Embed Size (px)
Citation preview

Chemoselective reaction of a sulfonate ester with methoxide ions in preference to benzyl mercaptide anions
RICHARD FRANCIS LANGLER' A N D NANCY ANN MORRISON Department of Chetnistry and Chetnical Engineering, Florida Instilule of Technology, Melbourne, FL 32901-6988, U.S.A.
Received October 10, 1986' 1 This paper is dedicated lo Professor Douglas E. Rya11 on the occasion of his 651h birthday
RICHARD FRANCIS LANGLER and NANCY ANN MORRISON. Can. J. Chem. 65, 2385 (1987).
1 Trifluoroethyl benzyl sulfide was prepared (in 2-propanol) and subsequently chlorinated as a further test of substituent electronegativity based regiochemical predictions. The initlal attempt to prepare that sulfide by reaction of 2,2,2-trifluoroethyl methanesulfonate and benzyl mercaptide anions (in methanol) furnished benzyl methyl sulfide. Mechanistic possibilities are discussed in detail and some synthetic consequences of this novel reaction are presented.
1 RICHARD FRANCIS LANGLER et NANCY ANN MORRISON. Can. J. Chem. 65, 2385 (1987). On a prCparC le sulfure de trifluorotthyle et de benzyle (dans le propanol-2) et, dans le but de confirmer la corrClation qui existe
entre I'ClectronCgativitC des substituants et les prkdictions rkgiochimiques, on I'a ulterieurement soumis 2 une chloration. L'essai initial de prtparer le sulfure par reaction du methanesulfonate de trifluoro-2,2,2 ethyle avec les anions sulfures de benzyle (dans le methanol) a conduit 2 la formation de sulfure de mCthyle et de benzyle. On discute des diverses possibilitCs concernant le mCcanisme et on prCsente quelques consCquences au niveau de la synthese de cette nouvelle reaction.
[Traduit par la revue]
Introduction CH30Na [2] CF3CH20SOrCH3 + PhCH2SH
Our longstanding interest in the chlorination of organosulfur CH30H compounds encompasses the regiochemical problem associated 4 PhCH2SCH3 + PhCH2SCH2CF3 with sulfide chlorinations (vide eq. [ l ] .) We have developed an
27% 3 (12.4%)
approach (1, 2) that utilizes substituent electronegativities to successfully predict regiochemical result^.^ Our initial rationale (1) for employing substituent electronegativities to make such predictions was discredited by our own theoretical study (5). Upon completion of that work, we were left with the following positions: ( i ) according to our M N D O study of intermediate thionium ions 2, phenyl was the best stabilizer and CF3 was a
destabilizer (relative to hydrogen at the sp2 carbon), s o that chlorination of 2,2,2-trifluoroethyl benzyl sulfide 3, (R1 = CF3, R2 = Ph in 1, eq. [ I ] ) should occur at the benzylic ~ a r b o n , ~ and (ii) according to substituent electronegativity predictions,5 reaction should g o exclusively into the trifluoroethyl group.
- -
W e resolved, at that time, to undertake the chlorinolysis of trifluoroethyl benzyl sulfide 3, which we intended to prepare from trifluoroethyl methanesulfonate 4.
. ... . Results and discussion . .... I .. ..
-:. . : . -:. . . < . i Electrophilicity of 4 a n d the preparation of 3 . I Our previous experience (8, 9) led us to treat 2,2,2-trifluoro- ) ethyl methanesulfonate 4 with the benzyl mercaptide anion in / methanol as our first attempt to obtain 3.
'Author to whom correspondence may be addressed. 'Revision received May 24, 1987. 3For recent reviews of this area see refs. 3 and 4. 4rThis expectation assumes that the favored pathway goes through
the most stable thionium ion. 5~aul ing electronegativity of phenyl is 2.41 (6) and Pauling
electronegativity of trifluoromethyl is 3.46 (7).
