Embed Size (px)
Citation preview

i
COMBINATION THERAPY IN MALARIA
Orientations and Options for the
African Region
Thomas Sukwa, Wilson M. Were
Yao Kassankogno, Antoine B. Kabore
WORLD HEALTH ORGANIZATION Regional Office for Africa
Brazzaville

ii
COMBINATION THERAPY IN MALARIA
Orientations and Options for the
African Region
Dr Thomas Sukwa Division of Prevention and Control
of Communicable Diseases,
WHO Regional Office for Africa
Dr Wilson M. Were Rubaga Hospital, Kampala, Uganda
Dr Yao Kassankogno WHO Representative for Chad,
Ndjamena, Chad
Dr Antoine B. Kabore Director, Division of Prevention and Control
of Communicable Diseases,
WHO Regional Office for Africa
WORLD HEALTH ORGANIZATION Regional Office for Africa
Brazzaville • 2004

iii
©
WHO Regional Office for Africa (2004)
Publications of the World Health Organization enjoy copyright protection in accordance with the provisions of Protocol 2 of the Universal Copyright Convention. All rights reserved. The designations employed and the presentation of the material in this publication do not imply the expression of any opinion whatsoever on the part of the Secretariat of the World Health Organization concerning the legal status of any country, territory, city or area or of its authorities or concerning the delimitation of its frontiers or boundaries. The mention of specific companies or of certain manufacturers’ products does not imply that they are endorsed or recommended by the World Health Organization in preference to others of a similar nature that are not mentioned. Errors and omissions excepted, the names of proprietary products are distinguished by initial capital letters.
Printed in the Republic of South Africa

iv
Contents
Pages
Abbreviations ................................................................................................................................ i
Acknowledgements ...................................................................................................................... ii
Preface .......................................................................................................................................... iii
Executive Summary .................................................................................................................... iv
1. INTRODUCTION .........................................................................................................................1
2. CURRENT STATUS OF ANTIMALARIAL DRUG RESISTANCE ....................................................3
2.1 Development of Resistance ..............................................................................................3 2.2 Assessment of Antimalarial Drug Efficacy .....................................................................4 2.3 Plasmodium falciparum Resistance .................................................................................5 2.4 Plasmodium vivax Resistance ..........................................................................................9 2.5 Regional Responses to Antimalarial Drug Resistance .....................................................9
3. COMBINATION THERAPY IN TREATMENT POLICY ................................................................14
3.1 Purpose of Drug Policy ..................................................................................................14 3.2 Combination Therapy of Antimalarial Drugs ................................................................15 3.3 Rationale for the Use of Combination Therapy in the African Region .........................17
4. AVAILABLE ANTIMALARIAL COMBINATION DRUGS .............................................................19
4.1 Artemisinin-based Combination Therapy ......................................................................19 4.2 Non-Artemisinin-based Combinations ..........................................................................24
5. COMBINATION DRUGS IN DEVELOPMENT .............................................................................28
5.1 Chloroproguanil-Dapsone-Artesunate ...........................................................................28 5.2 Pyronaridine-Artemisinin Derivative .............................................................................28 5.3 Piperaquine-Dihydroartemisinin ....................................................................................29
6. FORMULATING AND IMPLEMENTING COMBINATION THERAPY POLICY .............................30
6.1 Combination Therapy Drug Policy Formulation ...........................................................30 6.2 Drug Characteristics and Cost ........................................................................................31 6.3 Accessing Artemisinin-based Combination Treatment .................................................33 6.4 Policy Review and Update Towards Combination Therapy ..........................................34 6.5 Introduction of Combination Therapy ...........................................................................36 6.6 Technical Process of Combination Therapy Introduction .............................................37 6.7 Combination Therapy and Community Treatment of Malaria ......................................39
7. FUTURE RESEARCH ACTIVITIES ............................................................................................40
7.1 Research and Development for Combination Therapy ..................................................40 7.2 Research Priorities .........................................................................................................40
8. CONCLUSION ...........................................................................................................................42

v
Pages
References ....................................................................................................................................43
Annexes 1. Malaria Life Cycle and How Antimalarial Drugs Work ........................................................51 2. Drug Dosage Tables ................................................................................................................52 3. Characteristics of Common Antimalarial Drugs .....................................................................55
List of Tables
1. Current antimalarial drug resistance and antimalarial treatment policies in selected African countries ................................................................................................................................ 9 2. Current antimalarial drug resistance and antimalarial treatment policies in selected Asian countries .................................................................................................................. 11 3. Current antimalarial drug resistance and antimalarial treatment policies in selected oceanic countries ............................................................................................................... 12 4. Current antimalarial drug resistance and antimalarial treatment policies in selected countries in South America .............................................................................................. 13 5. Factors concerning artemisinin-based combination therapy in Africa ...................................... 18
List of Figures
1. Quality of antimalarial drugs dissolution ..................................................................................4 2. A global picture of reduced susceptibility of P. falciparum to various antimalarial drugs ......................................................................................................................6 3. In-vivo chloroquine therapeutic clinical efficacy in the African Region, 1995–2001 ..............7 4. Sulfadoxine-pyrimethamine therapeutic efficacy patterns in the African Region, 1995–2001 ...................................................................................................................8 5. Balancing prompt treatment against minimizing drug resistance ...........................................15 6. Cost of monotherapy versus combination therapy ..................................................................32 7. Options for replacing sulfadoxine-pyrimethamine as first-line drug ......................................35 8. Options for replacing chloroquine as first-line drug ..............................................................36 9. Phased introduction of combination therapy within a country ...............................................37

vi
Abbreviations
ACR adequate clinical response ACT artemisinin-based combination therapy AIDS acquired immunodeficiency syndrome AM artemether ART artesunate ASU artesunate ATM artemether AQ amodiaquine CD clindamycin CDA chlorproguanil-dapsone-artesunate CQ chloroquine CT combination therapy D doxycycline DHA dihydroartemisinin DHFR dihydrofolate reductase DHPS dihydropteroate synthetase DRA Drug Regulatory Authority ETF early treatment failure GMP Good Manufacturing Practice HIV human immunodeficiency virus IDA International Dispensary Association LapDap chlorproguanil-dapsone LTF late treatment failure LUM lumefantrine MQ mefloquine NGO nongovernmental organization PAHO Pan American Health Organization PQ primaquine Q quinine RBM Roll Back Malaria RDT rapid diagnostic test SP sulfadoxine-pyrimethamine T tetracycline UNICEF United Nations Children’s Fund US United States WHO World Health Organization WHO/AFRO World Health Organization, Regional Office for Africa

vii
Acknowledgements
We are grateful to all colleagues within the World Health Organization (WHO) for having dedicated their time to review and give input towards the finalization of this publication. The draft was reviewed by participants in two intercountry workshops held in Dar es Salaam, Tanzania and Cotonou, Benin in June 2002 to deliberate on issues regarding combination therapy in the African Region. We are thankful to them all for their individual and collective contributions.
The support from the secretarial staff of the Malaria Unit at the Regional Office for Africa
is greatly appreciated.

viii
Preface
Malaria is endemic in 42 of the 46 countries of the WHO African Region and ranks in the top five causes of illness and death. Malaria causes 300 million to 500 million episodes of acute illness and 1.2 million deaths per year globally. It is the leading cause of death in children under 5 years in sub-Saharan Africa and, in some countries, accounts for one-quarter of all such deaths. Malaria is a disease of poverty: 58% of all malaria deaths are concentrated in the world’s poorest 20%, the highest association of any disease with poverty.
Prompt diagnosis and treatment of cases is one major strategy for controlling malaria.
However, development and spread of resistance to commonly used antimalarial drugs is a major impediment to implementation of this strategy. Since 1996, the WHO Regional Office for Africa has provided technical support to a large number of countries to promote rational use of antimalarial drugs, monitor drug efficacy and update treatment policy according to agreed frameworks through networks of consultants and intercountry consultations. During recent years, an increasing number of African governments have implemented new malaria treatment guidelines. The high levels of resistance of Plasmodium falciparum to chloroquine have been the major factor for replacing this inexpensive and relatively safe antimalarial drug with alternative first-line treatments. Currently, 13 countries have replaced chloroquine as first-line treatment: eight have adopted sulfadoxine-pyrimethamine (SP) monotherapy, and five have adopted SP in combination with either chloroquine or amodiaquine.
WHO currently recommends that countries faced with the problem of antimalarial drug
resistance should adopt combination therapies, particularly those containing an artemisinin-based compound. Evidence from other WHO regions supports this recommendation. However, the following hindrances have been recognized: (i) cost of combination therapies, (ii) lack of post-marketing surveillance data on the new therapies and (iii) safety in very young children and during pregnancy. It is important to note that these should be seen as challenges and not deterrents from introducing combination therapies for treating malaria in the Region.
This document aims at guiding countries on making appropriate choices about what
combination drugs would be most suitable in their environment. It is targeted at policy-makers, programme managers, researchers, cooperating partners, nongovernmental organizations and others involved in malaria control. This publication should be used in conjunction with other documents on drug policy change developed by the Regional Office for Africa. Introducing combination therapy in the African Region should improve efficacy and effectiveness of first-line antimalarial treatment as well as delay the development and selection of resistant parasites to the few currently available and efficacious drugs.
Antoine Kaboré Director, Division of
Prevention and Control of
Communicable Diseases

ix
Executive Summary
Malaria remains the single biggest killer of young children in Africa and is a major public health problem in many other parts of the world. One of the important pillars of the Malaria Control Strategy in the African Region is case management. This is based on the principle of early recognition of disease with prompt treatment, using appropriate and effective antimalarial drugs.
In order to implement this strategy rationally, programmes must provide communities with
sensitive, affordable and efficacious drugs. However, the appearance of drug-resistant Plasmodium falciparum in Africa in 1978 made it difficult to choose first-line antimalarial drugs for widespread use in malaria control programmmes. This is because there are few available antimalarial drugs as optional choices for the current first-line drugs, the cost of the potential alternatives are beyond what the average person in the communities can afford, and some of the newer compounds may have side effects that preclude them from wide-scale use in public health care. In addition, the rate of emergence of resistant strains of malaria parasites to some of these new possible alternative first-line antimalarial compounds is very rapid compared to what obtained in the past when chloroquine was commonly used as the first-line drug. Chloroquine was deployed in endemic areas for treatment and prophylaxis for more than thirty years before the first resistant strains occurred almost simultaneously in south-east Asia and South America. On the other hand, within a few years of the deployment of sulfadoxine-pyrimethamine and mefloquine, albeit at different times, resistant strains became common in the areas where they were deployed. Unfortunately, all the factors that contribute to the emergence of resistance in any particular area are still not known.
In 2001, a World Health Organization consultation recommended that countries in Africa
should adopt combination therapy as a way of limiting the development and spread of resistance to antimalarial drugs. Combination therapy in malaria is defined as the simultaneous use of two or more blood schizonticidal drugs with independent modes of action and different biochemical targets in the parasite. The concept of combination therapy is based on the synergistic or additive potential of two or more drugs to improve therapeutic efficacy and also delay the development of resistance to the individual components of the combination. The aim is to improve efficacy and retard the development of resistance to the individual components of the combination. This concept has been realized in multiple-drug therapy for leprosy, tuberculosis and cancer, and, more recently, in antiretroviral treatments. Based on available safety and efficacy data, the consultation identified the following therapeutic options as available and having potential for deployment (in prioritized order) if costs were not an issue: (i) artemether-lumefantrine, (ii) artesunate plus amodiaquine, (iii) artesunate plus sulfadoxine-pyrimethamine in areas where SP efficacy remains high and (iv) SP plus amodiaquine in areas where the efficacy of both amodiaquine and SP remains high, which is mainly in countries of west Africa.
Access to these combinations was previously constrained by high prices; scarcity of
quality, fully validated co-formulated products; the relatively extensive regimens of some of the treatments; and the scarcity of post-marketing surveillance (Phase IV) safety data, particularly for pregnant women. The recommendation for combination therapy has brought a new dimension to the process of policy change at country level. The Regional Office for Africa,

x
therefore, felt it necessary to provide countries with orientations on the WHO recommendations for treatment of uncomplicated malaria with combination therapy.
This publication provides orientations on combination therapy, options available for
countries in the Region, policy formulation and implementation, key research priorities and general information on most antimalarial drugs, including dosage tables. It is intended for use by programme managers, researchers, cooperating partners, nongovernmental organizations and others involved in malaria control. It should be used in conjunction with other documents on drug policy change developed by the Regional Office for Africa. These orientations will guide countries in making appropriate choices about what drug combinations would be most suitable in their environment. Introducing combination therapy in the African Region should improve efficacy and effectiveness of first-line antimalarial treatment as well as delay the development and selection of resistant parasites to the few currently available and efficacious drugs.

1
1. INTRODUCTION
Malaria, the most important parasitic infection of humans, affects about 5% of the world’s population. It is estimated that the incidence of malaria in the world is between 300 million and 500 million clinical cases per year. Of the estimated 1.5 million to 2.7 million annual deaths from malaria worldwide, about one million occur among children under 5 years of age in Africa south of the Sahara (1). Malaria is endemic in 42 of the 46 countries in the WHO African Region; 90% of the malaria burden is estimated to exist in sub-Saharan Africa, most of it due to Plasmodium falciparum. In addition to the morbidity associated with acute symptoms, there is the burden of anaemia and chronic disease attributed to malaria parasites. In first and second pregnancies, malaria is associated with low birth weight, anaemia and both perinatal and maternal mortality (2). Using macroeconomic methods, the reduction in the growth of gross domestic product per capita due to malaria has been shown to vary substantially from 0.25% to 1.3% per year. The economic loss due to malaria in Africa in 1989 was estimated at US$ 800 million; in the year 2000, it was US$ 12 billion per annum (3).
The emergence and rapid spread of P. falciparum resistance to commonly used antimalarial
drugs poses a serious challenge to the effectiveness of early diagnosis and prompt treatment as a priority within current regional strategy for malaria control efforts (4–6). The effectiveness of this intervention is highly dependent on antimalarial drugs which should not only be safe and effective, but also available, affordable and acceptable to the population at risk. The rational use of an effective antimalarial drug reduces the risk of severe disease and death; it shortens the duration of the illness and at the same time hinders the development of parasite resistance.
In November 2000, an informal consultation on the use of antimalarial drugs was convened
by WHO in Geneva. The meeting reviewed and updated recommendations on the use of antimalarial drugs for chemoprophylaxis and treatment, based on the information available (7). Participants acknowledged the limited number of treatment options in countries to improve treatment policies, especially in sub-Saharan Africa which has the most resource constraints. Inadequate resources in the African Region have contributed to the continued use of drugs which have become ineffective and compromised by drug resistance. The potential value of malaria therapy using combinations of drugs was identified as a strategic and viable option in improving efficacy, and delaying development and selection of resistant parasites (8–12). However, the systematic review of existing data on combination therapy (CT) for malaria and identification of specific candidate drugs, especially for Africa, was beyond the scope of the November meeting.
As a result, in April 2001, Roll Back Malaria (RBM) convened a WHO technical
consultation on antimalarial combination therapy in Geneva, Switzerland. Participants reflected a wide range of expertise from Africa and other regions in the development and use of antimalarial drugs (13). The meeting reviewed current evidence on CT with antimalarial drugs; recommended the minimal criteria for selection and use of CT in different epidemiological settings; selected the appropriate combinations for use, particularly in African countries; and identified priority research, product development and production needs to facilitate the implementation of antimalarial CT. It was observed that artemisinin-based combinations have several distinct advantages in that they produce rapid clinical and parasitological cure, reduce gametocyte

2
carriage rate, and are well tolerated; there was no documented parasite resistance at that time. However, these artemisinin-based combination therapies (ACTs) are at an early stage in Africa and there is a need for post-marketing surveillance data.
Based on available safety and efficacy data, the meeting identified the following
therapeutic options as currently available and having potential for deployment (in prioritized order) if costs were not an issue: (i) artemether-lumefantrine; (ii) artesunate plus amodiaquine; (iii) artesunate plus sulfadoxine-pyrimethamine in areas where SP efficacy remains high and (iv) SP plus amodiaquine in areas where efficacy of both amodiaquine and SP remains high which is mainly in countries of west Africa.
However, two practical issues were raised. First, there has been limited clinical experience for many of the combinations being considered. It was acknowledged that full safety and efficacy needed to be demonstrated on a case-by-case basis involving appropriate prospective studies, including regulatory and Phase IV studies, and appropriate surveillance, particularly of adverse events. Secondly, in the case of artemisinin-based combinations, the cost of treatment is up to ten times more than traditional monotherapies such as chloroquine or SP. In most African countries where health care systems cannot afford the introduction of combination therapies, other alternatives may need to be considered.
This is the dilemma facing national malaria control programmes in the African Region, and increased global funding will be required to facilitate the appropriate exploration of the use and purchase of optimal combination therapy antimalarial drugs. As a follow-up to the April meeting, the WHO Regional Office for Africa found it necessary to give orientations to countries on the use of combination therapy. It is therefore the purpose of this publication to assist national programmes to critically review what combination therapy would be most appropriate for the level of drug resistance and epidemiological settings existing in the individual countries.
Other possible artemisinin-based combination therapies were recommended for accelerated development. They include piperaquine-DHA combinations (piperaquine-dihydroartemisinin-trimethoprim; piperaquine-dihydroartemisinin-trimethoprim-primaquine), LapDap-artesunate and pyronaridine-artesunate. On the other hand, some options were not recommended for antimalarial treatment policy in the African Region (for reasons explained in Chapter 4). These include (i) chloroquine-based combinations (CQ + SP and CQ + artesunate), (ii) one-day treatment of artesunate + SP, (iii) mefloquine-based combinations (e.g. mefloquine + artesunate) in areas of high malaria transmission. The reasons are discussed in Chapter 4 under each drug.
This publication is intended for use by malaria control programme managers, malaria drug
policy committees, international donor organizations, nongovernmental organizations involved in malaria control, and research and training institutions. It is meant to be a reference document to help define different national policies and guidelines on the use of combination therapy in the Region. However, it should be used in conjunction with other documents on drug policy change and diagnosis or treatment of malaria. It is essential that all health care providers, public or private, be fully cognizant of national antimalarial drug policies and their rationale.

3
2. CURRENT STATUS OF ANTIMALARIAL DRUG
RESISTANCE
2.1 Development of Resistance
Resistance to antimalarial drugs arises as a result of spontaneously occurring mutations that affect the structure and activity at the molecular level of the drug target in the malaria parasite or affect the access of the drug to that target (15). Mutant parasites are selected if antimalarial drug concentrations are sufficient to inhibit multiplication of susceptible parasites but are inadequate to inhibit the mutants, a phenomenon known as drug selection (9,16).
The evolution of drug resistance in Plasmodium is not fully understood although the molecular basis for resistance is becoming clearer. Available evidence indicates that for P. falciparum some of these mutations occur in a transporter-like gene on the surface of the parasite food vacuole, and probably different sets of mutations are involved in chloroquine resistance for P. vivax (17). The molecular basis for resistance to antifolates such as sulfadoxine-pyrimethamine (SP) has been well characterized. P. falciparum resistance to SP is primarily conferred by successive single-point mutations in parasite dhfr, the gene that encodes the target enzyme dihydrofolate reductase (DHFR), and by additional mutations in dhps which encodes for the enzyme dihydropteroate synthetase (DHPS) (18).
Various factors relating to drug, parasite and human host interactions contribute to the development and spread of drug resistance. The molecular mechanism of drug action is a critical element in the speed at which resistance develops. In addition, drugs with a long terminal elimination half-life enhance the development of resistance, particularly in areas of high transmission. Similarly, increased drug pressure is a significant contributor to drug resistance. As increased amounts of a drug are used, the likelihood that parasites will be exposed to inadequate drug levels rises and resistant mutants are more readily selected (19). Parasite factors associated with resistance include the Plasmodium species concerned and the intensity of transmission. Human host factors include the widespread incorrect use or irrational use of antimalarial drugs and possibly the level of host immunity. In 2001, WHO Essential Drugs and Medicines undertook studies on the quality of antimalarial drugs in eight countries. The study indicated that a substantial percent of chloroquine and sulfadoxine-pyrimethamine tablets were of low quality (Figure 1).
Antimalarial drug resistance is the ability of a parasite strain to survive or multiply despite the administration and absorption of a drug given in doses equal to or higher than those
usually recommended, but within the limits of tolerance of the subject (14).

