Embed Size (px)
Citation preview

Crystallographic Analysis and Structure-GuidedEngineering of NADPH-Dependent Ralstonia sp.Alcohol Dehydrogenase Toward NADHCosubstrate Specificity
Alexandra Lerchner, Alexander Jarasch, Winfried Meining, Andre Schiefner, Arne Skerra
Munich Center for Integrated Protein Science, CIPS-M, and Lehrstuhl f€ur Biologische
Chemie, Technische Universitat M€unchen, 85350, Freising-Weihenstephan, Germany;
telephone: þ49 8161 71 4351; fax: þ49 8161 71 4352; e-mail: [email protected]
ABSTRACT: The NADPþ-dependent alcohol dehydrogenasefrom Ralstonia sp. (RasADH) belongs to the proteinsuperfamily of short-chain dehydrogenases/reductases(SDRs). As an enzyme that accepts different types ofsubstrates—including bulky–bulky as well as small–bulkysecondary alcohols or ketones—with high stereoselectivity, itoffers potential as a biocatalyst for industrial biotechnology.To understand substrate and cosubstrate specificities ofRasADH we determined the crystal structure of the apo-enzyme as well as its NADPþ-bound state with resolutionsdown to 2.8 8A. RasADH displays a homotetrameric quater-nary structure that can be described as a dimer ofhomodimers while in each subunit a seven-stranded parallelb-sheet, flanked by three a-helices on each side, forms aRossmann fold-type dinucleotide binding domain. Dockingof the well-known substrate (S)-1-phenylethanol clearlyrevealed the structural determinants of stereospecificity.To favor practical RasADH application in the context ofestablished cofactor recycling systems, for example, thoseinvolving an NADH-dependent amino acid dehydrogenase,we attempted to rationally change its cosubstrate specificityfrom NADPþ to NADþ utilizing the structural informationthat NADPþ specificity is largely governed by the residuesAsn15, Gly37, Arg38, and Arg39. Furthermore, an extensivesequence alignment with homologous dehydrogenases thathave different cosubstrate specificities revealed a modifiedgeneral SDR motif ASNG (instead of NNAG) at positions86–89 of RasADH. Consequently, we constructed mutantenzymes with one (G37D), four (N15G/G37D/R38V/R39S),and six (N15G/G37D/R38V/R39S/A86N/S88A) amino acidexchanges. RasADH (N15G/G37D/R38V/R39S) was betterable to accept NADþ while showing much reduced catalyticefficiency with NADPþ, leading to a change in NADH/NADPH specificity by a factor of �3.6 million.
Biotechnol. Bioeng. 2013;110: 2803–2814.
� 2013 Wiley Periodicals, Inc.
KEYWORDS: enzymology; protein crystallography; proteinengineering; RasADH; Rossmann fold
Introduction
Dehydrogenases are currently the most promising class ofenzymes for the synthesis of chiral compounds and polymersas well as degradation of pollutants (Berenguer-Murcia andFernandez-Lafuente, 2010; Liu and Wang, 2007). Theseenzymes require a soluble redox cofactor to perform theirfunction, most commonly nicotinamide adenine dinucleo-tide (NADþ). Since cofactors are generally expensive,industrial implementation necessitates efficient recycling(Bond and Schüttelkopf, 2009). Several biocatalytic processesinvolving regeneration systems have been established in thebiotechnological industry, for example, comprising lactatedehydrogenase/NADH/formate dehydrogenase (FDH), alcoholdehydrogenase (ADH)/NADH/FDH, or ADH/NADH/glucosedehydrogenase (Liese et al., 2006), which all are linked toNADH.A dehydrogenase with promising catalytic potential is
the NADPþ-dependent ADH from Ralstonia sp. DSM 6428(RasADH), which accepts a variety of useful oxidizedsubstrates, such as bulky–bulky a-keto ester derivatives orsmall–bulky ketones as well as bulky–bulky alkyl/arylketones, and often shows very high stereoselectivity(Ferreira-Silva et al., 2010; Lavandera et al., 2008). Becauseof its broad substrate spectrum this ADH appears of generalinterest in asymmetric synthesis (Musa and Phillips, 2011).In a recent study, RasADH has shown high catalytic activitiesand stereoselectivities in the preparation of a wide rangeof 1,2-diols starting from chiral 2-hydroxy ketones (e.g.,(R)-2-hydroxy-1-phenylpropane-1-one and (R)-1-hydroxy-1-phenylpropan-2-one; Kulig et al., 2012). Also, RasADH-catalyzed reduction of a-substituted b-keto esters has been
Correspondence to: Arne Skerra
Contract grant sponsor: Bundesministerium f€ur Bildung und Forschung, Germany
Contract grant number: 0316044B
Received 14 February 2013; Revision received 21 April 2013; Accepted 29 April 2013
Accepted manuscript online 18 May 2013;
Article first published online 1 July 2013 in Wiley Online Library
(http://onlinelibrary.wiley.com/doi/10.1002/bit.24956/abstract).
DOI 10.1002/bit.24956
ARTICLE
� 2013 Wiley Periodicals, Inc. Biotechnology and Bioengineering, Vol. 110, No. 11, November, 2013 2803

successfully accomplished, yielding the correspondingb-hydroxy esters, in many cases with excellent enantio-and diastereoselectivities (Cuetos et al., 2012). Furtherintegration of RasADH into industrial processes shouldinclude coupling to an established NADH regenerationsystem but would require changing of its cofactor specificityfrom NADPþ to NADþ.
RasADH belongs to the superfamily of short-chaindehydrogenases/reductases (SDRs). These enzymes spanseveral E.C. classes, from oxidoreductases, which form themajority, over lyases to isomerases (Jörnvall et al., 1995b).The pair-wise sequence identity between different membersof the SDR family is usually just 15–30%, but all availablestructures exhibit a highly similar a/b supersecondarytopology including a Rossmann fold, which comprises acentral b-sheet flanked by a-helices (Oppermann et al., 2003;Rossmann et al., 1974). Most SDR enzymes have a corestructure of about 250–350 residues wherein the conservedsequence covers a variable N-terminal TGXXXGXGmotif, aspart of the nucleotide-binding region, as well as the active sitewith a tetrad of the catalytically important Asn, Ser, Tyr, andLys residues (Kavanagh et al., 2008) (Fig. 1). Additionalcommon sequence features include an NNAG motif, asingular Asp residue at position 60 in RasADH, and an IRVNsequence preceding a PG motif that is followed by aconserved Thr residue (Filling et al., 2001, 2002; Jörnvallet al., 1995b; Oppermann et al., 1997).
Enzymes of the SDR family are either NADþ- or NADPþ-specific or can utilize both cosubstrates (Cho et al., 2003).NADPþ structurally differs from NADþ merely by thepresence of an additional phosphate group esterified with the20-hydroxyl group of its AMP moiety. While NADþ is usedalmost exclusively in oxidative catabolic reactions yieldingATP, NADPH is confined, with few exceptions, to theanabolic pathways of reductive biosynthesis. Crystallographicstudies of numerous NADþ- and NADPþ-dependentenzymes have revealed a common bab fold in theirnucleotide-binding domains wherein the first three b-strandsand two a-helices comprise the AMP-binding region(Rossmann et al., 1974). Interestingly, a significant differencebetween NADþ- and NADPþ-dependent enzymes wasobserved right in this region (Scrutton et al., 1990):NADþ-binding enzymes specifically carry a negativelycharged Asp residue at the C-terminus of the secondb-strand. This residue forms hydrogen bonds with boththe 20-hydroxyl and 30-hydroxyl groups of the adenosineribose of NADþ (Wierenga et al., 1985). In most of theNADPþ-specific binding domains (Cho et al., 2003; Scruttonet al., 1990; Tanaka et al., 1996), this residue is missing,whereas a positively charged Arg residue is found in thefollowing position (cf. Fig. 1) which seems to form anelectrostatically favorable interaction with the 20-phosphategroup.
In the past years, there were many reports of enzymeredesign for altered cofactor specificity using a combinationof structural analysis with site-directed and saturationmutagenesis as well as computational modeling. Summaries
of the most successful mutations for changing cofactorspecificity from NAD(H) to NADP(H) and vice versa havebeen published (Ehrensberger et al., 2006; Khouryet al., 2009). The alteration of cosubstrate specificity fromNADPþ to NADþ, which would be of interest for RasADH,has been described so far for isocitrate dehydrogenase fromEscherichia coli (Chen et al., 1995) as well as from Thermusthermophilus (Yaoi et al., 1996), for aldehyde dehydrogenasefrom Vibrio harveyi (Zhang et al., 1999) and for ferredoxin-NADPþ reductase from Anabaena sp. (Medina et al., 2001).
To alter the NADPþ specificity of RasADH on a rationalbasis we determined the crystal structures of the apo-enzymeas well as of its complex with the natural cosubstrate. Byintroducing different sets of mutations, we succeeded indeveloping an engineered enzyme that carries four aminoacid exchanges and is able to utilize NADþ with tenfoldimproved efficiency.
