Embed Size (px)
DESCRIPTION
A paper on RAAS
Citation preview

Dysregulated Renin–Angiotensin–Aldosterone SystemContributes to Pulmonary Arterial Hypertension
Frances S. de Man1,2,3,4, Ly Tu1,2, M. Louis Handoko3,4, Silvia Rain3, Gerrina Ruiter3,Charlene Francois1,2, Ingrid Schalij3, Peter Dorfmuller1,2, Gerald Simonneau1,2,5, Elie Fadel1,2,Frederic Perros1,2, Anco Boonstra3, Piet E. Postmus3, Jolanda van der Velden4, Anton Vonk-Noordegraaf3,Marc Humbert1,2,5, Saadia Eddahibi1,2, and Christophe Guignabert1,2
1INSERM UMR 999, LabEx LERMIT, Centre Chirurgical Marie Lannelongue, Le Plessis-Robinson, France; 2School of Medicine,Universite Paris-Sud, Kremlin-Bicetre, France; 3Department of Pulmonology and 4Department of Physiology, VU University Medical
Center/Institute of Cardiovascular Research, Amsterdam, The Netherlands; and 5AP-HP, Service de Pneumologie, Centre de
Reference de l’Hypertension Pulmonaire Severe, DHU Thorax Innovation, Hopital de Bicetre, Le Kremlin-Bicetre, France
Rationale: Patients with idiopathic pulmonary arterial hypertension(iPAH) often have a low cardiac output. To compensate, neurohor-monal systems such as the renin–angiotensin–aldosterone system(RAAS) and the sympathetic nervous system are up-regulated, butthismayhave long-termnegativeeffectson theprogressionof iPAH.Objectives: Assess systemic and pulmonary RAAS activity in patientswith iPAH and determine the efficacy of chronic RAAS inhibition inexperimental PAH.Methods: We collected 79 blood samples from 58 patients with iPAHin the VUUniversityMedical Center Amsterdam (between 2004 and2010) to determine systemic RAAS activity.Measurements and Main Results: We observed increased levels of re-nin, angiotensin (Ang)I, and AngII, which were associated with dis-ease progression (P , 0.05) and mortality (P , 0.05). To determinepulmonary RAAS activity, lung specimens were obtained frompatientswith iPAH (during lung transplantation, n¼ 13) and controlsubjects (during lobectomy or pneumonectomy for cancer, n¼ 14).Local RAAS activity in pulmonary arteries of patients with iPAH wasincreased, demonstrated by elevated angiotensin-converting en-zyme activity in pulmonary endothelial cells and increased AngIItype 1 (AT1) receptor expression and signaling. In addition, localRAASup-regulationwas associatedwith increasedpulmonary arterysmooth muscle cell proliferation via enhanced AT1 receptor signal-ing in patients with iPAH compared with control subjects. Finally, todetermine the therapeutic potential of RAAS activity, we assessed
the chronic effects of an AT1 receptor antagonist (losartan) in themonocrotaline PAH ratmodel (60mg/kg). Losartan delayed diseaseprogression, decreased right ventricular afterload and pulmonaryvascular remodeling, and restored right ventricular–arterial cou-pling in rats with PAH.Conclusions: Systemic andpulmonary RAASactivities are increased inpatients with iPAH and are associated with increased pulmonaryvascular remodeling. Chronic inhibition of RAAS by losartan is ben-eficial in experimental PAH.
Keywords: idiopathic pulmonary arterial hypertension; renin angioten-
sin system; endothelial cells; smooth muscle cells; monocrotaline
Idiopathic pulmonary arterial hypertension (iPAH) is a fatal dis-ease with an average mortality rate of 58% within 3 years afterdiagnosis (1). The disease is characterized by excessive pulmo-nary vascular remodeling leading to elevated pressures in thepulmonary arterial system and the right heart (2). As a conse-quence, patients with iPAH often have a low cardiac output(2). To compensate, neurohormonal systems like the renin-angiotensin-aldosterone system (RAAS) and the sympatheticnervous system (SNS) are up-regulated, but this may havelong-term negative effects on the progression of iPAH (3).
In contrast to the increasing knowledge about the role of al-tered SNS signaling in the development of iPAH (4–7), only little
(Received in original form March 7, 2012; accepted in final form July 13, 2012)
Supported by a European Respiratory Society/Marie Curie Joint Research Fellow-
ship MC 1120-2009 (F.S.d.M.). The research leading to these results has received
funding from the European Respiratory Society and the European Community’s
Seventh Framework Program FP7/2007–2013 Marie Curie Actions grant agree-
ment RESPIRE, PCOFUND-GA-2008-229571. Also supported by the Netherlands
Organization for Scientific Research, grant Mozaıek 017.002.122 (M.L.H.) and
Vidi 917.96.306 (A.V.-N.), The Hague, The Netherlands.
Author Contributions: F.S.d.M., L.T., M.L.H., A.V.-N., M.H., S.E., and C.G. con-
tributed to conception and design of the present study. F.S.d.M., L.T., S.R., G.R.,
C.F., I.S., P.D., G.S., E.F., F.P., A.B., and C.G. contributed to the acquisition of
data. F.S.d.M., L.T., M.L.H., P.E.P., J.v.d.V., A.V.-N., M.H., S.E., and C.G. contrib-
uted to analysis or interpretation of the data. F.S.d.M., L.T., M.L.H., and C.G.
drafted the manuscript, and S.R., G.R., C.F., I.S., P.D., G.S., E.F., F.P., A.B., P.E.P.,
J.v.d.V., A.V.-N., M.H., and S.E. subsequently revised the article critically for
intellectual content. All authors gave their final approval of this version of the
manuscript to be published.
Correspondence and requests for reprints should be addressed to Frances S. de
Man, Ph.D., VU University Medical Center, Department of Pulmonology, De
Boelelaan 1117, 1081 HV Amsterdam, The Netherlands. E-mail: fs.deman@
vumc.nl
This article has an online supplement, which is accessible from this issue’s table of
contents at www.atsjournals.org
Am J Respir Crit Care Med Vol 186, Iss. 8, pp 780–789, Oct 15, 2012
Copyright ª 2012 by the American Thoracic Society
Originally Published in Press as DOI: 10.1164/rccm.201203-0411OC on August 2, 2012
Internet address: www.atsjournals.org
AT A GLANCE COMMENTARY
Scientific Knowledge on the Subject
Accumulating evidences suggest increased renin–angiotensin–aldosterone system (RAAS) activity in idiopathic pulmonaryarterial hypertension (iPAH). However, the functional con-sequences of these abnormalities to iPAH pathogenesis re-main obscure.
What This Study Adds to the Field
In this translational study we have demonstrated that: (1)systemic RAAS activation is increased in patients withiPAH and associated with worse prognosis; (2) due to theincreased ACE activity, pulmonary endothelial cells ofpatients with iPAH produce more angiotensin II, whichinduces significant pulmonary artery smooth muscle cellproliferation of patients with iPAH via AT1 receptorsignaling; (3) AT1 receptor expression and signaling areup-regulated in patients with iPAH, without changes in AT2
receptor expression; and (4) the use of an AT1 receptorblocker could have potential clinical implications due to itspositive effects on pulmonary vascular remodeling.

is known about alterations in RAAS signaling in iPAH. Forfiaand colleagues demonstrated an association between the occur-rence of hyponatremia (an indirect indicator of RAAS activity),right heart failure, and poor survival in PAH (8). Furthermore,two independent studies have reported that certain polymorphismsof the angiotensin-converting enzyme (ACE) (the microsatelliteinsertion/deletion polymorphism) and angiotensin II type 1 (AT1)receptor (the 1166C polymorphism) are associated with diseaseprogression in patients with iPAH, suggesting involvementof RAAS (9, 10). Finally, Orte and colleagues demonstrated in-creased immunostaining of ACE in the endothelium of intraaci-nar peripheral pulmonary arteries in lung tissue of patients withPAH (11). Taken together, these studies suggest involvement ofRAAS, but its pathophysiological mechanism remains unclear.To gain further insights into the role of RAAS in iPAH, we haveinvestigated whether systemic RAAS activity (in serum of patientswith iPAH) is associated with disease progression. Furthermore,we studied in detail the influence of pulmonary RAAS activity onpulmonary vascular remodeling, using cultured human pulmonaryendothelial (P-ECs) and smooth muscle cells (PA-SMCs) derivedfrom patients with iPAH and control patients.