4
Figure 1: Quality of Figure 1: Quality of antimalarialantimalarial drugs dissolutiondrugs dissolution
FF FF
0102030405060708090
100
Gabon
Ghana
Kenya
Mali
Mozambique
Sudan
Zimbabwe
Percent failure (%)
Chloroquine
Tablets
SP Tablets
Source: WHO/EDM/HQ, 2002
The increase in chloroquine resistance in east Africa has led to a rise in malaria mortality
(5). Similarly, a significant rise in malaria mortality in children under 5 years of age has been observed in Senegal in west Africa, coinciding with the emergence of chloroquine resistance in the area (20). In Democratic Republic of the Congo and Malawi, there was increased incidence of severe malaria (21). Antimalarial drug resistance has also been implicated in the increasing frequency and severity of epidemics (4).
2.2 Assessment of Antimalarial Drug Efficacy
A major purpose of assessing the therapeutic efficacy of antimalarial drugs in confirmed
malaria patients is to monitor efficacy over time, especially in vulnerable groups in highly endemic areas, and to guide treatment policy. Antimalarial drug responses are assessed clinically from rates of symptom resolution (e.g. fever clearance, coma recovery) or parasitologically from parasite clearance and overall cure rates.
Until the end of the 1980s, most in vivo studies focused on the parasitological response to a given drug, and infections were classified as sensitive (S) or resistant (R) at one of three levels: RI, RII or RIII. An RI response corresponds to an initial clearance of parasitaemia and then recrudescence 8 or more days after treatment; an RII response is the clearance or substantial reduction of parasitaemia with recrudescence of parasitaemia on days 4–7; and an RIII response refers to a situation in which there is no initial reduction of asexual parasitaemia and the levels may actually increase (13). Follow-up of treated patients for evidence of recurrence of

5
parasitaemia may continue for 7, 14 or 28 days, depending on the investigators’ interest in detecting lower levels of resistance and on budgetary limitations (22–24).
Protocols have been modified and simplified to facilitate their use in high-transmission
areas in Africa, where populations may have asymptomatic parasitaemia in the absence of clinical manifestation. The generally accepted objective of malaria treatment in these areas is not so much the clearance of parasitaemia but the resolution of clinical symptoms and acute febrile illness as measured by the adequate clinical response (ACR) and early and late treatment failure (ETF and LTF) (25). The therapeutic response is classified as ETF if the patient has aggravation or persistence of clinical symptoms in the presence of parasitaemia during the first 3 days of follow-up; and as LTF if there is reappearance of symptoms in the presence of parasitaemia during days 4–14 of follow-up, without previously meeting the criteria for ETF. ACR is either the absence of parasitaemia on day 14 (irrespective of axillary temperature), or the absence of clinical symptoms on day 14 (irrespective of parasitaemia) in patients who did not meet the criteria of ETF or LTF before. Although the measurement of clinical response is of value in areas of high transmission, the impact of asymptomatic residual parasitaemia on other malaria-related conditions, such as anaemia and malnutrition, has not been examined (4).
Experience in malaria control programmes has shown that in vitro tests of parasite
susceptibility to antimalarial drugs cannot substitute for in vivo observations on malaria therapy. However, they are useful research tools which provide background information for the development and evaluation of drug policies and can provide an early warning of the appearance of drug resistance. The application and usefulness of in vitro tests is restricted by the need for trained personnel and their labour-intensive nature.
2.3 Plasmodium falciparum Resistance
Chloroquine
Strains of P. falciparum resistant to chloroquine were first suspected in Thailand in 1957
and found in patients in Colombia and Thailand in 1960. Since then, resistance to this drug has spread widely (Figure 2), and there is now high-level resistance to chloroquine in south Asia, south-east Asia, Oceania, the Amazon Basin and some coastal areas of South America. In Africa, chloroquine resistance was first documented in Tanzania in 1979 and has spread and intensified in the last 20 years. Strains of P. falciparum remain sensitive to chloroquine in Central America north of the Panama Canal, the island of Hispaniola (Haiti and the Dominican Republic) and in El Faiyûm governorate in Egypt.

6
Figure 2: A global picture of reduced susceptibility of P. falciparum to various antimalarial
drugs
Chloroquine resistance
SP resistance
Multidrug resistance
Source: WHO/HQ, 2003
Chloroquine resistance in the African Region
WHO has been supporting countries to monitor drug efficacy since 1995, and so far data
available have shown increasing malaria parasite resistance to chloroquine moving generally across the continent. In most countries of east Africa and in Ethiopia, more than 50% of patients currently experience a recurrence of parasitaemia with symptoms by day 14 after treatment. Moderate levels of resistance are found in central and southern Africa. In west Africa, reported rates of resistance vary widely but tend to be lower than in central and southern Africa (Figure 3).

7
Figure 3: In vivo chloroquine therapeutic clinical efficacy in the African Region, 1995–2001
< 25%
≥≥≥≥ 25%
< 25%
≥≥≥≥ 25%
Source: WHO/AFRO database, 2001
Amodiaquine
Although amodiaquine is generally more effective than chloroquine against chloroquine-
resistant strains of P. falciparum (26), there is cross-resistance, and moderate-to-high levels of amodiaquine resistance have been reported from Papua New Guinea, east Africa and the Amazon Basin. This drug continues to be efficacious as a single drug in most of west and central Africa and on the northern Pacific coast of South America where, in some countries, it is used in combination with sulfadoxine-pyrimethamine.
Sulfadoxine-pyrimethamine (SP) High levels of resistance to sulfadoxine-pyrimethamine are found in the Amazon Basin and
throughout south-east Asia, with the possible exception of some areas in eastern Cambodia and northern Viet Nam. In east Africa, resistance rates are variable, ranging from 10% to 50% in 14-day therapeutic efficacy trials. Low levels of resistance (< 10% ETF + LTF) are found on the Indian subcontinent, in central and southern Africa and in coastal areas of South America.

8
Figure 4: Sulfadoxine-pyrimethamine therapeutic efficacy patterns in the African Region,
1995–2001
<25%< 25%
Source: WHO/AFRO database, 2001
Quinine
Decreasing sensitivity to quinine has been detected in areas of south-east Asia where it has
been extensively used as the first-line treatment for malaria and in some parts of South America. Patient adherence to a 7-day regimen as a single drug or in combination with other drugs such as tetracyclines is low, leading to incomplete treatment and parasite recrudescences. This may have led to the selection of resistant parasites. There is some cross-resistance between quinine and mefloquine, suggesting that the wide use of quinine in Thailand might have influenced the development of resistance to mefloquine in that country (27). Strains of P. falciparum from Africa are generally highly sensitive to quinine.
Mefloquine
Recurrences of parasitaemia in over 50% of the patients treated with mefloquine alone have
been reported from border areas between Cambodia, Myanmar and Thailand. Mefloquine resistance is uncommon in the remainder of south-east Asia. In the Amazon Basin, mefloquine resistance has been reported only from Brazil, where clinical failure rates remain below 5% (28). Existing data indicate that, in some endemic areas, mefloquine-resistant parasites may be found prior to the introduction of the drug. For example, isolates with reduced sensitivity to mefloquine have been reported from several sites in west and central Africa, although the drug has never been widely used there (29). In such cases, there is a potential for resistance to spread if mefloquine monotherapy is used on a large scale.

9
Artemisinin and its derivatives
The recrudescence rate is high when artemisinin and its derivatives are used in
monotherapy, depending on the dose administered, the duration of treatment and the severity of disease but not at present on parasite resistance (30–34). Treatment regimens of less than 7 days gave unacceptably high recrudescence rates (35). In spite of reports of decreasing in vitro susceptibility so far, there is no confirmed in vivo evidence of resistance of P. falciparum to artemisinin and its derivatives.
2.4 Plasmodium vivax Resistance
Chloroquine
Plasmodium vivax resistance to chloroquine was first reported from Irian Jaya (Indonesia)
and Papua New Guinea in 1989. Nearly 50% of strains from these areas currently show evidence of reduced susceptibility in 28-day in vivo tests (36). Well-documented reports of resistance in individual patients or small case series have also appeared from Brazil, Guatemala, Guyana, India and Myanmar, but the resistance appears to be focal and much less intense. In Africa, P. vivax is rare, and efficacy studies from both Eritrea and Ethiopia have not demonstrated any resistance yet.
2.5 Regional Responses to Antimalarial Drug Resistance
Africa
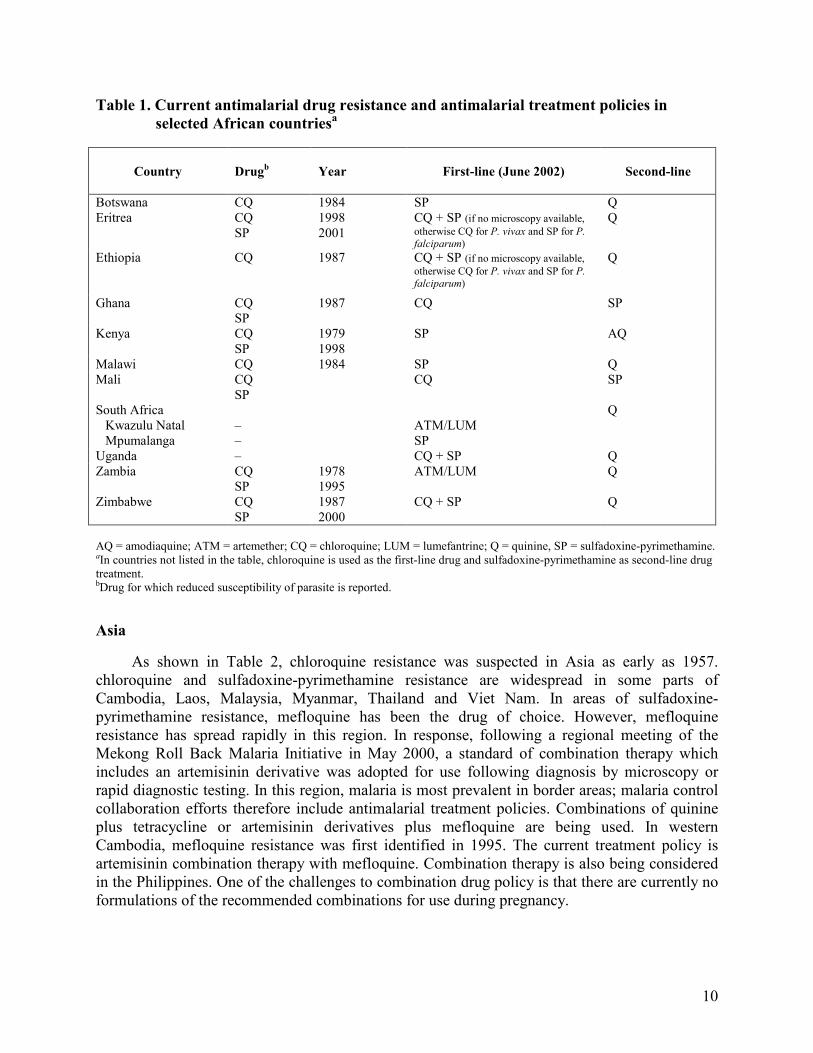
The current situation of antimalarial drug resistance in Africa is summarized in Table 1.
Since 1995, WHO and national malaria control programmes in the African Region have responded to the spread and intensification of chloroquine-resistant P. falciparum by strengthening national capacity in conducting 14-day in vivo drug efficacy studies in 37 countries south of the Sahara.
On the basis of the results from these studies and other available information, 17 countries have changed their antimalarial treatment policies: Botswana, Burundi, Cameroon, Democratic Republic of Congo, Eritrea, Ethiopia, Gabon, Kenya, Malawi, Mozambique, Rwanda, Senegal, South Africa, Tanzania (mainland and Zanzibar), Uganda, Zambia and Zimbabwe.
10
Table 1. Current antimalarial drug resistance and antimalarial treatment policies in
selected African countriesa
Country
Drugb
Year First-line (June 2002) Second-line
Botswana CQ 1984 SP Q Eritrea CQ
SP 1998 2001
CQ + SP (if no microscopy available,
otherwise CQ for P. vivax and SP for P. falciparum)
Q
Ethiopia
CQ 1987 CQ + SP (if no microscopy available,
otherwise CQ for P. vivax and SP for P. falciparum)
Q
Ghana
CQ SP
1987 CQ SP
Kenya
CQ SP
1979 1998
SP AQ
Malawi CQ 1984 SP Q Mali
CQ SP
CQ SP
South Africa Kwazulu Natal Mpumalanga
– –
ATM/LUM SP
Q
Uganda – CQ + SP Q Zambia CQ
SP 1978 1995
ATM/LUM Q
Zimbabwe CQ SP
1987 2000
CQ + SP Q
AQ = amodiaquine; ATM = artemether; CQ = chloroquine; LUM = lumefantrine; Q = quinine, SP = sulfadoxine-pyrimethamine. aIn countries not listed in the table, chloroquine is used as the first-line drug and sulfadoxine-pyrimethamine as second-line drug treatment. bDrug for which reduced susceptibility of parasite is reported.
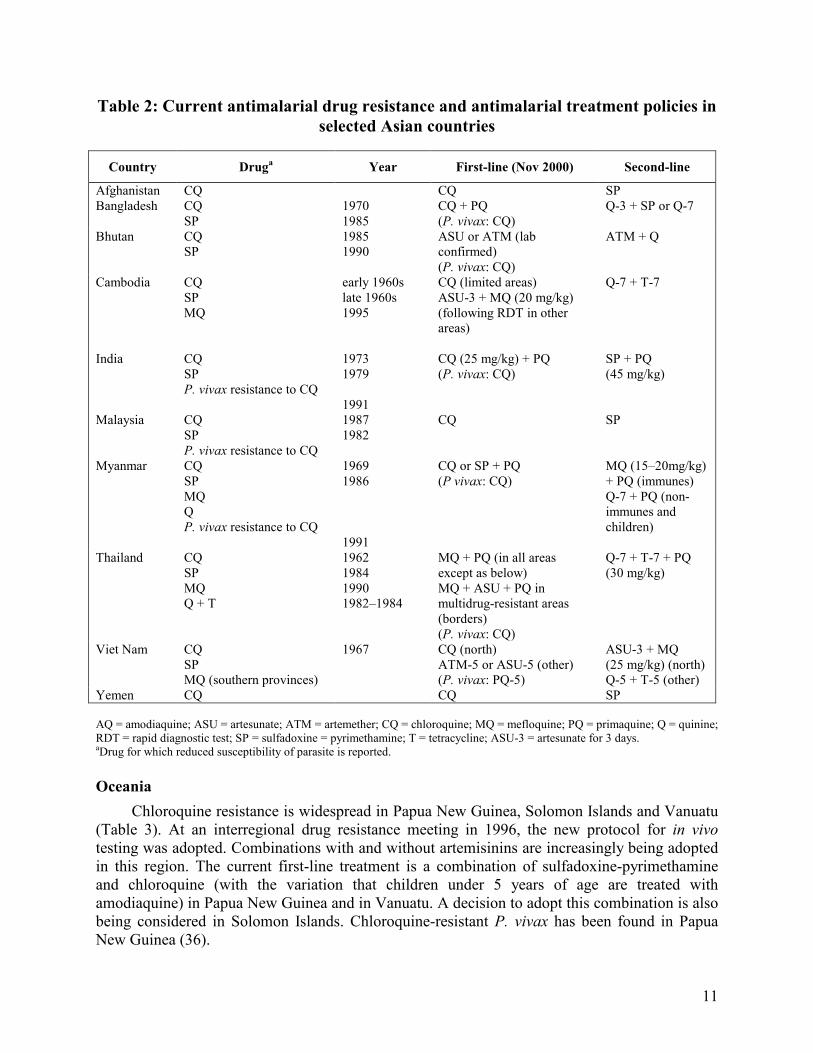
Asia
As shown in Table 2, chloroquine resistance was suspected in Asia as early as 1957. chloroquine and sulfadoxine-pyrimethamine resistance are widespread in some parts of Cambodia, Laos, Malaysia, Myanmar, Thailand and Viet Nam. In areas of sulfadoxine-pyrimethamine resistance, mefloquine has been the drug of choice. However, mefloquine resistance has spread rapidly in this region. In response, following a regional meeting of the Mekong Roll Back Malaria Initiative in May 2000, a standard of combination therapy which includes an artemisinin derivative was adopted for use following diagnosis by microscopy or rapid diagnostic testing. In this region, malaria is most prevalent in border areas; malaria control collaboration efforts therefore include antimalarial treatment policies. Combinations of quinine plus tetracycline or artemisinin derivatives plus mefloquine are being used. In western Cambodia, mefloquine resistance was first identified in 1995. The current treatment policy is artemisinin combination therapy with mefloquine. Combination therapy is also being considered in the Philippines. One of the challenges to combination drug policy is that there are currently no formulations of the recommended combinations for use during pregnancy.
11
Table 2: Current antimalarial drug resistance and antimalarial treatment policies in
selected Asian countries
Country Druga Year First-line (Nov 2000) Second-line
Afghanistan CQ CQ SP Bangladesh CQ
SP 1970 1985
CQ + PQ (P. vivax: CQ)
Q-3 + SP or Q-7
Bhutan CQ SP
1985 1990
ASU or ATM (lab confirmed) (P. vivax: CQ)
ATM + Q
Cambodia CQ SP MQ
early 1960s late 1960s 1995
CQ (limited areas) ASU-3 + MQ (20 mg/kg) (following RDT in other areas)
Q-7 + T-7
India CQ SP P. vivax resistance to CQ
1973 1979 1991
CQ (25 mg/kg) + PQ (P. vivax: CQ)
SP + PQ (45 mg/kg)
Malaysia CQ SP P. vivax resistance to CQ
1987 1982
CQ SP
Myanmar CQ SP MQ Q P. vivax resistance to CQ
1969 1986 1991
CQ or SP + PQ (P vivax: CQ)
MQ (15–20mg/kg) + PQ (immunes) Q-7 + PQ (non-immunes and children)
Thailand CQ SP MQ Q + T
1962 1984 1990 1982–1984
MQ + PQ (in all areas except as below) MQ + ASU + PQ in multidrug-resistant areas (borders) (P. vivax: CQ)
Q-7 + T-7 + PQ (30 mg/kg)
Viet Nam CQ SP MQ (southern provinces)
1967 CQ (north) ATM-5 or ASU-5 (other) (P. vivax: PQ-5)
ASU-3 + MQ (25 mg/kg) (north) Q-5 + T-5 (other)
Yemen CQ CQ SP AQ = amodiaquine; ASU = artesunate; ATM = artemether; CQ = chloroquine; MQ = mefloquine; PQ = primaquine; Q = quinine; RDT = rapid diagnostic test; SP = sulfadoxine = pyrimethamine; T = tetracycline; ASU-3 = artesunate for 3 days. aDrug for which reduced susceptibility of parasite is reported.
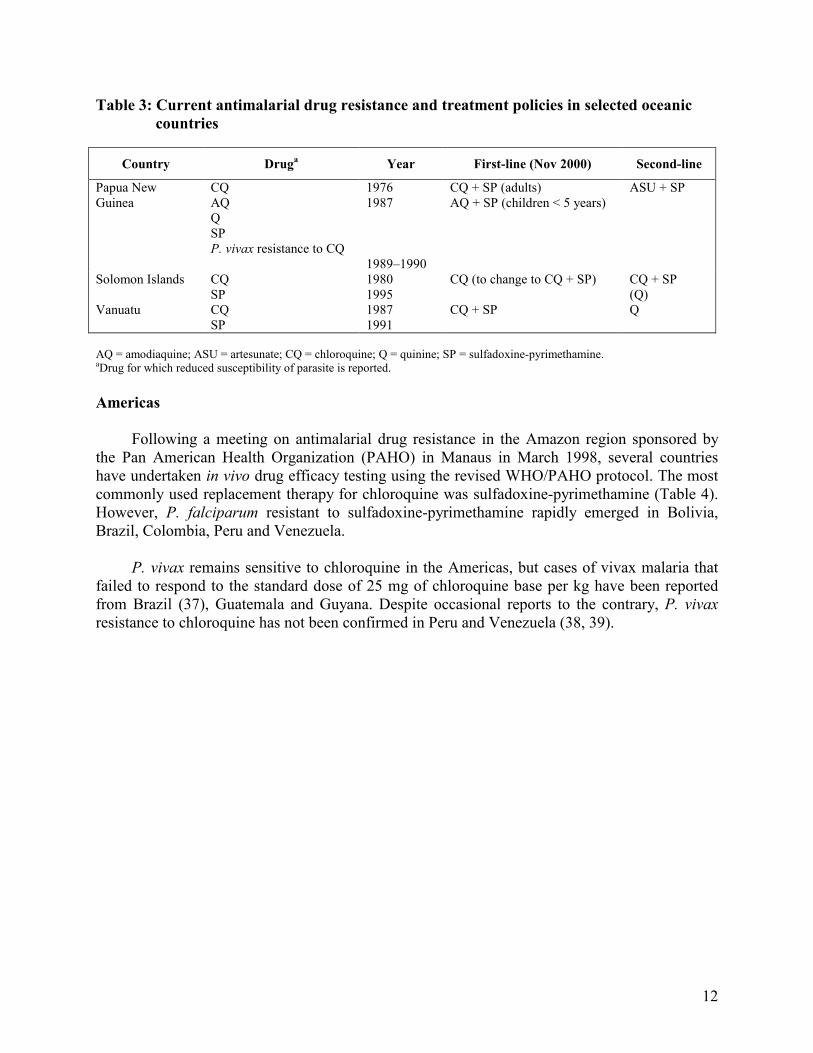
Oceania
Chloroquine resistance is widespread in Papua New Guinea, Solomon Islands and Vanuatu (Table 3). At an interregional drug resistance meeting in 1996, the new protocol for in vivo testing was adopted. Combinations with and without artemisinins are increasingly being adopted in this region. The current first-line treatment is a combination of sulfadoxine-pyrimethamine and chloroquine (with the variation that children under 5 years of age are treated with amodiaquine) in Papua New Guinea and in Vanuatu. A decision to adopt this combination is also being considered in Solomon Islands. Chloroquine-resistant P. vivax has been found in Papua New Guinea (36).
12
Table 3: Current antimalarial drug resistance and treatment policies in selected oceanic
countries
Country Druga Year First-line (Nov 2000) Second-line
Papua New Guinea
CQ AQ Q SP P. vivax resistance to CQ
1976 1987 1989–1990
CQ + SP (adults) AQ + SP (children < 5 years)
ASU + SP
Solomon Islands CQ SP
1980 1995
CQ (to change to CQ + SP) CQ + SP (Q)
Vanuatu CQ SP
1987 1991
CQ + SP Q
AQ = amodiaquine; ASU = artesunate; CQ = chloroquine; Q = quinine; SP = sulfadoxine-pyrimethamine. aDrug for which reduced susceptibility of parasite is reported.
Americas
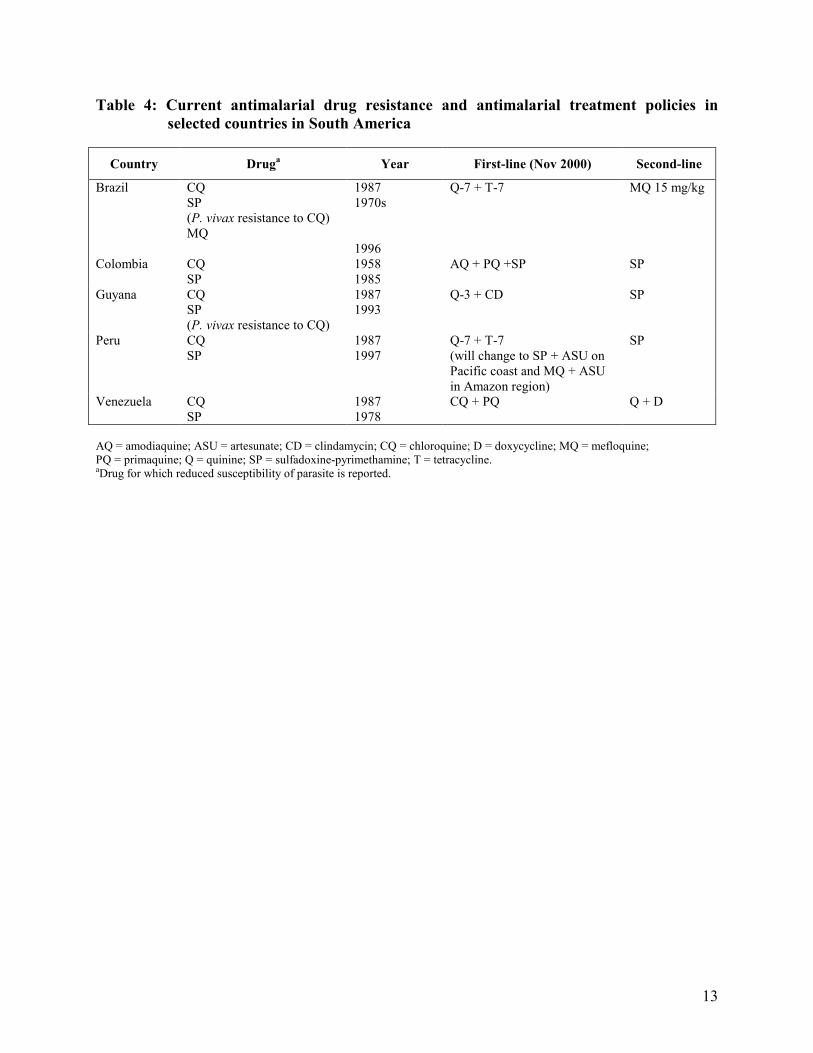
Following a meeting on antimalarial drug resistance in the Amazon region sponsored by the Pan American Health Organization (PAHO) in Manaus in March 1998, several countries have undertaken in vivo drug efficacy testing using the revised WHO/PAHO protocol. The most commonly used replacement therapy for chloroquine was sulfadoxine-pyrimethamine (Table 4). However, P. falciparum resistant to sulfadoxine-pyrimethamine rapidly emerged in Bolivia, Brazil, Colombia, Peru and Venezuela.
P. vivax remains sensitive to chloroquine in the Americas, but cases of vivax malaria that failed to respond to the standard dose of 25 mg of chloroquine base per kg have been reported from Brazil (37), Guatemala and Guyana. Despite occasional reports to the contrary, P. vivax resistance to chloroquine has not been confirmed in Peru and Venezuela (38, 39).
13
Table 4: Current antimalarial drug resistance and antimalarial treatment policies in
selected countries in South America
Country Druga Year First-line (Nov 2000) Second-line
Brazil CQ SP (P. vivax resistance to CQ) MQ
1987 1970s 1996
Q-7 + T-7 MQ 15 mg/kg
Colombia CQ SP
1958 1985
AQ + PQ +SP SP
Guyana CQ SP (P. vivax resistance to CQ)
1987 1993
Q-3 + CD SP
Peru CQ SP
1987 1997
Q-7 + T-7 (will change to SP + ASU on Pacific coast and MQ + ASU in Amazon region)
SP
Venezuela CQ SP
1987 1978
CQ + PQ Q + D
AQ = amodiaquine; ASU = artesunate; CD = clindamycin; CQ = chloroquine; D = doxycycline; MQ = mefloquine; PQ = primaquine; Q = quinine; SP = sulfadoxine-pyrimethamine; T = tetracycline. aDrug for which reduced susceptibility of parasite is reported.