Materials and Methods
Subcloning, Expression and Purification of RasADH
The coding sequence for RasADH (starting with Tyr2according to the UniProt data bank accession numberC0IR58) was amplified from the plasmid pEam-RasADH(Lavandera et al., 2008) via PCR using primers 50-GCC TATCGA CTA TTA AAC AAA ACA GCC-30 and 50-ACT GATGGT ACC TTA GAC CTG GGT CAA TCC-30. The PCRproduct was digested on one side with KpnI (Fermentas, St.Leon-Rot, Germany) and ligated with the expression vectorpASK-IBA35plus, a derivative of pASK75 (Skerra, 1994),which had been cut with KpnI and EheI (Fermentas).The resulting plasmid pASK-IBA35plus-RasADH, whichencodes the enzyme with an N-terminal His6-tag(MASRGSHHHHHHGA), was verified by restriction digestas well as double-stranded dideoxy-sequencing (ABI PRISM310 Genetic Analyzer; Applied Biosystems, Darmstadt,Germany).
Recombinant RasADH was produced in the E. coli strainBL21 (Studier et al., 2009) as a soluble protein. Shake flaskcultures were grown in 2 L LB medium (Sambrooket al., 2001) supplemented with 100mg/L ampicillin at30�C. Gene expression was induced at a cell density ofOD550¼ 0.5 by adding 0.2mg/L anhydrotetracycline(Skerra, 1994). After further shaking for 3 h the cells wereharvested by centrifugation, resuspended in 40mM HEPES/NaOH pH 7.5 and homogenized in a French pressure cell(SLM Aminco, Urbana, IL). Cell debris were removed bycentrifugation and, after supplementation with 0.5M NaCl,the cleared supernatant was applied onto an IDA-Sepharosecolumn (Chelating Sepharose Fast Flow; GE Healthcare,Munich, Germany) charged with ZnSO4 for immobilizedmetal affinity chromatography (IMAC). After washing, theHis6-tagged protein was eluted with a linear imidazoleconcentration gradient from 0 to 500mM in 40mMHEPES/NaOH, 0.5M NaCl, pH 7.5. Elution fractions wereconcentrated by ultrafiltration (30 kDa cut-off Amicon
2804 Biotechnology and Bioengineering, Vol. 110, No. 11, November, 2013
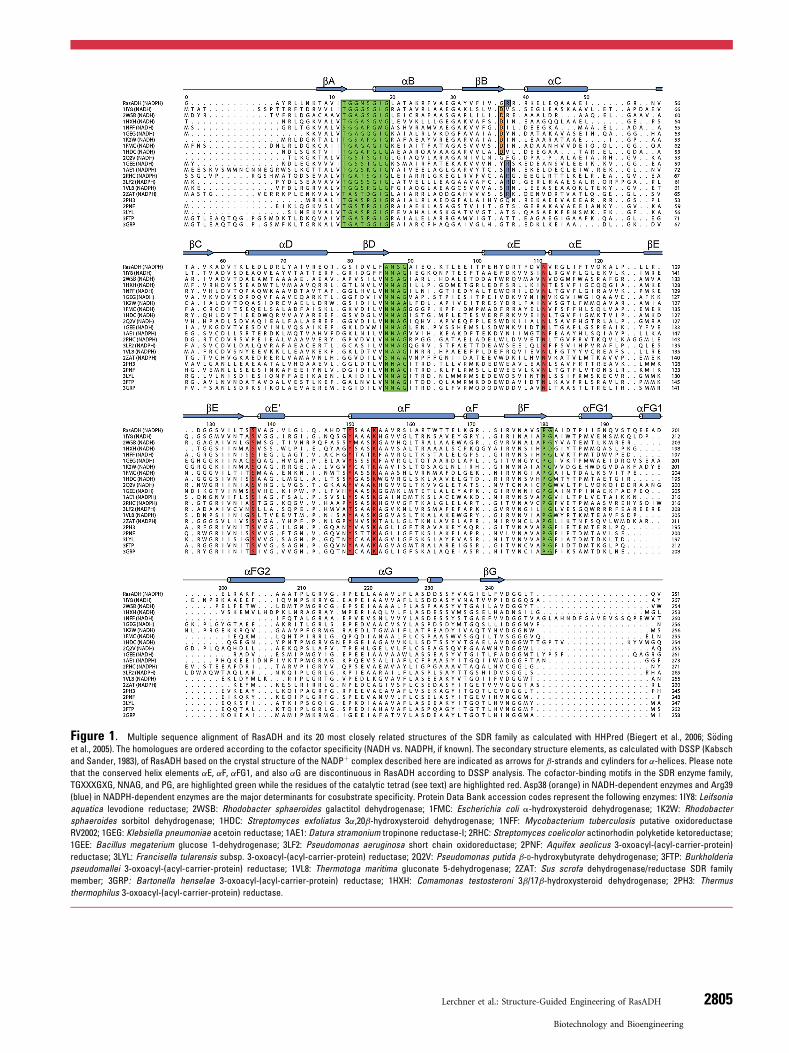
Figure 1. Multiple sequence alignment of RasADH and its 20 most closely related structures of the SDR family as calculated with HHPred (Biegert et al., 2006; S€oding
et al., 2005). The homologues are ordered according to the cofactor specificity (NADH vs. NADPH, if known). The secondary structure elements, as calculated with DSSP (Kabsch
and Sander, 1983), of RasADH based on the crystal structure of the NADPþ complex described here are indicated as arrows for b-strands and cylinders for a-helices. Please note
that the conserved helix elements aE, aF, aFG1, and also aG are discontinuous in RasADH according to DSSP analysis. The cofactor-binding motifs in the SDR enzyme family,
TGXXXGXG, NNAG, and PG, are highlighted green while the residues of the catalytic tetrad (see text) are highlighted red. Asp38 (orange) in NADH-dependent enzymes and Arg39
(blue) in NADPH-dependent enzymes are the major determinants for cosubstrate specificity. Protein Data Bank accession codes represent the following enzymes: 1IY8: Leifsonia
aquatica levodione reductase; 2WSB: Rhodobacter sphaeroides galactitol dehydrogenase; 1FMC: Escherichia coli a-hydroxysteroid dehydrogenase; 1K2W: Rhodobacter
sphaeroides sorbitol dehydrogenase; 1HDC: Streptomyces exfoliatus 3a,20b-hydroxysteroid dehydrogenase; 1NFF: Mycobacterium tuberculosis putative oxidoreductase
RV2002; 1GEG: Klebsiella pneumoniae acetoin reductase; 1AE1: Datura stramonium tropinone reductase-I; 2RHC: Streptomyces coelicolor actinorhodin polyketide ketoreductase;
1GEE: Bacillus megaterium glucose 1-dehydrogenase; 3LF2: Pseudomonas aeruginosa short chain oxidoreductase; 2PNF: Aquifex aeolicus 3-oxoacyl-(acyl-carrier-protein)
reductase; 3LYL: Francisella tularensis subsp. 3-oxoacyl-(acyl-carrier-protein) reductase; 2Q2V: Pseudomonas putida b-D-hydroxybutyrate dehydrogenase; 3FTP: Burkholderia
pseudomallei 3-oxoacyl-(acyl-carrier-protein) reductase; 1VL8: Thermotoga maritima gluconate 5-dehydrogenase; 2ZAT: Sus scrofa dehydrogenase/reductase SDR family
member; 3GRP: Bartonella henselae 3-oxoacyl-(acyl-carrier-protein) reductase; 1HXH: Comamonas testosteroni 3b/17b-hydroxysteroid dehydrogenase; 2PH3: Thermus
thermophilus 3-oxoacyl-(acyl-carrier-protein) reductase.
Lerchner et al.: Structure-Guided Engineering of RasADH 2805
Biotechnology and Bioengineering

Ultra-15 centrifugal filter units; Merck Millipore, Billerica,MA) and subjected to size exclusion chromatography (SEC)in the presence of 25mM HEPES/NaOH pH 8.3, 150mMNaCl, 200mM betaine on a HiLoad 16/60 Superdex 200column (GE Healthcare) using an Äkta purifier system(GE Healthcare). The addition of betaine was instrumentalto reduce the aggregation tendency of RasADH (Wanget al., 2010). His6-tagged RasADH was eluted in ahomogeneous peak with an apparent molecular size of113 kDa for the tetramer, precisely as calculated. The purifiedenzyme was analyzed by SDS–PAGE using CoomassieBrilliant Blue R-250 staining. Enzyme concentration wasmeasured via absorption at 280 nm using a calculatedextinction coefficient of 14,440M�1 cm�1 (Gasteigeret al., 2005). The yield of purified His6-RasADH was 5mg/L bacterial culture.
