Embed Size (px)
Citation preview

REVIEWARTICLE
Chemoprevention of pancreatic cancer—one step closer
Volker Fendrich
Received: 24 January 2012 /Accepted: 26 January 2012 /Published online: 15 February 2012# Springer-Verlag 2012
AbstractBackground Since for pancreatic cancer the mortality rateapproaches the incidence rate with only 1–4% of all patientssurviving 5 years, it would be would be of great value toprovide chemopreventive treatment for high-risk individuals.Discussion The preclinical study of pancreatic intraepithelialneoplasia (PanINs) has recently been made possible by thegeneration of genetically modified animal models, whichrecapitulate human PanINs and invasive pancreatic canceron a genetic and histomorphologic level. Very recently, sev-eral groups have reported first evidence of chemopreventionof pancreatic cancer.
Keywords Pancreatic cancer . PanINs . Chemoprevention .
ACE inhibitor . Aspirin
Introduction
Pancreatic ductal adenocarcinoma (PDAC) is a devastatingand almost uniformly lethal malignancy that accountsfor approximately 33,000 deaths in the USA every year,
rendering it the fourth most common cause of cancer-related mortality [1, 2]. Though the past decades have seenintense research efforts aimed at a better understanding ofthe underlying etiologic and pathophysiological mecha-nisms, this increased knowledge could so far not success-fully be translated into better clinical treatment strategiesand improved patient survival. In fact, during the past30 years, the overall median 5-year survival rates for pan-creatic cancer have improved only marginally and are cur-rently around 5%, which is among the worst of all humanmalignancies [1, 2]. As with other solid tumours, the cura-tive potential for pancreatic cancer is dependent on the stageof this disease at diagnosis. Unfortunately, for the majorityof patients diagnosed with pancreatic cancer, symptoms donot develop until it is either unresectable or metastatic, andtherefore incurable. Survival in this setting is typically mea-sured in months. Moreover, even when patients present withan early-stage disease, the likelihood of cure remains verylow with the currently available therapies. Although recentevidence suggests that screening strategies may reduce mor-tality for other common cancers such as colorectal, breastand cervical cancers, the currently available screeningapproaches for PDAC are unlikely to produce significantreductions in mortality associated with the disease. Inaddition, since the incidence rate of pancreatic cancer issubstantially lower than the rates for the more commonmalignancies, screening large segments of the adultpopulation for pancreatic cancer would require a hugeexpenditure per life saved. Because of this, emerging geneti-cally engineered mouse models of pancreatic cancer play akey role for chemopreventive studies. The goal of this reviewwas to provide current knowledge on the in vitro, in vivo andclinical studies conducted about this disease using variouschemopreventive agents.
Review criteria The data for this review were compiled by searching thePubMed and MEDLINE databases for articles published from 1 January2000 to 31 December 2011. Older key publications in the field ofchemoprevention for pancreatic cancer were also included. The search term‘chemoprevention’ was used in combination with the terms ‘pancreaticcancer’, ‘PanINs’, ‘mouse models’ and ‘agents’. The bibliography of eachrelevant article was screened for further relevant studies.
V. Fendrich (*)Department of Surgery, Philipps University Marburg,Baldingerstrasse,35043 Marburg, Germanye-mail: [email protected]
Langenbecks Arch Surg (2012) 397:495–505DOI 10.1007/s00423-012-0916-x

Genetic and molecular events in human pancreaticcarcinogenesis
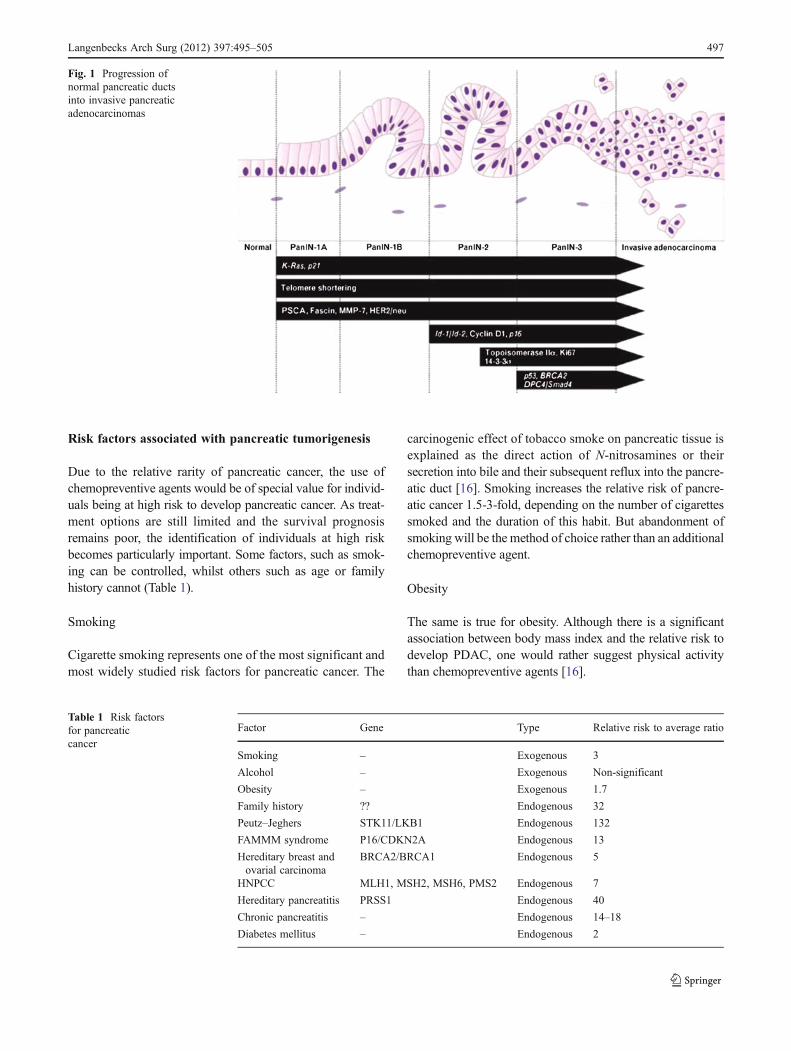
Pancreatic carcinogenesis comprises an array of geneticchanges finally culminating in fully invasive cancer [3–5].Today, pancreatic cancer is regarded as a genetic disease whicharises from morphologically and genetically clearly definedprecursor lesions through a stepwise accumulation of geneticalterations [6]. These precursor lesions comprise pancreaticintraepithelial neoplasia (PanIN), which can be subclassifiedinto PanIN-1, PanIN-2 and PanIN-3 lesions depending on thedegree of cytological and architectural atypia. Pancreatic can-cer may develop from these precursor lesions (Fig. 1). For acomprehensive review on the molecular pathology of precur-sor lesions of pancreatic cancer, see [7]. Genetic modificationsoccur at several levels during the multistep process of PDACpathogenesis. These include mutations of encoding oncogenesand tumour suppressor genes as well as epigenetic changes andaberrations of transcriptional patterns. Genetic changes mayalso affect caretaker genes such as genes of the Fanconi’sanaemia DNA repair pathway that normally function to min-imize genetic alterations during DNA replication, thus leadingto the accumulation of additional genetic mutations and facil-itating progression towards invasive carcinoma. Mutations ofmitochondrial DNA are also frequently observed in PDAC andnon-malignant precursor lesions [3]. The hallmark mutationsacquired along the pathogenic cascade of pancreatic cancerdevelopment are briefly discussed below.
