Embed Size (px)
Citation preview

Einführung in die Physikalische ChemieTeil 1: Mikrostruktur der Materie
Einführung in die Physikalische Chemie: Übersicht
Kapitel 1:Quantenmechanik
Mathematische GrundlagenSchrödingergleichungEinfache Beispiele
Kapitel 2:Atome
H-AtomSpinMehrelektronen-Atome und Spektroskopie
Kapitel 3:Moleküle
MolekülorbitaltheorieBorn-Oppenheimer-Potential
Kapitel 5:ZwischenmolekulareKräfte
Elektrostatische Eigenschaften von MolekülenZwischenmolekulare WechselwirkungenStruktur von Biomolekülen
Kapitel 6:Struktur der Materie
Reale GaseKondensierte PhasenMoleküldynamik
Mikrokosmos
Makrokosmos
Kapitel 4:Molekülspektroskopie
Bewegungsformen eines Moleküls:Rotationen,Schwingungen, elektron. BewegungMikrowellen-, Infrarot- und optische Spektroskopie
Kapitel 3: Molekülstruktur
3.1 Chemische Bindung3.2 Molekülorbitaltheorie3.3 Die Born-Oppenheimer-Potentialfläche
Übersicht:
Literatur:
Atkins, de Paula, Physikalische Chemie (4. Aufl.), Kapitel 11 Atkins, de Paula, Kurzlehrbuch Physikalische Chemie (4. Aufl.), Kap. 14
Kapitel 3: Molekülstruktur

3.1 Chemische Bindung
Ein Molekül ist eine stabile Ansammlung von Atomen, die durch Bindungen miteinander verknüpft sind.
Eine chemische Bindung zwischen Atomen entsteht, wenn diese Elektronen miteinander teilen, so dass dadurch die Gesamtenergie im Vergleich zu den isolierten Atomen verkleinert wird.
Historisch unterscheidet man eine Reihe von chemischen Bindungstypen:
• Kovalente Bindung: zwei Atome werden durch ein gemeinsames Elektronenpaar verbunden
• Ionische Bindung: zwei entgegengesetzt geladene Atome werden durch elektrostatische Kräfte gebunden
• Wasserstoffbrücken-Bindung: ein kovalent gebundenes Wasserstoffatom wird durch elektrostatische Wechselwirkung an ein weiteres Atom gebunden (s. Kapitel 5)
• Van-der-Waals- und Dispersionswechselwirkungen: Bindung durch induzierte elektrostatische Momente (s. Kapitel 5)
A B
A+ B-
A BH
A B
3.1 Chemische Bindung
Alle diese Bindungstypen lassen sich im Rahmen der Quantenmechanik mit einem einheitlichen Formalismus beschreiben. Die “klassischen” Bindungstypen sind als “Karikatur” der tatsächlichen Bindungsverhältnisse aufzufassen und treten als Grenzfälle in der Beschreibung der Elektronenstruktur von Molekülen auf.

3.2 Molekülorbitaltheorie
Die Molekülorbital (MO)- Theorie ist das heute am weitesten verbreitete Modell zur Beschreibung der Elektronenstruktur von Molekülen.
(3.2.1)
Ähnlich wie bei Atomen wird die gesamte elektronische Wellenfunktion eines Moleküls als Produkt von Ein-Elektronenwellenfunktionen, sog. Molekülorbitelen ψmol,i, formuliert:
Bem.: Genauso wie in Atomen muss auch diese Wellenfunktion das Pauli-Prinzip erfüllen, sodass Gl. (3.2.1) noch “antisymmetrisiert” werden muss (s. Vorlesung PC II, Prof. J.P. Maier). Wir schreiben die Antisymmetrisierung im Folgenden jedoch nicht explizit an.
3.2.1 Molekülorbitale aus Linearkombination von Atomorbitalen
�el(⌅r1,⌅r2, ..,⌅rN) = �mol,1(⌅r1)�mol,2(⌅r2)..�mol,N(⌅rN)
Jedes Molekülorbital kann zwei Elektronen mit antiparallelem Spin fassen. Die LCAO erstreckt sich - insbesondere bei mehratomigen Molekülen - über mehrere Atome. Die MO-Theorie bricht also mit der Vorstellung, dass bindende Elektronenpaare jeweils zwischen zwei Atomen lokalisiert sind.
