Embed Size (px)
Citation preview

Dandy-Walker Syndrome Associated with Congenital Heart Defects: Report of Three Cases
T. T. HUONG E. GOLDBATT D. A . SIMPSON
Introduction The Dandy-Walker syndrome may best be defined as a developmental posterior-fossa
cyst representing an expansion of the fourth ventricle and associated with partial or com- plete absence of the cerebellar vermis. Since it is a congenital defect, the syndrome may be expected to be associated with other congenital anomalies. Julh and Wesenberg (1966) listed a high incidence of associated anomalies, especially agenesis of the corpus callosum, but no cardiac anomalies were noted. We have found only three cases reported in the world literature since 1966 in which cardiac defects probably were associated with the Dandy- Walker syndrome (Blackburn and Belliveau 1971, Yarom and Fried 1971, Hart er al. 1972). In two of these cases the presence of the brain anomaly was unequivocal; in the third there were cerebral anomalies which were possibly related to the Dandy-Walker syndrome.
Between 1956 and 1974 we have encountered three cases of the Dandy-Walker syndrome with associated cardiac lesions out of a total of 10 children with Dandy-Walker anomalies treated at the Adelaide Children’s Hospital. The practical importance of such a rare association is that a ventriculo-peritoneal rather than a ventriculo-atrial shunt may be indicated when there is a major cardiac malformation. The association also throws some light on the embryology of the Dandy-Walker syndrome.
Case Reports CASE 1
This boy was delivered by forceps after 37 weeks gestation. Birthweight was 1.79kg and there was evidence of placental insufficiency. The family history was not significant. He was admitted to Adelaide Children’s Hospital when 10 days old because of congestive heart failure. Cyanotic episodes had been noted during gavage feeds. There was tachypnoea, tachycardia, and oedema of the feet. A grade 3 pansystolic murmur and third heart-sound were heard at the left sternal edge. Over the pulmonary area there were systolic and early diastolic murmurs. Head circumference was 31cm. Examination of the genitalia revealed testes in the groins, a median scrota1 cleft and a degree of chordee. Results of chromosomal studies were normal.
At two months, when the child had a second episode of congestive heart-failure, the head cir- cumference was 37cm and the fontanelle was large. Cardiac catheterisation at four months of age revealed a large ventricular septa1 defect associated
with pulmonary valvular and infundi bular stenosis. Signs of hydrocephalus became prominent. Air
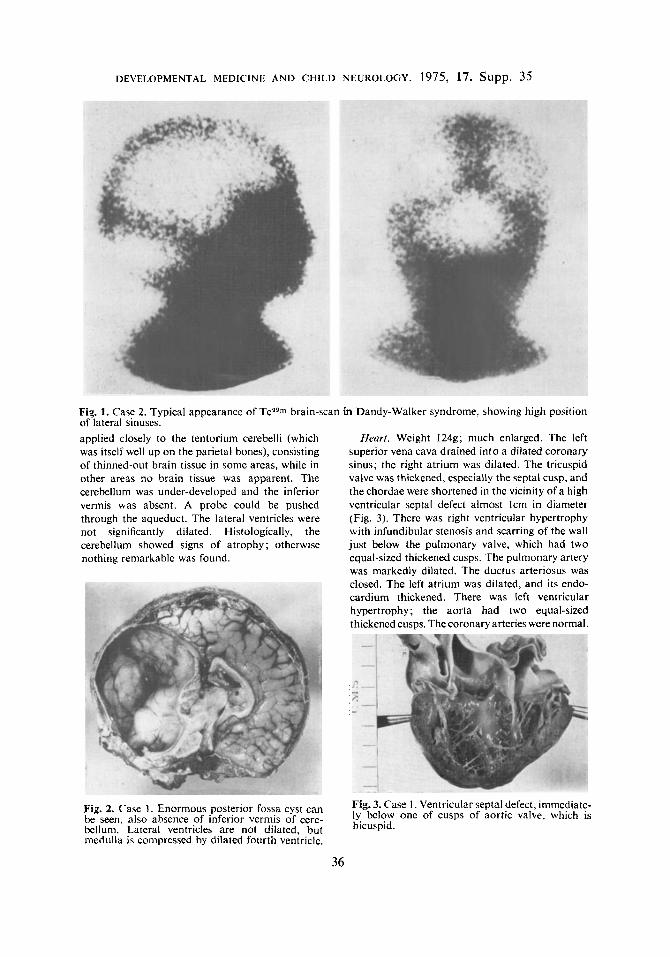
encephalography and ventriculography demon- strated dilated lateral ventricles and a grossly dilated fourth ventricle which was confluent with a huge posterior fossa cyst. Technetium brain-scan also showed grossly elevated lateral sinuses (Fig. 1 ).