Activating mutations of oncogenes in PDAC
KRAS
Activating point mutations of the oncogene KRAS occur in80–90% of all PDAC cases.Most commonly, they affect codon12, 13 or 61 and result in the abrogation of the intrinsic GTPaseactivity of the RAS protein, which leads to the constitutiveactivation of the intracellular signalling cascade [3]. Acquisi-tion of constitutively active KRAS is among the first geneticchanges observed in PDAC and is already present in non-malignant precursor lesions. About 30% of PanIN-1 lesionsharbour an activating point mutation of the KRAS gene [8].
CMYC, AKT2 and EGFR
Other oncogenes involved in the molecular pathogenesis ofpancreatic cancer are CMYC, AKT2 and epidermal growthfactor receptor (EGFR). Although mutations of those onco-genes are less frequent, they represent potential therapeutictargets, and experimental evaluation is currently underway[3]. CMYC is amplified and subsequently overexpressed in50–60% of pancreatic cancers, whilst amplifications of AKT2are detected in <5% of cases [3]. Nevertheless, the Akt
signalling pathway is activated in 30–60% of pancreatic can-cers, indicating additional activating mechanisms and under-scoring the potential therapeutic relevance of Akt targeting [9,10]. Constitutive EGFR pathway activation by amplificationof the EGFR gene and other mechanisms has likewise beendescribed in pancreatic cancer [3].
Mutations of tumour suppressor genes
CDKN2A/p16
The CDKN2A/p16 gene is located on the short arm of chro-mosome 9 (9p) and encodes the cell cycle regulator proteinp16INK4A, which exerts function via the p16/Rb pathway andinhibits cell cycle progression through the G1-S checkpoint.In >90% of PDAC, it is inactivated by either homozygousdeletion (~40% of cases), intragenic mutation (40%) or epige-netically by hypermethylation of its promoter (10–15%) [11].The loss-of-function mutation of the CDKN2a gene is thoughtto be an early event during the multistep process towardsinvasive carcinoma and can already be observed in 30% ofPanIN-1, 55% of PanIN-2 and 71% of PanIN-3 lesions [12]. Ofnote is that in mouse models of pancreatic cancer, bothp16INK4A as well as p19Arf (corresponding to p14Arf inhumans) inactivation seem to contribute to PDAC development[13, 14].
DPC4/SMAD4/MADH4
Deleted in pancreatic carcinoma 4 (DPC4/SMAD4/MADH4)is located on chromosome 18q21, and loss-of-function muta-tions of DPC4 can be observed in ~55% of all PDAC cases,whilst they occur only very rarely in other malignancies [3].Inactivation is mediated by the mutation of one allele withconcomitant loss of the second allele (25% of cases) or ho-mozygous mutation (30% of cases), resulting in reducedgrowth inhibition and increased proliferation via impairedTGF-b signalling. Loss of DPC4 constitutes a rather late eventin pancreatic cancer development and can only be observed ina minority of PanIN-3 lesions [15].
TP53
The oncogene TP53 on chromosome 17p usually inducescell cycle arrest and apoptosis in response to DNA damageand is inactivated due to intragenic mutation with loss of thesecond allele in 50–75% of pancreatic cancers. Loss offunction of TP53 facilitates the acquisition of additionalgenetic mutations and is considered to be a late event inpancreatic cancer development as the nuclear accumulationof mutated TP53 can only be observed in advanced PanIN-3lesions [15].
496 Langenbecks Arch Surg (2012) 397:495–505
Risk factors associated with pancreatic tumorigenesis
Due to the relative rarity of pancreatic cancer, the use ofchemopreventive agents would be of special value for individ-uals being at high risk to develop pancreatic cancer. As treat-ment options are still limited and the survival prognosisremains poor, the identification of individuals at high riskbecomes particularly important. Some factors, such as smok-ing can be controlled, whilst others such as age or familyhistory cannot (Table 1).
Smoking
Cigarette smoking represents one of the most significant andmost widely studied risk factors for pancreatic cancer. The
carcinogenic effect of tobacco smoke on pancreatic tissue isexplained as the direct action of N-nitrosamines or theirsecretion into bile and their subsequent reflux into the pancre-atic duct [16]. Smoking increases the relative risk of pancre-atic cancer 1.5-3-fold, depending on the number of cigarettessmoked and the duration of this habit. But abandonment ofsmoking will be the method of choice rather than an additionalchemopreventive agent.
Obesity
The same is true for obesity. Although there is a significantassociation between body mass index and the relative risk todevelop PDAC, one would rather suggest physical activitythan chemopreventive agents [16].
Fig. 1 Progression ofnormal pancreatic ductsinto invasive pancreaticadenocarcinomas
Table 1 Risk factorsfor pancreaticcancer
Factor Gene Type Relative risk to average ratio
Smoking – Exogenous 3
Alcohol – Exogenous Non-significant
Obesity – Exogenous 1.7
Family history ?? Endogenous 32
Peutz–Jeghers STK11/LKB1 Endogenous 132
FAMMM syndrome P16/CDKN2A Endogenous 13
Hereditary breast andovarial carcinoma
BRCA2/BRCA1 Endogenous 5
HNPCC MLH1, MSH2, MSH6, PMS2 Endogenous 7
Hereditary pancreatitis PRSS1 Endogenous 40
Chronic pancreatitis – Endogenous 14–18
Diabetes mellitus – Endogenous 2
Langenbecks Arch Surg (2012) 397:495–505 497

Alcohol
An analysis of 14 prospective studies has not confirmed anyassociation between alcohol consumption and a higher riskof pancreatic cancer [17].
Diabetes mellitus
Diabetes mellitus (DM) is widely considered to be as-sociated with risk of PDAC; however, whether DM is acause or a consequence of PDAC is still controversial.Ben et al. [18] conducted a detailed meta-analysis ofcohort studies. They included a total of 35 cohort stud-ies. DM was associated with an increased risk of PDAC(the summary RRs01.94, 95% CI01.66–2.27), with sig-nificant evidence of heterogeneity among these studies(p<0.001). In addition, the relative risk of PDAC wascorrelated negatively with the duration of DM, with thehighest risk of PDAC found among patients diagnosedwithin less than 1 year. The authors concluded thatthese findings strongly support that diabetes is associat-ed with an increased risk of PDAC in both males andfemales and that DM is both an early manifestation andan etiologic factor of pancreatic cancer.
Chronic inflammation/pancreatitis
Although no more than 5% of diagnosed cases of pancreaticcancer can be explained by recurrent attacks of chronicpancreatitis, the same genetic changes have been detectedin individuals with chronic inflammation of the pancreasand pancreatic cancer. Chronic inflammation is thought toinduce genetic alterations in tissue, whilst the ongoing heal-ing process exposes defective cells to growth factors; theresult is a pathological microenvironment in which stromalelements facilitate the neoplastic process in epithelial cells[19]. Chronic pancreatitis has long been suspected as a riskfactor for the development of pancreatic cancer. In 1993,Lowenfels et al. [20] contributed tremendously in thisfield. In total, 2,015 patients with chronic pancreatitis wererecruited and the incidence of pancreatic cancer comparedwith the expected incidence from population data. Thestandardised incidence ratio was 14.4 and the risk of devel-oping pancreatic cancer, 20 years after diagnosis, was ashigh as 4%. A smaller study from Italy demonstrated astandardised incidence ration of 18.5 using a similar meth-odology [21]. Because many patients with chronic pancrea-titis also smoke, it has been difficult to define the relativecontribution of smoking and chronic pancreatitis to pancre-atic cancer risk. A study in 497 patients revealed a twofoldincrease in pancreatic cancer risk in smokers compared withnon-smokers [22].