Die Molekülorbitale werden näherungsweise als eine Linearkombination von Atomorbitalen ψj (linear combination of atomic orbitals, LCAO) der beteiligten Atome formuliert:
�mol,i =�
j
cj�j (3.2.2)
3.2 Molekülorbitaltheorie
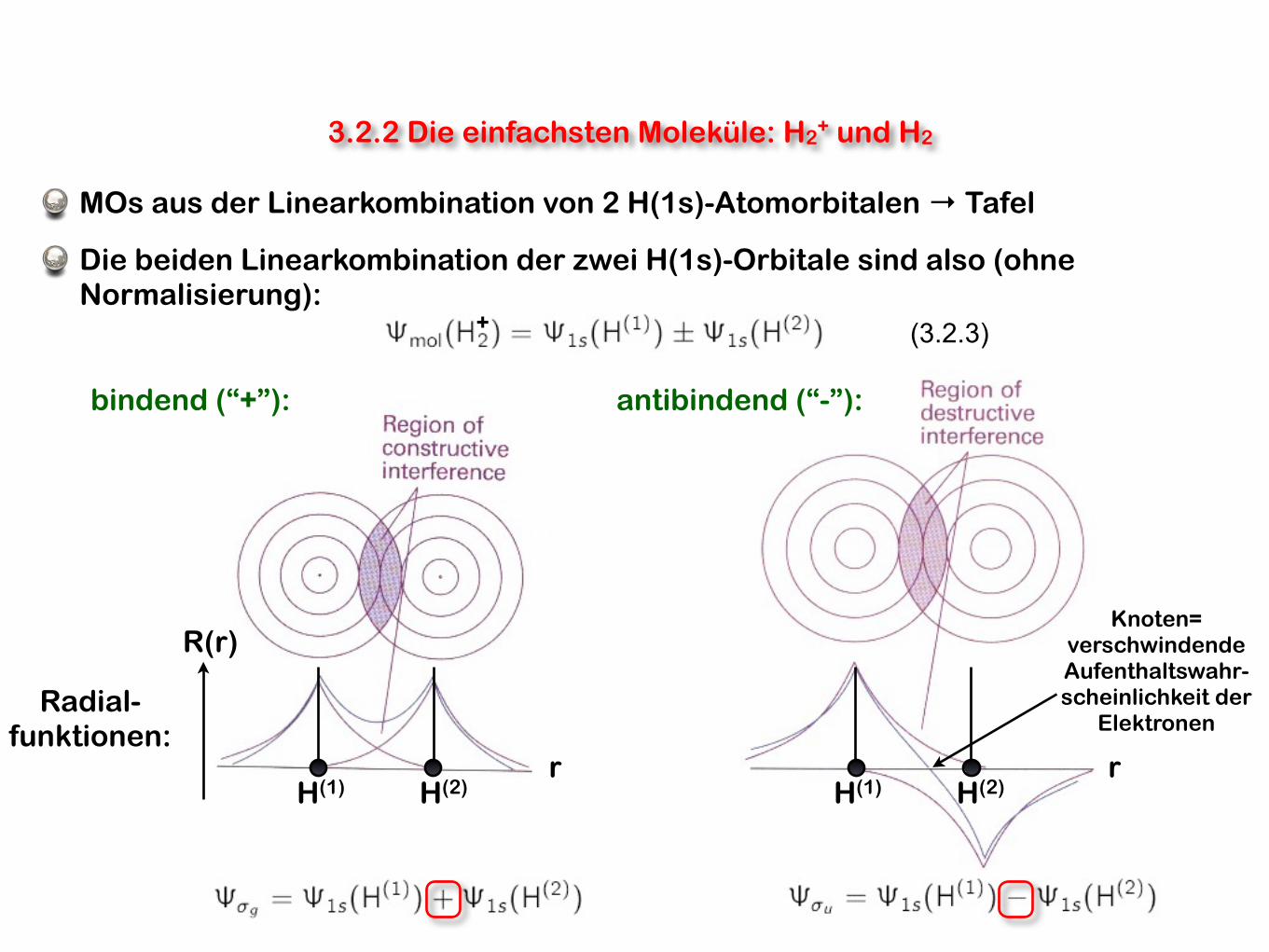
3.2.2 Die einfachsten Moleküle: H2+ und H2
MOs aus der Linearkombination von 2 H(1s)-Atomorbitalen → Tafel
3.2 Molekülorbitaltheorie
(3.2.3)
Die beiden Linearkombination der zwei H(1s)-Orbitale sind also (ohne Normalisierung):
H(1) H(2)r r
H(1) H(2)
R(r)
Radial-funktionen:
bindend (“+”): antibindend (“-”):
+
Knoten=verschwindende Aufenthaltswahr-scheinlichkeit der
Elektronen
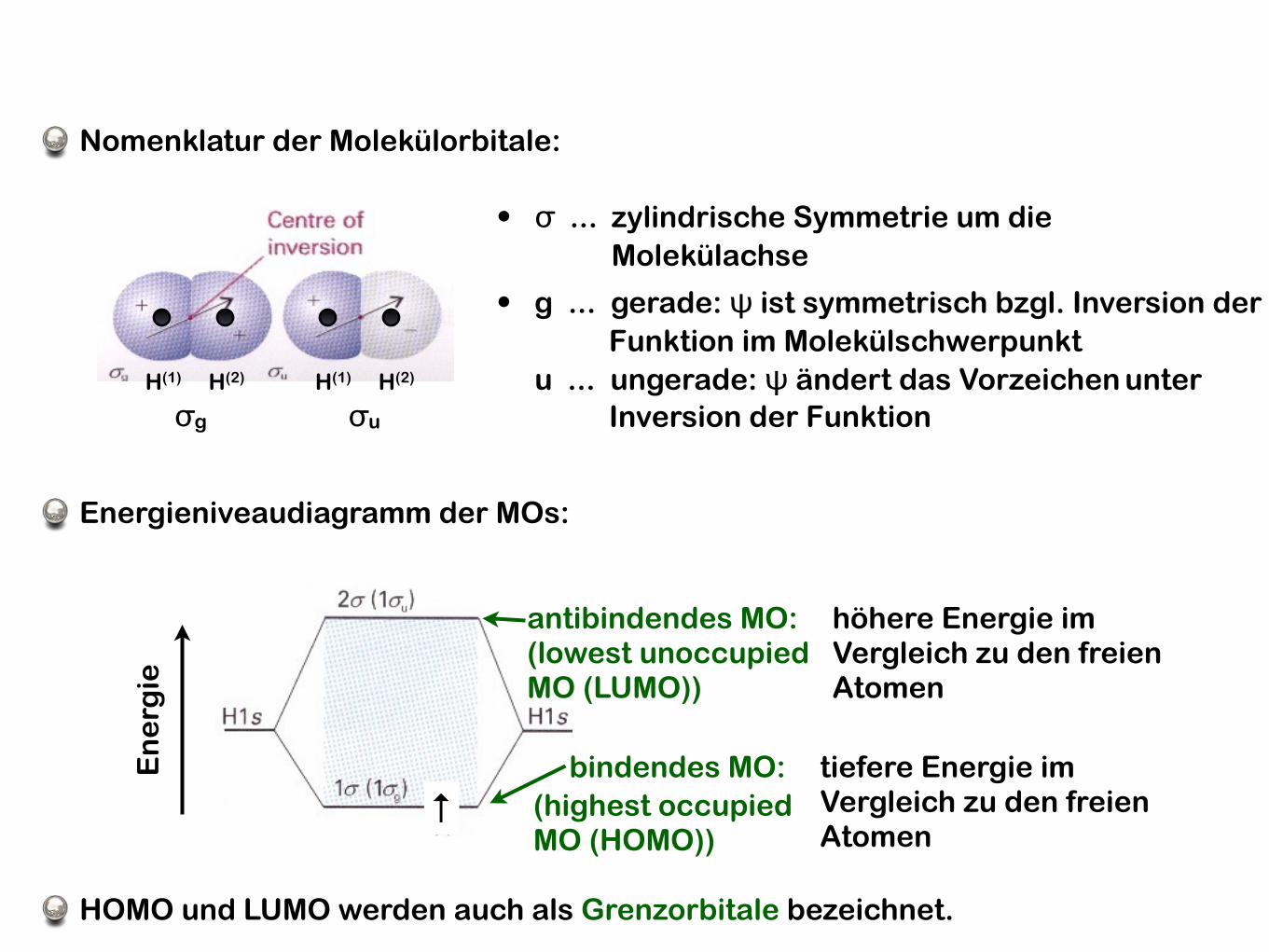
• σ ... zylindrische Symmetrie um die Molekülachse
• g ... gerade: ψ ist symmetrisch bzgl. Inversion der Funktion im Molekülschwerpunktu ... ungerade: ψ ändert das Vorzeichen unter Inversion der Funktionσg σu
H(1) H(2) H(1) H(2)
Nomenklatur der Molekülorbitale:
3.2 Molekülorbitaltheorie
tiefere Energie im Vergleich zu den freien Atomen
bindendes MO:
höhere Energie im Vergleich zu den freien Atomen
antibindendes MO:
En
erg
ie
Energieniveaudiagramm der MOs:
(lowest unoccupiedMO (LUMO))
(highest occupiedMO (HOMO))
↑
HOMO und LUMO werden auch als Grenzorbitale bezeichnet.
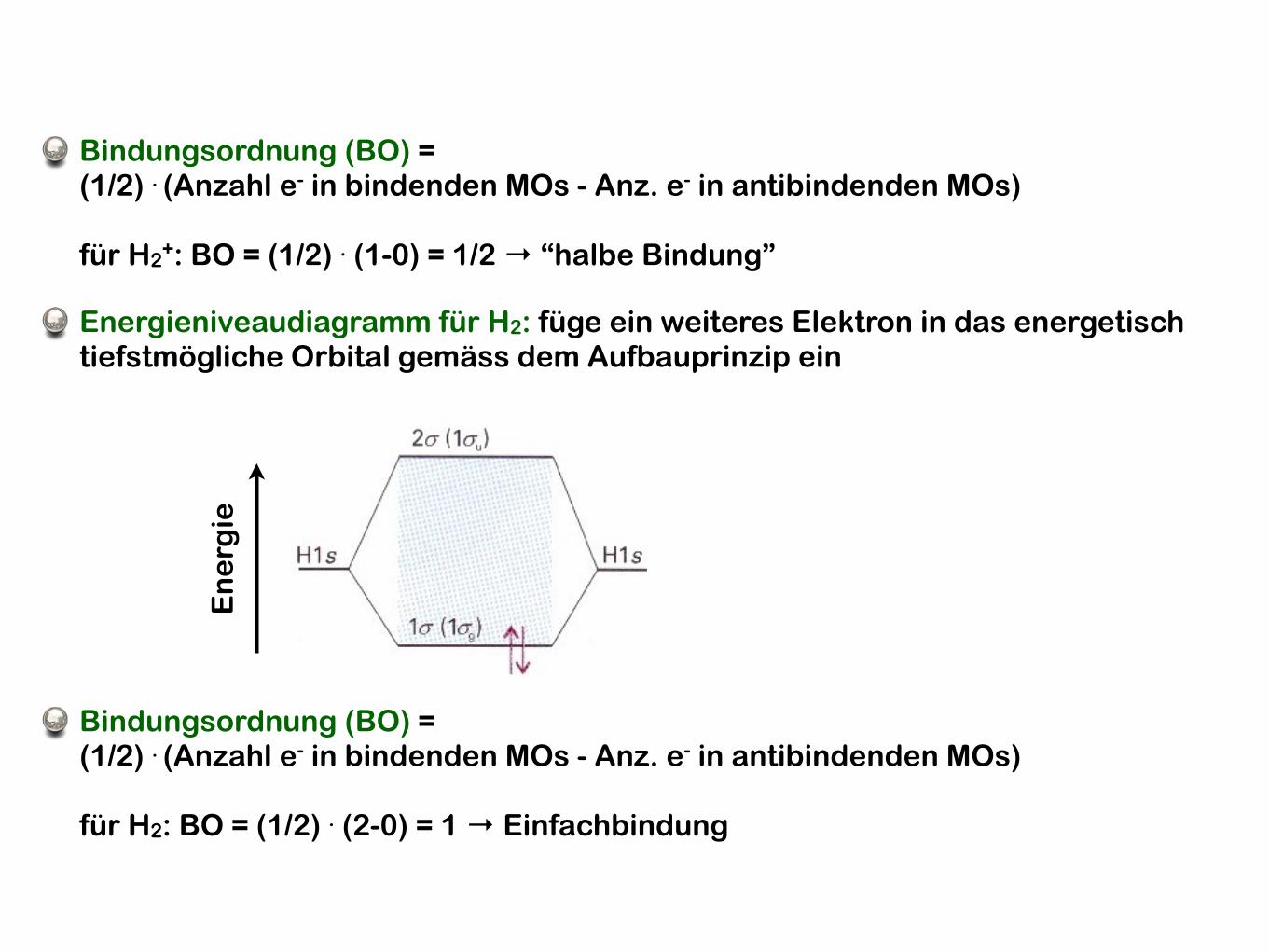
En
erg
ie
Energieniveaudiagramm für H2: füge ein weiteres Elektron in das energetisch tiefstmögliche Orbital gemäss dem Aufbauprinzip ein
Bindungsordnung (BO) = (1/2) . (Anzahl e- in bindenden MOs - Anz. e- in antibindenden MOs)
für H2: BO = (1/2) . (2-0) = 1 → Einfachbindung
Bindungsordnung (BO) = (1/2) . (Anzahl e- in bindenden MOs - Anz. e- in antibindenden MOs)
für H2+: BO = (1/2) . (1-0) = 1/2 → “halbe Bindung”
3.2 Molekülorbitaltheorie
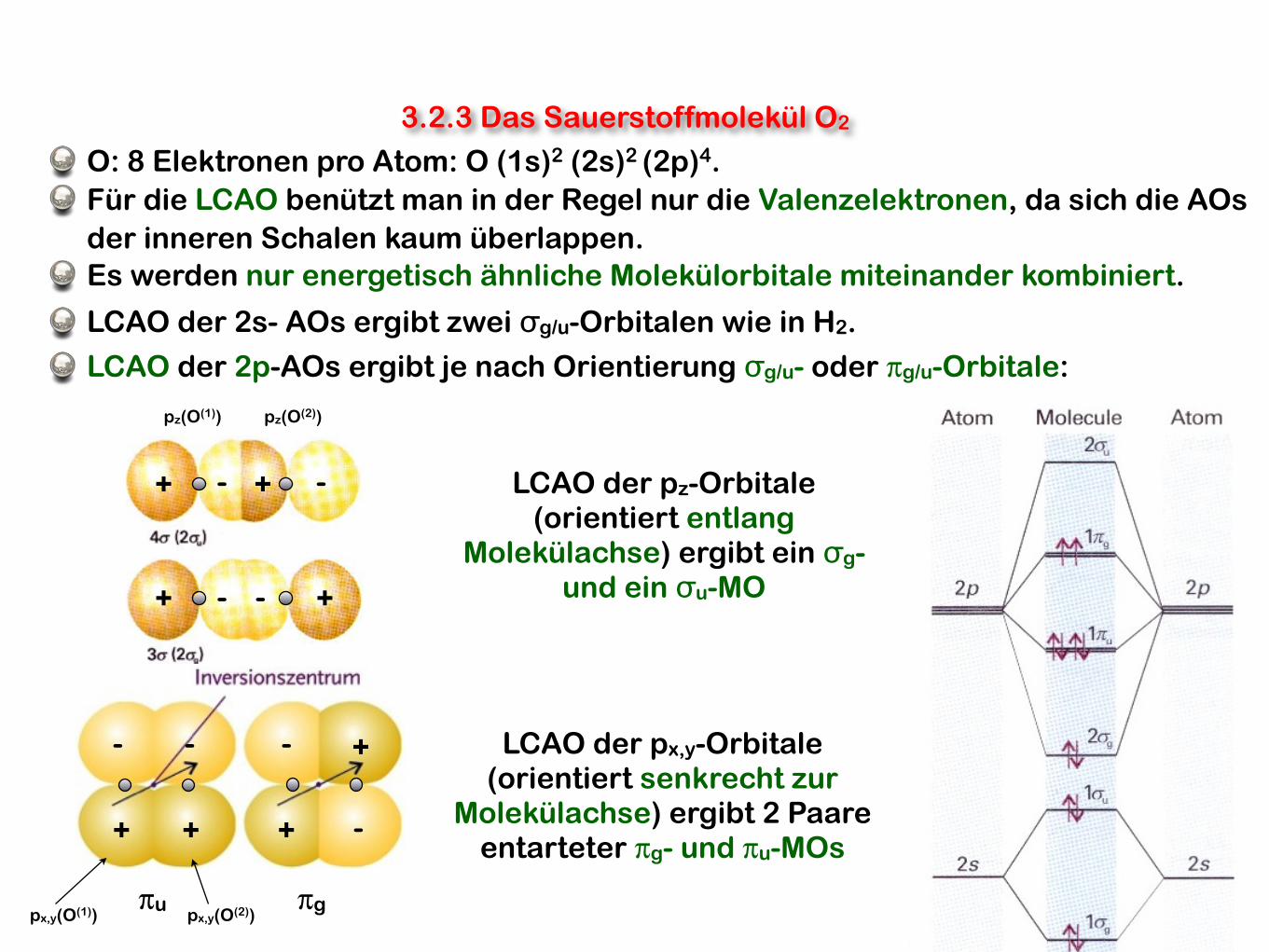
O: 8 Elektronen pro Atom: O (1s)2 (2s)2 (2p)4.
Für die LCAO benützt man in der Regel nur die Valenzelektronen, da sich die AOs der inneren Schalen kaum überlappen.Es werden nur energetisch ähnliche Molekülorbitale miteinander kombiniert.
LCAO der 2s- AOs ergibt zwei σg/u-Orbitalen wie in H2.
LCAO der 2p-AOs ergibt je nach Orientierung σg/u- oder πg/u-Orbitale:
LCAO der pz-Orbitale(orientiert entlang
Molekülachse) ergibt ein σg- und ein σu-MO
LCAO der px,y-Orbitale(orientiert senkrecht zur
Molekülachse) ergibt 2 Paare entarteter πg- und πu-MOs
πgπu
3.2.3 Das Sauerstoffmolekül O2
pz(O(1)) pz(O(2))
px,y(O(1)) px,y(O(2))
+ +- -
+ +- -
+ + +
+- - -
-
3.2 Molekülorbitaltheorie
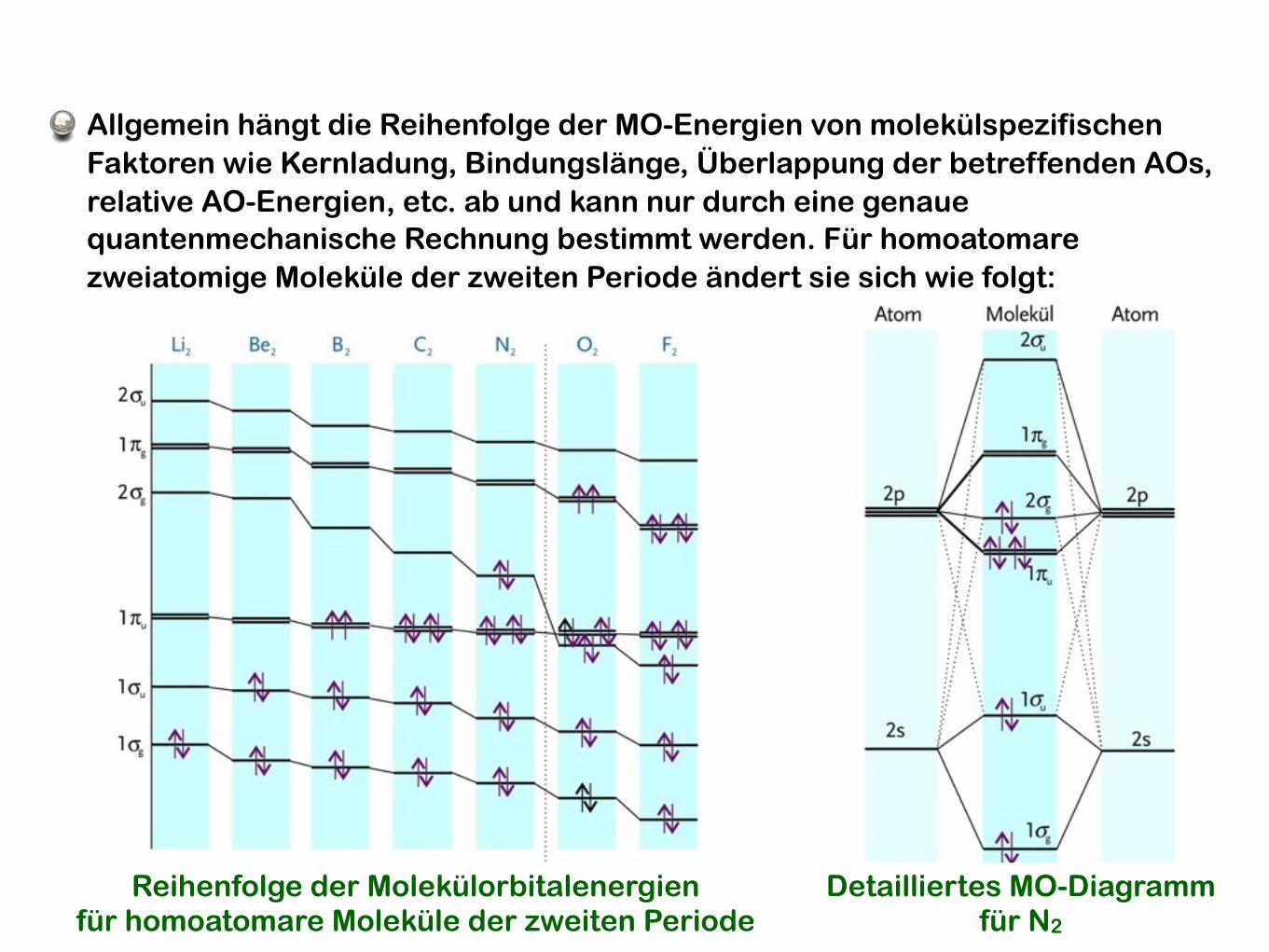
Reihenfolge der Molekülorbitalenergienfür homoatomare Moleküle der zweiten Periode
Detailliertes MO-Diagrammfür N2
Allgemein hängt die Reihenfolge der MO-Energien von molekülspezifischen Faktoren wie Kernladung, Bindungslänge, Überlappung der betreffenden AOs, relative AO-Energien, etc. ab und kann nur durch eine genaue quantenmechanische Rechnung bestimmt werden. Für homoatomare zweiatomige Moleküle der zweiten Periode ändert sie sich wie folgt:
3.2 Molekülorbitaltheorie

