Embed Size (px)
Citation preview

Linköping University Medical Dissertations No. 1249
Cryptogenic Polyneuropathy Clinical, Environmental, And Genetic Studies
Jonas Lindh
Department of Clinical and Experimental Medicine, Faculty of Health Sciences
Linköping University, SE-‐581 85 Linköping, Sweden
Department of Internal Medicine, Ryhov County Hospital, SE-‐55185 Jönköping, Sweden
Linköping 2011

© Jonas Lindh, 2011
Printed by LIU-tryck, Linköping, Sweden, 2011
ISBN: 978-91-7393-118-2
ISSN: 0345-0082

To Ulrika, Andrea & Philip

4
List of Papers The thesis is based on the following papers, which will be referred to in the
text by their roman numerals.
I. Lindh J, Tondel M, Österberg A, Vrethem M. Cryptogenic
polyneuropathy: clinical and neurophysiological findings. J Peripher Nerv
Syst. 2005; 10(1): 31-7.
II. Tondel M, Lindh J, Jonsson P, Vrethem M, Persson B. Occupational
determinants of cryptogenic polyneuropathy. Neuroepidemiology. 2006;
26(4): 187-94.
III. Lindh J, Tondel M, Persson B, Vrethem M. Health-related quality of
life in patients with cryptogenic polyneuropathy compared with the
general population. Disabil Rehabil. 2011; 33(7): 617-23
IV. Lindh J, Söderkvist P, Fredriksson M, Hosseininia S, Tondel M,
Persson B, Vrethem M. Polymorphism of GSTT1, GSTM1 and epoxide
hydrolase in cryptogenic polyneuropathy. Accepted By Brain and
Behavior. The articles are reproduced with the kind permission of the respective publisher.

5
Contents
LIST OF PAPERS 4
ABBREVIATIONS 7
INTRODUCTION 9
Cryptogenic polyneuropathy 11 Definition 11 Background 19 Pathology 21 Neurophysiology 24 Epidemiology 26 Sensory symptoms and findings 28 Motor symptoms and findings 29 Prognosis 30 Treatment 31
Effects of environmental toxins on the peripheral nervous system 31
Metabolism of hexacarbon solvents 33
Genetic Factors 37
Health Related Quality Of Life (HR-‐QoL) 41 SF-‐36 44 EQ-‐5D (EuroQol) 46 Other dysfunction scores 48
Diagnostic approach to peripheral neuropathies 49 Careful history 52 Detailed physical and neurological examination 53 Electrophysiological studies 53 Laboratory studies 54
HYPOTHESIS & AIMS 59
PATIENTS AND METHODS 60
Patients 60
Data collection 64 Medical records 64 Neurophysiology 65 Questionnaires 66 Blood samples 66
RESULTS 67

6
Study I: Clinical and neurophysiological findings 67
Study II: Occupational determinants 69
Study III: Health related quality of life 70 SF-‐36 71 EQ-‐5D 74 EQ barometer 74
Study IV: Genetic polymorphisms 75
Additional results 76 Evaluation of symptoms 76 Quality of life 79 Diabetes risk evaluation 80
DISCUSSIONS AND CONCLUSIONS 81
Clinical picture 81
Health related quality of life 84
Exposures in work and leisure time 87
Genetic polymorphisms and risk of cryptogenic polyneuropathy 91
Conclusions 93
ACKNOWLEDGEMENTS 96
REFERENCES 99
7
Abbreviations 2,5-HD 2,5-Hexandione
BMI Body Mass Index
CI Confidence Interval
CIAP Chronic Idiopathic Axonal Polyneuropathy
CIDP Chronic Inflammatory Demyelinating Polyneuropathy
CMAP Compound Muscle Action Potential
CMT Charcot-Marie-Tooth disease
COR Crude Odds Ratio
CSPN Cryptogenic Sensory and sensorimotor PolyNeuropathy
CV Conduction Velocity
CYP2E1 Cytochrome P450 2E1
DADS Distal Acquired Demyelinating Symmetric
Polyneuropathy
DML Distal Motor Latency
EMG ElectoMyoGraphy
EPHX Epoxide HydroXylase
EQ-5D EuroQol
ESR Erythrocyte Sedimentation Rate
GST Glutathione S-Transferase
GSTM Glutathione S-Transferase Mu (µ)
GSTT Glutathione S-Transferase Theta (ϑ)
HIV Human Immunodeficiency Virus
HR-QoL Health Related Quality of Life
ICD International Classification Of Diseases
IGT Impaired Glucose Tolerance
LOR Logistic Odds Ratio
MAG Myelin Associated Glycoprotein

8
MBK Methyl n-Butyl Ketone
mcs Mental Component Summary
mEPHX microsomal Epoxide HydroXylase
MGUS Monoclonal Gammopathy of Uncertain Significance
OGTT Oral Glucose Tolerance Test
OR Odds Ratio
PAH Polycyclic Aromatic Hydrocarbons
pcs Physical Component Summary
QOL Quality Of Life
QST Quantitative Sensory Testing
SF-36 Short Form 36
SNAP Sensory Nerve Action Potential
TSH Thyroid Stimulating Hormone
VAS Visual Analogue Scale

9
Introduction Cryptogenic polyneuropathy is characterized by a dying-back neuropathy and
patients present with symmetrical, distal loss of sensory and motor function in
the lower extremities that extends proximally in a graded manner. The result is
sensory loss in a stocking-like pattern, distal muscle weakness and atrophy, and
loss of ankle reflexes. Peripheral neuropathies are common neurological problems. They are caused by
disordered function and structure of peripheral motor, sensory, and autonomic
nerves. The overall prevalence in western communities, and also among Parsis
in Bombay, is about 2,400 per 100,000 population (2.4%), but in individuals
older than 55 years, the prevalence rises to about 8,000 per 100,000 (8%) [14,
17]. Other studies show a highly variable prevalence depending on the criteria
for polyneuropathy and the population studied (e.g., general population, primary
care, hospital, university hospital, neuropathy center). The prevalence in poor
populations is not known, but leprous neuritis is still highly prevalent in
Southeast Asia, India, Africa, and Central and South America [41] and it can be
expected that polyneuropathy is at least as common in these areas as in wealthy
populations.
There are many disparate known causes of polyneuropathy, such as diabetes
mellitus, alcohol, heredity, inflammatory disorders, ischemia, paraneoplastic
conditions, deficiency states, infections, toxins, and others. Leprosy (Hansen’s
disease) is a major cause of neuropathy globally, especially in tropical and
subtropical regions [41, 69], but is extremely rare in the Nordic countries. In an
Italian primary care population, the prevalence of polyneuropathy was highest in
patients with diabetes (18.3%), followed by alcoholism (12.5%), non-alcoholic
liver disease (10.9%) and tumor (7.1%) [13]. The most common clinical

10
condition in the patients with cryptogenic polyneuropathy in the same study was
hypertension.
Different terms for the condition are used in the literature. Chronic Idiopathic
Axonal Polyneuropathy (CIAP) [45, 75, 82, 121, 138, 146, 170, 180, 183, 189]
is used by several authors. Other names of the same or closely related conditions
are: chronic polyneuropathy of undetermined cause [107], and Cryptogenic
Sensory Polyneuropathy (which also includes sensory-motor neuropathies) [67,
98, 190], painful sensory neuropathy [133], sensory-predominant painful
idiopathic neuropathy [91], burning feet syndrome [36, 161], and distal small
fiber neuropathy [62, 77, 160]. The two latter conditions are, however, most
likely a separate condition. In some cases the same author uses different terms
for the same condition in separate articles [189-191]
It remains unclear how thorough the investigation of the patient’s
polyneuropathy should be before calling it cryptogenic. Cryptogenic
polyneuropathy should probably not be considered as a distinct disease, but
rather a clinical syndrome with different mechanisms leading up to the same
clinical picture. Nevertheless, the syndrome is clinically useful as the patients
share the same clinical findings and prognosis [67].
11
Cryptogenic polyneuropathy
Definition
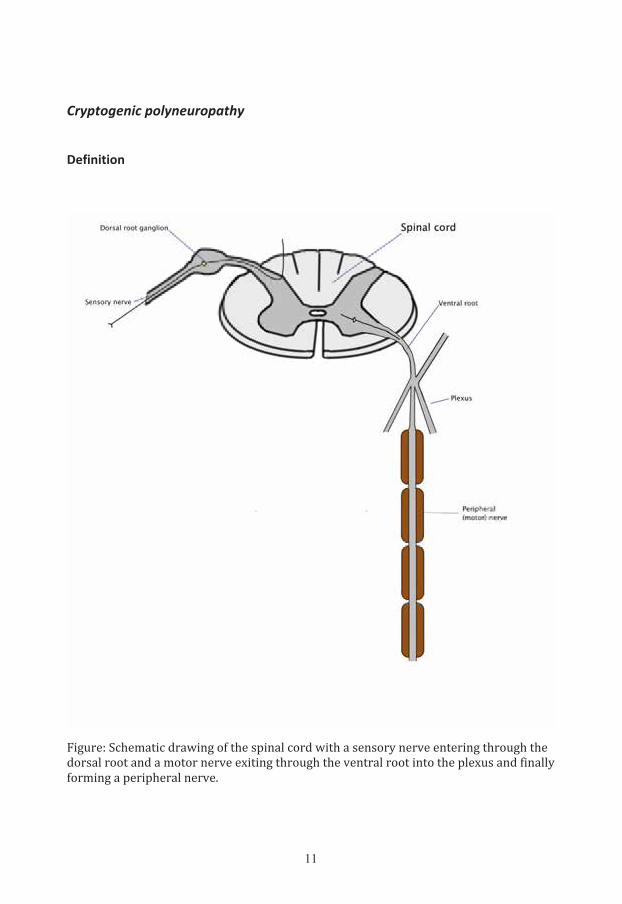
Figure: Schematic drawing of the spinal cord with a sensory nerve entering through the dorsal root and a motor nerve exiting through the ventral root into the plexus and finally forming a peripheral nerve.

12
By common definition, the peripheral nervous system includes the cranial
nerves, the spinal nerves with their roots and rami, the peripheral nerves, and the
peripheral components of the autonomic nervous system. The dorsal and the
ventral roots are attached to the spinal cord by a series of filaments. After
passing the subarachnoid space, each root enters a dural sac and the dural
sheath. Immediately peripheral to the spinal ganglion of the dorsal root,
corresponding dorsal and ventral roots join to form a spinal nerve. The dural
sheaths become confluent at the ganglion, and then merge with the epineurium
of the spinal nerve. Each spinal nerve quickly divides into a dorsal and a ventral
ramus. The dorsal rami supply the back; the ventral rami supply the limbs and
ventrolateral part of the body wall. In the cervical and lumbosacral regions, the
ventral rami intermingle and form plexuses from which the major peripheral
nerves emerge. As a general principle, each ramus entering a plexus contributes
to several peripheral nerves, and each peripheral nerve contains fibers derived
from several rami [38].

13
Figure: Schematic drawing of a nerve cell with its myelinated axon connecting to a muscle fiber.
Peripheral nerve fibers are divided into two different groups, myelinated and
unmyelinated nerve fibers. A nerve fiber is defined as an axon with its
associated Schwann cell, which creates the myelin sheath. The nerve fibers are
then arranged together in bundles or fascicles and then grouped into nerves. The
nerve sheaths contain the intrinsic blood and lymphatic vessels as well as the
nervi nervorum, which supply the connective tissue and vessels with sensory
and autonomic fibers [38].
The terms "polyneuropathy," "peripheral neuropathy," and "neuropathy" are
frequently used interchangeably, but are distinct. Polyneuropathy is a specific

14
term that refers to a generalized, relatively homogeneous process affecting many
peripheral nerves, with the distal nerves usually affected most prominently.
Peripheral neuropathy is a less precise term that is frequently used
synonymously with polyneuropathy, but can also refer to any disorder of the
peripheral nervous system including radiculopathies and mononeuropathies.
Neuropathy, which again is frequently used synonymously with peripheral
neuropathy and/or polyneuropathy, can refer even more generally to disorders of
the central and peripheral nervous system. Symmetric distal sensory loss,
burning, or weakness typically characterizes polyneuropathy. The
polyneuropathies must be distinguished from other diseases of the peripheral
nervous system, including the mononeuropathies and mononeuropathy
multiplex (multifocal neuropathy), and from some disorders of the central
nervous system [38].
Cryptogenic polyneuropathy is in essence a diagnosis of exclusion, established
after a careful medical, family, and social history, neurologic examination, and
directed laboratory testing. Patients must have a slowly progressive distal
symmetric sensory or sensorimotor polyneuropathy on neurological clinical
examination, and axonal degeneration on neurophysiological examination.
Different authors have dealt with this in different ways. Richard Hughes defined
CIAP as patients having a late onset symmetrical peripheral neuropathy of
undetermined cause. The patients must have a had previous investigations
including at least a clinical history and examination, urine analysis, blood count,
erythrocyte sedimentation rate (ESR), renal, liver and thyroid profiles, random
glucose, hemoglobin, vitamin B12 and folic acid concentrations, serum protein
electrophoresis, antinuclear factor and chest radiograph [82]. Peter Erdmann
made the diagnosis if the patients had a slowly progressive distal symmetric
sensory or sensorimotor polyneuropathy on neurological clinical examination,
and axonal degeneration on neurophysiological examination. Erdmann and

15
coworkers required normal values for hemoglobin, hematocrit, leukocytes,
platelets, ESR, serum glucose, renal function and electrolytes, liver enzymes,
serum calcium and phosphorous, creatinine kinase, serum protein, transketolase,
vitamins B1, B6 and B12, thyroid function, immunoelectrophoresis, antinuclear
antibodies, cryoglobulins, and rheumatoid factor. All their patients had
undergone a routine chest X-ray [45]. Hoffman-Snyder used another definition
of CIAP. Their inclusion criteria were (1) a documented history of positive
sensory complaints more than 3 months in duration, with or without neuropathic
pain; (2) a detailed neurological examination; (3) a fasting, non-gestational 2h-
Oral Glucose Tolerance Test (OGTT) using a 75-g oral D-glucose (dextrose)
load; (4) nerve conduction studies; and (5) a diagnosis of CIAP. Patients were
excluded if they had documented evidence for a known cause of chronic axonal
polyneuropathy, such as presence of a family history of neuropathy and
“hammer” or “claw” toe deformities. Patients were also excluded in they had
documentation of a toxic or pharmacological exposure or coexisting medical
conditions associated with neuropathy such as chronic alcoholism; metabolic
disturbances; diabetes mellitus; hypothyroidism; and autoimmune conditions
such as connective tissue diseases, including sicca syndrome, malignancies, or
human immunodeficiency virus (HIV) or other active infections (e.g., Lyme
disease, Hansen disease, hepatitis C); general weakness except for distal leg
muscle weakness; abnormal results on complete blood cell count, electrolyte
levels, liver function studies, vitamin B12 levels, Thyroid Stimulating Hormone
(TSH) levels, and serum protein electrophoresis with serum immunofixation.
HIV testing was not routinely performed in this low-risk population, and nerve
conduction studies excluded features of demyelination [75].
Gil Wolfe and Richard Barohn instead used the term Cryptogenic Sensory and
Sensorimotor Polyneuropathy (CSPN). They defined CSPN on the basis of pain,
numbness, and/or tingling in the distal extremities without symptoms of

16
weakness. Sensory symptoms had to occur in a roughly symmetrical pattern in
the distal lower extremities or upper extremities or both and evolve over weeks
to months. On examination, patients had to demonstrate distal sensory deficits
not confined to an individual peripheral nerve. Slight weakness in foot or hand
muscles were permitted [191]. In a second article Wolfe and colleagues added
information about laboratory testing. Routine laboratory tests consisted of a
complete chemistry panel and blood cell count, ESR, antinuclear antibodies,
rheumatoid factor, vitamin B12 level, thyroid function tests, syphilis serologic
screening, and serum protein electrophoresis with immunofixation
electrophoresis. Patients with monoclonal proteins were included in the study
population only if plasma cell dyscrasias were ruled out after evaluation by an
oncologist and a diagnosis of monoclonal gammopathy of uncertain significance
(MGUS) was made. Patients with identifiable causes of neuropathy such as
diabetes, chronic alcohol use, metabolic disturbances, endocrine abnormalities,
connective tissue diseases including sicca complex, malignant neoplasms, HIV
or other infections, pertinent toxic or pharmacological exposures, hereditary
neuropathy or amyloidosis, and primary amyloidosis were excluded by history
and laboratory testing [190]. Their definition was criticized by Peter James
Dyck for being too broad [33].
It is still unclear if idiopathic small fiber neuropathy is a specific entity or a
subgroup of cryptogenic polyneuropathy [30, 67]. In our studies we decided to
exclude patients with an isolated small fiber neuropathy.
A diagnostic problem is the “normal” neurological deterioration, both clinically
and neurophysiologically, in older people. For example, in healthy people older
than 60 years, sural responses may be absent [188]. Loss of proprioception has
been reported to occur in 28% and hyporeflexia, in 12% of healthy men between

17
64 and 73 years of age [153]. One way to handle this in studies is to add an
upper age limit for the diagnosis.
There is, as previously mentioned, no strict definition or even consensus about
which disorders have to be ruled out to diagnose the patient with a cryptogenic
polyneuropathy. It is very important to rule out diabetes, which has been
reported to account for more than one-third of polyneuropathy cases [177].
Nevertheless, many cases of impaired glucose metabolism can be found in a
patient series of cryptogenic polyneuropathy. A high prevalence of impaired
glucose metabolism has been found in those with neuropathic pain [117].
However, the relationship between impaired glucose metabolism and
polyneuropathy has been questioned [35].
Peripheral neuropathy is one of the most common reactions of the nervous
system to toxic chemicals. Many industrial, environmental, and biological
agents, heavy metals, and pharmaceutical agents are known to cause toxic
neuropathies. Medications, most notably anticancer drugs, are the leading
offenders in clinical practice today. All forms of neuropathies may be caused by
toxic agents [69]; however, in most cases, it is difficult to assess the individual
patient exposures and which substances are relevant.
Some studies have dealt with the contribution of “hereditary factors” in patients
with cryptogenic polyneuropathy. Singleton et al. [152] showed that 5.6% of
patients had first-degree relatives with foot sensory loss, weakness, or
deformities. Hughes et al. [82] reported that 12% of patients had relatives with
foot abnormalities. Hereditary neuropathies can present at all ages [177].
Other causes that need to be ruled out are inflammatory disease, chronic
alcoholism, metabolic disturbances, endocrine abnormalities, vitamin B12/folic

18
acid deficiency, critical illness, and acute inflammatory demyelinating
neuropathies, HIV, borreliosis or other infections, monoclonal gammopathy and
malignancies (especially myeloma and small cell carcinoma of the lung).
To summarize, different authors have used different terms for the same
condition and authors using the same term are still using different definitions.
There is no consensus document about how to define cryptogenic
polyneuropathy or CIAP known to the author. If a document was available, it
would have to be updated regularly as new information about the mechanisms of
the genesis of polyneuropathy become available. Both in clinical practice and in
research, however, a choice has to be made about how many investigations to
perform, before calling the disorder idiopathic or cryptogenic.
We chose to use a broad definition based on clinical grounds. We defined
polyneuropathy as one or more typical symptoms (numbness, pain, postural
instability, paresthesias, distal weakness, burning sensation, muscle cramps,
icing sensation, hypersensitivity to touch) with a distal distribution and at least
two of three clinical findings (distal deficit of sensation, reduced distal muscle
strength, and impaired or lost deep tendon reflexes). We regarded the diagnosis
as probable if patients had one or more of the symptoms above and only one of
three clinical findings as well as a chronic slowly progressive clinical course.
Patients whose diagnosis was based on neurophysiological findings alone were
not included.
A more detailed discussion about the diagnostic workup is presented in the
chapter “Diagnostic approach to peripheral neuropathies”. In our studies the
following laboratory investigations had to have been performed with normal
results: hemoglobin, fasting blood glucose concentration, vitamin B12
(cobalamin), folic acid, and thyroid function.

19
Background
The cause of cryptogenic polyneuropathy is, as the term implies, not known.
One suggestion has been that it is an inflammatory disorder. Clonal expansions
of T cells were strikingly high in patients with CIAP, as compared with elderly
normal controls, elderly controls with degenerative neurologic diseases, and
elderly patients with idiopathic chronic inflammatory demyelinating
polyneuropathy [54]. The relevance of T-cell clone expansion in relation to the
pathogenesis of idiopathic sensory neuropathies is still not clear, but some cases
can improve with treatment with steroids. In a recent study a significant increase
in C reactive protein (CRP) was found in patients with CIAP compared to
controls [142].
Another hypothesis is that the metabolic syndrome gives rise to chronic
ischemia, which causes the polyneuropathy. The metabolic syndrome is a
constellation of entities including impaired glucose tolerance, truncal obesity,
hypertension, and dyslipidemia [142]. Different mechanisms for polyneuropathy
secondary to the metabolic syndrome have been proposed. Patients with chronic
idiopathic axonal neuropathy more often have manifest cardiovascular disease
and cardiovascular risk factors than controls [80, 169]. In developed countries,
type 2 diabetes is the most common defined cause of axonal neuropathy in
middle and old age. Neuropathy is a common complication of chronic
hyperglycemia: the overall prevalence in patients with diabetes is 45–60%, and
in more than half of patients followed longitudinally, clinical symptoms of
neuropathy developed within 25 years of diagnosis [152]. As a consequence,
screening for diabetes is appropriate in evaluating patients with idiopathic
neuropathy. It is still controversial whether impaired glucose tolerance or

20
impaired fasting glucose gives rise to polyneuropathy. Some reports have found
an increased frequency of impaired glucose tolerance [75, 122, 152, 165],
though others have found contradicting results [82, 117]. Other studies have
found both impaired glucose tolerance and hypertriglyceridemia in increased
frequencies [81, 138, 139, 154]. Hughes and co-workers found using a logistic
regression analysis that environmental toxin exposure and hypertriglyceridemia,
but not glucose intolerance or alcohol overuse, were significant risk factors that
deserve further investigation as possible causes of cryptogenic polyneuropathy.
These findings, on the other hand, did not support that mild/moderate
hypertriglyceridemia was an independent risk factor for development of
neuropathy.
Another hypothesis is that chronic pain, stress and depression are common in
cryptogenic polyneuropathy patients and may predispose these patients to the
metabolic syndrome and impaired glucose tolerance [142]. If impaired glucose
tolerance is identified, the progression to diabetes is slow; frank diabetes
develops in only 20-35% of patients with impaired glucose tolerance during 5-
years of follow up [49]. However, the metabolic syndrome is an important factor
to consider as it is possible to treat and successful treatment prevents other
complications for the patient.
It is often believed that toxic substances in the environment cause many cases of
cryptogenic polyneuropathy. Hughes found that this was one of the main causes
in CIAP [82].
21
Pathology
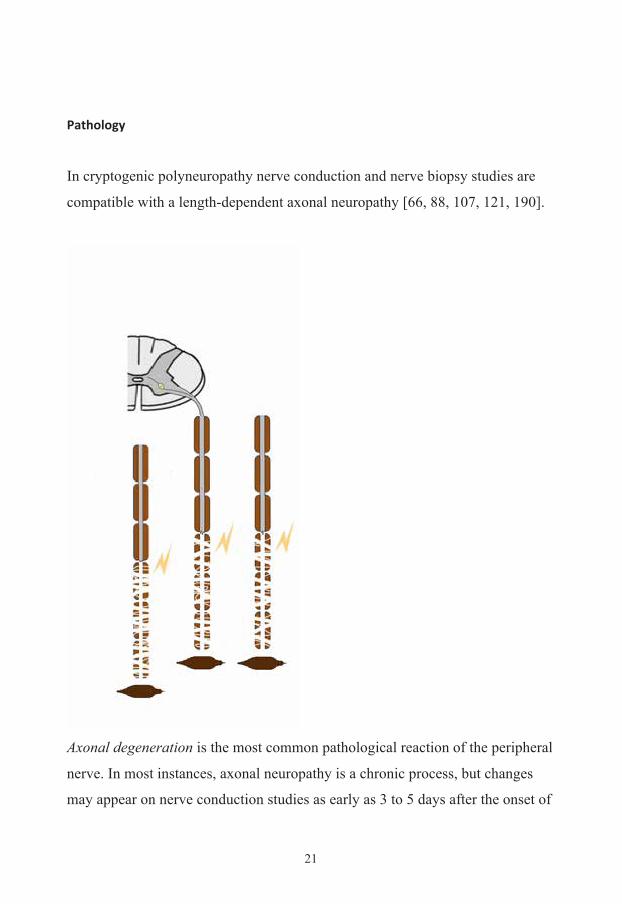
In cryptogenic polyneuropathy nerve conduction and nerve biopsy studies are
compatible with a length-dependent axonal neuropathy [66, 88, 107, 121, 190].
Axonal degeneration is the most common pathological reaction of the peripheral
nerve. In most instances, axonal neuropathy is a chronic process, but changes
may appear on nerve conduction studies as early as 3 to 5 days after the onset of

22
acute axonopathy caused by the rapid pace of Wallerian degeneration [61].
Systemic metabolic disorders, toxin exposure, and some inherited neuropathies
are the usual causes of axonal degeneration [69]. The myelin sheath breaks
down concomitantly with the axon in a process that starts at the most distal part
of the nerve fiber and progresses toward the nerve cell body, hence the term
dying-back or length-dependent neuropathy [69]. The selective length-
dependent vulnerability of distal axons could result from the failure of the
perikaryon to synthesize enzymes or structural proteins, from alterations in
axonal transport, or from regional disturbances of energy metabolism [69].
After nerve transection, axons and their myelin sheaths regenerate. This process
begins in the distal end of the proximal stump. Axons then form growth cones
and begin regenerating, and at the same time Schwann cells divide rapidly [38].

