Embed Size (px)
Citation preview

American Journal of Medical Genetics 44:754-756 (1992)
Choanal Atresia: Evidence for Autosomal Recessive Inheritance
Ruth Gershoni-Baruch Division of Genetics, Rambam Medical Center, and the Bruce Rappaport Faculty of Medicine, Technion-Israel Institute of Technology, Haifa, Israel
Three individuals with choanal atresia were born in an inbred kindred. This report shows that nonsyndromal choanal atresia can be transmitted as an autosomal recessive trait. 0 1992 Wiley-Liss, Inc.
KEY WORDS: choanal atresia, autosomal re- cessive inheritance
INTRODUCTION Nonsyndromal choanal atresia usually is sporadic
and probably multifactorially determined. Familial cases, suggestive of both autosomal dominant and auto- soma1 recessive modes of inheritance, have been re- ported [Lang, 1912; Wright, 1922; Phelps, 1926; Stew- art, 1931; Schweckendiek, 1937; Wilkerson and Cayce, 1948; McGovern, 1950; Becker et al., 1957; Ransome, 1964; Dirlewanger, 1966; Grahne and Kaltiokallio, 1966; Qazi et al., 19821.
Here, we report on a small inbred Arab Moslem kin- dred showing choanal atresia in 2 sibs and their pater- nal uncle.
CLINICAL REPORTS Patient 1
M.H., (Fig. 1, V-2), a boy, is the second child of a 31- year-old father and a 26-year-old mother, of Moslem Arab origin, who were healthy and consanguineous (first and second cousins). Pregnancy and delivery were normal. Birth weight (BW) was 2,780 g (3rd centile). Apgar scores were 9 and 10 at 1 and 5 minutes, respec- tively. Dyspnea was noted at birth and aggravated by feeding. Attempts to pass a catheter through both nares were unsuccessful. A choanogram showed no passage of contrast to the nasopharynx on the left and minimal passage on the right. At 3 days the infant was trans- ferred to our hospital for surgery. The choanae (osseo-
Received for publication November 20, 1991; revision received June 2, 1992.
Address reprint requests to Ruth Gershoni-Baruch, MD, De- partment of Pediatrics, Division of Genetics, Rambam Medical Center, POB 9649, 31096 Haifa, Israel.
membranous) were opened and enlarged, and venting tubes were installed. These were removed 1 month later, following which dyspnea recurred necessitating repeat of surgery. At 2 months, venting tubes were reinstalled for another 3 weeks.
Clinical signs at examination (Fig. 2) include a reced- ing forehead, mild proptosis, epicanthic folds, appar- ently low-set ears, low nasal bridge, short nose, ante- verted nostrils, short philtrum, prominent alveolar ridges, bifid uvula and micrognathia. Weight was 3,150 g (< 3rd centile), length 54 cm (< 3rd centile), and OFC 38 cm (3rd centile).
Follow-up examination has shown restenosis and re- occlusion of choanae warranting a third choanoplasty.
The child, currently 10 months old, is mentally nor- mal, weighs 7,400 g (< 3rd centile), and is awaiting surgery.
Patient 2 M.H. (Fig, 1, V-1) is the elder sister of patient 1.
Pregnancy was uneventful and delivery was at term, spontaneous, and uncomplicated. BW was 2,830 g (10th centile) and Apgar scores were 9 and 10 at 1 and 5 minutes, respectively. At 1 day this infant girl was oper- ated on for bilateral membranous choanal stresia.
M.H. is currently 39/iz years old (Fig. 3). She is well and thriving. Her psychomotor development is normal.
Patient 3 F.H. (Fig. 1, IV-3) is the 30-year-old paternal uncle of
patients 1 and 2. He was born at home following an apparently normal pregnancy to consanguineous (first cousins) parents. Dyspnea was noted at age 3 days and was aggravated by feeding. He later suffered from rhinitis, snoring, stress dyspnea, and nasal speech.
At 16 years, bilateral occlusion of choanae was docu- mented by a choanogram showing no passage of lipiodol on the left and minimal passage on the right (few drops). The patient underwent bilateral choanoplasty and the diagnosis of bilateral osseomembranous choanal atresia was confirmed.