Preparation of RasADH Variants
The mutations G37D, N15G/G37D/R38V/R39S and N15G/G37D/R38V/R39S/A86N/S88A were introduced into thecloned RasADH gene using the QuikChange Site-DirectedMutagenesis Kit (Agilent, Waldbronn, Germany). For G37D,we used the wild type expression plasmid pASK-IBA35plus-RasADH as template and the primer pair 50-GTA TTC ATTGTCGATCGCCGGCGGAAGG-30 and 50-CCT TCC GCCGGC GATCGACAATGA ATAC-30. The m4 variant (N15G/G37D/R38V/R39S) was generated in two steps. First, aplasmid coding for the N15G/G37D double mutant wasconstructed from the G37D plasmid template using theprimer pair 50-GTC ATA ACC GGT GGA GGC AGC GGCATT GGC-30 and 50-GCC AAT GCC GCT GCC TCC ACCGGT TAT GAC-30. Second, the resulting plasmid was furthermutated with the primer pair 50-GTA TTC ATT GTC GATGTC AGC CGG AAG GAACTC G-30 and 50-CGAGTTCCTTCC GGC TGA CAT CGA CAA TGA ATA C-30. The m6variant (N15G/G37D/R38V/R39S/A86N/S88A) was pre-pared on the basis of the m4 plasmid using the primerpair 50-CAT CGA CGT ACT ATT TAA TAA TGC CGG CGCAAT CGA G-30 and 50-CTC GAT TGC GCC GGC ATT ATTAAA TAG TAC GTC GAT G-30. All mutant enzymes wereexpressed and purified using the same procedure as for thewild type RasADH.
Crystallization of RasADH
Protein crystals were grown in hanging drops using the vapordiffusion technique. Drops mixed from 1ml of the purifiedprotein solution (7.1mg/ml enzyme in 25mM Tris/HCl pH8.0, 150mM NaCl, 200mM betaine), containing either10mM NADþ or 5mM NADPþ (sodium salt), and 1ml ofthe reservoir solution were equilibrated against 1ml reservoirsolution on siliconized glass cover slips. After 2 days at 20�C,crystals were obtained in the presence of 26% (w/v) PEG3,000, 100mMTris/HCl pH 9.0, 200mMLi2SO4 for RasADHwith NADPþ and in the presence of 24% (w/v) PEG 3,000,100mM Tris/HCl pH 8.5, 200mM Li2SO4 for RasADH with
NADþ. Protein crystals were transferred to conditions of thereservoir solution containing 30% (v/v) glycerol using Litho-loops (Molecular Dimensions, Newmarket, UK) and frozenin liquid nitrogen.
For crystallization of RasADHm4, drops were mixed from1ml protein solution (5.0mg/ml enzyme in 25mM HEPES/NaOH pH 8.3, 150mM NaCl, 200mM betaine, 10mMNADþ), 1ml distilled water and 1ml reservoir solution. After1 day at 20�C several crystals were obtained in the presence of11% (w/v) PEG 2,000, 50mM HEPES/NaOH pH 8.0. ForRasADHm6 drops were mixed from 1ml protein solution(6.8mg/ml enzyme in 25mM HEPES/NaOH pH 8.3,150mM NaCl, 200mM betaine, 5mM NADþ) and 1mlreservoir solution. After 2 days at 20�C several crystals wereobtained in the presence of 12% (w/v) PEG 2,000, 50mMHEPES/NaOH pH 8.0, 100mM LiCl.
X-Ray Data Collection and Refinement
Diffraction data were collected at BESSY beamlines 14.1 and14.2 of the Helmholtz-Zentrum Berlin (HZB) (Muelleret al., 2012). The data were indexed, integrated, and scaledwith the XDS software package (Kabsch, 2010) in spacegroups P1 or P21 (Table I). The structure of the wild typeRasADH with bound NADPþ was solved by molecularreplacement with the closely homologous b-ketoacyl-acylcarrier protein reductase of Streptomyces coelicolor (PDB code:2NM0, residues 2–233), which has 39% amino acid sequenceidentity. The resulting model was subjected to refinementwith REFMAC5 (Murshudov et al., 2011) and manuallyrebuilt in an iterative manner using Coot (Emsleyet al., 2010). At early stages of refinement, non-crystallo-graphic symmetry restraints were applied for the mutuallyequivalent chains A/B/C/D and E/F/G/H. TLS segments weredetermined with TLSMD (Painter and Merritt, 2006) andsubsequently refined with REFMAC5. The quality of theresulting structure was validated with MolProbity (Daviset al., 2007).
The crystal structures of the apo-enzyme as well as ofRasADHm4 and RasADHm6 were solved by molecularreplacement using the coordinates for RasADH�NADPþ andrefined accordingly. The coordinates and structure factorshave been deposited under accession codes 4I5D (RasADH),4I5E (RasADH�NADPþ), 4I5F (RasADHm4) and 4I5G(RasADHm6) at the Protein Data Bank at ResearchCollaboratory for Structural Bioinformatics (RCSB). Molec-ular graphics were generated with UCSF Chimera (Pettersenet al., 2004) and amino acid sequence alignments wereprepared with ALINE (Bond and Schüttelkopf, 2009).
Enzyme Kinetics
All assays were performed in the presence of 25mM HEPES/NaOH pH 8.3, 150mM NaCl. SEC-purified enzymes wereused at concentrations ranging from 35 nM to 9mM. Whenassaying catalytic activity with varied cosubstrate concentra-tion (NADþ or NADPþ), the concentration of the substrate
2806 Biotechnology and Bioengineering, Vol. 110, No. 11, November, 2013
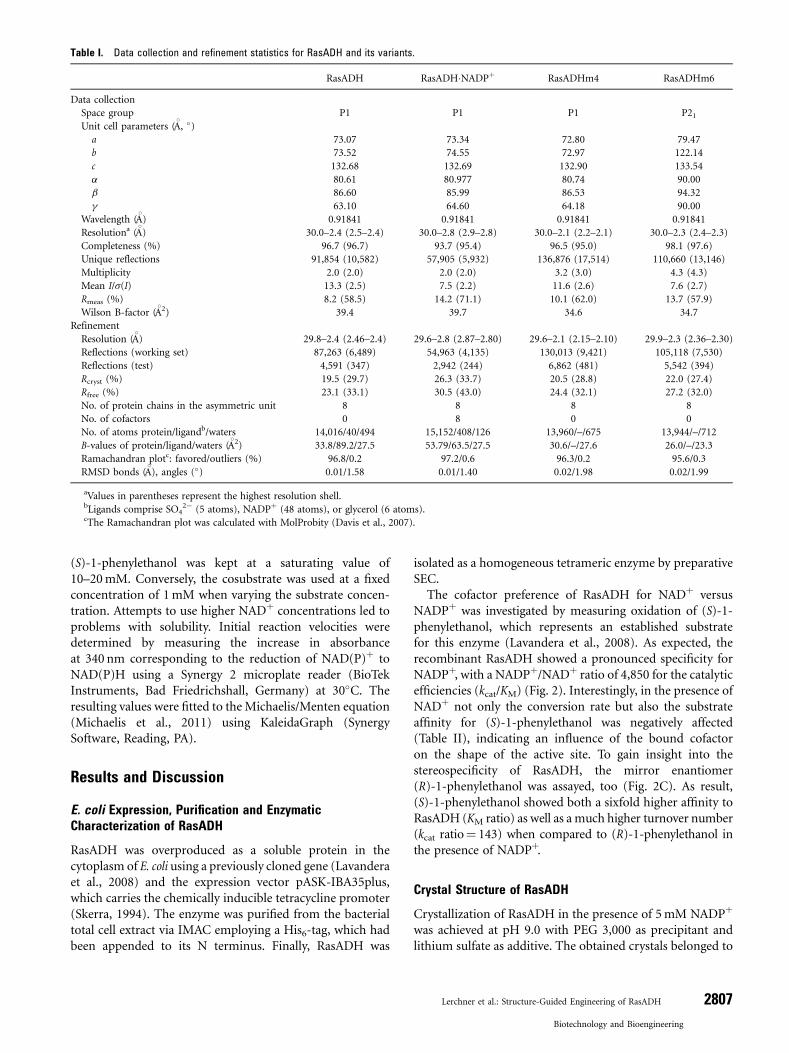
(S)-1-phenylethanol was kept at a saturating value of10–20mM. Conversely, the cosubstrate was used at a fixedconcentration of 1mM when varying the substrate concen-tration. Attempts to use higher NADþ concentrations led toproblems with solubility. Initial reaction velocities weredetermined by measuring the increase in absorbanceat 340 nm corresponding to the reduction of NAD(P)þ toNAD(P)H using a Synergy 2 microplate reader (BioTekInstruments, Bad Friedrichshall, Germany) at 30�C. Theresulting values were fitted to the Michaelis/Menten equation(Michaelis et al., 2011) using KaleidaGraph (SynergySoftware, Reading, PA).
Results and Discussion
E. coli Expression, Purification and EnzymaticCharacterization of RasADH
RasADH was overproduced as a soluble protein in thecytoplasm of E. coli using a previously cloned gene (Lavanderaet al., 2008) and the expression vector pASK-IBA35plus,which carries the chemically inducible tetracycline promoter(Skerra, 1994). The enzyme was purified from the bacterialtotal cell extract via IMAC employing a His6-tag, which hadbeen appended to its N terminus. Finally, RasADH was
isolated as a homogeneous tetrameric enzyme by preparativeSEC.The cofactor preference of RasADH for NADþ versus
NADPþ was investigated by measuring oxidation of (S)-1-phenylethanol, which represents an established substratefor this enzyme (Lavandera et al., 2008). As expected, therecombinant RasADH showed a pronounced specificity forNADPþ, with a NADPþ/NADþ ratio of 4,850 for the catalyticefficiencies (kcat/KM) (Fig. 2). Interestingly, in the presence ofNADþ not only the conversion rate but also the substrateaffinity for (S)-1-phenylethanol was negatively affected(Table II), indicating an influence of the bound cofactoron the shape of the active site. To gain insight into thestereospecificity of RasADH, the mirror enantiomer(R)-1-phenylethanol was assayed, too (Fig. 2C). As result,(S)-1-phenylethanol showed both a sixfold higher affinity toRasADH (KM ratio) as well as a much higher turnover number(kcat ratio¼ 143) when compared to (R)-1-phenylethanol inthe presence of NADPþ.
