Embed Size (px)
Citation preview

Brain (2001), 124, 2383–2392
Cerebral amyloid angiopathy is a pathogenic lesionin Alzheimer’s disease due to a novel presenilin 1mutationB. Dermaut,1 S. Kumar-Singh,1 C. De Jonghe,1 M. Cruts,1 A. Lofgren,1 U. Lubke,2 P. Cras,2 R. Dom,5
P. P. De Deyn,4 J. J. Martin3 and C. Van Broeckhoven1
1Department of Molecular Genetics, Flanders Correspondence address: Professor Dr Christine VanInteruniversity Institute for Biotechnology, 2Laboratory of Broeckhoven, Department of Molecular Genetics,Neurobiology and 3Neuropathology, Department of University of Antwerp (UIA), Department of Biochemistry,Medicine, 4Department of Neurology, General Hospital Universiteitsplein 1, B-2610 Antwerpen, BelgiumMiddelheim, Born-Bunge Foundation, University of E-mail: [email protected], Antwerpen and 5Laboratory of Neuropathology,University of Leuven, Faculty of Medicine, Leuven,Belgium
SummaryThe dense-cored plaques are considered the pathogenictype of amyloid deposition in Alzheimer’s disease brainsbecause of their predominant association with dystrophicneurites. Nevertheless, in >90% of cases of Alzheimer’sdisease amyloid is also deposited in cerebral blood vesselwalls (congophilic amyloid angiopathy; CAA) but its rolein Alzheimer’s disease pathogenesis remains enigmatic.Here, we report a family (family GB) in which early-onset Alzheimer’s disease was caused by a novel presenilin1 mutation (L282V). This was unusually severe CAAreminiscent of the Flemish amyloid precursor protein(A692G) mutation we reported previously, which causesAlzheimer’s disease and/or cerebral haemorrhages. Infamily GB, however, the disease presented as typicalprogressive Alzheimer’s disease in the absence of strokes
Keywords: familial Alzheimer’s disease; clinicopathological study; presenilin; amyloid angiopathy
Abbreviations: Ab � antibody; Aβ � amyloid β; APP � amyloid precursor protein; CAA � congophilic amyloid angiopathy;MMSE � Mini-Mental State Examination; PSEN � presenilin; SPECT � single-photon emission computed tomography
IntroductionA proteolytic fragment of the amyloid precursor protein(APP), called amyloid β (Aβ), is deposited as extracellularamyloid plaques in the brain of Alzheimer’s disease patients.Amyloid plaques range from non-congophilic diffuse plaquesto congophilic compact and often core-containing plaques.The strong association of amyloid cored plaques and onlyexceptionally diffuse plaques with dystrophic neurites hassuggested that the dense-cored plaque is the pathogeniclesion in Alzheimer’s disease (Wisniewski and Terry, 1973;Wisniewski et al., 1989). Besides its deposition in diffuse
© Oxford University Press 2001
or stroke-like episodes. Similarly, neuroimaging studiesand neuropathological examination favoured a degener-ative over a vascular dementia. Interestingly, animmunohistochemical study revealed that, similar tocausing dense-cored amyloid plaques, CAA also appearedcapable of instigating a strong local dystrophic andinflammatory reaction. This was suggested by theobserved neuronal loss, the presence of tau- and ubiquitin-positive neurites, micro- and astrogliosis, and comple-ment activation. Together, these data suggest that, like thedense-cored neuritic plaques, CAA might represent apathogenic lesion that contributes significantly to theprogressive neurodegeneration that occurs in Alzheimer’sdisease.
and cored plaques, Aβ is also present in the walls of cerebralcapillaries, arterioles and arteries, and is called congophilicamyloid angiopathy (CAA). Although CAA is found in�90% of autopsied Alzheimer’s disease brains (Glenneret al., 1981), it is highly variable in severity and quantity.
The extent to which CAA plays a role in the progressionof typical Alzheimer’s disease is not known, but severe CAAis classically considered an important cause of intracerebralhaemorrhage, a prevailing feature in carriers of the Flemishand Dutch variants of APP. In the brains of patients with

2384 B. Dermaut et al.
hereditary cerebral haemorrhage with amyloidosis of theDutch type, caused by Dutch APP (E693Q) (Van Broeckhovenet al., 1990), severe CAA is present abundantly and causesrecurrent cerebral haemorrhages. In addition to CAA andcerebral haemorrhage, Flemish APP (A692G) patients(Hendriks et al., 1992) also present with Alzheimer’s diseasewith large cored plaques (Cras et al., 1998). However,cerebral haemorrhage is not limited to Dutch and FlemishAPP. Some presenilin (PSEN) mutations also appear to becapable of causing haemorrhagic strokes occasionally. In theVolga-German Alzheimer’s disease family (PSEN2 N141I)(Levy et al., 1995), severe CAA is a consistent feature andcerebral haemorrhage has been reported in one mutationcarrier (Nochlin et al., 1998).
However, most cases of hereditary and sporadicAlzheimer’s disease present with progressive dementia andCAA in the absence of its drastic consequences, such aslarge cerebral haemorrhages and infarcts. Nevertheless, it isdifficult to assess how and to what extent CAA per se maycontribute to the gradual progressive neurodegeneration seenin Alzheimer’s disease. Detailed clinical and neuro-pathological descriptions of typical cases of Alzheimer’sdisease with unusually severe CAA are therefore likely tocontribute to our understanding of the role of CAA inAlzheimer’s disease patients. Here we describe family GB,in which clinically typical early-onset Alzheimer’s diseasewith unusually severe CAA is associated with a novel PSEN1mutation (L282V). This paper reports its clinical, moleculargenetic, biochemical, neuropathological and immuno-histochemical characteristics.