23
The pathological features in sural nerve biopsy specimens consist of axonal de-
and regenerative changes without evidence of inflammation. At the
ultrastructural level an increased thickness of endoneurial vessel basal lamina
can be observed indicating that ischemia plays a role in the development of the
polyneuropathy, but this is a non-specific finding [19, 171]. Sural nerve biopsy
shows an unspecific, axonal type neuropathy, mostly with secondary
demyelination [66]. In skin biopsies a reduction in intraepidermal nerve fibers is
observed [117].
24
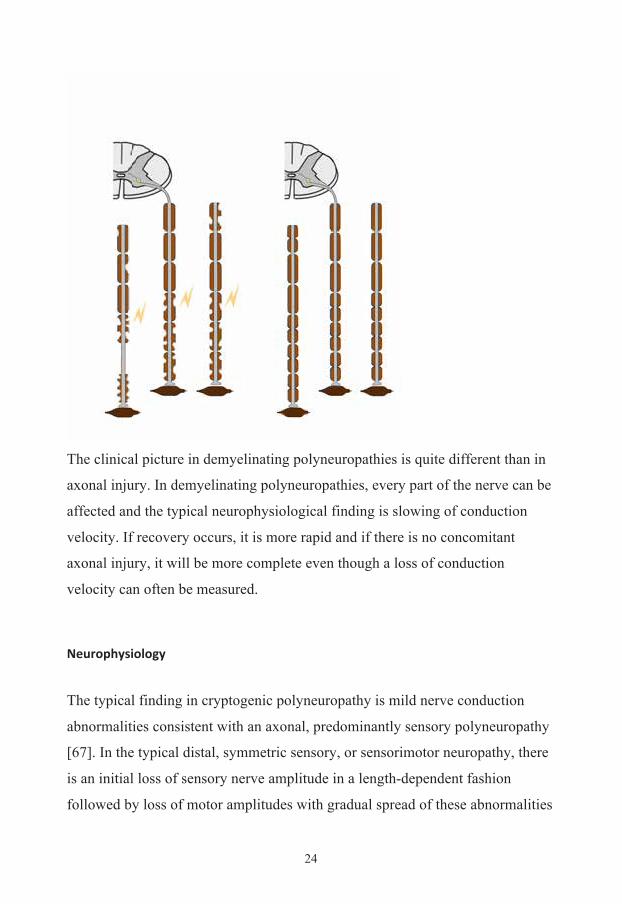
The clinical picture in demyelinating polyneuropathies is quite different than in
axonal injury. In demyelinating polyneuropathies, every part of the nerve can be
affected and the typical neurophysiological finding is slowing of conduction
velocity. If recovery occurs, it is more rapid and if there is no concomitant
axonal injury, it will be more complete even though a loss of conduction
velocity can often be measured.
Neurophysiology
The typical finding in cryptogenic polyneuropathy is mild nerve conduction
abnormalities consistent with an axonal, predominantly sensory polyneuropathy
[67]. In the typical distal, symmetric sensory, or sensorimotor neuropathy, there
is an initial loss of sensory nerve amplitude in a length-dependent fashion
followed by loss of motor amplitudes with gradual spread of these abnormalities

25
to the shorter nerve segments in the upper extremities [61]. This is largely
because the more distal nerve segments are farther from their cell bodies. In
some axonopathies, alterations in axon caliber, either axonal atrophy or axonal
swelling, may precede distal axonal degeneration [69]. Later in the course of
severe axonal disorders, conduction velocities may become abnormal because of
secondary demyelination or loss of the fastest conducting fibers [61]. Because
axonal regeneration proceeds at a maximal rate of 2 to 3 mm per day, recovery
may be delayed and is often incomplete [69].
Axonopathies result in low-amplitude sensory nerve action potentials (SNAPs)
and compound muscle action potentials (CMAPs), but they affect conduction
velocities only slightly [69]. In cryptogenic polyneuropathy the findings are
usually mild [107]. A problem with nerve conduction studies is, however, that
nerve conduction velocity and amplitudes decrease with increasing age and are
dependent on height and sex. The effect of height is greater than that of age
[143]. Absent sural SNAPs combined with spontaneous muscle fiber activity in
the anterior tibialis muscle support the diagnosis of neuropathy, since such
abnormalities are rarely found in healthy, older individuals [180]. There is also
evidence that finger circumference alters the amplitude of sensory recordings as
does the patient’s Body Mass Index (BMI). The sensory and mixed nerve
amplitudes are significantly lower in obese persons, but there are no differences
in nerve conduction velocities [23]. An index based on 12 electrophysiological
parameters has been suggested, which enables detection of slight impairments of
nerve conduction. The relatively low variability between recordings of the index
makes it suitable to follow the progression of a polyneuropathy with repeated
measurements over time [157].
In patients with prominent symptoms, demyelinating features on nerve
conduction studies are often found, giving rise to a combined axonal and

26
demyelinating polyneuropathy [107]. Sensory involvement is, in most cases,
more profound than motor involvement [191]. Sensory and motor nerve
conduction abnormalities typically consist of reduced amplitudes with normal or
minimal distal latency and conduction velocity changes [191]. In many of the
patients who presented with burning, painful paresthesias from resumed
idiopathic distal small-fiber neuropathy, the nerve conduction studies are normal
[62].
In a recent Dutch study of CIAP patients with pain, quantitative sensory
threshold and autonomic tests showed more frequent abnormal test results
compared to the healthy control group. The cold threshold and heat pain test in
patients with CIAP were both affected. The RR interval variation of deep
breathing tests and spectral analysis of RR intervals showed a significant
decrease in the high-frequency power [30]. It remains unclear if all patients with
cryptogenic polyneuropathy have small-fiber neuropathy or only those with
pain.
An electromyography (EMG) of distal muscles shows acute, chronic, or both
kinds of changes. Denervation activity such as fibrillation potentials or sharp
waves are found in approximately two-thirds of patients [191]. Patients with
cryptogenic polyneuropathy who have only sensory signs commonly have motor
involvement on electrophysiological studies.
Epidemiology The age of onset is predominantly in late middle age, with a median age of
symptom onset between 50 and 60 years and a range of 12 years and up [62, 66,
67, 91, 107, 119, 190]. It can, however, be argued that in early presenting
polyneuropathy a cause is likely to be found and that hereditary or metabolic

27
reasons are the most plausible. Men are overrepresented by as much as a 3:1 to
4:1 ratio [107, 119, 136].
Symptoms have usually been present for many years before the patient presents
to the neurologist [67]. Most reports are from western countries, but some
reports are available from developing countries indicating that it is a common
condition there as well [17, 85].
Community studies have reported that symptomatic peripheral neuropathy may
be seen in approximately 3% of the elderly [13, 14], but other studies have
found a prevalence up to 8%, which is likely due to patient recruitment methods
and wider age limits. The prevalence of idiopathic polyneuropathy in the Parsi
community of Bombay (Mumbai) in India was 0.21%. They classified 20% of
noncompressive neuropathies as idiopathic [17]. Diabetes is said to be the most
common cause of peripheral neuropathy in the elderly [52, 80], and alcoholic
and nutritional neuropathies are also common [52, 80]. In a study from London,
all incident cases of neurological disorders were ascertained prospectively in an
unselected urban population based in 13 general practices over an 18-month
period. The age- and sex-adjusted incidence rates were calculated and they
found 54 cases of diabetic polyneuropathy and 15 cases of peripheral
neuropathies per 100,000 persons per annum [103].
In early studies the cryptogenic group was thought to comprise as much as 50-
70% of polyneuropathy [47, 52, 80, 95, 106, 111, 145]. However, later studies
have revised the frequency downward to 10-25% of patients despite a thorough
medical investigation [13, 17, 107, 115, 119, 146]. In these cases the
polyneuropathy is called cryptogenic. The reason for the lower percentage in
more recent series is probably because of improved understanding of the causes
of neuropathy and diagnostic advances. However, cryptogenic polyneuropathy

28
still remains a common clinical problem both for general practitioners and
neurologists at secondary and tertiary centers.
Sensory symptoms and findings
Most patients with cryptogenic polyneuropathy have a predominantly sensory
disease [67]. It is most commonly characterized by symmetrical, distal motor
and sensory deficits that have a graded increase in severity distally and by distal
attenuation of reflexes. The sensory deficits generally follow a length-dependent
stocking-glove pattern. By the time sensory disturbances of the longest nerves in
the body (lower limbs) have reached the level of the knees, paresthesias are
noted in the distribution of the second longest nerves (i.e., those in the upper
limbs) at the fingertips. When the sensory impairment reaches the mid-thigh,
involvement of the third longest nerves, anterior intercostal and lumbar
segmental nerves, gives rise to a tent-shaped area of hypoesthesia on the anterior
chest and abdomen. The involvement of recurrent laryngeal nerves may occur at
this stage with hoarseness [69]. The clinical picture is usually a mixed motor
and sensory neuropathy; however, in some cases it is a prominently sensory
neuropathy and in fewer cases, a predominantly motor neuropathy [107].
Discomfort or pain is a very common presenting symptom, reported in 65 to
80% of patients [62, 66, 119, 190]. The description of pain varies from patient to
patient, but the most common description is a nagging pain [45]. Other common
symptoms are numbness or tingling with or without pain, heavy feeling, or
weakness in the distal limbs [119, 191]. These patients complain of tingling,
prickling, numbness, or burning of the feet, and, often, stiffness of the toes.
Worsening of sensory symptoms with heat or cold exposure, activity, or fatigue
is commonly reported by patients [62].

29
On physical examination there is loss of pinprick sensation, together with loss of
vibratory sensation in the feet, absent ankle reflexes, and mild toe-extensor
weakness. Ability to sense vibration is the primary modality most likely to be
abnormal in the feet. Vibration sensation was reduced in all patients in
Notermans’ study, typically below the knees [119]. Pinprick and light touch is
also reduced in most of the patients and position sense is least affected on
sensory examination [119, 191]. These abnormal signs must be distinguished
from normal manifestations of the aging in the peripheral nervous system. Loss
of vibratory sense that is restricted to the toes can be a normal finding in healthy
elderly controls (e.g., present in 28% of individuals age 65 years and older).
Absent ankle reflexes are found in 38% of healthy controls older than 65 years.
In an Australian population study of persons 75 years or older, 26% and 28%
had impaired vibration sense and 20% and 21% absent ankle jerks on the right
and left side, respectively. The study also had a subgroup free of neurological
disease and in that group only, impaired vibration sense in the thumbs and gait
instability significantly worsened with increasing age [184]. Gait instability and
other symptoms of large fiber dysfunction are less commonly affected than
those of small fibers [67]. Joint position sense is also only affected in a minority
of patients [67].
Motor symptoms and findings Approximately two-thirds of patients with sensorimotor polyneuropathy can be
expected to have distal weakness and wasting [107, 119]. Motor weakness is
greater in extensor muscles than in corresponding flexors. For example, walking
on heels is affected earlier than toe walking in most polyneuropathies. It is
helpful to determine the relative extent of sensory, motor, and autonomic neuron
involvement, although most polyneuropathies produce mixed sensorimotor

30
deficits and some degree of autonomic dysfunction [69]. The typical finding on
examination is distal muscle wasting and weakness in the lower limbs, in some
cases quite pronounced with bilateral foot drop [107]. Muscle cramps occur, but
they are seldom severe [67].
The average time for symptoms to spread from the lower to the upper
extremities appears to be about 5 years [119]. It is rare for patients to report
symptoms restricted to the upper extremities. Autonomic and cranial nerve
findings are also rare in these patients [62].
Loss of reflexes is a common finding, most often found in the ankle and less
often in the upper extremities [191]. Total areflexia is rare.
Prognosis
In an early study of cryptogenic polyneuropathy from the Neurological Unit at
the Manchester Royal Infirmary, 31 patients were followed-up from 1940 to
1950, of whom 16 had a negative outcome, and 7 of the patients died of the
disease [106]. Luckily, a great deal has improved since then. Today, both
idiopathic sensorimotor and sensory polyneuropathies pursue a very slowly
progressive course or reach a stable plateau [78, 107, 121]. The first more
modern patient series was published in 1973, but it includes cases that today
would be classified, even as Chronic Inflammatory Demyelinating
Polyneuropathy (CIDP) [78]. In McLeod’s series from the 1980s over 80% of
patients were unchanged or improved at a mean follow up of 3 years, and only
13% experienced significant disability from their neuropathy [107]. In more
recent studies, even after a course of more than 10 years, severe disability rarely
occurs. In most cases patients remain ambulatory without severe disability or
handicap [66, 88].

31
Spontaneous remissions have been reported at different rates. Grahmann
reported a complete or significant remission in 4 of 29 cryptogenic cases on
reevaluation and an even higher rate among cases that were initially classified as
cryptogenic but were later solved [66]. One possible explanation might be that
these were toxic neuropathies, which usually recover when the exposure ends,
but it cannot be excluded that even non-toxic cases might improve
spontaneously.
Treatment
There is no specific therapy for cryptogenic polyneuropathy. The management
of these common neuropathies instead centers on the treatment of neuropathic
pain, provision of braces in some cases, and patient education and reassurance
about the favorable long-term outcome [191]. Options for the treatment of pain
based on clinical experience and studies in other neuropathies include tricyclic
antidepressants, gabapentin, carbamazepine, nonsteroidal anti-inflammatory
drugs, opioids, capsaicin [67] and lidocaine medicated plaster [11]. Treatment
response is often unsatisfactory.
Effects of environmental toxins on the peripheral nervous system Exposure to neurotoxins may lead to dysfunction of any part of the central,
peripheral, or autonomic nervous system and the neuromuscular apparatus.
Neurotoxic disorders, especially those with an iatrogenic basis, are well
described. Hence, in any neurological condition, such as a peripheral
neuropathy, neurotoxins should be considered as possible cause. Peripheral
neuropathy is, in fact, one of the most common reactions of the nervous system
to toxic chemicals. Industrial, environmental, and biological agents; heavy
metals, and pharmaceutical agents are known to cause toxic neuropathies.

32
Medications, most notably anticancer drugs, are the leading offenders in clinical
practice today. Examples include cisplatin, taxanes, vincristine, metronidazole,
hydralazine, nitrous oxide, thalidomide, and phenytoin [69].
Neurotoxic disorders are occurring increasingly as a result of occupational or
environmental exposure to chemical agents and often go unrecognized.
Neurotoxic disorders are recognized readily if a close temporal relationship
exists between the clinical onset and prior exposure to a chemical agent,
especially one known to be neurotoxic. However, it is much more difficult to
identify neurotoxic agents when a group of persons are exposed to several
agents over a long period of time and the effects do not appear until several
years later [7]. This is common in industrial settings as well as in the home
environment. Suicide attempts with chemical agents and recreational drugs are
other possible causes of peripheral nerve damage.
For many agents only single case reports are available, but they may be
unreliable, especially when the neurological symptoms are frequent in the
general population. Epidemiological studies may be helpful in establishing a
neurotoxic basis for symptoms. However, it is difficult to perform good studies.
One major problem is finding adequate controls. A good study requires
matching of exposed subjects and unexposed controls, not only for age, gender,
and race, but also for social, and cultural background; and alcohol, recreational
drugs, and medication use. Laboratory test results are often not helpful in
confirming that the neurological syndrome is caused by a specific agent, either
because the putative neurotoxin cannot be measured in body tissues or because
the interval since exposure makes such measurements meaningless [7].
Most toxins produce symmetrical axonal degeneration in a dying-back (length-
dependent) pattern, eventually spreading proximally with continued exposure. A

33
number of toxic axonopathies also affect the central nervous system, showing
evidence of concurrent degeneration of dorsal column projections of sensory
neurons and optic nerve axons. Central axon involvement has been linked to
incomplete clinical recovery. Agents such as n-hexane cause simultaneous
degeneration of peripheral nerves, dorsal column axons, and corticospinal
pathways, often resulting in spasticity that may become apparent following
recovery from the peripheral axonopathy. Electrophysiological investigations
typically disclose an axonal pattern [69].
Metabolism of hexacarbon solvents Solvents are examples of foreign and potentially toxic, generally lipophilic,
substances absorbed into the body. They are therefore difficult to excrete as they
will be reabsorbed in the kidney or from the gastrointestinal tract after biliary
excretion and may remain in the body for long periods. The process of
detoxifying and excreting a substance is called biotransformation, which is a
complex process. The primary results of biotransformation are that the parent
molecule is transformed into a more polar metabolite, molecular weight and size
are often increased, excretion is facilitated, and elimination of the compound
from the body is increased. However, biotransformation may also underlie the
toxicity of a compound, of which n-hexane and methyl n-butyl ketone are both
examples [173].
n-Hexane, present in many commonly used organic solvents, is known to cause
primary axonal degeneration with secondary demyelination [24]. It is derived
from cracking of petroleum and from natural gas liquids. It is typically present
in motor fuels at 1 – 9 volume % and is currently employed in a variety of
industrial and commercial processes, including rubber, adhesive, ink, and paint
manufacturing, and in the extraction of vegetable oils for human consumption

34
[158]. Inhalation is the primary route of entry for n-hexane as well as methyl n-
butyl ketone (MBK). These solvents are also absorbed through the skin. Dermal
absorption is affected by the duration of exposure, and the size and condition of
any exposed area of skin [48]. Both n-hexane and MBK are lipophilic and easily
cross the blood-brain-barrier and rapidly reach an equilibrium in the brain [48].
n-Hexane concentration also increases rapidly within peripheral nerve fibers
after exposure. n-Hexane and its metabolites accumulate during chronic
exposures and are released from the liver when the exposure ends [48]. The
persisting effects of n-hexane and MBK are related to the ability of their mutual
metabolite 2,5-hexanedione (2,5HD) to cross-link axonal neurofilaments
chemically and to interrupt axonal transport mechanisms [48].
The clinical pictures of neuropathy as well as the histopathology of n-hexane
and MBK are identical [48]. Results of a nerve biopsy in a severe case of n-
hexane polyneuropathy showed giant axonal swelling due to accumulation of
neurofilaments, myelin sheath attenuation, and widening of nodal gaps [24]. n-
hexane induces hepatic cytochrome P-450 (CYP2E1, CYP2B6 and alcohol
dehydrogenases) [84] and is metabolized to the neurotoxic agents MBK and 2,5-
HD [158]. It is possible that individual differences in this specific metabolism
may account for the difference in susceptibility. Nerve conduction studies reveal
diminished amplitude of sensory nerve action potentials and slowed sensory and
motor nerve conduction velocities in the distal extremities of n-hexane-exposed
individuals [48]. Partial conduction block may also occur [128]. Acute
inhalation exposure may produce feelings of euphoria associated with
hallucinations, headache, unsteadiness, and mild narcosis. Thus, inhalation of
certain glues for recreational purposes causes pleasurable feelings of euphoria in
the short term but may lead to a progressive, predominantly motor neuropathy
and symptoms of dysautonomia after high-dose exposure and a more insidious
sensorimotor polyneuropathy following chronic use [6, 7, 63, 162]. Despite

35
cessation of exposure, progression of the neurological deficit may continue for
several weeks or rarely months before the downhill course is arrested and
recovery begins. Severe involvement is followed by incomplete recovery of the
peripheral neuropathy. When the polyneuropathy does resolve, previously
masked signs of central dysfunction, such as spasticity, may become evident [7].
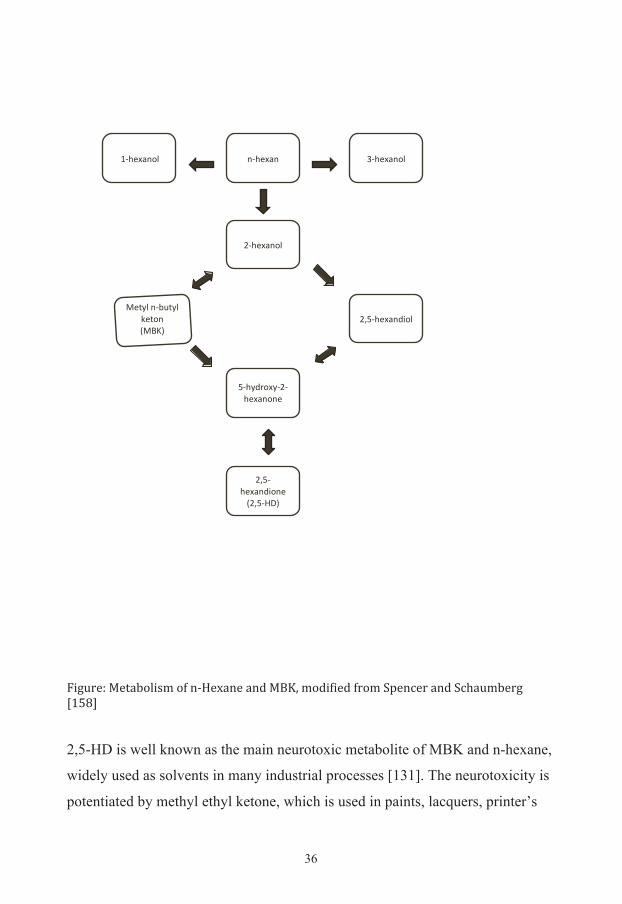
36
Figure: Metabolism of n-‐Hexane and MBK, modified from Spencer and Schaumberg [158] 2,5-HD is well known as the main neurotoxic metabolite of MBK and n-hexane,
widely used as solvents in many industrial processes [131]. The neurotoxicity is
potentiated by methyl ethyl ketone, which is used in paints, lacquers, printer’s
!!
"#$%&'"!(#$%&'")*! +#$%&'")*!
,#$%&'")*!
,-.#$%&'"/0)*!1%23*!"#4523*!
6%2)"!7189:!
.#$3/;)&3#,#$%&'")"%!
,-.#$%&'"/0)"%!7,-.#<=:!