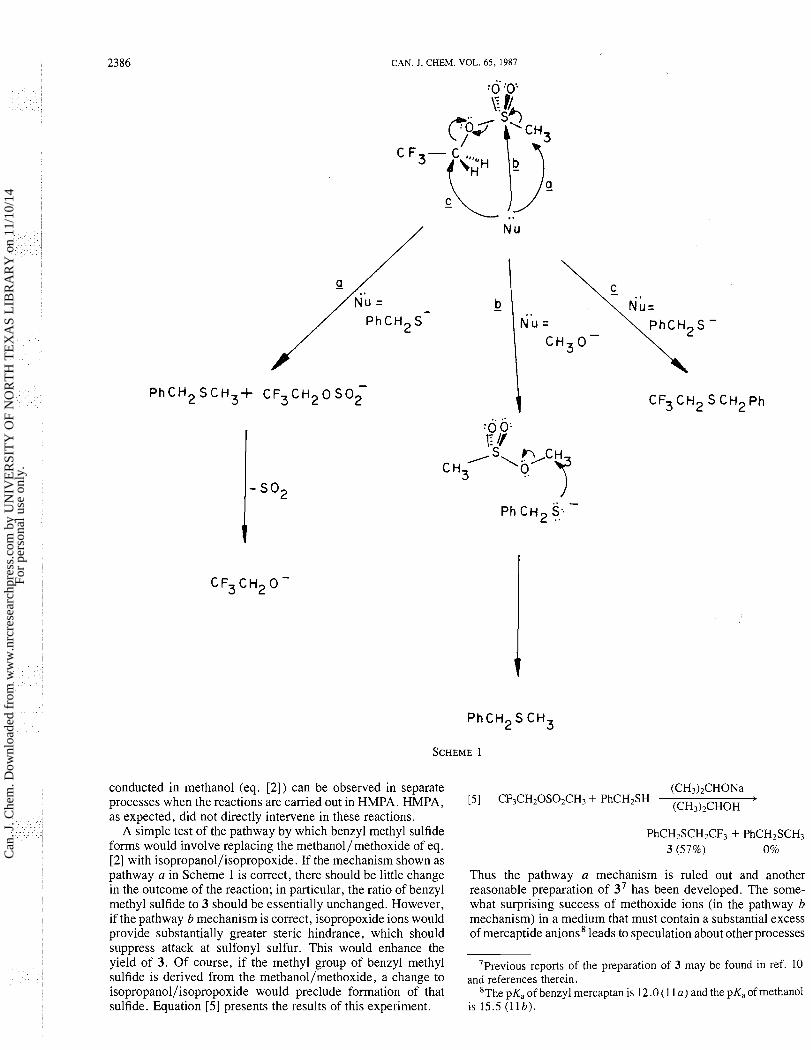
Several points arise from the results shown in eq. [ 2 ] . Firstly, this is clearly an unacceptable method for the preparation of 3. However, the surprising formation of benzyl methyl sulfide does raise an interesting mechanism problem. Morever, in conjunction with the mechanism problem, the mass balance is very poor, suggesting the possibility of quantitative loss of a major product. W e deal first with the origin of the benzyl methyl sulfide.
The simplest explanation for the formation of benzyl methyl sulfide, which involves what might be called an "ipso" attack, is shown as pathway a in Scheme 1. While we are unaware of any published precedent for nucleophilic attack at the carbon bearing sulfur in a sulfonate ester, we have observed analogous reactivity for some sulfonyl chlorides (8). Extension of this behavior to sulfonate esters would not appear to be a major step.
An alternative explanation for the formation of benzyl methyl sulfide involves attack by methoxide ions at sulfonyl sulfur (shown as pathway b in the scheme). Facile nucleophilic attack by an oxy-nucleophile at the sulfonyl sulfur of a sulfonate ester is readily illustrated by the following result.
HMPA [3] O~N-(&OSO~CH~ + PhONa - PhOS02CH3
u
Under the same conditions, mercaptide anions attack the nitromesylate at carbon.
HMPA 141 O ~ N ~ O S O ~ C H ~ + PhCH2SNa -
O I N * ~ H ~ P ~
Thus, the high chemoseiectivity6 postulated for the reactions
60f course, nucleophilic attack at the methylene carbon in 4 proceeds by an SN2 process (see pathway c in the scheme). In eq. [4], the corresponding nucleophilic attack at the sp2 carbon results in substitution by an addition-elimination sequence.