Hereditary pancreatitis
Hereditary pancreatitis is a rare autosomal dominant condi-tion with 80% penetrance. In patients with hereditary pan-creatitis, trypsin becomes activated whilst still in thepancreas. This accounts for the partial digestion of the pan-creatic tissue, which causes irritation and inflammation. Astrong genetic association exists with mutations found in thePRSS1, SPINK1 and CFTR genes and is associated with acumulative risk of pancreatic cancer by age 70 years of some40% [23].
Hereditary tumour syndromes
The existence of hereditary PDAC was initially suggestedby several case reports of a familial aggregation of ductalpancreatic adenocarcinomas. Lynch et al. [24] was the firstto systematically describe a larger cohort of families withfamilial pancreatic cancer (FPC) in 1989. Two prospectivestudies from Sweden and Germany demonstrated a familialaggregation of PDAC of only 2.7% and 1.9%, respectively,if the strict criteria of confirmation by histology and medicalrecords were used [25, 26]. An inherited predisposition toPDAC is currently believed to occur in three distinct clinicalsettings. First, it occurs in hereditary tumour predispositionsyndromes that are defined primarily by a clinical phenotypeother than PDAC, but are known to be associated with anincreased risk of PDAC. These syndromes include Peutz–Jeghers syndrome, melanoma pancreatic cancer syndromeor familial atypical multiple mole melanoma (FAMMM),hereditary breast and ovarian cancer, and others, respectively(see Table 1). The second setting is hereditary pancreatitis andcystic fibrosis in which genetically determined early changesof the pancreas can predispose to the development of PDAC.The third setting is FPC in which two or more first-degreerelatives have PC without fulfilling the criteria of anotherinherited tumour syndrome. Several studies have shown thatfamily members of patients with PC have an increased risk forthe development of PC [27–29]. The risk increases with thenumber of family members affected. Individuals with three ormore affected first-degree relatives have an estimated 32-foldincreased risk [28].
Mouse models of pancreatic cancer for chemopreventiveresearch
Various mouse models of pancreatic cancer have been de-veloped in the past few years, providing a crucial platformfor the investigation of chemopreventive agents andtheir impact on cancer development and tumour biology[30]. Years ago, the most often used mouse models forPDAC were xenograft mouse models where tumours in
498 Langenbecks Arch Surg (2012) 397:495–505

immunodeficient mice were generated by subcutaneous in-jection or orthotopic transplantation of PDAC cell lines. Ahuge advantage of these models is that they are simple to setup and human cancer cells can be assessed in the murine invivo environment. However, the biggest disadvantage is themissing immunological reaction of the cancer cells to themurine environment, making conclusions about the tumour–stroma relation impossible, something which is very impor-tant for PDAC [31]. Nowadays, genetically engineeredmouse models (GEMM) are an elegant alternative to in vitroand xenotransplantation studies, although their breeding andhousing is both very time-consuming as well as expensive.For PDAC studies, GEMM possess the potential whichrecapitulates human PanINs and invasive pancreatic canceron a genetic and histomorphologic level in a syngenicsystem with a fully intact host immune system [30]. There-fore, these models might at least, to some extent, also carrythe potential to predict the response of human pancreaticcancer to therapeutic intervention or chemoprevention. TwoGEMM were recently developed that bear striking resem-blance to human PDAC. The first is based on mutation ofthe endogenous murine Kras gene specifically in pancreaticprogenitor cells by crossing mice with a conditionally acti-vated Kras allele (LSL-KrasG12D) to transgenic strains thatexpress Cre recombinase in pancreatic lineages (PdxCre orp48Cre). These ‘KC’ mice develop murine PanIN lesionswith 100% penetrance, but only a small subset of theseanimals progress to PDAC at an advanced age, suggestingthat additional genetic alterations are necessary for tumourformation [32]. To accelerate the process of tumorigenesis,PdxCre-expressing compound mutant mice were generatedwith conditional mutations in both Kras and Trp53 in anal-ogy to the genetic alterations in human PDA. These ‘KPC’mice develop advanced PDA with 100% penetrance at anearly age, thus recapitulating human PDAC including his-topathological similarities in neoplastic cells, tumour–stro-ma reaction, metastasis and comorbidities such as cachexia[33]. ‘KC’ mice are considered a very valuable tool to studyPanIN biology since it mimics a rather slow progressionfrom PanIN-1 over PanIN-2 and PanIN-3 lesions to invasivecancer in around 12–15 months. Furthermore, ‘KPC’mice manifest widely metastatic pancreatic ductal ade-nocarcinoma that recapitulates the human spectrum. Theuse of these models provides now the opportunity toconduct chemopreventive studies on pancreatic cancer.Further important work was done by Guerra et al. [34],providing evidence that temporal expression of endoge-nous Kras in acinar cells of adult mice results in PanINsand invasive PDAC only in the context of caerulein-inducedpancreatitis. Since then, more and more GEMM forpancreatic cancer were constructed (overview in [35]), pro-viding now perfect abilities to perform preclinical chemopre-ventive studies.
Chemopreventive agents
As mentioned above, our ability to identify patients at risk ofdeveloping pancreatic cancer has improved, although we haveno interventions that can reduce this risk other than partial ortotal pancreatectomy. Clearly, surgical resection is a radicalintervention for patients whose lifetime risk of developingpancreatic cancer may be only elevated slightly over thebaseline risk in the general population. As shown in Fig. 1,PDAC is thought to evolve through non-malignant precursorlesions to fully invasive carcinoma. The following agents haveshown first evidence that this process can be slowed down, ifnot even stopped.
Aspirin, NSAIDs and other selective cyclooxygenaseinhibitors
Many in chemoprevention are beginning to think that perhapsthe best way to catch cancer is to target inflammation. Chronicinflammation appears to encourage tumours by prompting thegrowth of new blood vessels and a remodelling of the extra-cellular matrix, creating a prime setting for normal cell growthto turn malignant. This theory led chemoprevention research-ers to turn to two drugs—celecoxib and aspirin—that targetthe cyclooxygenase enzymes (COX-1 and COX-2), whichplay key roles in inflammation and pain. Both are non-steroidal anti-inflammatory drugs (NSAIDs), which epidemi-ological studies suggest may reduce the risk of colon and othercancers.