37
ink, and certain glues [7]. Several studies of the pathophysiology of
neuropathies due to 2,5-HD and identification of the parts of the nervous system
where 2,5-HD mostly exerts its toxic effect have been carried out [97, 159]. 2,5-
HD is mainly a product of intermediate metabolism in the human body and only
a minimal part could derive from n-hexane as a ubiquitous micropollutant [132].
Genetic Factors Biotransformation of exogenous and endogenous compounds may play a role in
individual susceptibility. The metabolism of biotransformation can be divided
into two phases. In phase 1, the original foreign molecule is altered by adding a
functional group, which can then be conjugated in phase 2. The conjugated
molecule can then be excreted. Normally, these steps lead to a less toxic
molecule, but in some cases, the opposite occurs. Glutathione is one of the most
important molecules in the cellular defense against toxic compounds. This
protective function is due in part to its involvement in conjugation reactions
[173].
The glutathione S-transferases (GST) are a family of enzymes responsible for
the metabolism of a broad range of xenobiotics and carcinogens [105]. The
enzymes catalyze the conjugation of glutathione with a wide variety of organic
compounds to form thioethers, a reaction that is sometimes a first step in a
detoxification process leading to mercapturic acid formation, which is a classic
excretion product of xenobiotics [105]. The GST enzymes have been shown to
protect organisms from reactive oxygen compound damage through their
abilities to bind with glutathione [72]. Two distinct superfamilies of GST
isoenzymes exist. The larger superfamily comprises cytosolic, or soluble,
dimeric enzymes that are principally, but not exclusively, involved in
biotransformation of toxic xenobiotics. The other superfamily is composed of
microsomal proteins primarily involved in arachidonic acid metabolism [72].

38
The GST family of soluble (cytosolic) enzymes is grouped into seven classes
based on structure, substrate specificities, and immunologic properties: alpha,
mu, kappa, pi, sigma, theta, and zeta. These classes are abbreviated in Roman
capitals with class members distinguished by Arabic numerals. GST isozymes
within a class share at least 40% homology of amino acid sequence, whereas
between classes there is less than 30% common identity. Within the M class,
five human isozymes were identified (M1 through M5), while two isozymes
were categorized in the GST T class (T1 and T2) [56]. By contrast with other
members of this superfamily, the class kappa GST is mitochondrial and, while
soluble, it is probably not located in cytoplasm [72]. Although different
transferases may exhibit overlapping substrate specificities, no common
substrate exists that is metabolized by all isoenzymes [72]. All GST enzymes are
dimers containing two subunits, with the identity of these subunits determining
the GST present [56]. In recent years, a wide array of GST functions has
received increasing attention, including the GST role in (1) conjugating
endogenous electrophiles, (2) maintaining intracellular redox status, and (3)
synthesizing and modifying leukotrienes and prostaglandins. Their ability to
conjugate electrophiles makes these enzymes critical in the detoxification of a
wide range of epoxides and certain other agents of environmental concern,
including pesticides, therapeutic drugs such as chemotherapeutic agents, or
dietary components [56]. Glutathione S-Transferase Mu-1 (GSTM1) and
Glutathione S-Transferase Theta-1 (GSTT1) are both polymorphic in humans
and deletions in the genes result in virtual absence of enzyme activity,
particularly with deletions in both genes (null genotype) [3]. The genetic
variations can change an individual's susceptibility to carcinogens and toxins as
well as affect the toxicity and efficacy of certain drugs. GSTT1-mediated
conjugation of halogenated solvents including bromobenzene,
bromodichloromethane, methylene chloride, and trichloroethylene may lead to
metabolic activation rather than detoxification [56].

39
The genes encoding the mu class of enzymes are organized in a gene cluster on
chromosome 1p13.3 and are known to be highly polymorphic [193]. The mu
class of enzymes functions in the detoxification of electrophilic compounds,
including carcinogens, therapeutic drugs, environmental toxins and products of
oxidative stress, by conjugation with glutathione. GSTM1 isoenzymes are
expressed predominantly in liver, followed by the testes, brain, and adrenal
glands, with low levels in lung [55]. GSTM1 null occurs as a large deletion,
which leads to complete loss of activity in homozygous variants [55]. It has
been reported that individuals with GSTM1 null genotype and high exposure to
solvents are at increased risk of developing solvent-induced chronic toxic
encephalopathy [166] and Parkinson’s disease [32].
The GSTT1 gene is situated on chromosome 22q11.2 [192]. One variant
involves deletion of the entire gene, GSTT1 null [130], and this variant lacks
enzyme activity. GSTT1 levels are highest in liver and kidney with low levels in
a variety of other organs [55]. The GSTT1 null genotype has, for example, been
associated with an about four-fold increased risk of myelodysplastic syndrome
[25]. In both the GSTM1 and GSTT1 genes, the null genotype has been
associated with an increased risk of optic neuropathies [3], adverse events,
including cognitive impairment after therapy, in patients with medulloblastoma
[10], but not in patients with Leber’s Hereditary Optic Neuropathy [86] nor
neuropathy in patients receiving oxiplatin-based chemotherapy [99].
Epoxide hydrolases play an important role in both the activation and
detoxification of a wide range of exogenous chemicals such as polycyclic
aromatic hydrocarbons (PAHs) [125]. Epoxides are three-membered oxirane
rings containing an oxygen atom and may be metabolized by the enzyme
epoxide hydrolase. This enzyme adds water to the epoxide to yield a dihydrodiol

40
[31]. Epoxides are often intermediates produced by the oxidation of various
substances. The enzyme exists in multiple forms with a broad range of substrate
selectivity against a diverse group of epoxides [31]. It is found mainly in the
endoplasmic reticulum in close proximity to cytochromes P450 [173].
Epoxides are metabolized via complex enzymatic mechanisms involving both
activation and detoxification reactions. Reactive and toxic epoxides are
frequently generated during PAH oxidative metabolism. Epoxides can be
detoxified partly by microsomal epoxide hydroxylase (mEPHX), which
catalyzes their hydrolysis, thereby yielding the corresponding dihydrodiols
[124]. Although this hydrolysis is generally considered to represent a
detoxification reaction because less toxic chemicals are produced, some
dihydrodiols generated from PAHs are substrates for additional metabolic
changes to highly toxic, mutagenic, and carcinogenic polycyclic hydrocarbon
diol epoxides. Thus, epoxide hydrolase plays the same dual role in
detoxification and activation of procarcinogens as found in some cytochromes
P450 and, as a consequence, may also play an important role in neurotoxicity
[68]. Epidemiological studies show that mEPHX activity in the liver, lung and
peripheral blood leucocytes varies as much as 50-fold in white populations
[125].
In humans, the gene is mapped to chromosome 1q42.1 [70], and is composed of
eight introns and nine exons, of which exons 2-9 are coding [31]. Two amino
acid polymorphisms have been identified in the coding region of exon three
(EPHX1 exon 3), the tyrosine 113 histidine (Y113H) exchange, results in a low
activity form of the enzyme [71], which may influence epoxide deactivation in
the cell. A decreased risk for lung cancer among African-Americans with the
low activity form of the enzyme has been found in Los Angeles, whereas the
risk did not differ among Caucasians in the same population [102]. Patients with

41
Leber’s Hereditary Optic Neuropathy who were homozygous for histidine 113
developed the disease earlier than those without this genotype [86]. The
polymorphism in exon four, histidine 139 arginine (H139R) has been suggested
as a high activity isoform of mEPHX [15, 155].
Furthermore, epoxide hydrolase plays an important role in the detoxification of
procarcinogens activated by some cytochrome P450s [15] and as a consequence,
may also play an important role in adverse drug responses. It should be noted
that some epoxides serve as important signaling molecules, regulating a large
variety of physiological functions, ranging from the regulation of vascular tone,
to inflammation, angiogenesis, and pain. Human soluble epoxide hydrolase,
which is expressed throughout the body, is, at present, regarded as the primary
enzyme in the metabolism of such endogenous epoxides [31].
The cytochrome P450 seems to be the most important phase 1 enzyme system
by which most drugs and other oxidants are metabolized [127, 173] and one of
these enzymes, cytochrome P450 2E1, is expressed in different tissues such as
liver, and nerve tissue [101]. To date, three CYP2E1 polymorphisms have been
identified in which the Rsa Ι has greater transcriptional and enzyme activity
compared to the wild-type allele [186], and was associated with higher risk of
developing alcoholic liver disease among Caucasians with alcohol abuse [135].
Health Related Quality Of Life (HR-‐QoL) Traditionally, the physician’s evaluation of the patient’s symptoms and clinical
findings has been a primary focus in the practice of medicine. Over time,
outcomes such as survival, the ability to walk without aid, neurologic deficits,
dysfunctions, neurophysiological findings and anatomical findings like number
of nerve endings have been evaluated to help in medical decision making.

42
However, during the last ten years it has become increasingly important to
evaluate the patient’s own subjective experience and to estimate the cost in
relation to the improvement. This is particularly important in chronic conditions
for which there is no cure and where therapeutic goals are to relieve symptoms
and improve function and quality of life (QOL).
Quality of life is an often used but usually ill-defined term. It has been the
subject of attention across a range of disciplines, including medicine,
psychology, sociology, philosophy, economics, and geography leading to an
array of definitions [22]. One way to define QOL is that there is an upper level
with an overall assessment of well-being, which can include life satisfaction and
general perceptions of well-being. Lower levels incorporate broad dimensions
(i.e., physical, psychological, economic, social) and take account of the
individual components of each domain and allow for variations in their content
[22]. The World Health Organization in 1980 defined quality of life as an
individual’s perception of their position in life in the context of culture and
value systems in which they live and in relation to their goals, expectations,
standards, and concerns [187]. It includes the person’s physical health,
psychological state, personal benefits, social relationships and other factors as
well [1]. The range of dimensions is broad, and deciding which aspects to
measure depends on what you want to study.
QOL is also becoming an important component in the discussion between health
care providers and politicians, who, in part, will determine how limited
resources are allocated. Measuring QOL helps describe the nature and extent of
functional, psychological and psychosocial problems experienced by patients.
Furthermore, and of much relevance in an era of cost containment, a thorough
examination of an intervention’s effect on outcomes such as QOL and over-all
well-being is a key component in the evaluation of both effectiveness and cost-

43
effectiveness [22]. It is also important for the treating physician to have a good
understanding of the patient’s QOL before deciding how to treat the patient,
especially if expensive or potentially life threatening treatments are being
considered.
The state of being healthy is not only the absence of disease, but also a concept
that incorporates notions of well-being or wellness in all areas of life (physical,
mental, emotional, social, and spiritual) [1]. Health-related (HR)-QoL may be
described as the patient’s perception of disease impact on well-being. The three
dimensions of physical (impairment), mental (emotional status), and social well-
being are usually the most important ones. The experience of health is also an
important dimension, which can be divided into subgroups such as mental
health, etc.
There are two types of HR-QoL measures, generic and condition-specific. An
advantage of generic measures is that they can be used in people with a diverse
range of illnesses or health problems, therefore enabling comparisons between
groups. Generic measures are also useful when patients have several concurrent
conditions. However, these measures are often unable to focus on the specific
problems of a given condition [22]. Condition-specific scales are more sensitive
to changes within and differences between individuals with the specific
condition and are, therefore, suitable for treatment studies.
The severity of a patient’s polyneuropathy is the sum of a patient’s symptoms,
neurological signs, test abnormalities, dysfunctions, and other adverse
outcomes. An ideal scale or set of scales should provide a comprehensive and
sensible evaluation of the activities of daily living, walking, running and
climbing stairs, and measure motor, autonomic, or sensory functions as well as
the psychological effects of the disease. The benefit of these scales is that they

44
provide additional characterizing information on the intervention of the disease
in patients’ lives. In the case of treatment studies, the results indicate whether
the treatment makes a real difference in the lives of the patients and from their
own perspective. It is, therefore, an important and independent measure of the
meaningfulness of the intervention and of the consequences of the disease.
Several scales are available that quantitate neuropathic symptoms, impairments
and outcomes [34].
Most studies on HR-QoL in polyneuropathy have been performed on patients
with diabetic neuropathy. Increasing severity of polyneuropathy in diabetes is
related to decreasing HR-QoL with patients without symptoms having an
average 0.81 in EQ-5Dindex to 0.25 in diabetic patients with severe symptoms
[29]. The QOL scores of diabetic patients, who had polyneuropathy with mixed
pathogenesis and sensorimotor type, became worse with time, even if the
patients did not have any clinical symptoms of polyneuropathy [126]. The
presence and severity of neuropathic pain in different types of neuropathy has
been found to be associated with a lower reported HR-QoL in the domains of
physical and emotional functioning, sleep, role functioning and global QOL
[89]. Two independent studies have shown conflicting data whether patients
with neuropathic pain in CIAP have a poorer HR-QoL or not [45, 82].
SF-‐36 A Short Form 36 (SF-36) has been used to evaluate quality of life. The SF-36,
which was developed by Ware and Sherbourne [185], assesses eight health
concepts during the last 4 weeks: physical functioning (PF), role limitations
because of physical problems (RP), social functioning (SF), role limitations due
to emotional problems (RE), mental health (MH), vitality (VT), bodily pain
(BP), and general health (GH) perception. Two norm-based (physical and

45
mental) scores can be calculated as summary scores. The score of the subgroups
as well as the final global score of the SF-36 changes between 0 and 100,
respectively, and higher scores better QOL. Different language versions of the
SF-36 are available, including Swedish [163, 164]. SF-36 is designed for self-
administration, telephone administration, or administration during a personal
interview. Normal values depend on sex, age, and socioeconomic status [18].
SF-36 does not cover all important health concepts, like health distress, family
functioning, sexual functioning, cognitive functioning, and sleep disorders. It
describes a general health status and is not specifically designed for
neuropathies or neurological disorders. SF-36 has, however, previously been
shown to be applicable to inflammatory neuropathies. Merkies showed that the
SF-36 scores of 113 stable patients with Guillain-Barré Syndrome, chronic
inflammatory demyelinating polyneuropathy (CIDP), or paraproteinemic
demyelinating neuropathies were lower than those of 1742 members of the
Dutch population and that neurological disability was related to lower scores in
PF and RE domains [108]. Abresch et al. measured QOL in people with
Charcot-Marie-Tooth (CMT) disease. They found that these patients had
significant bodily pain and that this was a much larger problem than had been
reported in the literature, with physical role and energy/vitality considerably
affected [2]. Ruhland and co-workers compared home exercise intervention with
a non-exercise control group in patients with chronic polyneuropathy, using the
SF-36 to assess impact on QOL. The exercise group improved on the role
limitations scales [148]. The SF-36 was chosen in our study because of its
brevity and its extensive use in clinical studies making it possible to compare
peripheral neuropathy with other medical conditions.
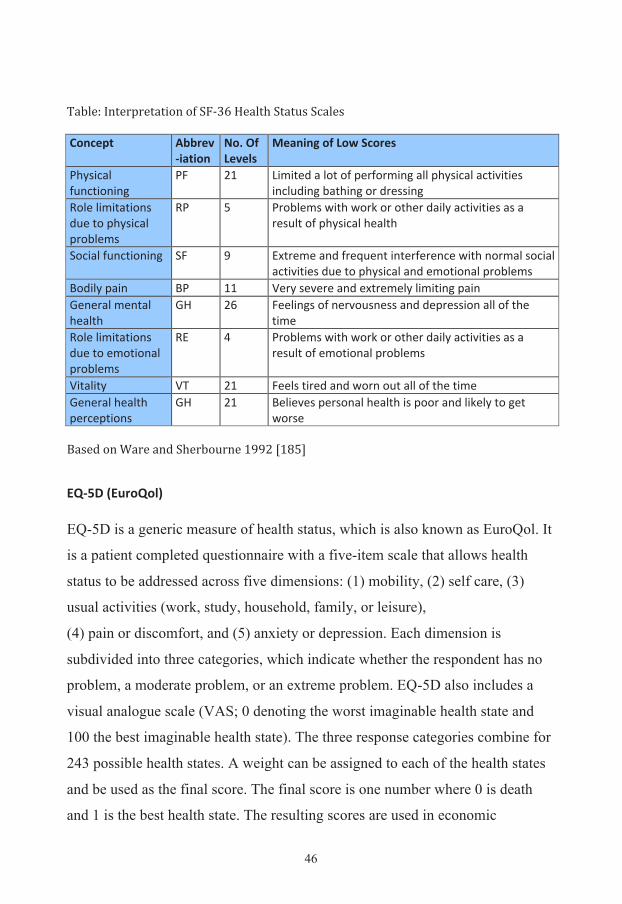
46
Table: Interpretation of SF-‐36 Health Status Scales Concept Abbrev
-‐iation No. Of Levels
Meaning of Low Scores
Physical functioning
PF 21 Limited a lot of performing all physical activities including bathing or dressing
Role limitations due to physical problems
RP 5 Problems with work or other daily activities as a result of physical health
Social functioning SF 9 Extreme and frequent interference with normal social activities due to physical and emotional problems
Bodily pain BP 11 Very severe and extremely limiting pain General mental health
GH 26 Feelings of nervousness and depression all of the time
Role limitations due to emotional problems
RE 4 Problems with work or other daily activities as a result of emotional problems
Vitality VT 21 Feels tired and worn out all of the time General health perceptions
GH 21 Believes personal health is poor and likely to get worse
Based on Ware and Sherbourne 1992 [185]
EQ-‐5D (EuroQol) EQ-5D is a generic measure of health status, which is also known as EuroQol. It
is a patient completed questionnaire with a five-item scale that allows health
status to be addressed across five dimensions: (1) mobility, (2) self care, (3)
usual activities (work, study, household, family, or leisure),
(4) pain or discomfort, and (5) anxiety or depression. Each dimension is
subdivided into three categories, which indicate whether the respondent has no
problem, a moderate problem, or an extreme problem. EQ-5D also includes a
visual analogue scale (VAS; 0 denoting the worst imaginable health state and
100 the best imaginable health state). The three response categories combine for
243 possible health states. A weight can be assigned to each of the health states
and be used as the final score. The final score is one number where 0 is death
and 1 is the best health state. The resulting scores are used in economic

47
appraisals (such as cost utility analyses), in the construction of quality-adjusted
life years for the calculation of cost per quality of life year gained [12]. EQ-5D
has been widely used as a measure of QoL as well as in cost utility analyses.
The validity and reliability are high [20]. The Swedish version of EQ-5D was
used with permission from the EuroQol Group and the responses were analyzed
according to the manual [46].
Although EQ-5D is one of the most frequently used HR-QoL scales in medicine,
its use in polyneuropathy has been limited. It has, for instance, been used as a
secondary measure in an open-label, non-randomized study comparing
venflaxine and gabapentin as monotherapy or adjuvant therapy for neuropathic
pain in peripheral neuropathy. There were no significant improvements in EQ-
5D scores, EQ-5D domains of EQ-5D health status scores for any monotherapy
or adjuvant therapy group, but other scales showed reduction in pain and anxiety
[40]. In another drug therapy study using pregabalin, a modest reduction in pain
was found as well as improvements in anxiety and sleep, but no difference was
found in EQ-5D [112]. The effect of a home exercise program in persons with
chronic peripheral neuropathies mainly consisting of CIDP was tested. The
patients improved in the average muscle score and the RP, RE and SF scales of
the SF-36, but there were no significant changes in EQ-5D [148]. On the other
hand, a large study of pregabalin treatment of neuropathic pain containing more
than 1,000 patients showed statistically significant improvements in all
dimensions of EQ-5D except pain (p=0.059), which was markedly reduced in
other scales [116]. A criticism of EQ-5D is that it is not sensitive to change in
neuropathies as illustrated by these studies, but a strength is that it makes it
possible to make comparisons between different patient groups and that it can be
used to calculate cost-effectiveness.

48
From the patients’ perspective, most consider both SF-36 and EQ-5D as
questionnaires equally suitable. Among those who were more satisfied with a
short questionnaire (EQ-5D), several still preferred a longer and more
comprehensive questionnaire (SF-36). Health outcome assessment seems to be
acceptable, and even appreciated, by patients [118].
Other dysfunction scores Since the planning of our studies, several polyneuropathy scores have been
published [64, 65, 109]. They have the advantage of being more specific for
problems in polyneuropathies, but they are more focused on motor functions and
do not describe the whole clinical picture of the disease. They are also affected
by concomitant disorders (which may be the cause of the neuropathy), poor
volition, medicolegal gain, and psychological changes. There are also tools
measuring neuropathic pain [50]. Ideally, in future trials, one or more generic
QOL scales would be combined with at least one polyneuropathy scale
depending of what kind of neuropathy is being studied and the design of the
study. These scales should be supplemented with objective measures such as
nerve conduction studies, summed scores of neurological signs, and test of
muscle strength.

49
Diagnostic approach to peripheral neuropathies Cryptogenic polyneuropathy is in essence a diagnosis of exclusion. The
following is a guideline to rule out other relevant polyneuropathies and medical
conditions, especially treatable ones, and also to find possible exposures to toxic
agents, orally or in the environment and to identify hereditary cases.
The percentage of cases of neuropathies of undetermined cause has been
steadily declining from a past prevalence of 50–70% [47, 52] to approximately
10% [37, 107, 146] more recently. This decline reflects the recognition of new
disease entities as well as the availability of better diagnostic techniques,
including genetic studies. Intensive evaluation of these neuropathies with
unrecognized causes shows that most of these cases are either inflammatory–
demyelinating or hereditary in origin. When patients are reevaluated a few years
later, it is possible to find a cause in up to more than half of the cases [66, 146].
For the majority of these patients, this could have been known at the time of the
diagnosis of CIAP, because firstly, not all the necessary tests according to the
diagnostic guideline had been carried out, secondly, some test results had been
misinterpreted, and thirdly, in some patients the neurological examination had
been misinterpreted. In other patients there had been insufficient questioning on
the medical history or family history or the diagnosis CIAP was established at
an unusually young age. In the remaining patients it was the clinical course and
the repeated neurological examinations that changed the diagnosis [146]. Two
other studies found that in 29% and 36% of patients, respectively, a cause can be
found at a follow up of 2 to 3 years. These studies identified cases of alcohol
abuse, monoclonal gammopathy, malignancy, and vitamin B12 deficiency [66,
107]. In a more recent study a cause could only be identified in 4 of 75 patients
after a 5-year follow-up (CMT, CIDP and alcohol abuse) [121]. It can be argued
that a treatable cause of the polyneuropathy is not likely to be found and that

50
reevaluation is not cost effective. On the other hand, these studies were
performed in centers with special interest in polyneuropathies and the yield for
reevaluation is probably higher than in an everyday setting. At a minimum,
patients who deteriorate should be reevaluated.
51

52
Careful history As in all medical conditions, the medical history is of extreme importance.
Information regarding onset, duration, and evolution of symptoms provides
important clues. Knowledge of the temporal profile of disease (acute, subacute,
or chronic), distribution of symptoms (symmetric or asymmetric, sensory or
motor or both, distal or proximal) and the course (monophasic, relapsing, or
progressive) points to different specific neuropathies. In the case of cryptogenic
polyneuropathy it should be a progressive chronic, symmetric, sensory or
sensorimotor polyneuropathy.
It is important to rule out toxic exposures at work or during leisure time. Many
pharmaceutical agents have been reported to increase the risk of
polyneuropathy, for example, chemotherapeutical agents (vincristine, taxanes,
and cisplatin), antibiotics (isoniazid, nitrofurantoin, metronidazole), and
phenytoin [81, 177].
In many cases the history reveals that the neuropathy is hereditary without the
need for genetic testing [146]. It is also important to ask specific questions about
current and previous medical history, as the neuropathy can be secondary to
other medical conditions. Alcohol intake must be evaluated in a systematic way.
Other causes that should be ruled out are metabolic disturbances, endocrine
abnormalities, connective tissue diseases, amyloidosis, critical illness, and acute
inflammatory demyelinating neuropathies, HIV, borreliosis, or other infections,
monoclonal gammopathy and malignancies (especially myeloma and small cell
carcinoma of the lung).