There are several therapeutic strategies to interfere in theRAAS signaling: renin inhibitors reduce the conversion of angio-tensinogen into angiotensin (Ang) I, ACE inhibitors reduce theconversion of AngI into AngII, and AT1 receptor antagonists in-hibit binding of AngII to AT1 receptor (12). Due to the unsatis-factory reduction in pulmonary vascular resistance and occurrenceof systemic hypotension in a small case study of seven patients withPAH, larger studies of the therapeutic potential of ACE inhibitorshave not been performed (13). In two recent preclinical studies itwas demonstrated that ACE2 supplementation prevented thedevelopment of MCT-induced PAH (14, 15). ACE2 functions asa carboxypeptidase and cleaves a single carboxylate-terminal res-idue from AngII to form Ang(1–7) (16). Ang(1–7) is known tohave vasodilatory effects via binding to the Ang(1–7) receptor(MAS receptor) and antagonizes the actions of AngII (16). How-ever, molecular biological investigations have shown substantialincrease in renin, angiotensinogen, ACE, and AT1 receptor expres-sion in lungs of these monocrotaline-treated rats but no changes inMAS receptor or ACE2 (14). Thus, the currently available AT1
receptor blockers that do not block AT2 receptor and ACE2 sig-naling now enable us to test the hypothesis that AT1 receptorantagonists could be a more relevant therapy for PAH (17, 18).
To our knowledge, there are only three studies that have usedAT1 receptor blockers to prevent consequences of different mod-els of experimental PAH (19–21), but no study that has studiedtheir efficacy in established PAH with right ventricular (RV) hy-pertrophy. Therefore, we evaluate the effect of losartan treatmentinitiated after ECG confirmation of PAH and RV hypertrophy.
The goals of this study were (1) to determine whether RAASactivity is increased in patients with iPAH, both systemically inserum and locally in pulmonary arteries (PA) and P-ECs ofpatients with iPAH and control subjects; (2) to investigatewhether local RAAS activity could induce proliferation ofhuman cultured PA-SMCs derived from patients with iPAHand control subjects; and (3) to elucidate the therapeutic poten-tial of RAAS, by evaluating the effects of chronic AT1 receptorinhibition in an experimental PAH model.
Some of the results of this study have been previously re-ported in the form of an abstract (22, 23).
METHODS
Part I: Systemic RAAS Activity
Between 2004 and 2010, 879 blood samples were collected from patientsadmitted to the Pulmonology Department of the VU University
Medical Center Amsterdam for standard clinical care. From these sam-ples, 79 blood samples were obtained from patients with iPAH with notreatment history with b blockers or RAAS inhibitors. Measurementsof serum RAAS were performed in the IJssellandziekenhuis (Capellea/d IJssel, The Netherlands) as described in the online supplement.
Part II: Local RAAS Activity in Pulmonary Vasculature
of Patients with iPAH
This part of the study was approved by the local ethics committee of theCPP Ile-de-France VII, Le Kremlin-Bicêtre (France). All patients gaveinformed consent before the study. Lung specimens were obtainedduring lung transplantation (iPAH, n ¼ 13) and during lobectomy orpneumonectomy for cancer (control subjects, n ¼ 14). In control sub-jects, pulmonary cells were isolated distant from tumor areas. PAs,P-ECs, and PA-SMCs of patients with iPAH and control subjects wereisolated as described before (24, 25). PAs were used for protein ana-lyses of AT1 receptor, AT2 receptor expression, and SRC and extracel-lular regulated kinase (ERK) activation. Localization of AT1 receptor,AngII production by P-ECs, and PA-SMC proliferation after exposure ofAngII were assessed as described in the online supplement.
Part III: Effects of Chronic AT1 Receptor Inhibition
in Experimental PAH
All animal experiments were approved by the Institutional AnimalCare and Use Committee of the VU University Amsterdam (theNetherlands).
Before the efficacy study, we performed a dose-finding study to de-termine the maximum tolerated dose of losartan, as described before(5). The maximum tolerated dose was defined as less than 10% reduc-tion in systemic blood pressure.
Subsequently, efficacy of chronic losartan treatment was tested in27 rats (no telemetry): 9 control rats and 18 rats with PAH (monocrotaline60 mg/kg). Ten days after monocrotaline injection (when rats have devel-oped PAH) (5), rats with PAH were randomized for losartan treatment(20 mg/kg dissolved in vanilla pudding) or vehicle (vanilla pudding) oncedaily (n ¼ 9/group). Rats were treated for maximal 25 days.
For hemodynamic assessment, longitudinal echocardiography andpressure-volume analyses were performed (5, 26). To determine mor-phological changes in heart and lungs, RV hypertrophy and muscula-rization of pulmonary arterioles were assessed (5, 26).
Statistical Analyses
All data were verified for normal distribution and log transformed whennecessary. Data are presented as mean6 SEM, unless stated otherwise.A P value less than 0.05 was considered statistically significant. Com-parisons of serum renin, AngI, and AngII, with their upper limit ofnormal reference values, were performed by one-sample t test. Prog-nostic relevance of serum renin, AngI, and AngII was determined byunivariate Cox regression analyses and subsequent correction for con-founding effects of sex, age, and time to diagnosis. All-cause mortalityand lung transplantation were recorded as an event in the Cox regres-sion analyses. Subgroup analyses of serum follow-up measurementswere performed by two-way analysis of variance for repeated measure-ments. AT1 receptor expression and activity were analyzed by unpairedt test. P-EC AngII production, PA-SMC proliferation, the effects oflosartan on disease progression, and pressure-volume analyses were an-alyzed by one-way analysis of variance with Bonferroni post hoc ana-lyses. Histological data were analyzed using multilevel analysis to correctfor nonindependence of successive measurements per animal (MLwiN2.02.03; Center for Multilevel Modeling, Bristol, UK) (5, 26–28).
RESULTS
Part I: Systemic RAAS Activity
Systemic RAAS activity was measured in 79 serum samples of 58patients with iPAH; 9 patients were naive of treatment on the dayof sampling, and from 21 patients follow-up serum samples wereavailable with a median follow-up of 39 months (interquartilerange, 19–48 mo). Patient characteristics are shown in Table 1.
de Man, Tu, Handoko, et al.: RAAS Activity in Patients with iPAH 781
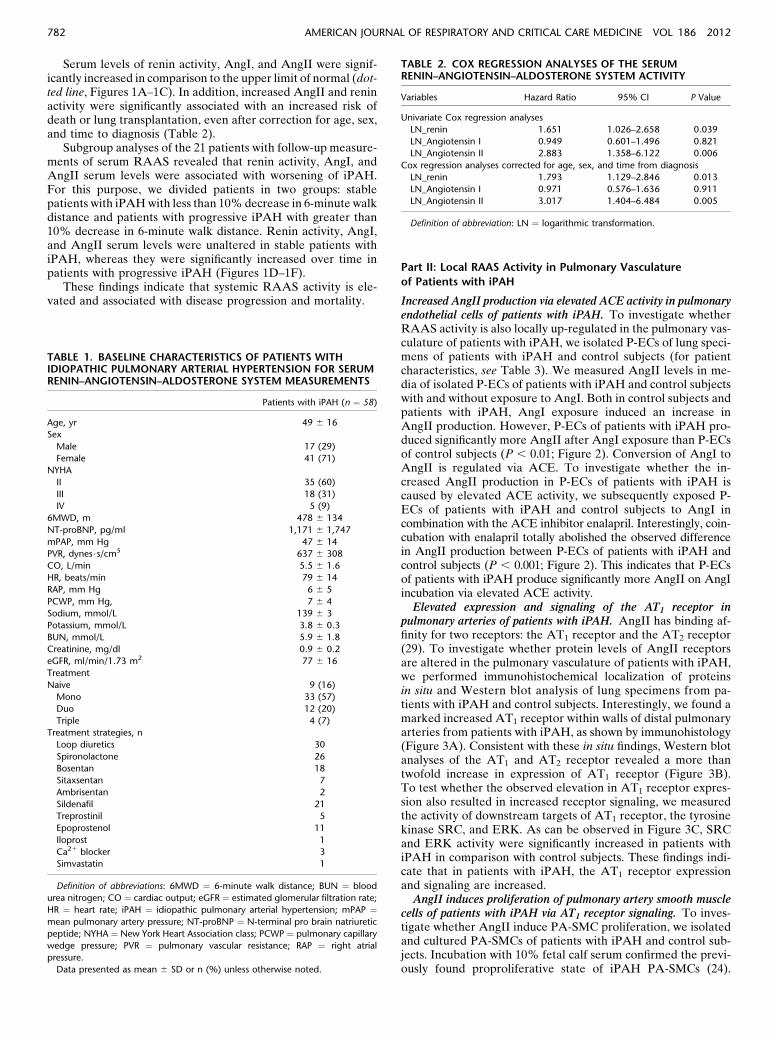
Serum levels of renin activity, AngI, and AngII were signif-icantly increased in comparison to the upper limit of normal (dot-ted line, Figures 1A–1C). In addition, increased AngII and reninactivity were significantly associated with an increased risk ofdeath or lung transplantation, even after correction for age, sex,and time to diagnosis (Table 2).