Subjects and methodsSubjects and family ascertainmentFamily GB is a three-generation family with two probands(Fig. 1). Blood for DNA extraction and detailed clinicalinformation was available for both probands (III-1 and III-4),who were diagnosed independently in two university hospitalsin Belgium. Molecular genetic screening was performedwithout knowledge of a familial relationship between thetwo individuals. Brain biopsy and limited clinical informationwas available for individual II-5. Autopsy and neuro-pathological examinations were performed in III-4. Forindividuals I-1, II-1 and II-2, information on the diagnosisof dementia and age at death were provided by the spouseof proband III-1.
Genetic analysisPCR (polymerase chain reaction)-based screening of thecoding exons of PSEN1 was performed on genomic DNAby SSCP (single-strand conformation polymorphism) analysisand direct sequencing as described previously (Cruts et al.,1998). To confirm the presence of PSEN1 L282V in patientsand to screen 80 Belgian healthy control individuals, a PCR-
Fig. 1 Pedigree structure of the Belgian family GB. Squaresrepresent males, circles represent females, filled symbols representhereditarily affected individuals, the half-filled symbol representsa sporadically affected individual, slashed symbols representdeceased individuals and arrows represent probands. Romannumerals indicate generations and Arabic numerals individuals.
based mismatch RFLP (restriction fragment lengthpolymorphism) analysis method was developed (forwardmismatch primer 5�-CTCAGGAGAGCAAATGCAACCG-3�; reverse primer 5�-AGCAATTTATCGGGCAACTT-3�),allowing the detection of PSEN1 L282V by MspI digestion.Allele sharing analysis was performed by genotyping thePSEN1 flanking microsatellite markers D14S1028, D14S77,D14S1004, D14S1025 and D14S999 using published primers,one of which was labelled fluorescently. The alleles wereseparated on 6% polyacrylamide gel containing 8 M ureausing an ABI373A automated DNA sequencer (AppliedBiosystems, Foster City, Calif., USA). Apolipoprotein E(APOE) genotyping was performed as described (Wenhamet al., 1991).
Aβ secretionSite-directed mutagenesis was performed on the full-lengthPSEN1 cDNA (De Jonghe et al., 1999) cloned in vectorpCDNA5/FRT (Invitrogen, Carlsbad, Calif., USA) using theQuikChange site-directed mutagenesis system (Stratagene,La Jolla, Calif., USA) with primers 5�-CTCAGGAGAGAAA-TGAAACGGTTTTTCCAGCTCTCATTTAC-3� and 5�-GTAAATGAGAGCTGGAAAAACCGTTTCATTTCTCTCC-TGAG-3� to introduce the L282V mutation. Humanembryonic kidney Flp-In (Flp-In-293; Invitrogen) cells wereco-transfected with wild-type or L282V mutant PSEN1 cDNAcloned in pCDNA5/FRT and recombinant Flp recombinase,cloned in pOG44 (Invitrogen) using Lipofectamine (LifeTechnologies, Gaithersburg, Md., USA) according to themanufacturer’s instructions. Cells were selected for stableintegration of the recombinant plasmid in the cell genomeby resistance to 400 µg/ml hygromycin. Aβ42 concentrationsin eightfold concentrated 24-h conditioned media weremeasured by sandwich-type ELISA (enzyme-linkedimmunosorbent assay), using the Innotest β-amyloid 1–42

Presenilin 1 L282V and amyloid angiopathy 2385
(Innogenetics, Zwijnaarde, Belgium) and Aβ40 concentra-tions were measured in the same media with the humanβ-amyloid 1–40 ELISA (Biosource, Camarillo, Calif., USA).A two-tailed unpaired t-test was used to compare the Aβ42/Aβ40 ratio produced by the wild-type and mutanttransfectants.
HistopathologyA paraffin-embedded biopsy specimen of the frontal cortexwas available for case II-5. A post-mortem neuropathologicalstudy was performed on case III-4. Macroscopically, frontalcortical atrophy was noted in this case. Tissue was obtainedfrom the superior frontal gyrus, superior temporal gyrus,hippocampus, area striata and cerebellum. After fixation in4% formaldehyde, tissue was dehydrated and embedded inparaffin. From paraffin-embedded blocks, sections 10 µmthick were sliced and stained with classical histological stains,such as haematoxylin–eosin, cresyl violet, Bodian, Congored and thioflavin S.
ImmunohistochemistryImmunohistochemistry was performed on serial 4 µm sectionsfrom the temporal and frontal cortical regions. The followingantibodies (Ab) were used: 4G8 (Senetek, Maryland Heights,Md., USA; directed against amino acid residues 18–24 ofAβ); 6E10 (Senetek; raised against Aβ 1–17, recognizes Aβ5–11), JRF/AβN/11 (specific for N-terminus of Aβ, generatedagainst Aβ residues 1–5), JRF/cAb40/10 (specific for Aβ40),β JRF/cAb42/12 (specific for Aβ42) (Kumar-Singh et al.,2000), 22C11 (against N-terminal APP; Roche), AT8[Innogenetics; against abnormally phosphorylated PHF(paired helical filament)-tau], ubiquitin (Dako, Glostrup,Denmark), anti-glial fibrillary acidic protein (GFAP; Dako),CD68 (Dako, for microglia). For Aβ immunohistochemistry,sections were preincubated with 98% formic acid for 5 min.APP (22C11) and GFAP immunohistochemistry wasperformed on sections retrieved in citrate buffer (pH 6) and,before CD68 staining, sections were preincubated with 0.1%pepsin for 20 min at 37°C. Blocking sera (rabbit and goatsera), link antibodies (biotinylated rabbit anti-mouse andgoat anti-rabbit) and horseradish peroxidase–avidin–biotincomplex were used at recommended dilutions (Dako).Sections were immersed in 0.03% hydrogen peroxide inmethanol and incubated for 0.5 h to block endogenousperoxidase, and were then preincubated with normal seradiluted 1 : 5. Sections were incubated overnight (16 h) at4°C with the primary Ab, followed by 0.5 h incubations withfirst biotinylated secondary Ab and then by horseradishperoxidase-conjugated avidin–biotin diluted 1 : 1 : 100.Sections were finally treated with peroxidase substratesolution containing 0.01% hydrogen peroxide and 0.05%DAB (diaminobenzidine tetrahydrochloride), counterstainedwith Harris haematoxylin and coverslipped in DPX. Alldilutions were made in 0.1 M PBS (phosphate-buffered
saline) containing 0.1% bovine serum albumin.Immunohistochemistry, involving the detection of more thanone antigen, was done using species-specific or IgG subtype-specific secondary Ab conjugated directly with biotin,horseradish peroxidase, alkaline phosphatase or galactosidase(Southern Biotechnology, Birmingham, Ala., USA). This wasfollowed by colour development using one of the followingchromogens (Roche): DAB, AEC (3-amino-9-ethylcarbazole), fast red, BCIP/NBT (5-bromo-4-chloro-3-indolyl phosphate/nitroblue tetrazolium solution) andX-gal (5-bromo-4-chloro-3-indolyl-β-D-galactopyranoside),as described elsewhere (Kumar-Singh et al., 2000).