Aspirin has been used to control pain and inflammationfor over a century. Epidemiological studies associated adecreased incidence of colorectal cancer with the long-term use of aspirin in the early 1980s. In subsequent years,the use of other NSAIDs, which inhibit COX enzymes, waslinked to reduced cancer risk in multiple tissues includingthose of the breast, prostate and lung [36, 37]. As in manyother types of malignant tissue, COX-2 is also overex-pressed in pancreatic carcinoma. Maitra et al. [38] foundan upregulation of COX-2 in a subset of PanINs, whichmakes it a potential target for chemoprevention with selec-tive COX-2 inhibitors. However, recent epidemiologic stud-ies investigating the preventive value of aspirin in pancreaticcarcinoma reached conflicting findings, perhaps due to thesubstantial differences in patient populations, exposure in-formation and outcome information [39–42]. Wang et al.[43] were the first to show that NF-κB activity is foundconstitutively in about 70% of pancreatic cancers, but not innormal pancreatic tissue. The findings of Sclabas et al. [44]showed that aspirin-mediated inhibition of NF-κB activa-tion in response to inflammation is a possible mechanism forthe cancer preventive effect of aspirin. They used an ortho-topic mouse model with human pancreatic carcinoma celllines to study the inhibitory effects of aspirin on pancreatic
Langenbecks Arch Surg (2012) 397:495–505 499

tumour formation. Animals given aspirin for 6 days beforetumour cell injection showed a lower incidence of tumourformation compared with those receiving aspirin 2 weeksafter inoculation. They suggested that aspirin-mediated anti-inflammatory approaches could be an effective strategy toprevent pancreatic carcinoma. These in vivo results are inline with several epidemiological studies [40, 41]. Recently,in a prospective study conducted by Anderson et al. [40], theuse of aspirin was associated with a reduced risk of pancre-atic cancer among 28,232 postmenopausal women. To fur-ther evaluate the association between aspirin, NSAID andacetaminophen use with pancreatic cancer risk, Tan et al.[45] used a clinic-based case–control study of 904 pancre-atic ductal adenocarcinoma cases and 1,224 age- and sex-matched healthy controls evaluated at Mayo Clinic from2004 to 2010. Aspirin use for 1 day/month or greater wasassociated with a significantly decreased risk of pancreaticcancer (OR00.74, 95% CI00.60–0.91, p00.005) comparedwith never or <1 day/month. This inverse association wasalso found for those who took low-dose aspirin for heartdisease prevention (OR00.67, 95% CI00.49–0.92, p00.013). Instead, they found no relationship between non-aspirin NSAID or acetaminophen use and risk of pancreaticcancer. The authors concluded that aspirin use, but not non-aspirin NSAID use, is associated with a lowered risk ofdeveloping pancreatic cancer.
In an elegant, preclinical study, Funahashi et al. [46] eval-uated the efficacy of the selective COX-2 inhibitor nimesulideto prevent the progression of PanINs in the KPC mousemodel. Mice were randomly allocated to a diet supplementedwith nimesulide or a control diet. After 10 months, the pan-creas of KPC mice was analysed for the presence of PanINs.Animals fed the COX-2 inhibitor had significantly fewerPanIN-2and PanIN-3 lesions than control animals (p<0.05).Ten per cent of all pancreatic ducts in the nimesulide-fedanimals showed PanIN-2 or PanIN-3 lesions, whereas 40%of the pancreatic ducts in the control animals had PanIN-2 orPanIN-3 lesions. Intrapancreatic prostaglandin E2 levels werereduced in nimesulide-fed animals. The authors concludedthat inhibition of COX-2 may represent an intriguing strategyto prevent pancreatic cancer in high-risk patients [46]. Recent-ly, our group extended these results and showed that aspirindelayed the progression of PanINs and partially inhibited theformation of invasive murine pancreatic cancer formation inKPC mice [47]. Aspirin delayed the progression from normalducts to PanIn1A and to PanIN1B. Because only PanIN3 areclassified as carcinoma in situ, delaying the progression to-wards PanIN3 would have a major impact on cancer devel-opment. Therefore, our study is strengthening the conclusionof Funahasi et al. [46] that aspirin or a COX-2 inhibitor mightbe an effective chemopreventive agent for pancreatic cancerprobably by the inhibition of NF-kappa B-driven Cox-2expression by blocking MAPK and Akt/protein kinase B
signalling [48]. NF-κB orchestrates the expression of genesthat encode key determinants in inflammation, tumorigenesis,and apoptosis and thus promotes the cardinal clinical featuresof pancreatic carcinoma of locally aggressive growth andmetastasis [49]. Constitutive activation of NF-κB is a frequentmolecular alteration in pancreatic carcinoma and is also foundin human pancreatic carcinoma cell lines, but not in immor-talized, non-tumorigenic pancreatic epithelial cells [50]. As-pirin inhibits NF-κB activation through the specific inhibitionof IKK-2 activity by binding to IKK-2 and reducing ATPbinding [51]. This could be an important step in pancreaticcancer because NF-κB is a critical downstream mediator ofcell transformation mediated by oncogenic ras [52]. In thiscontext, the transcriptional activity of RelA is significantlyincreased in Ras-transformed cells as compared with non-transformed cells [53]. It is thus reasonable to expect that theactivation of the Ras pathway in human tumours may yet beanother means to induce constitutive NF-κB activity and on-cogenesis. In agreement with this hypothesis, RelA has beenobserved to be constitutively activated in 67% of pancreaticadenocarcinomas and the human K-ras oncogene to be fre-quently mutated in pancreatic cancer [43].
Angiotensin I-converting enzyme inhibitors
Inhibitors of angiotensin I-converting enzyme (ACE inhibi-tors) inhibit stimulation by angiotensin II (AngII) by decreas-ing its conversion from AngI. ACE inhibitors (ACE-Is) weredeveloped and applied clinically as first-line drugs for hyper-tension. Interestingly, much evidence has accumulated toshowing that ACE-Is suppress the growth of a wide varietyof cultured cancer cells in vitro and inhibit tumorigenesis andangiogenesis induced in cancer animal models in vivo [54, 55].AngII is a multifunctional bioactive peptide, and recent reportshave suggested that it is a pro-angiogenic growth factor. It hasbeen shown that AngII selectively increases blood flow andthat ACE-Is decrease intratumoral blood flow without affect-ing blood flow in healthy organs. In experimental models,ACE-Is reduced the tumour cell growth rate and modulatedgene expression in vitro. In vivo, captopril was shown toinhibit tumour growth and angiogenesis [56].
Lever et al. [57] conducted a retrospective cohort study toassess the risk of cancer in hypertensive patients receivingACE-Is or other antihypertensive drugs. The relative risk ofcancer was lowest in women on ACE-Is. They concluded thatlong-term use of ACE-Is may protect against cancer. A retro-spective cohort study on 5,207 patients receiving ACE-I orother anti-hypertensive drugs with a 10-year follow-up dem-onstrated that ACE-I treatment may decrease the incidence ofadult cancer [57]. Furthermore, a study of 483,733 US veter-ans has found that ACE-Is can significantly reduce the risk ofpancreatic cancer (odds ratio00.48, p<0·01) [58]. Arafat et al.[59] analysed the expression and localization of ACE and
500 Langenbecks Arch Surg (2012) 397:495–505

AngII type 1 receptor (AT1) in relation to vascular endothelialgrowth factor (VEGF) in invasive human pancreatic cancer.VEGF is a crucial pro-angiogenic component in pancreaticductal adenocarcinoma, and its high expression levels havebeen correlated with poor prognosis and early postoperativerecurrence. They found an upregulation of ACE and AT1R in75% of analysed cancers when compared with matching con-trols. VEGF expression was significantly higher in tissues thatexpressed high levels of AT1 and ACE. The same groupexplored the signalling mechanisms involved in the AngII-mediated VEGF induction and correlated AT1 and VEGFexpression in noninvasive precursor lesions. An AT1 antago-nist inhibited the AngII-mediated induction of the VEGFmessenger RNA and protein in all PDA cell lines [60].