Subgroup analyses of the 21 patients with follow-up measure-ments of serum RAAS revealed that renin activity, AngI, andAngII serum levels were associated with worsening of iPAH.For this purpose, we divided patients in two groups: stablepatients with iPAHwith less than 10%decrease in 6-minute walkdistance and patients with progressive iPAH with greater than10% decrease in 6-minute walk distance. Renin activity, AngI,and AngII serum levels were unaltered in stable patients withiPAH, whereas they were significantly increased over time inpatients with progressive iPAH (Figures 1D–1F).
These findings indicate that systemic RAAS activity is ele-vated and associated with disease progression and mortality.
Part II: Local RAAS Activity in Pulmonary Vasculature
of Patients with iPAH
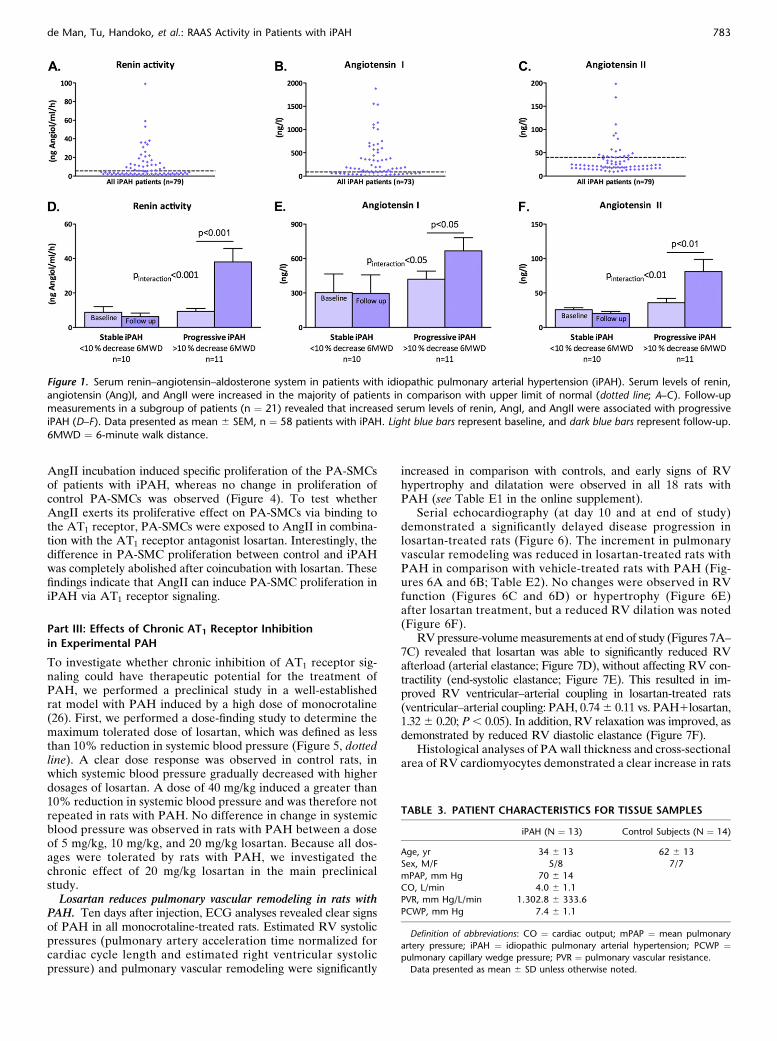
Increased AngII production via elevated ACE activity in pulmonaryendothelial cells of patients with iPAH. To investigate whetherRAAS activity is also locally up-regulated in the pulmonary vas-culature of patients with iPAH, we isolated P-ECs of lung speci-mens of patients with iPAH and control subjects (for patientcharacteristics, see Table 3). We measured AngII levels in me-dia of isolated P-ECs of patients with iPAH and control subjectswith and without exposure to AngI. Both in control subjects andpatients with iPAH, AngI exposure induced an increase inAngII production. However, P-ECs of patients with iPAH pro-duced significantly more AngII after AngI exposure than P-ECsof control subjects (P , 0.01; Figure 2). Conversion of AngI toAngII is regulated via ACE. To investigate whether the in-creased AngII production in P-ECs of patients with iPAH iscaused by elevated ACE activity, we subsequently exposed P-ECs of patients with iPAH and control subjects to AngI incombination with the ACE inhibitor enalapril. Interestingly, coin-cubation with enalapril totally abolished the observed differencein AngII production between P-ECs of patients with iPAH andcontrol subjects (P , 0.001; Figure 2). This indicates that P-ECsof patients with iPAH produce significantly more AngII on AngIincubation via elevated ACE activity.Elevated expression and signaling of the AT1 receptor in
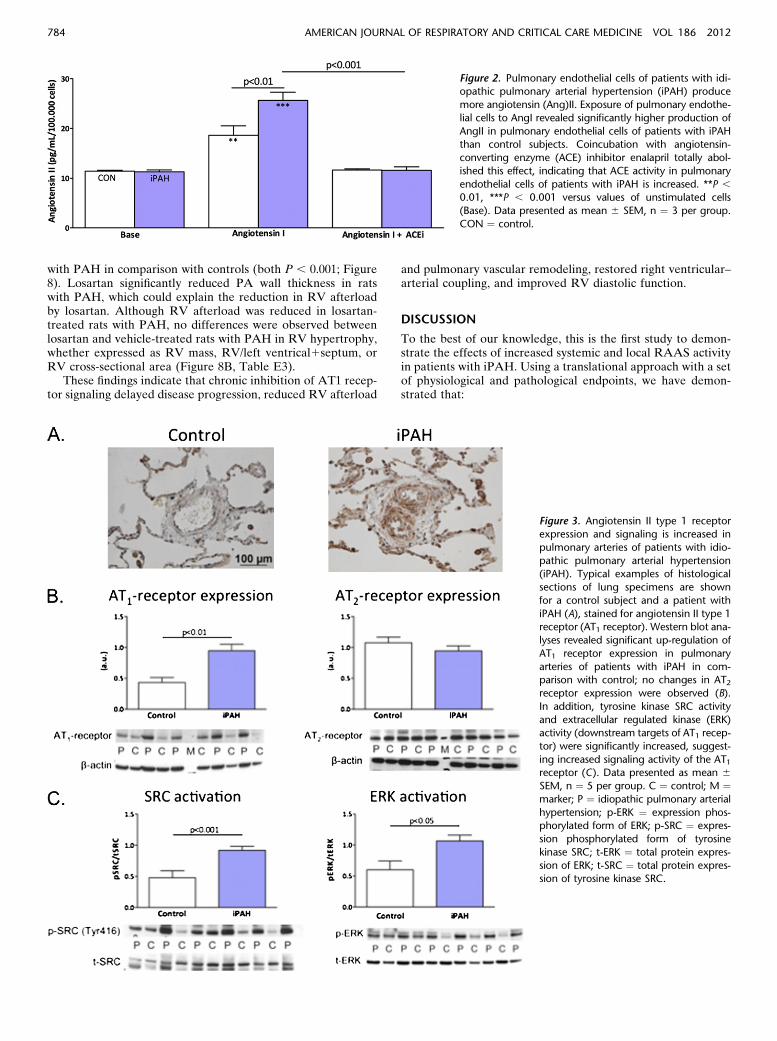
pulmonary arteries of patients with iPAH. AngII has binding af-finity for two receptors: the AT1 receptor and the AT2 receptor(29). To investigate whether protein levels of AngII receptorsare altered in the pulmonary vasculature of patients with iPAH,we performed immunohistochemical localization of proteinsin situ and Western blot analysis of lung specimens from pa-tients with iPAH and control subjects. Interestingly, we found amarked increased AT1 receptor within walls of distal pulmonaryarteries from patients with iPAH, as shown by immunohistology(Figure 3A). Consistent with these in situ findings, Western blotanalyses of the AT1 and AT2 receptor revealed a more thantwofold increase in expression of AT1 receptor (Figure 3B).To test whether the observed elevation in AT1 receptor expres-sion also resulted in increased receptor signaling, we measuredthe activity of downstream targets of AT1 receptor, the tyrosinekinase SRC, and ERK. As can be observed in Figure 3C, SRCand ERK activity were significantly increased in patients withiPAH in comparison with control subjects. These findings indi-cate that in patients with iPAH, the AT1 receptor expressionand signaling are increased.AngII induces proliferation of pulmonary artery smooth muscle
cells of patients with iPAH via AT1 receptor signaling. To inves-tigate whether AngII induce PA-SMC proliferation, we isolatedand cultured PA-SMCs of patients with iPAH and control sub-jects. Incubation with 10% fetal calf serum confirmed the previ-ously found proproliferative state of iPAH PA-SMCs (24).