ResultsClinical, neuropathological and molecular genetic results aresummarized in Table 1.
Case historiesPatient III-1 (proband 1)The patient had a known history of hypertension. At the ageof 45 years the first symptoms of memory impairment werenoticed. Five years later the patient was referred to aneurologist because both at home and at the workplacenegligent and inaccurate behaviour was observed. At thattime, the patient was also noticed to react more slowly andwas having difficulty in comprehension. At neurologicalexamination, an insecure, adynamic and apathetic man withcognitive impairment and aphasia was observed. A Mini-Mental State Examination (MMSE) score of 21 out of 30was calculated. Cognitive deficits were most pronounced inshort-term recall, calculation, word finding and compre-hension. Further clinical examination revealed a slightlypropulsive and insecure shuffling gait, sporadic myoclonusin the hands, mild rigidity in the upper limbs and positivefrontal release signs. On MRI and CT scanning, cortical andsubcortical atrophy were observed, together with a widenedcisterna magna. Single photon emission computedtomography (SPECT) revealed a diffuse pattern ofhypoperfusion. EEG was characterized by short paroxysmsof generalized slow-wave activity that increased withhyperventilation. Six months later, marked anosognosia wasobserved. A MMSE of 18 out of 30 was calculated and itwas mentioned that the patient had become severely short-tempered. The extrapyramidal syndrome, as reflected in upperlimb cogwheel rigidity, bradykinesia and propulsive shufflinggait, had become more marked and was most prominent onthe right side. Severe paratonia of the lower limbs wasobserved. The primitive reflexes were still provokable. Therewas no evidence of a cerebellar syndrome as tests oncoordination remained normal. Nine months after initialexamination, the MMSE had dropped to 12 out of 30 despitesymptomatic treatment with tacrine. Clinical neurologicalexamination remained largely unaltered: a prominent frontal
2386 B. Dermaut et al.
Table 1 Clinical, neuropathological and genetic characteristics of family GB
Patient Clinical features Neuroimaging Age at onset Age at death Neuropathology Molecular(years) (years) genetics
II-2 Dementia – 47 54 – –II-5 Dementia Cortical atrophy (PEG) �53 57 Neuritic CP, NFT, –
severe CAA (B)III-1 Dementia, frontal Cortical and subcortical 45 54 – APOE ε3ε4
signs, extrapyramidal atrophy (CT, MRI), late PSEN1 L282Vsyndrome, epilepsy, periventricular WML (CT),myoclonus late right parieto-occipital
and left hemispherehypoperfusion (SPECT)
III-4 Dementia, mild Cortical and subcortical 41 49 Neuritic CP, NFT, APOE ε3ε4cerebellar syndrome atrophy (CT, MRI), early severe cerebral and PSEN1 L282V
and severe left hemisphere cerebellar CAA (A)hypoperfusion (SPECT)
PEG � pneumoencephalography; CP � cored plaques; NFT � neurofibrillary tangles; B � biopsy; A � autopsy;APOE � apolipoprotein E gene.
and extrapyramidal syndrome was still observed and thepatient had become completely helpless. About 2 years laterthe patient was admitted to the hospital because of recurrentepileptic seizure-like episodes, for which valproic acid wasprescribed. By then the patient had progressed into a nearlyvegetative state with generalized rigidity, hyperactive deeptendon reflexes with outspoken diffusion, strongly positiveprimitive reflexes and generalized myoclonus. At this stagea CT scan showed cerebellar and cerebral atrophy as wellas periventricular ischaemic white-matter lesions. SPECTdemonstrated diffuse hypoperfusion of the brain with tworegions of clearly decreased perfusion, in the right parieto-occipital region and the left hemisphere. Nine months later,9 years after disease onset, the patient died at the ageof 54 years.
There was a family history of dementia as both parents(II-1 and II-2) were reported to have been demented. Thefather (II-1) died at the age of 83 years and the mother (II-2)at 54 years.
Patient III-4 (proband 2)The first symptoms of cognitive impairment were noticed atthe age of 41 years. Four years later symptoms of confusedbehaviour and disorientation, as a result of already severememory impairment, were observed. In her job as amathematics teacher, she started to give all students maximumscores. At home financial problems had arisen and atneurological consultation a depressed, aphasic, apractic andobese woman who was disorientated in time and space wasobserved. Further examination revealed dysmetria and anMMSE score of 15 out of 30. In the main, deficits inorientation, concentration and short-term recall wereobserved. CT and MRI of the brain showed marked corticaland subcortical atrophy. SPECT showed a marked relativedecrease in radionucleotide uptake in the left parietal and
temporal lobe, extending to the putamen and to a lesserextent the nucleus caudatus; the left frontal lobe exhibitedminimal hypoperfusion. In the right hemisphere there was arelative decrease in perfusion located in the parietofrontalregion. EEG showed frontotemporal slow-wave activity. Adiagnosis of probable Alzheimer’s disease with a positivefamily history was made. Around 1 year later the patient hadfurther deteriorated into complete mutism, showing importantapraxia, dysdiadochokinesia, acalculia and decreased abilityin imprinting. SPECT showed that the hypoperfusion defecthad extended to the entire left hemisphere but was lesspronounced in the occipital region (Fig. 2). CT and EEGremained unaltered. The patient died at the age of 49 yearsand an autopsy was performed.