14
3. COMBINATION THERAPY IN TREATMENT POLICY
A national antimalarial treatment policy is a set of recommendations and regulations concerning the availability and rational use of antimalarial drugs in a country (40). The policy should provide decision-makers with evidence-based recommendations in addition to giving health workers clear guidelines for providing early diagnosis and prompt treatment appropriate to the local context (41).
3.1 Purpose of Drug Policy
The objective of a national antimalarial treatment policy is to enable the population at risk of malaria infection to have access to safe, good quality, effective and acceptable antimalarial drugs in order to:
Figure 5 illustrates the main challenge facing antimalarial treatment policy development. The challenge is to achieve a balance between two essential but at times competing principles, thus ensuring prompt treatment of malaria and maximum useful therapeutic life of the antimalarial drug. The two essential parts of the balance, however, should be complementary. Ensuring adequate regulation and control of drug use should allow for equity and rational use of antimalarial drugs with the resultant reduction in mortality and at the same time reduce or delay drug resistance by the parasites.
1.1.1.1. Ensure a rapid and longEnsure a rapid and longEnsure a rapid and longEnsure a rapid and long----lasting clinical cure for individual malaria patients, lasting clinical cure for individual malaria patients, lasting clinical cure for individual malaria patients, lasting clinical cure for individual malaria patients,
2.2.2.2. Prevent progression of uncomplicated malaria to severe disease and death,Prevent progression of uncomplicated malaria to severe disease and death,Prevent progression of uncomplicated malaria to severe disease and death,Prevent progression of uncomplicated malaria to severe disease and death,
3.3.3.3. Shorten clinical episodes of malaria and reduce the occurrencShorten clinical episodes of malaria and reduce the occurrencShorten clinical episodes of malaria and reduce the occurrencShorten clinical episodes of malaria and reduce the occurrence of malariae of malariae of malariae of malaria----associated anaemia in populations associated anaemia in populations associated anaemia in populations associated anaemia in populations residing in areas of high malaria transmission,residing in areas of high malaria transmission,residing in areas of high malaria transmission,residing in areas of high malaria transmission,
4.4.4.4. Reduce consequences of placental malaria infection and maternal malariaReduce consequences of placental malaria infection and maternal malariaReduce consequences of placental malaria infection and maternal malariaReduce consequences of placental malaria infection and maternal malaria----associated anaemia through associated anaemia through associated anaemia through associated anaemia through chemoprophylaxis or preventive intermittent treatment during pchemoprophylaxis or preventive intermittent treatment during pchemoprophylaxis or preventive intermittent treatment during pchemoprophylaxis or preventive intermittent treatment during pregnancy,regnancy,regnancy,regnancy,
5.5.5.5. Delay the development and spread of resistance to antimalarial drugs.Delay the development and spread of resistance to antimalarial drugs.Delay the development and spread of resistance to antimalarial drugs.Delay the development and spread of resistance to antimalarial drugs.

15
Figure 5: Balancing prompt treatment against minimizing drug resistance
An effective first-line antimalarial treatment would have a greater impact on reducing malaria mortality than merely improving second-line treatment or the management of severe malaria. Therefore, combination therapies must be available and affordable for use as the first-line treatment of malaria.
3.2 Combination Therapy of Antimalarial Drugs
The concept of combination therapy is based on the synergistic or additive potential of two
or more drugs to improve therapeutic efficacy and also delay the development of resistance to the individual components of the combination. The aim is to improve efficacy and retard the development of resistance to the individual components of the combination. This concept has been realized in multiple-drug therapy for leprosy, tuberculosis and cancer and, more recently, in antiretroviral treatments. It has also already been used to some extent in the field of malaria with the development of such drugs as sulfadoxine-pyrimethamine, atovaquone-proguanil and mefloquine-sulfadoxine-pyrimethamine.
Definitions of combination therapy for antimalarial drugs
Combination therapyCombination therapyCombination therapyCombination therapy (CT) with antimalarial drugs is the simultaneous use of two or more blood schizonticidal drugs with independent modes of action and different biochemical targets in the parasite.Combination therapies can be either fixedfixedfixedfixed----combinationcombinationcombinationcombination medicinal products, in which the components are co-formulated in the same tablet or capsule, or multiplemultiplemultiplemultiple----drugdrugdrugdrug therapy, in which the components are co-administered in separate tablets or capsules.
ArtemisininArtemisininArtemisininArtemisinin----based combination therapybased combination therapybased combination therapybased combination therapy (ACT) is antimalarial combination therapy with an artemisinin derivative as one component of the combination.
Early Diagnosis and
prompt treatment
Minimise evolution of
drug resistance
•Goal: equity, reduce
morbidity and mortality
•Broad access to antimalarials
•Emphasis on community
and household management
•Requires high sensitivity
•Goal: reduce delay
resistance
•Restrictive access to a/m
•Emphasis on regulation
and control of drug use
•Requires high specificity

16
In the context of the above definitions, the following multiple-drug therapies are NOT considered to be combination therapy:
• the use of an antimalarial drug with a non antimalarial drug that may enhance its action (e.g. chloroquine plus chlorpheniramine),
• the use of a blood schizonticidal drug with a tissue schizonticidal or gametocytocidal drug (e.g. chloroquine plus primaquine).
Furthermore, products that fit the criteria of synergistic fixed-dose combinations are
operationally considered as single synergistic products in that neither of the individual components in itself would be given alone for antimalarial therapy, e.g. sulfadoxine-pyrimethamine, chlorproguanil-dapsone and atovaquone-proguanil.
Theoretical basis of combination therapy
The underlying theory of combination therapy in malaria is based on the fact that resistance
to antimalarial drugs arises from the selection of mutations. Provided that the constituent drugs administered in the combination have independent modes of action, the probability that a mutant will arise that is simultaneously resistant to both drugs is the product of the respective mutation rates multiplied by the number of parasite cells exposed to the drugs (8, 42, 43). For example, if two drugs are used, and for each one a single mutational event confers complete resistance and such events occur with a frequency of 1:1010 nuclear divisions, then the probability of a mutation resistant to both drugs is 1:1020. The number of asexual parasites (parasite biomass) during an acute malaria infection is usually between 109 and 1014 (43).
Antimalarial drugs which are to be combined should be pharmacokinetically and
dynamically compatible, and, ideally, resistance in the parasite should arise through different molecular mechanisms. The combination should include an effective short half-life drug and a longer half-life partner antimalarial drug which allows a reduction in the duration of antimalarial treatment while at the same time enhancing efficacy and reducing the likelihood of resistance development. In order to maximize the benefits of combination therapy, the resultant regimen must be amenable to good patient compliance; this is affected by dosing intervals, duration of therapy, toxicity profile and cost.
There is a growing interest in using antimalarial combinations containing an artemisinin
derivative as first-line treatment. The major immediate benefit and effect of the artemisinin component is the dramatic reduction in the parasite biomass and reduction in the production of gametocytes (10, 12). The residual biomass is exposed to maximum concentrations of the partner drug, well above its minimum inhibitory concentration, resulting in a lesser likelihood of resistant mutations breaking through.

17
3.3 Rationale for the Use of Combination Therapy in the African Region
It is currently estimated that 90% of global episodes of clinical malaria and 90% of global malaria mortality occur in sub-Saharan Africa. Malaria control efforts in the region have been greatly affected by the emergence and spread of chloroquine resistance. This was first recorded in 1979 in east Africa but has now been reported from almost all malaria-endemic countries of Africa (44). Sulfadoxine-pyrimethamine (SP) was, until recently, seen as the obvious successor to chloroquine. However, resistance to SP is developing quickly even with its current use (45, 46), thus reducing the useful therapeutic life of this drug.
The evolution of P. falciparum resistance in Africa follows trends observed elsewhere in
south-east Asia and the Americas where P. falciparum developed resistance to chloroquine and to SP in succession, and later to alternative monotherapies such as mefloquine and quinine. Most experts agree that it is a matter of time before SP, currently the first-line or second-line antimalarial drug in most countries, becomes ineffective for the treatment of malaria in many parts of Africa. The continued increase of resistance to antimalarial drugs in many countries in Africa has resulted in increased morbidity and mortality (20). It is essential to ensure rational deployment of the few remaining effective drugs to maximize their useful therapeutic life. This requirement has resulted in a re-examination of the potential of existing products and the development of new fixed combination drugs.
It is anticipated that, in some settings, CT could be introduced to protect the lifespan of a
still effective antimalarial monotherapy. In these cases, CT will be of long-term benefit to the community rather than of immediate benefit to the patient. However, if CT is to delay the development of resistance, existing monotherapy with either of its components should cease, though this has not been rigorously proven. Therefore, it is necessary to guarantee consistent access to the CT and restrict access to related drugs throughout the health sector, both private (formal and informal) and public. Fixed-combination products are preferred to multiple-drug therapy as these will improve the ease of use and compliance while minimizing the potential use of components of the combination as monotherapy.
In recent years, the artemisinin derivatives have been shown to be rapidly effective and well tolerated treatments for both uncomplicated and severe malaria (11). These drugs are now being used increasingly in south-east Asia and South America where multidrug resistant P. falciparum is prevalent. In Africa, these drugs are already available in many countries, particularly in the private sector, but their use is not well regulated. It has been argued that the artemisinin derivatives are so effective in the management of severe malaria that they should be withheld from use in uncomplicated malaria in areas where they are not needed in order to protect them from the development of resistance (see Table 5). This interest results from experience with the combination of artesunate and mefloquine on the Thai-Myanmar border (11, 12, 47–50). Following the introduction of the combination, there have been four principal clinical and epidemiological effects. The efficacy of the combination has exceeded 95% at a time when high-dose mefloquine was showing a failure rate of approximately 25% and has been sustained over the past 7 years, transmission of P. falciparum has been reduced and the in vitro sensitivity of mefloquine has increased, suggesting that the combination has reversed the previous decline in mefloquine sensitivity.

18
A change is needed in several countries where chloroquine already has poor efficacy and SP is becoming less efficacious. There are serious concerns that a change from chloroquine to SP monotherapy might affect the future utility of some ACTs, in particular SP plus artesunate, and chlorproguanil-dapsone (LapDap) plus artesunate.
Table 5: Factors concerning artemisinin-based combination therapy in Africa
Favourable factors Potentially prohibitive factors
• The need to replace inadequate drug regimens that are leading to increased malaria-related mortality and morbidity
• Potential avoidance of the loss of available effective and affordable antimalarial drugs
• Excellent efficacy (both clinical and parasitological clearance) of artemisinin derivatives
• Potential reduction in transmission (especially of resistant mutants) due to the gametocytocidal effect of artemisinin derivatives
• Higher cost
• Problems of adherence to non-fixed combinations and their rational use, particularly in the home
• Lack of extensive clinical experience with most of the combinations currently under investigation
• Lack of evidence of effectiveness in delaying the development of resistance
• Importance of not misusing artemisinin derivatives in view of their role in the treatment of severe malaria
The cost to the user and to the health system must be competitive with alternatives and
affordable to the poorest, otherwise the public health value of CT may be compromised. The substantially higher cost of CTs is probably the major obstacle to the implementation of this strategy, especially in sub-Saharan Africa. However, this should not affect the decision by the country to implement CT policy change considering the cost of treatment failures and death as an outcome. If countries showed a need for change to CT as a public health measure, subsidies could be justified, but assurance is needed that financial mechanisms will be sustainable. With support from external agencies, implementation of CT in the short term, in defined areas, may be feasible. However, greater efforts and resources are needed in isolated areas with poor services to ensure the sustainability of policies and programmes.

19
Why the urgency to change?
4. AVAILABLE ANTIMALARIAL COMBINATION DRUGS To increase understanding about how antimalarial drugs work, Annex 1 provides
information on the malaria life cycle. It also shows when and where the drugs exert their effects.
4.1 Artemisinin-based Combination Therapy
The advantages of artemisinin-based combination therapy (ACT) relate to the unique properties and mode of action of the artemisinin component. These which include the following:
• Rapid substantial reduction of the parasite biomass
• Rapid resolution of clinical symptoms
• Effective action against multidrug-resistant P. falciparum
• Reduction of gametocyte carriage which may reduce transmission of resistant alleles (parasite genes), particularly in areas with low or moderate malaria transmission
• No parasite resistance documented as yet
• Few reported adverse effects; however, pre-clinical toxicology data on artemisinin derivatives are limited.
Artemisinin (qinghaosu), artesunate, artemether and dihydroartemisinin have all been used
in combination with other antimalarial drugs for the treatment of malaria, but artesunate has the most documented clinical information (35). Because of the very short half-life of artemisinin derivatives, their use as monotherapy requires a multiple dose regimen of 7 days. Combination of one of these drugs with a longer half-life antimalarial drug allows a reduction in the duration of artemisinin treatment, while at the same time enhancing efficacy and reducing the development of resistance to the partner drug.
The urgency of changing to ACTs in Africa should be recognized in order to avert the foreseeable problems of:
• Cost of treatment failures including death as an outcome
• Increased use of artemisinin derivatives as monotherapy which could render them ineffective as candidates in combination therapy
• Limited availability of antimalarial drug options
• Need to preserve and extend the potential useful therapeutic life of antimalarial drugs by using them in combination rather than as monotherapy.

20
Furthermore, given the reassuring lack of artemisinin resistance to date, the rapid elimination of these drugs, such that sub-inhibitory blood concentrations occur for only hours, then it is reasonable to conclude that resistance to these drugs will develop relatively slowly. This will result in a considerably longer useful therapeutic life for both components in the combination than if the two drugs were deployed in sequence.
Artesunate used in combination therapy has been shown to delay the development of resistance to its partner drug (mefloquine) in low malaria transmission areas in south-east Asia, but this remains to be determined in high malaria transmission areas (11, 12, 48, 49). Following the introduction of artesunate-mefloquine on the Thai-Myanmar border, there were principal clinical and epidemiological effects (35, 45, 51–53). The efficacy of the combination has exceeded 95%, high efficacy has been sustained over the last 7 years, transmission of P. falciparum has been reduced and in vitro sensitivity of mefloquine has increased.