Da die Energien der Atomorbitale in H und F verschieden sind, treten diese nicht mehr mit gleicher Gewichtung in der LCAO auf. Ein MO in HF hat demnach die allgemeine Form
wobei ψH und ψF AOs von H bzw. F bezeichnen und die Koeffizienten cH und cF i.A. ungleich sind.
�HF = cH�H + cF�F (3.2.4)
Die chemische Bindung in HF erfolgt durch die LCAO der H(1s)- und F(2pz)-AOs. Man erhält daraus wieder ein bindendes und ein antibindendes MO.
In diesem Fall sind die MO-Energien und Koeffizienten cH und cF nur durch eine detaillierte numerische Rechnung bestimmbar (s. PC-Vertiefungsvorlesungen).
Im bindenden Orbital ist der Koeffizient am Fluoratom cF viel grösser als der Koeffizient am H-Atom cH, die Elektronendichte ist daher am F-Atom viel grösser als am H-Atom. Es ergibt sich eine asymmetrische Ladungsverteilung und somit eine polare Bindung.
3.2.4 Heteroatomare zweiatomige Moleküle: Fluorwasserstoff HF
3.2 Molekülorbitaltheorie

3.2.5 Mehratomige Moleküle
�MO =�
i
ci�i
Das Vorgehen zur Bildung von MOs in mehratomigen Molekülen ist analog zu dem in zweiatomigen Molekülen. Die Linearkombination verläuft i.A. über die Valenzorbitale ψi aller Atome im Molekül:
(3.2.5)
Die MOs sind also über das gesamte Molekül delokalisiert.
Ausser in den einfachsten Fällen lässt sich die MO-Struktur auch qualitativ nicht mehr ohne eine detaillierte quantenchemische Rechnung herleiten (mehr dazu in den PC-Vertiefungsvorlesungen).
Im folgenden soll die MO-Struktur einiger mehratomiger Moleküle diskutiert werden, ohne auf die entsprechenden Rechnungen im Detail eingehen zu wollen.
3.2 Molekülorbitaltheorie
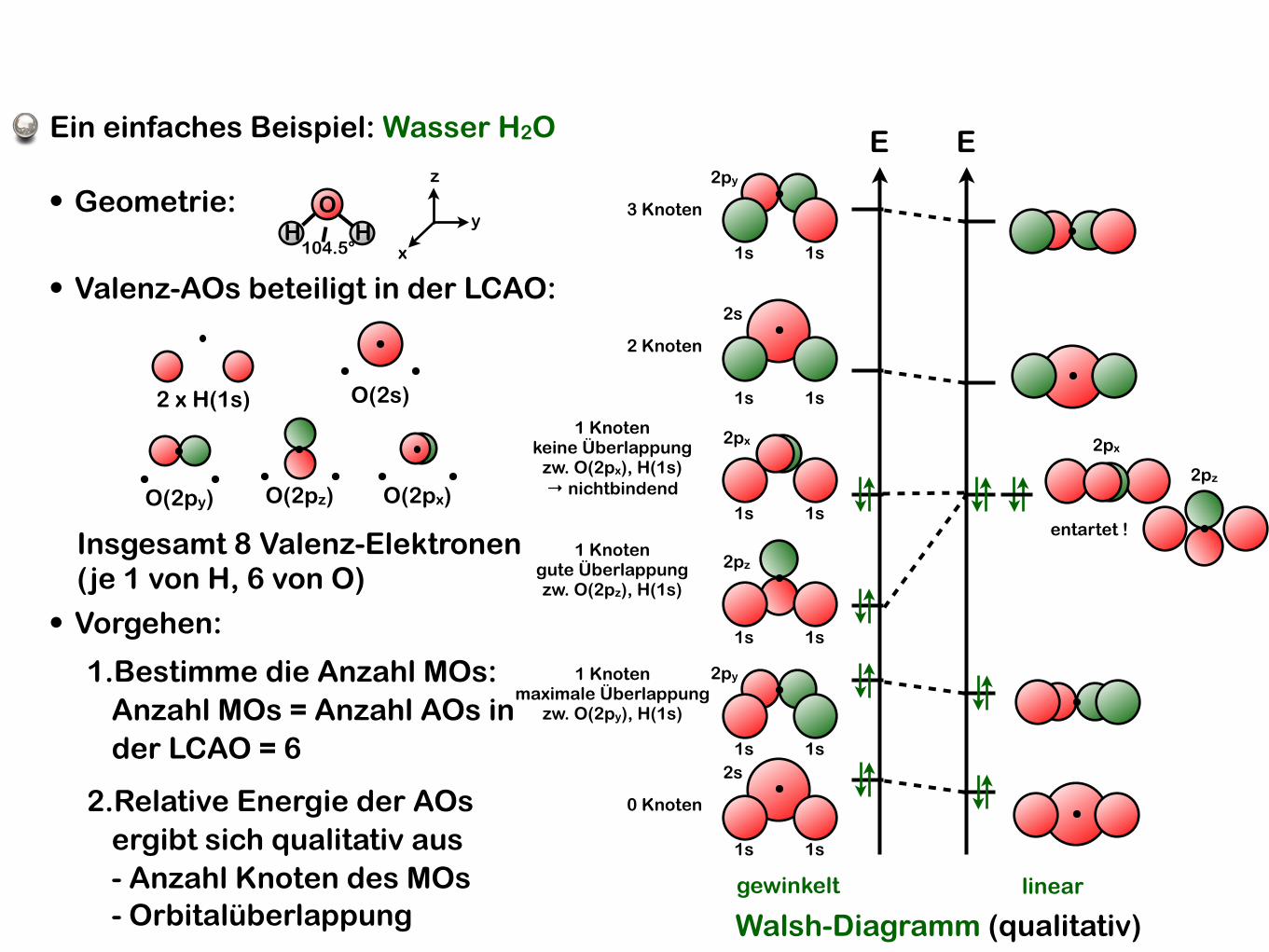
Ein einfaches Beispiel: Wasser H2O
• Geometrie: OH H
104.5° x
y
z
• Vorgehen:
1.Bestimme die Anzahl MOs:Anzahl MOs = Anzahl AOs in der LCAO = 6
2.Relative Energie der AOs ergibt sich qualitativ aus- Anzahl Knoten des MOs- Orbitalüberlappung
• Valenz-AOs beteiligt in der LCAO:
2 x H(1s) O(2s)
O(2py) O(2pz) O(2px)
Insgesamt 8 Valenz-Elektronen (je 1 von H, 6 von O)
E
1s 1s
2s1s 1s
2py
1s 1s
2pz
1s 1s
2px
1s 1s
2s
1s 1s
2py
0 Knoten
1 Knotenmaximale Überlappung
zw. O(2py), H(1s)
1 Knotengute Überlappungzw. O(2pz), H(1s)
1 Knotenkeine Überlappung
zw. O(2px), H(1s)→ nichtbindend
2 Knoten
3 Knoten
gewinkelt
E
2pz
entartet !
Walsh-Diagramm (qualitativ)linear
2px
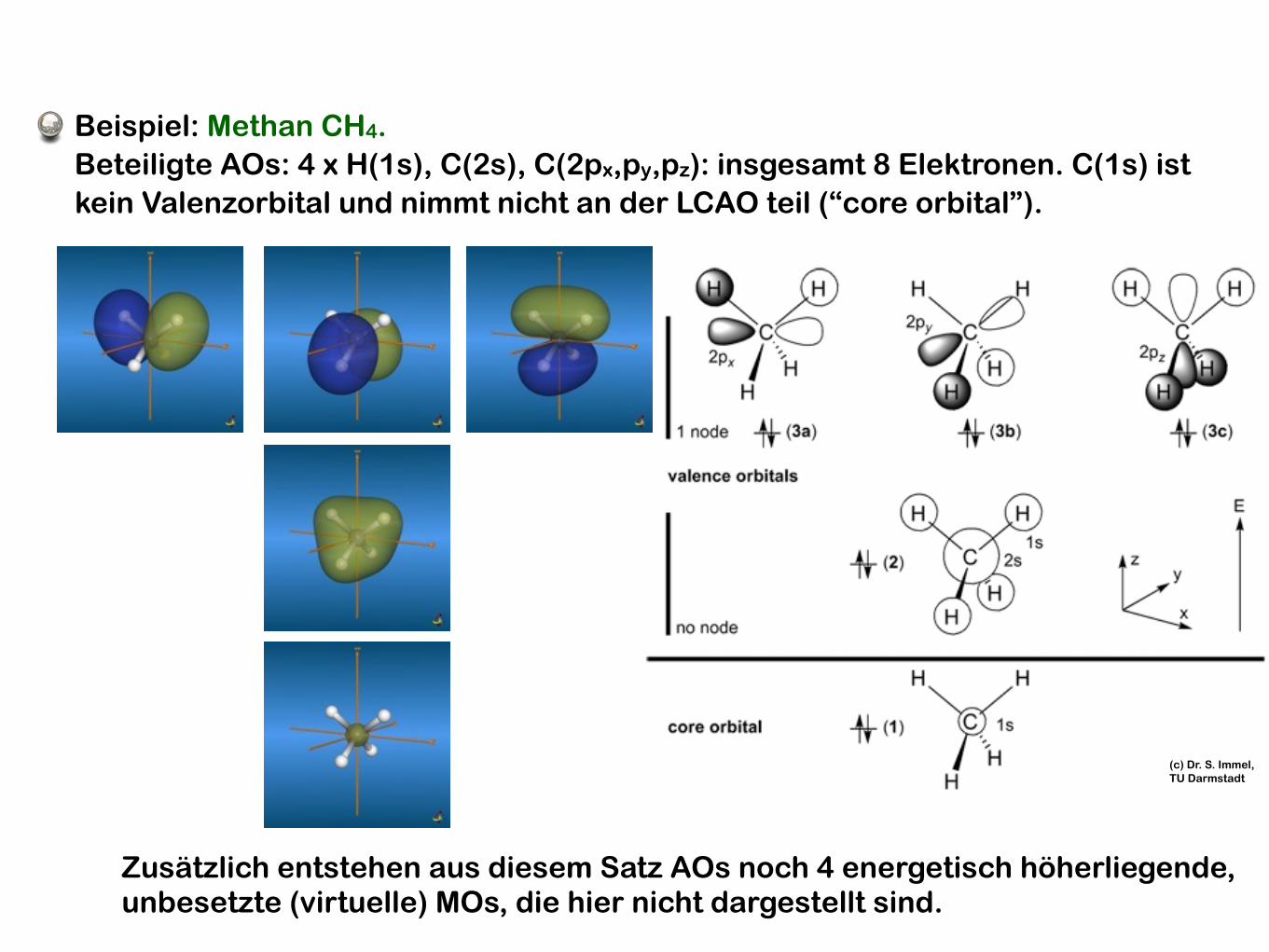
Beispiel: Methan CH4. Beteiligte AOs: 4 x H(1s), C(2s), C(2px,py,pz): insgesamt 8 Elektronen. C(1s) ist kein Valenzorbital und nimmt nicht an der LCAO teil (“core orbital”).
Zusätzlich entstehen aus diesem Satz AOs noch 4 energetisch höherliegende, unbesetzte (virtuelle) MOs, die hier nicht dargestellt sind.
(c) Dr. S. Immel, TU Darmstadt
3.2 Molekülorbitaltheorie
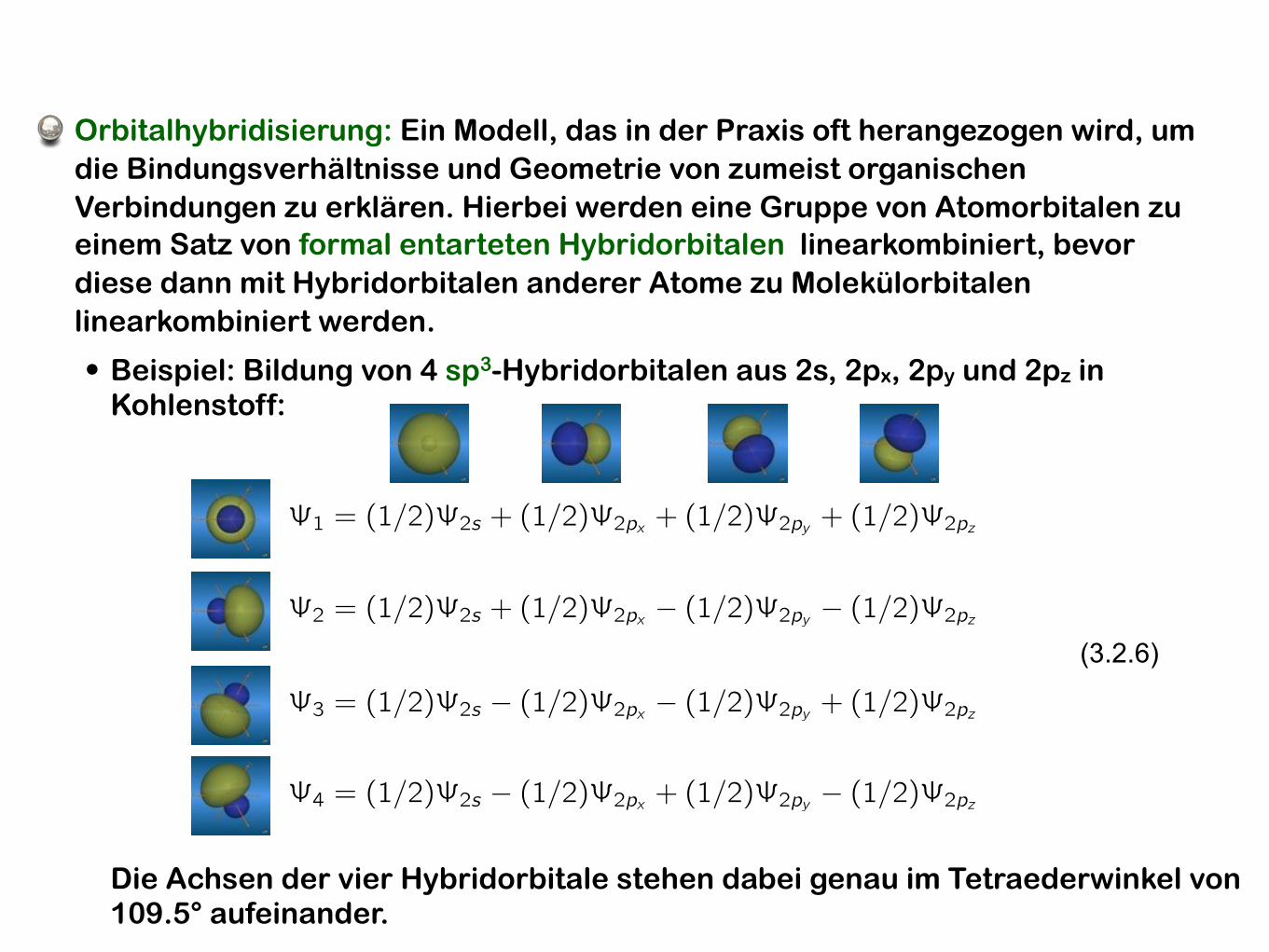
Orbitalhybridisierung: Ein Modell, das in der Praxis oft herangezogen wird, um die Bindungsverhältnisse und Geometrie von zumeist organischen Verbindungen zu erklären. Hierbei werden eine Gruppe von Atomorbitalen zu einem Satz von formal entarteten Hybridorbitalen linearkombiniert, bevor diese dann mit Hybridorbitalen anderer Atome zu Molekülorbitalen linearkombiniert werden.
• Beispiel: Bildung von 4 sp3-Hybridorbitalen aus 2s, 2px, 2py und 2pz in Kohlenstoff:
�1 = (1/2)�2s + (1/2)�2px + (1/2)�2py + (1/2)�2pz
�2 = (1/2)�2s + (1/2)�2px � (1/2)�2py � (1/2)�2pz
�3 = (1/2)�2s � (1/2)�2px � (1/2)�2py + (1/2)�2pz
�4 = (1/2)�2s � (1/2)�2px + (1/2)�2py � (1/2)�2pz
(3.2.6)
Die Achsen der vier Hybridorbitale stehen dabei genau im Tetraederwinkel von 109.5° aufeinander.
3.2 Molekülorbitaltheorie

Die tetraedrische Geometrie von Methan lässt sich dann durch LCAO der vier sp3-Hybrid-AOs mit den 1s-AOs der vier H-Atome erklären:
• Das Konzept der Orbital-Hybridisierung wird in der Praxis oft verwendet, es ist jedoch problematisch, da man energetisch unterschiedliche AOs nicht zu einem Satz von entarten Orbitalen kombinieren kann (z.B. 1x2s und 3x2p zu vier entarteten sp3-Hybridorbitalen). In der Realität findet man dann auch die tatsächliche MO-Struktur von Methan wie zwei Folien vorher dargestellt.
• Völlig analog können auch sp2-Hybridorbitale (aus 2s und 2px,2py) und sp-Hybridorbitale (aus 2s und 2px) bilden.
2 x sp 3 x sp2 4 x sp3
(c) Dr. S. Immel, TU Darmstadt
3.2 Molekülorbitaltheorie
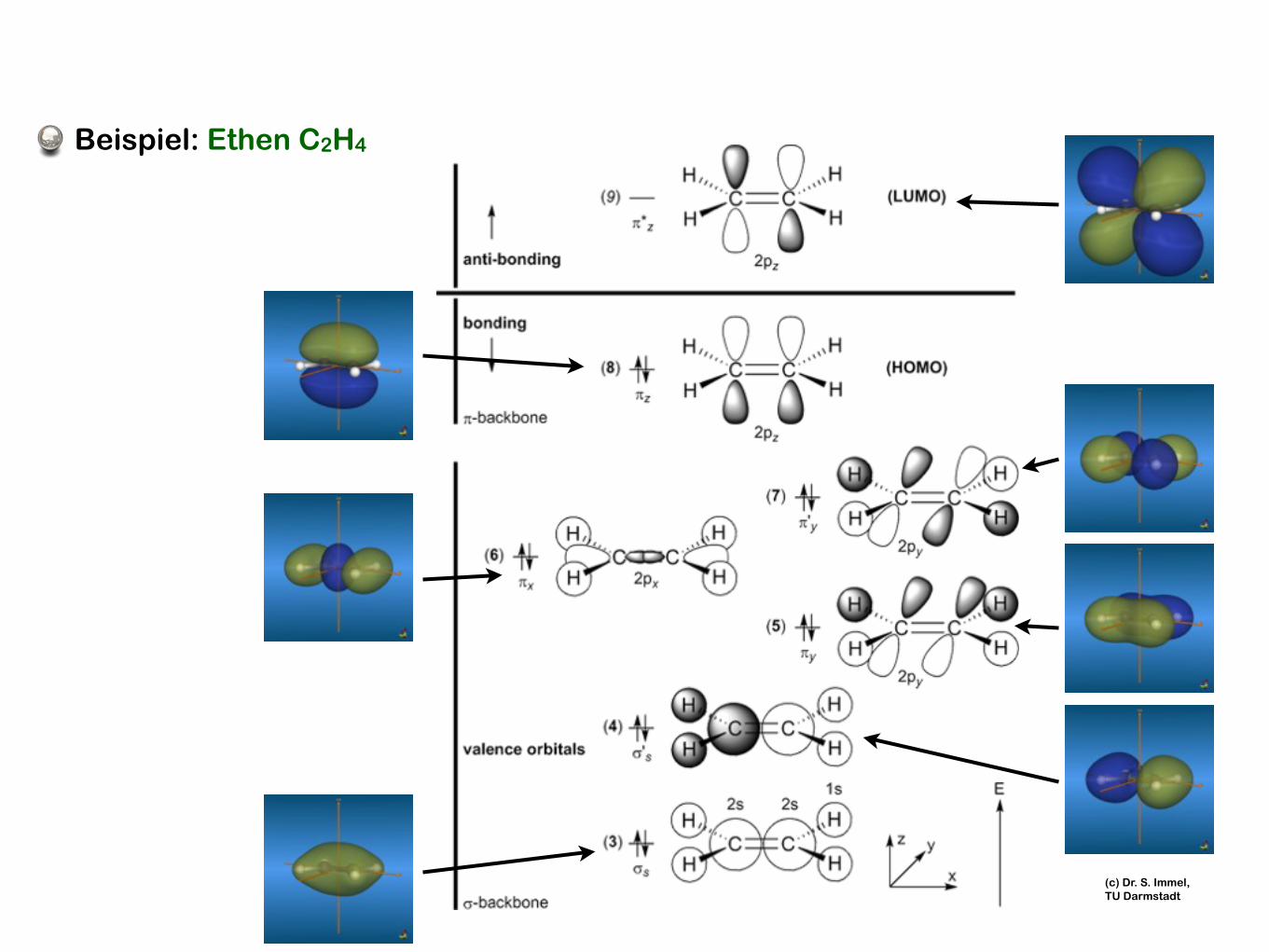
Beispiel: Ethen C2H4
(c) Dr. S. Immel, TU Darmstadt
3.2 Molekülorbitaltheorie

3.2 Molekülorbitaltheorie
3.2.6 π-MOs in mehratomigen Molekülen: die Hückel-Methode
Die Hückel-Methode ist ein einfaches Verfahren zur Beschreibung der Molekülorbitale von mehratomigen Molekülen mit Netzwerken von Doppelbindungen, vorwiegend Kohlenwasserstoffverbindungen wie Alkene, Kumulene und Aromaten.In diesen Verbindungen stellen die entsprechenden π-MOs die Grenzorbitale und bestimmen wesentlich deren chemischen und spektroskopischen Eigenschaften.
Doppelbindungen werden im Rahmen der MO-Theorie durch π-MOs dargestellt. In der Hückel-Näherung trifft man nun folgende Annahmen:• Die Molekülstruktur wird durch ein vorgegebenes, starres Gerüst von σ-MOs
festgelegt.• σ- und π-MOs werden getrennt behandelt.
Die π-MOs erhält man durch LCAO von p-AOs auf den Kohlenstoffen. • Jedes p-AO trägt ein Elektron bei.
Wir illustrieren die Hückel-Methode an einigen Beispielen: Ethen, Butadien und Benzol
Lit.: E. Heilbronner, H. Bock, “Das HMO-Modell und seine Anwendungen”, Verlag Chemie, Weinheim 1970