A year later the patient was reoperated on because of restenosis and occlusion of choanae. Follow-up examina- tion a few months later showed reocclusion of choanae warranting a third choanoplasty, which the patient re- fused to undergo.
0 1992 Wiley-Liss, Inc.
Familial Choanal Atresia 755
I
IV
V
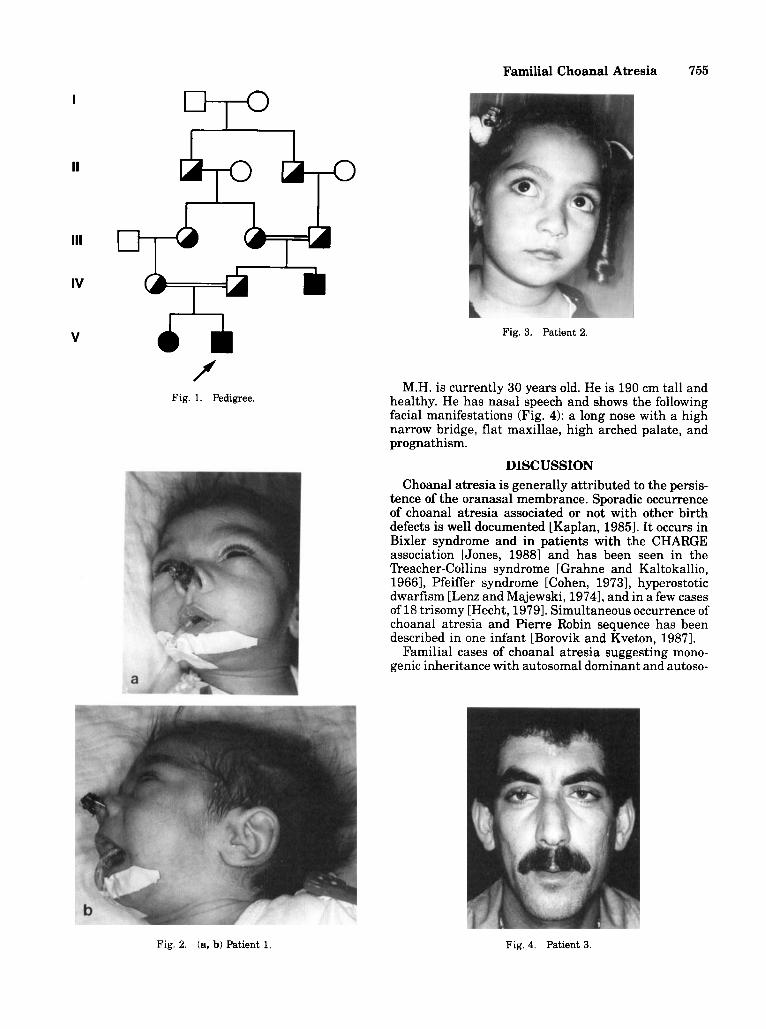
f Fig. 1. Pedigree.
Fig. 3. Patient 2.
M.H. is currently 30 years old. He is 190 cm tall and healthy. He has nasal speech and shows the following facial manifestations (Fig. 4): a long nose with a high narrow bridge, flat maxillae, high arched palate, and prognathism.
DISCUSSION Choanal atresia is generally attributed to the persis-
tence of the oranasal membrance. Sporadic occurrence of choanal atresia associated or not with other birth defects is well documented [Kaplan, 19851. It occurs in Bixler syndrome and in patients with the CHARGE association [Jones, 19881 and has been seen in the "reacher-Collins syndrome [Grahne and Kaltokallio, 19661, Pfeiffer syndrome [Cohen, 19731, hyperostotic dwarfism [Lenz and Majewski, 19741, and in a few cases of 18 trisomy [Hecht, 19791. Simultaneous occurrence of choanal atresia and Pierre Robin sequence has been described in one infant [Borovik and Kveton, 19871.
Familial cases of choanal atresia suggesting mono- genic inheritance with autosomal dominant and autoso-
Fig. 2. (a, b) Patient 1. Fig. 4. Patient 3.