53
Detailed physical and neurological examination
During the physical examination, the investigator should look for signs of other
medical conditions and especially signs of a malignancy. The skin should be
examined looking for signs of inflammatory diseases like sarcoidosis, Sjögren’s
syndrome or vasculitis. The extent of neuropathy should be evaluated including
sensory abnormalities (touch, vibration, heat, cold, pain, stereognosis and
proprioception), strength (proximal and distal), reflexes, and ataxia [21]. If
motor symptoms and findings dominate, then the diagnosis of hereditary motor
and sensory neuropathy type 2 should be considered, especially in younger
patients [170]. In patients with axonal neuropathy with MGUS, the arms were
more frequently affected and the disability was worse [120].
Electrophysiological studies Nerve conduction studies should, in principal, be performed on all patients with
clinical symptoms or signs of polyneuropathy [43, 181], with the possible
exception of patients with mild symptoms or very elderly patients. Cryptogenic
polyneuropathy is an exclusively axonal or combined axonal and demyelinating
polyneuropathy in which the axonal involvement is dominating. If the
demyelination is dominating, another cause should be sought. The
electrophysiological studies also characterize the distribution of involvement:
sensory only, motor only or both, and provide an objective picture of the parts of
the body that are involved.
Needle EMG plays a limited role in the evaluation of axonal neuropathy, but
remains important during initial diagnostic evaluation to exclude potential
clinical mimics (e.g., anterior horn cell disease, radiculopathy, myopathy).
Vrancken and colleagues investigated patients and controls older than 65 years
of age. They found that absence of the sural nerve sensory action potentials or

54
presence of spontaneous muscle fiber activity in the anterior tibial muscle was
common in patients, but exceptional in controls [180].
Important factors in patients that might influence the conduction velocities and
amplitudes are patient age and height [143], limb temperature, variants of
peripheral nervous system [61], and body mass index [23].
If nerve conduction studies and EMG are normal, quantitative sensory testing
(QST) for vibration and temperature detection thresholds should be considered.
An advantage of QST is that it assesses small fiber function. QST has been
reported to be a subjective test dependent on patients attention and cooperation
[191], while other authors argue that it has a high sensitivity at least for thermal
testing [67].
Laboratory studies
Several articles have been published recently discussing the laboratory work-up
of peripheral neuropathies [41, 42, 114]. Approaches vary from very limited
testing to the shotgun approach in which a large number of tests are performed
to rule out as many causes as possible. Saperstein has, for instance,
recommended that the initial laboratory testing should only include fasting
blood glucose, vitamin B12, thyroid function tests, and serum protein
electrophoresis including immunofixation electrophoresis [150]. The costs of the
test procedure, sensitivity, and specificity have to be considered. The cause of
most polyneuropathies is evident when the information obtained from the
medical history, neurological examination, and electrophysiological studies are
combined with simple screening laboratory tests. Laboratory test results must be
interpreted in the context of other clinical information since the etiologic yield

55
of laboratory testing alone is limited by the low specificity of many of the tests
[42].
If the history does not reveal the cause of the neuropathy, the following blood
tests are suggested: blood glucose (consider oral glucose tolerance testing),
complete blood count, ESR, renal function, liver function, vitamin B12, folic
acid, serum protein immunofixation electrophoresis, and TSH [41, 42, 114].
Whether or not OGTT should be performed is still under debate [138]. One
major issue is that the reproducibility of IGT (Impaired Glucose Tolerance) has
been found to be only fair to poor and the only study that used multiple OGTTs
was negative [138]. Vitamin B12 deficiency is relatively frequent in patients
with polyneuropathy, and the yield is greater when the metabolites of cobalamin
(methylmalonic acid and homocysteine) are tested. Serum methylmalonic acid
and homocysteine have been reported to be elevated in 5–10% of patients whose
serum B12 levels were in the low normal range of 200–500 pg/dL [42].
Homocysteine may also be elevated in pyridoxine deficiency and heterozygous
homocystinemia. Both homocysteine and methylmalonic acid may be elevated
in hypothyroidism, renal insufficiency, and hypovolemia [42].
IgM monoclonal gammopathies may be associated with autoantibody activity,
type I or II cryoglobulinemia, macroglobulinemia, or chronic lymphocytic
leukemia. IgG or IgA monoclonal gammopathies may be associated with
myeloma, POEMS (Polyneuropathy, organomegaly, endocrinopathy, M-band
and skin changes) syndrome, primary amyloidosis, or chronic inflammatory
conditions [42].
Lumbar puncture is only necessary if an inflammatory neuropathy is suspected
or in cases of asymmetric axonal polyneuropathy in which borreliosis can be
expected [114].

56
In cases of a chronic sensory polyneuropathy or a rapidly progressing motor
polyneuropathy, an asymptomatic cancer has to be suspected. An X-ray of the
chest is the first option and the result is normal, a computer tomography of the
chest and abdomen should be considered [114].
Genetic testing shall not be performed in routine cases. The majority of
genetically determined polyneuropathies are variants of Charcot–Marie–Tooth
(CMT) disease, and genetic testing is available for an increasing number of
these neuropathies [140]. The clinical phenotype of CMT is extremely variable
[42]. Muscle cramping in the legs and feet and the absence of paresthesias favor
a hereditary neuropathy over other etiologies [37]. For patients with a
cryptogenic polyneuropathy who exhibit a classic hereditary neuropathy
phenotype (autosomal dominant, primary demyelinating), routine genetic
screening may be useful for CMT1A duplication/deletion and Cx32 (causing an
X-linked neuropathy, CMTX) mutations in the appropriate phenotype. Further
genetic testing may be considered in selected cases guided by the clinical
question [42].
Tests for antibodies are normally not indicated. Antibodies against Myelin
Associated Glycoprotein (MAG) should be tested for in cases of a
demyelinating polyneuropathy with distal sensory loss as in DADS (distal
acquired demyelinating symmetric polyneuropathy) [113]. High levels of
antibodies against gangliosides (GM1) are positive in 20-80% of patients with
multifocal motor neuropathy and support the diagnosis [60], but are, like other
antibodies against nerve structures, of low value in unselected cases of
polyneuropathy [60, 190]. In patients with sicca syndrome, tests for Sjögren’s
syndrome should be performed. However, polyneuropathy is a rare
manifestation of primary Sjögren’s syndrome, although the most common

57
manifestation is sensory- or sensorimotor-neuropathy. [129]. Anti-nuclear
antigen (ANA), rheumatoid factor (RF) or Anti-neutrophil cytoplasmic
antibodies (ANCA) tests are only indicated when patients exhibit vasculitis or
rheumatic symptoms [114]. Tests for sarcoidosis should only be performed if
the patient is symptomatic. Celiac disease should be evaluated with antibody
testing in cases with diarrhea, malabsorption, or cerebellar ataxia [114].
Infectious diseases should also to be considered. HIV infection has been
reported to have a prevalence up to 16% of cases of symmetrical neuropathy, but
the risk depends on the population studied [81]. In the area where our studies
were performed, the risk of borreliosis is high [73, 92].
When heavy metal toxicity is suspected, blood, urine, hair, and nail samples can
be analyzed [41]. To decide which tests to perform, the following need to be
considered: the area the patient is living in, the patient’s occupation, family
history, and symptoms. Thus, to choose the appropriate test panel, a careful
history and examination must be performed.
Nerve biopsy is only indicated if vasculitis is suspected, i.e., in axonal
polyneuropathy with subacute onset [114]. Sural nerve biopsy is of limited value
as findings in sural nerves are similar in CIDP and CIAP [19, 191] and the
procedure may result in sensory loss and neuropathic pain. Skin biopsy can
confirm the presence of small fiber neuropathy, but should be done by clinicians
with expertise in the technique [41].
Several reports have described patients with a progressive and disabling
idiopathic axonal neuropathy responsive to treatment (prednisone, intravenous
immunoglobulin), and it has been suggested that these patients could be affected
by an immune-mediated neuropathy with the axon as the primary
immunological target. In the normal case of cryptogenic polyneuropathy,

58
treatment is not efficacious and not recommended. A progressive or intermittent
disease course, asymmetric or proximal deficits, neuropathic pain, moderate to
severe disability, and abnormal laboratory findings were prominent in patients
with progressive idiopathic axonal neuropathy and in patients with vasculitic
neuropathy. A wide range of other possible clinical features has been observed
including cranial neuropathy, sensory ataxia, and small fiber neuropathy. Such
diversity of clinical manifestations has also been described in vasculitic
neuropathy [182], but not in CIAP. There is, however, no need for nerve
biopsies in CIAP as Vrancken and co-workers found that there is a small chance
of finding sural nerve vasculitis upon scrutinizing biopsy examination in
progressive idiopathic axonal neuropathy [182]. TABLE: Routine Work Up Blood glucose Complete blood count ESR Serum electrophoresis Renal function Liver function Vitamin B12/folic acid TSH Chest X-‐ray Neurophysiology

59
Hypothesis & Aims
The previously published patient series on the clinical picture of cryptogenic
polyneuropathy are from the early 1990s or earlier. However, since then, the
diagnostic possibilities have improved considerably, especially for hereditary
and inflammatory neuropathies, and new causes of neuropathies have been
found. Despite the progress in determining an etiological cause for
polyneuropathy in many patients, there are still a considerable number of cases
with unknown etiology, thus regarded as cryptogenic. Even data published in the
late 1990s show that there are up to 10% of patients have MGUS in such
material [191]. This means that there is a need for descriptive studies on
cryptogenic polyneuropathy, both concerning symptoms, clinical and
neurophysiological findings, and QOL.
It is well known that many chemical compounds are toxic to peripheral nerves
and thereby can cause polyneuropathy. However, other new substances have
been found to cause neuropathy and we wanted to elucidate this field further.
Our hypothesis is that individuals with particular genotypes may have
difficulties in metabolizing certain environmental toxins and that this poor
metabolizer status make them susceptible to develop polyneuropathy following
exposure to such toxins.
60
Patients and Methods
Patients
!
!
!
!"#$%&''&&()*+,
-./01&&()232&
45"6..%&/(7.#$1$&&()288&
-./01&&()*89&
!"#$%&'&()*+9&
!"#$%&':&();<&
-./01=&8>?;,&%16@A&&()9;&
!"#$%&'''&()+2&

61
We collected data from three departments of neurology (University Hospital of
Linköping, Motala Hospital, and Ryhov County Hospital in Jönköping). The
hospital databases were searched for all outpatients between the ages of 40 and
79 years with a diagnosis of cryptogenic polyneuropathy from January 1, 1993
to December 31, 2000. Patients under the age of 40 years were excluded to
avoid including hereditary forms of polyneuropathy that had not yet been
diagnosed [121]. Older patients were not included in the study to exclude lesions
caused by the normal aging process in the nervous system and to avoid other
confounding diseases or medications. All patients were referred to the
departments of neurology from other physicians; usually a general practitioner,
and the neurologist continued the work up to exclude cases of known origin.
Patients with an ICD-9CM (International Classification of Diseases, 9th
Swedish edition) diagnosis of 356E, 356X, or 357X or an ICD-10 diagnosis of
G60.9, G61.9, or G62.9 (cryptogenic polyneuropathy) were included. The initial
inclusion was based on each neurologist’s judgment of the diagnosis because
there was no strict definition of cryptogenic polyneuropathy. In total, 255
patients fulfilled these criteria.
Population data obtained from the National Central Bureau of Statistics in
Sweden showed that the mean population aged 40–79 years in the study area
during that period was about 296,000. It is not possible to calculate incidence
during this period as most cases were probably diagnosed by general
practitioners and not evaluated by a neurologist.
In the study “Occupational determinants of cryptogenic polyneuropathy” (Study
II), all 232 patients who were still alive were included and were sent a

62
questionnaire. A total of 164 patients replied and were included in this study.
The reason for not excluding any patients was that we wanted the sample size to
be as large as possible so that power could be as high as possible.
In the other studies, we reevaluated the medical records, as we wanted to
ascertain that the polyneuropathy was cryptogenic/idiopathic. The following
laboratory investigations had to have been performed with normal results:
hemoglobin, fasting blood glucose concentration, vitamin B12 (cobalamin),
folic acid, and thyroid function. We defined polyneuropathy as one or more
typical symptoms (numbness, pain, postural instability, paresthesias, distal
weakness, burning sensation, muscle cramps, icing sensation, hypersensitivity to
touch) with a distal distribution and at least two of three clinical findings (distal
deficit of sensation, reduced distal muscle strength, and impaired or lost deep
tendon reflexes). We regarded the diagnosis as probable if there were one or
more of the symptoms above and one of three clinical findings. Patients whose
diagnosis was based on neurophysiological findings alone were excluded. We
also excluded patients when neurography showed a demyelinating neuropathy to
exclude inflammatory and hereditary neuropathies. These criteria were used to
reflect the clinical picture at a secondary center.
A total of 255 patients were retrieved from the databases, and of these, 168
fulfilled the criteria of cryptogenic polyneuropathy. A total of 136 patients
(81%) fulfilled the criteria of polyneuropathy and 32 (19%) had probable
polyneuropathy. The reasons for exclusion of 87 patients are listed in the table
below. Patients were not screened routinely for HIV because the prevalence in
the study area is low.

63
Reason for exclusion n Alcohol + diabetes 1 Alcohol abuse 5 Alcohol + heredity 1 Basedow's Disease 1 Diabetes 9 Dominantly demyelinating 14 Hereditary polyneuropathy 2 Hypothyroidism + Ankylosing Spondylitis 1 Jejuno-ileal by-pass surgery 1 Monoclonal gammopathy 8 Phenytoin exposure 3 Polymyalgia rheumatica 4 PMR + testicular cancer 1 Rheumatoid Arthritis 1 Renal Failure 1 Simvastatin treatment 1 Sjögren's Syndrome 1 SLE 1 Tumors 8 Vitamin B12 deficiency 18 Vitamin B12 deficiency + phenytoin 1 Non-symptomatic polyneuropathy 4
When the genetic study (Study IV) was performed, 158 of the initial 168
patients were still alive, and they were asked to participate. Blood samples were
collected and analyzed from 79 of the patients (response rate 50%). The QOL
study (Study III) was performed during the same time period, but only those
patients aged 50 to 74 years were included. The upper age limit was selected to
the match the reference material and the lower age limit was used, as there were
only two patients, who had not reached 50 years of age.
The studies used different control materials, however all control subjects lived
in the same area as the patients and were in the same age range.
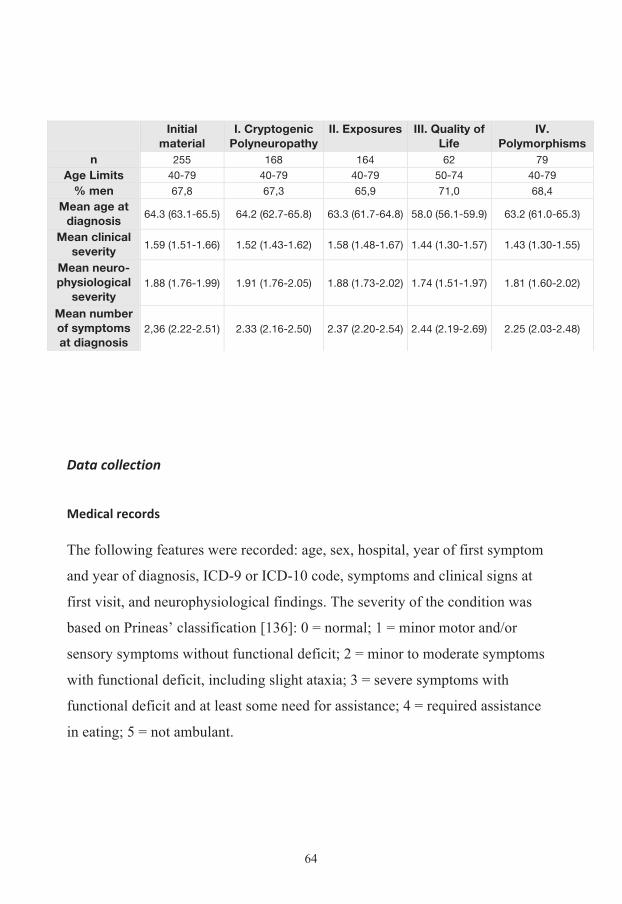
64
Data collection
Medical records The following features were recorded: age, sex, hospital, year of first symptom
and year of diagnosis, ICD-9 or ICD-10 code, symptoms and clinical signs at
first visit, and neurophysiological findings. The severity of the condition was
based on Prineas’ classification [136]: 0 = normal; 1 = minor motor and/or
sensory symptoms without functional deficit; 2 = minor to moderate symptoms
with functional deficit, including slight ataxia; 3 = severe symptoms with
functional deficit and at least some need for assistance; 4 = required assistance
in eating; 5 = not ambulant.
Initial material
I. Cryptogenic Polyneuropathy
II. Exposures III. Quality of Life
IV. Polymorphisms
n 255 168 164 62 79
Age Limits 40-79 40-79 40-79 50-74 40-79
% men 67,8 67,3 65,9 71,0 68,4
Mean age at diagnosis
64.3 (63.1-65.5) 64.2 (62.7-65.8) 63.3 (61.7-64.8) 58.0 (56.1-59.9) 63.2 (61.0-65.3)
Mean clinical severity
1.59 (1.51-1.66) 1.52 (1.43-1.62) 1.58 (1.48-1.67) 1.44 (1.30-1.57) 1.43 (1.30-1.55)
Mean neuro-physiological
severity 1.88 (1.76-1.99) 1.91 (1.76-2.05) 1.88 (1.73-2.02) 1.74 (1.51-1.97) 1.81 (1.60-2.02)
Mean number of symptoms at diagnosis
2,36 (2.22-2.51) 2.33 (2.16-2.50) 2.37 (2.20-2.54) 2.44 (2.19-2.69) 2.25 (2.03-2.48)

65
Neurophysiology
Of the 168 patients, 139 were examined with neurography and 117 with EMG.
Nerve conduction studies were performed with surface electrodes using standard
techniques. Skin temperature was maintained at 33°C. The following nerves
were studied: median and/or ulnar motor and sensory nerves, peroneal motor,
and sural sensory nerves. Distal motor latency (DML), motor and sensory
conduction velocity (CV), the compound muscle action potential (CMAP)
amplitude, and the sensory nerve action potential (SNAP) amplitude were
recorded. A conduction block was defined as reduction of more than 30% of the
proximal: distal amplitude ratio. Axonal polyneuropathy was diagnosed if
neurography showed decreased or absent amplitudes of CMAP or SNAP and
relatively preserved CV (>70% of the lower limit of normal) and absence of
conduction block in at least two nerves. Denervation activity on EMG was also
regarded as a sign of axonal degeneration. Demyelinating polyneuropathy was
diagnosed if CV was <70% of the lower limit of normal (<35 m/s in the arms
and <30 m/s in the legs), or prolonged DML >150% of the upper limit of normal
in at least two nerves. Conduction block was also regarded as a sign of
demyelinating polyneuropathy. All others were classified as mixed axonal and
demyelinating. Normal reference values from each laboratory were used as cut-
off values. EMG recordings from the anterior tibial muscle were regarded as
either normal or chronic neurogenic (decreased interference pattern or motor
unit potentials with increased amplitude and duration). Denervation activity
(sharp waves or fibrillation potentials) was also recorded. The
neurophysiological findings were graded as follows: 0 = normal findings; 1 =
slight decrease of CMAP, SNAP, or CV in at least two nerves; 2 = all findings
between 1 and 3; or 3 = loss of sensory or motor responses in at least two
nerves.

66
Questionnaires At two different time points, questionnaires were mailed to cases and referents.
The first questionnaire included questions about previous medical history,
symptoms and information on occupational and leisure time exposure to metal
dust, gases/smoke, solvents, engine exhausts, impregnating agents,
plastic/rubber, other chemicals, pesticides and vibrations. The subcategories
used in the questionnaire were chosen from substances suggested in the
literature to be neurotoxic.
In the second questionnaire the questions about symptoms were repeated and the
QOL instruments SF-36 and EQ-5D (EuroQol) were added.
Up to 4 reminders were sent to non-responders until they answered or refused
participation. For incomplete questionnaires, a telephone interview was
conducted with the subject.
Blood samples
Blood samples were collected from 79 of the patients fulfilling the criteria for
cryptogenic polyneuropathy (response rate 50%). The samples were analyzed
for the different genotypes of EPHX1 exon 3, GSTM1 and GSTT1. Whole
blood was collected and leukocyte DNA was isolated with Wizard Genome
DNA purification kit (Promega Inc., Madison, Wisconsin, U.S.A.). The GSTM1
and GSTT1 null genotypes were assessed in a multiplex polymerase chain
reaction (PCR) with β-globin as an internal control gene for a successful PCR-
amplification [9]. The amino acid polymorphisms in the mEPHX gene (EPHX1
exon 3) were determined by a PCR-RFLP (restriction fragment length
polymorphism) assay [96, 155]. For exon 3, there are 3 possible genotypes: YY,
YH and HH. The wild type normal activity allele is YY and the low activity
genotype is HH.