At five months a ventriculo-peritoneal shunt was inserted. Two weeks later the posterior cranial fossa was explored, and the diagnosis of Dandy- Walker syndrome was confirmed. An unsuccess- ful attempt was made to establish normal CSF circulation and the child remained shunt- dependent. His general health slowly deteriorated and he died suddenly at the age of 21 months.
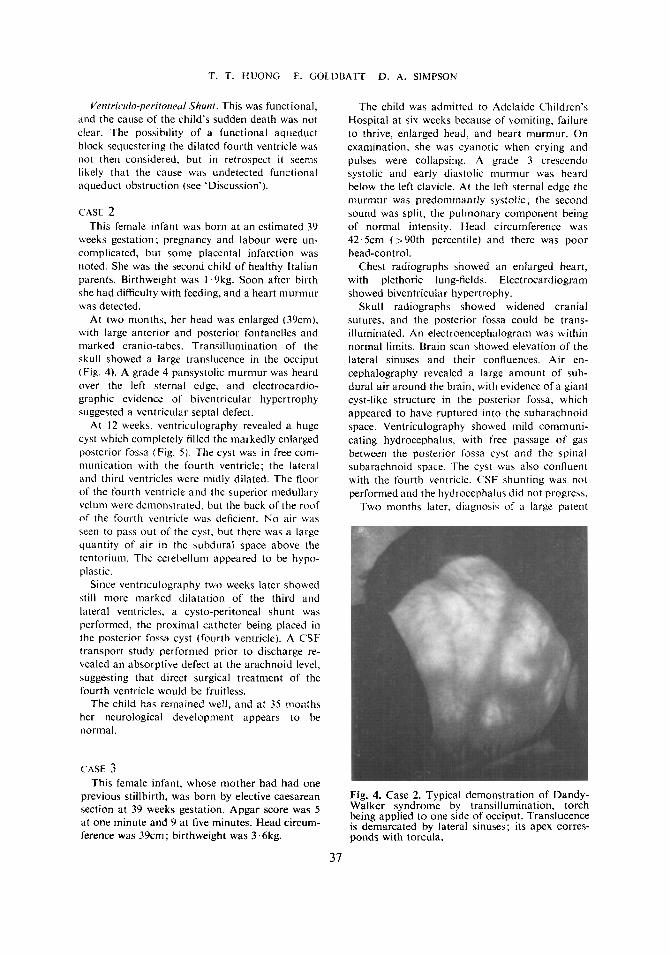
.4 utopss Bruin. A large cyst was found (9.5 r: 7’5cm).
representing a hugely dilated fourth ventricle (Fig. 2). The wall of the cyst was a thin membrane
Departments of Cardiology and Neurosurgery, Adelaide Children’s Hospital. 72 King William Road, North Adelaide, Australia, 5006.