Recently, our group used KP and KPC mice to evaluateenalapril as a chemopreventive agent for pancreatic cancer[47]. Drug treatment was initiated at the age of 5 weeks.LsL-KrasG12D; Pdx1-Cre or LsL-KrasG12D; LsL-Trp53R172H;Pdx1-Cre transgenic mice were randomly assigned to re-ceive either mock treatment or enalapril. After 3 and5 months of treatment, enalapril was able to significantlydelay the progression of PanINs in LsL-KrasG12D; Pdx1-Cremice. Furthermore, the development of invasive pancreaticcancer in LsL-KrasG12D; LsL-Trp53R172H; Pdx1-Cre trans-genic mice was partially inhibited by enalapril. Invasivepancreatic cancer was identified in 15 of 25 (60%) LsL-KrasG12D; LsL-Trp53R172H; Pdx1-Cre untreated controlmice, but in 4 of 17 (23.5%, p00.03) mice treated withenalapril alone. Using real-time PCR, we found a significantdownregulation of the target gene VEGF, demonstrating theability to achieve effective pharmacological levels of ena-lapril during pancreatic cancer formation in vivo.
Taken together, several studies suggest the involvement ofAT1 and AngII in PDAC progression and therefore mayprovide valuable chemopreventive targets.
Gefitinib
The chemopreventive efficacy of gefitinib, an EGFR inhib-itor, was evaluated against the progression of PanINs toPDAC in conditional LSL-KrasG12D/+ transgenic mice[61]. Mice were fed (AIN-76A) diets containing 0, 100and 200 ppm of gefitinib for 35 weeks. Dietary gefitinib at100 and 200 ppm significantly suppressed PDAC incidenceby 77% and 100%, respectively (p<0.0001) when comparedwith the control diet. Importantly, a significant inhibition ofcarcinoma and a dose-dependent suppression of PanINs[PanIN-1, 37–62% (p<0.002); PanIN-2, 38–41% (p<0.001); and PanIN-3, 7–34% (p<0.0141)] were observedin mice treated with gefitinib. Furthermore, mice treatedwith gefitinib exhibited 67.6–77.3% of the pancreas to befree from ductal lesions. Also, gefitinib reduced EGFR,proliferating cell nuclear antigen, cyclin D1, C2GNT,
RhoA, β-catenin, p38, phospho-extracellular signal-regulated kinase, caveolin-1, and mucin and increasedcyclin B1 in the pancreatic lesions/PDAC. These resultsshow that gefitinib can prevent the progression of pancreaticcancer precursor lesions to PDAC in a preclinical model[61].
Dietary cancer prevention
Citrus fruits
Flavonoids, one of the major classes of compounds in citrus,have been shown to have antiproliferative, antioxidant, anti-tumour, anti-inflammatory and pro-apoptotic activities [62].Carotenoids are also found in citrus fruits, and there areseveral studies that have shown that high carotenoid intakemay decrease the risk of cancer [63]. Several epidemiolog-ical studies have examined the effects of citrus fruits on therisk of developing pancreatic cancer (overview in [62]).Most of these studies show an inverse association betweenthe consumption of citrus fruit and the risk of developingpancreatic cancer. However, some of the studies are under-powered and show a huge variety in the comparison of theexposure level.
Curcumin
Curcumin is a fat-soluble polyphenolic compound that is themain and most active curcuminoid found in turmeric (Cur-cuma longa L.) [62]. It is commonly used as a spice in Asiancountries as well as a natural food colouring agent. Curcuminhas been a bioactive compound of interest for many types ofcancer, including breast, prostate, oral, ovarian and others [64].Its many pharmacological properties include antioxidant, anti-inflammatory, antimicrobial, antitumour, antidepressant andanti-atherogenic activities. The anticancer effects of curcuminon pancreatic cancer in vitro have been widely researched anddocumented (overview in [62]). Bar-Sela et al. [65] published adetailed review about the clinical trials that have been con-ducted or are currently ongoing that use curcumin as an anti-cancer agent. In these trails, curcumin was used in an adjuvantsetting rather than chemopreventive.
Flavonoids
Flavonoids are a large class of polyphenols, with over 5,000 ofthem described. They are water-soluble secondary metabolitesand are responsible of the flavour and colour of vegetables andfruits. In humans, flavonoids have a wide range of bioactivitiesincluding anticancer, anti-inflammatory and anti-hypertensiveproperties [62].
Langenbecks Arch Surg (2012) 397:495–505 501

Apigenin in vitro inhibits DNA synthesis and induces cellcycle arrest at G2/M phase through the downregulation ofcyclin A and B, phosphorylated cdc2, cdc25A and cdc25C inAsPC-1, MIA PaCa-2, CD18 and S2-013 cells [66]. In anotherstudy, it induces apoptosis and enhances the inhibitory andapoptotic effects of gemcitabine through the downregulation ofNFκB activity with the suppression of Akt activation and thereduction of Bcl-2 expression in MIA PaCa-2 and AsPC-1cells [67] and through the induction of both S and G2/Mphase cell cycle arrest [68]. Furthermore, it decreases glucoseuptake through the downregulation of GLUT-1 in CD18 andS2-013 cells and interferes with the PI3K/Akt pathway [69]. Invivo studies with Apigenin have shown that it potentiates theinhibitory effects of gemcitabine on tumour growth and abro-gates gemcitabine-induced activation of the Akt-NFκBpathway [67].
Kaempferol inhibits cell growth and induces apoptosis inMIA PaCa-2 and PANC-1 cells and provides an additive effecton the inhibition of MIA PaCa-2 cell proliferation when com-bined with 5-fluorouracil [70]. Quercetin shows antiprolifera-tive effects on BxPC-3, MIA PaCa-2 and PANC-1 cells andinduces apoptosis by upregulating caspase-3 and caspase-9and downregulating Hsp70 In nude mice, it decreases tumourgrowth and Hsp70) [71].
The isoflavone genistein inhibits cell growth, induces apo-ptosis and inhibits NFκB activity in BxPC-3 cells and poten-tiates the effects of cisplatin and docetaxel by inhibiting cellgrowth and inducing apoptosis inhibits [72]. It enhances thecell growth inhibition of gemcitabine on COLO 357 andL3.6pl cell lines, abrogates gemcitabine-induced activationof NFκB DNA-binding activity and sensitizes gemcitabine-treated cells to apoptosis by the induction of caspase-3-mediated PARP cleavage and the downregulation of Bcl-2,Bcl-XL and p-Akt. In the same study, it reduces tumour weightwhen combined with gemcitabine treatment and inhibitsgemcitabine-induced activation of NFκB activity [73]. It indu-ces apoptosis through the downregulation of Notch-1, whichleads to the inhibition of IKK protein and therefore reducedNFκB activity [74, 75]. It potentiates the growth inhibition oferlotinib-treated and increases the apoptotic effect of erlotinibin BxPC-3 cells through the downregulation of EGFR, pAkt,NFκB activation and survivin [76]. It inhibits cell proliferationin BxPC-3, HPAC, MIA PaCa-2 and PANC-28 cell lines anddownregulates the expression of FoxM1, thus leading to theinhibition of cdc25a, survivin, MMP-9 and VEGF, therebydecreasing the penetration of pancreatic cancer cells throughthe Matrigel-coated membrane [77].