Crystal Structure of RasADH
Crystallization of RasADH in the presence of 5mM NADPþ
was achieved at pH 9.0 with PEG 3,000 as precipitant andlithium sulfate as additive. The obtained crystals belonged to
Table I. Data collection and refinement statistics for RasADH and its variants.
RasADH RasADH�NADPþ RasADHm4 RasADHm6
Data collectionSpace group P1 P1 P1 P21Unit cell parameters (8A, �)a 73.07 73.34 72.80 79.47b 73.52 74.55 72.97 122.14c 132.68 132.69 132.90 133.54a 80.61 80.977 80.74 90.00b 86.60 85.99 86.53 94.32g 63.10 64.60 64.18 90.00
Wavelength (8A) 0.91841 0.91841 0.91841 0.91841Resolutiona (8A) 30.0–2.4 (2.5–2.4) 30.0–2.8 (2.9–2.8) 30.0–2.1 (2.2–2.1) 30.0–2.3 (2.4–2.3)Completeness (%) 96.7 (96.7) 93.7 (95.4) 96.5 (95.0) 98.1 (97.6)Unique reflections 91,854 (10,582) 57,905 (5,932) 136,876 (17,514) 110,660 (13,146)Multiplicity 2.0 (2.0) 2.0 (2.0) 3.2 (3.0) 4.3 (4.3)Mean I/s(I) 13.3 (2.5) 7.5 (2.2) 11.6 (2.6) 7.6 (2.7)Rmeas (%) 8.2 (58.5) 14.2 (71.1) 10.1 (62.0) 13.7 (57.9)Wilson B-factor (8A2) 39.4 39.7 34.6 34.7
RefinementResolution (8A) 29.8–2.4 (2.46–2.4) 29.6–2.8 (2.87–2.80) 29.6–2.1 (2.15–2.10) 29.9–2.3 (2.36–2.30)Reflections (working set) 87,263 (6,489) 54,963 (4,135) 130,013 (9,421) 105,118 (7,530)Reflections (test) 4,591 (347) 2,942 (244) 6,862 (481) 5,542 (394)Rcryst (%) 19.5 (29.7) 26.3 (33.7) 20.5 (28.8) 22.0 (27.4)Rfree (%) 23.1 (33.1) 30.5 (43.0) 24.4 (32.1) 27.2 (32.0)No. of protein chains in the asymmetric unit 8 8 8 8No. of cofactors 0 8 0 0No. of atoms protein/ligandb/waters 14,016/40/494 15,152/408/126 13,960/–/675 13,944/–/712B-values of protein/ligand/waters (8A2) 33.8/89.2/27.5 53.79/63.5/27.5 30.6/–/27.6 26.0/–/23.3Ramachandran plotc: favored/outliers (%) 96.8/0.2 97.2/0.6 96.3/0.2 95.6/0.3RMSD bonds (8A), angles (�) 0.01/1.58 0.01/1.40 0.02/1.98 0.02/1.99
aValues in parentheses represent the highest resolution shell.bLigands comprise SO4
2� (5 atoms), NADPþ (48 atoms), or glycerol (6 atoms).cThe Ramachandran plot was calculated with MolProbity (Davis et al., 2007).
Lerchner et al.: Structure-Guided Engineering of RasADH 2807
Biotechnology and Bioengineering
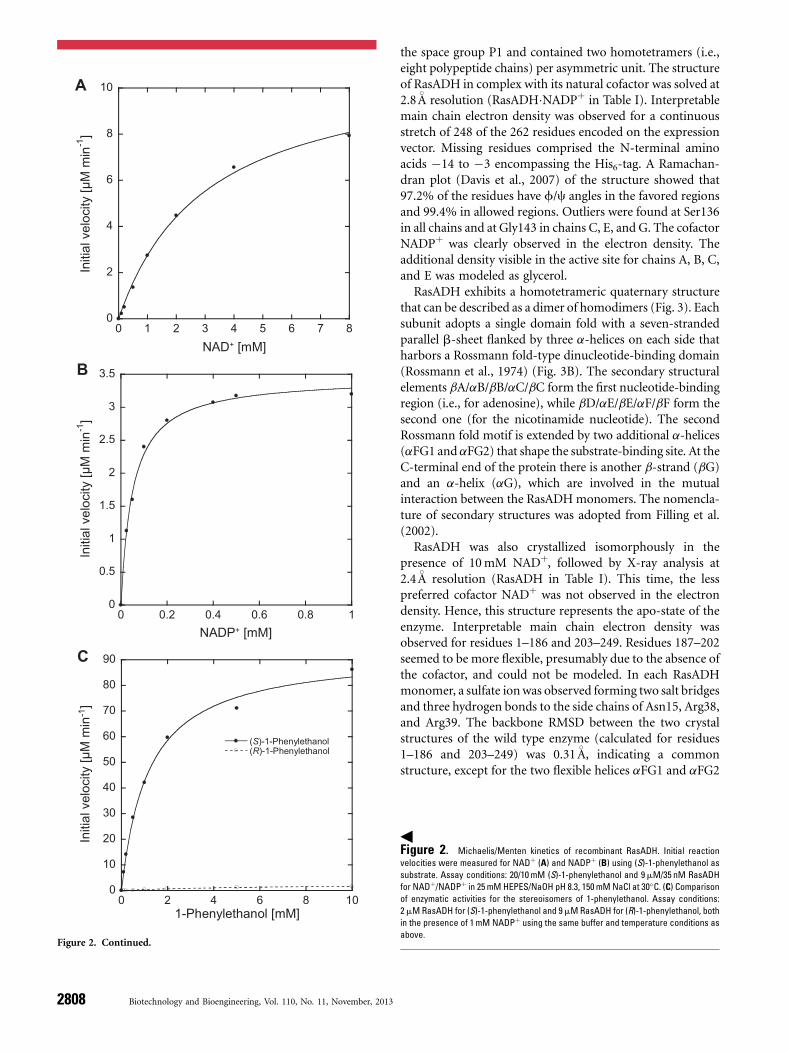
the space group P1 and contained two homotetramers (i.e.,eight polypeptide chains) per asymmetric unit. The structureof RasADH in complex with its natural cofactor was solved at2.8 8A resolution (RasADH�NADPþ in Table I). Interpretablemain chain electron density was observed for a continuousstretch of 248 of the 262 residues encoded on the expressionvector. Missing residues comprised the N-terminal aminoacids �14 to �3 encompassing the His6-tag. A Ramachan-dran plot (Davis et al., 2007) of the structure showed that97.2% of the residues have f/c angles in the favored regionsand 99.4% in allowed regions. Outliers were found at Ser136in all chains and at Gly143 in chains C, E, and G. The cofactorNADPþ was clearly observed in the electron density. Theadditional density visible in the active site for chains A, B, C,and E was modeled as glycerol.
RasADH exhibits a homotetrameric quaternary structurethat can be described as a dimer of homodimers (Fig. 3). Eachsubunit adopts a single domain fold with a seven-strandedparallel b-sheet flanked by three a-helices on each side thatharbors a Rossmann fold-type dinucleotide-binding domain(Rossmann et al., 1974) (Fig. 3B). The secondary structuralelements bA/aB/bB/aC/bC form the first nucleotide-bindingregion (i.e., for adenosine), while bD/aE/bE/aF/bF form thesecond one (for the nicotinamide nucleotide). The secondRossmann fold motif is extended by two additional a-helices(aFG1 and aFG2) that shape the substrate-binding site. At theC-terminal end of the protein there is another b-strand (bG)and an a-helix (aG), which are involved in the mutualinteraction between the RasADHmonomers. The nomencla-ture of secondary structures was adopted from Filling et al.(2002).
RasADH was also crystallized isomorphously in thepresence of 10mM NADþ, followed by X-ray analysis at2.4 8A resolution (RasADH in Table I). This time, the lesspreferred cofactor NADþ was not observed in the electrondensity. Hence, this structure represents the apo-state of theenzyme. Interpretable main chain electron density wasobserved for residues 1–186 and 203–249. Residues 187–202seemed to be more flexible, presumably due to the absence ofthe cofactor, and could not be modeled. In each RasADHmonomer, a sulfate ionwas observed forming two salt bridgesand three hydrogen bonds to the side chains of Asn15, Arg38,and Arg39. The backbone RMSD between the two crystalstructures of the wild type enzyme (calculated for residues1–186 and 203–249) was 0.31 8A, indicating a commonstructure, except for the two flexible helices aFG1 and aFG2
0
2
4
6
8
10
0 1 2 3 4 5 6 7 8
Initi
al v
eloc
ity [µ
M m
in-1
]
NAD+ [mM]
A
0
0.5
1
1.5
2
2.5
3
3.5
0 0.2 0.4 0.6 0.8 1
Initi
al v
eloc
ity [µ
M m
in-1
]
NADP+ [mM]
B
0
10
20
30
40
50
60
70
80
90
0 2 4 6 8 10
(S)-1-Phenylethanol(R)-1-Phenylethanol
Initi
al v
eloc
ity [µ
M m
in-1
]
1-Phenylethanol [mM]
C
Figure 2. Continued.