3.2 Molekülorbitaltheorie
Ethen: LCAO:
Man führt nun folgende weitere Näherungen ein:
• Alle Überlappungsintegrale werden gleich Null gesetzt.• Alle Kohlenstoffe werden als identisch betrachtet, sodass alle Coulomb-
Integrale gleich einem Wert α (α>0) gesetzt werden können.• Alle Resonanzintegrale zwischen nicht benachbarten Atomen werden gleich Null
gesetzt, die restlichen Resonanzintegrale sind alle gleich einem Wert β (β<0).• Die Parameter α und β können aus experimentellen Daten abgeschätzt werden.
Ethen:
p(1) p(2)
�� = c1�pz (1) + c2�pz (2)LCAO:
Die Wellenfunktionen ψπ können wieder mit dem Ritzschen Variationsprinzip berechnet werden. Die Rechnung ist völlig analog zum Beispiel des H2
+-Ions in Abschnitt 3.2.2. Man erhält wieder einen Satz von Sekulärgleichungen:
c1�H11 � E
�+ c2
�H12 � ES
�= 0
c1�H12 � ES
�+ c2
�H22 � E
�= 0
(3.2.7)
mit: H11, H22 ... CoulombintegraleH12
... ResonanzintegralS ... ÜberlappungsintegralE ... Energie
(3.2.8)
Man erhält damit aus Gl. (3.2.8):
(3.2.9)
Mit den Lösungen:
E+ = �+ ⇥ c1 = c2
E� = �� ⇥ c1 = �c2(3.2.10)
E+ besitzt die niedrigere Energie, da β<0.
c1(�� E) + c2⇥ = 0c1⇥ + c2(�� E) = 0
3.2: Molekülorbitaltheorie
(3.2.7)
Die Wellenfunktionen ψπ können wieder mit dem Ritzschen Variationsprinzip berechnet werden. Die Rechnung ist völlig analog zum Beispiel des H2
+-Ions in Abschnitt 3.2.2. Man erhält wieder einen Satz von Sekulärgleichungen:p(1) p(2)
Man führt nun folgende weitere Näherungen ein:
• Alle Überlappungsintegrale werden gleich Null gesetzt.• Alle Kohlenstoffe werden als identisch betrachtet, sodass alle Coulomb-
Integrale gleich einem Wert α (α>0) gesetzt werden können.• Alle Resonanzintegrale zwischen nicht benachbarten Atomen werden gleich Null
gesetzt, die restlichen Resonanzintegrale sind alle gleich einem Wert β (β<0).• Die Parameter α und β können aus experimentellen Daten abgeschätzt werden.
Ethen:
p(1) p(2)
�� = c1�pz (1) + c2�pz (2)LCAO:
Die Wellenfunktionen ψπ können wieder mit dem Ritzschen Variationsprinzip berechnet werden. Die Rechnung ist völlig analog zum Beispiel des H2
+-Ions in Abschnitt 3.2.2. Man erhält wieder einen Satz von Sekulärgleichungen:
c1�H11 � E
�+ c2
�H12 � ES
�= 0
c1�H12 � ES
�+ c2
�H22 � E
�= 0
(3.2.7)
mit: H11, H22 ... CoulombintegraleH12
... ResonanzintegralS ... ÜberlappungsintegralE ... Energie
(3.2.8)
Man erhält damit aus Gl. (3.2.8):
(3.2.9)
Mit den Lösungen:
E+ = �+ ⇥ c1 = c2
E� = �� ⇥ c1 = �c2(3.2.10)
E+ besitzt die niedrigere Energie, da β<0.
c1(�� E) + c2⇥ = 0c1⇥ + c2(�� E) = 0
3.2: Molekülorbitaltheorie
mit: H11, H22 ... Coulombintegrale H12 ... ResonanzintegralS ... ÜberlappungsintegralE ... Energie
(3.2.8)
Man führt nun folgende weitere Näherungen ein:• Alle Überlappungsintegrale werden gleich Null gesetzt. • Alle Kohlenstoffe werden als identisch betrachtet, sodass alle Coulomb-
Integrale gleich einem Wert α (α>0) gesetzt werden können. • Alle Resonanzintegrale zwischen nicht benachbarten Atomen werden gleich Null
gesetzt, die restlichen Resonanzintegrale sind alle gleich einem Wert β (β<0). • Die Parameter α und β können aus experimentellen Daten abgeschätzt werden.
Man erhält damit aus Gl. (3.2.8):
Man führt nun folgende weitere Näherungen ein:
• Alle Überlappungsintegrale werden gleich Null gesetzt.• Alle Kohlenstoffe werden als identisch betrachtet, sodass alle Coulomb-
Integrale gleich einem Wert α (α>0) gesetzt werden können.• Alle Resonanzintegrale zwischen nicht benachbarten Atomen werden gleich Null
gesetzt, die restlichen Resonanzintegrale sind alle gleich einem Wert β (β<0).• Die Parameter α und β können aus experimentellen Daten abgeschätzt werden.
Ethen:
p(1) p(2)
�� = c1�pz (1) + c2�pz (2)LCAO:
Die Wellenfunktionen ψπ können wieder mit dem Ritzschen Variationsprinzip berechnet werden. Die Rechnung ist völlig analog zum Beispiel des H2
+-Ions in Abschnitt 3.2.2. Man erhält wieder einen Satz von Sekulärgleichungen:
c1�H11 � E
�+ c2
�H12 � ES
�= 0
c1�H12 � ES
�+ c2
�H22 � E
�= 0
(3.2.7)
mit: H11, H22 ... CoulombintegraleH12
... ResonanzintegralS ... ÜberlappungsintegralE ... Energie
(3.2.8)
Man erhält damit aus Gl. (3.2.8):
(3.2.9)
Mit den Lösungen:
E+ = �+ ⇥ c1 = c2
E� = �� ⇥ c1 = �c2(3.2.10)
E+ besitzt die niedrigere Energie, da β<0.
c1(�� E) + c2⇥ = 0c1⇥ + c2(�� E) = 0
3.2: Molekülorbitaltheorie
Man führt nun folgende weitere Näherungen ein:
• Alle Überlappungsintegrale werden gleich Null gesetzt.• Alle Kohlenstoffe werden als identisch betrachtet, sodass alle Coulomb-
Integrale gleich einem Wert α (α>0) gesetzt werden können.• Alle Resonanzintegrale zwischen nicht benachbarten Atomen werden gleich Null
gesetzt, die restlichen Resonanzintegrale sind alle gleich einem Wert β (β<0).• Die Parameter α und β können aus experimentellen Daten abgeschätzt werden.
Ethen:
p(1) p(2)
�� = c1�pz (1) + c2�pz (2)LCAO:
Die Wellenfunktionen ψπ können wieder mit dem Ritzschen Variationsprinzip berechnet werden. Die Rechnung ist völlig analog zum Beispiel des H2
+-Ions in Abschnitt 3.2.2. Man erhält wieder einen Satz von Sekulärgleichungen:
c1�H11 � E
�+ c2
�H12 � ES
�= 0
c1�H12 � ES
�+ c2
�H22 � E
�= 0
(3.2.7)
mit: H11, H22 ... CoulombintegraleH12
... ResonanzintegralS ... ÜberlappungsintegralE ... Energie
(3.2.8)
Man erhält damit aus Gl. (3.2.8):
(3.2.9)
Mit den Lösungen:
E+ = �+ ⇥ c1 = c2
E� = �� ⇥ c1 = �c2(3.2.10)
E+ besitzt die niedrigere Energie, da β<0.
c1(�� E) + c2⇥ = 0c1⇥ + c2(�� E) = 0
3.2: Molekülorbitaltheorie
(3.2.9) (3.2.10)
E+ besitzt die niedrigere Energie, da β<0.
3.2 Molekülorbitaltheorie
Graphische Darstellung der Lösungen:Graphische Darstellung der Lösungen:
Monozyklische planare Ringsysteme:
Monozyklische Ring-Moleküle mit konjugierten Doppelbindungen besitzen im Hückel-Bild ein pz-Orbital an jedem C-Atom, aus denen die π-MOs durch LCAO gebildet werden. Für die π -Orbital-Energien erhält man die Formel:
Ek = �+ 2⇥ cos�2⇤k/N
�(3.2.11)
mit: N ... Anzahl pz-AOs in der LCAOk = 1,2,..,N
↵+ 2�
↵+ �
↵� �
↵� 2�
Bsp.: Benzol: N=6
E1 = E5 = �+ ⇥
E2 = E4 = �� ⇥E3 = �� 2⇥E6 = �+ 2⇥
Da α>0 und β<0 ist die Abfolge der Orbitalenergien: E6 < E1=E5 < E2=E4 < E3
3.2: Molekülorbitaltheorie
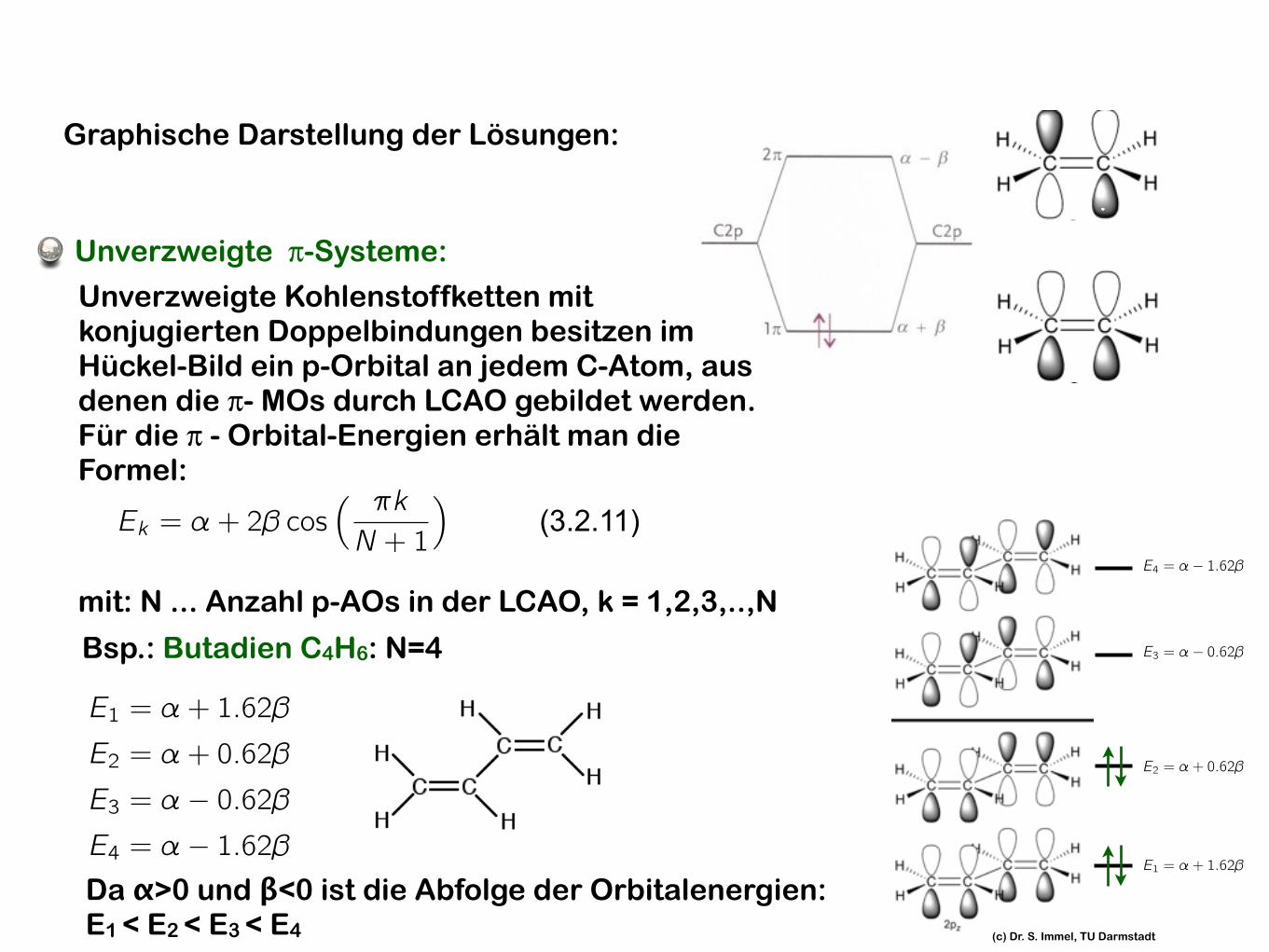
Bsp.: Butadien C4H6: N=4
E1 = ↵+ 1.62�
E2 = ↵+ 0.62�
E3 = ↵� 0.62�E4 = ↵� 1.62�Da α>0 und β<0 ist die Abfolge der Orbitalenergien: E1 < E2 < E3 < E4
E1 = ↵+ 1.62�
E2 = ↵+ 0.62�
E3 = ↵� 0.62�E4 = ↵� 1.62�
E1 = ↵+ 1.62�
E2 = ↵+ 0.62�
E3 = ↵� 0.62�E4 = ↵� 1.62�
E1 = ↵+ 1.62�
E2 = ↵+ 0.62�
E3 = ↵� 0.62�E4 = ↵� 1.62�
E1 = ↵+ 1.62�
E2 = ↵+ 0.62�
E3 = ↵� 0.62�E4 = ↵� 1.62�
(c) Dr. S. Immel, TU Darmstadt
Unverzweigte π-Systeme:
Unverzweigte Kohlenstoffketten mit konjugierten Doppelbindungen besitzen im Hückel-Bild ein p-Orbital an jedem C-Atom, aus denen die π- MOs durch LCAO gebildet werden. Für die π - Orbital-Energien erhält man die Formel:
Ek = ↵+ 2� cos⇣ ⇡kN + 1
⌘
mit: N ... Anzahl p-AOs in der LCAO, k = 1,2,3,..,N
(3.2.11)
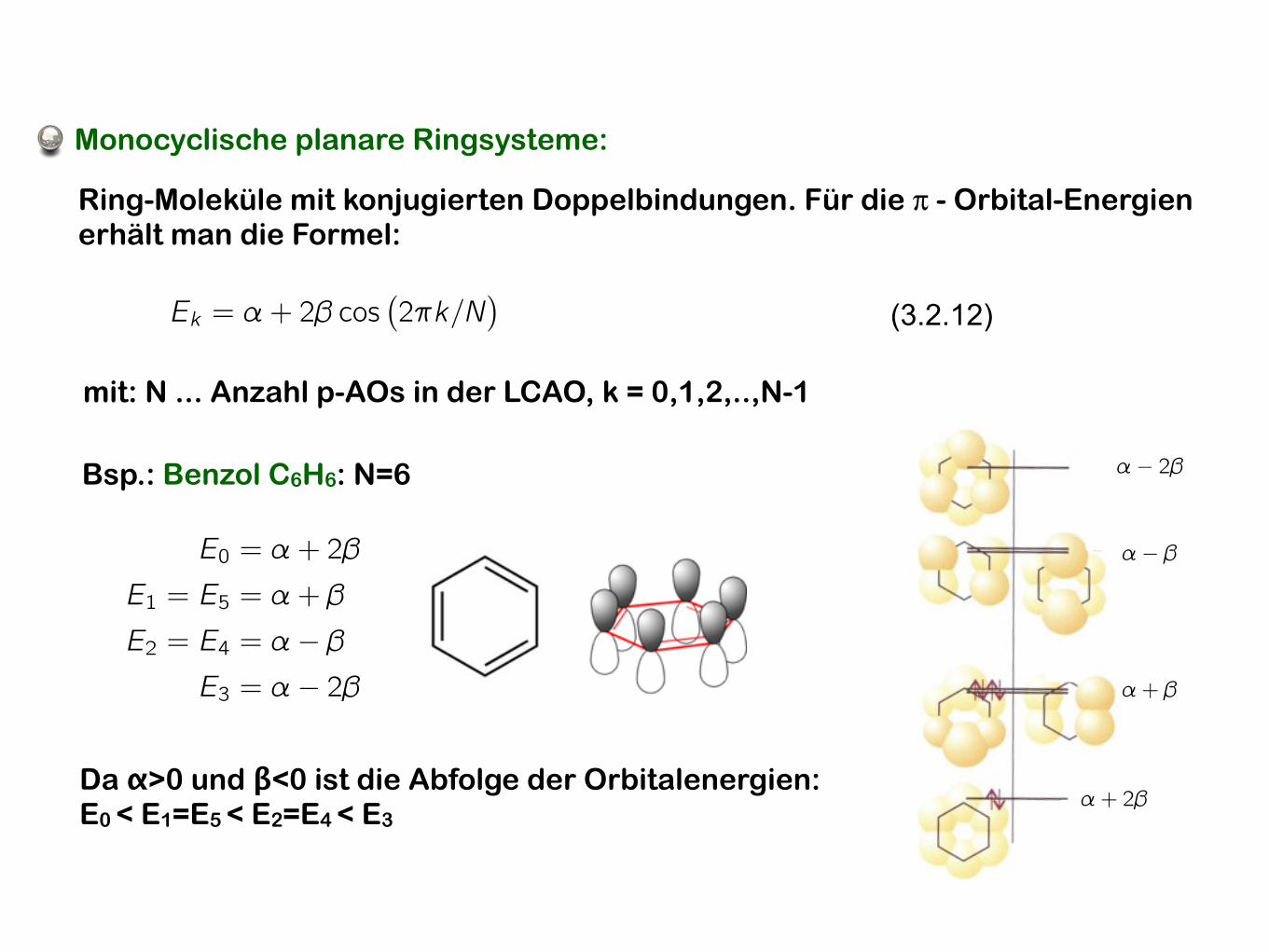
3.2 Molekülorbitaltheorie
Monocyclische planare Ringsysteme:
Ring-Moleküle mit konjugierten Doppelbindungen. Für die π - Orbital-Energien erhält man die Formel:
mit: N ... Anzahl p-AOs in der LCAO, k = 0,1,2,..,N-1
Bsp.: Benzol C6H6: N=6
Da α>0 und β<0 ist die Abfolge der Orbitalenergien: E0 < E1=E5 < E2=E4 < E3
Graphische Darstellung der Lösungen:
Monozyklische planare Ringsysteme:
Monozyklische Ring-Moleküle mit konjugierten Doppelbindungen besitzen im Hückel-Bild ein pz-Orbital an jedem C-Atom, aus denen die π-MOs durch LCAO gebildet werden. Für die π -Orbital-Energien erhält man die Formel:
Ek = �+ 2⇥ cos�2⇤k/N
�(3.2.11)
mit: N ... Anzahl pz-AOs in der LCAOk = 1,2,..,N
↵+ 2�
↵+ �
↵� �
↵� 2�
Bsp.: Benzol: N=6
E1 = E5 = �+ ⇥
E2 = E4 = �� ⇥E3 = �� 2⇥E6 = �+ 2⇥
Da α>0 und β<0 ist die Abfolge der Orbitalenergien: E6 < E1=E5 < E2=E4 < E3
3.2: Molekülorbitaltheorie
(3.2.12)
Graphische Darstellung der Lösungen:
Monozyklische planare Ringsysteme:
Monozyklische Ring-Moleküle mit konjugierten Doppelbindungen besitzen im Hückel-Bild ein pz-Orbital an jedem C-Atom, aus denen die π-MOs durch LCAO gebildet werden. Für die π -Orbital-Energien erhält man die Formel:
Ek = �+ 2⇥ cos�2⇤k/N
�(3.2.11)
mit: N ... Anzahl pz-AOs in der LCAOk = 1,2,..,N
↵+ 2�
↵+ �
↵� �
↵� 2�
Bsp.: Benzol: N=6
E1 = E5 = �+ ⇥
E2 = E4 = �� ⇥E3 = �� 2⇥E6 = �+ 2⇥
Da α>0 und β<0 ist die Abfolge der Orbitalenergien: E6 < E1=E5 < E2=E4 < E3
3.2: Molekülorbitaltheorie
E0 = ↵+ 2�
E1 = E5 = ↵+ �
E2 = E4 = ↵� �E3 = ↵� 2�
Graphische Darstellung der Lösungen:
Monozyklische planare Ringsysteme:
Monozyklische Ring-Moleküle mit konjugierten Doppelbindungen besitzen im Hückel-Bild ein pz-Orbital an jedem C-Atom, aus denen die π-MOs durch LCAO gebildet werden. Für die π -Orbital-Energien erhält man die Formel:
Ek = �+ 2⇥ cos�2⇤k/N
�(3.2.11)
mit: N ... Anzahl pz-AOs in der LCAOk = 1,2,..,N
↵+ 2�
↵+ �
↵� �
↵� 2�
Bsp.: Benzol: N=6
E1 = E5 = �+ ⇥
E2 = E4 = �� ⇥E3 = �� 2⇥E6 = �+ 2⇥
Da α>0 und β<0 ist die Abfolge der Orbitalenergien: E6 < E1=E5 < E2=E4 < E3
3.2: Molekülorbitaltheorie