Taken together, several flavonoids found in a variety offruits and vegetables have been proven to inhibit pancreaticcancer at various molecular targets including cell cycle, Akt,NFκB, extracellular signal-regulated kinase (ERK) and manyothers. The isoflavone genistein is one of the more studiedflavonoids in pancreatic cancer. It has been shown that it has
the ability to abolish the activation of NFκB induced by thechemotherapeutic drugs gemcitabine and cisplatin when usedas a pretreatment for either of them. Currently, there is onephase II clinical trial on the use of genistein for pretreatingpatients with resectable pancreatic cancer which was completedin 2011 (http://clinicaltrials.gov/ct2/show/NCT00882765).However, more clinical trials are needed to explore the efficacyand application of genistein in treating pancreatic cancer.
Capsaicin
Capsaicin is a major biologically active ingredient of chilipeppers. Studies indicate that capsaicin is a cancer-suppressing agent via blocking the activities of severalsignal transduction pathways including nuclear factor kap-paB, activator protein 1 and signal transducer and activatorof transcription 3. Very recently, the effect of capsaicin onpancreatitis and PanINs was determined in a mutant Kras-driven and caerulein-induced pancreatitis-associated carci-nogenesis in KPC mice [78]. KPC mice were subjected toone dose of caerulein at age 4 weeks to induce and synchro-nize the development of chronic pancreatitis and PanINlesions. One week after caerulein induction, animals weredistributed into three groups and fed with either AIN-76Adiet or AIN-76A diet containing 10 ppm capsaicin or20 ppm capsaicin for a total of 8 weeks. The results showedthat capsaicin significantly reduced the severity of chronicpancreatitis, as determined by evaluating the loss of acini,inflammatory cell infiltration and stromal fibrosis. Further-more, the progression of PanIN-1 to high-grade PanIN-2 andPanIN-3 were significantly inhibited by capsaicin. Furtherimmunochemical studies revealed that treatment with capsai-cin significantly reduced proliferating cell nuclear antigen-labelled cell proliferation and suppressed the phosphorylationof ERK and c-Jun as well blocked Hedgehog/GLI pathwayactivation. These results indicate that capsaicin could be apromising agent for the chemoprevention of pancreatic carci-nogenesis, possibly via inhibiting pancreatitis and mutantKras-led ERK activation [78].
Outlook—prevention strategies for pancreatic cancer
A chemoprevention agent that blocks the very first step wouldbe best. But most researchers would consider a drug success-ful if it could stop the disease progressing from any stage tothe next. That is the basic idea of cancer chemoprevention: toarrest or reverse the progression of premalignant cells towardsfull malignancy.
Complicating the problem is the fact that people feel finestarting a long-term drug regimen—one that can cause trou-blesome side effects or even put them at risk for anotherdisease. So we need to identify individuals or groups being
502 Langenbecks Arch Surg (2012) 397:495–505

at high risk for developing pancreatic cancer. Therefore, weshould not doing clinical chemoprevention trials in largepopulations of people at relatively low risk and instead focuson groups at the highest risk. There are many such groups:patients with hereditary pancreatitis, Peutz–Jeghers syndromeor familial pancreatic cancer. Chemoprevention trials on suchgroups will provide much more definitive results and withmuch less effort.
There are two major prevention strategies involved incancer research: cancer chemoprevention and dietary cancerprevention. Cancer chemoprevention is defined as the use ofnatural, synthetic or biologic chemical agents for pharmaco-logic intervention to prevent, inhibit or reverse carcinogenesis[79]. Dietary cancer prevention, on the other hand, means themodification or changes of food consumption patterns that canbe accompanied by lifestyle changes in order to decrease therisk [79].Which strategy will be the better one still needs to bedefined.
GEMM of pancreatic cancer are helping close the hugegap between in vitro results and clinical trials. The valuableinsights these mice provide will maybe help us designclinical trials leading to the prevention of this lethal diseasemalignancy.
Conflicts of interest None.
References
1. Jemal A, Siegel R, Ward E (2009) Cancer statistics, 2009. CACancer J Clin 59(4):225–249
2. ACS (2009) Cancer facts & figures 2009. American Cancer Society,Atlanta
3. Feldmann G, Maitra A (2008) Molecular genetics of pancreaticductal adenocarcinomas and recent implications for translationalefforts. J Mol Diagn 10(2):111–122
4. Maitra A, Kern SE, Hruban RH (2006) Molecular pathogenesis ofpancreatic cancer. Best Pract Res Clin Gastroenterol 20(2):211–226
5. Vogelstein B, Kinzler KW (2004) Cancer genes and the pathwaysthey control. Nat Med 10(8):789–799
6. Feldmann G, Rauenzahn S, Maitra A (2009) In vitro models ofpancreatic cancer for translational oncology research. Expert OpinDrug Discov 4(4):429–443
7. Feldmann G, Maitra A (2010) Molecular pathology of precursorlesions of pancreatic cancer. Pancreatic cancer. Springer, NewYork, pp 119–141
8. Moskaluk CA, Hruban RH, Kern SE (1997) p16 and K-ras genemutations in the intraductal precursors of human pancreatic ade-nocarcinoma. Cancer Res 57(11):2140–2143
9. Ruggeri BA, Huang L, Wood M (1998) Amplification and over-expression of the AKT2 oncogene in a subset of human pancreaticductal adenocarcinomas. Mol Carcinog 21(2):81–86
10. Asano T, Yao Y, Zhu J (2004) The PI 3-kinase/Akt signalingpathway is activated due to aberrant Pten expression and targetstranscription factors NF-kappaB and c-Myc in pancreatic cancercells. Oncogene 23(53):8571–8580
11. Schutte M, Hruban RH, Geradts J et al (1997) Abrogation of theRb/p16 tumor-suppressive pathway in virtually all pancreatic carci-nomas. Cancer Res 57(15):3126–3130
12. Wilentz RE, Geradts J, Maynard R et al (1998) Inactivation of thep16 (INK4A) tumor-suppressor gene in pancreatic duct lesions:loss of intranuclear expression. Cancer Res 58(20):4740–4744
13. Bardeesy N, Aguirre AJ, Chu GC et al (2006) Both p16(Ink4a) andthe p19(Arf)-p53 pathway constrain progression of pancreatic ade-nocarcinoma in the mouse. Proc Natl Acad Sci USA 103(15):5947–5952