3Figure 2. Michaelis/Menten kinetics of recombinant RasADH. Initial reaction
velocities were measured for NADþ (A) and NADPþ (B) using (S)-1-phenylethanol as
substrate. Assay conditions: 20/10 mM (S)-1-phenylethanol and 9mM/35 nM RasADH
for NADþ/NADPþ in 25mM HEPES/NaOH pH 8.3, 150 mM NaCl at 30�C. (C) Comparisonof enzymatic activities for the stereoisomers of 1-phenylethanol. Assay conditions:
2mMRasADH for (S)-1-phenylethanol and 9mMRasADH for (R)-1-phenylethanol, both
in the presence of 1mM NADPþ using the same buffer and temperature conditions as
above.
2808 Biotechnology and Bioengineering, Vol. 110, No. 11, November, 2013
(residues 187–202) that shape the substrate binding site(see above).
Binding of the Cofactor NADPþ
The cofactor-binding site in SDR enzymes is characterized bythe conserved TGXXXGXG motif (Fig. 1). The correspond-
ing TGGNSGIG sequence (positions 12–19) of RasADH islocated in the loop between the first b-strand (bA) and thefirst a-helix (aB). Further elements of the canonical cofactor-binding site are (i) the general SDR motif NNAG, located atthe C-terminal end of bD and (ii) the PG motif at theC-terminal end of bF. The NNAG motif corresponds to thesequence ANSG (positions 86–89) in RasADH, where Asn87
Table II. Kinetic parameters for engineered RasADH variants.
Substrate investigated/enzyme variant Cosubstrate KM (mM) kcat (min�1) kcat/KM (mM�1min�1)
NADþ
Wild type RasADH (S)-1-PhEtOH 3.1� 0.3 1.3� 0.1 0.42G37D (S)-1-PhEtOH 2.3� 0.2 2.3� 0.2 1.0N15G/G37D/R38V/R39S (S)-1-PhEtOH 1.1� 0.1 4.8� 0.3 4.4N15G/G37D/R38V/R39S/A86N/S88A (S)-1-PhEtOH 0.8� 0.1 1.8� 0.1 2.3
NADPþ
Wild type RasADH (S)-1-PhEtOH 0.051� 0.004 99� 2 1,940G37D (S)-1-PhEtOH 2.3� 0.5 0.15� 0.01 0.065N15G/G37D/R38V/R39S (S)-1-PhEtOH 0.28� 0.12 0.0017� 0.0002 0.0061N15G/G37D/R38V/R39S/A86N/S88A (S)-1-PhEtOH n.d. n.d. —
(S)-1-phenylethanolWild type RasADHa NADþ 9.9� 1.1 0.69� 0.03 0.069Wild type RasADH NADPþ 1.2� 0.1 47� 1 39N15G/G37D/R38V/R39Sa NADþ 8.3� 0.5 2.54� 0.08 0.31N15G/G37D/R38V/R39S/A86N/S88Aa NADþ 17.6� 1.3 1.38� 0.07 0.078
(R)-1-phenylethanolWild type RasADH NADPþ 5.5� 0.5 0.28� 0.01 0.051
n.d., not detectable.aApparent kinetic parameters are reported as NADþ could not be applied at saturating conditions (see Materials and Methods Section).
Figure 3. Crystal structure of RasADH. A: Tetrameric quaternary structure (chains A–D, one subunit colored) of RasADH with bound NADPþ (red). The two parts of the
Rossmann dinucleotide bindingmotif are highlighted in blue and greenwhile flanking helices are shown inmagenta.B: Close up view of one RasADHmonomerwith relevant features
labeled.
Lerchner et al.: Structure-Guided Engineering of RasADH 2809
Biotechnology and Bioengineering
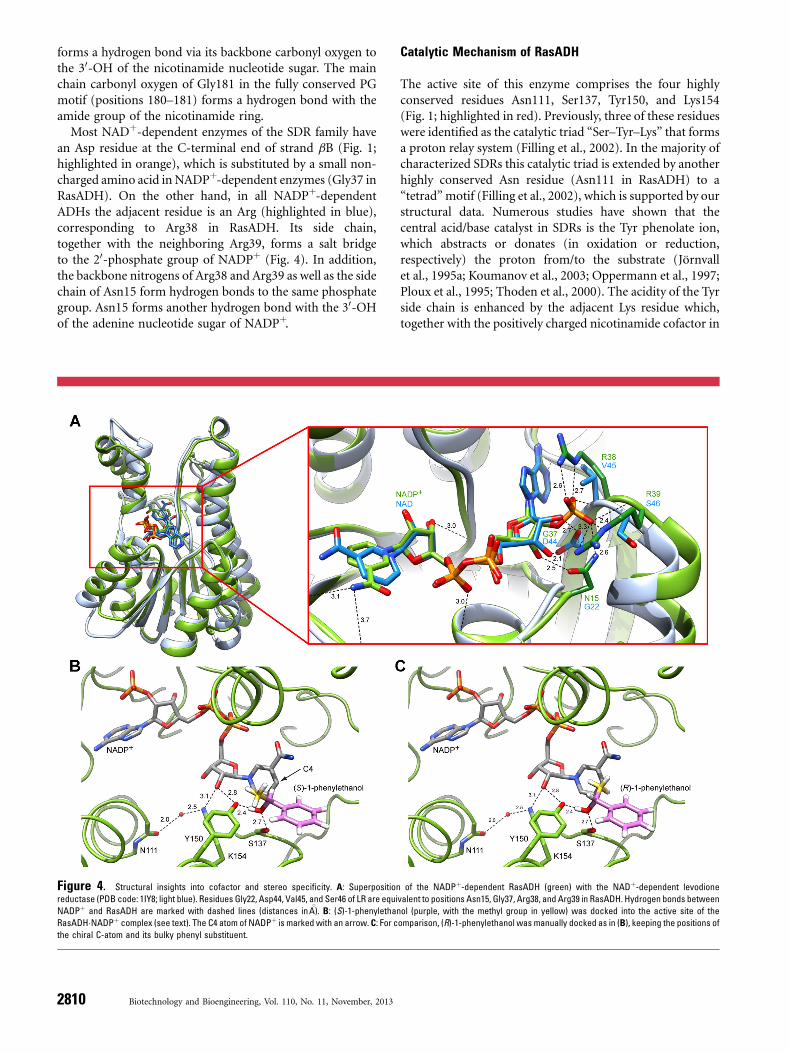
forms a hydrogen bond via its backbone carbonyl oxygen tothe 30-OH of the nicotinamide nucleotide sugar. The mainchain carbonyl oxygen of Gly181 in the fully conserved PGmotif (positions 180–181) forms a hydrogen bond with theamide group of the nicotinamide ring.
Most NADþ-dependent enzymes of the SDR family havean Asp residue at the C-terminal end of strand bB (Fig. 1;highlighted in orange), which is substituted by a small non-charged amino acid in NADPþ-dependent enzymes (Gly37 inRasADH). On the other hand, in all NADPþ-dependentADHs the adjacent residue is an Arg (highlighted in blue),corresponding to Arg38 in RasADH. Its side chain,together with the neighboring Arg39, forms a salt bridgeto the 20-phosphate group of NADPþ (Fig. 4). In addition,the backbone nitrogens of Arg38 and Arg39 as well as the sidechain of Asn15 form hydrogen bonds to the same phosphategroup. Asn15 forms another hydrogen bond with the 30-OHof the adenine nucleotide sugar of NADPþ.
Catalytic Mechanism of RasADH
The active site of this enzyme comprises the four highlyconserved residues Asn111, Ser137, Tyr150, and Lys154(Fig. 1; highlighted in red). Previously, three of these residueswere identified as the catalytic triad “Ser–Tyr–Lys” that formsa proton relay system (Filling et al., 2002). In the majority ofcharacterized SDRs this catalytic triad is extended by anotherhighly conserved Asn residue (Asn111 in RasADH) to a“tetrad”motif (Filling et al., 2002), which is supported by ourstructural data. Numerous studies have shown that thecentral acid/base catalyst in SDRs is the Tyr phenolate ion,which abstracts or donates (in oxidation or reduction,respectively) the proton from/to the substrate (Jörnvallet al., 1995a; Koumanov et al., 2003; Oppermann et al., 1997;Ploux et al., 1995; Thoden et al., 2000). The acidity of the Tyrside chain is enhanced by the adjacent Lys residue which,together with the positively charged nicotinamide cofactor in
Figure 4. Structural insights into cofactor and stereo specificity. A: Superposition of the NADPþ-dependent RasADH (green) with the NADþ-dependent levodione
reductase (PDB code: 1IY8; light blue). Residues Gly22, Asp44, Val45, and Ser46 of LR are equivalent to positions Asn15, Gly37, Arg38, and Arg39 in RasADH. Hydrogen bonds between
NADPþ and RasADH are marked with dashed lines (distances in 8A). B: (S)-1-phenylethanol (purple, with the methyl group in yellow) was docked into the active site of the
RasADH�NADPþ complex (see text). The C4 atom of NADPþ is marked with an arrow. C: For comparison, (R)-1-phenylethanol was manually docked as in (B), keeping the positions of
the chiral C-atom and its bulky phenyl substituent.