TABLE 1. BASELINE CHARACTERISTICS OF PATIENTS WITHIDIOPATHIC PULMONARY ARTERIAL HYPERTENSION FOR SERUMRENIN–ANGIOTENSIN–ALDOSTERONE SYSTEM MEASUREMENTS
Patients with iPAH (n ¼ 58)
Age, yr 49 6 16
Sex
Male 17 (29)
Female 41 (71)
NYHA
II 35 (60)
III 18 (31)
IV 5 (9)
6MWD, m 478 6 134
NT-proBNP, pg/ml 1,171 6 1,747
mPAP, mm Hg 47 6 14
PVR, dynes$s/cm5 637 6 308
CO, L/min 5.5 6 1.6
HR, beats/min 79 6 14
RAP, mm Hg 6 6 5
PCWP, mm Hg, 7 6 4
Sodium, mmol/L 139 6 3
Potassium, mmol/L 3.8 6 0.3
BUN, mmol/L 5.9 6 1.8
Creatinine, mg/dl 0.9 6 0.2
eGFR, ml/min/1.73 m2 77 6 16
Treatment
Naive 9 (16)
Mono 33 (57)
Duo 12 (20)
Triple 4 (7)
Treatment strategies, n
Loop diuretics 30
Spironolactone 26
Bosentan 18
Sitaxsentan 7
Ambrisentan 2
Sildenafil 21
Treprostinil 5
Epoprostenol 11
Iloprost 1
Ca21 blocker 3
Simvastatin 1
Definition of abbreviations: 6MWD ¼ 6-minute walk distance; BUN ¼ blood
urea nitrogen; CO ¼ cardiac output; eGFR ¼ estimated glomerular filtration rate;
HR ¼ heart rate; iPAH ¼ idiopathic pulmonary arterial hypertension; mPAP ¼mean pulmonary artery pressure; NT-proBNP ¼ N-terminal pro brain natriuretic
peptide; NYHA ¼ New York Heart Association class; PCWP ¼ pulmonary capillary
wedge pressure; PVR ¼ pulmonary vascular resistance; RAP ¼ right atrial
pressure.
Data presented as mean 6 SD or n (%) unless otherwise noted.
TABLE 2. COX REGRESSION ANALYSES OF THE SERUMRENIN–ANGIOTENSIN–ALDOSTERONE SYSTEM ACTIVITY
Variables Hazard Ratio 95% CI P Value
Univariate Cox regression analyses
LN_renin 1.651 1.026–2.658 0.039
LN_Angiotensin I 0.949 0.601–1.496 0.821
LN_Angiotensin II 2.883 1.358–6.122 0.006
Cox regression analyses corrected for age, sex, and time from diagnosis
LN_renin 1.793 1.129–2.846 0.013
LN_Angiotensin I 0.971 0.576–1.636 0.911
LN_Angiotensin II 3.017 1.404–6.484 0.005
Definition of abbreviation: LN ¼ logarithmic transformation.
782 AMERICAN JOURNAL OF RESPIRATORY AND CRITICAL CARE MEDICINE VOL 186 2012
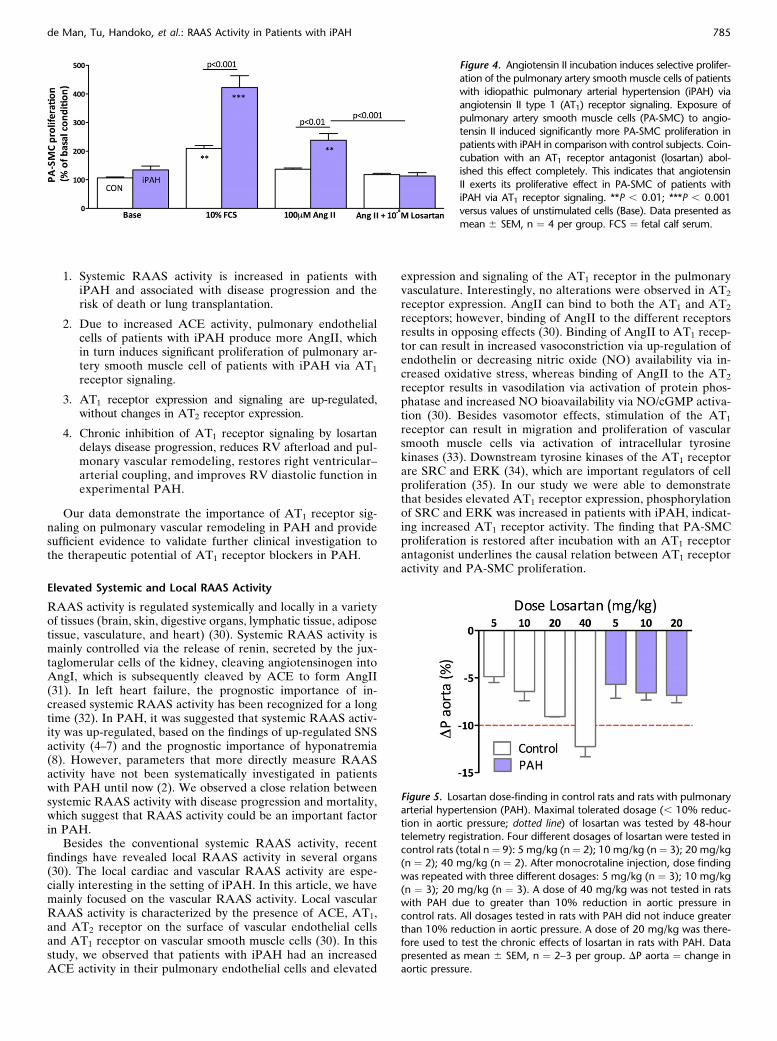
AngII incubation induced specific proliferation of the PA-SMCsof patients with iPAH, whereas no change in proliferation ofcontrol PA-SMCs was observed (Figure 4). To test whetherAngII exerts its proliferative effect on PA-SMCs via binding tothe AT1 receptor, PA-SMCs were exposed to AngII in combina-tion with the AT1 receptor antagonist losartan. Interestingly, thedifference in PA-SMC proliferation between control and iPAHwas completely abolished after coincubation with losartan. Thesefindings indicate that AngII can induce PA-SMC proliferation iniPAH via AT1 receptor signaling.
Part III: Effects of Chronic AT1 Receptor Inhibition
in Experimental PAH
To investigate whether chronic inhibition of AT1 receptor sig-naling could have therapeutic potential for the treatment ofPAH, we performed a preclinical study in a well-establishedrat model with PAH induced by a high dose of monocrotaline(26). First, we performed a dose-finding study to determine themaximum tolerated dose of losartan, which was defined as lessthan 10% reduction in systemic blood pressure (Figure 5, dottedline). A clear dose response was observed in control rats, inwhich systemic blood pressure gradually decreased with higherdosages of losartan. A dose of 40 mg/kg induced a greater than10% reduction in systemic blood pressure and was therefore notrepeated in rats with PAH. No difference in change in systemicblood pressure was observed in rats with PAH between a doseof 5 mg/kg, 10 mg/kg, and 20 mg/kg losartan. Because all dos-ages were tolerated by rats with PAH, we investigated thechronic effect of 20 mg/kg losartan in the main preclinicalstudy.Losartan reduces pulmonary vascular remodeling in rats with
PAH. Ten days after injection, ECG analyses revealed clear signsof PAH in all monocrotaline-treated rats. Estimated RV systolicpressures (pulmonary artery acceleration time normalized forcardiac cycle length and estimated right ventricular systolicpressure) and pulmonary vascular remodeling were significantly
increased in comparison with controls, and early signs of RVhypertrophy and dilatation were observed in all 18 rats withPAH (see Table E1 in the online supplement).