14. Sharpless NE, Ramsey MR, Balasubramanian P et al (2004) Thedifferential impact of p16(INK4a) or p19(ARF) deficiency on cellgrowth and tumorigenesis. Oncogene 23(2):379–385
15. Maitra A, Adsay NV, Argani P et al (2003) Multicomponentanalysis of the pancreatic adenocarcinoma progression model usinga pancreatic intraepithelial neoplasia tissue microarray. Mod Pathol16(9):902–912
16. Zavoral M, Minarikova P, Zavada F, Salek C, Minarik M (2011)Molecular biology of pancreatic cancer. World J Gastroenterol 17(24):2897–2908
17. Genkinger JM, Spiegelman D, Anderson KE et al (2009) Alcoholintake and pancreatic cancer risk: a pooled analysis of fourteencohort studies. Cancer Epidemiol Biomarkers Prev 18:765–776
18. Ben Q, Xu M, Ning X, Liu J, Hong S, Huang W, Zhang H, Li Z(2011) Diabetes mellitus and risk of pancreatic cancer: a meta-analysis of cohort studies. Eur J Cancer 47(13):1928–1937
19. McKay CJ, Glen P, McMillan DC (2008) Chronic inflammationand pancreatic cancer. Best Pract Res Clin Gastroenterol 22(1):65–73
20. Lowenfels AB, Maisonneuve P, Lankisch PG (1999) Chronicpancreatitis and other risk factors for pancreatic cancer. Gastro-enterol Clin North Am 28:673–685
21. Talamini G, Falconi M, Bassi C, Sartori N, Salvia R, Caldiron E,Frulloni L, Di Francesco V, Vaona B, Bovo P, Vantini I, PederzoliP, Cavallini G (1999) Incidence of cancer in the course of chronicpancreatitis. Am J Gastroenterol 94:1253–1260
22. Lowenfels AB, Maisonneuve P (2005) Risk factors for pancreaticcancer. J Cell Biochem 95(4):649–656
23. Lowes N, LerchMM, Charnley R et al (2002) Hereditary pancreatitis(HP) and the risk of pancreatic ductal adenocarcinoma (PDAC). Gut50:A43
24. Lynch HT, Lanspa SJ, Fitzgibbons RJ Jr et al (1989) Familialpancreatic cancer (part 1): genetic pathology review. Nebr Med J74:109–112
25. Bartsch DK, Kress R, Sina-Frey M et al (2004) Prevalence offamilial pancreatic cancer in Germany. Int J Cancer 110:902–906
26. Hemminki K, Li X (2003) Familial and second primary pancreaticcancers: a nationwide epidemiologic study from Sweden. Int JCancer 103:525–530
27. Tersmette AC, Petersen GM, Offerhaus GJ et al (2001) Increased riskof incident pancreatic cancer among first-degree relatives of patientswith familial pancreatic cancer. Clin Cancer Res 7:738–744
28. Klein AP, Brune KA, Petersen GM et al (2004) Prospective risk ofpancreatic cancer in familial pancreatic cancer kindreds. CancerRes 64:2634–2638
29. Rulyak SJ, Lowenfels AB, Maisonneuve P et al (2003) Risk factorsfor the development of pancreatic cancer in familial pancreatic cancerkindreds. Gastroenterol 124:1292–1299
30. Hruban RH, Adsay NV, Albores-Saavedra J et al (2006) Pathology ofgenetically engineered mouse models of pancreatic exocrine cancer:consensus report and recommendations. Cancer Res 66:95–106
31. Gopinathan A, Tuveson DA (2008) The use of GEM models forexperimental cancer therapeutics. Dis Model Mech 1:83–86
32. Hingorani SR, Petricoin EF, Maitra A et al (2003) Preinvasive andinvasive ductal pancreatic cancer and its early detection in the mouse.Cancer Cell 4:437–450
Langenbecks Arch Surg (2012) 397:495–505 503

33. Hingorani SR, Wang L, Multani AS et al (2005) Trp53R172H andKrasG12D cooperate to promote chromosomal instability andwidely metastatic pancreatic ductal adenocarcinoma in mice. CancerCell 7:469–483
34. Guerra C, Schuhmacher AJ, Canamero M et al (2007) Chronicpancreatitis is essential for induction of pancreatic ductal adeno-carcinoma by K-Ras oncogenes in adult mice. Cancer Cell 11:291–302
35. Schutte U, Bisht S, Brossart P, Feldmann G (2011) Recent devel-opments of transgenic and xenograft mouse models of pancreaticcancer for translational research. Expert Opin Drug Discov 6(1):33–48
36. Sandler RS, Halabi S, Baron JA, Budinger S, Paskett E, KeresztesR, Petrelli N, Pipas JM, Karp DD, Loprinzi CL, Steinbach G,Schilsky R (2003) A randomized trial of aspirin to prevent colorectaladenomas in patients with previous colorectal cancer. N Engl J Med348:883–890
37. Baron JA, Cole BF, Sandler RS, Haile RW, Ahnen D, Bresalier R,McKeown-Eyssen G, Summers RW, Rothstein R, Burke CA, SnoverDC, Church TR, Allen JI, Beach M, Beck GJ, Bond JH, Byers T,Greenberg ER, Mandel JS, Marcon N, Mott LA, Pearson L, Saibil F,van Stolk RU (2003) A randomized trial of aspirin to prevent colorec-tal adenomas. N Engl J Med 348:891–899
38. Maitra A, Ashfaq R, Gunn CR, Rahman A, Yeo CJ, Sohn TA,Cameron JL, Hruban RH, Wilentz RE (2002) Cyclooxygenase 2expression in pancreatic adenocarcinoma and pancreatic intraepithe-lial neoplasia: an immunohistochemical analysis with automatedcellular imaging. Am J Clin Pathol 118:194–201
39. Gridley G, McLaughlin JK, Ekbom A, Klareskog L, Adami HO,Hacker DG, Hoover R, Fraumeni JE (1993) Incidence of canceramong patients with rheumatoid arthritis. J Natl Cancer Inst 85:307–311
40. Anderson KE, Johnson TW, Lazovich D, Folsom AR (2002)Association between nonsteroidal anti-inflammatory drug useand the incidence of pancreatic cancer. J Natl Cancer Inst 94:1168–1171
41. Schernhammer ES, Kang JH, Chan AT, Michaud DS, Skinner HG,Giovannucci E, Colditz GA, Fuchs CS (2004) A prospective studyof aspirin use and the risk of pancreatic cancer in women. J NatlCancer Inst 96:22–28
42. Lara LF, Chari ST (2004) Does an aspirin a day keep pancreascancer away? Gastroenterology 127:1002–1004
43. Wang W, Abbruzzese JL, Evans DB, Larry L, Cleary KR, Chiao PJ(1999) The nuclear factor-κB RelA transcription factor is consti-tutively activated in human pancreatic adenocarcinomas cells. ClinCancer Res 5:119–127
44. Sclabas GM, Uwagawa T, Schmidt C, Hess KR, Evans DB,Abbruzzese JL, Chiao PJ (2005) Nuclear factor kappa B activationis a potential target for preventing pancreatic carcinoma by aspirin.Cancer 12:2485–2490
45. Tan XL, Lombard KM, Bamlet WR et al (2011) Aspirin, nonste-roidal anti-inflammatory drugs, acetaminophen, and pancreaticcancer risk: a clinic-based case–control study. Cancer Prev Res 4(11):1835–1841
46. Funahashi H, Satake M, Dawson D, Huynh NA, Reber HA, HinesOJ, Eibl G (2007) Delayed progression of pancreatic intraepithelialneoplasia in a conditional Kras(G12D) mouse model by a selectivecyclooxygenase-2 inhibitor. Cancer Res 67:7068–7071