67
Results
Study I: Clinical and neurophysiological findings Approximately twice as many men as women were affected with cryptogenic
polyneuropathy. The incidence of polyneuropathy increased with age. With
regard to clinical severity, 90 patients (54%) had slight symptoms and findings,
68 (40%) had moderately severe, and 10 patients (6%) had severe
polyneuropathy.
The mean age at first symptom was 61 years and at diagnosis, 64 years.
Regarding time from first symptom to diagnosis, 82% of the patients had a delay
from first symptom to diagnosis of less than 5 years, 14% had a delay of 6 to 10
years and 4% had a delay of more than 10 years. Patients with the longest delay
had more severe disease (clinical score 2.0 for >10 years compared with 1.5 for
<5 years), but there were no significant differences between the clinical or
neurophysiological scores in the groups.
The most common symptom was distal numbness, which was recorded in 115
case notes (65%). The patients with distal weakness or impairment of balance
had higher mean clinical severity scores. They also had significantly higher
neurophysiological severity scores. Patients who complained of burning
sensations had significantly lower neurophysiological severity scores. No other
differences were significant. Most patients (65%) had two or three symptoms,
with a mean of 2.3. There was no significant correlation between the number of
symptoms and age, clinical severity score, or neurophysiological severity score.
The shortest delay from first symptom to diagnosis was 1.8 years for burning
sensation and 2.8 years for hypersensitivity to touch, whereas the longest was

68
4.1 years for pain. The mean number of abnormal clinical findings (decreased or
lost responses or hypersensitivity) was 3.6.
Of the 168 patients, 139 had neurographic examinations. Those who were not
examined were older and had a lower mean clinical severity score although not
significant. The median or ulnar nerves and the peroneal and sural nerves on
both sides were examined in 132 patients. In 127 of these patients, at least 2 of 6
nerves showed abnormal values. Twelve patients had abnormal findings in only
one nerve or had normal findings, and these patients had significantly lower
mean clinical severity scores. Most patients had a combination of sensory and
motor nerve involvement with a mean clinical severity score of 1.7. The 6
patients with motor nerve involvement alone had the same clinical severity score
(1.7). The 29 patients with sensory nerve involvement alone had a lower mean
clinical severity score (1.3), which was significantly lower than the score of
those with sensorimotor involvement.
In 131 patients it was possible to state whether the nerve involvement was
demyelinating, axonal, or combined. Axonal polyneuropathy was the most
common (n=85) and these patients had a mean clinical severity score of 1.5. The
46 patients with the axonal and demyelinating form had a significantly higher
clinical severity score (1.8). The most common neurographic finding was that of
a mixed sensory-motor nerve involvement with axonal degeneration (n=62).
EMG examinations had been performed in 117 of the 168 patients. In 24
patients EMG in the anterior tibial muscle was normal. These patients had
significantly lower clinical severity scores than patients with abnormal EMG
(p<0.001). The 22 patients with chronic neurogenic findings and denervation
activity had significantly higher clinical severity scores (2.0) than the 70 patients

69
with chronic neurogenic findings alone (1.6) (p<0.05). One patient had
denervation activity alone.
Of the 139 patients who underwent neurography, 4 had neurophysiological
severity grade 0, 46 patients grade 1, 48 patients grade 2, and 41 patients grade
3. There was no difference in neurophysiological severity between men and
women. The patients in the most severe group were significantly older than
those in group 1 and 2 (p<0.01); and had significantly higher clinical severity
scores. Patients in the moderate group (grade 2) also had a higher clinical
severity score than those in the mild group (grade 1) (p<0.001). However, there
was no significant correlation.
Study II: Occupational determinants
Male sex increased the risk for cryptogenic polyneuropathy, COR (Crude Odds
Ratio) 2.33 (95% CI 1.61-3.38), and increasing age was a determinant for both
men and women showing significantly statistical trends. Fewer women reported
chemical or vibration exposure; 19% of female referents were exposed
compared to 64% of male referents. Exposure in men to Stoddard solvent, petrol
exhausts, herbicides, and hand and foot vibrations were significantly associated
with increased COR, respectively.
As individuals tended to have multiple exposures, logistic regression was
performed. In men the logistic regression showed particularly increased risks
(LOR, Logistic Odds Ratio>3.50) for occupational exposure to sulphur dioxide,
xylene, methyl ethyl ketone, and herbicides. Risks remained increased
(LOR>1.50) for occupational exposure to lead, hydrogen sulphide, Stoddard
solvent, petrol exhausts, fungicides, and vibration exposure to the feet,
respectively. For leisure time only exposure to gases/smoke and solvents

70
showed increased risks in the logistic regression, but only solvents was
significant, LOR 8.84 (95% CI 2.43-32.04). In women half of the CORs above
1.50 remained that high in LOR i.e., lead, mercury, nitrous oxide, and
insecticides. Comparing LORs in men and women, only the exposure to lead
showed increased risk in both sexes. However, our results do not contradict risks
in both sexes as the confidence intervals are overlapping, except for mercury
exposure.
In the second logistic model including exposure categories and without an
unexposed reference group, the results were identical with the former results
both for men and women.
Interaction between occupational and leisure time exposure could only be
analyzed in detail for men. Occupational pesticide exposure was the only
statistically significant determinant without a concomitant leisure time exposure,
COR 3.13 (95% CI 1.20-7.91). Comparing the results, interaction was
particularly strong for leisure time exposure to solvents especially in
combination, with occupational exposure to metal dust, gases/smoke, solvents,
engine exhausts and vibrations, respectively. However, even if the interactive
effect seems strong, the largest contribution to the risk estimates is from the
occupational exposures as sole leisure time exposure is rare.
Study III: Health related quality of life
This study comprised 62 patients with an average age of 64.9 years. The mean
time from diagnosis was 6.9 years (range 2-21 years). As reference, 1,429
women and 1,292 men from the general population were used, with a mean age
of 60.6 years, which was significantly lower than in the patients.

71
SF-‐36 When comparing the group of patients with cryptogenic polyneuropathy as a
whole with the general population, patients had lower QOL in all scales except
for MH (Mental Health) and mcs (Mental Component Summary). In the general
population, women had lower QOL than men for all scales. Women with
polyneuropathy tended to have lower QOL than men in all scales but the
differences did not reach significance. When patients and referents were
grouped according to age and sex, the physical scales PF (Physical Functioning),
RP (Role Physical), BP (Bodily Pain) and GH (General Health); and mental
scales VT (Vitality) and RE (Role Emotional) were decreased in most groups of
patients except for older women where only GH and VT were significantly
affected. On the other hand, no significant differences were observed in any
group in MH and mcs.
In the general population, the scores in PF, RP, GH, RE and both sum scales
decreased with increasing age. In polyneuropathy patients, the only statistically
significant difference was that 50- to 64-year old women had lower BP scores
than those who were 65 to 74 years old.
72
PF RP BP GH VT SF RE MH pcs mcs 50-‐59 (n=8) 70,8 57,4 49,4 51,8 56,2 76,5 58,8 75,5 39,9 45,8
60-‐69 (n=22) 59,9 54,6 60,1 61,9 59,3 80,6 72,8 80,4 38,3 50,9
0,0
10,0
20,0
30,0
40,0
50,0
60,0
70,0
80,0
90,0
SF-‐36 versus age in men
PF RP BP GH VT SF RE MH pcs mcs 50-‐64 (n=10) 52,0 37,5 42,3 48,2 43,0 81,2 51,7 75,2 32,8 47,2
65-‐74 (n=7) 55,7 53,6 60,0 39,5 51,2 76,8 76,2 83,4 34,5 51,5
0,0
10,0
20,0
30,0
40,0
50,0
60,0
70,0
80,0
90,0
SF-‐36 versus age in women
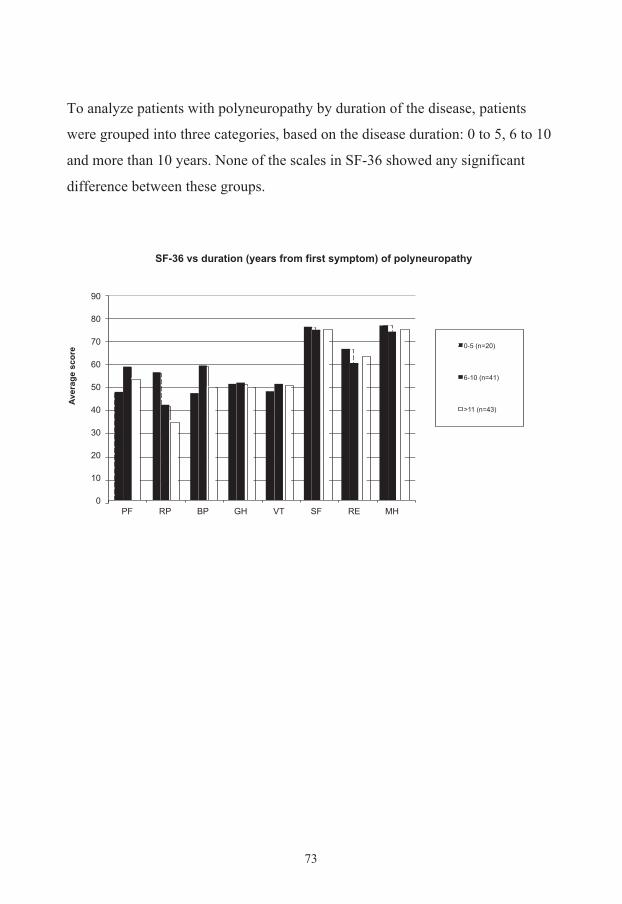
73
To analyze patients with polyneuropathy by duration of the disease, patients
were grouped into three categories, based on the disease duration: 0 to 5, 6 to 10
and more than 10 years. None of the scales in SF-36 showed any significant
difference between these groups.
0
10
20
30
40
50
60
70
80
90
PF RP BP GH VT SF RE MH
Aver
age
scor
e
SF-36 vs duration (years from first symptom) of polyneuropathy
0-5 (n=20)
6-10 (n=41)
>11 (n=43)

74
EQ-‐5D Because of the small number of patients in each group, all patients reporting any
problem have been analyzed together. Patients with cryptogenic neuropathy
reported significantly more problems in walking (mobility) (42%) than reference
subjects (14%). Problems with usual activities were reported by 31% of patients,
which was significantly more common than in the general population (8%). Six
percent of the patients reported problems with self-care (washing and dressing)
compared to 2% of the reference subjects. Pain was common in both patients
(85%) and the general population (56%). Anxiety or depression was reported by
35% of patients and 29% of reference subjects, which was the only not
significant difference. No significant differences were seen between men and
women. The results were not affected by disease duration for any of the five
questions.
EQ barometer Health state measured by the Visual Analogue Scale (VAS) was significantly
lower in male polyneuropathy patients older than 60 years and in women
younger than 70 years compared to the general population. The VAS scores
tended to decrease with increasing age, but there were no significant differences
between groups except between 50- to 59-year-old males and 60- to 69-year-old
males. Women with polyneuropathy tended to have lower VAS scores than men
in all age groups, but the difference was only significant for 50- to 59-year-old
patients. No relationship was seen between the QOL measured by the EQ
barometer and disease duration. However, patients reporting many symptoms
had lower EQ barometer values.

75
Study IV: Genetic polymorphisms In total, 79 cases with cryptogenic polyneuropathy and 398 controls were tested
for genetic polymorphisms in the GSTM1, GSTT1, and mEPHX genes. The OR
for the null genotype of GSTM1 and GSTT1 was 0.99 (0.61-1.61) and 1.86
(0.82-4.24), respectively and for EPHX*3 YY versus YH/HH the OR was 1.06
(0.65-1.71). Among the controls there were significantly more women with
GSTT1 null than in men, (p=0.04), and the homozygous HH variant in mEPHX
was more common in men (p<0.01). The other variants did not differ between
men and women. There were no statistically significant differences between
cases and controls in any group.
Regarding clinical findings, 24 patients were considered to have mild findings
and 39 patients had severe findings. No significant differences were found
between the groups in clinical or neurophysiological severity at diagnosis except
a tendency for GSTM1 null to have more severe clinical findings than GSTM1
positive cases (mean 1.55 vs. 1.31, p=0.064). Axonal neuropathy was observed
in 41 patients and combined axonal and demyelinating neuropathy in 19
patients. Regarding neurophysiological findings, 2 patients had pure motor
neuropathy, 13, pure sensory neuropathy, and 64, a mixed sensorimotor
neuropathy. Genetic polymorphisms were not significantly related to these
neurographic findings.
We also investigated the effects of different exposures. The frequencies are
presented in the table below.
76
Exposure Referents Cryptogenic
polyneuropathy
Smokers or previous
smokers
189 (47%) 43 (54%)
Solvents 132 (33%) 24 (30%)
Pesticides 29 (7%) 8 (10%)
Generalized anesthesia 312 (78%) 59 (74%)
Private water supply 29 (71%) 51 (65%)
The odds ratios for cryptogenic polyneuropathy among exposed individuals
were calculated. GSTT1 null among smokers reached the highest odds ratio
(3.72, p=0.08), and EPHX*3 HH vs. YY among solvent exposed had the lowest
odds ratio (0.30, p=0.14). A logistic regression analysis for the different
polymorphisms, sex, age, and exposures did not show any confounding effects
except that increasing age and male sex increased the risk of cryptogenic
polyneuropathy. Interactions between genes were analyzed and showed an
increased odds ratio for GSTT1, which was strongest if the patients had the HH
form of EPHX*3 (OR 2.37, p=0.234).
Additional results
Evaluation of symptoms In 2000 and again in2002, a questionnaire was sent to the patients, and 105
patients (77 men and 32 women) replied to both of the questionnaires. Of those
who responded, 87 fulfilled the criteria for cryptogenic polyneuropathy (61 men,
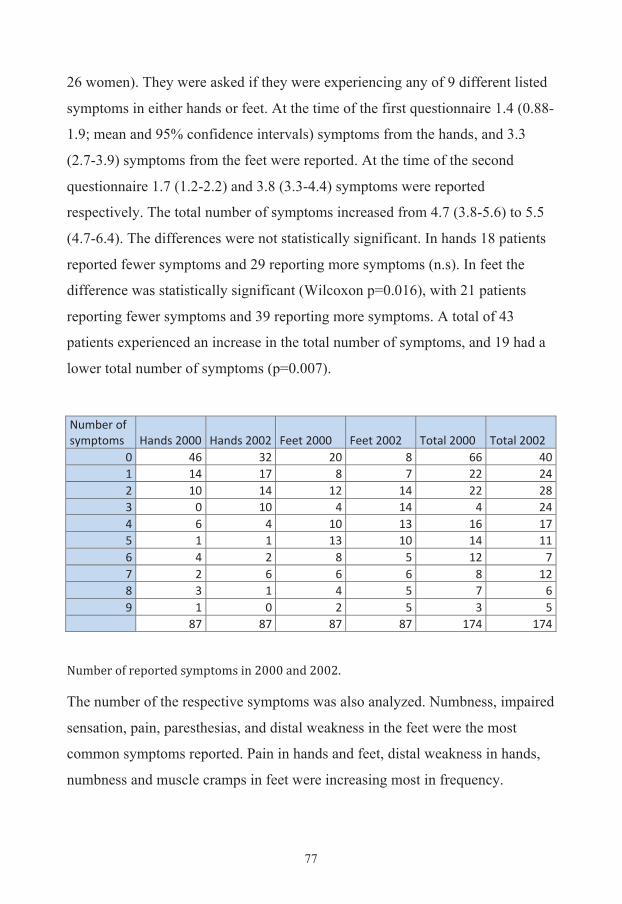
77
26 women). They were asked if they were experiencing any of 9 different listed
symptoms in either hands or feet. At the time of the first questionnaire 1.4 (0.88-
1.9; mean and 95% confidence intervals) symptoms from the hands, and 3.3
(2.7-3.9) symptoms from the feet were reported. At the time of the second
questionnaire 1.7 (1.2-2.2) and 3.8 (3.3-4.4) symptoms were reported
respectively. The total number of symptoms increased from 4.7 (3.8-5.6) to 5.5
(4.7-6.4). The differences were not statistically significant. In hands 18 patients
reported fewer symptoms and 29 reporting more symptoms (n.s). In feet the
difference was statistically significant (Wilcoxon p=0.016), with 21 patients
reporting fewer symptoms and 39 reporting more symptoms. A total of 43
patients experienced an increase in the total number of symptoms, and 19 had a
lower total number of symptoms (p=0.007).
Number of symptoms Hands 2000 Hands 2002 Feet 2000 Feet 2002 Total 2000 Total 2002
0 46 32 20 8 66 40 1 14 17 8 7 22 24 2 10 14 12 14 22 28 3 0 10 4 14 4 24 4 6 4 10 13 16 17 5 1 1 13 10 14 11 6 4 2 8 5 12 7 7 2 6 6 6 8 12 8 3 1 4 5 7 6 9 1 0 2 5 3 5
87 87 87 87 174 174
Number of reported symptoms in 2000 and 2002.
The number of the respective symptoms was also analyzed. Numbness, impaired
sensation, pain, paresthesias, and distal weakness in the feet were the most
common symptoms reported. Pain in hands and feet, distal weakness in hands,
numbness and muscle cramps in feet were increasing most in frequency.
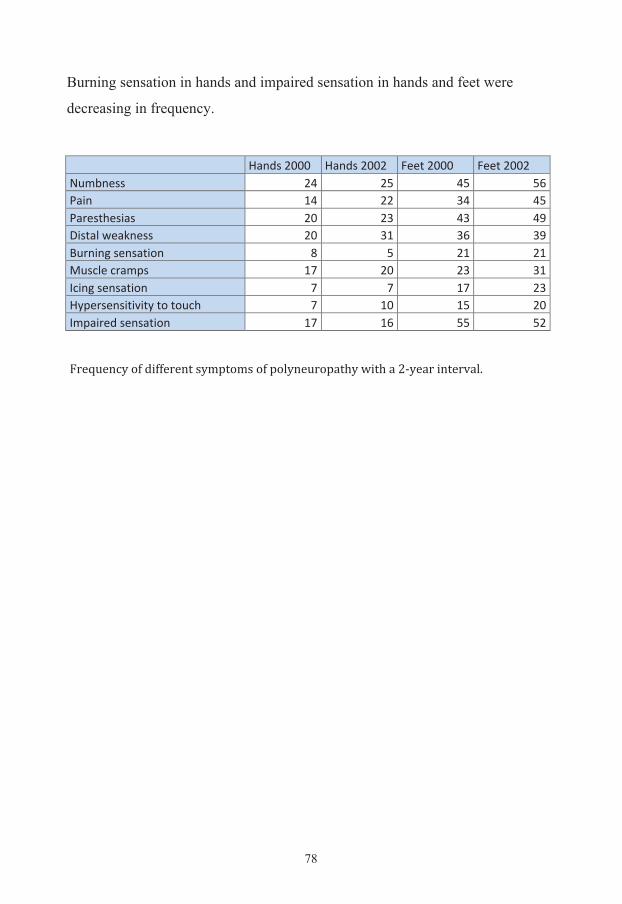
78
Burning sensation in hands and impaired sensation in hands and feet were
decreasing in frequency. Hands 2000 Hands 2002 Feet 2000 Feet 2002 Numbness 24 25 45 56 Pain 14 22 34 45 Paresthesias 20 23 43 49 Distal weakness 20 31 36 39 Burning sensation 8 5 21 21 Muscle cramps 17 20 23 31 Icing sensation 7 7 17 23 Hypersensitivity to touch 7 10 15 20 Impaired sensation 17 16 55 52 Frequency of different symptoms of polyneuropathy with a 2-‐year interval.
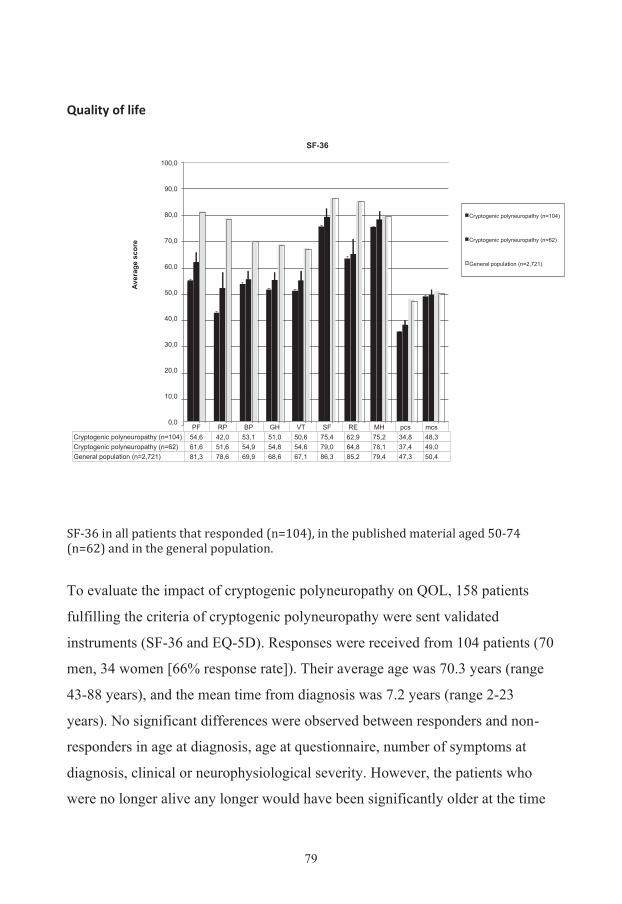
79
Quality of life
SF-‐36 in all patients that responded (n=104), in the published material aged 50-‐74 (n=62) and in the general population.
To evaluate the impact of cryptogenic polyneuropathy on QOL, 158 patients
fulfilling the criteria of cryptogenic polyneuropathy were sent validated
instruments (SF-36 and EQ-5D). Responses were received from 104 patients (70
men, 34 women [66% response rate]). Their average age was 70.3 years (range
43-88 years), and the mean time from diagnosis was 7.2 years (range 2-23
years). No significant differences were observed between responders and non-
responders in age at diagnosis, age at questionnaire, number of symptoms at
diagnosis, clinical or neurophysiological severity. However, the patients who
were no longer alive any longer would have been significantly older at the time
PF RP BP GH VT SF RE MH pcs mcs Cryptogenic polyneuropathy (n=104) 54,6 42,0 53,1 51,0 50,6 75,4 62,9 75,2 34,8 48,3 Cryptogenic polyneuropathy (n=62) 61,6 51,6 54,9 54,8 54,6 79,0 64,8 78,1 37,4 49,0 General population (n=2,721) 81,3 78,6 69,9 68,6 67,1 86,3 85,2 79,4 47,3 50,4
0,0
10,0
20,0
30,0
40,0
50,0
60,0
70,0
80,0
90,0
100,0
Aver
age
scor
e
SF-36
Cryptogenic polyneuropathy (n=104)
Cryptogenic polyneuropathy (n=62)
General population (n=2,721)

80
the questionnaire was sent out and were older at first symptom and diagnosis,
but did not differ in number of symptoms or clinical or neurophysiological
severity. As the control population had an upper age limit at 74 years and as
only two of the patients were younger than 50 years, the age limit in the final
study was changed to 50-74 years. The data of both the complete and published
materials are shown above. The older excluded patients had a lower HR-QoL in
most of the scales.
Diabetes risk evaluation
According to the information in the medical records as described in Study I, 168
patients were considered as cryptogenic. All of these patients were sent the
questionnaire described in Study II. This questionnaire also included questions
about the medical history. A total of 110 patients replied (76 men, 34 women),
58 did not reply (37 men, 21 women) and 604 controls replied (274 men, 330
women).
It has been described in the literature that patients with cryptogenic
polyneuropathy have an increased frequency of impaired glucose tolerance [75,
122, 152, 165] and probably are at risk of developing diabetes, though others
have found contradicting results [82, 117]. As the patients’ data were collected
during a 7-year period, one important question was if they still were
cryptogenic. We, therefore, asked specifically about diabetes. The patients had a
mean age of 68.2 years (66.2-70.1; 95% confidence interval) and the controls
were 62.2 (61.3-63.1) years old (p<0.001) at the time of the questionnaire. Three
patients, who initially were not diagnosed with diabetes, reported that they had
developed diabetes (2.7%) and 30 controls (5.0%) reported that they had
diabetes. The incidence of diabetes type 1 and 2 in Sweden for 60- to 69-years-
olds is 732 cases per 100,000 and year [172], therefore, the incidence in our
material is at the same level.