2810 Biotechnology and Bioengineering, Vol. 110, No. 11, November, 2013

its oxidized state, contributes Coulomb energy (Benachet al., 1999; Koumanov et al., 2003).The e-amino group of Lys is also involved in the binding of
the nicotinamide ribose group, whereas the role of the Serresidue is to stabilize and polarize the carbonyl group of theoxidized product resulting from the ADH reaction (Jörnvallet al., 1995a; Oppermann et al., 1997). The invariant Asnresidue at position 111 located in helix aE introduces acharacteristic helical kink (Oppermann et al., 2003), and itsmain-chain carbonyl group ligates a conserved watermolecule in hydrogen-bonding distance to the active siteLys side chain. In this manner, an extended proton relaysystem is established (Filling et al., 2002) that connects thebulk solvent reservoir to the active site Tyr residue.In order to gain insight into the mechanism of the
biocatalytic oxidation reaction, the model substrate (S)-1-phenylethanol was docked manually into the active site of theRasADH�NADPþ complex (Fig. 4B) using an orientationsimilar to the one observed for pseudotropine in the crystalstructure of tropinone reductase II from Datura stramonium(PDB code: 2AE2; Yamashita et al., 1999). As result, thehydroxyl group of (S)-1-phenylethanol is arranged betweenTyr150 and Ser137 while its hydride leaving group is orientedtoward the C4 atom of the oxidized nicotinamide ring ofthe cosubstrate. Obviously, Ser137 stabilizes the substrateorientation, and it appears likely that the oxidation reaction isinitiated by proton transfer from the hydroxyl group of thesubstrate to the Tyr150 phenolate, acting as catalytic base,followed by hydride transfer to NADPþ. The proton relaymechanism would further involve the 20-OH of thenicotinamide ribose, the basic side chain of Lys154 andthe backbone carbonyl oxygen of Asn111 which coordinatesthe conserved water molecule described above. With regardto the RasADH-catalyzed backward (reduction) reaction,the pro-(R)-hydride of NADPH would be in a position toattack the prochiral ketone from its re face to yield thecorresponding (S)-alcohol (Musa and Phillips, 2011). In fact,the majority of commercially available ADHs, including yeastADH and horse liver ADH, have this Prelog stereo-preference(Musa and Phillips, 2011).For comparison, (R)-1-phenylethanol was superimposed
to (S)-1-phenylethanol based on the common phenyl ring asthe sterically dominant substituent (Fig. 4C) and the hydroxylgroup was again arranged between Tyr150 and Ser137. Basedon this superposition the stereoselectivity of RasADH towardthe (S)-1-phenylethanol becomes immediately apparent.First, the hydride transfer from the (R)-substrate to theC4-atom of NADPþ is not possible since the leaving ion doesnot face this atom. Second, the distance between the methylsubstituent of the substrate and the C4-atom of NADPþ
would be sterically too close, which should even preventbinding of (R)-1-phenylethanol to the active site at first,regardless of catalytic conversion. This notion was confirmedby automated docking of (R)-1-phenylethanol to the activesite of RasADH using the Glamdock software tool of theChil2 platform (Tietze and Apostolakis, 2007). The best30 placements all showed inappropriate orientations of the
disfavored substrate, which is in agreement with thestereoselectivity of RasADH toward the (S)-enantiomer asexperimentally proven further above.
Protein Engineering of RasADH for CosubstrateSpecificity Toward NADH
In a structure-based multiple sequence alignment(Söding, 2005; Fig. 1), RasADH was compared with 20 ofits closest homologues. The NADþ-dependent levodionereductase from Leifsonia aquatica (LR; PDB code: 1IY8; Sogabeet al., 2003) appeared as the most similar three-dimensionalstructure, with an amino acid sequence identity of 34% andan RMSDof 0.54 8A (calculated for 151 pairs of equivalent Ca-atoms). Therefore, LR was used for a structural superposition(see Fig. 4A). When comparing the sequences of the 20NADþ- or NADPþ-dependent enzymes the conserved Aspresidue at the C-terminal end of strand bB, including LR, wasclearly indicative of the NADþ-specific dehydrogenases(highlighted in orange in Fig. 1) whereas RasADH carries aGly residue at this position.Consequently, Gly37 was chosen as starting point for the
construction of RasADH variants with altered cofactorspecificity. Initially, the substitution from Gly to Asp wasintroduced in order to allow hydrogen bonding betweenAsp37 and the 20- and 30-hydroxyl groups of the adenosineribose of NADþ, which at the same time sterically andelectrostatically prevented binding of the 20-phosphate groupof NADPþ to the active site (Fig. 4A). Compared with thewild type enzyme, this mutation dramatically decreased theaffinity for NADPþ while the KM value of NADþ was onlymoderately improved (Table II). Nevertheless, the specificityfor NADþ
—defined as ratio of the catalytic efficiencies(Eisenthal et al., 2007) between NADþ and NADPþ—wasclearly increased, with a value of 17 for this single site mutantversus 0.00021 for the wild type enzyme.Further inspection of the crystal structure of RasADH
revealed that the positively charged residues Arg38 andArg39, and also the side chain of Asn15, seem to hamperoptimal binding of NADþ (Fig. 4A). These residues areresponsible for the high NADPþ affinity by forming saltbridges and/or hydrogen bond interactions with thephosphate group as described above. Considering the aminoacids Val45, Ser46, and Gly22 at the equivalent positions inthe NADþ-specific homolog LR (Fig. 4A), the correspondingmutations R38V, R39S, and N15G were introduced furtherto the G37D substitution, leading to the mutant enzymeRasADHm4 (N15G/G37D/R38V/R39S). Indeed, this variantshowed improved specificity for NADþ (NADþ/NADPþ
ratio of 721 for kcat/KM) and an increased conversion rate withthis cofactor, leading to a tenfold higher catalytic efficiencyfor NADþ in comparison with the wild type enzyme(Table II).Finally, the conserved sequence motif NNAG at the
C-terminus of strand bD in SDR enzymes, which appearedmodified in RasADH with its sequence ANSG, was changed
Lerchner et al.: Structure-Guided Engineering of RasADH 2811
Biotechnology and Bioengineering
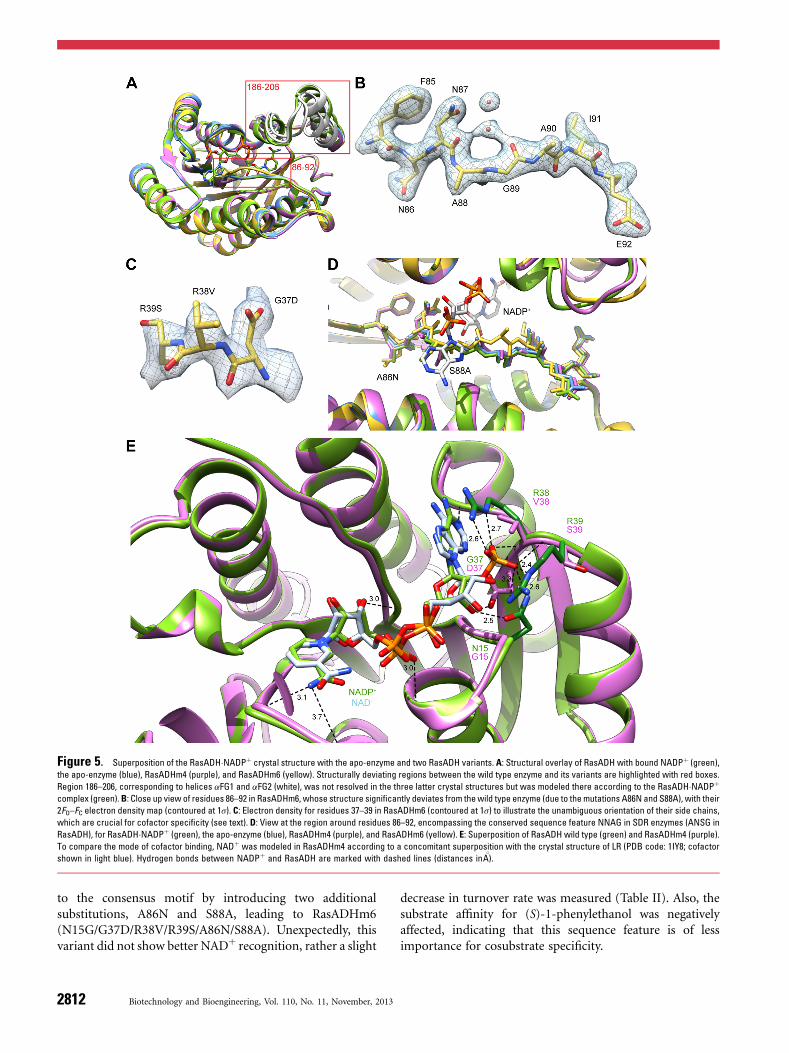
to the consensus motif by introducing two additionalsubstitutions, A86N and S88A, leading to RasADHm6(N15G/G37D/R38V/R39S/A86N/S88A). Unexpectedly, thisvariant did not show better NADþ recognition, rather a slight
decrease in turnover rate was measured (Table II). Also, thesubstrate affinity for (S)-1-phenylethanol was negativelyaffected, indicating that this sequence feature is of lessimportance for cosubstrate specificity.