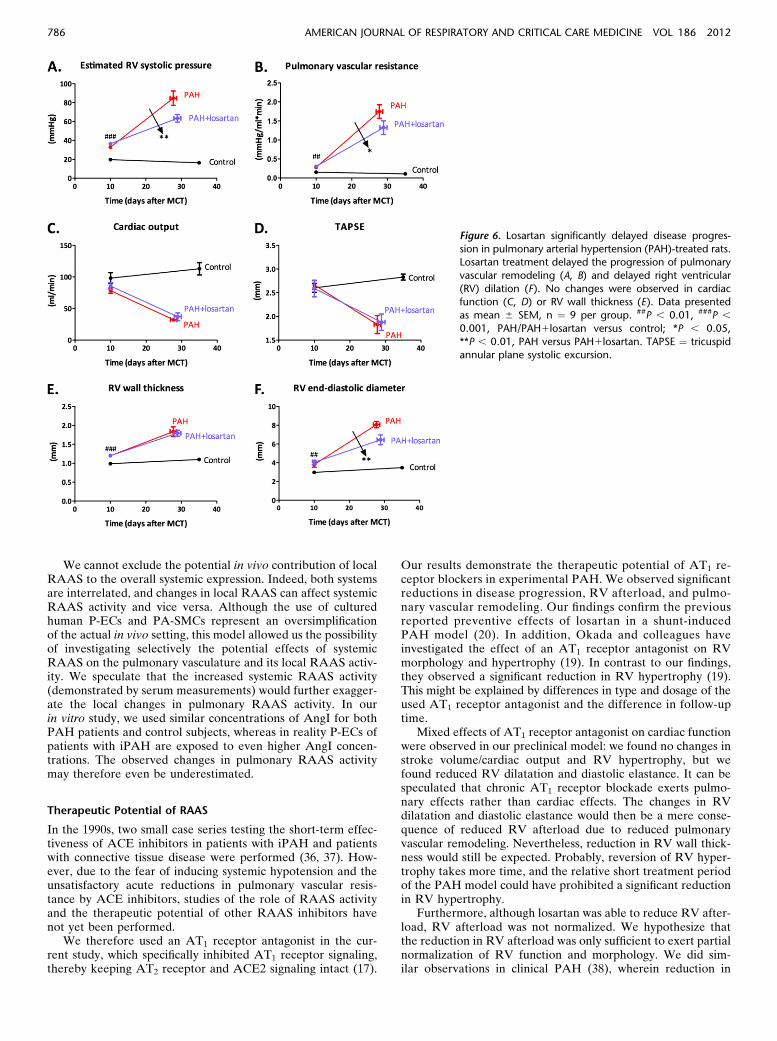
Serial echocardiography (at day 10 and at end of study)demonstrated a significantly delayed disease progression inlosartan-treated rats (Figure 6). The increment in pulmonaryvascular remodeling was reduced in losartan-treated rats withPAH in comparison with vehicle-treated rats with PAH (Fig-ures 6A and 6B; Table E2). No changes were observed in RVfunction (Figures 6C and 6D) or hypertrophy (Figure 6E)after losartan treatment, but a reduced RV dilation was noted(Figure 6F).
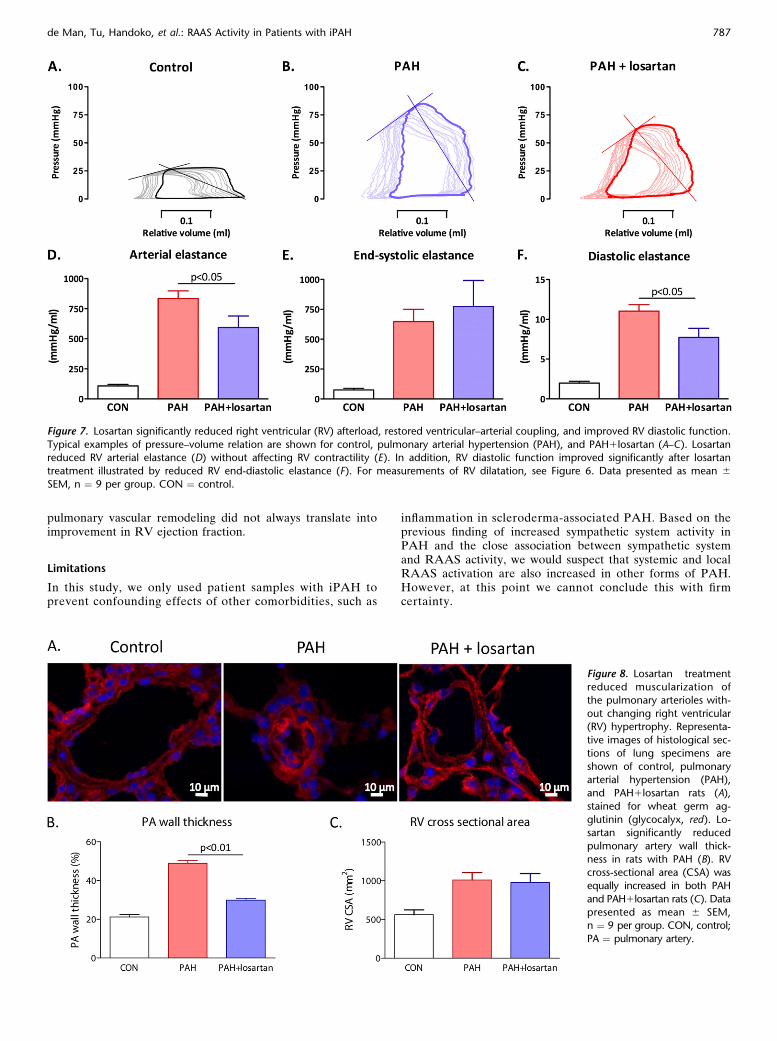
RVpressure-volumemeasurements at end of study (Figures 7A–7C) revealed that losartan was able to significantly reduced RVafterload (arterial elastance; Figure 7D), without affecting RV con-tractility (end-systolic elastance; Figure 7E). This resulted in im-proved RV ventricular–arterial coupling in losartan-treated rats(ventricular–arterial coupling: PAH, 0.746 0.11 vs. PAH1losartan,1.326 0.20; P, 0.05). In addition, RV relaxation was improved, asdemonstrated by reduced RV diastolic elastance (Figure 7F).
Histological analyses of PA wall thickness and cross-sectionalarea of RV cardiomyocytes demonstrated a clear increase in rats
Figure 1. Serum renin–angiotensin–aldosterone system in patients with idiopathic pulmonary arterial hypertension (iPAH). Serum levels of renin,
angiotensin (Ang)I, and AngII were increased in the majority of patients in comparison with upper limit of normal (dotted line; A–C). Follow-upmeasurements in a subgroup of patients (n ¼ 21) revealed that increased serum levels of renin, AngI, and AngII were associated with progressive
iPAH (D–F). Data presented as mean 6 SEM, n ¼ 58 patients with iPAH. Light blue bars represent baseline, and dark blue bars represent follow-up.
6MWD ¼ 6-minute walk distance.
TABLE 3. PATIENT CHARACTERISTICS FOR TISSUE SAMPLES
iPAH (N ¼ 13) Control Subjects (N ¼ 14)
Age, yr 34 6 13 62 6 13
Sex, M/F 5/8 7/7
mPAP, mm Hg 70 6 14
CO, L/min 4.0 6 1.1
PVR, mm Hg/L/min 1.302.8 6 333.6
PCWP, mm Hg 7.4 6 1.1
Definition of abbreviations: CO ¼ cardiac output; mPAP ¼ mean pulmonary
artery pressure; iPAH ¼ idiopathic pulmonary arterial hypertension; PCWP ¼pulmonary capillary wedge pressure; PVR ¼ pulmonary vascular resistance.
Data presented as mean 6 SD unless otherwise noted.
de Man, Tu, Handoko, et al.: RAAS Activity in Patients with iPAH 783
with PAH in comparison with controls (both P , 0.001; Figure8). Losartan significantly reduced PA wall thickness in ratswith PAH, which could explain the reduction in RV afterloadby losartan. Although RV afterload was reduced in losartan-treated rats with PAH, no differences were observed betweenlosartan and vehicle-treated rats with PAH in RV hypertrophy,whether expressed as RV mass, RV/left ventrical1septum, orRV cross-sectional area (Figure 8B, Table E3).
These findings indicate that chronic inhibition of AT1 recep-tor signaling delayed disease progression, reduced RV afterload
and pulmonary vascular remodeling, restored right ventricular–arterial coupling, and improved RV diastolic function.
DISCUSSION
To the best of our knowledge, this is the first study to demon-strate the effects of increased systemic and local RAAS activityin patients with iPAH. Using a translational approach with a setof physiological and pathological endpoints, we have demon-strated that:
Figure 2. Pulmonary endothelial cells of patients with idi-opathic pulmonary arterial hypertension (iPAH) produce
more angiotensin (Ang)II. Exposure of pulmonary endothe-
lial cells to AngI revealed significantly higher production of
AngII in pulmonary endothelial cells of patients with iPAHthan control subjects. Coincubation with angiotensin-
converting enzyme (ACE) inhibitor enalapril totally abol-
ished this effect, indicating that ACE activity in pulmonary
endothelial cells of patients with iPAH is increased. **P ,0.01, ***P , 0.001 versus values of unstimulated cells
(Base). Data presented as mean 6 SEM, n ¼ 3 per group.