Patient II-5Limited clinical information was available for this patientfrom a neurological consultation at the age of 57 years. Thereport mentioned progressive mental deterioration that hadstarted at least 4 years earlier. The patient was noted to beconfused, disorientated in time and space, and apathetic.Severe recent memory impairment characterized bydifficulties in text comprehension, arithmetic and imprintingwas observed. Constructional apraxia was also seen. However,no other neurological symptoms were noted to accompanythe dementia syndrome. Pneumoencephalography showedmoderate ventricular dilatation and cortical atrophy. EEGshowed frontotemporal slow-wave activity. The patient wasdiagnosed as having Alzheimer’s disease. At this time a brainbiopsy was taken; it confirmed the diagnosis of Alzheimer’sdisease and the patient died the same year. The family historyrevealed dementia with early onset in the father (I-1).
Genetics and Aβ secretionSSCP analysis of exon 8-containing PCR fragments of PSEN1from both probands (III-1 and III-4) showed aberrant patterns.

Presenilin 1 L282V and amyloid angiopathy 2387
Table 2 Genotypes of PSEN1 flanking microsatellite markers in the 2 PSEN1 L282Vcarriers (III-1 and III-4)
D14S1028 D14S77 D14S1004 D14S1025 D14S999
III-1 223–231 213–205 192–194 174–143 238–246III-4 223–231 213–205 190–192 143–145 240–246
Allele sizes are given in base pairs and shared alleles are underlined.
Fig. 2 [99mTc]HMPAO scan image of patient III-4 showingmultiple regions of reduced perfusion in the left frontal, parietal,temporal and parieto-occipital regions, with extension of theperfusion defect to the right temporoparietal cortex.
Direct sequencing of the PCR fragments demonstrated aC→G transversion in exon 8 resulting in a Leu (CTT) to Val(GTT) substitution at codon 282 of PSEN1 (L282V). TheAPOE genotype was ε3ε4 for both patients. Genotype analysisof the polymorphic microsatellite markers D14S1028,D14S77, D14S1004, D14S1025 and D14S999, located nearPSEN1 (Cruts et al., 1995), showed that the two probandsshared at least one allele for each of these markers (Table 2).The mutation was absent in 160 Belgian controlchromosomes. Measurement of Aβ42 and Aβ40 levels inmedium of HEK-293 cell lines stably expressing mutant andwild-type PSEN1 cDNA showed a twofold increase inthe Aβ42/Aβ40 ratio for L282V compared with wild type(6.6 � 0.5 versus 3.3 � 0.7%; P � 0.02).
Histopathology and immunohistochemistryMarked neuronal loss in all layers accompanied by corticalatrophy was noted in the superior temporal gyrus, superiorfrontal gyrus and area striata. Similarly, pyramidal cell lossand pycnotic neurones were evident in layer III of the
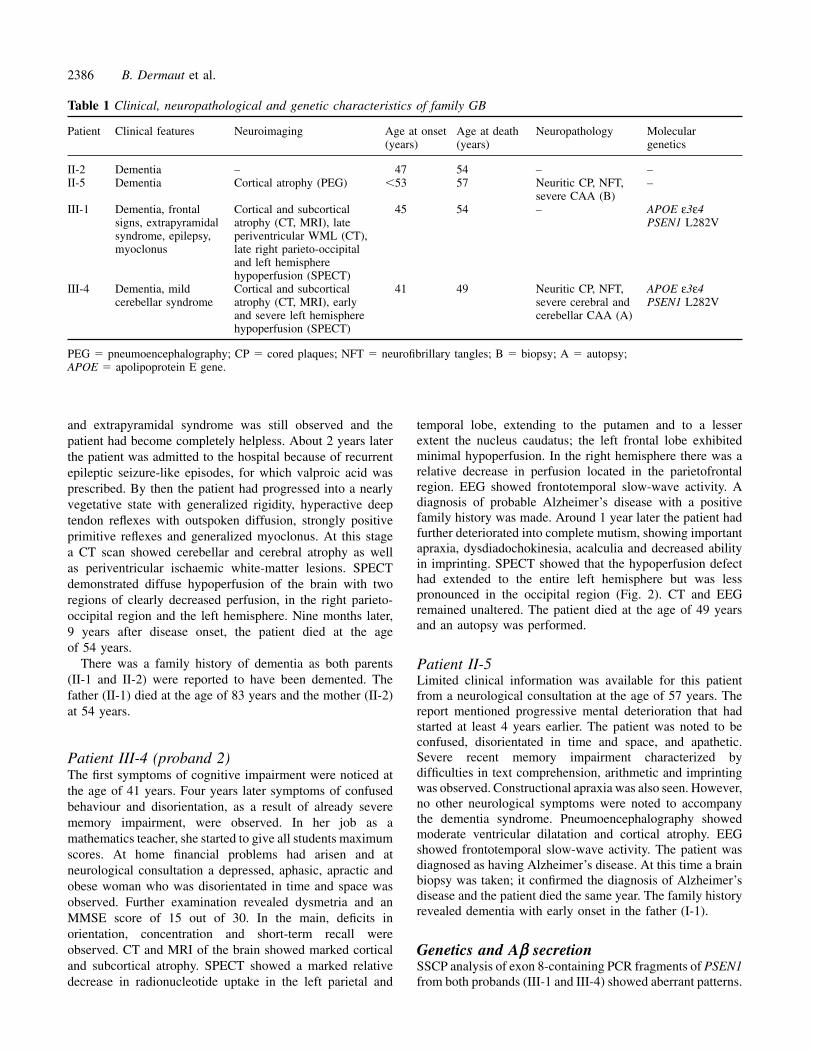
entorhinal cortex. Thioflavin S and Congo red staining of brainsections showed fluorescence and apple-green birefringenceunder polarized light, respectively, in the dense-cored plaquesand blood vessels. Neurofibrillary tangles, recognized bysilver stains and Ab for hyperphosphorylated tau, werenoted in the entorhinal cortex, fields CA1 and CA2 of thehippocampus and in the neocortex, mostly in associationwith neuritic amyloid plaques. Neuropil threads recognizedby AT8 were also abundant in all these regions, especiallyin the entorhinal cortex. Rarely, granulovacuolar degenerationwas evident in neocortical regions. In the molecular layer ofthe dentate gyrus, mild spongiosis was noticed together withamyloid plaques and CAA. The amyloid deposits wereabundantly present, both as dense-cored plaques and diffuseplaques, in all regions analysed. Double immuno-histochemistry showed that the dense-cored senile plaqueswere associated with astroglial proliferation and activatedmicroglia. Besides neuritic plaques, diffuse plaques(preamyloid) were also noted in all brain regions analysed,including the subpial region and cerebellum (Fig. 3).