21
Artesunate plus Sulfadoxine-Pyrimethamine
Formulation
The drugs are available as free individual artesunate and sulfadoxine-pyrimethamine (SP).
Efficacy
The efficacy and safety of artesunate plus SP have been evaluated in three randomized
double blind placebo-controlled clinical trials in Gambia (54), Kenya and Uganda. The 3-day regimen of artesunate was more efficacious than the one-day regimen. The combination of artesunate with SP may reduce the rate of emergence of SP resistance. In two trials in Gambia (54, 55) and Kenya, a 3-day artesunate regimen was necessary to optimize treatment efficacy.
Use
The increasing levels of resistance to SP may limit the use of artesunate plus SP,
particularly in the eastern parts of Africa. However, it may still be a viable option for some countries of west Africa and other areas where SP efficacy is not yet compromised by resistance.
It is important to decide whether SP resistance has progressed too far in some parts of
Africa to warrant the high cost of implementing artemisinin-based combination therapy that includes SP (SP + ACT). Sulfadoxine-pyrimethamine resistance is strongly associated with mutations in parasite dhfr, the gene that encodes parasite dihydrofolate reductase (DHFR) (56–58). While three mutations are now common in many parts of Africa (56, 59), a fourth mutation, providing complete resistance to SP, has not been reported (60). It is important to take into consideration the fact that it would be extremely difficult to eliminate SP monotherapy from the market as it is cheap, well-known and produced by many generic drug manufacturers.
Recommended treatment
Recent studies in Africa have demonstrated that combinations of artesunate (oral administration of 4 mg/kg daily for 3 days) plus a single dose of SP on the first day are highly efficacious, although efficacy appears to be reduced in areas with pre-existing moderate levels of SP resistance (54). Dosing schedule is in Table A2.3 in Annex 2.
Use in pregnancy
Use in pregnancy has not been evaluated and more data are required.
Drug disposition
The pharmacokinetics and distribution of these drug combinations has not been evaluated.

22
Contraindications
The contraindications are as for the individual drugs (SP and artesunate). Use in
chemoprophylaxis, severe hepatic or renal dysfunction, infants less than 2 months and first trimester of pregnancy is not recommended.
Artesunate plus Amodiaquine
Formulation
The drugs are available as free individual drugs. Paediatric blister packs containing
artesunate 25mg and amodiaquine 75mg or artesunate 50mg and amodiaquine 150mg are available from WHO. Adult blister packs contain artesunate 50mg and amodiaquine 150mg.
Efficacy
The efficacy and safety of artesunate plus amodiaquine have been evaluated in three
randomized, double blind placebo-controlled clinical trials conducted in Gabon, Kenya and Senegal. The combination was efficacious and well-tolerated. The level of efficacy coincides with low levels of AQ resistance in the study sites. The 14-day parasitological cure rate of the combination was > 90% in intent-to-treat analysis at all sites (61).
Use
Artesunate plus amodiaquine appears to be a viable option particularly in areas where CQ
efficacy is already compromised. However, continued monitoring of resistance to AQ and the impact of AQ resistance on the effectiveness of the combination would need to be carefully monitored. The rate of development and spread of amodiaquine resistance is unknown and cross-resistance with chloroquine may be a limiting factor for long-term efficacy.
Recommended treatment
The 3-day regimen of both components is currently co-administered although co-
formulation is feasible. In clinical trials of around 960 patients, amodiaquine as monotherapy or combined with 3 days of artesunate was well-tolerated. The WHO blister pack dosing schedule is provided in Table A2.4 in Annex 2.
Use in pregnancy
There are no data on the use of the artesunate-amodiaquine combination in pregnancy.
Drug disposition
The pharmacokinetics and distribution of the combination have not been evaluated.

23
Adverse effects
There was no evidence of significant hepatotoxicity. However, it is not yet known whether
hepatotoxicity may develop after repeated treatments.
Contraindications
Use in persons with hepatic disorders and as malaria prophylaxis is not recommended.
Artemether-Lumefantrine
Formulation
Tablets exist which contain 20 mg of artemether plus 120 mg of lumefantrine
(benflumetol).
Efficacy
A total of 16 clinical trials with more than 3 000 patients, including 600 children under 5
years of age, have been carried out in Europe, south-east Asia and Africa. A six-dose regimen gave cure rates of 95.1% to 97.5% in areas with multidrug-resistant malaria (62–65). A dose-finding trial in Thailand demonstrated the importance of the number of doses rather than the dose level for the efficacy of this combination drug. These studies also showed that the cure rate was 97% in patients receiving a total dose of lumefantrine greater than or equal to 50 mg/kg, regardless of the level of initial parasitaemia, but that cure rates were significantly lower with parasite densities greater than or equal to 20 000 per ml when the total dose was less than 50 mg/kg (66–69).
Use
Artemether-lumefantrine can be used for the treatment of uncomplicated infections with P.
falciparum, including strains from multidrug-resistant areas. WHO recommends a standard treatment of six doses for children and adults in the treatment of uncomplicated malaria irrespective of the malaria transmission pattern or the immune status of the individual.
Recommended treatment In areas with multidrug-resistant P. falciparum and in non-immune patients, an intensive
six-dose course consisting of doses at 0 hour and 8 hours, and twice daily doses on the next 2 days is recommended. See Table 2.5 in Annex 2. Thus, the course for an adult would be four tablets at 0 hour and 8 hours and four tablets twice a day on the second and third days. The total course for adults is 24 tablets which gives a total of 480 mg of artemether plus 2 680 mg of lumefantrine.

24
There is no evidence of increased toxicity with the six-dose as compared to the four-dose regimen. For simplicity of implementation, it may be advantageous to use the six-dose regimen in all areas.
Use in pregnancy
This drug combination should not be used in pregnant women. Safe use in pregnancy has
not yet been established.
Drug disposition Maximum blood levels are observed 6–12 hours after drug administration. Adequate
absorption of the lumefantrine component seems to be increased by taking a fatty meal. The elimination half-life is 88 hours in healthy subjects and about twice as long in malaria patients. The drug is excreted via the liver and faeces. There is no evidence of pharmacokinetic interaction between artemether and lumefantrine (70).
Adverse effects
The following adverse effects have been reported (71): dizziness and fatigue, anorexia,
nausea, vomiting, abdominal pain, palpitations, myalgia, sleep disorders, arthralgia, headache and rash.
In children and adults treated with this combination, the frequency and degree of QTc
prolongations was lower than with chloroquine, mefloquine or halofantrine (70). Studies show no indication of cardiotoxicity (72).
Contraindications Artemether-lumefantrine is contraindicated in pregnancy, lactating women, very young
children (less than 10 kg body weight) and those with known hypersensitivity to either of the components.
4.2 Non-Artemisinin-based Combinations
Non-artemisinin-based combinations are antimalarial drug combinations not involving artemisinin derivatives acting at different sites as blood schizoitocidal drugs used as free individual drugs or as fixed combination drugs. They have been shown to have higher cure rates than monotherapy (73). The available combinations that have been used so far involve chloroquine, SP and amodiaquine.
Chloroquine plus Sulfadoxine-Pyrimethamine
Chloroquine (CQ) and sulfadoxine-pyrimethamine (SP) are antimalarial drugs that are used
frequently in Africa as either first-line or second-line drug for the treatment of P. falciparum malaria. CQ is a 4-aminoquinoline while SP is a fixed-dose combination of two antifolate

25
compounds. Several physicians in Africa are already practising the combined use of CQ plus SP for the treatment of P. falciparum infection in individual patient care, and national health authorities have deployed the use of the combination in some countries (e.g. Eritrea and Ethiopia) where both P. falciparum and P. vivax are common. It is also being used as an interim antimalarial treatment policy in Uganda and Zimbabwe.
Formulation
Chloroquine plus SP is a co-administered combination of individual drugs since there is no
co-formulated tablet.
Efficacy
In Africa, P. falciparum resistance to CQ has increasingly spread and intensified since it
was first documented in 1979. Similarly, P. falciparum resistance to SP has also increased since the late 1980s, particularly in east Africa where it has been used on a larger scale as a first-line drug (45, 46, 74). Resistance to SP has also been demonstrated in parts of west Africa (75).
The pharmacokinetic properties of CQ and SP have, individually, been extensively studied
(76, 77). CQ and SP have reasonably similar pharmacokinetic profiles, with varied modes of action on different biochemical targets in the parasite and are therefore technically suitable candidates for combination therapy. However, as there is no published information on the in-vitro pharmacodynamic interactions of the combination, it is not known whether their activities are synergistic, antagonistic or additive (Warhurst D., personal communication). Studies in Gambia and Papua New Guinea, which compared the efficacy and safety of CQ plus SP to that of SP alone, show that the efficacy of the combination is dependant on the levels of resistance to the individual components (78, 79).
Use
In areas with high levels of P. falciparum resistance to CQ and moderate resistance to SP,
the combination of CQ plus SP would not be expected to achieve significantly better cure rates than SP alone. Moreover, it is unlikely that the use of CQ plus SP would retard the development and selection of resistance to SP. Despite parasite resistance, CQ still effects a significant anti-inflammatory action through modulation of the cytokine pathway, and therefore the use of CQ plus SP may achieve a more rapid resolution of symptoms than treatment with SP alone.
The combination has been in use in Vanuatu since 1994, and there has not been any
recorded malaria mortality since 1996, though this reduction cannot be entirely attributed to the
use of the combination. The combination of CQ plus SP has also been a standard first-line
treatment in peninsular Malaysia since 1997, and in Papua New Guinea since 2000; both areas
have P. falciparum and P. vivax infections. It was adopted as an interim antimalarial treatment
policy in 2000 in Uganda and Zimbabwe.

26
The available evidence has shown that the CQ plus SP combination is unlikely to have a
significant advantage over SP alone in areas of predominant P. falciparum transmission with
high levels of resistance to CQ. Since this reflects the current situation in most of sub-Saharan
Africa, a change to this combination as a first-line treatment is unlikely to give any significant
useful long-term advantage.
Recommended treatment
There are no clinical data on dosing for the combination of these drugs. Currently they are
available as individual drugs for co-administration using the same dosage schedules for chloroquine and SP. SP and first dose of chloroquine are given on day one and chloroquine continued on second and third day to complete 3 days.
Drug disposition
Pharmacokinetics are known only for the individual free drugs but not the combination.
Adverse effects
The adverse effects of CQ plus SP have not been evaluated but could be as in the free
individual drugs.
Contraindications
The contraindications for CQ plus SP are as for the individual drugs. Conditions such as
psoriasis, porphyria and megaloblastic anaemia preclude use of these drugs.
Amodiaquine plus Sulfadoxine-Pyrimethamine
Amodiaquine (AQ) and SP have reasonably similar pharmacokinetic profiles, with varied
modes of action on different biochemical targets in the parasite and are therefore technically suitable candidates for combination therapy.
Formulation
AQ and SP are available as individual drugs for co-administration.
Efficacy
A review of studies on the treatment of uncomplicated falciparum malaria conducted over
the past ten years in Africa showed a higher therapeutic efficacy of amodiaquine over chloroquine, with a tendency towards faster clinical recovery even in areas with mild to moderate parasite resistance to chloroquine (26, 80).

27
A meta-analysis on three published studies from China and Mozambique since the 1980s on the efficacy and safety of AQ plus SP compared to AQ alone showed that parasite clearance rates at 28 days tended to favour AQ plus SP (73, 81–83). A trial in Uganda showed a higher rate of clinical and parasitological cure with AQ plus SP than with SP alone (84). However, the combination was no more effective than AQ alone, which still had an efficacy of 95% when used as monotherapy. The safety data obtained in these clinical trials were limited but did not suggest that adverse reactions of AQ were increased by the co-administration of SP.
Use
In some countries in west and central Africa, where levels of resistance to AQ are generally less than those of CQ (26), a change to AQ plus SP would probably be a more cost-effective option with a longer useful therapeutic life than a change to monotherapy with SP. However, there are still some concerns over the safety of AQ for widespread unsupervised repeated treatment of malaria. More data on safety, including its use during pregnancy, are required.
Recommended treatment
Amodiaquine and sulfadoxine-pyrimethamine are co-administered as free individual drugs: Amodiaquine 10 mg/kg per day for 3 days and SP (25mg/kg sulfadoxine and 1.25mg/kg pyrimethamine) as single dose on day one. The dosing schedule for weight or age is given in Annex 3 as individual drugs.
Use in pregnancy
Use in pregnancy has not been evaluated and more data are required.
Drug disposition
The pharmacokinetics and distribution of the combination have not been evaluated.
Contraindications
Use in chemoprophylaxis, severe hepatic or renal dysfunction, infants less than 2 months
and first trimester of pregnancy is not recommended.

28
5. COMBINATION DRUGS IN DEVELOPMENT
It is possible that some of the antimalarial combination drugs currently under development will prove to be highly efficacious, safe and well-tolerated, with a potential for widespread use at prices affordable in most of the world. It should be noted that some of the components of these combinations would be “new chemical entities” that have not been used as monotherapy for the management of malaria. Resistance to these new combinations may therefore be slower to develop.
5.1 Chlorproguanil-Dapsone-Artesunate
The final product of chlorproguanil-dapsone-artesunate (CDA) will be a fixed-ratio co-
formulation of chlorproguanil-dapsone (LapDap) and artesunate. While data exist for LapDap and artesunate individually, there are no data yet on CDA. However, initial toxicology work is under way. Other on-going studies include Phase I pharmacokinetic work, Phase II artesunate dose-finding studies and Phase III safety and efficacy studies in adults. Safety and efficacy data are also required in young children, pregnant women and breastfeeding mothers.
LapDap is now entering the market in a number of African countries following its registration in the United Kingdom in 2003. While CDA provides a potential combination option, there are concerns that the deployment of LapDap as monotherapy for widespread use could affect the useful therapeutic life of CDA. Thus, the deployment of LapDap as monotherapy is not recommended in Africa in order to prevent a possible compromise of the useful therapeutic life of CDA. There is also an added advantage of using a combination partner that would not have been deployed previously as monotherapy.
5.2 Pyronaridine-Artemisinin Derivative
Pyronaridine (an acridine-type Mannich base) co-formulated with artesunate is being
developed as a fixed combination. The rate of development of resistance was high in the mouse model. If a dosage regimen can be designed with a maximum of three daily doses, this may become a practicable ACT regimen. There are no pharmacokinetic data on the combination, though studies of pyronaridine alone have been made on a limited scale. The toxicity profile of pyronaridine is comparable to that of chloroquine, but at high doses mutagenicity and fetal resorption in rats have been observed.
A study in Hainan (an area with P. falciparum resistance to pyronaridine due to the earlier
widespread use of the drug) showed a clinical response rate of 100% with the combination on a regimen of pyronaridine (400mg) plus dihydroartemisinin (DHA, 100mg) daily for 2 days (Liu Dequan, personal communication). A dosage regimen of pyronaridine (400mg) in combination

29
with artemether (150mg) or artesunate (150mg) daily for 2 days gave a clinical efficacy of 100%. There are no data for children under 5 years, pregnant women or drug excretion in breast milk.
5.3 Piperaquine-Dihydroartemisinin
Piperaquine-dihydroartemisinin is being developed as a fixed dose combination drug 1: 8
dihydroartemisinin: piperaquine (40mg: 320mg). Holleykin Pharmaceutical Co Ltd, Guangzhou, China, is currently producing it as Artekin. This combination is cheap, has fewer adverse effects and is available as a co-formulated tablet. Clinical studies have been carried out mainly in multidrug-resistant areas of Viet Nam. Available pre-clinical and pharmacokinetic data support its usefulness over that of single agents alone. It has also been demonstrated that the combination delays development of drug resistance in vitro. Toxicological data exist for the individual components of the combination.
The clinical experience is based on studies with piperaquine-dihydroartemisinin and other
combinations containing these components performed in selected regions in Viet Nam since 1993. This experience covers almost 800 patients, and studies are still on-going.
Efficacy
Piperaquine-dihydroartemisinin has an average parasite cure and fever clearance rates at 28 days of follow-up of 95%. It is encouraging that the combination is highly efficacious in Viet Nam in an area with multidrug resistance, and also in areas of Hainan with piperaquine resistance. However, the long half-life of piperaquine is of concern in relation to potential development of resistance in areas of intense malaria transmission.
Dosage
A fixed dosage combination of piperaquine-dihydroartemisinin is given for 3 days. The total dose for adults is eight tablets: initial dose of two tablets, then repeat after 6–8 hours, 24 hours and 36 hours.

30
6. FORMULATING AND IMPLEMENTING COMBINATION
THERAPY POLICY
6.1 Combination Therapy Drug Policy Formulation
During recent years, an increasing number of African governments have implemented or
are in the process of implementing new malaria treatment guidelines. The selection of a new recommended treatment should be guided by the various factors which influence the effectiveness of antimalarial treatment. These include magnitude of resistance; political environment; and drug characteristics such as therapeutic efficacy, simplicity of regimen, price or affordability, safety (including minor side effects affecting compliance), availability and potential for widespread use.
The implementation process involves the following:
• Policies which enable the private health sector to implement the treatment policy
• Incorporating new treatments into national treatment guidelines and national Essential Drugs Lists
• Training of health workers, including drug vendors
• Estimating the country-specific costs of policy change
• Drug procurement, i.e. sourcing, purchasing, storage and distribution
• Development, production and distribution of information, education and communication materials.
The framework document for developing, implementing and updating antimalarial treatment policy in Africa is available and should be used in changing to combination therapy. The essential components and actions are set out in the box below (41).
Essential components and actions for developing, evaluating and updatEssential components and actions for developing, evaluating and updatEssential components and actions for developing, evaluating and updatEssential components and actions for developing, evaluating and updating a national antimalarial drug ing a national antimalarial drug ing a national antimalarial drug ing a national antimalarial drug policypolicypolicypolicy
• The magnitude of malaria and resistance, potential interventions and the consequences of action or inaction; a clear analysis of the technical, social and economic issues related to malaria control and antimalarial drug resistance;
• Analysis of the political environment for decision-making;
• Consensus building and selection of options among policy-makers, malaria programme personnel, researchers, drug regulatory authorities, and other relevant stakeholders (e.g. donors and private providers, industry and user representatives);
• A supervisory body to oversee the development, implementation and revision of the policy.

31
6.2 Drug Characteristics and Cost
Therapeutic Efficacy
The therapeutic efficacy of the combination product is the most important criterion. This
compares the combinations in terms of clinical and parasitological cure and the rapidity of clinical recovery. Therapeutic efficacy (clinical cure) is currently proposed to guide the process of changing antimalarial treatment policy in the African Region (41). This could also be applied to the antimalarial combination drugs. Parasitological cure rate is also very important as this has been closely linked to the development of resistance by the parasites, particularly in areas of low transmission. The level of parasite resistance to a single drug, at which point it is no longer considered a useful component of a combination therapy, is not yet defined. It is, however, generally accepted that the lower the level of resistance, the higher the chances that the combination drug will have a longer useful therapeutic life. Thus the best candidates for combination therapy will be novel drugs that have not been previously used in monotherapy, have no demonstrable parasite resistance and are not going to be used for monotherapy.
Safety
The primary safety concern of combination products is the possibility of additive or
synergistic adverse interactions between the components. The fact that drugs have been registered for use as monotherapy does not preclude the need to gather toxicological data when using them in combination. These interactions can take the form of:
• Chemical interactions which can decrease efficacy, increase toxicity and diminish the shelf life of the product;
• Biological interactions with an additive or synergistic effect on adverse reactions;
• Pharmacokinetic interactions with an influence of one component on the absorption, bioavailability, distribution, biotransformation or elimination of the second component, resulting in either an increased or decreased potential for toxicity.
It is essential to establish the toxicology profile in animal studies prior to evaluation of
safety in the general human population. Safety profile is particularly required in special risk groups such as pregnant women, children under 5 years of age, lactating women (excretion of product in breast milk and its effect on the child) and in people with HIV/AIDS.
Consumer Acceptability and Compliance
Patient acceptance of any treatment strategy is a function of both the true characteristics of
a drug (efficacy, half-life and side effects) and the consumers’ perceived characteristics (e.g. product presentation—taste, colour, duration of treatment etc.). Other influential factors include the reputation of the drug or drug combination, capacity to induce rapid onset of symptomatic relief (antipyretic) irrespective of the rate of elimination of parasites and possible side effects. The acceptance of new treatment may also be influenced by the complexity of the dose regimen.
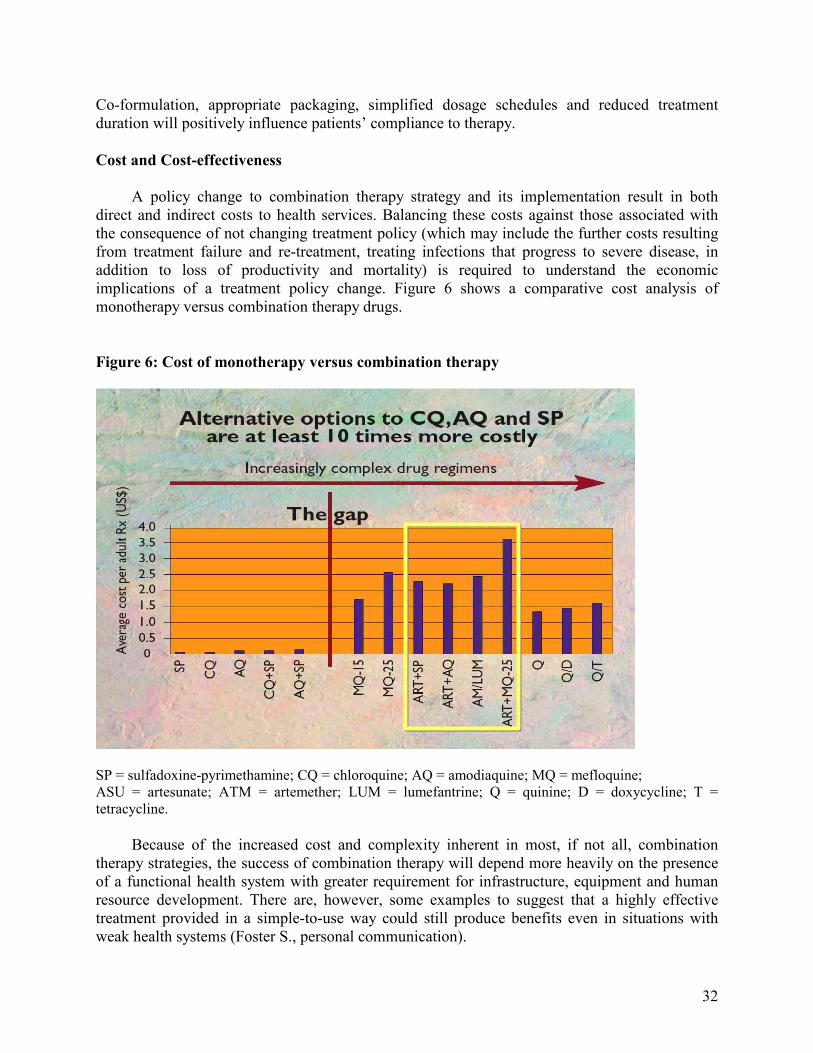
32
Co-formulation, appropriate packaging, simplified dosage schedules and reduced treatment duration will positively influence patients’ compliance to therapy.
Cost and Cost-effectiveness
A policy change to combination therapy strategy and its implementation result in both
direct and indirect costs to health services. Balancing these costs against those associated with the consequence of not changing treatment policy (which may include the further costs resulting from treatment failure and re-treatment, treating infections that progress to severe disease, in addition to loss of productivity and mortality) is required to understand the economic implications of a treatment policy change. Figure 6 shows a comparative cost analysis of monotherapy versus combination therapy drugs.
Figure 6: Cost of monotherapy versus combination therapy
SP = sulfadoxine-pyrimethamine; CQ = chloroquine; AQ = amodiaquine; MQ = mefloquine; ASU = artesunate; ATM = artemether; LUM = lumefantrine; Q = quinine; D = doxycycline; T = tetracycline.
Because of the increased cost and complexity inherent in most, if not all, combination
therapy strategies, the success of combination therapy will depend more heavily on the presence of a functional health system with greater requirement for infrastructure, equipment and human resource development. There are, however, some examples to suggest that a highly effective treatment provided in a simple-to-use way could still produce benefits even in situations with weak health systems (Foster S., personal communication).