47. Fendrich V, Chen NM, Neef M, Waldmann J, Bucholz M, FeldmannG, Slater EP, Maitra A, Bartsch DK (2010) The angiotensin-I-converting enzyme inhibitor enalapril and aspirin delay progressionof pancreatic intraepithelial neoplasia and cancer formation in agenetically engineered mouse model of pancreatic cancer. Gut59:630–637
48. Hwang DM, Kundu JK, Shin JW, Lee JC, Lee HJ, Surh YJ (2007)cis-9,trans-11-conjugated linoleic acid down-regulates phorbol
ester-induced NF-kappaB activation and subsequent COX-2 ex-pression in hairless mouse skin by targeting IkappaB kinase andPI3K-Akt. Carcinogenesis 28:363–371
49. Fujioka S, Sclabas GM, Schmidt C et al (2003) Inhibition ofconstitutive NF-kappa B activity by I kappa B alpha M suppressestumorigenesis. Oncogene 22:1365–1370
50. Dong QG, Sclabas GM, Fujioka S et al (2002) The function ofmultiple IkappaB: NF-kappaB complexes in the resistance of cancercells to Taxol-induced apoptosis. Oncogene 21:6510–6519
51. Kopp E, Ghosh S (1994) Inhibition of NF-kappa B by sodiumsalicylate and aspirin. Sci 265:956–959
52. Mayo MW, Norris JL, Baldwin AS (2001) Ras regulation of NF-kBand apoptosis. Methods Enzymol 333:73–87
53. Mayo MW, Wang CY, Cogswell PC et al (1997) Requirement ofNF-κB activation to suppress p53-independent apoptosis inducedby oncogenic Ras. Science 278:1812–1815
54. Reddy MK, Baskaran K, Molteni A (1995) Inhibitors ofangiotensin-converting enzyme modulate mitosis and gene expres-sion in pancreatic cancer cells. Proc Soc Exp Biol Med 210:221–226
55. Uemura H, Nakaigawa N, Ishiguro H, Kubota Y (2005) Antipro-liferative efficacy of angiotensin II receptor blockers in prostatecancer. Curr Cancer Drug Targets 5:307–323
56. Yoshiji H, Kuriyama S, Noguchi R, Fukui H (2004) Angiotensin-Iconverting enzyme inhibitors as potential anti-angiogenic agentsfor cancer therapy. Curr Cancer Drug Targets 4:555–567
57. Lever AF, Hole DJ, Gillis CR, McCallum IR, McInnes GT,MacKinnon PL, Meredith PA, Murray LS, Reid JL, Robertson JW(1998) Do inhibitors of angiotensin-I-converting enzyme protectagainst risk of cancer? Lancet 352:179–184
58. Khurana V, Caldito G, Barkin JS (2008) Angiotensin convertingenzyme inhibitors decrease the incidence of pancreatic cancer: astudy of half a million US veterans. Eur J Cancer 1:S47–S48
59. Arafat HA, Gong Q, Chipitsyna G, Rizvi A, Saa CT, Yeo CJ(2007) Antihypertensives as novel antineoplastics: angiotensin-I-converting enzyme inhibitors and angiotensin II type 1 receptorblockers in pancreatic ductal adenocarcinoma. J Am Coll Surg204:996–1005
60. Anandanadesan R, Gong Q, Chipitsyna G, Witkiewicz A, Yeo CJ,Arafat HA (2008) Angiotensin II induces vascular endothelialgrowth factor in pancreatic cancer cells through an angiotensin IItype 1 receptor and ERK1/2 signaling. J Gastrointest Surg 12:57–66
61. Mohammed A, Janakiram NB, Li Q et al (2011) The epider-mal growth factor receptor inhibitor gefitinib prevents theprogression of pancreatic lesions to carcinoma in a conditionalLSL-KrasG12D/+ transgenic mouse model. Cancer Prev Res3:1417–1426
62. Johnson J, de Mejia EG (2011) Dietary factors and pancreaticcancer: the role of food bioactive compounds. Mol Nutr FoodRes 55:58–73
63. Marti N, Mena P, Canovas J, Micol V, Saura D (2009) Vitamin Cand the role of citrus juices as functional food. Nat Prod Commun4:677–700
64. Goel A, Kunnumakkara A, Aggarwal B (2008) Curcumin as“curecumin”: from kitchen to clinic. Biochem Pharmacol 75:787–809
65. Bar-Sela G, Epelbaum R, Schaffer M (2010) Curcumin as an anti-cancer agent: review of the gap between basic and clinical applica-tions. Curr Med Chem 17:190–197
66. Ujiki M, Ding X, Salabat M, Bentrem D et al (2006) Apigenininhibits pancreatic cancer cell proliferation through G2/M cellcycle arrest. Mol Cancer 5:76
67. Lee S, Ryu J, Lee K, Woo S et al (2008) Enhanced anti-tumoreffect of combination therapy with gemcitabine and apigenin inpancreatic cancer. Cancer Lett 259:39–49
504 Langenbecks Arch Surg (2012) 397:495–505

68. Strouch M, Milam B, Melstrom L, McGill J et al (2009) Theflavonoid apigenin potentiates the growth inhibitory effects of gem-citabine and abrogates gemcitabine resistance in human pancreaticcancer cells. Pancreas 38:409–415
69. Melstrom L, Salabat M, Ding X, Milam B et al (2008) Apigenininhibits the GLUT-1 glucose transporter and the phosphoinositide3-kinase/Akt pathway in human pancreatic cells. Pancreas 37:426–431
70. Zhang Y, Chen A, Li M, Chen C, Yao Q (2008) Ginkgo bilobaextract kaempferol inhibits cell proliferation and induces apoptosisin pancreatic cancer cells. J Surg Res 148:17–23
71. Aghdassi A, Phillips P, Dudeja V, Dhaulakhandi D et al (2007) Heatshock protein 70 increases tumorigenicity and inhibits apoptosis inpancreatic adenocarcinoma. Cancer Res 67:616–625
72. Li Y, Ahmed F, Ali S, Philip P et al (2005) Inactivation of nuclearfactor κB by soy isoflavone genistein contributes to increasedapoptosis induced by chemotherapeutic agents in human cancercells. Cancer Res 65:6934–6942
73. Banerjee S, Zhang Y, Ali S, Bhuiyan M et al (2005) Molecularevidence for increased antitumor activity of gemcitabine by genistein
in vitro and in vivo using an orthotopic model of pancreatic cancer.Cancer Res 65:9064–9072
74. Wang Z, Zhang Y, Banerjee S, Li Y, Sarkar F (2006) Inhibition ofnuclear factor κB activity by genistein is mediated via Notch-1 sig-naling pathway in pancreatic cancer cells. Int J Cancer 118:1930–1936
75. Wang Z, Zhang Y, Li Y, Banerjee S et al (2006) Down-regulationof Notch-1 contributes to cell growth inhibition and apoptosis inpancreatic cancer cells. Mol Cancer Ther 5:483–493
76. El-Rayes B, Ali S, Ali I, Philip P et al (2006) Potentiation of theeffect of erlotinib by genistein in pancreatic cancer: the role of Aktand nuclear factor-κB. Cancer Res 66:10553–10559
77. Wang Z, Ahmad A, Banerjee S, Azmi A et al (2010) FoxM1 is anovel target of a natural agent of pancreatic cancer. Pharm Res 27:1159–1168
78. Bai H, Li H, Zhang W, Matkowskyj KA, Liao J, Srivastava SK,Yang GY (2011) Inhibition of chronic pancreatitis and pancreaticintraepithelial neoplasia (PanIN) by capsaicin in LSL-KrasG12D/Pdx1-Cre mice. Carcinogenesis 32:1689–1696
79. Tsao A, Kim E, Hong W (2004) Chemoprevention of cancer. CACancer J Clin 54:150–180
Langenbecks Arch Surg (2012) 397:495–505 505





