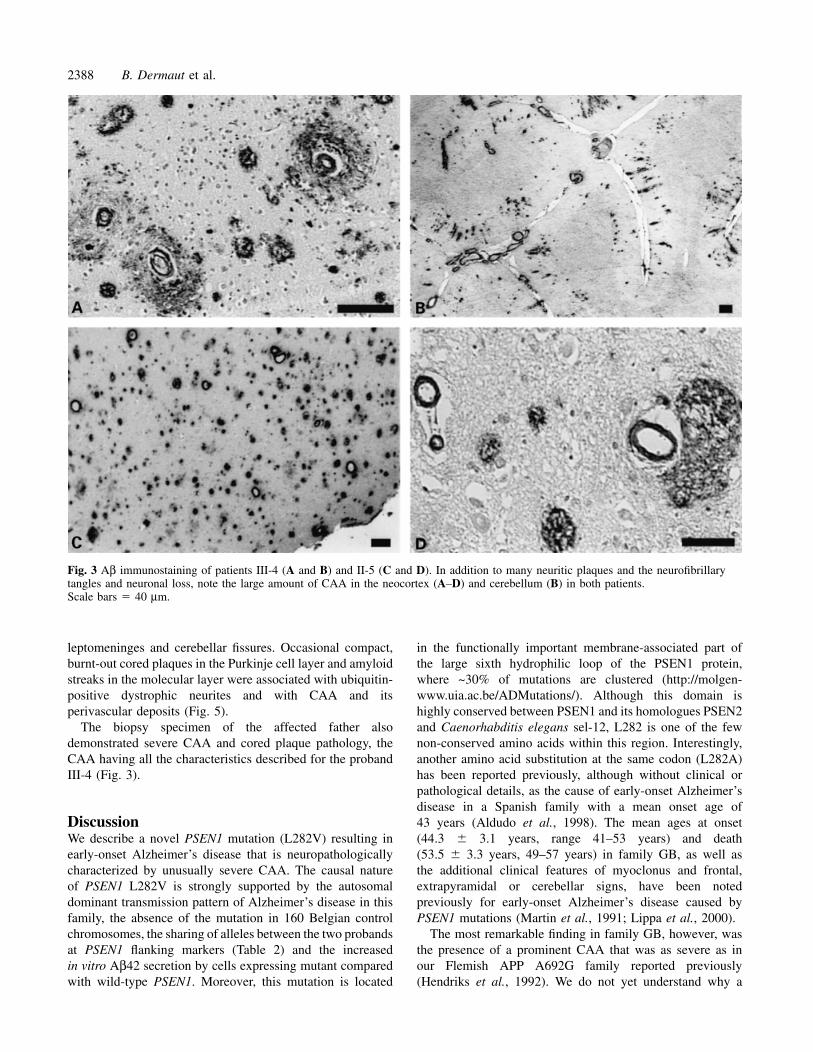
Staining serial neocortical and hippocampal sections forAb specific for C-termini Aβ40 and Aβ42, Aβ middleportion (4G8; Aβ 17–24) and Ab directed to the N-terminusdemonstrated that vascular amyloid and amyloid in thecompact cores was full-length Aβ40, whereas those in thediffuse plaques were N-truncated Aβ42 (Fig. 4).
Most interesting was the extensive CAA in all regionsanalysed, including the neocortex, hippocampus andcerebellum. 4G8 stained amyloid in thickened arteries andarterioles in a concentric fashion and most of the vesselswere associated with strong perivascular amyloid deposition.Staining for smooth-muscle actin demonstrated that smoothmuscle cells were lost from arteries. We noted all theassociated characteristics of dense-cored plaques with CAA,especially with its perivascular deposits. Such depositsshowed dystrophic neurites stained with ubiquitin and AT8as well as specific markers for astro- and microgliosis andcomplement activation. Co-staining sections with Congo redand microglial markers showed a strong association ofmicroglia with CAA. Severe to moderate white matter losswas present in the neocortical regions. Pathology in thecerebellum was severe and associated with focal loss ofPurkinje cells. A remarkable arrangement of linear streaksof amyloid arranged perpendicularly to the interfolial fissuresin the outer molecular layer of the cerebellum was associatedwith severe CAA that was present parenchymally and in the
2388 B. Dermaut et al.
Fig. 3 Aβ immunostaining of patients III-4 (A and B) and II-5 (C and D). In addition to many neuritic plaques and the neurofibrillarytangles and neuronal loss, note the large amount of CAA in the neocortex (A–D) and cerebellum (B) in both patients.Scale bars � 40 µm.