35
DEVELOPMENTAL MEDICINE AND CHILD NEUROLOGY. 1975, 17. Supp. 35
Fiq. 1. Case 2. Typical appearance of Tcg9" brain-scan in Dandy-Walker syndrome, showing high position of lateral sinuses. applied closely to the tentorium cerebelli (which H a r t . Weight 124g; much enlarged. The left was itself well up on the parietal bones), consisting superior vena cava drained into a dilated coronary of thinned-out brain tissue in some areas, while in sinus; the right atrium was dilated. The tricuspid other areas no brain tissue was apparent. The valve was thickened, especially the septal cusp, and cerebellum was under-developed and the inferior thechordae were shortened in the vicinity of a high vermis was absent. A probe could be pushed ventricular septal defect almost Icm in diameter through the aqueduct. The lateral ventricles were (Fig. 3). There was right ventricular hypertrophy not significantly dilated. Histologically, the with infundibular stenosis and scarring of the wall cerebellum showed signs of atrophy; otherwise just below the pulmonary valve, which had two nothing remarkable was found. equal-sized thickened cusps. The pulmonary artery
was markedly dilated. The ductus arteriosus was closed. The left atrium was dilated, and its endo- cardium thickened. There was left ventricular hypertrophy; the aorta had two equal-sized thickened cusps. Thecoronary arteries were normal.
Fig. 2. Case E~~~~~~~ posterior fossa cyst can be seen, also absence of inferior vermis of cere- bellum. Lateral ventricles are not dilated. but
Fig. 3. Case I . Ventricular septal defect, immediate- b' below one of CUSPS of aortic valve, which is bicuspid.
medulla is compressed by dilated fourth ventricle.
36
T. T. HUONG E. GOLDBATT D. A. SIMPSON
~ e n r r i c u l o - p ~ r i t o n ~ ~ ~ i l Shrcrit. This was functional, and the cause of the child’s sudden death was not clear. The possibility of a functional aqueduct block sequestering the dilated fourth ventricle was not then considered, but in retrospect i t seems likely that the cause was undetected functional aqueduct obstruction (see ‘Discussion’).
CASE 2 This female infant was born at an estimated 39
weeks gestation: pregnancy and labour were un- complicated, but some placental infarction was noted. She was the second child of healthy Italian parents. Birthweight was 1 .9kg. Soon after birth she had difficulty with feeding, and a heart murmur was detected.
At two months, her head was enlarged (39cm), with large anterior and posterior fontanelles and marked cranio-tabes. Transilluniination of the skull showed a large translucence in the occiput (Fig. 4). A grade 4 pansystolic murmur was heard over the left sternal edge, and electrocardio- graphic evidence of biventricular hypertrophy suggested a ventricular septa1 defect.
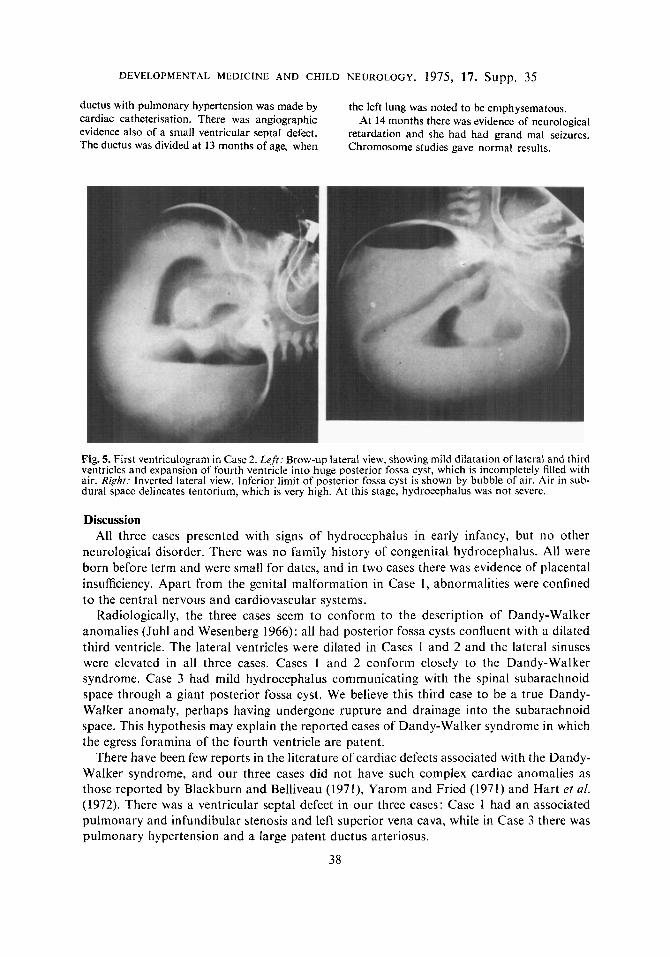
At 12 weeks, ventriculography revealed a huge cyst which conipletely filled the markedly enlarged posterior fossa (Fig. 5 ) . The cyst was in free com- munication with the fourth ventricle; the lateral and third ventricles were niidly dilated. The floor o f the four th ventricle and the superior medullary velum were demonstrated. hut the back of the roof of the fourth ventricle was deficient. N o air was seen to pass out of the cyst, but there was a large quantity of air in the subdural space above the tentoriuni. The cerebelluni appeared to be hypo- plastic.