1 Prinlcd in Canada 1 lmpnrni au C a n d a
Can
. J. C
hem
. Dow
nloa
ded
from
ww
w.n
rcre
sear
chpr
ess.
com
by
UN
IVE
RSI
TY
OF
NO
RT
H T
EX
AS
LIB
RA
RY
on
11/1
0/14
For
pers
onal
use
onl
y.
CAN. J. CHEM. VOL. 65, 1987
conducted in methanol (eq. [2]) can be observed in separate processes when the reactions are carried out in HMPA. HMPA, as expected, did not directly intervene in these reactions.
A simple test of the pathway by which benzyl methyl sulfide forms would involve replacing the methanol/methoxide of eq. [2] with isopropanol/isopropoxide. If the mechanism shown as pathway a in Scheme 1 is correct, there should be little change in the outcome of the reaction; in particular, the ratio of benzyl methyl sulfide to 3 should be essentially unchanged. However, if the pathway b mechanism is correct, isopropoxide ions would provide substantially greater steric hindrance, which should suppress attack at sulfonyl sulfur. This would enhance the yield of 3. Of course, if the methyl group of benzyl methyl sulfide is derived from the methanol/methoxide, a change to isopropanol/isopropoxide would preclude formation of that sulfide. Equation [5] presents the results of this experiment.
Thus the pathway a mechanism is ruled out and another reasonable preparation of 37 has been developed. The some- what surprising success of methoxide ions (in the pathway b mechanism) in a medium that must contain a substantial excess of mercaptide anions8 leads to speculation about other processes
'~revious reports of the preparation of 3 may be found in ref. 10 and references therein.
he pK, of benzyl mercaptan is 12.0 ( 1 l a ) and the pK,of methanol is 15.5 ( 1 1 b ) .
Can
. J. C
hem
. Dow
nloa
ded
from
ww
w.n
rcre
sear
chpr
ess.
com
by
UN
IVE
RSI
TY
OF
NO
RT
H T
EX
AS
LIB
RA
RY
on
11/1
0/14
For
pers
onal
use
onl
y.

LANGLER AND MORRISON 2387
in which they might be involved. Methoxide ions could attack (CH&CHONa the methyl methanesulfonate formed in sit14 (see pathway b) [81 PhCHzOH + CF3CH20SO2CH3 (CH3)zCHOH + to give dimethyl ether. The low-boiling ether would either be lost during the reaction or during work-up. The formation of (PhCH2)20
dimethyl ether was confirmed as shown below. 23%
4
Corresponding conversions of alkoxide ions into symmetrical
[9] PhCHzOH + PhSH + PhOS02CH3 (CH313COK (CH313COH '
ethers have been reported by other workers ( 1 2 ~ 7 b). Now it Further results will be communicated in due course. We note, is clear what processes actually occurred when we carried out in connection with one-pot methodologies, the highly effective the eq. [21 it was proceed in a method for the conversion of alcohols to sulfides developed by fashion similar to that shown in eq. [7] (taken from ref. 8). ~ ~ k ~ ~ ~ ~ ~ , ~ k i , and H~~~ 8).
One can begin to deal with an explanation for the unusual result shown in eq. [2] using Hard and Soft Acid and Base the01-y.~ The hard base (alkoxide ion) should attack preferen- tially at the hard acid site (sulfonyl sulfur) and the soft base (mercaptide anion) should attack preferentially at the soft acid sites (sp3 carbon), as observed. The unusual outcome of the reaction shown in eq. [2] would make sense if the rate of attack by mercaptide anion on the trifluoroethyl mesylate 4 was unusually slow (cf. eq. [7]). A rate retardation would neces- sarily be attributed to the trifluoromethyl group attached to the electrophilic carbon. Two possible origins for such an effect came to mind readily, viz: ( i) a steric effect, or (ii) a field effect in which the negatively charged fluorine atoms repel approach- ing anionic nucleophiles. However, trifluoromethyl groups are believed to be only a little larger than methyl groups (14), which appears to rule out a steric argument. Field effects have been argued for the sulfonyl group. We have published results (15) that are inconsistent with this view for the sulfonyl group. Consequently, it is even less appealing to us as an explanation for a trifluoromethyl substituent effect. In connection with this point, we note that our MNDO gas phase results,1° on the CF3 containing systems 5 and 6 , show that the partial charge on fluorine is rather small (- 0.17 in 5 and -0.23 in 6) and is, to a first approximation, insensitive to demand for electron density.
