Embed Size (px)
Citation preview

Journal f. txakt. Chemie. Band 329. Heft 3. 1987. S. 511-519 J. A. Rarth, Leipzig
Experimentelle und theoretische Untersuchungen an Kohlenwasserstoffen der Formel C,H, : Pyrolyse von Cyclopenta-I ,3-dien
E. GEU*
Zentralinstitut fur physikalische Chemie der Akademie der Wissenschaften der DDR, Berlin
B. ONDRUSCHKA, G. ZIMMERMANN Zentralinstitut fur organische Chemie der Akademie dcr Wissenschaften der DDR, Bereich Organische Grundstoffe Leipzig
Herrn Prof. Dr. Dr. H.G. 0. Becker zum 65. Geburtstag gewidnzet.
Experimental and Theoretical Investigations on Hydrocarbons of the Formula CjHa: Pyrolysis of 1,3-Cyclopentadiene
Abst rac t . The details of high temperature chemistry of the conversion of 1,3-cyclopentadiene (CPD) are unknown. Therefore the pyrolysis of CPD was studied under various reaction conditions in labscale dimension. The attempt to inteqret the composition of the reaction products only on the basis of convenient axiomes does not Seem satisfactory. For that reason heats of formation and relative stabilities for 2.5 closed and open shell hydrocarbons of the formula C,H, were calculated using the MINDO/3 procedure. These enthalpy values were used to estimate heats of reactions for selected start steps of the pyrolysis of CPD. The molecular-assisted hydrogen transfer under formation of cyclopentadienyl and cyclopentenyl radicals ought to bc onc of the most important reaction steps.
Ebfuhrung Das Reaktionsverhalten von Kohlenwasserstofferi der Eormel C‘,H, ist u. a. Gegen-
stand einer Reihe experimenteller Untersuchungen [I]. Im folgenden wird das Verhalten von Cyclopenta-lf5-dien (CPD) bei der Pyrolyse naher betrachtet. Hei Temperaturen unterhalb 670 K sollten unter homogenen Versuchsbedingungen neben Gerustumlagerun- gen bevorzugt sigmatrope Wasserstoff- und Alkylwanderungen ablaufen [ 21. ifber den Mechanismus der Wandlung voii CPD bei hoheren Temperat wen, die zur Bildnng von Aromaten fuhrt, ist dagegen bisher wenig bekannt [3]. Bei uniinolekularen VLW- Bedingungen, uriterhalb 1000 K und weniger als einer Sekunde Verweilzeit reagiert CPD praktisch nicht [4]. Bei StoBzahlerhohung wird inassenspektrometrisch m/e = (5 beobachtet ; hieraus folgt, dai3 das Cyclopentadienylradikal vorliegen sollte (vgl. [?)I). Unter industrienahen Pyrolysebedingungen wird CPD zu etwa 30;d umgesetzt [ t i ] . Die bisher in der Literatur diskutierten Vorstellungen zum Mechanismus der Pyrolyse von CPD im Temperaturbereich zwischen 800 und 1100 K [7 - 91 ermoglichen keine hefriedigende Deutung des Reaktionsablaufes und des Anteils der einzelnen Reaktioiix- produkte. Neben unimolekularem C - H-Bindungsbruch bzw. bimolekularer Wasser- stoffubertragung im Startschritt, die unter Bildung von Cyclopentadienyl- und C’y~lo-

512 J. prakt. Chem. 399 (1987) 3
@ cvclisch 1191
@ acvclisch 16)
Abb. 1 Struktur und Anztilil der I(ol~len\r.asserstoffe der Formel C,H, mit abgeschlossenen Elektro- nenschalen. Aiimerkung: In den vechteekigen KSstchen ist jeweils die Anzahl der Spezies drr ent- sprechenden Strukt III' angegeben.