Figure 5. Superposition of the RasADH�NADPþ crystal structure with the apo-enzyme and two RasADH variants. A: Structural overlay of RasADH with bound NADPþ (green),
the apo-enzyme (blue), RasADHm4 (purple), and RasADHm6 (yellow). Structurally deviating regions between the wild type enzyme and its variants are highlighted with red boxes.
Region 186–206, corresponding to helices aFG1 and aFG2 (white), was not resolved in the three latter crystal structures but was modeled there according to the RasADH�NADPþcomplex (green).B: Close up view of residues 86–92 in RasADHm6, whose structure significantly deviates from thewild type enzyme (due to themutations A86N and S88A), with their
2FO–FC electron density map (contoured at 1s). C: Electron density for residues 37–39 in RasADHm6 (contoured at 1s) to illustrate the unambiguous orientation of their side chains,
which are crucial for cofactor specificity (see text). D: View at the region around residues 86–92, encompassing the conserved sequence feature NNAG in SDR enzymes (ANSG in
RasADH), for RasADH�NADPþ (green), the apo-enzyme (blue), RasADHm4 (purple), and RasADHm6 (yellow). E: Superposition of RasADH wild type (green) and RasADHm4 (purple).
To compare the mode of cofactor binding, NADþ was modeled in RasADHm4 according to a concomitant superposition with the crystal structure of LR (PDB code: 1IY8; cofactor
shown in light blue). Hydrogen bonds between NADPþ and RasADH are marked with dashed lines (distances in 8A).
2812 Biotechnology and Bioengineering, Vol. 110, No. 11, November, 2013

X-Ray Structural Analysis of RasADH Variants
The crystal structures of RasADHm4 and RasADHm6 weresolved at 2.1 and 2.3 8A, respectively (Table I). Similar to thewild type apo-enzyme, the electron density for backbone andside chain atoms was well defined for positions 1–186 and203–249 while the region 187–202 was disordered. Unfortu-nately, although crystallized in the presence of NADþ, thecofactor was not visible in both X-ray structures.Whereas the overall conformations of the mutant enzymes
are very similar to the wild type RasADH the variants showeddeviations in the following two regions: (i) residues 86–92located near the pyrophosphate moiety of the cofactor,encompassing the conserved sequence feature NNAG in SDRenzymes (ANSG in RasADH, see above), and (ii) residues186–206 corresponding to the two a-helices aFG1 and aFG2involved in substrate binding (Fig. 5A). As described above,this second regionwas only resolved for the wild typeNADPþ
complex, whereas in the structures lacking the cofactor thetwo a-helices appear more flexible and show no distinctconformation (Fig. 5A).For the first region, a comparison between RasADH and
RasADH�NADPþ yielded a subtle backbone Ca RMSD of0.37 8A, which can be explained by the presence of thecofactor in one of the two structures. However, RasADHm4and RasADHm6 revealed a more pronounced structuraldifference (Fig. 5D) with Ca RMSDs from the wild type apo-enzyme of 0.76 and 1.23 8A, respectively. This significantdeviation could be explained by an increased flexibility in theloop region between strand bA and helix aB caused byintroduction of the substitutions N15G/G37D/R38V/R39Sin RasADHm4 and also in RasADHm6 (Fig. 5A and D).Due to the additional mutations A86N and S88A,
RasADHm6 showed another change in its backboneconformation in this region when compared to the wildtype apo-enzyme as well as to the variant m4 (Fig. 5A and D).In particular, substitution of Ala by the larger side chain ofAsn seemed to effect an altered backbone conformation. Thissignificant structural deviation should explain the detrimen-tal effect of the side chain replacements A86N and S88A inthis variant on the catalytic activity as described above.To investigate the mechanism of altered cofactor specificity,
the RasADH crystal structure with bound NADPþ wassuperimposed on that of RasADHm4 in its apo-state (Fig. 5E).The position of NADþ in RasADHm4 was approximated bysuperimposing the X-ray structure of LR with the boundcofactor NADþ (PDB code: 1IY8). Due to the replacementsof Arg38 and Arg39—which form salt bridges to the20-phosphate group of the NADPþ in the wild type RasADH,as explained above—by Val and Ser, respectively, the bindingof NADPþ is no longer favored (Fig. 5E). Instead, the newcofactor NADþ likely is able to form productive hydrogenbonds with the previously introduced Asp37 residue.
Conclusions
The crystallographic analysis of the biotechnologicallyrelevant enzyme RasADH as well as the docking simulation
with one of its relevant substrates has revealed insight into thestructural determinants of its biocatalytic activity. Inaddition, we have succeeded in engineering a mutant withdrastically reduced affinity toward the natural cofactorNADPH while showing a tenfold increased catalyticefficiency with NADH, overall leading to a change inNADH/NADPH cofactor specificity from 0.00021 to 721.Further improvement with regard to NADH acceptanceshould be possible but probably requires more subtle changesto the cofactor binding and/or catalytically active sites as wellas specific tailoring for the aspired substrate. Nevertheless,the mutated RasADH with its NADH-adapted catalyticactivity shows potential for applications in biotechnologicalprocesses in combination with well-established NADHregeneration systems.
The authors wish to thank Stefan Achatz for technical assistance andDr. Uwe Müller and Dr. Manfred Weiss for technical support atBESSY beamlines 14.1 and 14.2 of the HZB, Germany. We also thankEvonik Industries AG, Marl, Germany, in particular Dr. Jan Pfefferand Dr. Thomas Haas, for discussions and supply of materials as wellas the Bundesministerium für Bildung und Forschung, Germany, forfinancial support in frame of the project “Bioxamine” (Grant No.0316044B). The authors are grateful to Professor Dr. WolfgangKroutil (Karl-Franzens-Universität Graz, Austria) for providing theRasADH gene.
References
Benach J, Atrian S, Gonzalez-Duarte R, Ladenstein R. 1999. The catalyticreaction and inhibition mechanism of Drosophila alcohol dehydrogenase:Observation of an enzyme-bound NAD-ketone adduct at 1.4 8Aresolution by X-ray crystallography. J Mol Biol 289:335–355.
Berenguer-Murcia A, Fernandez-Lafuente R. 2010. New trends in therecycling of NAD(P)H for the design of sustainable asymmetricreductions catalyzed by dehydrogenases. Curr Org Chem 14:1000–1021.
Biegert A, Mayer C, Remmert M, Söding J, Lupas AN. 2006. The MPIBioinformatics Toolkit for protein sequence analysis. Nucleic Acids Res34:W335–W339.
Bond CS, Schüttelkopf AW. 2009. ALINE: A WYSIWYG protein-sequencealignment editor for publication-quality alignments. Acta Crystallogr DBiol Crystallogr 65:510–512.
Chen R, Greer A, Dean AM. 1995. A highly active decarboxylatingdehydrogenase with rationally inverted coenzyme specificity. Proc NatlAcad Sci USA 92:11666–11670.
Cho H, Oliveira MA, Tai HH. 2003. Critical residues for the coenzymespecificity of NADþ-dependent 15-hydroxyprostaglandin dehydroge-nase. Arch Biochem Biophys 419:139–146.
Cuetos A, Rioz-Martínez A, Bisogno FR, Grischek B, Lavandera I, de GonzaloG, Kroutil W, Gotor V. 2012. Access to enantiopure a-alkyl-b-hydroxyesters through dynamic kinetic resolutions employing purified/overexpressed alcohol dehydrogenases. Adv Synth Catal 354:1743–1749.
Davis IW, Leaver-Fay A, Chen VB, Block JN, Kapral GJ, Wang X, Murray LW,Arendall WB III, Snoeyink J, Richardson JS, Richardson DS. 2007.MolProbity: All-atom contacts and structure validation for proteins andnucleic acids. Nucleic Acids Res 35:W375–W383.
Ehrensberger AH, Elling RA,WilsonDK. 2006. Structure-guided engineeringof xylitol dehydrogenase cosubstrate specificity. Structure 14:567–575.
Eisenthal R, DansonMJ, HoughDW. 2007. Catalytic efficiency and kcat/KM: Auseful comparator? Trends Biotechnol 25:247–249.
Emsley P, Lohkamp B, ScottWG, Cowtan K. 2010. Features and developmentof COOT. Acta Crystallogr D Biol Crystallogr 66:486–501.
Lerchner et al.: Structure-Guided Engineering of RasADH 2813
Biotechnology and Bioengineering

Ferreira-Silva B, Lavandera I, Kern A, Faber K, Kroutil W. 2010.Chemo-promiscuity of alcohol dehydrogenases: Reduction of phenyl-acetaldoxime to the alcohol. Tetrahedron 66:3410–3414.