33
Increasingly, an effective drug supply system for the distribution of antimalarial drugs is considered an effective mix of public and private (profit and non-profit) involvement in the areas of procurement, storage and transport services as well as in dispensing the medication to the patient. The criterion of cost-effectiveness has been difficult to apply in most regulatory systems. However, with the increasing use of various tools of pharmaco-economics, a number of countries are starting to explore the possibility of incorporating cost-effectiveness with other traditional markers (safety, efficacy and quality) in assessing products.
Key Issues in Implementing Combination Therapy
It is paramount to consider various issues before implementing combination therapy. Some
of these issues are highlighted in the box below:
6.3 Accessing Artemisinin-based Combination Treatment
At present, only one artemisinin-based combination therapy (artemether-lumefantrine) is available as a fixed dose drug in which both compounds are co-formulated into a single tablet and as a prequalified product. Other artemisinin-based combination therapies are available as individual products that need to be co-administered. Co-packaging of the combination partner drugs in a blister is highly recommended in order to make them user-friendly and to increase adherence to the complete therapy. Artemisinin compounds are sensitive to moisture. The shelf-life of most products is around 2 years, and this is contingent on high standards of blister packaging and good storage. Fortunately, the WHO in collaboration with other UN agencies has taken several steps to assist Member countries to access and purchase quality assured artemisinin-based combination drugs. These are discussed below.
Quality Assurance
A prequalification and sourcing project has been set up at the World Health Organization
Headquarters. This is an initiative through which WHO, in collaboration with other UN agencies, will pre-qualify manufacturers of artemisinin compounds and artemisinin-based combination therapy on the basis of compliance with internationally recommended standards of manufacturing and quality. WHO has requested an expression of interest from pharmaceutical manufacturers to participate in this initiative for the following products: artesunate (oral preparations), dihydroartemisinin (tablets, capsules, paediatric granules, suppositories),
• Limited data available on safety of ACTs in young infants
• Lack of adequate safety and efficacy data on drug combinations in pregnant women
• Financing of drugs
• Implications of price on the country’s financing policies
• Complex treatment schedules pose challenge for ensuring compliance
• Complexity of regimens for treatment near the home.

34
artemether (intramuscular preparations), artemether-lumefantrine (oral preparations), artesunate-amodiaquine (oral preparation) and artesunate-sulfadoxine-pyrimethamine (oral preparation).
Negotiated Prices
In a joint effort to provide essential medicines at affordable prices, WHO and the Swiss pharmaceutical company, Norvatis (manufacturer of artemether-lumefantrine), have come to a special pricing agreement. Norvatis will provide the drug at cost price (US$ 2.40 per full adult treatment and US$ 0.70 for the child dose) for use in the public sector of malaria-endemic countries. WHO and Norvatis have jointly developed specially designed packs for artemether-lumefantrine to facilitate proper use of the drug. WHO reviews requests for supplies of the drug and distributes it through the governments or NGOs in malaria-endemic countries. Full details are available on the Roll Back Malaria website (http://rbm.who.int).
Centralized Procurement
WHO and UNICEF have issued a joint call for the supply of artesunate and amodiaquine. However, while the prequalification and sourcing project is underway, some countries have found themselves in immediate need of procuring the artemisinin-based combination therapy, artesunate and amodiaquine. Although quality products for each of the two drugs are available, it has not been possible to source this combination drug as a co-blister packed product which meets required standards. As an interim measure to meet the immediate requirement of some countries in the short term, WHO and UNICEF have called for a joint tender for the procurement of artesunate and amodiaquine as separate blister packs which will then be packed and dispensed together.
WHO will monitor the delivery of artesunate and amodiaquine to malaria control
programmes and provide technical assistance for the deployment of these products in the malaria-endemic countries. Efforts to procure co-blister packed products to Good Manufacturing Practice standards continues in parallel with this effort.
6.4 Policy Review and Update Towards Combination Therapy
The choice of drugs for treatment of malaria is difficult. Each country needs to consider the evidence available locally and internationally to make its choices, and each country should have systems of continual review of these choices. With respect to the process of updating treatment policy and the selection of a new recommended treatment, countries may need to replace their first-line treatment of malaria (85).
Replacing Sulfadoxine-Pyrimethamine as First-line Treatment
Countries that need to replace sulfadoxine-pyrimethamine as first-line treatment of malaria include countries which have switched from chloroquine to SP monotherapy as first-line treatment and are facing growing resistance. These countries are facing the need to change from SP monotherapy to more expensive drug combinations. Combinations with SP are not advisable

35
OPTIONS
because SP is failing in many of these countries and it may not have a useful therapeutic life. These countries have only the following option (Figure 7):
Figure 7: Options for replacing sulfadoxine-pyrimethamine as first-line drug
Artesunate plus amodiaquine
Artemether-lumefantrine
To replace SP directly with ACTs, artesunate plus amodiaquine or artemether-
lumefantrine, as a durable long-term drug combination.
Countries which decide to replace SP with a more durable ACT such as artesunate plus amodiaquine or artemether-lumefantrine are facing serious challenges of cost. Choice between the two will depend on therapeutic efficacy, cost, availability, safety in groups at risk (especially infants and pregnant women), potential for widespread use and high adherence to treatment. The deployment will probably require a phased introduction either through selected well-monitored districts or through a tiered treatment system, i.e. specific drugs for health facility use or specific groups (not for use in pregnancy). However, in most African countries a tiered deployment may be difficult to implement. Possibly, a phased introduction in selected districts could allow: (i) better monitoring of unexpected adverse drug reactions (pharmaco-vigilance), (ii) communication and other efforts to ensure good compliance, and (iii) evaluation of operational needs of large-scale deployment in regions with high resource constraints.
Replacing Chloroquine as First-line Treatment
Countries that need to replace chloroquine as first-line treatment include countries that
have not replaced chloroquine as first-line treatment of malaria but are facing growing resistance to it. These countries have two options according to levels of SP resistance (Figure 8).
(1) To replace chloroquine directly with ACTs, such as artesunate plus SP, artesunate
plus amodiaquine or artemether-lumefantrine (choice depending on cost-
effectiveness)
In terms of costs, artesunate plus SP is preferable to artemether-lumefantrine. In terms of therapeutic efficacy, it must be compared with artesunate plus amodiaquine, especially in areas with incipient or focal SP resistance. SP and amodiaquine plus artesunate and artemether-lumefantrine are currently in the WHO Essential Drugs List, and together with amodiaquine they are registered and available in most countries that need to consider a drug treatment policy

36
change. The concomitant use of these drugs is possible as drugs in the existing formulation can be co-packaged in conveniently designed presentations.
(2) To introduce in the short term drug combinations that do not contain an artemisinin
derivative, such as amodiaquine plus SP, while preparing for a more durable
deployment of ACTs
Combinations that do not contain an artemisinin derivative could be the preferred options for reasons of cost and accessibility in some countries. On the international price lists, amodiaquine plus SP is three times more expensive than SP monotherapy and (at least) five times less expensive than ACTs.
Figure 8: Options for replacing chloroquine as first-line drug
Artesunate plus SP
Artesunate plus amodiaquine
OPTIONS Artemether-lumefantrine
Amodiaquine plus SP
6.5 Introduction of Combination Therapy
In view of the fact that countries will have to pay more for combination therapy compared
to monotherapy or interim combination drugs, it is essential that consideration be given to the mechanisms used in introducing the new policy. This could be achieved by looking at the costs involved in the introduction versus the need to change for the whole country. A country may decide on a phased introduction of combination therapy by region, province or district, using the geographical basis. Another country may phase in countrywide by health system level in terms of facility or in terms of first-line and second-line treatment.
There are various advantages and disadvantages of each method of introduction, but the choice will depend on the country situation. Figure 9 summarizes these methods of introduction.
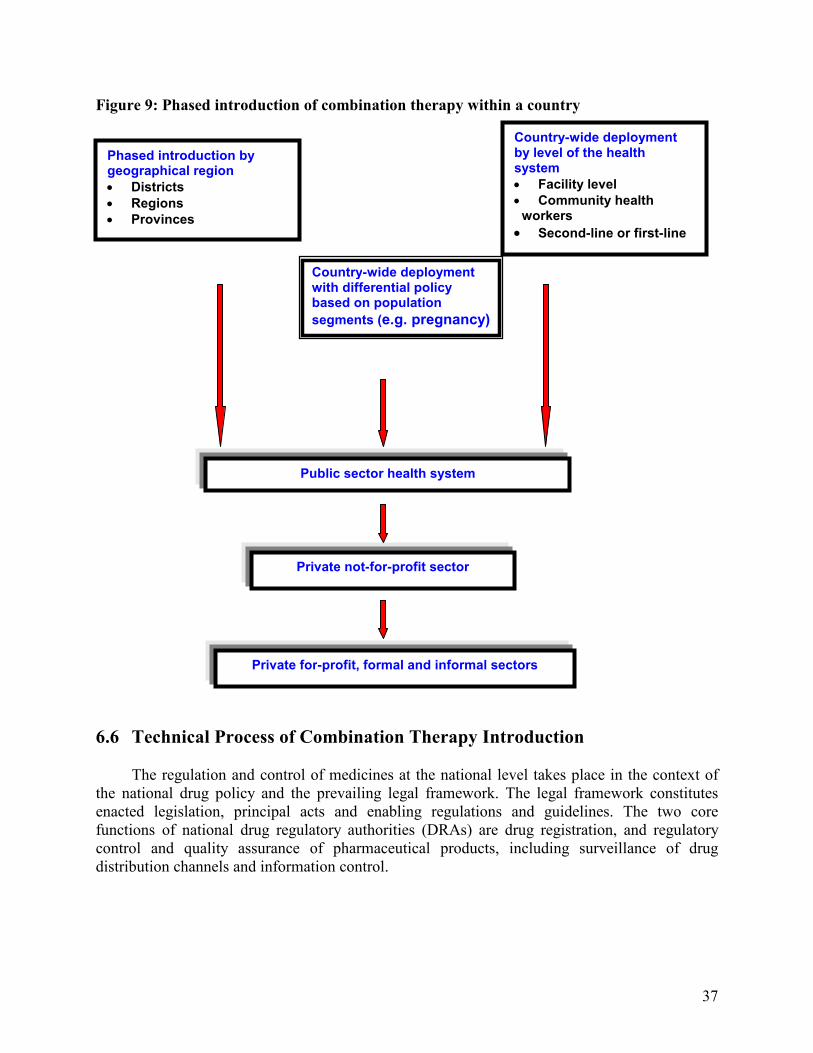
37
Figure 9: Phased introduction of combination therapy within a country
1.1
6.6 Technical Process of Combination Therapy Introduction
The regulation and control of medicines at the national level takes place in the context of the national drug policy and the prevailing legal framework. The legal framework constitutes enacted legislation, principal acts and enabling regulations and guidelines. The two core functions of national drug regulatory authorities (DRAs) are drug registration, and regulatory control and quality assurance of pharmaceutical products, including surveillance of drug distribution channels and information control.
Phased introduction by geographical region • Districts
• Regions • Provinces
Country-wide deployment by level of the health system
• Facility level • Community health workers
• Second-line or first-line
Public sector health system
Private not-for-profit sector
Private for-profit, formal and informal sectors
Country-wide deployment with differential policy based on population
segments (e.g. pregnancy)

38
Drug Registration
Drug registration is the first component in ensuring the quality of drugs available in the
market, and it is one of the main responsibilities of national DRAs. Most countries are already undertaking the exercise of registering drugs based on criteria of safety, efficacy, quality and, in some cases, access needs.
With the introduction of new antimalarial drugs, including innovative combinations of
already existing drugs, drug registration and marketing authorization will be critical. The co-administration of drugs already registered individually for monotherapy may not require any special regulatory action. However, co-packaging of such drugs could require the justification of need in addition to a demonstration of safety. It would also be necessary to develop new package inserts. Co-formulation of combination drugs would also require a full regulatory process, necessitating the establishment of a regulatory dossier. This technical documentation for the dossier should contain administrative details, formula, chemistry and pharmacy, pharmacology and toxicology, labelling, package insert and storage conditions of the product.
Regulatory Control and Quality Assurance
Drug Regulatory Authorities are also responsible for ensuring that drug information and
promotional materials meet the standards and ethical criteria of drug promotion. It is critical that all antimalarial drugs deployed are of good quality and are efficacious. In order to ensure that the antimalarial drugs available in the market are of the desired quality specifications, countries need to have competent pharmaceutical inspection programmes backed by well-equipped and functional national drug quality control laboratories. Rapid quality assessment tools, such as the mini-lab, can supplement the testing of drugs at points of entry and in peripheral settings.
Practical measures are needed to strengthen regulatory control to assure drug quality and
combat counterfeit drugs likely to be a major threat with the introduction of, and expected high demand for, more expensive combination antimalarial drugs. This needs to be done in collaboration and in cooperation with law enforcement agents. DRAs should approach overall quality assurance by combining inspection of pharmaceutical manufacturing plants with regard to Good Manufacturing Practice (GMP) as well as ensuring that the pharmaceutical products available on the market meet the prescribed quality specifications. It should be noted that DRAs have varying capacities for GMP inspection. In most of sub-Saharan Africa, this area of pharmaceutical regulation is not fully developed, and there may be a benefit in taking sub-regional approaches to building capacities.
The DRAs regulation of public and private drug distribution channels through issuing of
licences and permits should be strengthened. These distribution channels include wholesale and retail pharmacies, hospitals, clinics and the informal sector (e.g. drug vendors) where large amounts of medicines are dispensed.

39
6.7 Combination Therapy and Community Treatment of Malaria
Communities in the African Region are increasingly practising home treatment or self-
treatment for malaria. While it is most desirable for patients to be treated by trained personnel, it is recognized that an important component of primary health care is personal and community participation in decisions related to health. The diagnosis of malaria by mothers and caregivers should be based on recognition of the symptom of fever. Household and community level antimalarial drug use represents an important entry point for malaria control programmes in most African countries (86). In Africa, shops are the main source of antimalarial drugs (86–89). However, the course of treatment sold is often sub-optimal (90–92). The currently available antimalarial drugs have been cheap, safe and relatively affordable and available to the communities. It has also been cost-effective to treat all fevers or suspected cases of malaria with chloroquine or SP. However, with the increasing cost of the antimalarials, especially CT with artemisinin-based combinations, this policy should be reconsidered.
Because countries in the African Region are starting to use household and community
management of fever as a strategy, it is becoming increasingly important to look at combination therapy. A strategy could be employed whereby there is a two-tier system with the community using one of the available monotherapy drugs which is not part of the combination therapy and the health facilities using the CT. This will allow the health system to adjust to the increasing costs of the delivery of CT. Currently there is no information to indicate at what level of resistance an antimalarial drug will not be cost-effective to use at household or community level when it is available as an effective drug at the health facility.
Since even in highly malarious areas, studies have shown varying levels of parasitaemia
from as low as 13% to a maximum of about 45% among fever patients, another possible strategy would be to improve community diagnosis. There is need, therefore, to evaluate the role of rapid diagnostic tests for malaria in areas of high transmission in order to make malaria diagnosis more specific as the cost of antimalarial treatment increases.

40
7. FUTURE RESEARCH ACTIVITIES
7.1 Research and Development for Combination Therapy
All national malaria control programmes require the capability for operational research so
that programme activities can be made more effective and adapted to changing antimalarial treatment policy. Such research needs to be relevant to national programme objectives, addressing not only the efficacy of specific antimalarial drug combinations but also social, economic, cultural and behavioural factors that may affect their implementation. Many malaria-endemic countries are in the process of implementing health reforms in which individuals and communities are being requested to increase their contribution to health while the provision of free health care is being reduced. Health systems are also being decentralized to increase community involvement. There is need to consider how this process can be used to optimize the provision of effective combination therapy for treatment of malaria and deployment within community-based interventions.
Research on operational issues, including cost-effectiveness and willingness to pay, related
to the CT and possibly antigen detection tests and microscopy is of great interest in areas where there is need for improving the current practice of clinical diagnosis. There are a number of unanswered questions on the available combination therapy drugs. These concern their widespread use in hyper-endemic areas, their role in transmission reduction, toxicity of various drugs, and use in infancy and pregnancy. Some drugs under development also need post-marketing surveillance (Phase III–IV trials) data. This could be obtained alongside implementation while those that have not been widely used are constantly monitored in the early phases of implementation.
In this context, therefore, there are immediate, intermediate and long-term research
priorities to be undertaken by various malaria control programmes and partners in order to forge a way forward in CT implementation.
7.2 Research Priorities
Immediate
Immediate priorities need to be addressed within one year or so. The identified areas are as
follows:
1. The effectiveness of other combinations (not including an artemisinin derivative) with cheaper and already available antimalarial drugs such as 4-aminoquinoline and sulfa drugs should also be explored for use in Africa;
2. Amodiaquine toxicity and its potential cross-resistance with chloroquine require further evaluation as these characteristics may limit the useful therapeutic life of this antimalarial drug;

41
3. Safety and efficacy of recommended first-line artemisinin-based combination therapy, especially in infants, pregnant women and patients with HIV/AIDS;
4. Improving post-marketing surveillance in resource-constrained countries;
5. Pharmaceutical development studies on improved formulations and presentations of ACTs, particularly paediatric formulations;
6. Studies of phased or controlled implementation of ACT, including at community and home level;
7. Assessing the impact of ACTs on malaria transmission, in areas with high inoculation rates;
8. Assessing the impact of ACTs on delaying the emergence of resistance to component drugs in areas with high inoculation rates;
9. Determining the costs of introduction of ACT;
10. Evaluation of the accessibility of the population;
11. How to reduce the duration of the process of drug policy change;
12. How to ensure the quality of the combination therapy drugs entering a country;
13. Determining and standardizing the evaluation tools of ACTs;
14. Determining the survey tools for pharmaco-vigilance of ACTs.
Medium-term
The issues identified below need to be addressed in 2 to 3 years.
1. Updating the existing policy instruments to accelerate the process of treatment policy review and change;
2. Fast-track development and testing of drug combinations in the pipeline, particularly dihydroartemisinin-piperaquine, LapDap-artesunate, pyronaridine-artesunate;
3. Evaluating combinations of drugs that are currently available and have independent modes of action to confer mutual protection against the development of resistance to individual drugs;
4. Evaluating the major determinants of the effectiveness of antimalarial treatment, i.e. under large-scale operational use;
5. Evaluating the cost of drug resistance, modelling, cost-effectiveness and cost-benefit analysis of different strategies to delay resistance;
6. Evaluating the cost (health burden and economic and development costs) of changing antimalarial treatment policy;
7. Financing for combination therapy antimalarial drugs.

