Activated Nucleotidase
1 gene (CANT1) have been identified in individuals with DBQD
[Huber et al., 2009; Faden et al., 2010]. However,CANT1mutation
analysis was negative in a patient with Catel–Manzke syndrome
[Cormier-Daire, personal communication].
TThe occurrence of several affected individuals within families
suggests an underlying genetic cause but the mode of inheritance is
still unclear. The rarity of female patients initially reported
suggested an X-linked pattern of inheritance. However, an increase
in the number of reported female patients makes this unlikely.
Although most cases are sporadic, five familial cases with various
relationships have been reported so far [Manzke et al., 2008]. No
chromosomal abnormalities were described in the reported cases.
The present patient and his family also represent one of the familial
cases. The first-born baby of this couple had short extremities, cleft
palate, and vertebral anomaly. It is not known whether the first-
born had the typical finger anomalies but it is likely that she had the
Catel–Manzke syndrome, which would suggest an autosomal
recessive inheritance.
The second pregnancy of this couple, which resulted in utero
exitus at term, also had short extremities andkyphoscoliosis, butwe
have no information onwhether cleft palate or finger anomaly were
observed. In the earlier literature an infant with Catel–Manzke
syndrome was reported whose stillborn brother had Pierre Robin
sequence and atrial septal defect, but it was not known whether he
had the typical finger abnormality or not [Gewitz et al., 1978]. The
authors suggested that the latter sib also had the Catel–Manzke
FIG. 4. Digital abnormalities of both feet. Note deviation and
clinodactyly of the fifth toes of both feet. There is a wide gap
between the fourth and the fifth toes. Extrametatarsal
ossifications can be seen in the radiograph. [Color figure can be
seen in the online version of this article, available at http://
onlinelibrary.wiley.com/journal/10.1002/(ISSN)1552-4833]
2290 AMERICAN JOURNAL OF MEDICAL GENETICS PART A

syndrome, which would suggest autosomal or X-linked recessive
inheritance [Gewitz et al., 1978].
The family andpatientwe report onherewith consanguinity and
affected siblings of both genders support previous suggestions of
autosomal recessive mode of inheritance. Until the underlying
molecular mechanism is clear, we suggest caution when counseling
families with a single affected child.
REFERENCES
Clarkson JH, Homfray T, Heron CW, Moss AL. 2004. Catel–Manzkesyndrome: A case report of a female with severely malformed handsand feet. An extension of the phenotype or a new syndrome? ClinDysmorphol 13:237–240.
Desbuquois G, Grenier B, Michel J, Rossignol C. 1966. Nanisme chon-drodystrophique avec ossification anarchique et polymalformations chezdeux soeurs. Arch Fr Pediatr 23:573–587.
FadenM, Al-Zahrani F, Arafah D, Alkuraya FS. 2010. Mutation of CANT1causes desbuquois dysplasia. Am J Med Genet Part A 152A:1157–1160.
Faivre L, Cormier-Daire V, Young I, Bracq H, Finidori G, Padovani JP,Odent S, Lachman R, Munnich A, Maroteaux P, Le Merrer M. 2004.Long-term outcome in desbuquois dysplasia: A follow-up in four adultpatients. Am J Med Genet Part A 124A:54–59.
Gewitz M, Dinwiddie R, Yuille T, Hill E, Carter CO. 1978. Cleft palate andaccessory metacarpal of index finger syndrome: Possible familial occur-rence. J Med Genet 15:162–163.
Huber C, Benedicte O, Bertoli M, ChamiM, FradinM, Alanay Y, Al-GazaliLI, Ausems MGEM, Bitoun P, Cavalcanti DP, Krebs A, Merrer ML,Mortier G, Shafeghati Y, Superti-Furga A, Robertson SP, Goff CL, MudaAO, Paterlini-Brechot P, Munnich A, Cormier-Daire V. 2009. Identi-fication of CANT1mutations in desbuquois dysplasia. Am J HumGenet85:706–710.
Manzke H, Lehmann K, Klopocki E, Caliebe A. 2008. Catel–Manzkesyndrome: Two new patients and a critical review of the literature.Eur J Med Genet 51:452–465.
FIG. 5. A: Radiograph of frontal view of the lower extremities and spine. B: Radiograph of lateral view of the lower extremities and spine.
KIPER ET AL. 2291

Puri RD, Phadke SR. 2003. Catel–Manzke syndromewithout cleft palate: Acase report. Clin Dysmorphol 12:279–281.
Sheffield LJ, Reiss JA, Strohm K, Gilding M. 1987. A genetic follow-upstudy of 64 patients with the Pierre Robin complex. Am J Med Genet28:25–36.
Stevenson RE, Taylor HA, Burton OM, Hearn HB. 1980. A digito-palatalsyndrome with associated anomalies of the heart, face, and skeleton.J Med Genet 17:238–241.
Thompson EM, Winter RM, Williams MJ. 1986. A male infant with theCatel–Manzke syndrome anddislocatable knees. JMedGenet 23:271–274.
2292 AMERICAN JOURNAL OF MEDICAL GENETICS PART A





