81
Discussions and Conclusions
Clinical picture Polyneuropathy is one of the most common neurological disorders and patients
are seen by physicians with many different specialties. The clinical diagnosis is
based on the finding of a distal, fairly symmetrical impairment of sensation,
muscle strength, or muscular atrophy, and progression over a period of months
or years. Reflexes are decreased or lost in the affected parts, but particularly in
the ankles. Symptoms usually occur in the feet before the hands [179].
Neurophysiological investigations with nerve conduction studies and EMG can
support the diagnosis.
The 182 cases of polyneuropathy in our first study were diagnosed as
cryptogenic by the treating neurologist. To make certain that there was no
known cause of neuropathy, the medical records were reviewed according to our
protocol. In many cases the patients had been followed up after the initial visit.
The questionnaire that was sent to the patients also included questions about
disorders known to cause polyneuropathy. As the patients were diagnosed up to
7 years before the first questionnaire, it is likely that tumors presenting with
polyneuropathy, as the first symptom would have been diagnosed. 8 patients
actually were excluded due to tumors. The risk of developing diabetes was also
low, confirming that the cases were correctly diagnosed. The major problem
with the diagnosis is that it is always possible to do a more thorough
investigation. Unfortunately, neurophysiological examinations were performed
in only 139 of the patients. It is, however, unlikely that any of these patients
would have an inflammatory neuropathy such as CIDP, as patients who were
deteriorating would have been followed up or referred back to our clinics. There
are no other clinics in the study area treating progressive neuropathies. In fact,

82
the patients who were not examined neurophysiologically had a lower clinical
severity at diagnosis supporting that they were cryptogenic. The most common
reasons for not performing neurophysiological tests were old age and low
clinical severity. Our definition of cryptogenic polyneuropathy is relatively
broad, but has the advantage of reflecting the clinical picture that is seen by a
neurologist at a secondary or tertiary center, which was the aim of our study.
The study has the drawback of being retrospective.
Polyneuropathy was more common in elderly patients and about twice as many
men as women were affected. This difference is in agreement with most
previous studies [37, 66, 107, 119] except one [190]. A male predominance may
support our hypothesis that polyneuropathy could be caused by a chronic toxic
effect of occupational or environmental factors.
In our series two thirds of the patients complained of numbness and about one
third complained of paresthesias, pain, impairment of balance, or distal
weakness. In a smaller study of 29 cases of unclassified polyneuropathy, 28
patients had hypoesthesia or hyperalgesia, 26 paresthesias, and 10 pain [66].
Vrancken reported higher prevalences of symptoms in CIAP, this is, however,
probably due to study design [180]. In a study of chronic cryptogenic sensory
polyneuropathy, distal numbness was reported in 86% and pain in 72% of the
patients [190]. In our series impairment of balance and muscle weakness was as
common as pain, probably reflecting the fact that we also included patients with
motor or sensorimotor polyneuropathy.
Two symptoms brought patients more rapidly to the neurologist: a burning
sensation and hypersensitivity to touch. These symptoms are very disturbing to
patients and make them seek medical attention early. In contrast, patients with
pain and muscle cramps had the longest delay from first symptom to diagnosis.

83
Wolfe also found that patients with pain had their symptoms significantly longer
than those without pain, for which they had no clear explanation [190]. Wolfe
and colleagues also found that only 10% of their patients complained of pain
alone [190].
When patients were asked about symptoms with a 2-year interval, the number of
symptoms increased. All symptoms except burning sensation and impaired
sensation in hands or feet became more common. Unfortunately, we do not have
any data about the development of severity of the symptoms.
The most sensitive signs of polyneuropathy are decreased or lost distal
proprioception or sense of vibration, and ankle jerks, both with a sensitivity of
75-80%.
Our scale for clinical severity focused on functional deficits (particularly ataxia
and need for assistance), so it was not surprising to find that the symptoms
impairment of balance and distal weakness had the highest clinical severity
scores. However, other symptoms such as pain and burning or icing sensations
probably affect the QOL as much. We classified 93% of the patients in this
series as having mild or moderate disease; but they probably had a more severe
disease than patients in other studies. Some of the previous epidemiological
studies were based on people in primary care [14, 103] and there might be
important differences between those studies and ours. Because of the large
number of patients in primary care with mild symptoms, some patients might be
diagnosed as having cryptogenic polyneuropathy, but later turn out to have other
diseases or simply normal aging of the peripheral nerves.
Of the 84% of patients who had neurographic examinations performed, more
than half had a mixed motor and sensory nerve involvement and only 4% had

84
motor nerve involvement alone, which is in accordance with the results of
Notermans et al. [119], who found only 3% with motor involvement alone.
Several of their patients who initially had sensory neuropathy later developed
sensorimotor involvement [121].
The EMG studies showed chronic neurogenic signs with denervation in 23 of
123 patients, compatible with a mainly axonal polyneuropathy, which is in
agreement with others [119]. The EMG findings also suggest that even if most
cryptogenic polyneuropathies are mainly sensory or sensorimotor, there is also
an involvement of motor nerves, which is important when judging the relevance
of neurophysiological findings and their correlation with somatic neurological
findings [66, 119, 190].
We found a weak correlation between clinical and neurophysiological severity.
A larger prospective study with a better classification of neurophysiological
severity would probably find a stronger correlation. In some cases the
neurophysiological severity is greater than expected from the neurological
examination. Another reason for neurophysiological examination is that axonal
and demyelinating polyneuropathies can be distinguished. The latter includes
hereditary neuropathies and the treatable chronic inflammatory demyelinating
polyneuropathy (CIDP) [144].
Health related quality of life Cryptogenic polyneuropathy has usually been regarded as a relatively mild
neurological condition, which commonly does not severely affect the patient’s
ability to walk or perform activities of daily living (ADL). However, we found
that 42% of patients reported problems with walking and 31% had problems
with usual activities such as work, study, household work, family or leisure
activities. These problems were not affected by age. In another study focusing

85
on QOL in patients with CIAP, fatigue and walking disability were found to
interfere markedly with patients’ functioning in daily life [44].
Disease duration did not affect SF-36 or EQ-5D barometer scores in our study.
That finding might, in part, be explained by the study design as patients were
recruited over a 7-year period and some patients had short time of follow-up
from diagnosis to participation in our study. However, the results of a
prospective follow-up study of 40 patients with chronic polyneuropathy of
undetermined cause support that the disease is a stable or only slowly
progressive disorder in most cases [88].
Teunissen and co-workers showed that patients with CIAP have lower scores
than expected in all areas of the SF-36 except for ‘role limitations due to
emotional problems’. The differences in ‘social functioning’, ‘pain’, and
‘physical functioning’ were most marked [168]. Their patients and the reference
population were not sex and age matched. These results are in line with our
results, although we did not find any difference in ‘mental health’. Hughes
reports that QOL in CIAP was worse than norms from Australian Bureau of
Statistics in all SF-36 scales except ‘role emotional’. In the scale ‘mental
health’, they only found a difference among patients with pain [83]. The patients
reported by Hughes had lower scores in almost all scales most likely because
these patients were older or due to selection of the patient material. All three
studies support that chronic idiopathic or cryptogenic polyneuropathy impairs
physical functions and that the disease limits the ability to perform usual
activities in daily life.
In our study, 85% of the patients reported pain in EQ-5D. However, EQ-5D is a
very sensitive instrument for assessment of pain, which was reported by 56% of
females in the general population older than 50 years of age and at a slightly

86
lower frequency in men. In a study of Italian patients with chronic
polyneuropathy, the QOL was affected not only by the presence of neuropathy,
but also by the presence and intensity of pain. Moreover, the mental domains of
the SF-36 were only correlated with pain. Pain per se worsens the QOL of
neuropathic patients, regardless of disease severity [27]. Abresch and co-
workers have investigated the QOL in patients with different slowly progressive
neuromuscular diseases (myotonic muscular dystrophies, limb-girdle
syndromes, CMT disease, fascioscapulohumeral dystrophy, spinal muscular
atrophy and post-polio syndrome). Their results indicated that, except for adult
spinal muscular atrophy, the prevalence and severity of pain reported in slowly
progressive neuromuscular diseases were significantly greater than levels of
pain reported by the general US population. There was a significant correlation
between increased pain and lower levels of general health, vitality, social
function, and physical role, which confirms that pain is an essential symptom
affecting QOL in patients with diseases in the peripheral nervous system and
muscles [2]. They found the same results in patients with diabetic neuropathy,
which indicates that patients who have moderate or severe levels of pain may
benefit from a pain management program, rather than focusing on only physical
impairment [1]. These results were confirmed in patients with different kinds of
polyneuropathy by Liedberg and Vrethem [100].
In a study focusing on pain in CIAP it was found that non-neuropathic pain was
as common as neuropathic pain and that pain was strongly associated with the
physical functioning domain of the SF-36 in patients with mixed neuropathic
and non-neuropathic pain [45].
HR-QoL has been described in several studies of diabetic neuropathy showing
lower HR-QoL in patients with diabetes than in our group of patients with
cryptogenic polyneuropathy [8, 28, 74].

87
Exposures in work and leisure time Considering the magnitude of risk, the confidence intervals and the confounding
effect from other exposures, in our study, the independent determinants for
polyneuropathy in men seemed to be occupational exposure to lead, sulphur
dioxide, hydrogen sulphide, xylene, methyl ethyl ketone, herbicides, fungicides
and foot vibrations. Among leisure time exposures only solvents could be
regarded as a strong independent determinant. All other leisure time exposures,
except perhaps gases/smoke, seemed to be of minor importance for
polyneuropathy since the increased risk could be explained by concomitant
occupational exposures. In women the corresponding occupational determinants
were lead, mercury, nitrous oxide and insecticides, however no leisure time
determinant could be identified. Our study was limited by small numbers of
cases and by low frequency of exposure, especially in women, which was
reflected in broad confidence intervals, not always including unity.
Some individuals, especially in the older ages, might have found it difficult to
remember their life time exposures, but as cases were older than referents,
memory bias should work in the direction of lowering the risk estimates. None
of the referents answered that they had polyneuropathy. Evaluation of the
validity of information from subjects in case-referent studies has also failed to
find any recall bias of importance [90, 134]. In a study of 188 metal-exposed
subjects, exposure assessment performed by an industrial hygienist was
compared with self-reported exposures, showing an agreement in the
assessments from 83 to 94% for different metals. Interestingly, the hygienist
classified more workers to be exposed than the workers themselves [149].

88
Our study was too small to allow any further analyses of the exposure i.e.,
length or intensity, thus including only ever exposed. As a majority of the male
cases and referents were exposed to several potential determinants, a reference
category free of such exposures was used. Although Swedish women have a
relatively high employment rate, only a minority were exposed to occupational
chemicals or vibrations. This is the reason why the study was analyzed with men
and women separated.
Out of the initial list of well-established, but often rarely occurring, substances
that cause polyneuropathy, only lead, n-hexane and nitrous oxide showed an
increased risk in our study [158]. We also showed some synergistic effects with
increased risk estimates in various occupational and leisure time exposures
combined.
Occupational mercury exposure occurs in workers associated with glass cutting
and in the manufacture of scientific instruments, in dental personnel, and
following release of mercury vapor from amalgam tooth fillings. Methyl
mercury is used in the manufacture of paper, in the chloralkali industry, and in
other chemical production. General human exposure usually results from
ingestion of fish obtained from water contaminated with industrial effluents
[178]. Mercury was a determinant for cryptogenic polyneuropathy in women
according to our study. Organic mercury is clearly associated with
polyneuropathy [178]. However, there are case reports that elemental mercury
can also cause polyneuropathy in humans [4, 5, 26, 76, 79, 87, 151, 178]. Only
one study of workers exposed to mercury has failed to find any relationship with
polyneuropathy [176]. Unfortunately, our questionnaire did not distinguish
between organic and inorganic mercury.

89
Sulphur dioxide and hydrogen sulphide appeared to be strong determinants for
cryptogenic polyneuropathy in our study, and these associations have not been
previously published. A well-known mechanism of hydrogen sulphide is
respiratory paralysis and subsequent hypoxia to the central nervous system, but
there could also be a direct toxic effect on the brain [110]. Several enzymes in
the brain are inhibited by hydrogen sulphide i.e., carbonic anhydrase, an enzyme
involved in the neuronal lipid synthesis and/or re-organization of myelin
membranes [147]. Whether these mechanisms also are applicable to the
peripheral nervous system is less clear.
In our study toluene did not show an increased risk in the logistic regression
analysis although polyneuropathy has been described after abuse of toluene-
containing products [162]. Polyneuropathy has also occurred in different
occupational settings where toluene has been present [57, 104]. In contrast to
toluene, xylene was a relatively strong determinant in our study; a finding
supported by case reports on intoxication with products containing 5-10%
xylene [57]. Xylene (dimethylbenzene) is also chemically closely related to
toluene (methylbenzene) and can, therefore, be suggested as a potential agent to
cause polyneuropathy despite less documentation.
Methyl ethyl ketone was a relatively strong determinant in our study. In the
literature only case reports have been found, but the neurotoxic potential of
methyl ethyl ketone is difficult to evaluate as these cases have been exposed to a
mixture of solvents [6, 39, 63]. It has also been reported that methyl ethyl ketone
can act indirectly as an inducer of n-hexane-metabolizing enzymes [84].
7 male cases and 4 referents added exposure to jet fuel and jet engine exhausts
in the questionnaire, COR 6.73 (95% CI 1.54-33.21). There are only two
previous reports on this relationship in the literature [93, 94]. As both civilian

90
and military airports as well as an aircraft industry are located in southeast
Sweden, where our investigation was performed, this result seems reasonable.
In our study herbicide, and to some extent also fungicide, exposure in men and
insecticide exposure in women were determinants. Herbicides are not commonly
reported to cause peripheral polyneuropathy in the literature. In fact, only three
case reports describe polyneuropathy after occupational exposure to the
herbicide 2,4-D with predominant dermal uptake [16, 59, 174]. A case report
describes intoxication with weed killers containing phenoxyacetic acid that
resulted in a delayed peripheral neuropathy [123]. On the other hand, an
evaluation of soldiers exposed to Agent Orange (50:50 mixture of 2,4-D and
2,4,5-T) has concluded that there is inadequate or insufficient evidence for an
association with peripheral neuropathy [58]. Therefore, our results contribute to
the evidence that herbicides might cause polyneuropathy.
In contrast, exposure to organophosphates is a recognized cause of
polyneuropathy [7], but these compounds are no longer extensively used in
Sweden. Organophosphates are used mainly as pesticides and herbicides but are
also used as petroleum additives, lubricants, antioxidants, flame-retardants, and
plastic modifiers. Certain organophosphates cause a delayed polyneuropathy
that occurs approximately 2 to 3 weeks after acute exposure even in the absence
of the otherwise typical acute cholinergic toxicity [7]. Another insecticide,
dichlorodiphenyltrichloroethylene (DDT), was widely used in Sweden until the
ban in 1970. DDT belongs to the organochlorine pesticides known to cause
symptoms of the central nervous system at high doses, but the effects of chronic
low-level exposure are uncertain [7]. Lindane, another widely used insecticide,
has also been reported to cause polyneuropathy [175]. However, there were too
few exposed in our study to obtain a risk estimate. Pyrethroids are synthetic
insecticides that affect voltage-sensitive sodium channels and possibly also

91
voltage-sensitive calcium and chloride channels. Occupational exposure has led
to paresthesias that have been attributed to repetitive activity in sensory fibers as
a result of abnormal prolongation of the sodium current during membrane
excitation [156]. Convulsions may occur if substantial amounts are ingested
[137]. Fungicide exposure has not been suggested to be a determinant for
polyneuropathy, but in our study, the risk was a doubled in men.
Exposure to vibrating tools may cause a variety of symptoms depicted as the
hand–arm vibration syndrome. The symptoms may be of vascular, neural, and
muscular origin and may appear as digital vasospasm (vibration white fingers;
VWF), sensorineural disturbances, and/or as muscular weakness and fatigue
[53]. A negative relationship has been observed between nerve conduction
velocities in motor and sensory nerves in the arms, on the one hand, and age at
the time of the study or total number of working years with vibrating tools, on
the other [53]. Until now, there have been no reports that vibration of the feet
can cause a more generalized polyneuropathy, and therefore, in that respect our
finding should be interpreted with caution.
Genetic polymorphisms and risk of cryptogenic polyneuropathy The frequency of GSTM1 null is 42-60% in Caucasians [51]. The frequency of
homozygous null GSTT1 varies greatly with ethnicity and is 10-20% in
Caucasians [141]. The EPHX*3 gene can be found in three different forms, the
wild type/normal activity variant (YY), heterozygous (YH), or homozygous/low
activity (HH) genotypes. In a Caucasian population, approximately about 40%
of subjects are heterozygous and 12% are homozygous for the HH genotype
[51]. The frequency of these polymorphisms in our study population was
similar. No differences in allele frequencies by age or sex were seen in large
studies [51]. Unfortunately, we had an imbalance in our control group with 19%

92
of the women and 12% of the men having the GSTT1 null polymorphism and
9% of the women and 18% of the men having the EPHX*3 HH polymorphism.
Thus, it is not possible to draw any conclusions about differences in risks of
cryptogenic polyneuropathy among men and women separately. In Study IV of patients with cryptogenic polyneuropathy, we examined the
association of GSTM1 and GSTT1 null polymorphisms and EPHX1 exon 3 HH
polymorphism in relation to several environmental and chemical exposures.
Although we did not find any statistically significant increased risk, the GSTT1
null genotype was associated with an almost two-fold increased risk of
polyneuropathy. Our hypothesis is that the GSTT1 null polymorphism may be
related to an impaired metabolism of toxic substances and reactive oxygen that
could lead to nerve damage, involving multiple sites along motor and sensory
axons in the peripheral nervous system. This may result in axonal atrophy or
axonal swelling, leading to progressive distal axonal degeneration. The myelin
sheath may break down concomitantly with the axon. This could contribute to,
or directly result in, an axonal or combined axonal-demyelinating neuropathy.
Components of cigarette smoke are examples of exogenous substrates that are
toxic and, furthermore, are subject to bioactivation and may both directly and
indirectly be neurotoxic. We found a nearly four-fold increased risk of
polyneuropathy in GSTT1 null smokers that almost achieved statistical
significance. Teunissen and co-authors reported an odds ratio of 2.1 for current
smoking in patients with CIAP [169], and it has also been found that tobacco
use may predispose to earlier development and more severe symptoms of
diabetic neuropathy [167]. Our data indicate that this risk might be explained by
smokers carrying certain genetic polymorphisms leading to impaired
detoxification of the toxic compounds in cigarette smoke. The mechanism for
the toxicity of cigarette smoke on nerves is not known, but it has been

93
speculated that chemicals in the smoke mediate it, where PAHs are regarded as
the most important component. Impaired breakdown of PAHs in persons with
the null genotype of GSTs may lead to an increased exposure on nerves and
thereby increased loss of nerves. n-Hexane is another toxic substance that is
present in cigarette smoke and is a well-known cause of polyneuropathy [194].
We did not find a significant correlation between clinical or neurophysiological
severity and genotype except a small increase in the severity of clinical findings
in GSTM1 null patients that almost reached statistical significance. It is possible
that a correlation will be found if a more sensitive scale for clinical or
neurophysiological severity was used in a larger material.
There are several possible reasons why we did not find any significant
differences. The main reason is probably the small number of patients in our
study. However, in a Chinese study of 22 cases of polyneuropathy working in a
printing factory and 163 controls, an association was found with CYP2E1 Dra,
but not GSTM1 and GSTT1, indicating that either a more pronounced exposure
to a toxic agent is necessary or that other genes may have greater importance for
the development of polyneuropathy [194]. It is also possible that the referring
general practitioner or the diagnosing neurologist identified most cases of toxic
neuropathies.
Conclusions Cryptogenic polyneuropathy is a slowly progressive sensorimotor nerve lesion
with minor or moderate severity. Men are affected almost twice as often as
women. The typical patient is at least 60 years old and complains of numbness
in the feet. Neurological investigation reveals impaired proprioception and/or
sense of vibration and decreased or lost ankle tendon reflexes.

94
Neurophysiological examination shows sensorimotor polyneuropathy with
mainly axonal involvement.
We have found that patients with cryptogenic polyneuropathy have poorer HR-
QOL, except for mental health scores, than the general population. Pain seems
to be one of the main contributors to the poor HR-QoL. More than 3 out of 10
patients reported problems performing activities of daily living, and it was even
more common to have problems walking, which was more common than
expected. Age and disease duration did not seem to affect QOL, but this is
contradicted by the fact that patients reported more symptoms when the
questionnaires were repeated 2 years later. However, our data support that
cryptogenic polyneuropathy is a slowly progressive disorder.
Our study is the first case-referent study focusing on occupational and
environmental determinants for cryptogenic polyneuropathy. The study has
limitations in size and exposure assessment as we had to rely on self-reported
exposure. Our results suggested that not only known determinants could be
confirmed, but also some new ones appeared i.e., sulphur dioxide, hydrogen
sulphide, fungicides, and vibrations in the feet. Moreover, our results point to a
synergistic effect of various exposures. Men were more often exposed to
neurotoxic chemicals and vibrations than women, probably explaining most of
the increased risk for polyneuropathy in men.
No significant correlation was found between GSTM1, GSTT1, and EPHX1
polymorphisms in patients with cryptogenic polyneuropathy compared with
controls. A strong tendency, however, was seen for the GSTT1 null phenotype,
which was enhanced among smokers compared to controls (OR 3.7). The
GSTT1 null polymorphism may be related to an impaired metabolism of toxic
substances and reactive oxygen that could lead to nerve damage in the peripheral

95
nervous system. This could contribute to, or directly result in an axonal or
combined axonal-demyelinating neuropathy. A larger study of unselected
polyneuropathy cases and controls is warranted to confirm these data.
Future work should also include a larger community-based prospective study of
slowly progressive symmetric polyneuropathy in which all patients have a
thorough neurological examination, and laboratory tests including blood
glucose, complete blood count, ESR, serum electrophoresis, renal and liver
function, vitamin B12/folic acid, TSH, and chest X-ray. In addition, nerve
conduction velocities and amplitudes should be performed before diagnosis. It
would also be of value to assess QOL using both generic and neuropathy-
specific QOL scales and perform clinical evaluation of patients with repeated
functional assessment and neurophysiological evaluations to describe the
implications in patients with cryptogenic polyneuropathy and the prognosis over
a longer period.