CON ¼ control.
Figure 3. Angiotensin II type 1 receptorexpression and signaling is increased in
pulmonary arteries of patients with idio-
pathic pulmonary arterial hypertension(iPAH). Typical examples of histological
sections of lung specimens are shown
for a control subject and a patient with
iPAH (A), stained for angiotensin II type 1receptor (AT1 receptor). Western blot ana-
lyses revealed significant up-regulation of
AT1 receptor expression in pulmonary
arteries of patients with iPAH in com-parison with control; no changes in AT2receptor expression were observed (B).
In addition, tyrosine kinase SRC activity
and extracellular regulated kinase (ERK)activity (downstream targets of AT1 recep-
tor) were significantly increased, suggest-
ing increased signaling activity of the AT1receptor (C). Data presented as mean 6SEM, n ¼ 5 per group. C ¼ control; M ¼marker; P ¼ idiopathic pulmonary arterial
hypertension; p-ERK ¼ expression phos-phorylated form of ERK; p-SRC ¼ expres-
sion phosphorylated form of tyrosine
kinase SRC; t-ERK ¼ total protein expres-
sion of ERK; t-SRC ¼ total protein expres-sion of tyrosine kinase SRC.
784 AMERICAN JOURNAL OF RESPIRATORY AND CRITICAL CARE MEDICINE VOL 186 2012
1. Systemic RAAS activity is increased in patients withiPAH and associated with disease progression and therisk of death or lung transplantation.
2. Due to increased ACE activity, pulmonary endothelialcells of patients with iPAH produce more AngII, whichin turn induces significant proliferation of pulmonary ar-tery smooth muscle cell of patients with iPAH via AT1
receptor signaling.
3. AT1 receptor expression and signaling are up-regulated,without changes in AT2 receptor expression.
4. Chronic inhibition of AT1 receptor signaling by losartandelays disease progression, reduces RV afterload and pul-monary vascular remodeling, restores right ventricular–arterial coupling, and improves RV diastolic function inexperimental PAH.
Our data demonstrate the importance of AT1 receptor sig-naling on pulmonary vascular remodeling in PAH and providesufficient evidence to validate further clinical investigation tothe therapeutic potential of AT1 receptor blockers in PAH.
Elevated Systemic and Local RAAS Activity
RAAS activity is regulated systemically and locally in a varietyof tissues (brain, skin, digestive organs, lymphatic tissue, adiposetissue, vasculature, and heart) (30). Systemic RAAS activity ismainly controlled via the release of renin, secreted by the jux-taglomerular cells of the kidney, cleaving angiotensinogen intoAngI, which is subsequently cleaved by ACE to form AngII(31). In left heart failure, the prognostic importance of in-creased systemic RAAS activity has been recognized for a longtime (32). In PAH, it was suggested that systemic RAAS activ-ity was up-regulated, based on the findings of up-regulated SNSactivity (4–7) and the prognostic importance of hyponatremia(8). However, parameters that more directly measure RAASactivity have not been systematically investigated in patientswith PAH until now (2). We observed a close relation betweensystemic RAAS activity with disease progression and mortality,which suggest that RAAS activity could be an important factorin PAH.
Besides the conventional systemic RAAS activity, recentfindings have revealed local RAAS activity in several organs(30). The local cardiac and vascular RAAS activity are espe-cially interesting in the setting of iPAH. In this article, we havemainly focused on the vascular RAAS activity. Local vascularRAAS activity is characterized by the presence of ACE, AT1,and AT2 receptor on the surface of vascular endothelial cellsand AT1 receptor on vascular smooth muscle cells (30). In thisstudy, we observed that patients with iPAH had an increasedACE activity in their pulmonary endothelial cells and elevated
expression and signaling of the AT1 receptor in the pulmonaryvasculature. Interestingly, no alterations were observed in AT2
receptor expression. AngII can bind to both the AT1 and AT2
receptors; however, binding of AngII to the different receptorsresults in opposing effects (30). Binding of AngII to AT1 recep-tor can result in increased vasoconstriction via up-regulation ofendothelin or decreasing nitric oxide (NO) availability via in-creased oxidative stress, whereas binding of AngII to the AT2
receptor results in vasodilation via activation of protein phos-phatase and increased NO bioavailability via NO/cGMP activa-tion (30). Besides vasomotor effects, stimulation of the AT1
receptor can result in migration and proliferation of vascularsmooth muscle cells via activation of intracellular tyrosinekinases (33). Downstream tyrosine kinases of the AT1 receptorare SRC and ERK (34), which are important regulators of cellproliferation (35). In our study we were able to demonstratethat besides elevated AT1 receptor expression, phosphorylationof SRC and ERK was increased in patients with iPAH, indicat-ing increased AT1 receptor activity. The finding that PA-SMCproliferation is restored after incubation with an AT1 receptorantagonist underlines the causal relation between AT1 receptoractivity and PA-SMC proliferation.
Figure 4. Angiotensin II incubation induces selective prolifer-
ation of the pulmonary artery smooth muscle cells of patientswith idiopathic pulmonary arterial hypertension (iPAH) via
angiotensin II type 1 (AT1) receptor signaling. Exposure of
pulmonary artery smooth muscle cells (PA-SMC) to angio-
tensin II induced significantly more PA-SMC proliferation inpatients with iPAH in comparison with control subjects. Coin-
cubation with an AT1 receptor antagonist (losartan) abol-
ished this effect completely. This indicates that angiotensin
II exerts its proliferative effect in PA-SMC of patients withiPAH via AT1 receptor signaling. **P , 0.01; ***P , 0.001
versus values of unstimulated cells (Base). Data presented as
mean 6 SEM, n ¼ 4 per group. FCS ¼ fetal calf serum.
Figure 5. Losartan dose-finding in control rats and rats with pulmonaryarterial hypertension (PAH). Maximal tolerated dosage (, 10% reduc-
tion in aortic pressure; dotted line) of losartan was tested by 48-hour
telemetry registration. Four different dosages of losartan were tested incontrol rats (total n ¼ 9): 5 mg/kg (n¼ 2); 10 mg/kg (n¼ 3); 20 mg/kg
(n ¼ 2); 40 mg/kg (n ¼ 2). After monocrotaline injection, dose finding
was repeated with three different dosages: 5 mg/kg (n ¼ 3); 10 mg/kg
(n ¼ 3); 20 mg/kg (n ¼ 3). A dose of 40 mg/kg was not tested in ratswith PAH due to greater than 10% reduction in aortic pressure in
control rats. All dosages tested in rats with PAH did not induce greater
than 10% reduction in aortic pressure. A dose of 20 mg/kg was there-
fore used to test the chronic effects of losartan in rats with PAH. Datapresented as mean 6 SEM, n ¼ 2–3 per group. DP aorta ¼ change in
aortic pressure.
de Man, Tu, Handoko, et al.: RAAS Activity in Patients with iPAH 785
We cannot exclude the potential in vivo contribution of localRAAS to the overall systemic expression. Indeed, both systemsare interrelated, and changes in local RAAS can affect systemicRAAS activity and vice versa. Although the use of culturedhuman P-ECs and PA-SMCs represent an oversimplificationof the actual in vivo setting, this model allowed us the possibilityof investigating selectively the potential effects of systemicRAAS on the pulmonary vasculature and its local RAAS activ-ity. We speculate that the increased systemic RAAS activity(demonstrated by serum measurements) would further exagger-ate the local changes in pulmonary RAAS activity. In ourin vitro study, we used similar concentrations of AngI for bothPAH patients and control subjects, whereas in reality P-ECs ofpatients with iPAH are exposed to even higher AngI concen-trations. The observed changes in pulmonary RAAS activitymay therefore even be underestimated.
Therapeutic Potential of RAAS
In the 1990s, two small case series testing the short-term effec-tiveness of ACE inhibitors in patients with iPAH and patientswith connective tissue disease were performed (36, 37). How-ever, due to the fear of inducing systemic hypotension and theunsatisfactory acute reductions in pulmonary vascular resis-tance by ACE inhibitors, studies of the role of RAAS activityand the therapeutic potential of other RAAS inhibitors havenot yet been performed.
We therefore used an AT1 receptor antagonist in the cur-rent study, which specifically inhibited AT1 receptor signaling,thereby keeping AT2 receptor and ACE2 signaling intact (17).
Our results demonstrate the therapeutic potential of AT1 re-ceptor blockers in experimental PAH. We observed significantreductions in disease progression, RV afterload, and pulmo-nary vascular remodeling. Our findings confirm the previousreported preventive effects of losartan in a shunt-inducedPAH model (20). In addition, Okada and colleagues haveinvestigated the effect of an AT1 receptor antagonist on RVmorphology and hypertrophy (19). In contrast to our findings,they observed a significant reduction in RV hypertrophy (19).This might be explained by differences in type and dosage of theused AT1 receptor antagonist and the difference in follow-uptime.