Another view of this problem'would suppose a rate accelera- tion for nucleophilic attack at the sulfonyl sulfur of 4 by methoxide ions. This view is supported by the pK, of CF3CH20H, which is 12.4 (17). The pK, suggests that the trifluoroethoxy group is a significantly better leaving group than is an alkoxy group, thereby enhancing electrophilicity at the hard acid site.
We felt that the synthetic potential for pathway b chemistry was worth further exploration. Among many potential method- ologies, this chemistry could offer "one-pot" methodologies for symmetric ethers and for sulfides. Equations [8] and [9] give preliminary results for each of these methodologies.
1 or a discussion of sulfur nucleophiles and electrophiles in terms
1 of HSAB theory see ref. 13. 1 'O~hese calculations were carried out as part of a previously 1 published study. Details may be found in that paper (16).
Chlorinolysis of 3 We have attempted to chlorinate 3 with sulfuryl chloride
only to find no reaction. However, facile chlorination was accomplished with molecular chlorine.
The result depicted in eq. [ lo] is consistent with the outcome expected on the basis of substituent electronegativity differ- e n ~ e . ~ In accord with the conclusions reached from our earlier theoretical study ( 9 , this result suggests that the major regioisomeric thionium ion 2 is not always the most stable one and that "sulfides which have substituents with high electro- negativities follow an El,,-like mechanism". Equation [lo] brings the total to 51 asymmetric sulfide chlorinations for which substituent electronegativities have been successful in anticipating the regiochemistry.
Conclusions 1. CF3 is a better directing group than Ph in asymmetric
acyclic sulfide chlorination reactions, as expected on the basis of AXp (substituent electronegativity difference) arguments.
2. Some sulfonate esters can induce alcohols to alkylate mercaptide anions (see eq. [2]). The reaction appears to involve two highly chemoselective steps: (i) nucleophilic attack by alkoxide ions (particularly primary ones) at sulfonyl sulfur, then (ii) mercaptide anion attack at carbon of the intermediate sulfonate ester.
3. High chemoselectivity in the nucleophilic substitutions on sulfonate esters suggests a variety of possible one-pot synthetic methods. Preliminary results show potential for such syntheses of sulfides and symmetric ethers.
Experimental General
The ir spectra were recorded on a Perkin Elmer 237B grating spectrophotometer. The nmr spectra were obtained on a Varian EM360A instrument using TMS as the internal standard. Melting points were determined on a Mel-Temp capillary melting-point apparatus and are uncorrected.
Conversion of benzyl mercaptan to benzyl methyl sulfide Sodium metal (1.204 g, 52.3 mrnol) was dissolved in methanol
(100 mL) and benzyl mercaptan (6.2 mL, 50 rnrnol) added. 2,2,2- Trifluoroethyl methanesulfonate 4 (8.900 g, 50 mmol) (19) was added and the reaction mixture refluxed for 20 h. Water (100 mL) was added and the resultant mixture washed with methylene chloride (three 100-mL aliquots). The combined organic layers were dried (MgS04) and concentrated. The residue was rectified at reduced pressure affording a mixture (4.5579 g, bp 106-1 12"C/50 Torr) (1 Torr = 133.3 Pa).
Can
. J. C
hem
. Dow
nloa
ded
from
ww
w.n
rcre
sear
chpr
ess.
com
by
UN
IVE
RSI
TY
OF
NO
RT
H T
EX
AS
LIB
RA
RY
on
11/1
0/14
For
pers
onal
use
onl
y.