96
Acknowledgements
This work was carried out as a collaboration project between the Departments of
Neurology, Neurophysiology and Occupational and Environmental Medicine at
the University Hospital in Linköping; the Department of Public Health and
Community Medicine, Institute of Medicine, University of Gothenburg;
Division of Cellular Biology, Department of Biomedicine and Surgery,
Linköping University; the Department of Internal Medicine, Division of
Neurology, Motala Hospital, and The Department of Internal Medicine at Ryhov
County Hospital, Jönköping.
I wish to express my sincere gratitude to:
Assistant Professor Magnus Vrethem, main supervisor, co-author
for his immense knowledge of neurology and neurophysiology in general and of
polyneuropathies in particular,
for his never-ending enthusiasm and encouraging support during the long
project and for constructive criticism during the preparation of manuscripts
and most of all for being a very good friend
Professor Eva Svanborg, co-supervisor
for her extensive knowledge about neurophysiology,
for her guidance in the scientific world and bureaucracy.
Med dr Martin Tondel, co-author
for deep knowledge in environmental medicine and public health,
for always asking questions and raising new possible hypotheses.
Med dr Bodil Persson, co-author

97
for her knowledge about the metabolism of toxic substances.
Professor Peter Söderkvist, co-author
for introducing me into the world of genetic polymorphisms and their
importance in the metabolism of solvents and other toxic substances.
Assistant Professor Mats Fredriksson, co-author
for his expertise in statistical methods,
for sorting everything out
Med dr Anders Österberg, co-author
for recruitment of patients
Dr Shahrzad Hosseininia, co-author
for laboratory work
Pia Jönsson, co-author
for epidemiological expertise
Research nurses Miriam Carlsson and Gun Johansson
for their outstanding assistance in collecting blood samples and questionnaires,
for bringing joy and laughter into the work.
Professor Jan-Edvin Olsson
for advice and help
Associate Professor Raymond Press
for advice and encouragement

98
Professor Anna-Christina Ek
for advice and for sharing her expertise in quality of life study methodology
Professor Bo-Erik Malmwall
for supporting me to take the step to work as a researcher
Dr Margareta Hultgren
for her endurance in clinical neurology and cooperative support during these
years
Co-workers at the Section of Neurology at the Department of Internal Medicine
at Ryhov County Hospital: Med Dr Anna Budzianowska, Dr Anna Eklund, Dr
Helena Bruhn, Dr Greta Gustafsson, Dr Owe Lannemyr, Dr Robert Bielis, Med
Dr Sven Pålhagen, nurses Gudrun Carlsson, Ingvor Lindh and Ulla Andersson
for cooperative support during years of studies.
The heads of Internal Medicine at Ryhov County Hospital: Dr Leif Ockander,
Dr Staffan Schöön and Dr Agneta Ståhl
The staff at the medical library at Ryhov Hospital
for always being nice and helpful in the search for literature
And finally to my wife Ulrika and our children Andrea and Philip
for supporting me during these years of too much work.

99
References 1. Abresch RT, Jensen MP, Carter GT (2001) Health-‐related quality of life
in peripheral neuropathy. Phys Med Rehabil Clin N Am 12:461-‐472
2. Abresch RTC, Gregory T. Jensen, Mark P. Kilmer, David D. (2002)
Assessment of pain and health-‐related quality of life in slowly
progressive neuromuscular disease. American Journal of Hospice &
Palliative Care 19:39-‐48
3. Abu-‐Amero KK, Milcarek B, Bosley TM (2009) GSTM1 and GSTT1
deletion genotypes in various spontaneous optic neuropathies in
Arabs. Br J Ophthalmol 93:1101-‐1104
4. Albers JW, Cavender GD, Levine SP, Langolf GD (1982) Asymptomatic
sensorimotor polyneuropathy in workers exposed to elemental
mercury. Neurology 32:1168-‐1174
5. Albers JW, Kallenbach LR, Fine LJ, Langolf GD, Wolfe RA, Donofrio PD,
Alessi AG, Stolp-‐Smith KA, Bromberg MB (1988) Neurological
abnormalities associated with remote occupational elemental
mercury exposure. Ann Neurol 24:651-‐659
6. Altenkirch H, Mager J, Stoltenburg G, Helmbrecht J (1977) Toxic
polyneuropathies after sniffing a glue thinner. J Neurol 214:137-‐152
7. Aminoff MJ (2008) Effects of toxins and physical agents on the
nervous system. In: Bradley WG, Daroff RB, Fenichel MG, Jankovic J
(eds) Neurology In Clinical Practice. Butterworth Heinemann
Elsevier, Philalelphia, PA, pp 1657-‐1691
8. Apostolou TG, R. (1999) Neuropathy and quality of life in diabetic
continuous ambulatory peritoneal dialysis patients. Peritoneal
Dialysis International 19 Suppl 2:S242-‐247

100
9. Arand M, Muhlbauer R, Hengstler J, Jager E, Fuchs J, Winkler L, Oesch
F (1996) A multiplex polymerase chain reaction protocol for the
simultaneous analysis of the glutathione S-‐transferase GSTM1 and
GSTT1 polymorphisms. Anal Biochem 236:184-‐186
10. Barahmani N, Carpentieri S, Li XN, Wang T, Cao Y, Howe L, Kilburn L,
Chintagumpala M, Lau C, Okcu MF (2009) Glutathione S-‐transferase
M1 and T1 polymorphisms may predict adverse effects after therapy
in children with medulloblastoma. Neuro Oncol 11:292-‐300
11. Baron R, Mayoral V, Leijon G, Binder A, Steigerwald I, Serpell M
(2009) 5% lidocaine medicated plaster versus pregabalin in post-‐
herpetic neuralgia and diabetic polyneuropathy: an open-‐label, non-‐
inferiority two-‐stage RCT study. Curr Med Res Opin 25:1663-‐1676
12. Beaton DE, Schemitsch E (2003) Measures of health-‐related quality of
life and physical function. Clinical Orthopaedics & Related
Research:90-‐105
13. Beghi E, Monticelli ML (1998) Chronic symmetric symptomatic
polyneuropathy in the elderly: a field screening investigation of risk
factors for polyneuropathy in two Italian communities. Italian
General Practitioner Study Group (IGPST). J Clin Epidemiol 51:697-‐
702
14. Beghi E, Monticelli ML, (IGPSG) IGPSG (1995) Chronic symmetric
symptomatic polyneuropathy in the elderly: a field screening
investigation in two Italian regions. I. Prevalence and general
characteristics of the sample. Neurology 45:1832-‐1836
15. Benhamou S, Reinikainen M, Bouchardy C, Dayer P, Hirvonen A
(1998) Association between lung cancer and microsomal epoxide
hydrolase genotypes. Cancer Research 58:5291-‐5293

101
16. Berkley MC, Magee KR (1963) Neuropathy following exposure to a
dimethylamine salt of 2, 4-‐D. Arch Intern Med 111:351-‐352
17. Bharucha NE, Bharucha AE, Bharucha EP (1991) Prevalence of
peripheral neuropathy in the Parsi community of Bombay. Neurology
41:1315-‐1317
18. Bobak M, Kristenson M, Pikhart H, Marmot M (2004) Life span and
disability: a cross sectional comparison of Russian and Swedish
community based data. Bmj 329:767
19. Bosboom WM, van den Berg LH, Franssen H, Giesbergen PC, Flach HZ,
van Putten AM, Veldman H, Wokke JH (2001) Diagnostic value of
sural nerve demyelination in chronic inflammatory demyelinating
polyneuropathy. Brain 124:2427-‐2438
20. Brazier J, Jones N, Kind P (1993) Testing the validity of the Euroqol
and comparing it with the SF-‐36 health survey questionnaire. Quality
of life research : an international journal of quality of life aspects of
treatment, care and rehabilitation 2:169-‐180
21. Bromberg MB (2010) An approach to the evaluation of peripheral
neuropathies. Semin Neurol 30:350-‐355
22. Buck D, Jacoby A (2005) Health Outcomes and Quality of Life. In: Dyck
PJ, Thomas PK (eds) Peripheral Neuropathy. Elsevier Saunders,
Philadelphia, Pennsylvania, pp 1053-‐1062
23. Buschbacher RM (1998) Body mass index effect on common nerve
conduction study measurements. Muscle & Nerve 21:1398-‐1404
24. Chang CM, Yu CW, Fong KY, Leung SY, Tsin TW, Yu YL, Cheung TF,
Chan SY (1993) N-‐hexane neuropathy in offset printers. J Neurol
Neurosurg Psychiatry 56:538-‐542
25. Chen H, Sandler DP, Taylor JA, Shore DL, Liu E, Bloomfield CD, Bell DA
(1996) Increased risk for myelodysplastic syndromes in individuals

102
with glutathione transferase theta 1 (GSTT1) gene defect. Lancet
347:295-‐297
26. Chu CC, Huang CC, Ryu SJ, Wu TN (1998) Chronic inorganic mercury
induced peripheral neuropathy. Acta Neurol Scand 98:461-‐465
27. Cocito D, Paolasso I, Pazzaglia C, Tavella A, Poglio F, Ciaramitaro P,
Scarmozzino A, Cossa F, Bergamasco B, Padua L (2006) Pain affects
the quality of life of neuropathic patients. Neurological Sciences
27:155-‐160
28. Currie CJ, Poole CD, Woehl A, Morgan CL, Cawley S, Rousculp MD,
Covington MT, Peters JR (2007) The financial costs of healthcare
treatment for people with Type 1 or Type 2 diabetes in the UK with
particular reference to differing severity of peripheral neuropathy.
Diabet Med 24:187-‐194
29. Currie CJ, Poole CD, Woehl A, Morgan CL, Cawley S, Rousculp MD,
Covington MT, Peters JR (2006) The health-‐related utility and health-‐
related quality of life of hospital-‐treated subjects with type 1 or type
2 diabetes with particular reference to differing severity of peripheral
neuropathy. Diabetologia 49:2272-‐2280
30. de Schryver EL, van Schelven LJ, Notermans NC, de Valk HW, Oey PL
(2011) Small-‐fibre neuropathy can be detected in patients with
chronic idiopathic axonal polyneuropathy. European journal of
neurology : the official journal of the European Federation of
Neurological Societies 18:1003-‐1005
31. Decker M, Arand M, Cronin A (2009) Mammalian epoxide hydrolases
in xenobiotic metabolism and signalling. Arch Toxicol 83:297-‐318
32. Dick FD, De Palma G, Ahmadi A, Osborne A, Scott NW, Prescott GJ,
Bennett J, Semple S, Dick S, Mozzoni P, Haites N, Wettinger SB, Mutti
A, Otelea M, Seaton A, Soderkvist P, Felice A, on behalf of the

103
Geoparkinson Study G (2007) Gene-‐environment interactions in
parkinsonism and Parkinson's disease: the Geoparkinson study.
Occup Environ Med 64:673-‐680
33. Dyck PJ (1999) Cryptogenic sensory polyneuropathy.[comment].
Archives of Neurology 56:519-‐520
34. Dyck PJ, Hughes RAC, O'Brien (2005) Quantitating Overall
Neuropathic Symptoms, Impairments, and Outcoms. In: Dyck PJ,
Thomas PK (eds) Peripheral Neuropathy. Elsevier Saunders,
Philadelphia, Pennsylvania, pp 1031-‐1051
35. Dyck PJ, Klein CJ, Weigand SD (2007) Does impaired glucose
metabolism cause polyneuropathy? Review of previous studies and
design of a prospective controlled population-‐based study. Muscle
Nerve 36:536-‐541
36. Dyck PJ, Low PA, Stevens JC (1983) "Burning feet" as the only
manifestation of dominantly inherited sensory neuropathy. Mayo Clin
Proc 58:426-‐429
37. Dyck PJ, Oviatt KF, Lambert EH (1981) Intensive evaluation of
referred unclassified neuropathies yields improved diagnosis. Annals
of Neurology 10:222-‐226
38. Dyck PJ, Thomas PK (2005) Peripheral Neuropathy. Elsevier
Saunders, Philadelphia, Pennsylvania
39. Dyro FM (1978) Methyl ethyl ketone polyneuropathy in shoe factory
workers. Clin Toxicol 13:371-‐376
40. Eardley W, Toth C (2010) An open-‐label, non-‐randomized
comparison of venlafaxine and gabapentin as monotherapy or
adjuvant therapy in the management of neuropathic pain in patients
with peripheral neuropathy. J Pain Res 3:33-‐49

104
41. England JD, Asbury AK (2004) Peripheral neuropathy. Lancet
363:2151-‐2161
42. England JD, Gronseth GS, Franklin G, Carter GT, Kinsella LJ, Cohen JA,
Asbury AK, Szigeti K, Lupski JR, Latov N, Lewis RA, Low PA, Fisher
MA, Herrmann DN, Howard JF, Jr., Lauria G, Miller RG, Polydefkis M,
Sumner AJ (2009) Practice Parameter: Evaluation of distal symmetric
polyneuropathy: Role of laboratory and genetic testing (an evidence-‐
based review): Report of the American Academy of Neurology,
American Association of Neuromuscular and Electrodiagnostic
Medicine, and American Academy of Physical Medicine and
Rehabilitation. Neurology 72:185-‐192
43. England JD, Gronseth GS, Franklin G, Miller RG, Asbury AK, Carter GT,
Cohen JA, Fisher MA, Howard JF, Kinsella LJ, Latov N, Lewis RA, Low
PA, Sumner AJ (2005) Distal symmetric polyneuropathy: A definition
for clinical research: Report of the American Academy of Neurology,
the American Association of Electrodiagnostic Medicine, and the
American Academy of Physical Medicine and Rehabilitation.
Neurology 64:199-‐207
44. Erdmann P, Teunissen L, van Genderen F, Notermans N, Lindeman E,
Helders P, van Meeteren N (2007) Functioning of patients with
chronic idiopathic axonal polyneuropathy (CIAP). Journal of
Neurology 254:1204-‐1211
45. Erdmann PG, van Genderen FR, Teunissen LL, Notermans NC,
Lindeman E, van Wijck AJ, van Meeteren NL (2010) Pain in patients
with chronic idiopathic axonal polyneuropathy. Eur Neurol 64:58-‐64
46. EuroQol-‐Group (2009) EQ-‐5D User Guide. In: http://www.euroqol.org/fileadmin/user_upload/Document/User_Guide_v2_Mar
ch_2009.pdf.

105
47. Fagius J (1983) Chronic cryptogenic polyneuropathy. The search for a
cause. Acta Neurologica Scandinavica 67:173-‐180
48. Feldman RG (1999) n-‐Hexane and Methyl n-‐Butyl Keton. In:
Occupational and Environmental Neurotoxicology. Lippincott-‐Raven,
Philadelphia, pp 290-‐317
49. Fujimoto WY, Bergstrom RW, Boyko EJ, Kinyoun JL, Leonetti DL,
Newell-‐Morris LL, Robinson LR, Shuman WP, Stolov WC, Tsunehara
CH, et al. (1994) Diabetes and diabetes risk factors in second-‐ and
third-‐generation Japanese Americans in Seattle, Washington. Diabetes
Res Clin Pract 24 Suppl:S43-‐52
50. Galer BS, Jensen MP (1997) Development and preliminary validation
of a pain measure specific to neuropathic pain: the Neuropathic Pain
Scale. Neurology 48:332-‐338
51. Garte S, Gaspari L, Alexandrie AK, Ambrosone C, Autrup H, Autrup JL,
Baranova H, Bathum L, Benhamou S, Boffetta P, Bouchardy C,
Breskvar K, Brockmoller J, Cascorbi I, Clapper ML, Coutelle C, Daly A,
Dell'Omo M, Dolzan V, Dresler CM, Fryer A, Haugen A, Hein DW,
Hildesheim A, Hirvonen A, Hsieh LL, Ingelman-‐Sundberg M, Kalina I,
Kang D, Kihara M, Kiyohara C, Kremers P, Lazarus P, Le Marchand L,
Lechner MC, van Lieshout EM, London S, Manni JJ, Maugard CM,
Morita S, Nazar-‐Stewart V, Noda K, Oda Y, Parl FF, Pastorelli R,
Persson I, Peters WH, Rannug A, Rebbeck T, Risch A, Roelandt L,
Romkes M, Ryberg D, Salagovic J, Schoket B, Seidegard J, Shields PG,
Sim E, Sinnet D, Strange RC, Stucker I, Sugimura H, To-‐Figueras J,
Vineis P, Yu MC, Taioli E (2001) Metabolic gene polymorphism
frequencies in control populations. Cancer Epidemiol Biomarkers
Prev 10:1239-‐1248

106
52. George J, Twomey JA (1986) Causes of polyneuropathy in the elderly.
Age Ageing 15:247-‐249
53. Gerhardsson L, Balogh I, Hambert PA, Hjortsberg U, Karlsson JE
(2005) Vascular and nerve damage in workers exposed to vibrating
tools. The importance of objective measurements of exposure time.
Appl Ergon 36:55-‐60
54. Gherardi RK, Farcet JP, Creange A, Claudepierre P, Malapert D,
Authier FJ, Delfau-‐Larue MH (1998) Dominant T-‐cell clones of
unknown significance in patients with idiopathic sensory
neuropathies. Neurology 51:384-‐389
55. Ginsberg G, Guyton K, Johns D, Schimek J, Angle K, Sonawane B
(2010) Genetic polymorphism in metabolism and host defense
enzymes: implications for human health risk assessment. Crit Rev
Toxicol 40:575-‐619
56. Ginsberg G, Smolenski S, Hattis D, Guyton KZ, Johns DO, Sonawane B
(2009) Genetic Polymorphism in Glutathione Transferases (GST):
Population Distribution of GSTM1, T1, and P1 Conjugating Activity.
Journal of Toxicology and Environmental Health, Part B: Critical
Reviews 12:389 -‐ 439
57. Glaser J, Dessauer M, Proksch E (1985) Akute Polyneuropathie durch
Inhalation von Lösemittelgemischen in handelsüblichen
Lackentfernern und Verdünnern. Dtsch Med Wochenschr
110:1374,1377
58. Goetz CG, Bolla KI, Rogers SM (1994) Neurologic health outcomes and
Agent Orange: Institute of Medicine report. Neurology 44:801-‐809
59. Goldstein NP, Jones PH, Brown JR (1959) Peripheral neuropathy after
exposure to an ester of dichlorophenoxyacetic acid. J Am Med Assoc
171:1306-‐1309

107
60. Gooch CL, Amato AA (2010) Are anti-‐ganglioside antibodies of clinical
value in multifocal motor neuropathy? Neurology 75:1950-‐1951
61. Gooch CL, Weimer LH (2007) The electrodiagnosis of neuropathy:
basic principles and common pitfalls. Neurol Clin 25:1-‐28
62. Gorson KC, Ropper AH (1995) Idiopathic distal small fiber
neuropathy. Acta Neurol Scand 92:376-‐382
63. Goto I, Matsumura M, Inoue N, Murai Y, Shida K (1974) Toxic
polyneuropathy due to glue sniffing. J Neurol Neurosurg Psychiatry
37:848-‐853
64. Graham RC, Hughes RA (2006) Clinimetric properties of a walking
scale in peripheral neuropathy. J Neurol Neurosurg Psychiatry
77:977-‐979
65. Graham RC, Hughes RAC (2006) A modified peripheral neuropathy
scale: the Overall Neuropathy Limitations Scale. J Neurol Neurosurg
Psychiatry 77:973-‐976
66. Grahmann F, Winterholler M, Neundorfer B (1991) Cryptogenetic
polyneuropathies: an out-‐patient follow-‐up study. Acta Neurologica
Scandinavica 84:221-‐225
67. Grant IA (2005) Cryptogenic Sensory Polyneuropathy. In: Dyck PJ,
Thomas PK (eds) Peripheral Neuropathy. Elsevier Saunders,
Philadelphia, Pennsylvania, pp 2321-‐2333
68. Guengerich FP (1982) Epoxide hydrolase: properties and metabolic
roles. Reviews of Biochemical Toxicology 4:4-‐30
69. Harati Y, Bosch EP (2008) Disorders of Peripheral Nerves. In: Bradley
WG, Daroff RB, G.M. F, Jankovic J (eds) Neurology in Clinical Practice.
Butterworth Heinemann Elsevier, Philadelphia, PA, pp 2249-‐2355
70. Hartsfield JK, Jr., Sutcliffe MJ, Everett ET, Hassett C, Omiecinski CJ,
Saari JA (1998) Assignment of microsomal epoxide hydrolase

108
(EPHX1) to human chromosome 1q42.1 by in situ hybridization.
Cytogenet Cell Genet 83:44-‐45
71. Hassett C, Aicher L, Sidhu JS, Omiecinski CJ (1994) Human
microsomal epoxide hydrolase: genetic polymorphism and functional
expression in vitro of amino acid variants.[erratum appears in Hum
Mol Genet 1994 Jul;3(7):1214]. Human Molecular Genetics 3:421-‐428
72. Hayes JD, Strange RC (2000) Glutathione S-‐transferase
polymorphisms and their biological consequences. Pharmacology
61:154-‐166
73. Henningsson AJ, Malmvall BE, Ernerudh J, Matussek A, Forsberg P
(2010) Neuroborreliosis-‐-‐an epidemiological, clinical and healthcare
cost study from an endemic area in the south-‐east of Sweden. Clin
Microbiol Infect 16:1245-‐1251
74. Hoffman DL, Sadosky A, Alvir J (2009) Cross-‐national burden of
painful diabetic peripheral neuropathy in Asia, Latin America, and the
Middle East. Pain Pract 9:35-‐42
75. Hoffman-‐Snyder C, Smith BE, Ross MA, Hernandez J, Bosch EP (2006)
Value of the oral glucose tolerance test in the evaluation of chronic
idiopathic axonal polyneuropathy.[see comment]. Archives of
Neurology 63:1075-‐1079
76. Hohage H, Otte B, Westermann G, Witta J, Welling U, Zidek W,
Heidenreich S (1997) Elemental mercurial poisoning. South Med J
90:1033-‐1036
77. Holland NR, Crawford TO, Hauer P, Cornblath DR, Griffin JW,
McArthur JC (1998) Small-‐fiber sensory neuropathies: clinical course
and neuropathology of idiopathic cases. Ann Neurol 44:47-‐59

109
78. Hopf HC, Althaus HH, Vogel P (1973) An evaluation of the course of
peripheral neuropathies based on clinical and neurographical re-‐
examinations. Eur Neurol 9:90-‐104
79. Hsu LF, Lee HS, Chia SE, Lam KN (1999) Acute mercury vapour
poisoning in a shipyard worker-‐-‐a case report. Ann Acad Med
Singapore 28:294-‐298
80. Huang CY (1981) Peripheral neuropathy in the elderly: a clinical and
electrophysiologic study. J Am Geriatr Soc 29:49-‐54
81. Hughes R (2010) Investigation of peripheral neuropathy. Bmj
341:c6100
82. Hughes R, Umapathi T, Gray IA, Gregson NA, Noori M, Pannala AS,
Proteggente A, Swan AV (2004) A controlled investigation of the
cause of chronic idiopathic axonal polyneuropathy. Brain 127:1723-‐
1730
83. Hughes RAC, Umapathi T, Gray IA, Gregson NA, Noori M, Pannala AS,
Proteggente A, Swan AV (2004) A controlled investigation of the
cause of chronic idiopathic axonal polyneuropathy. Brain 127:1723-‐
1730
84. Iba MM, Fung J, Gonzalez FJ (2000) Functional Cyp2e1 is required for
substantial in vivo formation of 2,5-‐hexanedione from n-‐hexane in
the mouse. Arch Toxicol 74:582-‐586
85. IndINeP SG (2008) Burden of Neuropathic Pain in Indian Patients
Attending Urban, Specialty Clinics: Results from a Cross Sectional
Study. Pain Practice 8:362-‐378
86. Ishikawa K, Funayama T, Ohde H, Inagaki Y, Mashima Y (2005)
Genetic variants of TP53 and EPHX1 in Leber's hereditary optic
neuropathy and their relationship to age at onset. Jpn J Ophthalmol
49:121-‐126