Since ventriculography two weeks later showed still more marked dilatation of the third and lateral ventricles, a cysto-peritoneal shunt was performed, the proximal catheter being placed in the posterior fossa cyst (fourth ventricle). A CSF transport study performed prior to discharge re- vealed an absorptive defect at the arachnoid level, suggesting that direct surgical treatment of the fourth ventricle would be fruitless.
The child has remained well, and at 35 months her neurological development appears to be normal.
CASE 3 This female infant, whose mother had had one
previous stillbirth. was born by elective caesarean section at 39 weeks gestation. Apgar score was 5 at one minute and 9 at five minutes. Head circum- ference was 39cm; birthweight was 3 ‘6kg.
The child was admitted to Adelaide Children’s Hospital at six weeks because of vomiting. failure to thrive, enlarged head, and heart murmur. On examination, she was cyanotic when crying and pulses were collapsing. A grade 3 crescendo systolic and early diastolic murmur was heard below the left clavicle. At the left sternal edge the murmur was predominantly systolic; the second sound was split, the pulmonary component being of normal intensity. Head circumference was 42.5cm (>90th percentile) and there was poor head-control.
Chest radiographs showed an enlarged heart, with plethoric lung-fields. Electrocardiogram showed biventricular hypertrophy.
Skull radiographs showed widened cranial sutures, and the posterior fossa could be trans- illuminated. An electroencephalogram was within normal limits. Brain scan showed elevation of the lateral sinuses and their confluences. Air en- cephalography revealed a large amount of sub- dural air around the brain, with evidence of a giant cyst-like structure in the posterior fossa. which appeared to have ruptured into the subarachnoid space. Ventriculography showed mild communi- cating hydrocephalus, with free passage of gas between the posterior fossa cyit and the spinal subarachnoid space. The cyst was also confluent with the fourth ventricle. CSF shunting was not performed and the hydrocephalus did not progress.
Two months later, diagnosis of a large patent
Fig. 4. Case 2. Typical demonstration of Dandy- Walker syndrome by transillumination, torch being applied to one side of occiput. Translucence is demarcated by lateral sinuses; its apex corres- ponds with torcula.
DEVELOPMENTAL MEDICINE AND CHILD NEUROLOGY. 1975, 17. SUpp. 35
ductus with pulmonary hypertension was made by cardiac catheterisation. There was angiographic evidence also of a small ventricular septal defect. The ductus was divided at 13 months of age, when
the left lung was noted to be emphysematous. At 14 months there was evidence of neurological
retardation and she had had grand ma1 seizures. Chromosome studies gave normal results,
Fig. 5. First ventriculogram in Case 2. Left: Brow-up lateral view, showing mild dilatation of lateral and third ventricles and expansion of fourth ventricle into huge posterior fossa cyst, which is incompletely filled with air. Righf: Inverted lateral view. lnferior limit of posterior fossa cyst is shown by bubble of air. Air in sub- dural space delineates tentorium, which is very high. At this stage, hydrocephalus was not severe.
Discussion All three cases presented with signs of hydrocephalus in early infancy, but no other
neurological disorder. There was no family history of congenital hydrocephalus. All were born before term and were small for dates, and in two cases there was evidence of placental insufficiency. Apart from the genital malformation in Case I , abnormalities were confined to the central nervous and cardiovascular systems.