756 Gershoni-Baruch
ma1 recessive modes of transmission have been reported [Lang, 1912; Wright, 1922; Phelps, 1926; Stewart, 1931; Schweckendiek, 1937; Wilkerson and Cayce, 1948; McGovern, 1950; Becker et al., 1957; Ransome, 1964; Dirlewanger, 1966; Grahne Kaltiokallio, 1966; Qazi et al., 19821. However, most cases of familial choanal at- resia have occurred in sibs born to unrelated and normal parents [Wright, 1922; Stewart, 1931; Schweckendiek, 1937; Wilkerson and Cayce, 1948; Becker et al., 1957; Ransome, 1964; Grahne and Kaltiokallio, 19661. On one occasion, choanal atresia was reported in 2 sibs and their paternal aunt, all products of consanguineous mat- ings [Qazi et al., 19821.
This report documents recurrence of choanal atresia in 3 close relatives. Patients 1 and 2 have an F of 7.1% and the pedigree (Fig. 1) is consistent with autosomal recessive inheritance.
REFERENCES Becker W, Matzker J , Schiffer KH (1957): Uber familiares Vorkommen
verschiedener Formen von Choanalatresie. Acta Otolaryngol 47: 377-382.
Borovik HR, Kveton JF (1987): Pierre Robin syndrome combined with unilateral choanal atresia. Otolaryngol Head Neck Surg 9667-70.
Cohen MM Jr (1973): An etiological and nosologic overview of crani- osynostosis syndromes. In Bergsma D (ed): “The Infant at Risk.” Miami: Symposia Specialists for The National Foundation - March of Dimes, BD:OAS X(2):137-189.
Dirlewanger A (1966): Hereditares Vorkommen von Choanalatresien. Pract Oto Rhino Laryngol 28:211-218.
Grahne B, Kaltiokallio K (1966): Congenital choanal atresia and its heredity. Acta Otolaryngol 62193-200.
Hecht F (1979): Chromosome eighteen trisorny syundrome. In Bergsma D (4): “Birth Defects Compendium,” 2nd New York: Alan R. Liss for The National Foundation - March of Dimes, pp 201-202.
Jones KL (1988): “Smith’s Recognizable Patterns of Human Malforma- tion,” 4th ed. Philadelphia: Saunders, pp 286-289.
Kaplan LC (1985): Choanal atresia and its associated anomalies: Fur- ther support for the charge association. Int J Pediatr Otorhino- laryngol 8:237-242.
Lang J (1912): Choanenatresie (Hereditat derselben). Mschr Ohren- heilk 46:970-1001.
Lenz WD, Majewski F (1974): A generalized disorder of the connective tissues with progeria, choanal atresia, symphangism, hypoplasia of dentine and craniodiaphyseal hyperostosis. In Bergsma D (ed): “Skeletal Dysplasia.” New York Excerpta Medica for The National Foundation - March of Dimes, BD:OAS X(12):133-136.
McGovern FH (1950): Congenital choanal atresia. Laryngoscope 60:815-831.
Phelps KA (1926): Congenital occlusion of choanae. Ann Otol 35: 143-151.
Qazi QH, Kanchanapoomi R, Beller E, Collins R (1982): Inheritance of posterior choanal atresia. Am J Med Genet 13:413-416.
Ransome J (1964): Familial incidence of posterior choanal atresia. J Laryngol78:551-554.
Schweckendiek H (1937): Transpalatine Behandlung angeborener Choanalatresien. Z Hals Nas Ohrenheilk 42:367-.
Stewart JP (1931): Congenital atresia of the posterior nares. Arch Otolaryngol 13:570-583.
Wilkerson WW Jr , Cayce JF (1948): Congenital choanal occlusion. ‘ham Am Acad Ophthalmol Otolaryngol52:234-236.
Wright AJ (1922): Congenital occlusion of the posterior choanae. J Laryngol37:556-569.
NOTE ADDED IN PROOF Since the submission of the manuscript, another sib,
sister of patients 1 and 2, with bilateral osse- omembranous choanal atresia was born. She is cur- rently 7 months old and was operated on twice. Follow up examination shows reocclusion of choanal warranting a third choanoplasty.





