110
87. Iyer K, Goodgold J, Eberstein A, Berg P (1976) Mercury poisoning in a
dentist. Arch Neurol 33:788-‐790
88. Jann S, Sandro Beretta Manuela Bramerio Carlo Alberto Defanti
(2001) Prospective follow-‐up study of chronic polyneuropathy of
undetermined cause. Muscle & Nerve 24:1197-‐1201
89. Jensen MP, Chodroff MJ, Dworkin RH (2007) The impact of
neuropathic pain on health-‐related quality of life: Review and
implications. Neurology 68:1178-‐1182
90. Järvholm B, Sanden A (1987) Estimating asbestos exposure: a
comparison of methods. J Occup Med 29:361-‐363
91. Kelkar P, McDermott WR, Parry GJ (2002) Sensory-‐predominant,
painful, idiopathic neuropathy: inflammatory changes in sural nerves.
Muscle Nerve 26:413-‐416
92. Kindstrand E, Nilsson BY, Hovmark A, Nennesmo I, Pirskanen R,
Solders G, Asbrink E (2000) Polyneuropathy in late Lyme borreliosis -‐
a clinical, neurophysiological and morphological description. Acta
Neurol Scand 101:47-‐52
93. Knave B, Olson BA, Elofsson S, Gamberale F, Isaksson A, Mindus P,
Persson HE, Struwe G, Wennberg A, Westerholm P (1978) Long-‐term
exposure to jet fuel. II. A cross-‐sectional epidemiologic investigation
on occupationally exposed industrial workers with special reference
to the nervous system. Scand J Work Environ Health 4:19-‐45
94. Knave B, Persson HE, Goldberg JM, Westerholm P (1976) Long-‐term
exposure to jet fuel: an investigation on occupationally exposed
workers with special reference to the nervous system. Scand J Work
Environ Health 2:152-‐164
95. König F, Neundörfer B, Kömpf D (1984) Polyneuropatien im höheren
Lebensalter. Dtsch Med Wochenschr 109:735-‐737

111
96. Lancaster JM, Brownlee HA, Bell DA, Futreal PA, Marks JR, Berchuck
A, Wiseman RW, Taylor JA (1996) Microsomal epoxide hydrolase
polymorphism as a risk factor for ovarian cancer. Molecular
Carcinogenesis 17:160-‐162
97. Lapadula DM, Suwita E, Abou-‐Donia MB (1988) Evidence for multiple
mechanisms responsible for 2,5-‐hexanedione-‐induced neuropathy.
Brain Research 458:123-‐131
98. Lauria G, Pareyson D, Sghirlanzoni A (2000) Chronic cryptogenic
sensory polyneuropathy.[comment]. Archives of Neurology 57:759-‐
760
99. Lecomte T, Landi B, Beaune P, Laurent-‐Puig P, Loriot M-‐A (2006)
Glutathione S-‐Transferase P1 Polymorphism (Ile105Val) Predicts
Cumulative Neuropathy in Patients Receiving Oxaliplatin-‐Based
Chemotherapy. Clin Cancer Res 12:3050-‐3056
100. Liedberg GM, Vrethem M (2009) Polyneuropathy, with and without
neurogenic pain, and its impact on daily life activities -‐ A descriptive
study. Disabil Rehabil 31:1-‐7
101. Lindros KO (1997) Zonation of cytochrome P450 expression, drug
metabolism and toxicity in liver. General Pharmacology 28:191-‐196
102. London SJ, Smart J, Daly AK (2000) Lung cancer risk in relation to
genetic polymorphisms of microsomal epoxide hydrolase among
African-‐Americans and Caucasians in Los Angeles County. Lung
Cancer 28:147-‐155
103. MacDonald BK, Cockerell OC, Sander JW, Shorvon SD (2000) The
incidence and lifetime prevalence of neurological disorders in a
prospective community-‐based study in the UK.[see comment]. Brain
123:665-‐676

112
104. Magnussen Z, Fossan GO (1983) [Neurasthenia and polyneuropathy.
The dangers of chronic toluene poisoning]. Tidsskr Nor Laegeforen
103:2039-‐2041
105. Mannervik B (1985) The isoenzymes of glutathione transferase. Adv
Enzymol Relat Areas Mol Biol 57:357-‐417
106. Matthews WB (1952) Cryptogenic polyneuritis. Proc R Soc Med
45:667-‐669
107. McLeod JG, Tuck RR, Pollard JD, Cameron J, Walsh JC (1984) Chronic
polyneuropathy of undetermined cause. Journal of Neurology,
Neurosurgery & Psychiatry 47:530-‐535
108. Merkies IS, Schmitz PI, van der Meche FG, Samijn JP, van Doorn PA,
Group INCaT (2002) Quality of life complements traditional outcome
measures in immune-‐mediated polyneuropathies. Neurology 59:84-‐
91
109. Merkies IS, van Nes SI, Hanna K, Hughes RA, Deng C (2010)
Confirming the efficacy of intravenous immunoglobulin in CIDP
through minimum clinically important differences: shifting from
statistical significance to clinical relevance. J Neurol Neurosurg
Psychiatry 81:1194-‐1199
110. Milby TH, Baselt RC (1999) Hydrogen sulfide poisoning: clarification
of some controversial issues. Am J Ind Med 35:192-‐195
111. Mitsumoto H, Wilbourn AJ (1994) Causes and diagnosis of sensory
neuropathies: a review. J Clin Neurophysiol 11:553-‐567
112. Moon DE, Lee DI, Lee SC, Song SO, Yoon DM, Yoon MH, Kim HK, Lee
YW, Kim C, Lee PB (2010) Efficacy and tolerability of pregabalin using
a flexible, optimized dose schedule in korean patients with peripheral
neuropathic pain: a 10-‐week, randomized, double-‐blind, placebo-‐
controlled, multicenter study. Clin Ther 32:2370-‐2385

113
113. Mygland A (2007) Approach to the patient with chronic
polyneuropathy. Acta Neurol Scand 115 (Suppl. 187):15-‐21
114. Mygland A (2007) Kronisk polynevropati -‐ utredning og diagnostikk
[Chronic polyneuropathy-‐-‐evaluation and diagnosis]. Tidsskr Nor
Laegeforen 127:291-‐295
115. Mygland A, Monstad P (2001) Chronic polyneuropathies in Vest-‐
Agder, Norway. European Journal of Neurology 8:157-‐165
116. Navarro A, Saldana MT, Perez C, Torrades S, Rejas J (2010) Patient-‐
reported outcomes in subjects with neuropathic pain receiving
pregabalin: evidence from medical practice in primary care settings.
Pain Med 11:719-‐731
117. Nebuchennykh M, Loseth S, Jorde R, Mellgren SI (2008) Idiopathic
polyneuropathy and impaired glucose metabolism in a Norwegian
patient series. Eur J Neurol 15:810-‐816
118. Nilsson E, Wenemark M, Bendtsen P, Kristenson M (2007)
Respondent satisfaction regarding SF-‐36 and EQ-‐5D, and patients'
perspectives concerning health outcome assessment within routine
health care. Quality of life research : an international journal of
quality of life aspects of treatment, care and rehabilitation 16:1647-‐
1654
119. Notermans NC, Wokke JH, Franssen H, van der Graaf Y, Vermeulen M,
van den Berg LH, Bar PR, Jennekens FG (1993) Chronic idiopathic
polyneuropathy presenting in middle or old age: a clinical and
electrophysiological study of 75 patients. Journal of Neurology,
Neurosurgery & Psychiatry 56:1066-‐1071
120. Notermans NC, Wokke JH, van den Berg LH, van der Graaf Y, Franssen
H, Teunissen LL, Lokhorst HM (1996) Chronic idiopathic axonal

114
polyneuropathy. Comparison of patients with and without
monoclonal gammopathy. Brain 119 ( Pt 2):421-‐427
121. Notermans NC, Wokke JH, van der Graaf Y, Franssen H, van Dijk GW,
Jennekens FG (1994) Chronic idiopathic axonal polyneuropathy: a
five year follow up. Journal of Neurology, Neurosurgery & Psychiatry
57:1525-‐1527
122. Novella SP, Inzucchi SE, Goldstein JM (2001) The frequency of
undiagnosed diabetes and impaired glucose tolerance in patients
with idiopathic sensory neuropathy. Muscle Nerve 24:1229-‐1231
123. O'Reilly JF (1984) Prolonged coma and delayed peripheral
neuropathy after ingestion of phenoxyacetic acid weedkillers.
Postgrad Med J 60:76-‐77
124. Oesch F (1973) Mammalian epoxide hydrases: inducible enzymes
catalysing the inactivation of carcinogenic and cytotoxic metabolites
derived from aromatic and olefinic compounds. Xenobiotica 3:305-‐
340
125. Omiecinski CJ, Aicher L, Holubkov R, Checkoway H (1993) Human
peripheral lymphocytes as indicators of microsomal epoxide
hydrolase activity in liver and lung. Pharmacogenetics 3:150-‐158
126. Ovayolu N, Akarsu E, Madenci E, Torun S, Ucan O, Yilmaz M (2008)
Clinical characteristics of patients with diabetic polyneuropathy: the
role of clinical and electromyographic evaluation and the effect of the
various types on the quality of life. Int J Clin Pract 62:1019-‐1025
127. Parkinson A (2001) Biotranformation of xenobiotics. In: Klaasen CD
(ed) Casarett and Doull's toxicology. The basic science of poisons.
McGraw-‐Hill, pp 133-‐224

115
128. Pastore C, Izura V, Marhuenda D, Prieto MJ, Roel J, Cardona A (2002)
Partial conduction blocks in N-‐hexane neuropathy. Muscle Nerve
26:132-‐135
129. Pavlakis PP, Alexopoulos H, Kosmidis ML, Stamboulis E, Routsias JG,
Tzartos SJ, Tzioufas AG, Moutsopoulos HM, Dalakas MC (2011)
Peripheral neuropathies in Sjogren syndrome: a new reappraisal.
Journal of neurology, neurosurgery, and psychiatry 82:798-‐802
130. Pemble S, Schroeder KR, Spencer SR, Meyer DJ, Hallier E, Bolt HM,
Ketterer B, Taylor JB (1994) Human glutathione S-‐transferase theta
(GSTT1): cDNA cloning and the characterization of a genetic
polymorphism. Biochem J 300 ( Pt 1):271-‐276
131. Perbellini L, Brugnone F, Pavan I (1980) Identification of the
metabolites of n-‐hexane, cyclohexane, and their isomers in men's
urine. Toxicology and Applied Pharmacology 53:220-‐229
132. Perbellini L, Pezzoli G, Brugnone F, Canesi M (1993) Biochemical and
physiological aspects of 2,5-‐hexandione: endogenous or exogenous
product? Int Arch Occup Environ Health 65:49-‐52
133. Periquet MI, Novak V, Collins MP, Nagaraja HN, Erdem S, Nash SM,
Freimer ML, Sahenk Z, Kissel JT, Mendell JR (1999) Painful sensory
neuropathy: prospective evaluation using skin biopsy. Neurology
53:1641-‐1647
134. Pershagen G, Axelson O (1982) A validation of questionnaire
information on occupational exposure and smoking. Scand J Work
Environ Health 8:24-‐28
135. Pirmohamed M, Kitteringham NR, Quest LJ, Allott RL, Green VJ,
Gilmore IT, Park BK (1995) Genetic polymorphism of cytochrome
P4502E1 and risk of alcoholic liver disease in Caucasians.
Pharmacogenetics 5:351-‐357

116
136. Prineas J (1970) Polyneuropathies of undetermined cause. Acta
Neurologica Scandinavica Supplementum 44:3-‐72
137. Proudfoot AT (2005) Poisoning due to pyrethrins. Toxicol Rev
24:107-‐113
138. Rajabally YA (2010) Neuropathy and impaired glucose tolerance: an
updated review of the evidence. Acta Neurol Scand
139. Rajabally YA, Shah RS (2011) Dyslipidaemia in chronic acquired distal
axonal polyneuropathy. J Neurol
140. Rautenstrauss B (2011) Targeting inherited peripheral neuropathies
in the postgenomic era. Neurology 77:518-‐519
141. Rebbeck TR (1997) Molecular epidemiology of the human glutathione
S-‐transferase genotypes GSTM1 and GSTT1 in cancer susceptibility.
Cancer Epidemiol Biomarkers Prev 6:733-‐743
142. Rezania K, Soliven B, Rezai KA, Roos RP (2011) Impaired glucose
tolerance and metabolic syndrome in idiopathic polyneuropathy: The
role of pain and depression. Med Hypotheses
143. Rivner MH, Swift TR, Malik K (2001) Influence of age and height on
nerve conduction. Muscle & Nerve 24:1134-‐1141
144. Robinson LR (2000) Role of neurophysiologic evaluation in diagnosis.
Journal of the American Academy of Orthopaedic Surgeons 8:190-‐199
145. Rose FC (1960) Peripheral neuropathy. Proc R Soc Med 53:51-‐53
146. Rosenberg NR, Vermeulen M (2004) Chronic idiopathic axonal
polyneuropathy revisited. J Neurol 251:1128-‐1132
147. Roth SH, Skrajny B, Bennington R, Brookes J (1997) Neurotoxicity of
hydrogen sulfide may result from inhibition of respiratory enzymes.
Proc West Pharmacol Soc 40:41-‐43

117
148. Ruhland JL, Shields RK (1997) The effects of a home exercise program
on impairment and health-‐related quality of life in persons with
chronic peripheral neuropathies. Phys Ther 77:1026-‐1039
149. Rybicki BA, Johnson CC, Peterson EL, Kortsha GX, Gorell JM (1997)
Comparability of different methods of retrospective exposure
assessment of metals in manufacturing industries. Am J Ind Med
31:36-‐43
150. Saperstein DS (2008) Selecting Diagnostic Tests In Peripheral
Neuropathy Patients. Educational Program Syllabus 2FC.003. In:
American Academy Of Neurology 60th Annual Meeting. Chicago, pp
36-‐74
151. Shapiro IM, Cornblath DR, Sumner AJ, Uzzell B, Spitz LK, Ship, II, Bloch
P (1982) Neurophysiological and neuropsychological function in
mercury-‐exposed dentists. Lancet 1:1147-‐1150
152. Singleton JR, Smith AG, Bromberg MB (2001) Increased prevalence of
impaired glucose tolerance in patients with painful sensory
neuropathy. Diabetes Care 24:1448-‐1453
153. Skre H (1972) Neurological signs in a normal population. Acta
Neurologica Scandinavica 48:575-‐606
154. Smith AG, Rose K, Singleton JR (2008) Idiopathic neuropathy patients
are at high risk for metabolic syndrome. J Neurol Sci 273:25-‐28
155. Smith CAD, Harrison DJ (1997) Association between polymorphism in
gene for microsomal epoxide hydrolase and susceptibility to
emphysema. The Lancet 350:630-‐633
156. Soderlund DM, Clark JM, Sheets LP, Mullin LS, Piccirillo VJ, Sargent D,
Stevens JT, Weiner ML (2002) Mechanisms of pyrethroid
neurotoxicity: implications for cumulative risk assessment.
Toxicology 171:3-‐59

118
157. Solders G, Andersson T, Borin Y, Brandt L, Persson A (1993)
Electroneurography index: a standardized neurophysiological
method to assess peripheral nerve function in patients with
polyneuropathy. Muscle Nerve 16:941-‐946
158. Spencer PS, Schaumburg HH (2000) Experimental and Clinical
Neurotoxicology. Oxford University Press
159. Spencer PS, Schaumburg HH (1975) Experimental neuropathy
produced by 2,5-‐hexanedione-‐-‐a major metabolite of the neurotoxic
industrial solvent methyl n-‐butyl ketone. Journal of neurology,
neurosurgery, and psychiatry 38:771-‐775
160. Stewart JD, Low PA, Fealey RD (1992) Distal small fiber neuropathy:
results of tests of sweating and autonomic cardiovascular reflexes.
Muscle Nerve 15:661-‐665
161. Stogbauer F, Young P, Kuhlenbaumer G, Kiefer R, Timmerman V,
Ringelstein EB, Wang JF, Schroder JM, Van Broeckhoven C, Weis J
(1999) Autosomal dominant burning feet syndrome. J Neurol
Neurosurg Psychiatry 67:78-‐81
162. Streicher HZ, Gabow PA, Moss AH, Kono D, Kaehny WD (1981)
Syndromes of toluene sniffing in adults. Ann Intern Med 94:758-‐762
163. Sullivan M, Karlsson J (1998) The Swedish SF-‐36 Health Survey III.
Evaluation of criterion-‐based validity: results from normative
population. Journal of Clinical Epidemiology 51:1105-‐1113
164. Sullivan M, Karlsson J, Ware JE, Jr. (1994) Hälsoenkät: Svensk manual
och tolkningsguide (Swedish Manual and Interpretation Guide).
Sahlgrenska University Hospital, Gothenburg
165. Sumner CJ, Sheth S, Griffin JW, Cornblath DR, Polydefkis M (2003) The
spectrum of neuropathy in diabetes and impaired glucose tolerance.
Neurology 60:108-‐111

119
166. Söderkvist P, Ahmadi A, Akerback A, Axelson O, Flodin U (1996)
Glutathione S-‐transferase M1 null genotype as a risk modifier for
solvent-‐induced chronic toxic encephalopathy. Scandinavian Journal
of Work, Environment & Health 22:360-‐363
167. Tesfaye S, Chaturvedi N, Eaton SE, Ward JD, Manes C, Ionescu-‐
Tirgoviste C, Witte DR, Fuller JH (2005) Vascular risk factors and
diabetic neuropathy. N Engl J Med 352:341-‐350
168. Teunissen LL, Eurelings M, Notermans NC, Hop JW, van Gijn J (2000)
Quality of life in patients with axonal polyneuropathy. Journal of
Neurology 247:195-‐199
169. Teunissen LL, Franssen H, Wokke JH, van der Graaf Y, Linssen WH,
Banga JD, Laman DM, Notermans NC (2002) Is cardiovascular disease
a risk factor in the development of axonal polyneuropathy? Journal of
Neurology, Neurosurgery & Psychiatry 72:590-‐595
170. Teunissen LL, Notermans NC, Franssen H, van der Graaf Y, Oey PL,
Linssen WH, van Engelen BG, Ippel PF, van Dijk GW, Gabreels-‐Festen
AA, Wokke JH (1997) Differences between hereditary motor and
sensory neuropathy type 2 and chronic idiopathic axonal neuropathy.
A clinical and electrophysiological study. Brain 120 ( Pt 6):955-‐962
171. Teunissen LL, Notermans NC, Jansen GH, Banga JD, Veldman H,
Wokke JH (2000) Thickness of endoneurial vessel basal lamina area
in chronic idiopathic axonal polyneuropathy. Acta Neuropathologica
100:445-‐450
172. Thunander M, Petersson C, Jonzon K, Fornander J, Ossiansson B, Torn
C, Edvardsson S, Landin-‐Olsson M (2008) Incidence of type 1 and type
2 diabetes in adults and children in Kronoberg, Sweden. Diabetes Res
Clin Pract 82:247-‐255

120
173. Timbrell J (2000) Factors affecting toxic responses: Metabolism. In:
Francies T (ed) Principles of biochemical toxicology., London, pp 65-‐
110
174. Todd RL (1962) A case of 2-‐4D intoxication. J Iowa State Med Soc
52:663-‐664
175. Tolot F, Lenglet JP, Prost G, Bertholon J (1969) [Polyneuritis due to
lindane]. J Med Lyon 50:747-‐754
176. Triebig G, Schaller KH (1982) Neurotoxic effects in mercury-‐exposed
workers. Neurobehav Toxicol Teratol 4:717-‐720
177. Verghese J, Bieri PL, Gellido C, Schaumburg HH, Herskovitz S (2001)
Peripheral neuropathy in young-‐old and old-‐old patients. Muscle
Nerve 24:1476-‐1481
178. Verity MA, Sarafian TA (2000) Mercury and mercury compounds. In:
Spencer PS, Schaumburg HH (eds) Experimental and Clinical
Neurotoxology. Oxford University Press, New York, pp 763-‐770
179. Victor M, Ropper AH (2000) Diseases of the Peripheral Nerves. In:
Victor M, Ropper AH (eds) Adam's and Victor's Principles of
Neurology. McGraw-‐Hill, New York, pp 1370-‐1378
180. Vrancken AF, Franssen H, Wokke JH, Teunissen LL, Notermans NC
(2002) Chronic idiopathic axonal polyneuropathy and successful
aging of the peripheral nervous system in elderly people. Archives of
Neurology 59:533-‐540
181. Vrancken AF, Kalmijn S, Buskens E, Franssen H, Vermeulen M, Wokke
JH, Notermans NC (2006) Feasibility and cost efficiency of a
diagnostic guideline for chronic polyneuropathy: a prospective
implementation study.[erratum appears in J Neurol Neurosurg
Psychiatry. 2006 May;77(5):710]. Journal of Neurology,
Neurosurgery & Psychiatry 77:397-‐401

121
182. Vrancken AF, Notermans NC, Jansen GH, Wokke JH, Said G (2004)
Progressive idiopathic axonal neuropathy-‐-‐a comparative clinical and
histopathological study with vasculitic neuropathy. J Neurol 251:269-‐
278
183. Vrancken AF, van Schaik IN, Hughes RA, Notermans NC (2004) Drug
therapy for chronic idiopathic axonal polyneuropathy. Cochrane
Database of Systematic Reviews:CD003456
184. Waite LM, Broe GA, Creasey H, Grayson D, Edelbrock D, O'Toole B
(1996) Neurological signs, aging, and the neurodegenerative
syndromes. Arch Neurol 53:498-‐502
185. Ware JE, Jr., Sherbourne CD (1992) The MOS 36-‐item short-‐form
health survey (SF-‐36). I. Conceptual framework and item selection.
Medical Care 30:473-‐483
186. Watanabe J, Hayashi S, Kawajiri K (1994) Different regulation and
expression of the human CYP2E1 gene due to the RsaI polymorphism
in the 5'-‐flanking region. Journal of Biochemistry 116:321-‐326
187. WHO (1980) World Health Organization (WHO): International
Classification of Impairments, Disabilities and Handicaps. A Manual of
Classification relating to the Consequences of Diseases., Geneva,
Switzerland
188. Wilbourn AJ (1993) Diabetic neuropathies. In: Brown WF, Bolton CG
(eds) Electomyography. Butterworth-‐Heineman, Boston, pp 477-‐515
189. Wolfe GI (2002) Chronic idiopathic axonal polyneuropathy and
successful aging of the peripheral nervous system. Archives of
Neurology 59:520-‐522
190. Wolfe GI, Baker NS, Amato AA, Jackson CE, Nations SP, Saperstein DS,
Cha CH, Katz JS, Bryan WW, Barohn RJ (1999) Chronic cryptogenic

122
sensory polyneuropathy: clinical and laboratory characteristics.[see
comment]. Archives of Neurology 56:540-‐547
191. Wolfe GI, Barohn RJ (1998) Cryptogenic sensory and sensorimotor
polyneuropathies. Semin Neurol 18:105-‐111
192. Wormhoudt LW, Commandeur JNM, Vermeulen NPE (1999) Genetic
Polymorphisms of Human N-‐Acetyltransferase, Cytochrome P450,
Glutathione-‐S-‐Transferase, and Epoxide Hydrolase Enzymes:
Relevance to Xenobiotic Metabolism and Toxicity. Critical Reviews in
Toxicology 29:59 -‐ 124
193. Xu S, Wang Y, Roe B, Pearson WR (1998) Characterization of the
human class Mu glutathione S-‐transferase gene cluster and the
GSTM1 deletion. J Biol Chem 273:3517-‐3527
194. Zhang Y, Liu Q, Duan H, Cheng J, Jiang S, Huang X, Leng S, He F, Zheng
Y (2006) Association between metabolic gene polymorphisms and
susceptibility to peripheral nerve damage in workers exposed to n-‐
hexane: a preliminary study. Biomarkers 11:61-‐69





