Radiologically, the three cases seem to conform to the description of Dandy-Walker anomalies (Juhl and Wesenberg 1966): all had posterior fossa cysts confluent with a dilated third ventricle. The lateral ventricles were dilated in Cases I and 2 and the lateral sinuses were elevated in all three cases. Cases 1 and 2 conform closely to the Dandy-Walker syndrome. Case 3 had mild hydrocephalus communicating with the spinal subarachnoid space through a giant posterior fossa cyst. We believe this third case to be a true Dandy- Walker anomaly, perhaps having undergone rupture and drainage into the subarachnoid space. This hypothesis may explain the reported cases of Dandy-Walker syndrome in which the egress foramina of the fourth ventricle are patent.
There have been few reports in the literature of cardiac defects associated with the Dandy- Walker syndrome, and our three cases did not have such complex cardiac anomalies as those reported by Blackburn and Belliveau (1971), Yarom and Fried (1971) and Hart el a/. (1972). There was a ventricular septal defect in our three cases: Case 1 had an associated pulmonary and infundibular stenosis and left superior vena cava, while in Case 3 there was pulmonary hypertension and a large patent ductus arteriosus.
38

T. T. HUONG E. GOLDBATT D. A. SIMPSON
There has been speculation about the embryological association between brain anomalies and the Dandy-Walker syndrome. Dandy (1921 ), and Taggart and Walker (1942), attributed cystic enlargement of the fourth ventricle and consequent failure of development of the vermis to atresia of the foramina of Luschka and Magendie. Many other authors have supported this concept, but Benda (1954) thought that the atresia of the developing fourth ventricular foramina was only a non-essential part of the syndrome. He favoured a relation- ship between the anomaly and the group of rachischisis malformations. Brodal and Hauglie- Hanssen (1959) proposed that these anomalies of the cerebellum and roof of the fourth ventricle arise at a fetal stage considerably earlier than the formation of the foramina of Luschka and Magendie. Hart et a/. (1972) thought that that agenesis of the corpus callosum, which is a very common association in the Dandy-Walker syndrome, must be due to a factor operating very early in utero, since the corpus callosum appears at about three months gestational age.
However, studies of the normal development of the human fourth ventricle (Brocklehurst 1969) suggest that the original concept of a primary atresia of the foramina of Luschka and Magendie may still be tenable. Brocklehurst has shown convincingly that communica- tion between the fourth ventricle and the developing subarachnoid space is established as early as the eighth week: therefore it can be argued that both the agenesis of the cerebellar vermis and the atresia of the egress foramina might result from i: single embryological defect or noxious agent. At this early stage, such an agent could strike the lateral rudiment of the cerebellar hemisphere (in fact we have seen agenesis of the whole of one cerebellar hemisphere in an otherwise typical Dandy-Walker syndrome).
The origin of the ventricular septal defect may give some information about the relation- ship between the Dandy-Walker syndrome and cardiac defects. The development of the muscular intraventricular septum begins early: by the sixth week it is well established (Hamilton and Mossman 1972). However, most ventricular septal defects are located in the membranous part of the septum (e.y. high ventricular septal defect seen a t autopsy in Case I ) . The process of formation of the membranous septum is complex. Division of the atrio-ventricular canal into right and left orifices by sub-endocardial cushions may be seen even earlier than the muscular septum. Fusion of these sub-endocardial cushions with the top of the muscular intraventricular septum occiirs during the sixth week and is complete by seven weeks. The complete formation of the membranous septum can be seen by about eight weeks. Presumably an embryological defect or noxious agent could induce a ventricular septal defect at this age: and the co-incidental anomalies in our three cases might therefore arise simultaneously. On the other hand, multiple errors in development may not happen at the same time; a teratogen could well continue to affect the embryo for a considerable period of time.
I t is curious that, despite only rare reports in the literature, we have seen three cases of cardiac lesions associated with Dandy-Walker anomalies. A hypothesis that deserves serious consideration is that some environmental factors operating in South Australia contributed to the occurrence of these three cases.