2388 CAN. J . CHEM. VOL. 6 5 , 1987
A portion of the distillate (1.983 g) was chromatographed on silica gel (200 g) employing hexanes elution (100-mL fractions). Fraction 10 was concentrated, affording clean trifluoroethyl benzyl sulfide 3 (0.068 g, 0.33 mmol). Fractions 1 1- 13 were combined and concen- trated, affording more 3 (0.496 g) contaminated with benzyl mercap- tan. Fractions 16-24 were combined and concentrated, affording benzyl methyl sulfide (0.8209 g, 6.09 mmol).
Chromatographed benzyl methyl sulfide (0.8209 g, 6.09 mmol) was oxidized by chromium trioxide (1.525 g, 15.2 mmol) in glacial acetic acid (50 mL) in the usual way (8). Benzyl methyl sulfone (0.362 g, 2.12 mmol) was obtained and recrystallized from methanol. It was identical to authentic material by ir, nmr, tlc, mp (123.5-124"C, lit. (20) mp 127"C), and mixture mp.
Preparation of p-nitrophenyl rnethanesulfonate A solution of dry triethylamine (14.538 g, 143.9 mmol) and
p-nitrophenol (19.999 g, 143.8 mmol) in dry pyridine (200 mL) was reacted with methanesulfonyl chloride (16.388 g, 143.7 mmol) in the usual way. The residue from routine work-up was recrystallized from methanol, affording clean p-nitrophenyl rnethanesulfonate (20.60 g, 94.9 mmol, 66%, mp 88-89°C). The sulfonate ester had Rf 0.81 on analytical tlc plates (0.5 mm silica gel thickness) when developed with chloroform; ir (CHC13): 1540, 1390, 1360, and 1170 cm-I; uv (CH30H): 261 nm ( E 8 955); nmr (CDCl,) 6: 8.30 (2H, d, J = 9 Hz), 7.36 (2H, d, J = 9 Hz), and 3.25 (3H, s). Anal. calcd. for C7H7N05S: C 38.70, H 3.24; found: C 38.66, H 3.19.
Reaction of p-nitrophenyl methanesulfonate with phenoxide ions Phenol (2.1573 g, 22.9 mmol) in HMPA" (10 mL) was added
dropwise to a slurry of sodium hydride (0.61 11 g, 25.4 mmol) in HMPA (30 mL). Upon completion of the 5-min addition, the reaction mixture was stirred at ambient temperature for 1 h to permit complete evolution of hydrogen. p-Nitrophenyl rnethanesulfonate (5.0229 g, 23.1 mmol) was added and the reaction mixture stirred at ambient temperature for 18 h.
The reaction mixture was poured into water (400 mL) and 10% HC1 (5 mL). The resultant mixture was washed with diethyl ether (three 100-mL portions). The ether was concentrated and the residue poured into water (100 mL). The aqueous layer was washed with diethyl ether (three 100-mL portions). The combined ether layers were washed with 2.5% sodium hydroxide (two 100-mL aliquots). The organic layer was dried (MgS04) and the solvent evaporated, affording crude phenyl rnethanesulfonate (3.3708 g, 19.5 mmol, 84%).
The crude mesylate was chromatographed on silica gel (400 g) employing chloroform elution (100-mL fractions). Fractions 12 and 13 were combined and concentrated, affording clean phenyl methane- sulfonate (2.5932 g, 15.0 mmol). After recrystallization the methane- sulfonate was shown to be identical to authentic material by ir, nmr, double-spotted tlc, mp (61-62"C), and mixture mp (60-61.3"C). Fractions 14 and 15 were combined, concentrated, and rechromato- graphed on silica gel (80 g) employing 1:l chloroform - carbon tetrachloride elution (50-mL fractions). Fractions 8- 12 were combined and concentrated, yielding more clean phenyl rnethanesulfonate (0.3802 g, 2.2 mmol).