3.3 Die Born-Oppenheimer-Potentialfläche
3.3.1 Born-Oppenheimer-Näherung und molekulare Potentialflächen
Wir haben bisher bei der Diskussion der Bewegung der Elektronen die Bewegung der Kerne vernachlässigt und das Molekülgerüst als starr angenommen. Tatsächlich ist sogar der leichteste Atomkern (H) ca. 1800 mal schwerer als ein Elektron und bewegt sich dementsprechend langsamer.
Die Bewegungen der Elektronen und der Kerne können daher in sehr guter Näherung voneinander getrennt werden (Born-Oppenheimer-Näherung). Die schnellen Elektronen “sehen” daher immer ein starres Kerngerüst und ihre Wellenfunktionen passen sich instantan an, wenn sich die Geometrie der Kerne ändert, um die Energie zu minimisieren.
3.3 Die Born-Oppenheimer-Potentialfläche
Im Rahmen der Orbitalnäherung ist die gesamte elektronische Energie Eel eines Moleküls die Summe der Molekülorbital-Energien Ei aller N Elektronen im Molekül*:
Eel =N�
i=1
Ei (3.3.1)
Die Orbitalenergien - und damit die gesamte elektronische Energie - hängen von der Kernkonfiguration ab.
* Es handelt sich hierbei um eine sehr grobe, qualitative Näherung, da die Wechselwirkung zwischen den Elektronen vernachlässigt wird. Für eine genauere Behandlung siehe PC-Vertiefungsvorlesungen.
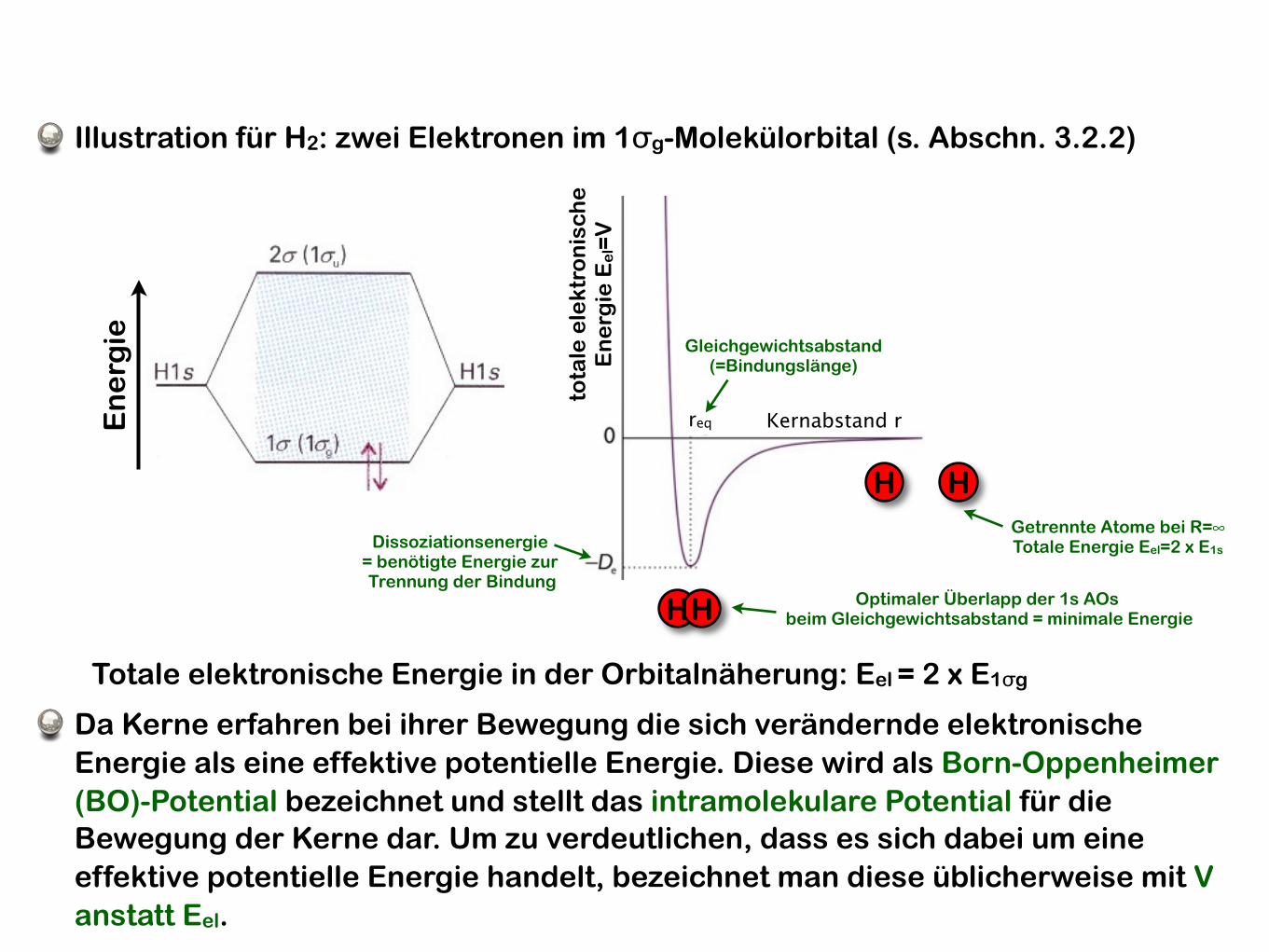
Illustration für H2: zwei Elektronen im 1σg-Molekülorbital (s. Abschn. 3.2.2)
HH
HH
Gleichgewichtsabstand(=Bindungslänge)
Optimaler Überlapp der 1s AOs beim Gleichgewichtsabstand = minimale Energie
Getrennte Atome bei R=∞Totale Energie Eel=2 x E1s
En
erg
ie
Dissoziationsenergie= benötigte Energie zur Trennung der Bindung
tota
le e
lekt
ron
isch
e
En
erg
ie E
el=
V
Totale elektronische Energie in der Orbitalnäherung: Eel = 2 x E1σg
Da Kerne erfahren bei ihrer Bewegung die sich verändernde elektronische Energie als eine effektive potentielle Energie. Diese wird als Born-Oppenheimer (BO)-Potential bezeichnet und stellt das intramolekulare Potential für die Bewegung der Kerne dar. Um zu verdeutlichen, dass es sich dabei um eine effektive potentielle Energie handelt, bezeichnet man diese üblicherweise mit V anstatt Eel.
req Kernabstand r
3.3 Die Born-Oppenheimer-Potentialfläche

Für mehratomige Moleküle ist das Born-Oppenheimer-Potential eine mehrdimensional Funktion der Koordinaten aller Kerne im Molekül. Man spricht daher von einer mehrdimensionalen Born-Oppenheimer-Potentialhypefläche.
1. Ein Atom kann sich in alle drei räumlichen Dimensionen (x,y,z) bewegen
→ 3 FG für die Bewegung in x,y,z
3.3.2 Interne und externe Bewegungsfreiheitsgrade eines Moleküls
Für die weitere Diskussion des BO-Potentials müssen wir zwischen internen und externen Bewegungsfreiheitsgraden (FG) eines Moleküls unterscheiden. Jedem FG entspricht eine Koordinate der Bewegung.
Analyse der möglichen Bewegungen:
2. Ein Molekül bestehend aus K Atomen kann als Ansammlung seiner Atome
betrachtet werden → 3K FG für die Bewegung jedes Atoms in x,y,z
Ein stabiles Molekül entspricht einem Minimum der BO-Hyperfläche. Die entsprechende Kernkonfiguration wird als Gleichgewichtsgeometrie des Moleküls bezeichnet.
3.3 Die Born-Oppenheimer-Potentialfläche