700
650
600
550
500
450
400
350
300
250
200
150
650
600
550
500
=C-C-C=C f 232 I 4.50
400

E. GREY 11. a., Pyrolyse von Cyclopenta-l,?,-dien 5 13
pentenylradikalen ablaufeti, sind noch Isomerisierungsreaktionen zu berucksichtigen. Als Intermediate treten vermutlich auch Carbene und Biradikale auf, die gewohnlich nur mit grol3em Aufwaiid experimentell untersucht werdeti konnen. Weniger aufwendig sirid im Vergleich dazu quantenchemische Berechnungen, die z. B. in jungster Zeit an wasserstoffarmen Kohlenwasserstoffen der Formeln C,H, [ 10, 111, C,H, [ 121, C4H, [MI, C,H, [14, 131 und C,H, [lG] durchgefiihrt wurden. Auf diese Weise koririte eine .Hriicke zum Verstandnis ihrer Chemie geschlagen werden. Fur die hier iriteressierenderi Kohlenwaseerstoffe der Formel C5H, liegen zwar teilweise quantenchemische Angaheii vor [ l T - 2.21, doch steht, eine syst.ematisehe Untersuchung aller in Abh. 1 dargestellten closed shell-Systeme und der entsprechenden open shell-Systeme noch aus. Wie ari unge- sattigten Kohlenwasserstoffradikalen gezeigt werderi konnte [ 2 5 ] , ist die MIND0/3- Methode [ 261 als ein relativ wenig aufwendiges halbempirisches Berechnutigsverfahren fiir eirie derartige Untersuchung geeignet, wobei im Falle von open-shell-Systemen die Half Electron (HE)-Naherung [27] Anwendung fand. Die fiir 25 ausgewahlte Spezies der Bruttoformel C,H, mit dieser Methode berechneten Bildungsenthalpien fur den Staridardzustaiid (Abb. 2) werden z w Diskussion der Stabilitat dieser Spezies und fiir die Bestimniung der Reaktionsenthalpien AH:Ys ausgewahlter Schritte der Pyro- lyse von CPD herangezogen. Fur die betrachteten closed shell-Systeme wurden die d HfY8-Werte alternat iv nach einem von LEROY [ 281 vorgeschlagenen Inkrementver- fahren abgeschatzt, wobei im Falle cyclischer Verbindungen die entsprechenden Ring- spannungsenergien (SE) [29, 301 beriicksichtigt. wurderi. Soferri i n der Literatur Daten aus ab initio- Herechnurigen vorlagen, wurden diese ebenfalls zum Vergleich herange- zogen. Unter Benutzung der Rechenergebnisse werden Hetrachtungeri uber die Wahr- scheinlichkeit des Ablaufs ausgewahlter Elemetitarschritte der Pyrolyse von CPD ange- stellt.
Ergebnisse und Diskussion
B i Id un g s e n t ha 1 p ie r i A H f" u Iid S t a b i 1 i t a t e ti Wie aus Abb. 1 ersichtlich, existieren insgesamt 25 closed shell-Spezies der Formel