Filling C, Nordling E, Benach J, Berndt KD, Ladenstein R, Jörnvall H,Oppermann U. 2001. Structural role of conserved Asn179 in theshort-chain dehydrogenase/reductase scaffold. Biochem Biophys ResCommun 289:712–717.
Filling C, Berndt KD, Benach J, Knapp S, Prozorovski T, Nordling E,Ladenstein R, Jörnvall H, Oppermann U. 2002. Critical residues forstructure and catalysis in short-chain dehydrogenases/reductases. J BiolChem 277:25677–25684.
Gasteiger E, Hoogland C, Gattiker A, Duvaud S, Wilkins MR, Appel RD,Bairoch A. 2005. Protein identification and analysis tools on the ExPASyserver. In: Walker JM, editor. The proteomics protocols handbook.New York, NY: Humana Press. p 571–607.
Jörnvall H, Danielsson O, Hjelmqvist L, Persson B, Shafqat J. 1995a. Thealcohol dehydrogenase system. Adv Exp Med Biol 372:281–294.
Jörnvall H, Persson B, Krook M, Atrian S, Gonzalez-Duarte R, Jeffery J,Ghosh D. 1995b. Short-chain dehydrogenases/reductases (SDR).Biochemistry 34:6003–6013.
Kabsch W. 2010. XDS. Acta Crystallogr D Biol Crystallogr 66:125–132.Kabsch W, Sander C. 1983. Dictionary of protein secondary structure:
Pattern recognition of hydrogen-bonded and geometrical features.Biopolymers 22:2577–2637.
Kavanagh KL, Jörnvall H, Persson B, Oppermann U. 2008. The SDRsuperfamily: Functional and structural diversity within a family ofmetabolic and regulatory enzymes. Cell Mol Life Sci 65:3895–3906.
Khoury GA, Fazelinia H, Chin JW, Pantazes RJ, Cirino PC, Maranas CD.2009. Computational design of Candida boidinii xylose reductase foraltered cofactor specificity. Protein Sci 18:2125–2138.
Koumanov A, Benach J, Atrian S, Gonzalez-Duarte R, Karshikoff A,Ladenstein R. 2003. The catalytic mechanism of Drosophila alcoholdehydrogenase: Evidence for a proton relay modulated by the coupledionization of the active site lysine/tyrosine pair and a NADþ ribose OHswitch. Proteins 51:289–298.
Kulig J, Simon RC, Rose CA, Husain SM, Häckh M, Lüdeke S, Zeitler K,Kroutil W, Pohl M, Rother D. 2012. Stereoselective synthesis of bulky1,2-diols with alcohol dehydrogenases. Catal Sci Technol 2:1580–1589.
Lavandera I, Kern A, Ferreira-Silva B, Glieder A, de Wildeman S, Kroutil W.2008. Stereoselective bioreduction of bulky-bulky ketones by a novelADH from Ralstonia sp. J Org Chem 73:6003–6005.
Liese A, Seelbach K, Wandrey C. 2006. Industrial biotransformations.Weinheim: Wiley-VCH.
Liu W, Wang P. 2007. Cofactor regeneration for sustainable enzymaticbiosynthesis. Biotechnol Adv 25:369–384.
Medina M, Luquita A, Tejero J, Hermoso J, Mayoral T, Sanz-Aparicio J,Grever K, Gomez-Moreno C. 2001. Probing the determinants ofcoenzyme specificity in ferredoxin-NADPþ reductase by site-directedmutagenesis. J Biol Chem 276:11902–11912.
Michaelis L, Menten ML, Johnson KA, Goody RS. 2011. The originalMichaelis constant: Translation of the 1913 Michaelis-Menten paper.Biochemistry 50:8264–8269.
Mueller U, Darowski N, Fuchs MR, Forster R, Hellmig M, Paithankar KS,Puhringer S, Steffien M, Zocher G, Weiss MS. 2012. Facilitiesfor macromolecular crystallography at the Helmholtz-Zentrum Berlin.J Synchrotron Radiat 19:442–449.
Murshudov GN, Skubak P, Lebedev AA, Pannu NS, Steiner RA, Nicholls RA,Winn MD, Long F, Vagin AA. 2011. REFMAC5 for the refinement ofmacromolecular crystal structures. Acta Crystallogr D Biol Crystallogr67:355–367.
Musa MM, Phillips RS. 2011. Recent advances in alcohol dehydrogenase-catalyzed asymmetric production of hydrophobic alcohols. Catal SciTechnol 1:1311–1323.
Oppermann UC, Filling C, Berndt KD, Persson B, Benach J, Ladenstein R,Jörnvall H. 1997. Active site directed mutagenesis of 3b/17b-
hydroxysteroid dehydrogenase establishes differential effects on short-chain dehydrogenase/reductase reactions. Biochemistry 36:34–40.
Oppermann U, Filling C, Hult M, Shafqat N, Wu X, Lindh M, Shafqat J,Nordling E, Kallberg Y, Persson B, Jörnvall H. 2003. Short-chaindehydrogenases/reductases (SDR): The 2002 update. Chem Biol Interact143–144:247–253.
Painter J, Merritt EA. 2006. TLSMD web server for the generation of multi-group TLS models. J Appl Cryst 39:109–111.
Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM,Meng EC,Ferrin TE. 2004. UCSF chimera—Avisualization system for exploratoryresearch and analysis. J Comput Chem 25:1605–1612.
Ploux O, Lei Y, Vatanen K, Liu HW. 1995. Mechanistic studies on CDP-6-deoxy-delta 3,4-glucoseen reductase: The role of cysteine residues incatalysis as probed by chemical modification and site-directedmutagenesis. Biochemistry 34:4159–4168.
Rossmann MG, Moras D, Olsen KW. 1974. Chemical and biologicalevolution of nucleotide-binding protein. Nature 250:194–199.
Sambrook J, Fritsch, EF, Maniatis, T. 2001. Molecular cloning: A laboratorymanual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press.
Scrutton NS, Berry A, Perham RN. 1990. Redesign of the coenzymespecificity of a dehydrogenase by protein engineering. Nature 343:38–43.
Skerra A. 1994. Use of the tetracycline promoter for the tightly regulatedproduction of a murine antibody fragment in Escherichia coli. Gene151:131–135.
Söding J. 2005. Protein homology detection by HMM-HMM comparison.Bioinformatics 21:951–960.
Söding J, Biegert A, Lupas AN. 2005. The HHpred interactive server forprotein homology detection and structure prediction. Nucleic Acids Res33:W244–W248.
Sogabe S, Yoshizumi A, Fukami TA, Shiratori Y, Shimizu S, Takagi H,Nakamori S,WadaM. 2003. The crystal structure and stereospecificity oflevodione reductase from Corynebacterium aquaticum M-13. J Biol Chem278:19387–19395.
Studier FW, Daegelen P, Lenski RE, Maslov S, Kim JF. 2009. Understandingthe differences between genome sequences of Escherichia coli B strainsREL606 and BL21(DE3) and comparison of the E. coli B and K-12genomes. J Mol Biol 394:653–680.
Tanaka N, Nonaka T, Nakanishi M, Deyashiki Y, Hara A, Mitsui Y. 1996.Crystal structure of the ternary complex of mouse lung carbonylreductase at 1.8 8A resolution: The structural origin of coenzymespecificity in the short-chain dehydrogenase/reductase family. Structure4:33–45.
Thoden JB, Wohlers TM, Fridovich-Keil JL, Holden HM. 2000. Crystallo-graphic evidence for Tyr 157 functioning as the active site base in humanUDP-galactose 4-epimerase. Biochemistry 39:5691–5701.
Tietze S, Apostolakis J. 2007. GlamDock: Development and validation of anew docking tool on several thousand protein-ligand complexes. J ChemInf Model 47:1657–1672.
Wang W, Nema S, Teagarden D. 2010. Protein aggregation—Pathways andinfluencing factors. Int J Pharm 390:89–99.
Wierenga RK, Demaeyer MCH, Hol WGJ. 1985. Interaction of pyrophos-phate moieties with a-helixes in dinucleotide binding-proteins.Biochemistry 24:1346–1357.
Yamashita A, Kato H, Wakatsuki S, Tomizaki T, Nakatsu T, Nakajima K,Hashimoto T, Yamada Y, Oda J. 1999. Structure of tropinone reductase-II complexed with NADPþ and pseudotropine at 1.9 8A resolution:Implication for stereospecific substrate binding and catalysis. Biochem-istry 38:7630–7637.
Yaoi T, Miyazaki K, Oshima T, Komukai Y, Go M. 1996. Conversion ofthe coenzyme specificity of isocitrate dehydrogenase by modulereplacement. J Biochem 119:1014–1018.
Zhang L, Ahvazi B, Szittner R, Vrielink A, Meighen E. 1999. Change ofnucleotide specificity and enhancement of catalytic efficiency in singlepoint mutants of Vibrio harveyi aldehyde dehydrogenase. Biochemistry38:11440–11447.
2814 Biotechnology and Bioengineering, Vol. 110, No. 11, November, 2013





