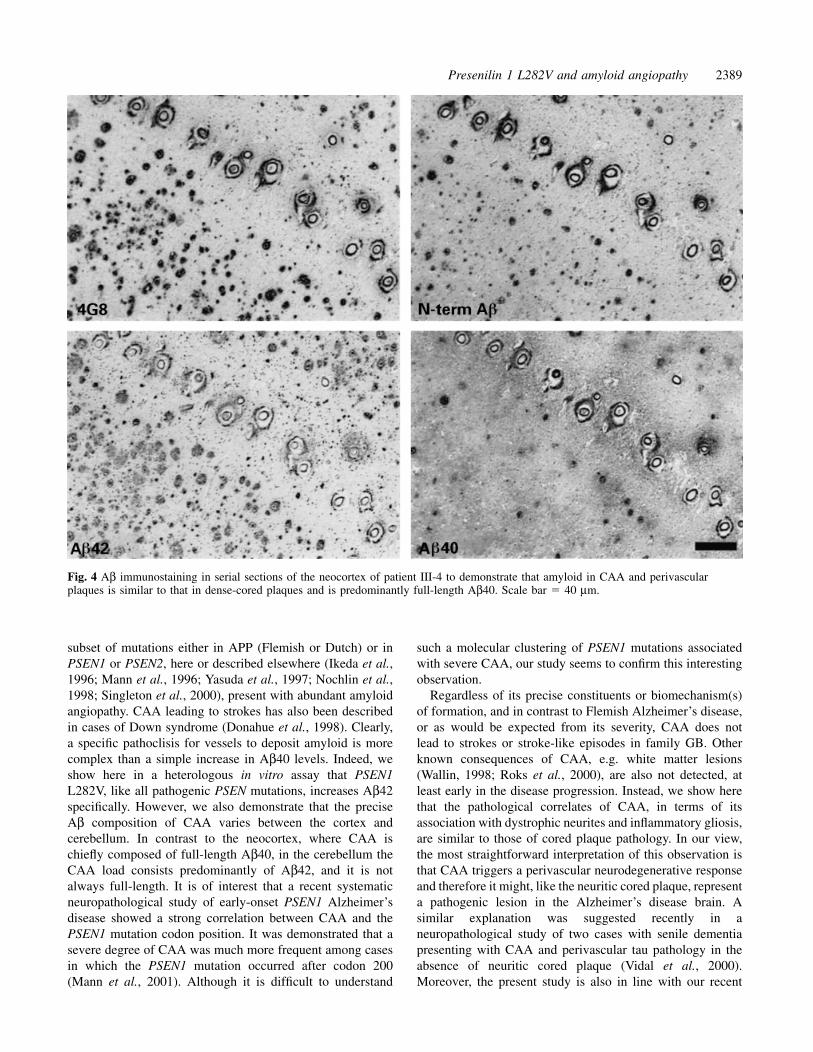
leptomeninges and cerebellar fissures. Occasional compact,burnt-out cored plaques in the Purkinje cell layer and amyloidstreaks in the molecular layer were associated with ubiquitin-positive dystrophic neurites and with CAA and itsperivascular deposits (Fig. 5).
The biopsy specimen of the affected father alsodemonstrated severe CAA and cored plaque pathology, theCAA having all the characteristics described for the probandIII-4 (Fig. 3).
DiscussionWe describe a novel PSEN1 mutation (L282V) resulting inearly-onset Alzheimer’s disease that is neuropathologicallycharacterized by unusually severe CAA. The causal natureof PSEN1 L282V is strongly supported by the autosomaldominant transmission pattern of Alzheimer’s disease in thisfamily, the absence of the mutation in 160 Belgian controlchromosomes, the sharing of alleles between the two probandsat PSEN1 flanking markers (Table 2) and the increasedin vitro Aβ42 secretion by cells expressing mutant comparedwith wild-type PSEN1. Moreover, this mutation is located
in the functionally important membrane-associated part ofthe large sixth hydrophilic loop of the PSEN1 protein,where ~30% of mutations are clustered (http://molgen-www.uia.ac.be/ADMutations/). Although this domain ishighly conserved between PSEN1 and its homologues PSEN2and Caenorhabditis elegans sel-12, L282 is one of the fewnon-conserved amino acids within this region. Interestingly,another amino acid substitution at the same codon (L282A)has been reported previously, although without clinical orpathological details, as the cause of early-onset Alzheimer’sdisease in a Spanish family with a mean onset age of43 years (Aldudo et al., 1998). The mean ages at onset(44.3 � 3.1 years, range 41–53 years) and death(53.5 � 3.3 years, 49–57 years) in family GB, as well asthe additional clinical features of myoclonus and frontal,extrapyramidal or cerebellar signs, have been notedpreviously for early-onset Alzheimer’s disease caused byPSEN1 mutations (Martin et al., 1991; Lippa et al., 2000).
The most remarkable finding in family GB, however, wasthe presence of a prominent CAA that was as severe as inour Flemish APP A692G family reported previously(Hendriks et al., 1992). We do not yet understand why a
Presenilin 1 L282V and amyloid angiopathy 2389
Fig. 4 Aβ immunostaining in serial sections of the neocortex of patient III-4 to demonstrate that amyloid in CAA and perivascularplaques is similar to that in dense-cored plaques and is predominantly full-length Aβ40. Scale bar � 40 µm.
subset of mutations either in APP (Flemish or Dutch) or inPSEN1 or PSEN2, here or described elsewhere (Ikeda et al.,1996; Mann et al., 1996; Yasuda et al., 1997; Nochlin et al.,1998; Singleton et al., 2000), present with abundant amyloidangiopathy. CAA leading to strokes has also been describedin cases of Down syndrome (Donahue et al., 1998). Clearly,a specific pathoclisis for vessels to deposit amyloid is morecomplex than a simple increase in Aβ40 levels. Indeed, weshow here in a heterologous in vitro assay that PSEN1L282V, like all pathogenic PSEN mutations, increases Aβ42specifically. However, we also demonstrate that the preciseAβ composition of CAA varies between the cortex andcerebellum. In contrast to the neocortex, where CAA ischiefly composed of full-length Aβ40, in the cerebellum theCAA load consists predominantly of Aβ42, and it is notalways full-length. It is of interest that a recent systematicneuropathological study of early-onset PSEN1 Alzheimer’sdisease showed a strong correlation between CAA and thePSEN1 mutation codon position. It was demonstrated that asevere degree of CAA was much more frequent among casesin which the PSEN1 mutation occurred after codon 200(Mann et al., 2001). Although it is difficult to understand
such a molecular clustering of PSEN1 mutations associatedwith severe CAA, our study seems to confirm this interestingobservation.