The surgical management of the Dandy-Walker syndrome has been well discussed by Raimondi et a/. (1969). Of course some cases never require surgical treatment, and a few (especially in older children) may successfully be treated by opening the fourth ventricle into the spinal subarachnoid space (although we have had n o success with this operation). Most commonly, a s h u n t will be necessary. Many writers have discussed the complications of ventriculo-atrial shunts, which include septicaemia, endocarditis, pulmonary embolism,
39

DEVELOPMENTAL M E D I C N E A N D CHILD NEUROLOGY. 1975, 17. SUpP. 35
superior vena cava obstruction and thrombo-embolism. Cardiac lesions such as ventricular septa1 defect could well precipitate such complications; therefore we chose peritoneal drainage for two of our cases, with satisfactory results. Our experience in these and other cases also suggests that drainage from the cyst (dilated fourth ventricle) may be preferable to drainage from the lateral ventricle, both to avoid blockage by choroid plexus and because secondary aqueduct obstruction is less likely to develop (Taggart and Walker 1942, Raimondi et a(. 1969).
ArknowledRernents: We thank Dr. R. F. Carter, Director of the Department of Histopathology, Adelaide Children’s Hospital, for the autopsy report quoted in Case 1 . One of us (T.H.) was in receipt of a Colombo Plan Scholarship for the Commonwealth of Australia during the preparation of this study, which was also assisted by the Neurosurgical Research Foundation of South Australia, Inc. (Mollie O’Leary Bequest).
SUMMARY
Three cases of Dandy-Walker syndrome associated with congenital heart defects are reported, and their management is described. The various theories concerning the aetiology of the Dandy-Walker syndrome are discussed and it is suggested that the theory of primary developmental atresia of the foramina of Magendie and Luschka is still acceptable.
If the child with Dandy-Walker syndrome requires treatment by extracranial CSF drainage, it is proposed that a ventriculo-peritonea1 shunt is preferable when there is any suspicion of cardiac anomaly. The proximal catheter should be placed in the posterior fossa cyst (dilated fourth ventricle) rather than in the lateral ventricle.
RESUME SJndrome de Dandy- Walker associk ri des cardiopathies congknitales: trois cas
Trois cas de Dandy-Walker associts A une cardiopathie congtnitale sont rapportes et leur traitement est dkcrit. Les dif€trentes theories concernant l’ttiologie du syndrome de Dandy-Walker sont discutees et il est suggtrt que la theorie d’une atrtsie du dtveloppement primaire des trous de Magendie et Luschka est toujours acceptable.
Si l’enfant prtsentant un syndrome de Dandy-Walker necessite un traitement par drainage extra-crinien du LCR, les auteurs pensent qu’un shunt ventriculo-ptritontal est prtfirable chaque fois qu’une anomalie cardiaque est suspectie. Le catheter proximal devrait Ctre place dans la citerne de la fosse posttrieure (quatrikme ventricule dilate) plut8t que dans le ventricule lateral.
ZUSAMMENFASSUNG Dandy- Walker Syndrom verbunden mit angeborenen Herzfehlern: Berirht iiber drei Falle
Es wird uber drei Falle mit Dandy-Walker Syndrom und angeborenen Herzfehlern berichtet und die Therapie beschrieben. Eine Diskussion uber die verschiedenen Theorien uber die Atiologie des Dandy-Walker Syndroms schlieat sich an mit dem Ergebnis, dap die Theorie uber die primare Atresie der Foramina Magendie und Luschka noch weiterhin Bestand hat.
Wenn ein Kind mit einem Dandy-Walker Syndrom mit einer Liquordrainage behandelt werden mup, sollte man bei dem geringsten Verdacht auf einen Herzfehler einen ventriculo- peritonealen Shunt implantieren. Der proximale Katheter sollte besser in die Cyste der Fossa posterior (erwieterter vierter Ventrikel) eingelegt werden als in den Seitenventrikel.