42
Long-term
Long-term issues need to be addressed in the time frame of 4 to 5 years. These are:
1. Development of other strategies for extending the therapeutic life of antimalarial drugs;
2. Discovery and development of new drugs, particularly those with novel modes of action;
3. Development and validation of new tools for monitoring drug resistance.
8. CONCLUSION
The development and spread of resistance to antimalarial drugs is a major impediment to
the delivery of prompt, effective and safe treatment in countries of the African Region as prompt diagnosis and early disease management remain fundamental and indispensable elements of malaria control. It is in this regard that WHO currently recommends that countries faced with the problem of antimalarial drug resistance should adopt combination therapies, particularly those containing an artemisinin derivative.
This publication provides orientations on combination therapy, options available for
countries in the Region, policy formulation and implementation, and key research priorities. It is intended for use by programme managers, researchers, cooperating partners, nongovernmental organizations and others involved in malaria control. It should be used in conjunction with other documents on drug policy change developed by the Regional Office for Africa. These orientations will guide countries in making appropriate choices about what combinations drugs would be most suitable in their environment. Introducing combination therapy in the African Region should improve efficacy and effectiveness of first-line antimalarial treatment as well as delay the development and selection of resistant parasites to the few currently available and efficacious drugs.

43
REFERENCES
1. World Health Organization (1999). Expert Committee on Malaria. Twentieth Report. WHO, Geneva.
2. Garner P, Brabin B (1994). A review of randomized controlled trials of routine antimalarial
drug prophylaxis during pregnancy in endemic malarious areas. Bulletin of the World Health Organization 72(1): 89–99.
3. Sachs J (2000). Economic analyses indicate that the burden of malaria is great. Paper
presented at the African Summit on Roll Back Malaria, Abuja, Nigeria. 4. Bloland PB, Lackritz EM, Kazembe PN, Were JB, Steketee R and Campbell CC (1993).
Beyond chloroquine: implications of drug resistance for evaluating malaria therapy efficacy and treatment policy in Africa. Journal of Infectious Diseases 167(4): 932–937.
5. Bloland PB, Ettling M (1999). Making malaria treatment policy in the face of drug
resistance. Annals of Tropical Medicine and Parasitology 93(1): 5–23. 6. Marsh K (1998). Malaria disaster in Africa. Lancet 352: 924. 7. World Health Organization (2000). The use of antimalarial drugs. Report of a WHO informal
consultation. WHO, Geneva, WHO/CDS/RBM/2001.33. 8. White N (1999). Antimalarial drug resistance and combination therapy. Philosophical
Transactions of the Royal Society of London (354): 739–749. 9. White NJ (1999). Averting a malaria disaster. Lancet 353: 1965–1967. 10. White NJ (1998). Preventing antimalarial drug resistance through combinations. Drug
Resistance Updates 1:3–9. 11. World Health Organization (1998). The use of artemisinin and its derivatives as antimalarial
drugs: report of a joint CTD/DMP/TDR informal consultation. WHO, Geneva, WHO/MAL/98.1086.
12. Price RN, Nosten F, Luxemburger C, ter Kuile FO, Paiphun L, Chongsuphajaisiddhi T and
White NJ (1996). Effects of artemisinin derivatives on malaria transmissibility. Lancet 347: 1654–1658.
13. World Health Organization (2001). Antimalarial drug combination therapy. Report of a
WHO technical consultation. WHO, Geneva, WHO/CDS/RBM/2001.35. 14. World Health Organization (1973). Chemotherapy of malaria and resistance to antimalarials.
Report of a WHO scientific group. WHO, Geneva, WHO Technical Report Series, No. 529.

44
15. Peters W (1987). Chemotherapy and Drug Resistance in Malaria. London, Academic Press. 16. Peters W (1990). The prevention of antimalarial drug resistance. Pharmacology and
Therapeutics 47: 497–508. 17. Su X, Kirkman LA, Fujioka H and Wellems TE (1997). Complex polymorphisms in an
approximately 330-kDa protein are linked to chloroquine resistant P. falciparum in south-east Asia and Africa. Cell 91(5): 593–603.
18. Triglia T, Menting JG, Wilson C and Cowman AF (1997). Mutations in DHFS are
responsible for sulfone and sulfonamide resistance in P. falciparum. Proceedings of the National Academy of Science USA 94(25): 13944–13949
19. Watkins WM, Mosobo M (1993). Treatment of Plasmodium falciparum malaria with
pyrimethamine-sulphadoxine: selective pressure is a function of long elimination half-life. Transactions of the Royal Society of Tropical Medicine and Hygiene 87: 75–78.
20. Trape JF, Pison G, Preziosi MP, Enel C, Desgrees du Lou A, Delaunay V, et al. (1998).
Impact of chloroquine resistance on malaria mortality. Comptes rendu de l’Académie des Sciences 321(Series III): 689–697.
21. Greenberg AE, Ntumbanzondo M, Ntula N, Mawa L, Howell J and Davacl F (1998).
Hospital based surveillance of malaria related paediatric morbidity and mortality in Kinshasa, Zaire. Bulletin of the World Health Organization 67(2): 189–196.
22. Bruce-Chwatt LJ (1987). Chemotherapy of Malaria, second ed., World Health Organization,
Geneva. 23. Prasad RN, Prasad H, Virk KJ and Sharma VP (1990). Application of simplified in vivo test
system for determining chloroquine resistance in Plasmodium falciparum. Bulletin of the World Health Organization 68: 755–758.
24. Rieckman KH (1990). Monitoring the response of malaria infections to treatment. Bulletin of
the World Health Organization 68: 759–760.
25. World Health Organization (1996). Assessment of therapeutic efficacy of antimalarial drugs for uncomplicated malaria in areas with intense transmission. WHO, Geneva.
26. Olliaro P, Nevill C, Le Bras J, Ringwald P, Mussano P, Garner P and Brasseur P (1996).
Systematic review of amodiaquine treatment in uncomplicated malaria. Lancet 348: 1196–1201.
27. Suebsaeng L, Wernsdorfer WH and Rooney W (1986). Sensitivity to quinine and mefloquine of Plasmodium falciparum in Thailand. Bulletin of the World Health Organization 64(5): 759–765.

45
28. Cerutti C Jr, Durlacher RR, de Alencar FE, Segurado AA and Pang LW (1999). In vivo efficacy of mefloquine for the treatment of falciparum malaria in Brazil. Journal of Infectious Diseases 180: 2077–2080.
29. Basco LK, Ringwald P, Thor R, Doury JC and Le Bras J (1995). Activity in vitro of
chloroquine, cycloguanil and mefloquine against African isolates of Plasmodium falciparum: presumptive evidence for chemoprophylactic efficacy in central and west Africa. Transactions of the Royal Society of Tropical Medicine and Hygiene 89: 657–658.
30. Bunnag D, Viravan C, Looareesuwan S, Karbwang J and Harinasuta T (1991). Double blind
randomised clinical trial of oral artesunate at once or twice daily dose in falciparum malaria. Southeast Asian Journal of Tropical Medicine and Public Health 22(4): 539–543.
31. Bunnag D, Viravan C, Looareesuwan S, Karbwang J and Harinasuta (1991). Double blind
randomised clinical trial of two different regimens of oral artesunate in falciparum malaria. Southeast Asian Journal of Tropical Medicine and Public Health 22(4): 534–538.
32. Bunnag D, Karbwang J, Chitamas S and Harinasuta T (1993). Intramuscular artemether in
female patients with uncomplicated falciparum malaria. Southeast Asian Journal of Tropical Medicine and Public Health 24(1): 49–52.
33. Bunnag D, Karbwang J and Harinasuta T (1992). Artemether in the treatment of multiple
drug resistant falciparum malaria. Southeast Asian Journal of Tropical Medicine and Public Health 23(4): 762–767.
34. Luxemburger C, Nosten F, Raimond SD, Chongsuphajaisiddhi T and White NJ (1995). Oral
artesunate in the treatment of uncomplicated hyperparasitic falciparum malaria. American Journal of Tropical Medicine and Hygiene 53(5): 522–535.
35. Hien TT (1994). An overview of the clinical use of artemisinin and its derivatives in the
treatment of falciparum malaria in Viet Nam. Transactions of the Royal Society of Tropical Medcine and Hygiene 88(Suppl. 1): S7–S8.
36. Murphy GS, Basri H, Purnomo B, Andersen EM, Bangs MJ, Mount DL et al. (1993). Vivax
malaria resistant to treatment and prophylaxis with chloroquine. Lancet 341: 96–100. 37. Alecrim Mdos G, Alecrim WD, Macedo V, Korves CT, Roberts DR, Li J et al. (1999).
Description of a possible clonal expansion of Plasmodium vivax in Manaus-Amazonas, Brazil. Revista da Sociadade Brasiliera de Medicina Tropical 32: 303–305.
38. Rodriguez RM (1999). Eficacia terapéutica de la cloroquina en la malaria por Plasmodium
vivax. Parroquia El Dorado, Municipio Sifontes, estado Bolívar, Venezuela. Escuela de Medicina. Bolivar, Universidad de Oriente.

46
39. Paez E (2000). Evaluation of in vivo response of Plasmodium vivax to chloroquine and primaquine in Sifontes, Bolivar State, Venezuela. In: XV International Congress of Tropical Medicine and Malaria, Venezuela.
40. World Health Organization (1994). Antimalarial drug policies: data requirements, treatment
of uncomplicated malaria and the management of malaria in pregnancy. WHO, Geneva, WHO/MAL/94.1070.
41. WHO Regional Office for Africa. Framework for developing, implementing and updating
antimalarial treatment policy in Africa. A guide for country malaria control programmes (in press).
42. White NJ (1999). Delaying antimalarial drug resistance with combination therapy.
Parassitologia 41: 301–308. 43. White NJ, Olliaro PL (1996). Strategies for the prevention of antimalarial drug resistance:
rationale for combination therapy for malaria. Parasitology Today 12(10): 399–401. 44. World Health Organization (2000). The use of essential drugs. WHO, Geneva, WHO
Technical Report Series No. 895. 45. Ogutu RB, Smoack BL, Nduati RW, Mbori-Ngacha DA, Mwathe F and Shanks GD (2000).
The efficacy of pyrimethamine-sulphadoxine resistance of Plasmodium falciparum malaria in Kenyan children. Transactions of the Royal Society of Tropical Medicine and Hygiene 94: 83–84.
46. Trigg JK, Mbwana H, Chambo O, Hills E, Watkins B and Curtis CF (1997). Resistance to
pyrimethamine-sulfadoxine in Plasmodium falciparum in 12 villages in northeast Tanzania and a test of chlorproguanil-dapsone. Acta Tropica 63: 185–189.
47. Price RN, Nosten F, Luxemburger C, van Vugt M, Phaipun L Chogsuphajaisiddhi T and
White NJ (1997). artesunate-mefloquine treatment of multi-drug resistant falciparum malaria. Transactions of the Royal Society of Tropical Medicine and Hygiene 91: 574–577.
48. White NJ (1997). Minireview: assessment of the pharmacodynamic properties of antimalarial
drugs in vivo. Antimicrobial Agents and Chemotherapy 41(7): 1413–1422. 49. World Health Organization (1998). Informal consultation on the neurological investigations
required for patients treated with artemisinin compounds and derivatives. WHO, Geneva. 50. Brockman A, Price RN, van Vugt M, Heppner DG, Walsh D, Sookto P et al. (2000).
Plasmodium falciparum antimalarial drug susceptibility on the northwestern border of Thailand during five years of extensive use of artesunate-mefloquine. Transactions of the Royal Society of Tropical Medicine and Hygiene 94: 537–544.

47
51. Rieckmann KH, Suebsaeng L and Rooney W (1987). Response of Plasmodium falciparum infections to pyrimethamine-sulphadoxine in Thailand. American Journal of Tropical Medicine and Hygiene 37: 211–216.
52. White NJ (1987). Combination treatment for falciparum prophylaxis. Lancet 1: 680–681. 53. Looareesuwan S, Vanijanonta S, Viravan C, Wilairatana P, Charoenla P et al. (1994).
Randomised trials of mefloquine-tetracycline and quinine-tetracycline for acute uncomplicated falciparum malaria. Acta Tropica 57: 47–53.
54. von Seidlein L, Milligan P, Pinder M, Bojang K, Anyalebechi C et al. (2000). Efficacy of
artesunate plus pyrimethamine-sulphadoxine for uncomplicated malaria in Gambian children: a double blind, randomised, controlled trial. Lancet 355: 352–357.
55. Doherty JF, Sadiq AD, Bayo L, Alloueche A, Oliaro P et al. (1999). A randomised safety and
tolerability trial of artesunate plus sufladoxine-pyrimethamine versus sulfadoxine-pyrimethamine alone for the treatment of uncomplicated malaria in Gambian children. Transactions of the Royal Society of Tropical Medicine and Hygiene 93(5): 543–546.
56. Nzila MA, Mberu EK, Sulo J, Dayo H, Winstanley PA, Sibley CH et al. (2000). Towards an
understanding of the mechanism of pyrimethamine/sulfadoxine resistance in Plasmodium falciparum: the genotyping of dihydrofolate reductase and dihydropteroate synthase of Kenyan parasites. Antimicrobial Agents and Chemotherapy 44(4): 991–996.
57. Reeder JC, Rieckmann KH, Genton B, Lorry K, Wines B and Cowman AT (1996). Point
mutations in the dihydrofolate reductase and dihydropteroate synthetase genes and in vitro susceptibility to pyrimethamine and cycloguanil of Plasmodium falciparum isolates from Papua New Guinea. American Journal of Tropical Medicine and Hygiene 55: 209–213.
58. Diourte Y, Djimde A, Doumbo OK, Sagara I, Coulibaly Y et al. (1999). Pyrimethamine-
sulfadoxine efficacy and selection for mutations in Plasmodium falciparum dihydrofolate reductase and dihydropteroate synthase in Mali. American Journal of Tropical Medicine and Hygiene 60: 475–478.
59. Basco LK, Ringwald P (2000). Molecular epidemiology of malaria in Yaounde, Cameroon.
Vl. Sequence variations in the Plasmodium falciparum dihydrofolate reductase-thymidylate synthase gene and in vitro resistance to pyrimethamine and cycloguanil. American Journal of Tropical Medicine and Hygiene 62: 271–276.
60. Mokherjee S, Howard V, Nzila-Mouanda A, Watkins W and Sibley CH (1999).
Identification and analysis of dihydrofolate reductase alleles from Plasmodium falciparum present at low frequency in polyclonal patient samples. American Journal of Tropical Medicine and Hygiene 61: 131–140.

48
61. Adjuik M, Agnamey P, Babiker A, Borrmann S, Brasseur P, Cisse M et al. (2002). Amodiaquine-artesunate versus amodiaquine for uncomplicated Plasmodium falciparum malaria in African children: a randomised, multicentre trial. Lancet 359: 1365–1372.
62. Dost FH, Gladtke E (1969). Pharmacokinetics of 2-sulfanilamido-3-methoxy pyrazine in
children (elimination, enteral absorption, distribution and dosage). Arzneimittelforschung 19(8): 1304–1307 (in German).
63. Looareesuwan S, Wilairatana P, Chokejindachai W, Chalermrut K, Wernsdorfer W et al.
(1999). A randomized, double blind, comparative trial of a new oral combination of artemether and benflumetol (CGP 56697) with mefloquine in the treatment of acute Plasmodium falciparum malaria in Thailand. American Journal of Tropical Medicine and Hygiene 60(2): 238–243.
64. Van Vugt M, Brockman A, Gemperli B, Luxemburger C, Gathmann I et al. (1998).
Randomized comparison of artemether-benflumetol and artesunate-mefloquine in treatment of multidrug-resistant falciparum malaria. Antimicrobial Agents and Chemotherapy 42(1): 135–139.
65. Von Seidlein L, Jaffar S, Pinder M, Haywood M, Snounou G, et al. (1997). Treatment of
African children with uncomplicated falciparum malaria with a new antimalarial drug, CGP 56697. Journal of Infectious Diseases 176(4): 1113–1116.
66. Norvatis Pharma AG (1997). A randomised, double-blind, parallel group trial comparing
efficacy, safety and pharmacokinetics of the standard schedule (4-4 tablets over 48 hours) with two higher dose schedules of co-artemether in the treatment of acute Plasmodium falciparum malaria in adults and children in Thailand. Basle.
67. Van Vugt M, Ezzet F, Phaipun L, Nosten F and White NJ (1998). The relationship between
capillary and venous concentrations of the antimalarial drug lumefantrine (benflumetol). Transactions of the Royal Society of Tropical Medicine and Hygiene 92: 564–565.
68. Tanariya P, Tippawangkoso P, Karbwang J, Na-Bangchang K and Wernsdorfer WH (2000).
In vitro sensitivity of Plasmodum falciparum and clinical response to lumefantrine (benflumetol) and artemether. British Journal of Clinical Pharmacology 49(5): 437–444.
69. Vugt MV, Wilairatana P, Gemperli B, Gathmann I, Phaipun L et al. (1999). Efficacy of six
doses of artemether-lumefantrine (benflumetol) in the treatment of multi-drug resistant falciparum malaria. American Journal of Tropical Medicine and Hygiene 60(6): 936–942.
70. White NJ, van Vugt M and Ezzet F (1999). Clinical pharmacokinetics and
pharmacodynamics of artemether-lumefantrine. Clinical Pharmacokinetics 37(2): 105–125. 71. Norvatis Pharma AG (1998). Coartem® tablets (CGP 56697, co-artemether). Integrated
summary of safety (ISS). Basle.

49
72. Van Vugt M, Ezzet F, Nosten F, Gathmann I, Wilairatana P et al. (1999). No evidence of cardiotoxicity during antimalarial treatment with artemether-lumefantrine. American Journal of Tropical Medicine and Hygiene 61(6): 964–967.
73. McIntosh HM and Greenwood BM (1998). Chloroquine or amodiaquine combined with
sulphadoxine-pyrimethamine as a treatment for uncomplicated malaria: A systematic review. Annals of Tropical Medicine and Parasitology 93(3): 265–270.
74. Ronn AM, Msangeni HA, Mhina J, Wernsdorfer WH and Bygbjerg B (1996). High level of
resistance of Plasmodium falciparum to sulfadoxine-pyrimethamine in children in Tanzania. Transactions of the Royal Society of Tropical Medicine and Hygiene 90(2): 179–181.
75. Onyiorah E, van Hensbroek MB, Jah MS and Greenwood B (1996). Early clinical failures
after pyrimethamine-sulfadoxine treatment of uncomplicated malaria. Transactions of the Royal Society of Tropical Medicine and Hygiene 90: 307–308.
76. World Health Organization (1984). Advances in malaria chemotherapy. Report of a WHO
scientific group. WHO, Geneva, WHO Technical Report Series No. 711. 77. World Health Organization (1990). Practical chemotherapy of malaria. Report of a WHO
scientific group. WHO, Geneva, WHO Technical Report Series, No. 805. 78. Bojang KA, Schneider G, Forck S, Obaro SK, Jaffar S et al. (1998). A trial of Fansidar TM
plus chloroquine or Fansidar’ alone for the treatment of uncomplicated malaria in Gambian children. Transactions of the Royal Society of Tropical Medicine and Hygiene 92: 73–76.
79. Darlow B, Vrbova H, Gibney S, Jolley D, Stace J and Alpers M (1982). Sulfadoxine-
pyrimethamine for the treatment of acute malaria in children in Papua New Guinea. I. Plasmodium falciparum. American Journal of Tropical Medicine and Hygiene 31: 1–9.
80. Brasseur P, Guiguemde R, Diallo S, Guiyedi V, Kombila M et al. (1999). Amodiaquine
remains effective for treating uncomplicated malaria in west and central Africa. Transactions of the Royal Society of Tropical Medicine and Hygiene 93(6): 645–650.
81. Huang Q et al. (1988). Effectiveness of amodiaquine, sulfadoxine-pyrimethamine and
combinations of these drugs for treating chloroquine-resistant falciparum malaria in Hainan Island, China. Bulletin of the World Health Organization 66: 353–358.
82. Schapira A, Schwalbach JFL (1988). Evaluation of four therapeutic regimens for falciparum
malaria in Mozambique. Bulletin of the World Health Organization 66: 219–226. 83. Dinis DV, Schapira A (1990). Comparative study of the efficacy and side-effects of two
therapeutic regimens against chloroquine-resistant falciparum malaria in Maputo, Mozambique. Bulletin de la Société de Pathologie Exotique 83: 521–528.