Regardless of its precise constituents or biomechanism(s)of formation, and in contrast to Flemish Alzheimer’s disease,or as would be expected from its severity, CAA does notlead to strokes or stroke-like episodes in family GB. Otherknown consequences of CAA, e.g. white matter lesions(Wallin, 1998; Roks et al., 2000), are also not detected, atleast early in the disease progression. Instead, we show herethat the pathological correlates of CAA, in terms of itsassociation with dystrophic neurites and inflammatory gliosis,are similar to those of cored plaque pathology. In our view,the most straightforward interpretation of this observation isthat CAA triggers a perivascular neurodegenerative responseand therefore it might, like the neuritic cored plaque, representa pathogenic lesion in the Alzheimer’s disease brain. Asimilar explanation was suggested recently in aneuropathological study of two cases with senile dementiapresenting with CAA and perivascular tau pathology in theabsence of neuritic cored plaque (Vidal et al., 2000).Moreover, the present study is also in line with our recent
2390 B. Dermaut et al.
Fig. 5 Double immunohistochemistry suggesting a neurodegenerative role of CAA in the cognitive decline in Alzheimer’s disease.Neurites accumulating (A) phosphorylated tau stained by AT8 (brown) and (B) ubiquitin, in association with anti-Aβ antibody (blue) inCAA and perivascular plaques, indicating a ‘neuritic’ role for CAA. Staining (C) microglia and (D) astroglia for specific Ab (brown)shows the same pattern of glial reactivity with CAA (anti-Aβ antibody, blue) as for dense-cored plaques (arrow in C). (E) Similarly,complement C1q reactivity was present in both CAA and dense-cored plaques (arrow). In the cerebellum, ubiquitin-positive dystrophicneurites (brown) showed an equal magnitude of association with both dense-cored plaques (F) and CAA (blue) (G), and amyloid streaksin the molecular layer (blue) (H) paralleled and were associated with blood vessels (CD31/34, brown). Scale bars � 20 µm.

Presenilin 1 L282V and amyloid angiopathy 2391
demonstration that the unusually large dense cored plaquesin Flemish Alzheimer’s disease (Cras et al., 1998), are mostprobably derived from stenosed vessels (Kumar-Singh et al.,submitted for publication). A role of concomitant minorvascular lesions that cumulatively lead to the progressivedecline in Alzheimer’s disease might explain, in part, theincreasingly recognized close overlap between Alzheimer’sdisease and vascular dementia (Kalaria and Ballard, 1999),especially the linear progressive variant of vascular dementia(Pantoni et al., 1996). However, this overlap betweenAlzheimer’s disease and vascular dementia can also beexplained by hypoxic injury due to amyloidogenic stenosingvessels or by direct toxicity incurred by the accumulation offull-length Aβ in CAA, similar to that deposited in dense-cored plaques. Moreover, the close link between CAA and aneurodegenerative-like decline shown in our study and theknown effects of cerebrovascular events, such as strokes inAlzheimer’s disease (Snowdon et al., 1997), should promptthe reconsideration of the clinical diagnosis of Alzheimer’sdisease, as such events are normally considered to beexclusion criteria.
In conclusion, we suggest that, besides the classicalpathological Alzheimer’s disease hallmark of dense-coredplaques with neuritic pathology, the presence of CAA,although not necessary, should be considered a contributingfactor in the progressive cognitive decline in Alzheimer’sdisease.
AcknowledgementsWe wish to thank Dr F. Van Genechten and Dr L. Swerts forsharing clinical information. Financial support was receivedfrom the Fund for Scientific Research Flanders (FWO-F),DWTC Interuniversity Attractionpoles (IUAP) and theInternational Alzheimer’s Research Foundation (IARF). M.C.and C.De J. are postdoctoral fellows and B.D. is a Ph.D.fellow of the FWO-F.
ReferencesAldudo J, Bullido MJ, Arbizu T, Oliva R, Valdivieso F. Identificationof a novel mutation (Leu282Arg) of the human presenilin 1 genein Alzheimer’s disease. Neurosci Lett 1998; 240: 174–6.
Cras P, van Harskamp F, Hendriks L, Ceuterick C, van Duijn CM,Stefanko SZ, et al. Presenile Alzheimer dementia characterized byamyloid angiopathy and large amyloid core type senile plaques inthe APP 692Ala→Gly mutation. Acta Neuropathol (Berl) 1998; 96:253–60.
Cruts M, Backhovens H, Theuns J, Clark RF, Le Paslier D,Weissenbach J, et al. Genetic and physical characterization of theearly-onset Alzheimer’s disease AD3 locus on chromosome 14q24.3.Hum Mol Genet 1995; 4: 1355–64.
Cruts M, van Duijn CM, Backhovens H, van den Broeck M,Wehnert A, Serneels S, et al. Estimation of the genetic contributionof presenilin-1 and -2 mutations in a population based study ofpresenile Alzheimer disease. Hum Mol Genet 1998; 7: 43–51.
De Jonghe C, Cruts M, Rogaeva EA, Tysoe C, Singleton A,Vanderstichele H, et al. Aberrant splicing in the presenilin-1 intron4 mutation causes presenile Alzheimer’s disease by increasedabeta42 secretion. Hum Mol Genet 1999; 8: 1529–40.