40

T. T. HUONG E. GOLDBATT D. A . SIMPSON
RESUMEN
Sinclronw cle Dundjj- Wnlk er asociudo con malformaciones congkriitas cardinens: aportacicin lie tres cusos
Se aportan tres casos de sindrome de Dandy-Walker asociados con malformaciones congenitas cardiacas y se describe su tratamiento. Se discuten varias teorias respecto a la etiologia del sindrome de Dandy-Walker y se sugiere que la teoria de una atresia primaria en el desarrollo de 10s agujeros de Magendie y Luschka es todavia aceptable.
Si el nifio con el sindrome de Dandy-Walker requiere tratamiento por medio de u n drenaje extracraneano de liquido cefalorraquideo, se propone que es preferible una deri- vxion ventr-iculoperitoneal, si se sospecha una anomalia cardiaca. El catCter proximal debe ser colocado en el quiste de la fosa posterior (cuarto ventriculo dilatado) mis bien que en el bentriculo lateral.
REFERENCES Benda, C . E. ( 1954) ‘Dandy-Walker syndrome or so-called atresia of the foramen of Magendie.’ Journal of
Neirroporholo.qi. otid .&perit?ientd Neurolog~’, 13, 14. Blackburn, M. G. , Relliveau. R. E. (1971) ‘Ellis-van Creveld syndrome. A report of previously undescrihed
anomalies in two siblings.’ American Jourtral o/ the Diseases of Childreti, 122, 267. Brocklehurct, G . ( 1969) ’The development of the human cerebrospinal fluid pathway with particular refer-
ence to the roof of the fourth ventricle.’ Journu/ of Anatomy, 105,467. Brodal, A,, Hauglie-Hanssen. E. (1959) ’Congenital hydrocephalus with defective development of the
cerebellar vermis (Dandy-Walker syndrome): clinical and anatomical findings in two cases with particular reference to the so-called atresia of the foramina of Magendie and Luschka.’ fournrrl of Ncwvlogj , , Neiiro.siir~qer>.. t r t d Psychiiitrj~, 22, 99.
Dandy, W. E. ( 1921) ‘The diagnosis and treatment of hydrocephalus due to occlusions of the foramina of Magendie and Luschka.’ Siwyery. G v n c m h ~ ~ ~ ~ rmd Ohtetr ics , 32, I 12.
Hamilton. W. J . , Mossman, H. W. (1972) Hiinion Ovbrjwlogj~, Prenatal Dedopi t ient of Fortti onrl Function, 4th erl. Cambridge: Heffer.
Hart, M . N., Malamud, N.. Ellis, W. G . (1972) ‘The Dandy-Walker syndrome. A clinicopathologic study based on 28 cases.’ Neuro/ogv, 22, 771.
Juhl, J . H.. Wesenherg, R. 1. (1966) ‘Radiological findings in congenital and acquired occlusions of the foramina of Magendie and Luschka.’ RadioloKv, 86, 801.
Raimondi, A. .I., Saniuelson, G., Yarzagaray. 1.. Norton, T. (1969) ‘Atresia of the foramina of Luschka and hfagendie: the Dandy-Walker cyst.’ Journal o/ Nrwro.wrger.v, 31, 202.
Taggart. J . K.. Walker. A. E. (1942) ‘Congenital atresia of the foramens of Luschka and Magendie.’ Archive.\ of Neiirolo,i~i. nnd Pqlzchiritr!.. 48, 583.
Yarom, R.. Fried, K. (1971) ‘Hypertrophy of heart with outflow tract obstruction and myocardial necrosis i n an infant mith multiple congenital malformations.’ British Heart Journal, 33, 420.
41



![Hospital-based Birth Defects · PDF fileHospital-based birth defects surveillance: ... Birth defects are one type of congenital conditions. [Slides 5-6] ... • Congenital malformations:](https://img.pdfslide.net/doc/110x75/5ab1551c7f8b9a7e1d8c4658/hospital-based-birth-defects-birth-defects-surveillance-birth-defects-are-one.jpg)

