50
84. Staedke SG, Kamya MR, Dorsey G, Gasasira A, Ndeezi G, et al. (2001). Amodiaquine, sulfadoxine-pyrimethamine, and combination therapy for treatment of uncomplicated falciparum malaria in Kampala, Uganda: a randomised trial. Lancet 358: 368–374.
85. World Health Organization (2002). Implementation of new antimalarial treatment policies in
Africa. WHO Secretariat to the RBM Partnership. Discussion paper for the RBM Partners Meeting.
86. Foster S (1995). Treatment of malaria outside the formal health services. Journal of Tropical
Medicine and Hygiene 98(1): 29–34. 87. Ndyomugyenyi R, Neema S and Magnussen P (1998). The use of formal and informal
services for antenatal care and malaria treatment in rural Uganda. Health Policy and Planning 1: 94–102.
88. Ongore D, Nyabola L (1996). Role of shops and shopkeepers in malaria control. East African
Medical Journal 6: 390–394. 89. Snow RW, Peshu N, Forster D, Mwenesi H and Marsh K (1992). The role of shops in the
treatment and prevention of childhood malaria on the coast of Kenya. Transactions of the Royal Society of Tropical Medicine and Hygiene 86(3): 237–239.
90. Nyamongo IK (1999). Home case management of malaria: an ethnographic study of lay
people’s classification of drugs in Suneka division, Kenya. Tropical Medicine and International Health 4: 736–743.
91. Nsimba SE (1999). A household survey of source, availability, and use of antimalarials in a
rural area of Tanzania. Drug Information Journal 33: 1025–1032. 92. Mwenesi HT, Harpham T and Snow RW (1995). Child malaria treatment practices among
mothers in Kenya. Social Science and Medicine 40: 1271–1277.
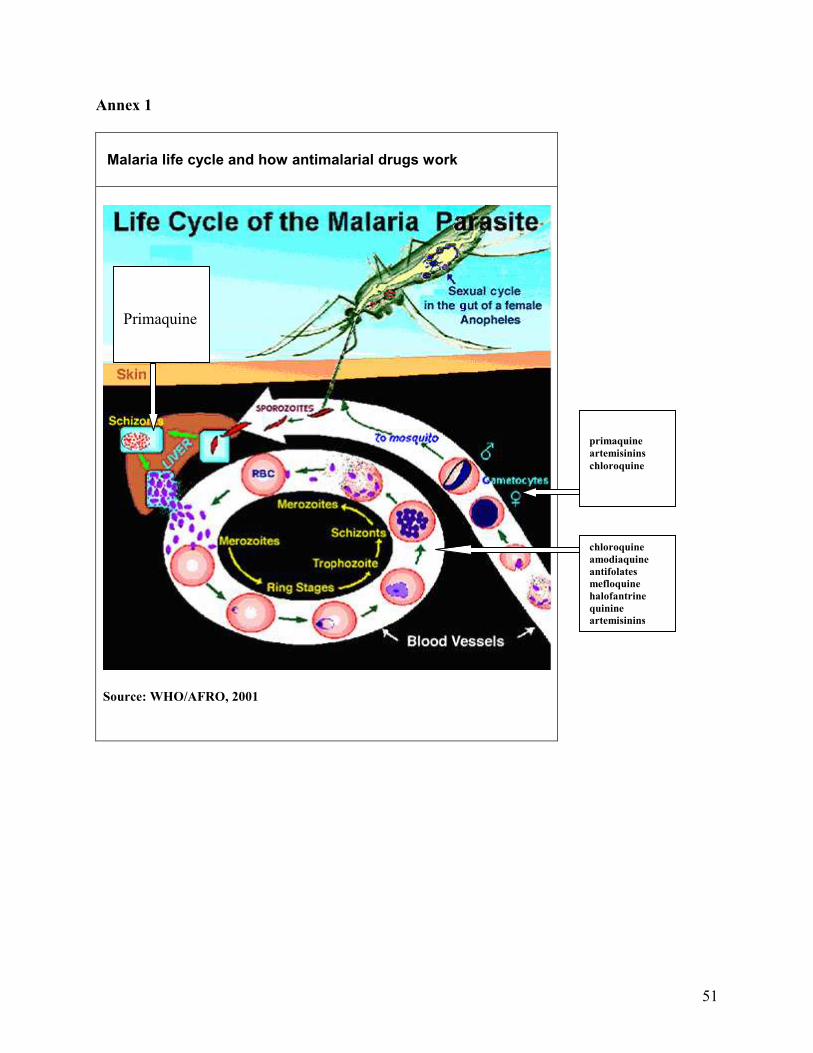
51
Annex 1
Malaria life cycle and how antimalarial drugs work
Source: WHO/AFRO, 2001
primaquine
artemisinins
chloroquine
chloroquine
amodiaquine
antifolates
mefloquine
halofantrine
quinine
artemisinins
Primaquine
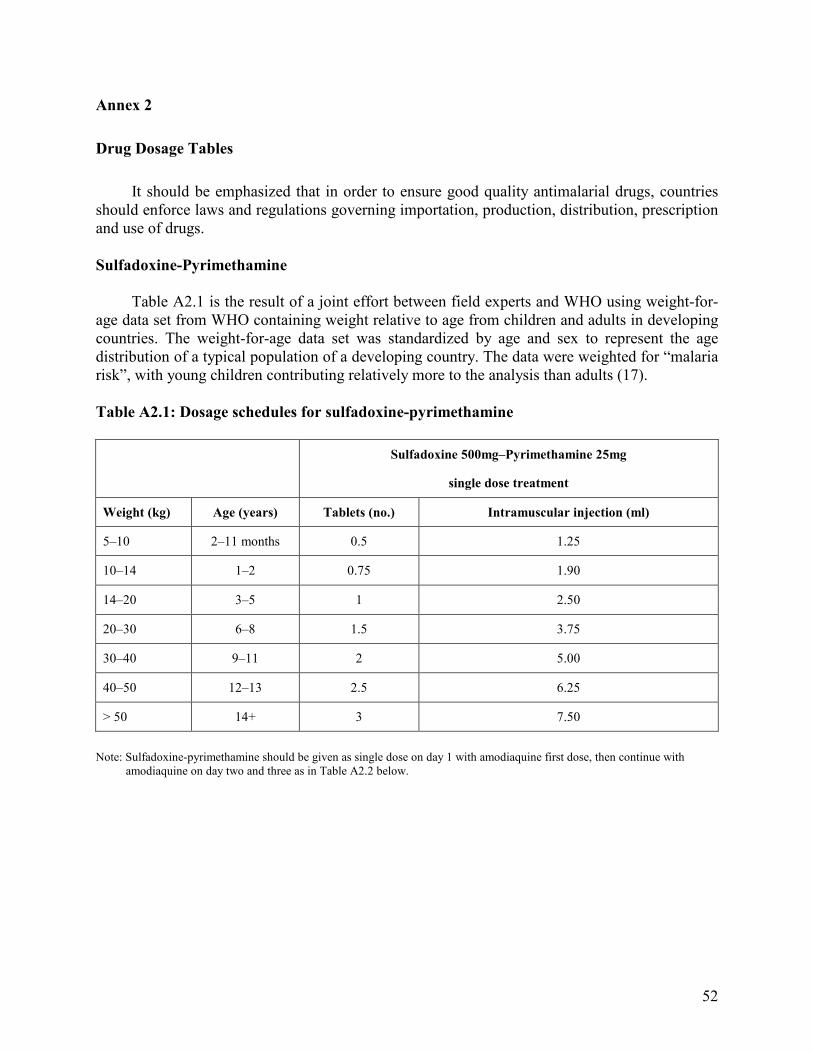
52
Annex 2
Drug Dosage Tables
It should be emphasized that in order to ensure good quality antimalarial drugs, countries should enforce laws and regulations governing importation, production, distribution, prescription and use of drugs.
Sulfadoxine-Pyrimethamine
Table A2.1 is the result of a joint effort between field experts and WHO using weight-for-
age data set from WHO containing weight relative to age from children and adults in developing countries. The weight-for-age data set was standardized by age and sex to represent the age distribution of a typical population of a developing country. The data were weighted for “malaria risk”, with young children contributing relatively more to the analysis than adults (17).
Table A2.1: Dosage schedules for sulfadoxine-pyrimethamine
Sulfadoxine 500mg–Pyrimethamine 25mg
single dose treatment
Weight (kg) Age (years) Tablets (no.) Intramuscular injection (ml)
5–10 2–11 months 0.5 1.25
10–14 1–2 0.75 1.90
14–20 3–5 1 2.50
20–30 6–8 1.5 3.75
30–40 9–11 2 5.00
40–50 12–13 2.5 6.25
> 50 14+ 3 7.50
Note: Sulfadoxine-pyrimethamine should be given as single dose on day 1 with amodiaquine first dose, then continue with
amodiaquine on day two and three as in Table A2.2 below.
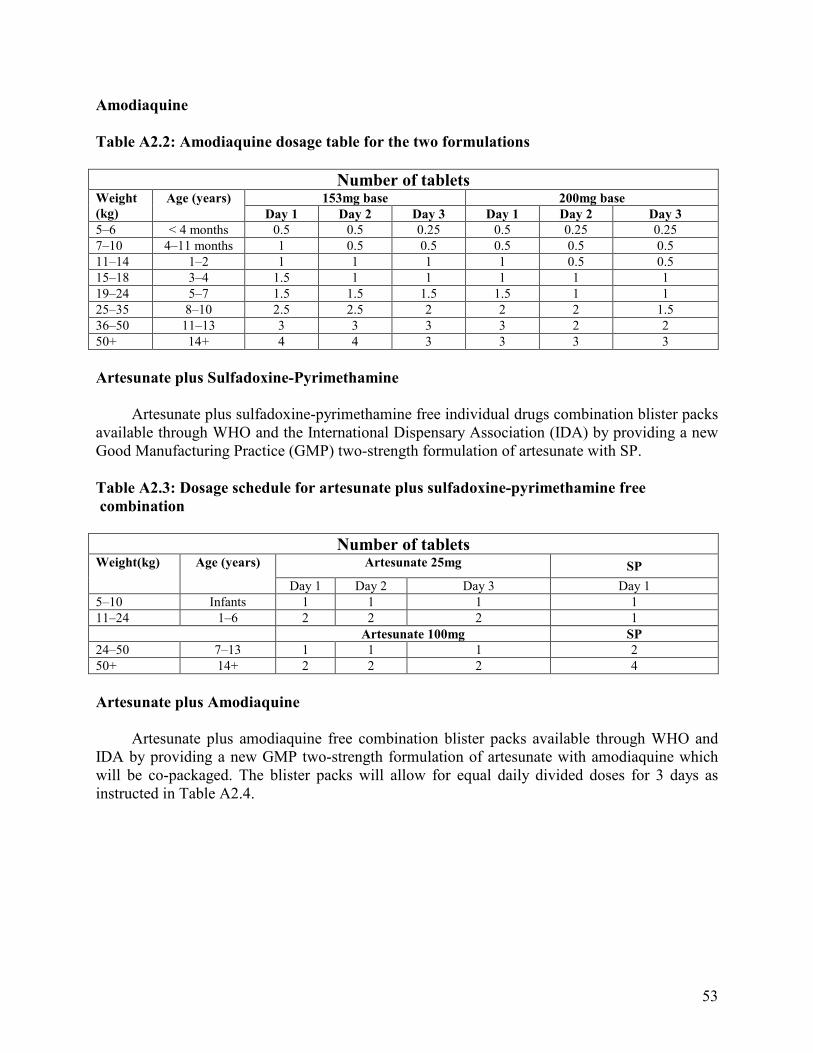
53
Amodiaquine
Table A2.2: Amodiaquine dosage table for the two formulations
Number of tablets 153mg base 200mg base Weight
(kg)
Age (years)
Day 1 Day 2 Day 3 Day 1 Day 2 Day 3
5–6 < 4 months 0.5 0.5 0.25 0.5 0.25 0.25
7–10 4–11 months 1 0.5 0.5 0.5 0.5 0.5
11–14 1–2 1 1 1 1 0.5 0.5
15–18 3–4 1.5 1 1 1 1 1
19–24 5–7 1.5 1.5 1.5 1.5 1 1
25–35 8–10 2.5 2.5 2 2 2 1.5
36–50 11–13 3 3 3 3 2 2
50+ 14+ 4 4 3 3 3 3
Artesunate plus Sulfadoxine-Pyrimethamine
Artesunate plus sulfadoxine-pyrimethamine free individual drugs combination blister packs available through WHO and the International Dispensary Association (IDA) by providing a new Good Manufacturing Practice (GMP) two-strength formulation of artesunate with SP.
Table A2.3: Dosage schedule for artesunate plus sulfadoxine-pyrimethamine free
combination
Number of tablets Artesunate 25mg SP Weight(kg) Age (years)
Day 1 Day 2 Day 3 Day 1
5–10 Infants 1 1 1 1
11–24 1–6 2 2 2 1
Artesunate 100mg SP
24–50 7–13 1 1 1 2
50+ 14+ 2 2 2 4
Artesunate plus Amodiaquine
Artesunate plus amodiaquine free combination blister packs available through WHO and IDA by providing a new GMP two-strength formulation of artesunate with amodiaquine which will be co-packaged. The blister packs will allow for equal daily divided doses for 3 days as instructed in Table A2.4.
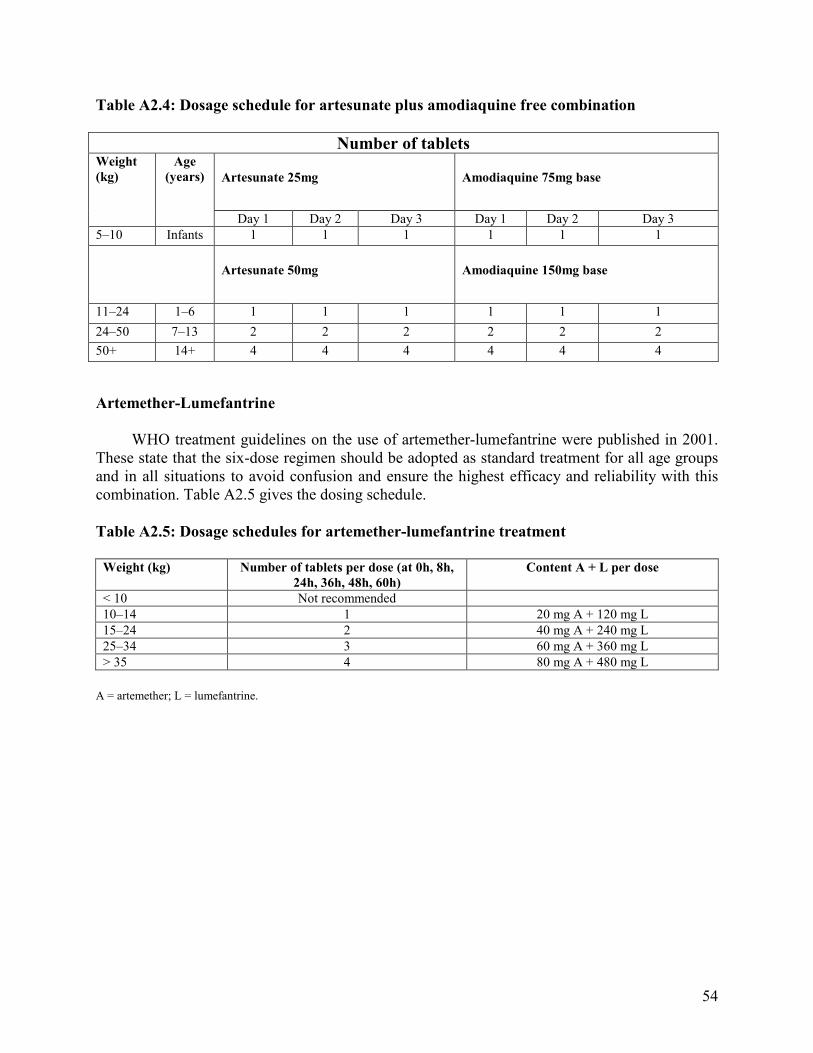
54
Table A2.4: Dosage schedule for artesunate plus amodiaquine free combination
Number of tablets
Artesunate 25mg Amodiaquine 75mg base
Weight
(kg)
Age
(years)
Day 1 Day 2 Day 3 Day 1 Day 2 Day 3
5–10 Infants 1 1 1 1 1 1
Artesunate 50mg Amodiaquine 150mg base
11–24 1–6 1 1 1 1 1 1
24–50 7–13 2 2 2 2 2 2
50+ 14+ 4 4 4 4 4 4
Artemether-Lumefantrine
WHO treatment guidelines on the use of artemether-lumefantrine were published in 2001. These state that the six-dose regimen should be adopted as standard treatment for all age groups and in all situations to avoid confusion and ensure the highest efficacy and reliability with this combination. Table A2.5 gives the dosing schedule.
Table A2.5: Dosage schedules for artemether-lumefantrine treatment
Weight (kg) Number of tablets per dose (at 0h, 8h,
24h, 36h, 48h, 60h)
Content A + L per dose
< 10 Not recommended
10–14 1 20 mg A + 120 mg L
15–24 2 40 mg A + 240 mg L
25–34 3 60 mg A + 360 mg L
> 35 4 80 mg A + 480 mg L
A = artemether; L = lumefantrine.
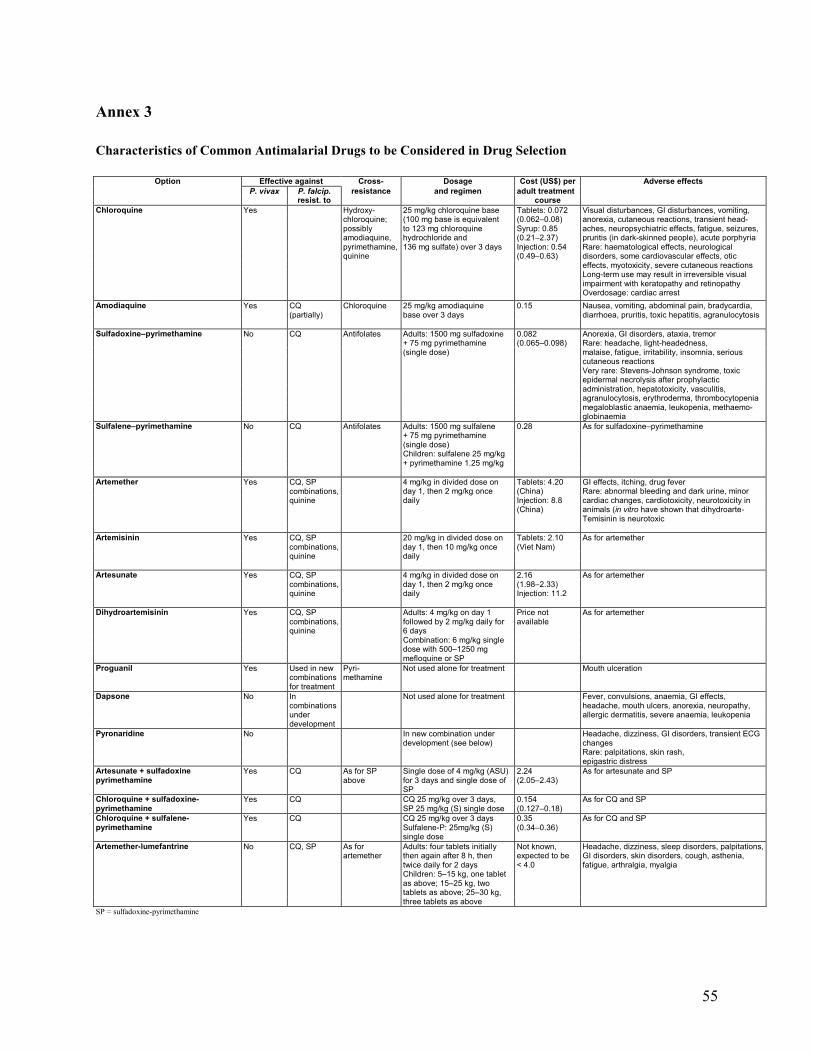
55
Annex 3
Characteristics of Common Antimalarial Drugs to be Considered in Drug Selection
Option Effective against Cross- Dosage Cost (US$) per Adverse effects
P. vivax P. falcip. resistance and regimen adult treatment resist. to course
Chloroquine Yes
Hydroxy- chloroquine; possibly amodiaquine, pyrimethamine, quinine
25 mg/kg chloroquine base (100 mg base is equivalent to 123 mg chloroquine hydrochloride and 136 mg sulfate) over 3 days
Tablets: 0.072 (0.062–0.08) Syrup: 0.85 (0.21–2.37) Injection: 0.54 (0.49–0.63)
Visual disturbances, GI disturbances, vomiting, anorexia, cutaneous reactions, transient head- aches, neuropsychiatric effects, fatigue, seizures, pruritis (in dark-skinned people), acute porphyria Rare: haematological effects, neurological disorders, some cardiovascular effects, otic effects, myotoxicity, severe cutaneous reactions Long-term use may result in irreversible visual impairment with keratopathy and retinopathy Overdosage: cardiac arrest
Amodiaquine Yes Chloroquine 0.15
CQ (partially)
25 mg/kg amodiaquine base over 3 days
Nausea, vomiting, abdominal pain, bradycardia, diarrhoea, pruritis, toxic hepatitis, agranulocytosis
Sulfadoxine–pyrimethamine No CQ Antifolates
Adults: 1500 mg sulfadoxine + 75 mg pyrimethamine (single dose)
0.082 (0.065–0.098)
Anorexia, GI disorders, ataxia, tremor Rare: headache, light-headedness, malaise, fatigue, irritability, insomnia, serious cutaneous reactions Very rare: Stevens-Johnson syndrome, toxic epidermal necrolysis after prophylactic administration, hepatotoxicity, vasculitis, agranulocytosis, erythroderma, thrombocytopenia megaloblastic anaemia, leukopenia, methaemo- globinaemia
Sulfalene–pyrimethamine No CQ 0.28 As for sulfadoxine–pyrimethamine
Antifolates
Adults: 1500 mg sulfalene + 75 mg pyrimethamine (single dose) Children: sulfalene 25 mg/kg + pyrimethamine 1.25 mg/kg
Artemether Yes CQ, SP combinations, quinine
4 mg/kg in divided dose on day 1, then 2 mg/kg once daily
Tablets: 4.20 (China) Injection: 8.8 (China)
GI effects, itching, drug fever Rare: abnormal bleeding and dark urine, minor cardiac changes, cardiotoxicity, neurotoxicity in animals (in vitro have shown that dihydroarte- Temisinin is neurotoxic
Artemisinin Yes As for artemether
CQ, SP combinations, quinine
20 mg/kg in divided dose on day 1, then 10 mg/kg once daily
Tablets: 2.10 (Viet Nam)
Artesunate Yes As for artemether
CQ, SP combinations, quinine
4 mg/kg in divided dose on day 1, then 2 mg/kg once daily
2.16 (1.98–2.33) Injection: 11.2
Dihydroartemisinin Yes
CQ, SP combinations, quinine
Adults: 4 mg/kg on day 1 followed by 2 mg/kg daily for 6 days Combination: 6 mg/kg single dose with 500–1250 mg mefloquine or SP
Price not available
As for artemether
Proguanil Yes Mouth ulceration
Used in new combinations for treatment
Pyri- methamine
Not used alone for treatment
Dapsone No
In combinations under development
Not used alone for treatment
Fever, convulsions, anaemia, GI effects, headache, mouth ulcers, anorexia, neuropathy, allergic dermatitis, severe anaemia, leukopenia
Pyronaridine No
In new combination under development (see below)
Headache, dizziness, GI disorders, transient ECG changes Rare: palpitations, skin rash, epigastric distress