Mixed effects of AT1 receptor antagonist on cardiac functionwere observed in our preclinical model: we found no changes instroke volume/cardiac output and RV hypertrophy, but wefound reduced RV dilatation and diastolic elastance. It can bespeculated that chronic AT1 receptor blockade exerts pulmo-nary effects rather than cardiac effects. The changes in RVdilatation and diastolic elastance would then be a mere conse-quence of reduced RV afterload due to reduced pulmonaryvascular remodeling. Nevertheless, reduction in RV wall thick-ness would still be expected. Probably, reversion of RV hyper-trophy takes more time, and the relative short treatment periodof the PAH model could have prohibited a significant reductionin RV hypertrophy.
Furthermore, although losartan was able to reduce RV after-load, RV afterload was not normalized. We hypothesize thatthe reduction in RV afterload was only sufficient to exert partialnormalization of RV function and morphology. We did sim-ilar observations in clinical PAH (38), wherein reduction in
Figure 6. Losartan significantly delayed disease progres-
sion in pulmonary arterial hypertension (PAH)-treated rats.Losartan treatment delayed the progression of pulmonary
vascular remodeling (A, B) and delayed right ventricular
(RV) dilation (F). No changes were observed in cardiacfunction (C, D) or RV wall thickness (E). Data presented
as mean 6 SEM, n ¼ 9 per group. ##P , 0.01, ###P ,0.001, PAH/PAH1losartan versus control; *P , 0.05,
**P , 0.01, PAH versus PAH1losartan. TAPSE ¼ tricuspidannular plane systolic excursion.
786 AMERICAN JOURNAL OF RESPIRATORY AND CRITICAL CARE MEDICINE VOL 186 2012
pulmonary vascular remodeling did not always translate intoimprovement in RV ejection fraction.
Limitations
In this study, we only used patient samples with iPAH toprevent confounding effects of other comorbidities, such as
inflammation in scleroderma-associated PAH. Based on theprevious finding of increased sympathetic system activity inPAH and the close association between sympathetic systemand RAAS activity, we would suspect that systemic and localRAAS activation are also increased in other forms of PAH.However, at this point we cannot conclude this with firmcertainty.
Figure 7. Losartan significantly reduced right ventricular (RV) afterload, restored ventricular–arterial coupling, and improved RV diastolic function.
Typical examples of pressure–volume relation are shown for control, pulmonary arterial hypertension (PAH), and PAH1losartan (A–C). Losartanreduced RV arterial elastance (D) without affecting RV contractility (E). In addition, RV diastolic function improved significantly after losartan
treatment illustrated by reduced RV end-diastolic elastance (F). For measurements of RV dilatation, see Figure 6. Data presented as mean 6SEM, n ¼ 9 per group. CON ¼ control.
Figure 8. Losartan treatmentreduced muscularization of
the pulmonary arterioles with-
out changing right ventricular(RV) hypertrophy. Representa-
tive images of histological sec-
tions of lung specimens are
shown of control, pulmonaryarterial hypertension (PAH),
and PAH1losartan rats (A),
stained for wheat germ ag-
glutinin (glycocalyx, red). Lo-sartan significantly reduced
pulmonary artery wall thick-
ness in rats with PAH (B). RVcross-sectional area (CSA) was
equally increased in both PAH
and PAH1losartan rats (C). Data
presented as mean 6 SEM,n ¼ 9 per group. CON, control;
PA ¼ pulmonary artery.
de Man, Tu, Handoko, et al.: RAAS Activity in Patients with iPAH 787

A less dramatic increase in AngII levels in comparison withrenin and AngI levels was observed in serum samples of patientswith iPAH. This could be partially explained by the likely varia-tions in AngII metabolizing systems including ACE2 (16). Nev-ertheless, AngII was closely associated with disease progressionand prognosis, suggesting that, although a less dramatic increasewas observed, the observed increase was clinically relevant.
To assess AT1 receptor activity, we measured phosphorylationof the tyrosine kinase SRC and ERK1/2, because there is no clearreadout for AT1 receptor signaling. However, other signalingpathways could also affect their relative phosphorylation levels.Nevertheless, the abolishing effect of AT1 receptor antagonist onPA-SMC proliferation indicates that increased AT1 receptor ac-tivity is important in the pathophysiology of iPAH.
In our study we could not observe a significant reduction inAT2 receptor expression in pulmonary arteries. This may bedue to a low power of the analyses. Although the differencebetween control and iPAH is small, we cannot exclude thatreduced AT2 receptor expression could further exaggerate theproliferative effects on PA-SMCs. However, this further un-derlines the importance of selective AT1 receptor blockers, be-cause ACE inhibitors would affect both AT1 and AT2 receptorexpression.
To translate our findings, we used the monocrotaline ratmodel to mimic PAH in vivo. Increased RAAS signaling hasalready been demonstrated in lungs of monocrotaline-injectedrats (14). Furthermore, the monocrotaline model is character-ized by profound RV dysfunction and is thereby the most suitedmodel to test whether AT1 receptor blockers are not detrimen-tal for the right ventricle. To circumvent the shortcomings of(any) animal model, we have investigated the importance ofthe RAAS system in three different ways: (1) PH rat model,(2) pulmonary endothelial and smooth muscle cell culture ofpatients with iPAH, and (3) serum samples of patients withiPAH.
Our pressure-volume analyses do not allow us to determinewhether treatment has altered diastolic and systolic volumes.However, in combination with our ECG-derived RV end-diastolic diameters, we were able to conclude that AT1 receptorantagonist could reduce RV dilatation in PAH-treated rats.
Conclusions
In this translational study, we have demonstrated systemic andlocal up-regulation of RAAS activity in pulmonary arteries ofpatients with iPAH. In addition, local RAAS up-regulation wasassociated with increased pulmonary artery smooth muscle cellproliferation via enhanced angiotensin II type 1 (AT1) receptorsignaling in patients with iPAH compared with control subjects.Chronic treatment with an AT1 receptor antagonist in experi-mental PAH revealed reduced disease progression, decreasedRV afterload, and pulmonary vascular remodeling and restoredright ventricular–arterial coupling. Our findings should encouragefurther clinical investigation and reevaluation of the use ofRAAS inhibitors for treatment of patients with iPAH.
Author disclosures are available with the text of this article at www.atsjournals.org.
Acknowledgment: The authors thank Dr. Lianne Boesten for quantifying systemicRAAS activity in serum of patients with iPAH.
References
1. Humbert M, Sitbon O, Chaouat A, Bertocchi M, Habib G, Gressin V, Yaici
A, Weitzenblum E, Cordier JF, Chabot F, et al. Survival in patients with
idiopathic, familial, and anorexigen-associated pulmonary arterial hy-
pertension in the modern management era. Circulation 2010;122:156–163.
2. Handoko ML, de Man FS, Allaart CP, Paulus WJ, Westerhof N, Vonk-
Noordegraaf A. Perspectives on novel therapeutic strategies for right
heart failure in pulmonary arterial hypertension: Lessons from the left
heart. Eur Respir Rev 2010;19:72–82.
3. Eichhorn EJ, Bristow MR. Medical therapy can improve the biological
properties of the chronically failing heart. A new era in the treatment
of heart failure. Circulation 1996;94:2285–2296.
4. Ciarka A, Doan V, Velez-Roa S, Naeije R, van de Borne P. Prognostic
significance of sympathetic nervous system activation in pulmonary
arterial hypertension. Am J Respir Crit Care Med 2010;181:1269–
1275.
5. de Man FS, Handoko ML, van Ballegoij JJ, Schalij I, Bogaards SJ,
Postmus PE, van der Velden J, Westerhof N, Paulus WJ, Vonk-
Noordegraaf A. Bisoprolol delays progression towards right heart
failure in experimental pulmonary hypertension. Circ Heart Fail 2012;
5:97–105.
6. Nootens M, Kaufmann E, Rector T, Toher C, Judd D, Francis GS, Rich
S. Neurohormonal activation in patients with right ventricular failure
from pulmonary hypertension: relation to hemodynamic variables and
endothelin levels. J Am Coll Cardiol 1995;26:1581–1585.
7. Velez-Roa S, Ciarka A, Najem B, Vachiery JL, Naeije R, van de Borne
P. Increased sympathetic nerve activity in pulmonary artery hyper-
tension. Circulation 2004;110:1308–1312.