Reaction of p-nitrophenyl methanesulfonate with benzyl mercaptide anions
Sodium hydride (0.3326 g, 13.8 mmol) was added to HMPA (30 mL). A solution of benzyl mercaptan (1.7 mL, 13.7 mmol) in HMPA (10 mL) was added dropwise over 10 min. The reaction mixture was stirred for an additional 10 min to permit complete evolution of Hz. The nitromesylate (2.9813 g, 13.7 mmol) was added and almost immediately the reaction mixture turned very deep red-almost black. The reaction mixture was stirred at ambient temperature for 2 h. Crude work-up followed the procedure used for reaction with phenoxide ions.
The residue was recrystallized from methanol, affording clean p-nitrophenyl benzyl sulfide (1.7561 g, 7.1 mmol, 52%, mp 119-
"HMPA is hexamethylphosphoramide [(CH3)zN]3P0. HMPA is toxic and carcinogenic (21).
119.5"C). The sulfide had Rf 0.46 on analytical tlc plates when developed with 3:l carbon tetrachloride - chloroform; ir (CHCI,): 1520 and 1350 cm-'; nmr (CDC13) 6: 8.06 (2H, d), 7.30 (SH, s), 7.25 (2H, d), and4.23 (2H, s). Anal. calcd. forC13HII02NS: C 63.65, H 4.51; found: C 63.46, H 4.48.
Preparation of 2,2,2-tr~jluoroethyl benzyl suljde 3 Small scale Sodium metal (0.1233 g, 5.3 mmol) was reacted with 2-propanol
(20 mL). Phenylmethanethiol (0.7 mL, 5.6 mmol) and the mesylate 4 (1.000 g, 5.6 mmol) (19) were added and the reaction mixture refluxed for 4 days. A standard extraction procedure furnished crude product. The crude was chromatographed on silica gel (100 g) employing hexanes for elution (100-mL fractions). Fractions 5-7 were combined and concentrated, furnishing benzylic sulfide 3 (0.660 g, 3.2 mmol, 57%) with known properties (10).
Large scale without chromatographic work-up ~ e a c t i o n was carried out as outlined above using sodium metal
(1.285 g, 55.8 mmol), phenylmethanethiol(7 mL, 56.4 mmol), and the trifluorosulfonate 4 (10.0145 g, 56.2 mmol) in 2-propanol(200 mL). The reaction mixture was refluxed for only 2 h. Upon completion of reflux, 3% sodium hydroxide (100 mL) was added and the reaction mixture refluxed for an additional hour. An extraction procedure furnished crude, which was added to pyridine (25 mL) and iodine (1 g) and stirred at ambient temperature for 5 min. Standard extraction furnished an organic layer, which was washed with 10% w/v sodium thiosulfate (two 50-mL aliquots). The solvent was evaporated and the residue rectified at reduced pressure, yielding 3 (6.131 g, 29.7 mmol, 52%, bp 1 12-12O0C/35 Torr). The sulfide obtained this way is free of benzyl mercaptan and trifluoromesylate 4 but has a yellowish color.
Preparation of tr~jluoroethyl benzyl sulfone The trifluorosulfide 3 (0.728 g, 3.5 mmol) in glacial acetic acid
(5 mL) was added dropwise to a mixture of chromium trioxide (0.3556 g, 3.55 mmol) in glacial acetic acid (5 mL) and the oxidation carried out in the usual way (8). The crude sulfone (0.371 g, 1.5 mmol, 44%) was recrystallized from methanol, yielding clean trifluoroethyl benzyl sulfone (0.2601 g, mp 139-141°C). The sulfone had ir (CHC13): 1360 and 1145 cm-I; nmr (CDC13) 6: 7.30 (SH, s), 4.26 (2H, s), and 3.50 (2H, q, J = 9 Hz). Anal. calcd. for C9H9F302S: C 45.37, H 3.80; found: C 45.59, H 3.76.
Reaction of trifluoroethyl mesylate 4 with methoxide ions Sodium metal (1.0998 g) was dissolved in methanol (100 mL) and
the trifluoroethyl mesylate (4.458 g) added. The reaction was refluxed for 0.5 h. Effluent gases were passed into a trap cooled with isopropanol/Dry Ice. Upon completion of the reflux, the trap con- tained dimethyl ether (0.7 mL), which was identical with authentic material by nrnr.