Donahue JE, Khurana JS, Adelman LS. Intracerebral hemorrhagein two patients with Down’s syndrome and cerebral amyloidangiopathy. Acta Neuropathol (Berl) 1998; 95: 213–16.
Glenner GG, Henry JH, Fujihara S. Congophilic angiopathy in thepathogenesis of Alzheimer’s degeneration. Ann Pathol 1981; 1:120–9.
Hendriks L, van Duijn CM, Cras P, Cruts M, Van Hul W, vanHarskamp F, et al. Presenile dementia and cerebral haemorrhagelinked to a mutation at codon 692 of the beta-amyloid precursorprotein gene. Nat Genet 1992; 1: 218–21.
Ikeda M, Sharma V, Sumi SM, Rogaeva EA, Poorkaj P,Sherrington R, et al. The clinical phenotype of two missensemutations in the presenilin I gene in Japanese patients. Ann Neurol1996; 40: 912–17.
Kalaria RN, Ballard C. Overlap between pathology of Alzheimerdisease and vascular dementia. [Review]. Alzheimer Dis AssocDisord 1999; 13 Suppl 3: S115–23.
Kumar-Singh S, De Jonghe C, Cruts M, Kleinert R, Wang R,Mercken M, et al. Nonfibrillar diffuse amyloid deposition due togamma(42) secretase site mutation points to an essential role forN-truncated abeta(42) in Alzheimer’s disease. Hum Mol Genet2000; 9: 2589–98.
Levy-Lahad LE, Wasco W, Poorkaj P, Romano DM, Oshima J,Pettingell WH, et al. Candidate gene for the chromosome 1 familialAlzheimer’s disease locus. Science 1995; 269: 973–7.
Lippa CF, Swearer JM, Kane KJ, Nochlin D, Bird TD, Ghetti B,et al. Familial Alzheimer’s disease: site of mutation influencesclinical phenotype. Ann Neurol 2000; 48: 376–9.
Mann DM, Iwatsubo T, Cairns NJ, Lantos PL, Nochlin D, SumiSM, et al. Amyloid beta protein (Abeta) deposition in chromosome14-linked Alzheimer’s disease: predominance of Abeta42(43). AnnNeurol 1996; 40: 149–56.
Mann DM, Pickering-Brown SM, Takeuchi A, Iwatsubo T. Amyloidangiopathy and variability in amyloid beta deposition is determinedby mutation position in presenilin-1-linked Alzheimer’s disease.Am J Pathol 2001; 158: 2165–75.
Martin JJ, Gheuens J, Bruyland M, Cras P, Vandenberghe A, MastersCL, et al. Early-onset Alzheimer’s disease in 2 large Belgianfamilies. Neurology 1991; 41: 62–8.
Nochlin D, Bird TD, Nemens EJ, Ball MJ, Sumi SM. Amyloidangiopathy in a Volga German family with Alzheimer’s disease anda presenilin-2 mutation (N141I). Ann Neurol 1998; 43: 131–5.
Pantoni L, Garcia JH, Brown GG. Vascular pathology in three casesof progressive cognitive deterioration. J Neurol Sci 1996; 135:131–9.
Roks G, van Harskamp F, De Koning I, Cruts M, De Jonghe C,Kumar-Singh S, et al. Presentation of amyloidosis in carriers of thecodon 692 mutation in the amyloid precursor protein gene (APP692).Brain 2000; 123: 2130–40.

2392 B. Dermaut et al.
Singleton AB, Hall R, Ballard CG, Perry RH, Xuereb JH,Rubinsztein DC, et al. Pathology of early-onset Alzheimer’s diseasecases bearing the Thr113-114ins presenilin-1 mutation. Brain 2000;123: 2467–74.
Snowdon DA, Greiner LH, Mortimer JA, Riley KP, Greiner PA,Markesbery WR. Brain infarction and the clinical expressionof Alzheimer disease. The Nun study. JAMA, 1997; 277:813–17.
Van Broeckhoven C, Haan J, Bakker E, Hardy JA, Van Hul W,Wehnert A, et al. Amyloid beta protein precursor gene and hereditarycerebral hemorrhage with amyloidosis (Dutch). Science 1990; 248:1120–2.
Vidal R, Calero M, Piccardo P, Farlow MR, Unverzagt FW,Mendez E, et al. Senile dementia associated with amyloid betaprotein angiopathy and tau perivascular pathology but not neuriticplaques in patients homozygous for the APOE-epsilon4 allele. ActaNeuropathol (Berl) 2000; 100: 1–12.
Wallin A. The overlap between Alzheimer’s disease and vasculardementia: the role of white matter changes. [Review]. DementGeriatr Cogn Disord 1998; 9 Suppl 1: 30–5.
Wenham PR, Price WH, Blandell G. Apolipoprotein E genotypingby one-stage PCR. Lancet 1991; 337: 1158–9.
Wisniewski HM, Terry RD. Reexamination of the pathogenesisof the senile plaque. In: Zimmerman HM, editor. Progress inneuropathology, Vol. II. New York: Grune & Stratton; 1973. p. 1–26.
Wisniewski HM, Bancher C, Barcikowska M, Wen GY, Currie J.Spectrum of morphological appearance of amyloid deposits inAlzheimer’s disease. Acta Neuropathol (Berl) 1989; 78: 337–47.
Yasuda M, Maeda K, Ikejiri Y, Kawamata T, Kuroda S, Tanaka C.A novel missense mutation in the presenilin-1 gene in a familialAlzheimer’s disease pedigree with abundant amyloid angiopathy.Neurosci Lett 1997; 232: 29–32.
Received April 18, 2001. Revised June 18, 2001.Accepted July 7, 2001





