Yes CQ As for SP 2.24 Artesunate + sulfadoxine pyrimethamine above
Single dose of 4 mg/kg (ASU) for 3 days and single dose of SP
(2.05–2.43) As for artesunate and SP
Yes CQ Chloroquine + sulfadoxine- pyrimethamine
CQ 25 mg/kg over 3 days, SP 25 mg/kg (S) single dose
0.154 (0.127–0.18)
As for CQ and SP
Yes CQ Chloroquine + sulfalene- pyrimethamine
CQ 25 mg/kg over 3 days Sulfalene-P: 25mg/kg (S) single dose
0.35 (0.34–0.36)
As for CQ and SP
Artemether-lumefantrine No CQ, SP Not known, expected to be < 4.0
Headache, dizziness, sleep disorders, palpitations, GI disorders, skin disorders, cough, asthenia, fatigue, arthralgia, myalgia
As for artemether
Adults: four tablets initially then again after 8 h, then twice daily for 2 days Children: 5–15 kg, one tablet as above; 15–25 kg, two tablets as above; 25–30 kg, three tablets as above
SP = sulfadoxine-pyrimethamine
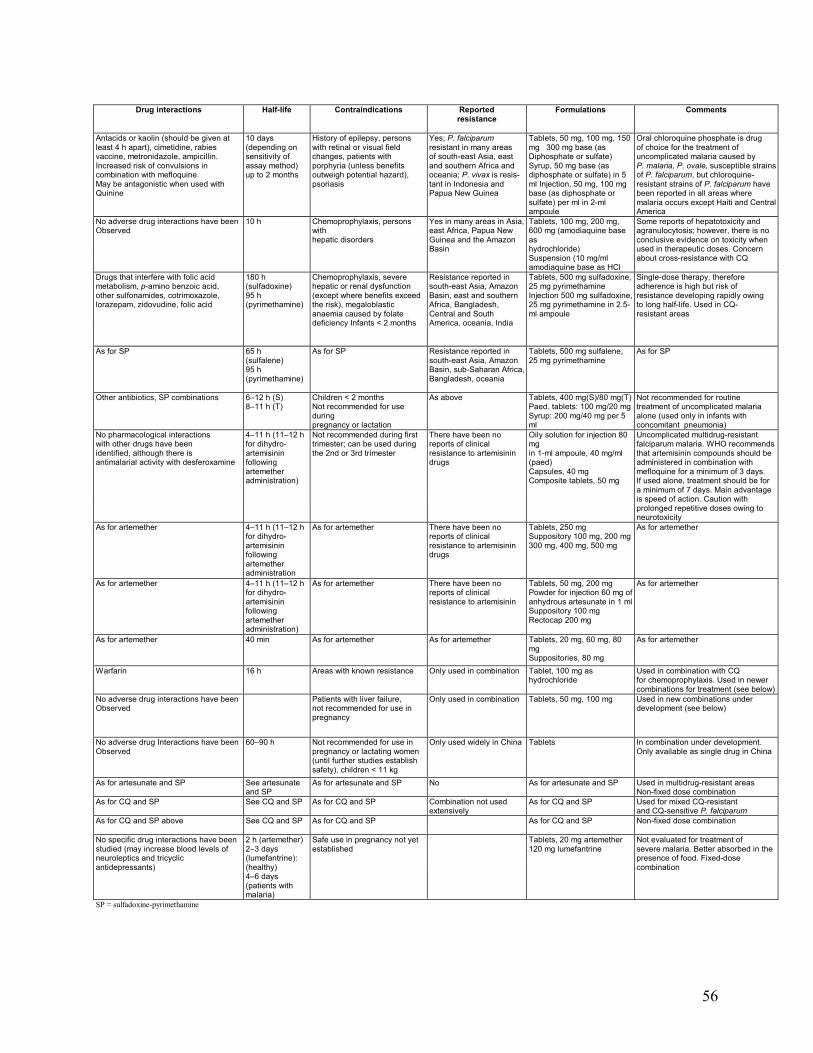
56
Drug interactions Half-life Contraindications Reported Formulations Comments resistance
10 days (depending on sensitivity of assay method) up to 2 months
History of epilepsy, persons with retinal or visual field changes, patients with porphyria (unless benefits outweigh potential hazard), psoriasis
Antacids or kaolin (should be given at least 4 h apart), cimetidine, rabies vaccine, metronidazole, ampicillin. Increased risk of convulsions in combination with mefloquine. May be antagonistic when used with Quinine
Yes; P. falciparum resistant in many areas of south-east Asia, east and southern Africa and oceania; P. vivax is resis- tant in Indonesia and Papua New Guinea
Tablets, 50 mg, 100 mg, 150 mg 300 mg base (as Diphosphate or sulfate) Syrup, 50 mg base (as diphosphate or sulfate) in 5 ml Injection, 50 mg, 100 mg base (as diphosphate or sulfate) per ml in 2-ml ampoule
Oral chloroquine phosphate is drug of choice for the treatment of uncomplicated malaria caused by P. malaria, P. ovale, susceptible strains of P. falciparum, but chloroquine- resistant strains of P. falciparum have been reported in all areas where malaria occurs except Haiti and Central America
10 h
No adverse drug interactions have been Observed
Chemoprophylaxis, persons with hepatic disorders
Yes in many areas in Asia, east Africa, Papua New Guinea and the Amazon Basin
Tablets, 100 mg, 200 mg, 600 mg (amodiaquine base as hydrochloride) Suspension (10 mg/ml amodiaquine base as HCl
Some reports of hepatotoxicity and agranulocytosis; however, there is no conclusive evidence on toxicity when used in therapeutic doses. Concern about cross-resistance with CQ
180 h (sulfadoxine) 95 h (pyrimethamine)
Resistance reported in south-east Asia, Amazon Basin, east and southern Africa, Bangladesh, Central and South America, oceania, India
Drugs that interfere with folic acid metabolism, p-amino benzoic acid, other sulfonamides, cotrimoxazole, lorazepam, zidovudine, folic acid
Chemoprophylaxis, severe hepatic or renal dysfunction (except where benefits exceed the risk), megaloblastic anaemia caused by folate deficiency Infants < 2 months
Tablets, 500 mg sulfadoxine, 25 mg pyrimethamine Injection 500 mg sulfadoxine, 25 mg pyrimethamine in 2.5-ml ampoule
Single-dose therapy, therefore adherence is high but risk of resistance developing rapidly owing to long half-life. Used in CQ- resistant areas
As for SP 65 h (sulfalene) 95 h (pyrimethamine)
As for SP Resistance reported in south-east Asia, Amazon Basin, sub-Saharan Africa, Bangladesh, oceania
Tablets, 500 mg sulfalene, 25 mg pyrimethamine
As for SP
Other antibiotics, SP combinations
6–12 h (S) 8–11 h (T)
Children < 2 months Not recommended for use during pregnancy or lactation
As above Tablets, 400 mg(S)/80 mg(T) Paed. tablets: 100 mg/20 mg Syrup: 200 mg/40 mg per 5 ml
Not recommended for routine treatment of uncomplicated malaria alone (used only in infants with concomitant pneumonia)
Oily solution for injection 80 mg in 1-ml ampoule, 40 mg/ml (paed) Capsules, 40 mg Composite tablets, 50 mg
4–11 h (11–12 h for dihydro- artemisinin following artemether administration)
There have been no reports of clinical resistance to artemisinin drugs
No pharmacological interactions with other drugs have been identified, although there is antimalarial activity with desferoxamine
Not recommended during first trimester; can be used during the 2nd or 3rd trimester
Uncomplicated multidrug-resistant falciparum malaria. WHO recommends that artemisinin compounds should be administered in combination with mefloquine for a minimum of 3 days. If used alone, treatment should be for a minimum of 7 days. Main advantage is speed of action. Caution with prolonged repetitive doses owing to neurotoxicity
As for artemether 4–11 h (11–12 h for dihydro- artemisinin following artemether administration
As for artemether There have been no reports of clinical resistance to artemisinin drugs
Tablets, 250 mg Suppository 100 mg, 200 mg 300 mg, 400 mg, 500 mg
As for artemether
As for artemether 4–11 h (11–12 h for dihydro- artemisinin following artemether administration)
As for artemether There have been no reports of clinical resistance to artemisinin
Tablets, 50 mg, 200 mg Powder for injection 60 mg of anhydrous artesunate in 1 ml Suppository 100 mg Rectocap 200 mg
As for artemether
As for artemether 40 min As for artemether As for artemether As for artemether
Tablets, 20 mg, 60 mg, 80 mg Suppositories, 80 mg
Warfarin 16 h Areas with known resistance Only used in combination
Tablet, 100 mg as hydrochloride
Used in combination with CQ for chemoprophylaxis. Used in newer combinations for treatment (see below)
No adverse drug interactions have been Observed
Patients with liver failure, not recommended for use in pregnancy
Only used in combination Tablets, 50 mg, 100 mg Used in new combinations under development (see below)
60–90 h Only used widely in China Tablets
No adverse drug Interactions have been Observed
Not recommended for use in pregnancy or lactating women (until further studies establish safety), children < 11 kg
In combination under development. Only available as single drug in China
As for artesunate and SP As for artesunate and SP No As for artesunate and SP
See artesunate and SP
Used in multidrug-resistant areas Non-fixed dose combination
As for CQ and SP See CQ and SP As for CQ and SP As for CQ and SP
Combination not used extensively
Used for mixed CQ-resistant and CQ-sensitive P. falciparum
As for CQ and SP above See CQ and SP As for CQ and SP As for CQ and SP Non-fixed dose combination
No specific drug interactions have been studied (may increase blood levels of neuroleptics and tricyclic antidepressants)
Safe use in pregnancy not yet established
Tablets, 20 mg artemether 120 mg lumefantrine
Not evaluated for treatment of severe malaria. Better absorbed in the presence of food. Fixed-dose combination
2 h (artemether) 2–3 days (lumefantrine): (healthy) 4–6 days (patients with malaria)
SP = sulfadoxine-pyrimethamine
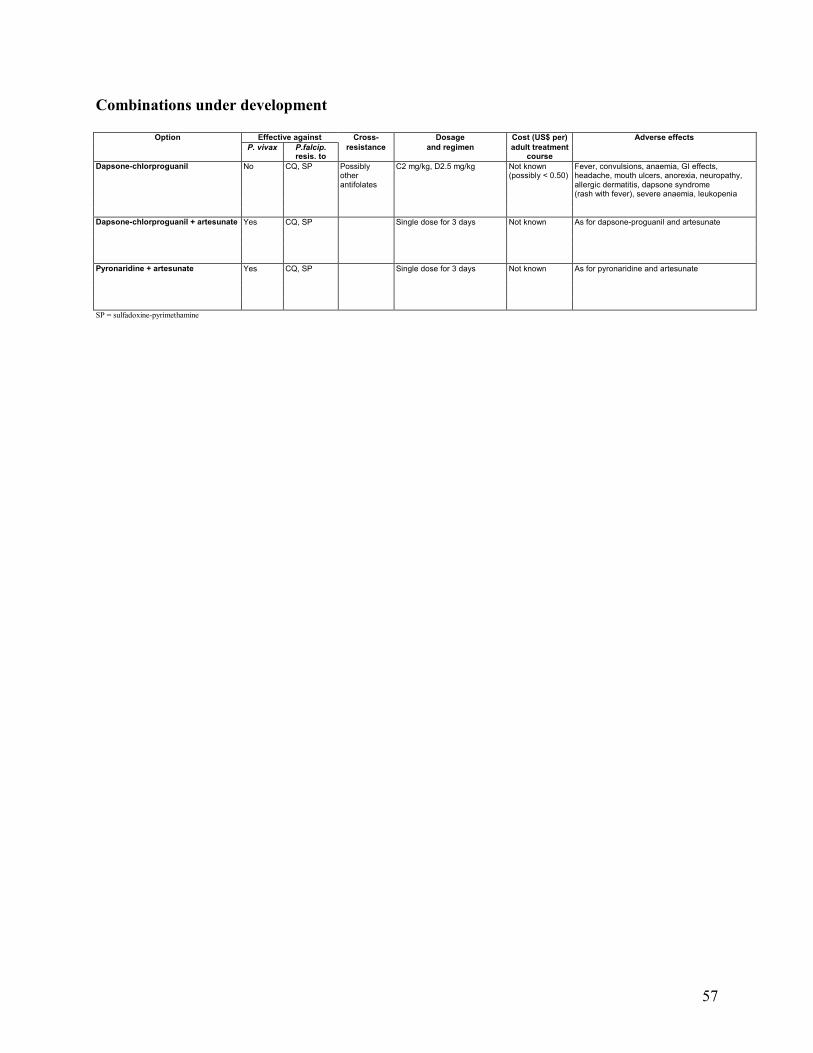
57
Combinations under development
Option Effective against Cross- Dosage Cost (US$ per) Adverse effects
P. vivax P.falcip. resistance and regimen adult treatment resis. to course
Dapsone-chlorproguanil No CQ, SP Possibly C2 mg/kg, D2.5 mg/kg Not known Fever, convulsions, anaemia, GI effects, other (possibly < 0.50) headache, mouth ulcers, anorexia, neuropathy, antifolates allergic dermatitis, dapsone syndrome (rash with fever), severe anaemia, leukopenia
Dapsone-chlorproguanil + artesunate Yes CQ, SP Single dose for 3 days Not known As for dapsone-proguanil and artesunate
Pyronaridine + artesunate Yes CQ, SP Single dose for 3 days Not known As for pyronaridine and artesunate
SP = sulfadoxine-pyrimethamine
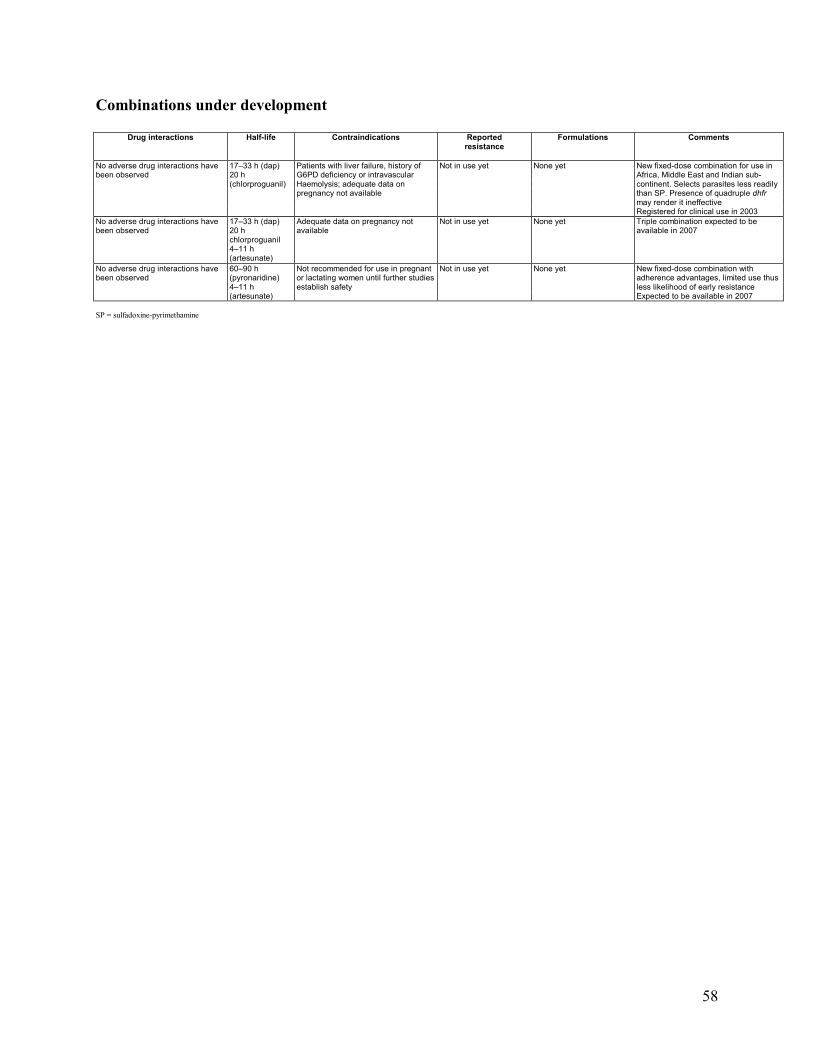
58
Combinations under development
Drug interactions Half-life Contraindications Reported Formulations Comments
resistance
None yet
No adverse drug interactions have been observed
17–33 h (dap) 20 h (chlorproguanil)
Patients with liver failure, history of G6PD deficiency or intravascular Haemolysis; adequate data on pregnancy not available
Not in use yet
New fixed-dose combination for use in Africa, Middle East and Indian sub- continent. Selects parasites less readily than SP. Presence of quadruple dhfr may render it ineffective Registered for clinical use in 2003
No adverse drug interactions have been observed
17–33 h (dap) 20 h chlorproguanil 4–11 h (artesunate)
Adequate data on pregnancy not available
Not in use yet None yet Triple combination expected to be available in 2007
Not in use yet
No adverse drug interactions have been observed
60–90 h (pyronaridine) 4–11 h (artesunate)
Not recommended for use in pregnant or lactating women until further studies establish safety
None yet New fixed-dose combination with adherence advantages, limited use thus less likelihood of early resistance Expected to be available in 2007
SP = sulfadoxine-pyrimethamine