Preparation of dibenzyl ether Sodium metal (1.2953 g, 56.3 mmol) was dissolved in 2-propanol
(100 mL) and benzyl alcohol (3.0337 g, 28.0 mmol) was added. 2,2,2-Trifluoroethyl rnethanesulfonate (4.9968 g, 28.0 mmol) (19) was added and the resultant mixture refluxed for 0.5 h. Acidification and extraction afforded a residue that was chromatographed on silica gel (400 g) employing chloroform elution (100-mL fractions). Fraction 12 was concentrated, affording a mixture of dibenzyl ether (0.056 g, 0.2 mmol) and benzyl trifluoroethyl ether (0.087 g, 0.4 mmol). Fractions 13- 17 were concentrated and combined, affording clean dibenzyl ether (0.5775 g, 2.9 mmol). Dibenzyl ether had noOH stretch in the ir, and nmr (CDC13) 6: 7.33 (SH, s) and 4.53 (2H, s). Fractions 18-20 were concentrated and combined, yielding 2-propyl methane- sulfonate (0.0754 g, 0.5 mmol). Further elution provided unchanged benzyl alcohol (1.0792 g, 9.9 mmol).
Preparation of benzyl phenyl sulfide Potassium metal (1.0807 g, 27.7 mmol) was dissolved in tert-
butanol(100 mL). Benzenethiol (1 SO98 g, 13.7 mmol), benzyl alco- hol (1.4935 g, 13.8 mmol), and phenyl methanesulfonate (2.3659 g, 13.7 mmol) were added. The reaction mixture was refluxed for 2 h.
Can
. J. C
hem
. Dow
nloa
ded
from
ww
w.n
rcre
sear
chpr
ess.
com
by
UN
IVE
RSI
TY
OF
NO
RT
H T
EX
AS
LIB
RA
RY
on
11/1
0/14
For
pers
onal
use
onl
y.

LANGLER AND MORRISON 2389
Standard extraction gave crude, which was a mixture of ca. 2: 1 benzyl phenyl sulfide - benzyl alcohol.
Half of the crude product was added to a mixture of chromium trioxide (1.9989 g, 19.9 mmol) in glacial acetic acid (50 mL). The reaction mixture was maintained at 90-100°C for 0.5 h. Standard extraction yielded clean benzyl phenyl sulfone (0.7770 g, 3.3 mmol). Upon recrystallization from methanol, the product was shown to be identical to authentic material by mp (147-14g°C, lit. (ref. 20, p. 425) mp 14g°C), ir, and nmr.
Chlorinolysis of 3 Trifluoroethyl benzyl sulfide 3 (0.502 g, 2.4 mmol) was dissolved in
carbon tetrachloride (5 mL), and C12 (ca. 200 mL/min) was bubbled into the solution for 1 min. An nmr of the crude indicated that the reaction mixture was now ca. 28% monochlorosulfide and 72% dichlorosulfide 7. C12 was bubbled into the reaction mixture for another minute at the same rate. An nmr12 of the crude indicated complete conversion of the starting material into the dichlorosulfide 7.
The solvent was evaporated and the residue oxidized with chromium trioxide (0.6121 g, 6.1 mrnol) in glacial acetic acid (30 mL) in theusual way (8). After recrystallization of crude dichlorotrifluorosulfone from methanol, clean sulfone (0.3497 g, 1.4 mmol, 58% after two steps, mp 78-79.5"C) was obtained. The product sulfone had R f 0.26 on analytical tlc plates when developed with carbon tetrachloride; ir (CHC13): 1370 and 1170 cm-I; nmr (CDC13) 6: 7.43 (5H, s) and 4.70 (2H, s). Anal. calcd. for C9H7C12F302S: C 35.19, H 2.29, C123.08, S 10.44; found: C 35.27, H 2.28, C1 23.11, S 10.28.
Acknowledgements The authors are grateful to the Florida Institute of Technology
for financial support.