8. Forfia PR, Mathai SC, Fisher MR, Housten-Harris T, Hemnes AR,
Champion HC, Girgis RE, Hassoun PM. Hyponatremia predicts right
heart failure and poor survival in pulmonary arterial hypertension.
Am J Respir Crit Care Med 2008;177:1364–1369.
9. Abraham WT, Raynolds MV, Badesch DB, Wynne KM, Groves BM,
Roden RL, Robertson AD, Lowes BD, Zisman LS, Voelkel NF, et al.
Angiotensin-converting enzyme DD genotype in patients with pri-
mary pulmonary hypertension: Increased frequency and association
with preserved haemodynamics. J Renin Angiotensin Aldosterone Syst
2003;4:27–30.
10. Chung WK, Deng L, Carroll JS, Mallory N, Diamond B, Rosenzweig
EB, Barst RJ, Morse JH. Polymorphism in the angiotensin II type 1
receptor (AGTR1) is associated with age at diagnosis in pulmonary
arterial hypertension. J Heart Lung Transplant 2009;28:373–379.
11. Orte C, Polak JM, Haworth SG, Yacoub MH, Morrell NW. Expression
of pulmonary vascular angiotensin-converting enzyme in primary
and secondary plexiform pulmonary hypertension. J Pathol 2000;192:
379–384.
12. Bader M. Tissue renin-angiotensin-aldosterone systems: targets for
pharmacological therapy. Annu Rev Pharmacol Toxicol 2010;50:
439–465.
13. Leier CV, Bambach D, Nelson S, Hermiller JB, Huss P, Magorien RD,
Unverferth DV. Captopril in primary pulmonary hypertension. Cir-
culation 1983;67:155–161.
14. Ferreira AJ, Shenoy V, Yamazato Y, Sriramula S, Francis J, Yuan L,
Castellano RK, Ostrov DA, Oh SP, Katovich MJ, et al. Evidence for
angiotensin-converting enzyme 2 as a therapeutic target for the pre-
vention of pulmonary hypertension. Am J Respir Crit Care Med 2009;
179:1048–1054.
15. Shenoy V, Ferreira AJ, Qi Y, Fraga-Silva RA, Diez-Freire C, Dooies A,
Jun JY, Sriramula S, Mariappan N, Pourang D, et al. The angiotensin-
converting enzyme 2/angiogenesis-(1–7)/Mas axis confers cardiopul-
monary protection against lung fibrosis and pulmonary hypertension.
Am J Respir Crit Care Med 2011;182:1065–1072.
16. Lambert DW, Hooper NM, Turner AJ. Angiotensin-converting enzyme
2 and new insights into the renin-angiotensin system. Biochem
Pharmacol 2008;75:781–786.
17. Burnier M. Angiotensin II type 1 receptor blockers. Circulation 2001;
103:904–912.
18. Johnston CI. Angiotensin receptor antagonists: focus on losartan. Lancet
1995;346:1403–1407.
19. Okada M, Harada T, Kikuzuki R, Yamawaki H, Hara Y. Effects of
telmisartan on right ventricular remodeling induced by monocrotaline
in rats. J Pharmacol Sci 2009;111:193–200.
20. Rondelet B, Kerbaul F, Van Beneden R, Hubloue I, Huez S, Fesler P,
Remmelink M, Brimioulle S, Salmon I, Naeije R. Prevention of
pulmonary vascular remodeling and of decreased BMPR-2 expression
by losartan therapy in shunt-induced pulmonary hypertension. Am J
Physiol Heart Circ Physiol 2005;289:H2319–H2324.
788 AMERICAN JOURNAL OF RESPIRATORY AND CRITICAL CARE MEDICINE VOL 186 2012

21. Rouleau JL, Kapuku G, Pelletier S, Gosselin H, Adam A, Gagnon C,
Lambert C, Meloche S. Cardioprotective effects of ramipril and los-
artan in right ventricular pressure overload in the rabbit: Importance
of kinins and influence on angiotensin II type 1 receptor signaling
pathway. Circulation 2001;104:939–944.
22. de Man FS, Guignabert C, Tu L, Francois C, Simonneau G, Fadel E,
Humbert M, Vonk-Noordegraaf A, Eddahibi S. Increased renin-
angiotensin-aldosterone system activity in lungs of patients with idiopathic
pulmonary arterial hypertension [abstract]. Eur Respir J 2011;274S:P1516.
23. de Man FS, Guignabert C, Tu L, Handoko ML, Rain S, Ruiter G, Francois
C, Schalij I, Simonneau G, Fadel E, et al. The therapeutic potential of
the renin-angiotensin-aldosteron system in idiopathic pulmonary arterial
hypertension [abstract]. Am J Respir Crit Care Med 2012;185:A2494.
24. Izikki M, Guignabert C, Fadel E, Humbert M, Tu L, Zadigue P,
Dartevelle P, Simonneau G, Adnot S, Maitre B, et al. Endothelial-
derived FGF2 contributes to the progression of pulmonary hyper-
tension in humans and rodents. J Clin Invest 2009;119:512–523.
25. Tu L, Dewachter L, Gore B, Fadel E, Dartevelle P, Simonneau G,
Humbert M, Eddahibi S, Guignabert C. Autocrine fibroblast growth
factor-2 signaling contributes to altered endothelial phenotype in
pulmonary hypertension. Am J Respir Cell Mol Biol 2011;45:311–322.
26. Handoko ML, de Man FS, Happe CM, Schalij I, Musters RJ, Westerhof
N, Postmus PE, Paulus WJ, van der Laarse WJ, Vonk-Noordegraaf A.
Opposite effects of training in rats with stable and progressive pul-
monary hypertension. Circulation 2009;120:42–49.
27. de Man FS, Handoko ML, Groepenhoff H, van ’t Hul AJ, Abbink J,
Koppers RJ, Grotjohan HP, Twisk JW, Bogaard HJ, Boonstra A,
et al. Effects of exercise training in patients with idiopathic pulmonary
arterial hypertension. Eur Respir J 2009;34:669–675.
28. de Man FS, van Hees HW, Handoko ML, Niessen HW, Schalij I,
Humbert M, Dorfmuller P, Mercier O, Bogaard HJ, Postmus PE,
et al. Diaphragm muscle fiber weakness in pulmonary hypertension.
Am J Respir Crit Care Med 2011;183:1411–1418.
29. Mogi M, Iwai M, Horiuchi M. Emerging concepts of regulation of an-
giotensin II receptors: new players and targets for traditional recep-
tors. Arterioscler Thromb Vasc Biol 2007;27:2532–2539.
30. Paul M, Poyan MA, Kreutz R. Physiology of local renin-angiotensin
systems. Physiol Rev 2006;86:747–803.
31. Castrop H, Hocherl K, Kurtz A, Schweda F, Todorov V, Wagner C.
Physiology of kidney renin. Physiol Rev 2010;90:607–673.
32. Packer M, Lee WH, Kessler PD, Gottlieb SS, Bernstein JL, Kukin
ML. Role of neurohormonal mechanisms in determining survival
in patients with severe chronic heart failure. Circulation 1987;75:
IV80–IV92.
33. Mehta PK, Griendling KK. Angiotensin ii cell signaling: physiological
and pathological effects in the cardiovascular system. Am J Physiol
Cell Physiol 2007;292:C82–C97.
34. Griendling KK, Ushio-Fukai M, Lassegue B, Alexander RW. Angio-
tensin II signaling in vascular smooth muscle. New concepts. Hyper-
tension 1997;29:366–373.
35. Li L, Zhou Y, Wang C, Zhao YL, Zhang ZG, Fan D, Cui XB, Wu LL.
SRC tyrosine kinase regulates angiotensin II–induced protein kinase
Czeta activation and proliferation in vascular smooth muscle cells.
Peptides 2010;31:1159–1164.
36. Alpert MA, Pressly TA, Mukerji V, Lambert CR, Mukerji B. Short- and
long-term hemodynamic effects of captopril in patients with pulmo-
nary hypertension and selected connective tissue disease. Chest 1992;
102:1407–1412.
37. Ikram H, Maslowski AH, Nicholls MG, Espiner EA, Hull FT. Haemo-
dynamic and hormonal effects of captopril in primary pulmonary
hypertension. Br Heart J 1982;48:541–545.
38. van de Veerdonk M, Kind T, Marcus JT, Mauritz GJ, Heymans MW,
Bogaard HJ, Boonstra A, Marques KM, Westerhof N, Vonk-
Noordegraaf A. Progressive right ventricular dysfunction in patients
with pulmonary arterial hypertension responding to therapy. J Am
Coll Cardiol 2011;58:2511–2519.
de Man, Tu, Handoko, et al.: RAAS Activity in Patients with iPAH 789





