• Das Molekül kann sich als ganzes bewegen: Translation → 3 FG
Die Bewegungen der Atome im Molekül sind jedoch nicht unabhängig voneinander:
• Das Molekül kann als ganzes rotieren: Rotation: lineare Moleküle → 2 FG
nicht-lineare Mol. → 3 FG
• Die restlichen 3K-3-2=3K-5 (lineare Mol.) bzw. 3K-6 (nicht-lineare Mol.) FG
entsprechen den Schwingungen (Vibrationen) des Moleküls
Translation und Rotation werden als externe Bewegungsfreiheitsgrade bezeichnet, da sie die Bewegung des Moleküls als ganzes betreffen.
Die Molekülschwingungen entsprechen den internen Bewegungsfreiheitsgraden, da sich dabei die relative Lage der Kerne zueinander ändert:
Lineare Moleküle: 3K-5 interne FG = interne KoordinatenNicht-lineare Moleküle: 3K-6 interne FG = interne Koordinaten
3.3 Die Born-Oppenheimer-Potentialfläche
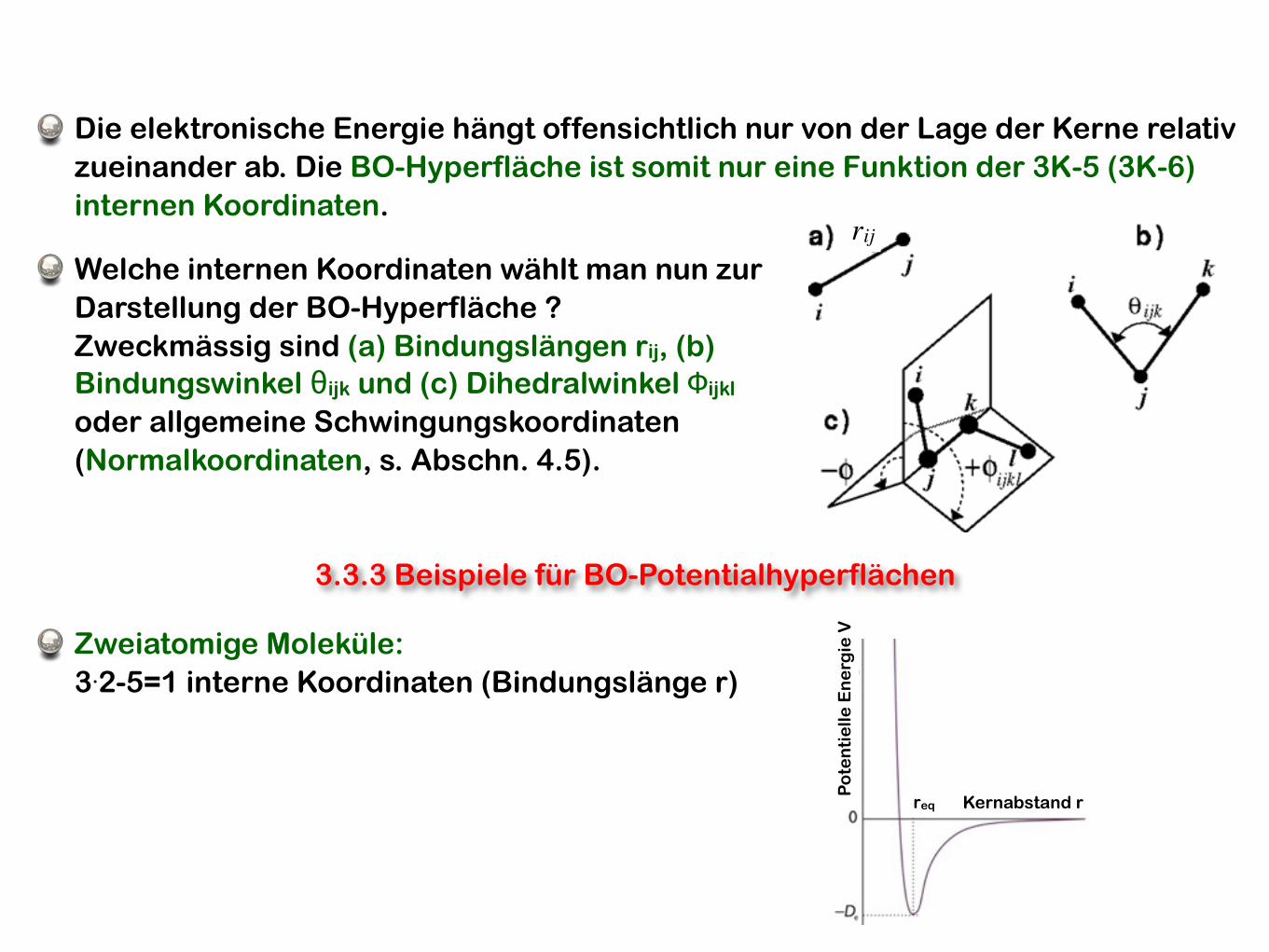
Die elektronische Energie hängt offensichtlich nur von der Lage der Kerne relativ zueinander ab. Die BO-Hyperfläche ist somit nur eine Funktion der 3K-5 (3K-6) internen Koordinaten.
Welche internen Koordinaten wählt man nun zur Darstellung der BO-Hyperfläche ? Zweckmässig sind (a) Bindungslängen rij, (b) Bindungswinkel θijk und (c) Dihedralwinkel Φijkl oder allgemeine Schwingungskoordinaten (Normalkoordinaten, s. Abschn. 4.5).
rij
3.3.3 Beispiele für BO-Potentialhyperflächen
Po
ten
tie
lle E
ne
rgie
VZweiatomige Moleküle:3.2-5=1 interne Koordinaten (Bindungslänge r)
Kernabstand rreq
3.3 Die Born-Oppenheimer-Potentialfläche
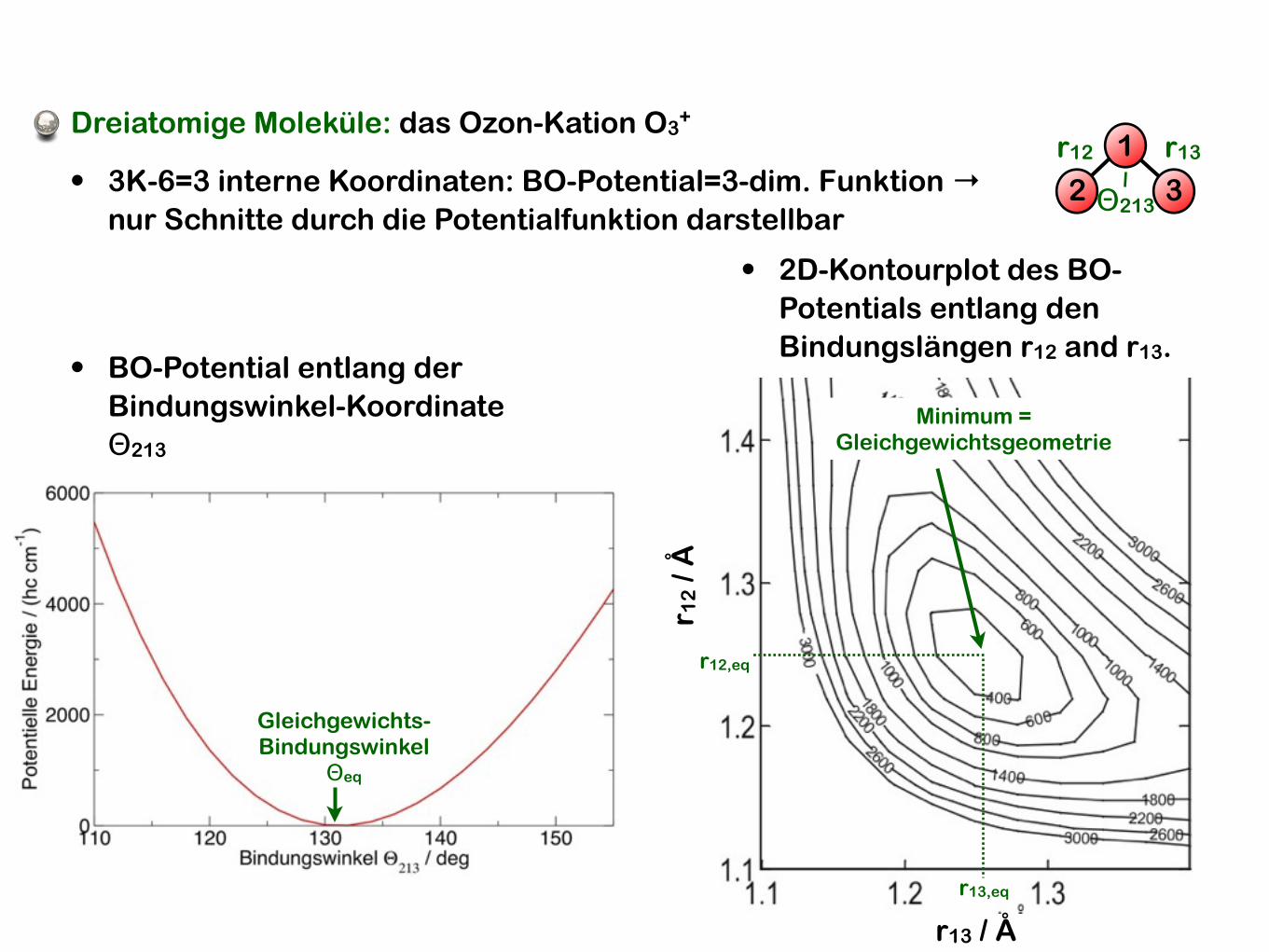
Gleichgewichts-Bindungswinkel
Θeq
• BO-Potential entlang der Bindungswinkel-Koordinate Θ213
• 3K-6=3 interne Koordinaten: BO-Potential=3-dim. Funktion →nur Schnitte durch die Potentialfunktion darstellbar
• 2D-Kontourplot des BO-Potentials entlang den Bindungslängen r12 and r13.
r 12 /
Å
r13 / Å
Minimum =Gleichgewichtsgeometrie
r12,eq
r13,eq
Dreiatomige Moleküle: das Ozon-Kation O3+
r12 r13
Θ2132 3
1
3.3 Die Born-Oppenheimer-Potentialfläche
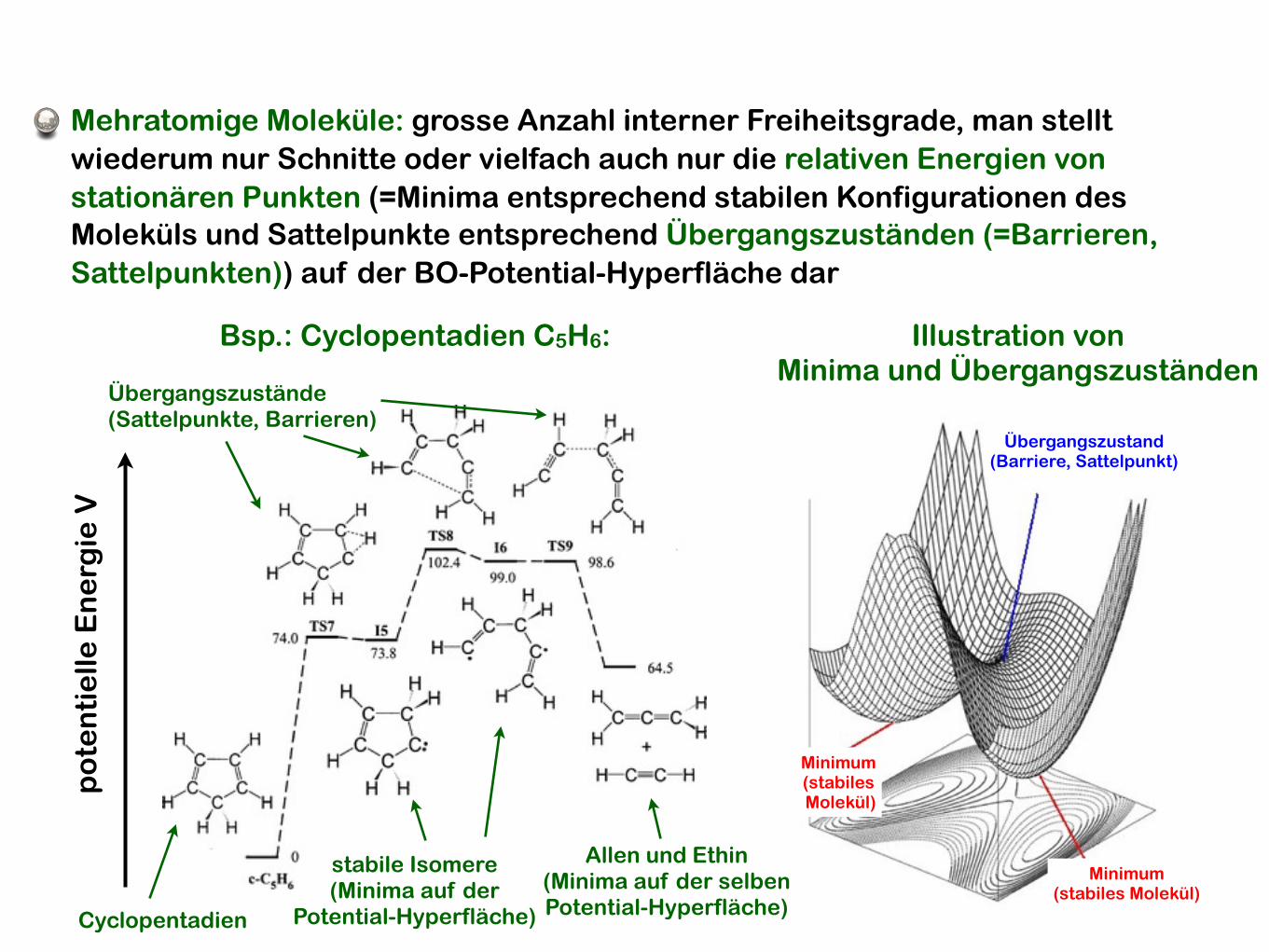
Mehratomige Moleküle: grosse Anzahl interner Freiheitsgrade, man stellt wiederum nur Schnitte oder vielfach auch nur die relativen Energien von stationären Punkten (=Minima entsprechend stabilen Konfigurationen des Moleküls und Sattelpunkte entsprechend Übergangszuständen (=Barrieren, Sattelpunkten)) auf der BO-Potential-Hyperfläche dar
Bsp.: Cyclopentadien C5H6:
po
ten
tie
lle E
ne
rgie
V
Cyclopentadien
Übergangszustände(Sattelpunkte, Barrieren)
stabile Isomere(Minima auf der
Potential-Hyperfläche)
Allen und Ethin(Minima auf der selbenPotential-Hyperfläche)
Übergangszustand(Barriere, Sattelpunkt)
Minimum(stabiles Molekül)
Minimum(stabiles Molekül)
Illustration vonMinima und Übergangszuständen
3.3 Die Born-Oppenheimer-Potentialfläche
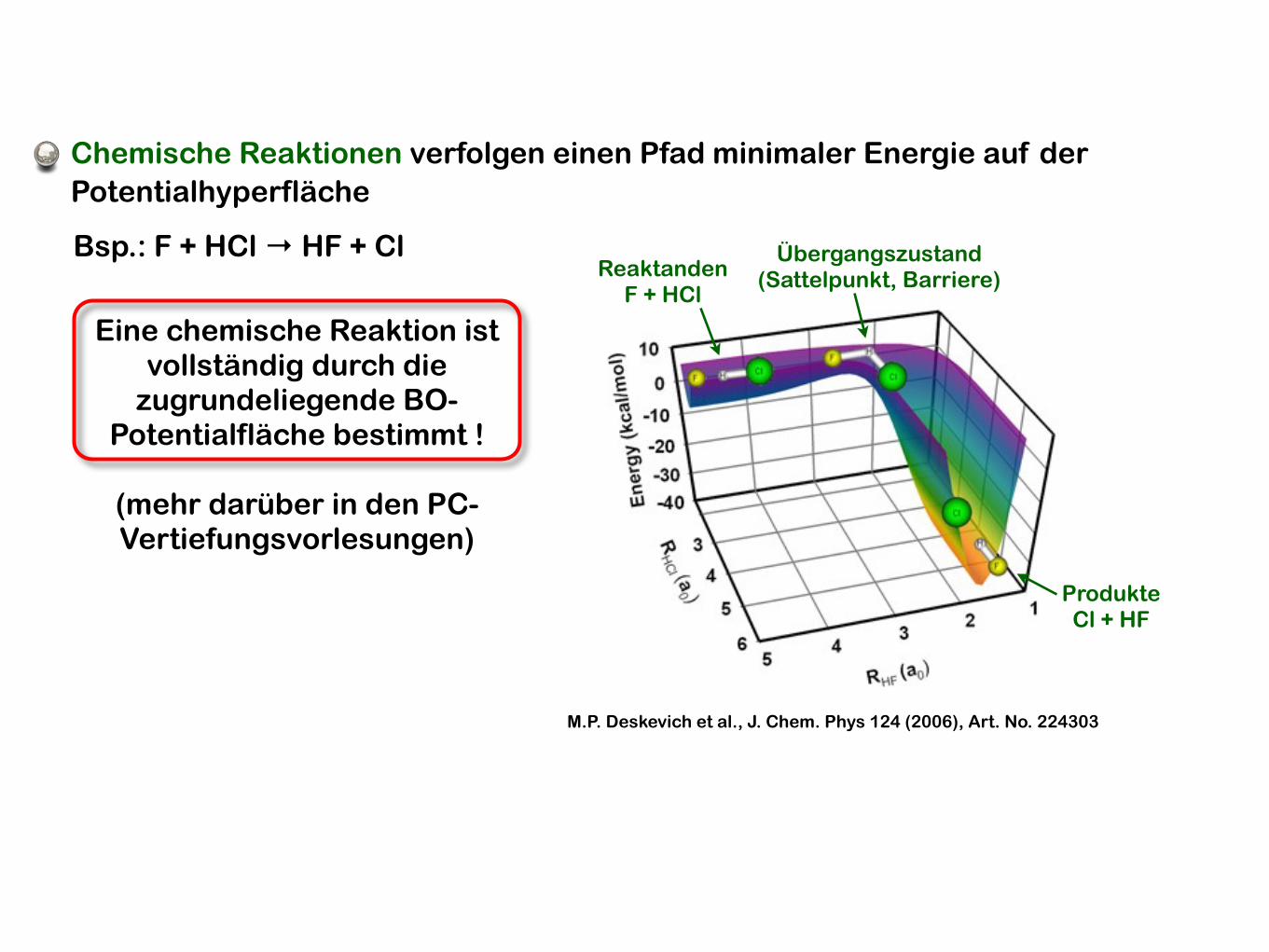
M.P. Deskevich et al., J. Chem. Phys 124 (2006), Art. No. 224303
Chemische Reaktionen verfolgen einen Pfad minimaler Energie auf der Potentialhyperfläche
Bsp.: F + HCl → HF + ClReaktanden
F + HCl
ProdukteCl + HF
Übergangszustand(Sattelpunkt, Barriere)
Eine chemische Reaktion ist vollständig durch die
zugrundeliegende BO-Potentialfläche bestimmt !
(mehr darüber in den PC-Vertiefungsvorlesungen)
3.3 Die Born-Oppenheimer-Potentialfläche

3.3.4 Intramolekulare Modellpotentiale
Die Berechnung einer vollständigen BO-Hyperfläche mit quantenchemischen Methoden ist extrem (!) aufwändig und derzeit nur für maximal vieratomige Moleküle machbar. Man verwendet daher häufig analytische Modellpotentiale um Bereiche der BO-Hyperfläche - in der Regel eindimensionale Schnitte entlang bestimmter internen Koordinaten - zu approximieren.
harmonisch
BO
• einfachste mögliche Potentialfunktion:
(3.3.2)
k ... Kraftkonstanter ... irgendeine interne Koordinate
• gute Näherung an das tatsächliche BO-Potential nahe an der Gleichgewichtsgeometrie req
• wird bei grossen Auslenkungen aus der Gleichgewichtslage (r-req) unphysikalisch, da es keine Dissoziation des Moleküls erlaubt
Harmonische Potentialfunktion:
Harmonische Näherung des BO-Potentials für ein zweiatomiges Molekül
3.3 Die Born-Oppenheimer-Potentialfläche