1. T. P. AHERN, D. G. KAY, and R. F. LANGLER. Can. J. Chem. 56, 2422 (1978).
2. J. R. HANCOCK, W. R. HARDSTAFF, P. A. JOHNS, R . F. LANGLER, and W. S. MANTLE. Can. J. Chem. 61, 1472 (1983).
3. W. R. HARDSTAFF and R. F. LANGLER. In Sulfur in organic and -
I2It is important to minimize the chlorination time, since cleavage of benzylic sulfides similar to 7 is an amply precedented process (ref. 3, p. 199).
inorganic Chemistry. Vol. 4. Edited by A. Senning. M. Dekker, Inc., New York. 1982. pp. 193-281.
4. G. E. WILSON, JR. Tetrahedron, 38, 2597 (1982). 5. J. L. GINSBURG and R. F. LANGLER. Can. J. Chem. 61, 589
(1983). 6. W. K. JARD~NE, R. F. LANGLER, and J. A. MACGREGOR. Can. J.
Chem. 60, 2069 (1982). 7. J. E. HUHEEY. J. Phys. Chem. 69, 3284 (1965). 8. H. 0 . FONG, W. R. HARDSTAFF, D. G. KAY, R. F. LANGLER,
R. H. MORSE, and D. N. SANDOVAL. Can. J . Chem. 57, 1206 ( 1979).
9. T. P. AHERN, H. 0. FONG, R. F. LANGLER, and P. M. MASON. Can. J. Chem. 58, 878 (1980).
10. C. BUNYAGIDJ, H. PIOTROWSKA, and M. H. ALDRIDGE. J. Org. Chem. 46, 3335 (1981).
11. (a) R. F. LANGLER and J. A. PINCOCK. Can. J. Chem. 55, 2316 (1977); (b) A. STREITWIESER, JR. and C. H. HEATHCOCK. Introduction to organic chemistry. 3rd ed. MacMillan, New York. 1985. p. 1156.
12. (a) F. G. BORDWELL, B. M. PITT, and M. KNELL. J. Am. Chem. Soc. 73,5004 (1951); (b) J. FERNS and A. LAPWORTH. J. Chem. SOC. 101, 273 (1912).
13. T. L. Ho. Hard and soft acids and bases principle in organic chemistry. Academic Press, New York. 1977. pp. 126-146.
14. Y. KOBAYASHI and I. KUMADAKI. ACC. Chem. Res. 14, 76 (1981).
15. D. G. KAY, R. F. LANGLER, and J. E. TRENHOLM. Can. J. Chem. 57, 2185 (1979).
16. A. M. AISSANI, J . C. BAUM, R. F. LANGLER, and J. L. GINSBURG. Can. J. Chem. 64, 532 (1986).
17. L. MUKHERJEE and E. GRUNWALD. J . Phys. Chem. 62, 131 1 (1958).
18. I. NAKAGAWA, K. AKI, and T. HATA. J. Chem. Soc. Perkm Trans. 1, 1315 (1983).
19. R. K. CROSSLAND and K. L. SERVIS. J . Org. Chem. 35, 3195 (1 970).
20. Z. RAPPOPORT. Handbook of tables for organic compound identification. 3rd ed. Chemical Rubber Co., Cleveland, OH. 1967. p. 421.
21. N. I. SAX. Dangerous properties of industrial materials. Van Nostrand Rheinhold Co., New York. 1979. p. 721.
Can
. J. C
hem
. Dow
nloa
ded
from
ww
w.n
rcre
sear
chpr
ess.
com
by
UN
IVE
RSI
TY
OF
NO
RT
H T
EX
AS
LIB
RA
RY
on
11/1
0/14
For
pers
onal
use
onl
y.





























![Catalyst-Controlled Chemoselective All-Alkene [2 + 2 + 2 ...tangyong.sioc.ac.cn/upfile/2019.2-acs.orglett.9b00209.pdf · Catalyst-Controlled Chemoselective All-Alkene [2 + 2 + 2]](https://img.pdfslide.net/doc/110x75/5f07e3ca7e708231d41f4418/catalyst-controlled-chemoselective-all-alkene-2-2-2-catalyst-controlled.jpg)