• Die Kraftkonstante k kann aus der zweiten Ableitung des BO-Potentials V(r) bei der Gleichgewichtsgeometrie req berechnet werden
(3.3.3)k =�2V (r)
�r2
����r=req
Anharmonische Potentialfunktion:
(3.3.4)
kubischeAnharmonizität
quartischeAnharmonizität
• Die harmonische Näherung kann als erstes Glied einer Taylorreihen-Approximation des BO-Potentials um die Gleichgewichtsgeometrie aufgefasst werden, die durch die Berücksichtigung von Termen höherer Ordnung (=Anharmonizitäten) systematisch verbessert werden kann:
• Diese Form von Reihenentwicklung ist der in der Praxis am häufigsten gebrauchte Ansatz, da sie eine beliebig genaue Approximation des BO-Potentials erlaubt.
3.3 Die Born-Oppenheimer-Potentialfläche
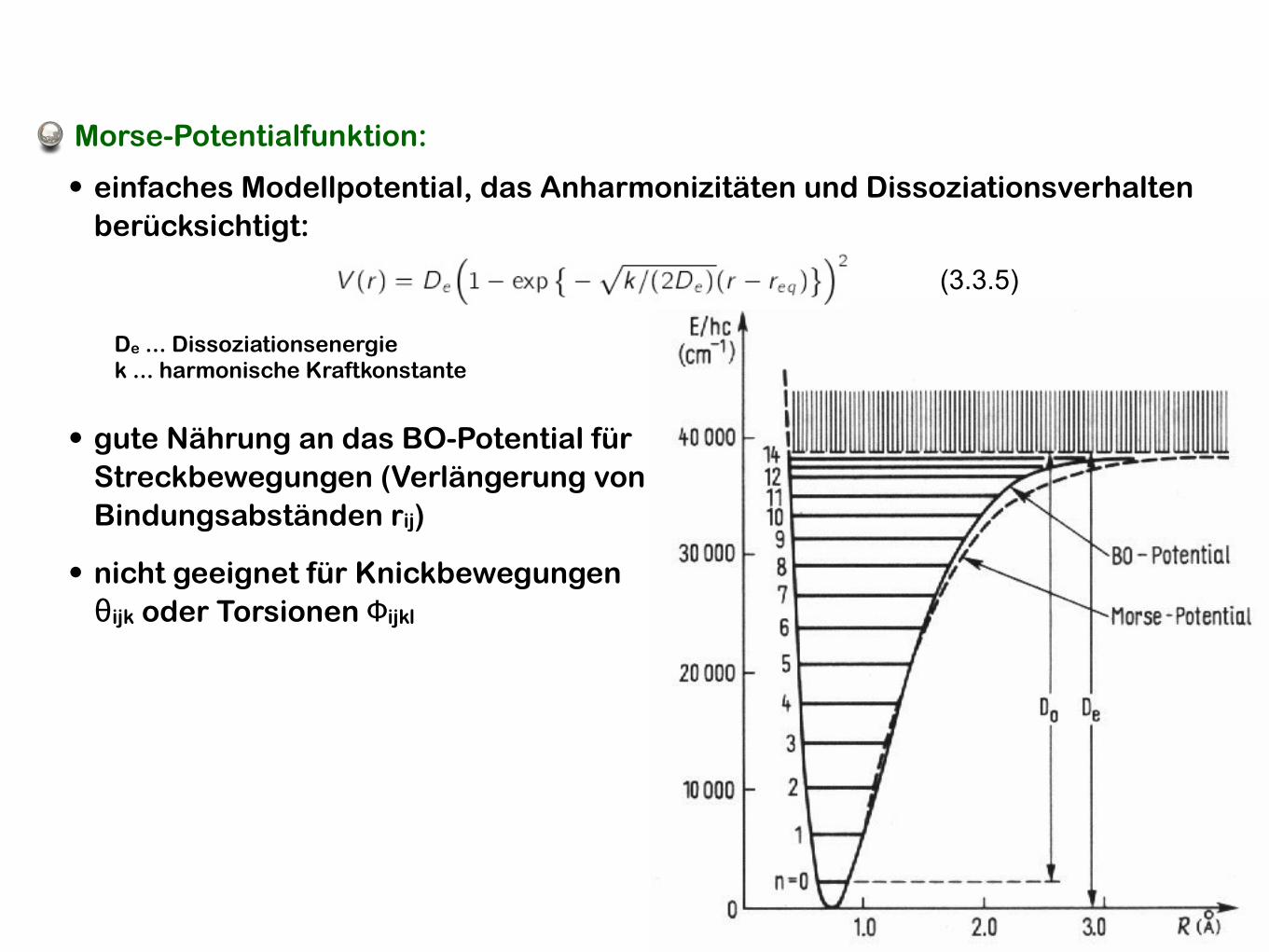
Morse-Potentialfunktion:
De ... Dissoziationsenergiek ... harmonische Kraftkonstante
• nicht geeignet für Knickbewegungen θijk oder Torsionen Φijkl
• einfaches Modellpotential, das Anharmonizitäten und Dissoziationsverhalten berücksichtigt:
(3.3.5)
• gute Nährung an das BO-Potential für Streckbewegungen (Verlängerung von Bindungsabständen rij)
3.3 Die Born-Oppenheimer-Potentialfläche
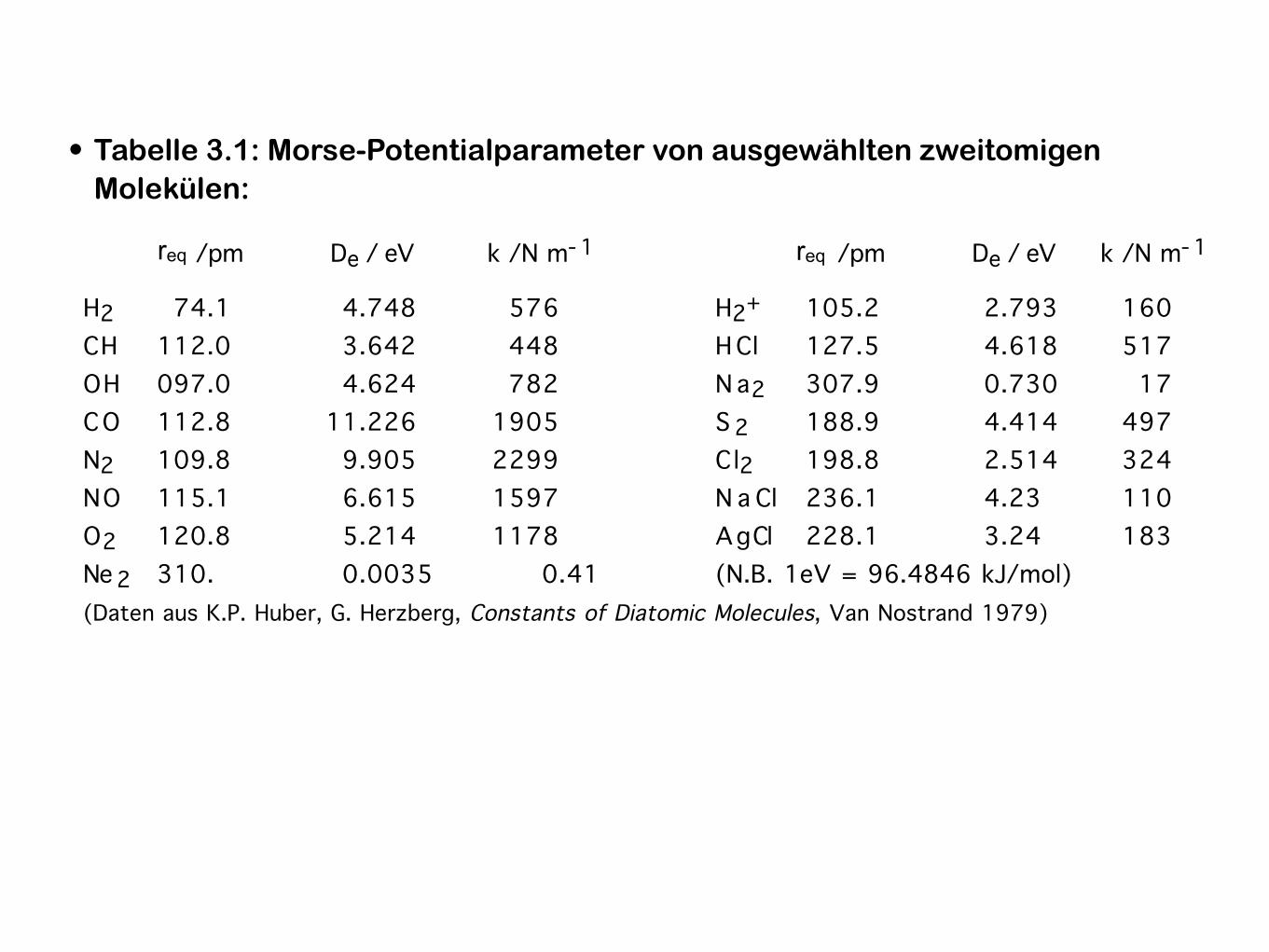
• Tabelle 3.1: Morse-Potentialparameter von ausgewählten zweitomigen Molekülen:
Empirische Potentialfunktion
#
V R( ) = De 1$e$ k 2De( ) % R$Re( )[ ]{ }
2
$De
Re = Gleichgewichtsabstand (Minimum)
De = Dissoziationsenergie
k = Kraftkonstante (Krümmung bei Re)
Die Figur zeigt den typischen Verlauf eines
Morse-Potentials; der Energie-Nullpunkt ist
(entgegen der obigen Definition) auf der
Höhe des Potentialminimums gewählt. Die
Figur zeigt auch eine Parabel mit demselben
Minimum und derselben Krümmung.
Die Parametrisierung erlaubt die
Konstruktion von V(R) nach Messung
dreier Grössen.
Die Morse-Funktion eignet sich vorzüglich
zur Modellierung des Potentialverlaufs in
chemischen Bindungen. Sie wird zur Be-
(Figur aus C.N. Banwell, Fundamentals of schreibung von zweiatomigen, aber auch
Molecular Spectroscopy, McGraw-Hill 1972) von mehratomigen Molekülen verwendet.
_______________________________________________________________________________
Parameter für die Morse-Potentialfunktion (Beispiele)
Gleichgewichtsabstände Re, Dissoziationsenergien De, Kraftkonstanten k zweiatomiger Moleküle.
Re /pm De / eV k /N m- 1 Re /pm De / eV k /N m- 1
H2 74.1 4.748 576 H2+ 105.2 2.793 160
C H 112.0 3.642 448 H Cl 127.5 4.618 517
O H 097.0 4.624 782 N a 2 307.9 0.730 17
C O 112.8 11.226 1905 S 2 188.9 4.414 497
N2 109.8 9.905 2299 Cl2 198.8 2.514 324
N O 115.1 6.615 1597 N a Cl 236.1 4.23 110
O 2 120.8 5.214 1178 A g Cl 228.1 3.24 183
Ne 2 310. 0.0035 0.41 (N.B. 1eV = 96.4846 kJ/mol)
(Daten aus K.P. Huber, G. Herzberg, Constants of Diatomic Molecules, Van Nostrand 1979)
E i n f ü h r u n g i n d i e P h y s i k a l i s c h e C h e m i e
K3-3 Struktur der Moleküle
Morse - Funktion
req req
3.3 Die Born-Oppenheimer-Potentialfläche
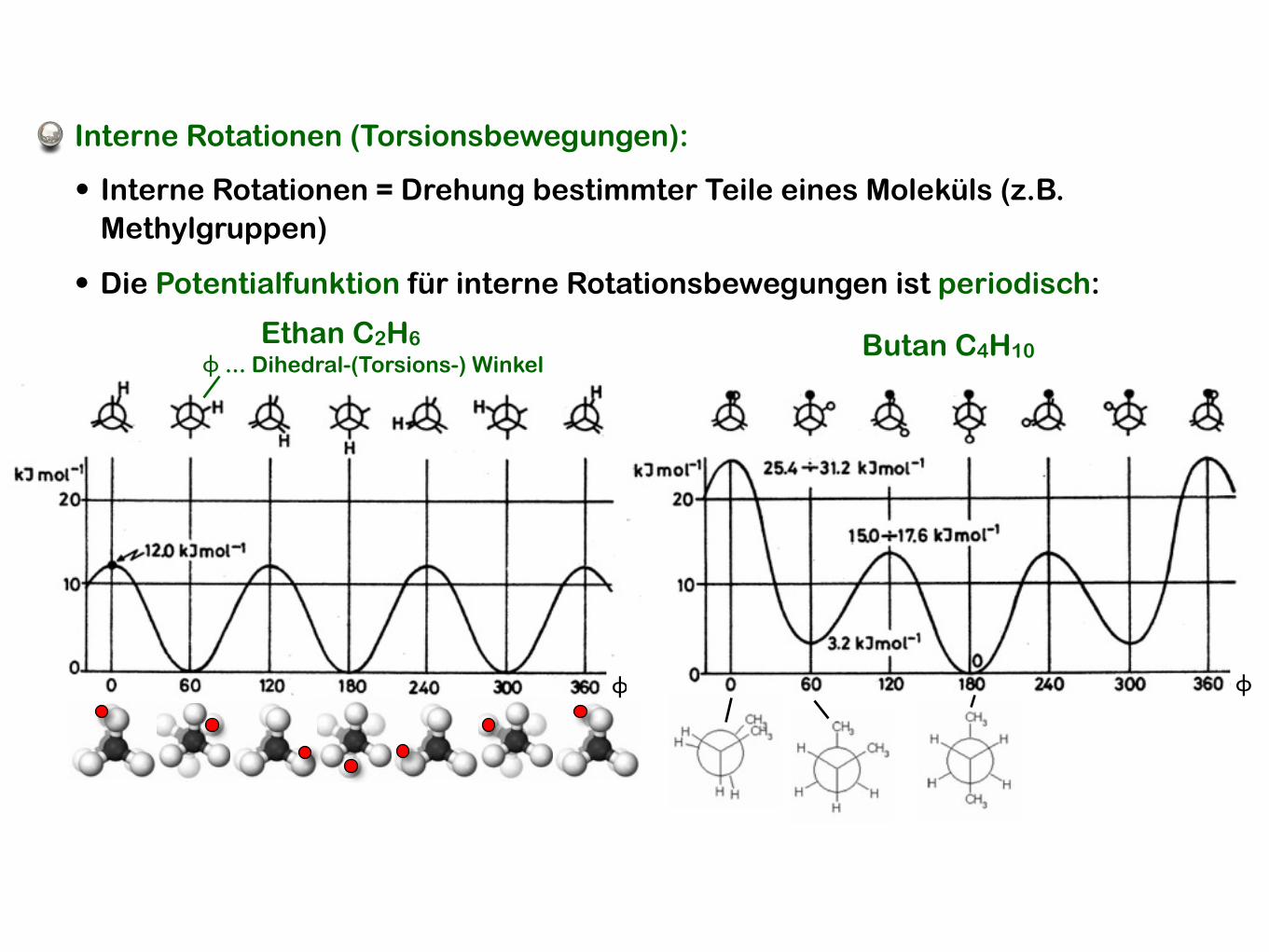
Interne Rotationen (Torsionsbewegungen):
• Die Potentialfunktion für interne Rotationsbewegungen ist periodisch:
Ethan C2H6ϕ ... Dihedral-(Torsions-) Winkel
• Interne Rotationen = Drehung bestimmter Teile eines Moleküls (z.B. Methylgruppen)
ϕ
Butan C4H10
ϕ
3.3 Die Born-Oppenheimer-Potentialfläche
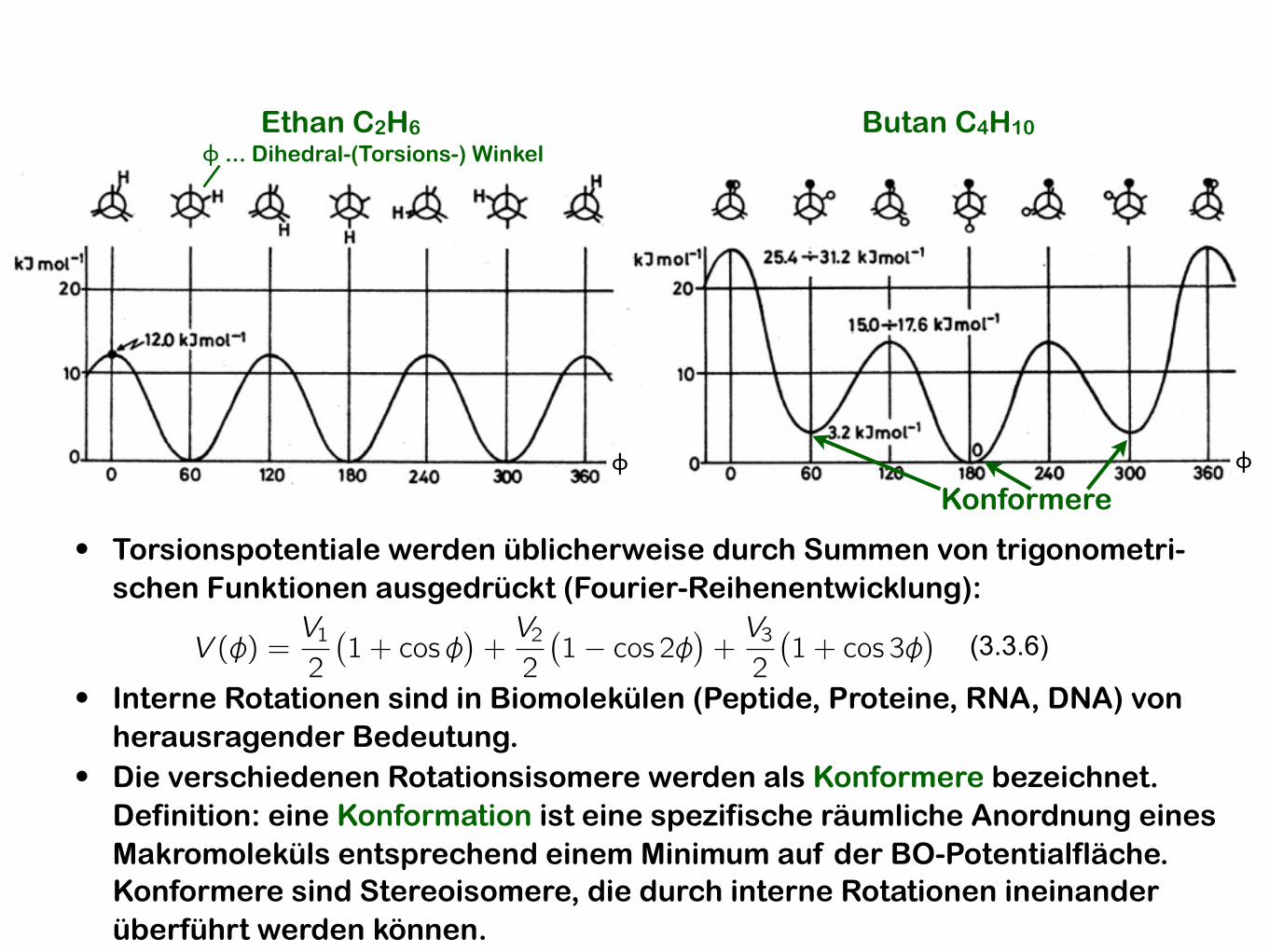
• Torsionspotentiale werden üblicherweise durch Summen von trigonometri-schen Funktionen ausgedrückt (Fourier-Reihenentwicklung):
(3.3.6)
• Interne Rotationen sind in Biomolekülen (Peptide, Proteine, RNA, DNA) von herausragender Bedeutung.
• Die verschiedenen Rotationsisomere werden als Konformere bezeichnet. Definition: eine Konformation ist eine spezifische räumliche Anordnung eines Makromoleküls entsprechend einem Minimum auf der BO-Potentialfläche. Konformere sind Stereoisomere, die durch interne Rotationen ineinander überführt werden können.
Konformere
Ethan C2H6ϕ ... Dihedral-(Torsions-) Winkel
Butan C4H10
ϕ ϕ
V (�) =V12
�1 + cos�
⇥+V22
�1� cos 2�
⇥+V32
�1 + cos 3�
⇥
3.3 Die Born-Oppenheimer-Potentialfläche
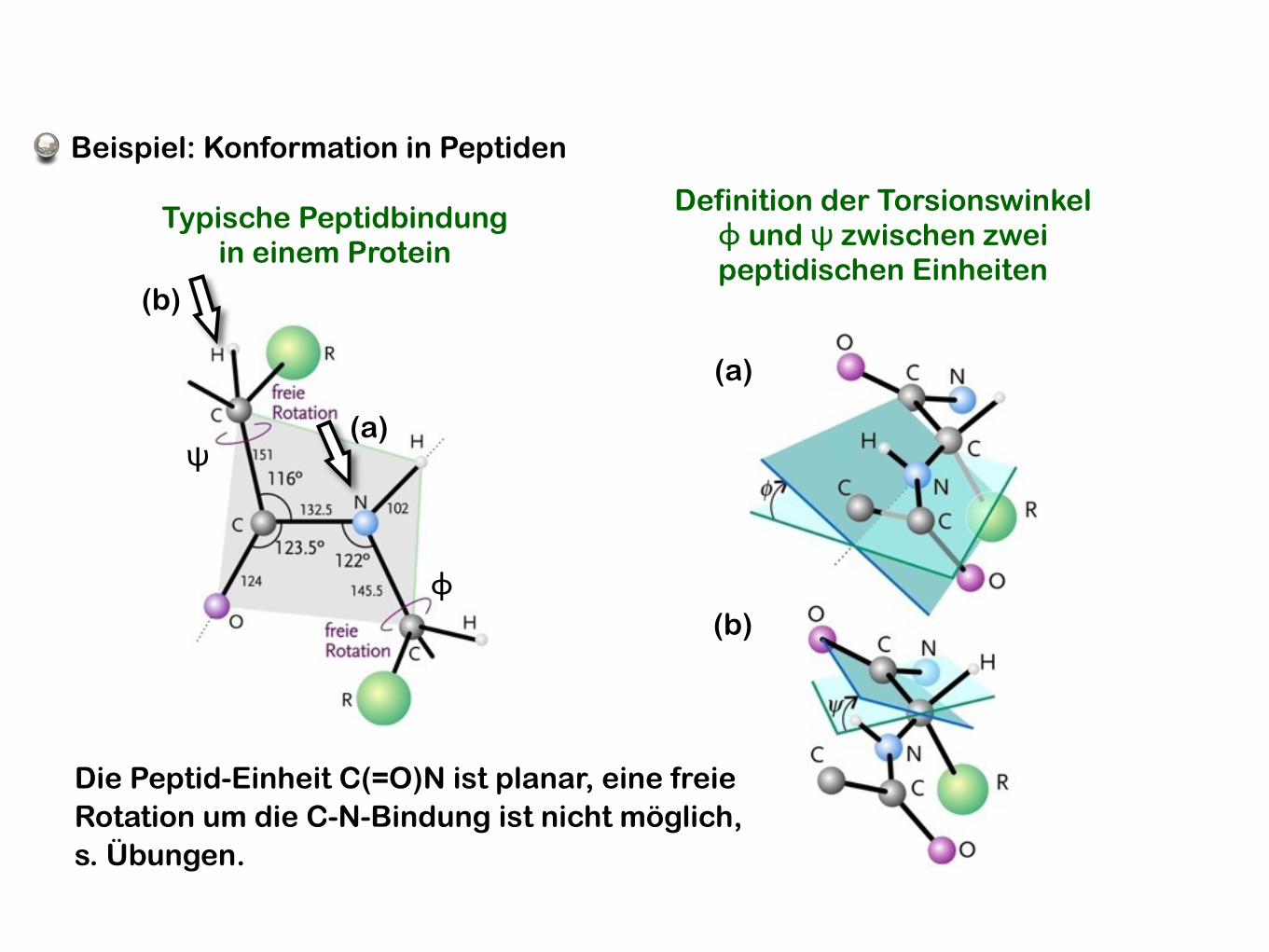
Beispiel: Konformation in Peptiden
Typische Peptidbindungin einem Protein
Definition der Torsionswinkelϕ und ψ zwischen zweipeptidischen Einheiten
Die Peptid-Einheit C(=O)N ist planar, eine freie Rotation um die C-N-Bindung ist nicht möglich,s. Übungen.
ϕ
ψ
(b)
(a)
(a)
(b)
3.3 Die Born-Oppenheimer-Potentialfläche

3.3.5 Molekülmechanik (MM) - Rechnungen
Der verwendete Satz intramolekularer Modellpotentiale wird als Kraftfeld bezeichnet. Die Werte der in den Modellpotentialen enthaltenen Parametern werden dabei durch Vergleich mit experimentellen oder quantenchemischen Daten angepasst.
Molekülmechanik (MM) - Rechnungen werden verwendet um die Potentialhyperflächen von grossen Molekülen zu modellieren. Dabei wird die totale potentielle Energie als Summe von Beiträgen aus Modellpotentialen für alle internen Koordinaten berechnet.
Heute sind eine Vielfalt verschiedener Kraftfelder sowie MM-Computerprogrammpakete in Gebrauch (CHARM, AMBER, GROMOS,..., s. PC-Vertiefungsvorlesungen). Molekülmechanik-Modelle stellen den heute wichtigsten Ansatz zur theoretischen Modellierung von Biomolekülen dar.
Beispiel: das MM2-Kraftfeld (nach N. Allinger, J. Am. Chem. Soc. 99 (1977), 8127). Die das Kraftfeld konstituierenden Modellpotentiale sind:
• Kompressionsenergie EC (Bindungsstreckbewegungen für alle gebundenen Atompaare i,j, vgl. mit anharmonischer Potentialfunktion Gl. (3.3.4)):
(3.3.7)
Parameter: ks,ij, rij,eq, CS i k
jrij
θijkrjk
ECi j =ks,ij2(ri j � ri j,eq)2
�1 + CSi j(ri j � ri j,eq)
�
3.3 Die Born-Oppenheimer-Potentialfläche

• Knickenergie EBijk (für alle Bindungswinkel θijk,vgl. mit anharmonischer Potentialfunktion Gl. (3.3.4)):
(3.3.8)
Parameter: kb,ijk, θijk,eq, SFijk
i k
jrij
θijkrjkEBi jk =
kb,i jk2(�i jk � �ijk,eq)2
�1 + SFi jk(�i jk � �ijk,eq)4
⇥
• Streck-Knickenergie ESBijk (für alle Bindungswinkel θijk): Knick- und Streckbewegungen sind in der Regel nicht unabhängig voneinander. Eine Änderung des Bindungswinkels führt meist auch zu einer Änderung der Bindungslänge. Der betreffende Energiebeitrag wird modelliert als
(3.3.9)
Parameter: ksb,ijk, θeq, rijk,eq
ESB = ksb,i jk(�i jk � �i jk,eq)�(ri j � ri j,eq) + (rjk � rjk,eq)
⇥
• Zudem gibt es weitere Beiträge für inter-molekulare Wechselwirkungen (van-der-Waals-WW, Dipol-Dipol-WW, s. Kapitel 5).
• Torsionsenergie ETijkl (für alle Dihedralwinkel ϕijkl=ϕα, α=ijkl, vgl. mit Gl. (3.3.6)):
ET� =V1,�2
�1 + cos��
⇥+V2,�2
�1� cos 2��
⇥+V3,�2
�1 + cos 3��
⇥(3.3.10)
Parameter: V1,α, V2,α, V3,α
3.3 Die Born-Oppenheimer-Potentialfläche
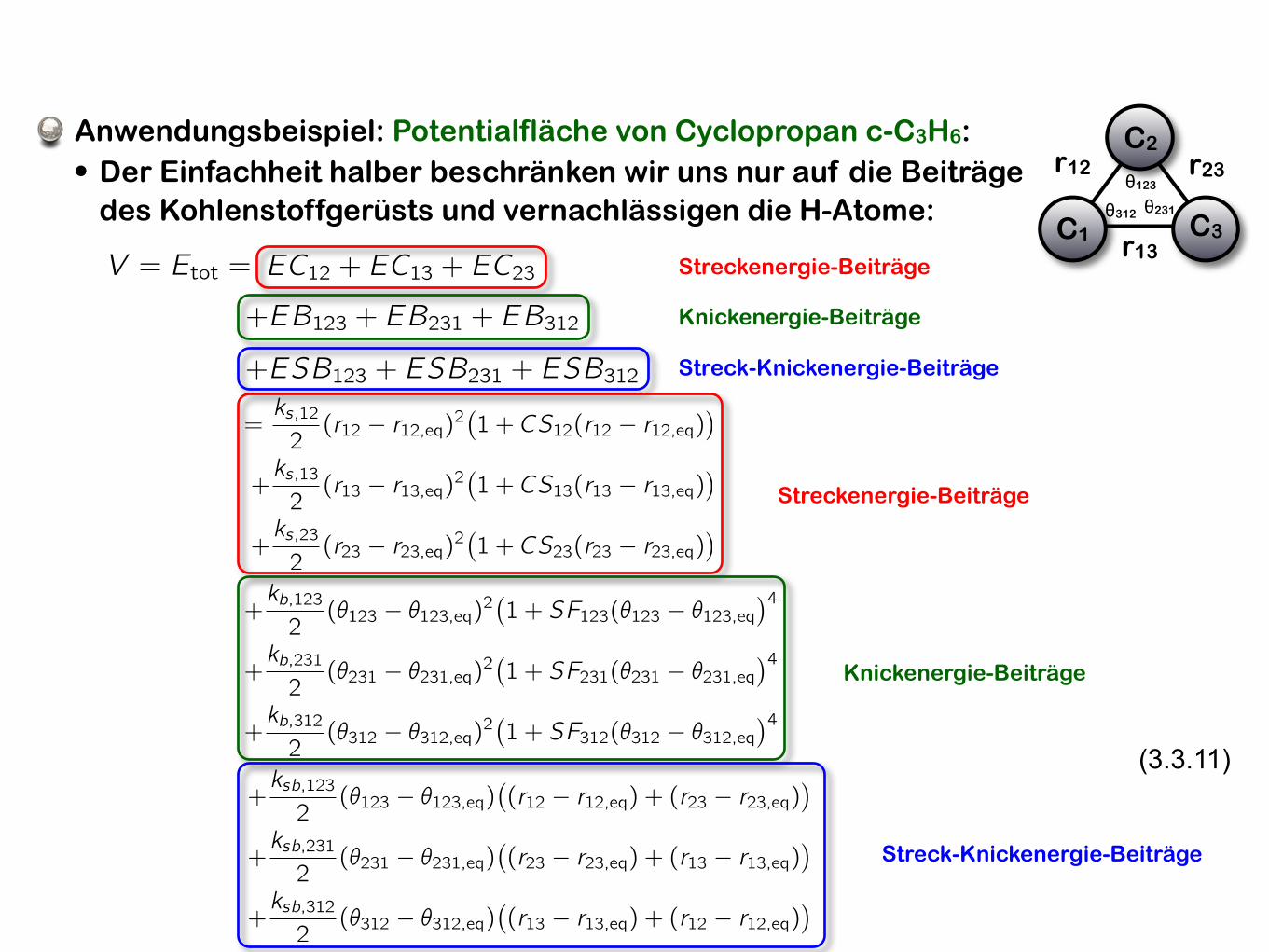
Anwendungsbeispiel: Potentialfläche von Cyclopropan c-C3H6:
EC12 + EC13 + EC23 Streckenergie-Beiträge
Streck-Knickenergie-Beiträge
θ123r23
C1 C3
C2r12
θ312 θ231
r13
Knickenergie-Beiträge
• Der Einfachheit halber beschränken wir uns nur auf die Beiträge des Kohlenstoffgerüsts und vernachlässigen die H-Atome:
V = Etot =
+ESB123 + ESB231 + ESB312
+EB123 + EB231 + EB312
=ks,122(r12 � r12,eq)2
�1 + CS12(r12 � r12,eq)
⇥
+ks,132(r13 � r13,eq)2
�1 + CS13(r13 � r13,eq)
⇥
+ks,232(r23 � r23,eq)2
�1 + CS23(r23 � r23,eq)
⇥
+kb,1232(�123 � �123,eq)2
�1 + SF123(�123 � �123,eq
⇥4
+kb,2312(�231 � �231,eq)2
�1 + SF231(�231 � �231,eq
⇥4
+kb,3122(�312 � �312,eq)2
�1 + SF312(�312 � �312,eq
⇥4
Streckenergie-Beiträge
Knickenergie-Beiträge
Streck-Knickenergie-Beiträge
(3.3.11)+ksb,1232(✓123 � ✓123,eq)
�(r12 � r12,eq) + (r23 � r23,eq)
�
+ksb,2312(✓231 � ✓231,eq)
�(r23 � r23,eq) + (r13 � r13,eq)
�
+ksb,3122(✓312 � ✓312,eq)
�(r13 � r13,eq) + (r12 � r12,eq)
�
3.3 Die Born-Oppenheimer-Potentialfläche
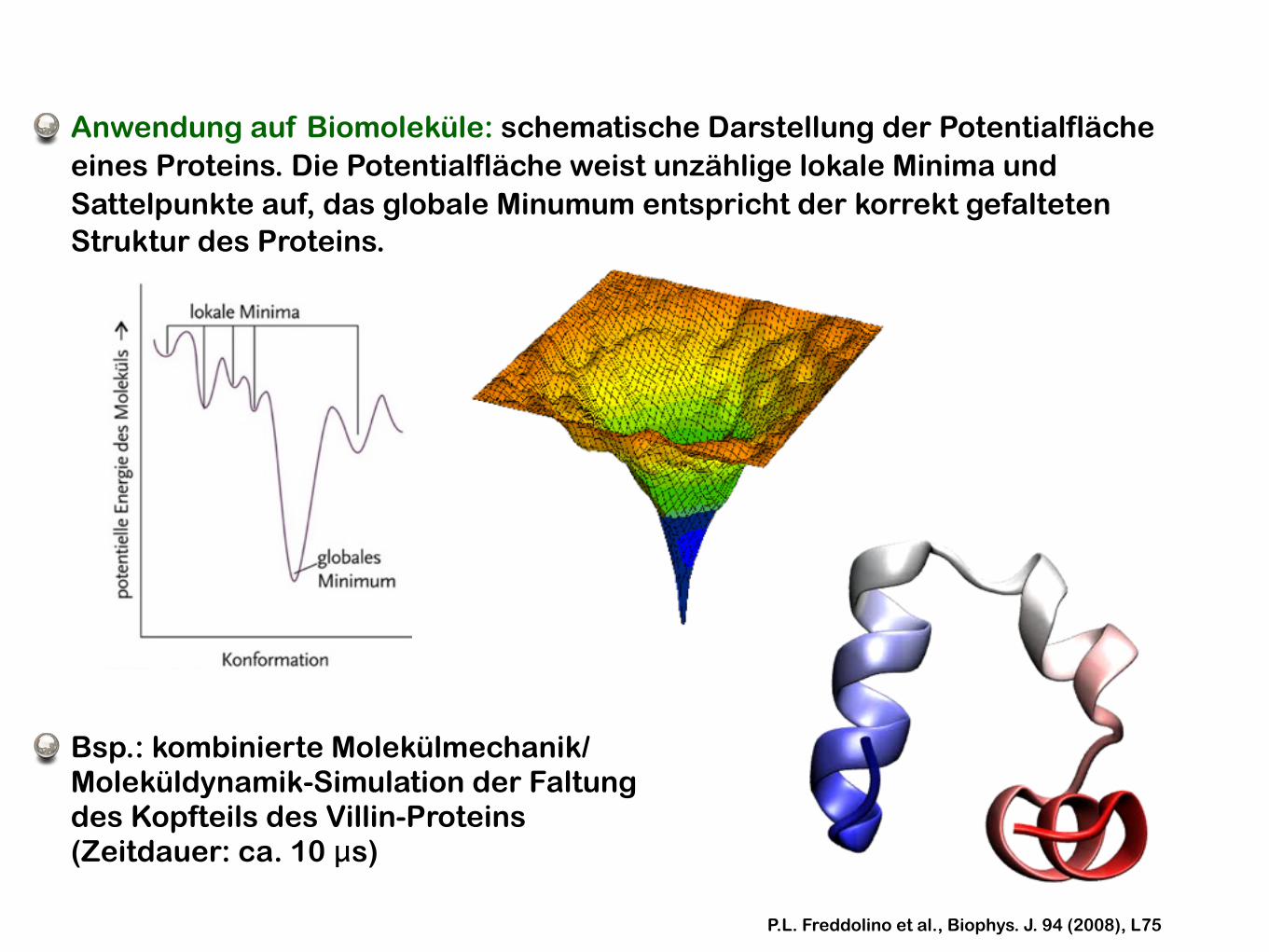
Anwendung auf Biomoleküle: schematische Darstellung der Potentialfläche eines Proteins. Die Potentialfläche weist unzählige lokale Minima und Sattelpunkte auf, das globale Minumum entspricht der korrekt gefalteten Struktur des Proteins.
Bsp.: kombinierte Molekülmechanik/Moleküldynamik-Simulation der Faltung des Kopfteils des Villin-Proteins (Zeitdauer: ca. 10 μs)
P.L. Freddolino et al., Biophys. J. 94 (2008), L75
3.3 Die Born-Oppenheimer-Potentialfläche
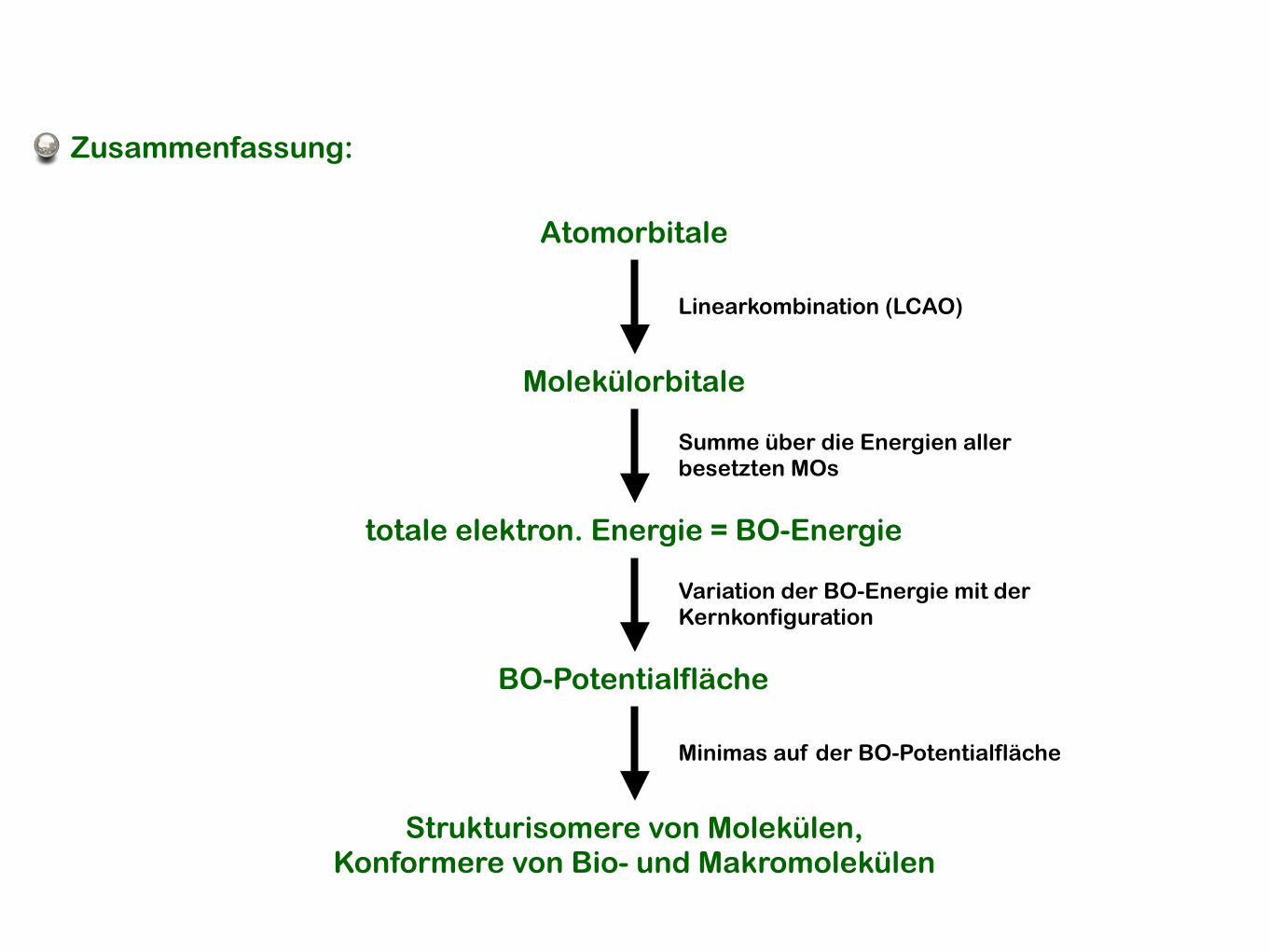
Atomorbitale
Molekülorbitale
totale elektron. Energie = BO-Energie
BO-Potentialfläche
Strukturisomere von Molekülen,Konformere von Bio- und Makromolekülen
Linearkombination (LCAO)
Summe über die Energien aller besetzten MOs
Variation der BO-Energie mit der Kernkonfiguration
Minimas auf der BO-Potentialfläche
Zusammenfassung:
3.3 Die Born-Oppenheimer-Potentialfläche