C5H,, davon 6 mit acyclischer und 19 mit cyclischer Struktur. Gem613 [19] wird fiir die acyclischen Spezies eine symbolische vierziffrige Kennzeichnung benutzt, die Reihen- folge iind Art der C-C-Hindungen charakterisiert, wobei 1, 2 bzw. 3 fur eine Einfach-, Doppel- bw. Dreifachbiridung steht. So entspricht z. R. die Kennzeichnung 2131 der Verbind ung Pent-1-en-3-in. Neben CPD sollten diese acyclischen Molekiile am stabilsteri sein. Die mit der MINDO/S-Methode erhalteneri ~ l H ; ~ ~ - W e r t e stiitzen diesc Annahme, erlauben allerdings wegen der dem Kechenverfahren atihaftenden Mange1 riicht in jedem eirizelnen Falle eirie zuverlgssige Aussage. Whhrend z. B. die Ergebnisse der Herechnung nach einem ab initio-SCF-Verfahren [ 17, 181 und nach dem Inkrementverfahren [28] eitideutig dafiir sprechen, da13 der ~lH;'~-?lrert fiir CPD am kleinsten und folglich diese Spezies an1 stabilsten seiri sollten, ware das nach der MIND0/3-Methode fur die acycli- sche Verbindung 2131 der Pall, wobei die berechriete Enthalpiedifferenz allerdings inner- halb der Fehlergrenie der Methode liegt. I r i Tab. 1 sind fiir alle uriverzweigten acyclischen closed shell-Spezies, die auf Verbindung 2131 bezogenen nach der MINDO/3-Methode [%I und dem Inkrementverfahren [28] von uns berechrieten sowie die fruher mit einem ab initio-?Terfahren [19] erhaltenen dAH,-Werte den entsprechenden experimentellerl AAG-Werten gegeniibergestellt. Wahrend das Inkrementverfahren nur geringfugig V O ~ den ab initio-Ergebnissen abweichende Werte liefert und in beiden Fallen die Stabilitat in der Reihenfolge 2131 > 1213 > 2212 > 2113 > 2221 abnimmt, treten bei den MINDO/3-Werten erhebliche Abweichungen auf, die in erster Linie daraus resultieren, da13 Systeme mit kumulierten Doppelbindungen zii instabil vorausgesagt werden. Die
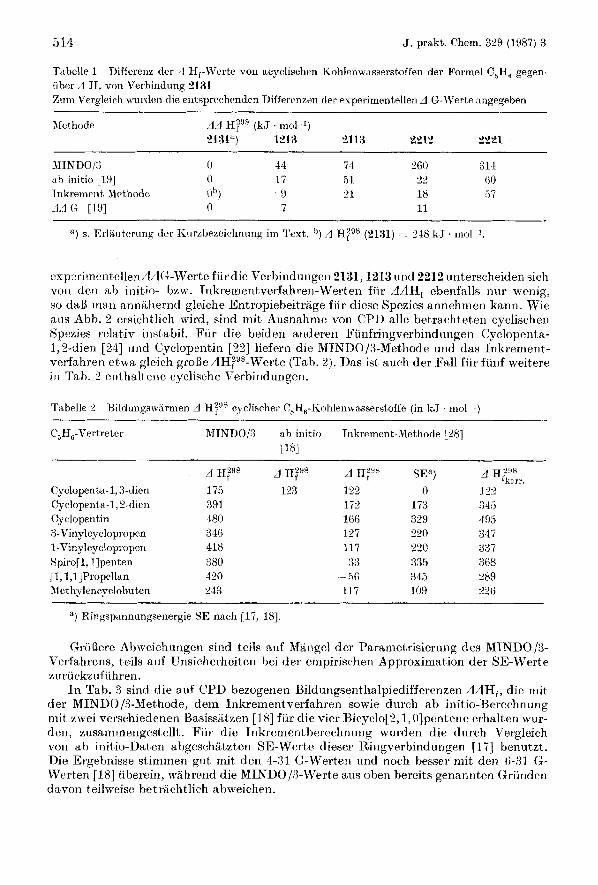
514 J. prakt. Chem. 364 (1987) 3
Tabelle 1 i iber A H, von Verbindung 2131 Zum Vergleich nnrden die ent3prechenden Difterrnzen der evperimentellen A G-Werte angegeben
Diffrrcnz der 3 Hf-Werte von acyclisclien ~ohlena-,isserstoffen der Formel C,Ho gegen-
Met hodc
'1) s. Erlarrterung dcr K~irzbrzeichnung im 'l'ext,. 'I) A H;98 (2131) = 248 k J . rnolkl.
exp-.rirnentellenAAG-Werte fiirdie Verbindungen 2131,12 13 und 2212 unterscheiden sich von den ah iriitio- bzw. Inkretnentverfahren-Werten fiw A H , ebeiifalls nur wenig, so daB m a n annahernd gleiche Entropiebeitrage f i i r diese Spezies annehmen kann. Wie ails Abh. 2 ersichtlich wird, sirid rnit Ausnahme von CPI) alle betrachteten cyclischen Spezies relativ instabil. Fur die beiden anderen Fiinfringverbindungeii Cyclopenta- I, 8-dien [24] iind Cycloperitin [ 8 2 ] liefern die MINl)O/3-Methode und das Iiikrement- vrrfahren etwa gleich groBedH,2Y8-Werte (Tab. 2). Das ist auch der Fall fiir funf weitere i n Tah. 2 enthaltene oyclische Verbindungen.
Tabelle 2
C,H,-Vertreter MINUO/Y ab iriitio InBrement-Methode [281
RildungswLrnicn A H:98 cyclischer C,H,-I.;ohlen\Vasserstoffe (in kJ . inol-I)
V81
Cyclopenta-l,3-dieii Cyclopenta-l,2--dien Cy clopentin 5-Vinylcyclopropen 1 -Vinyley clopropen Spirofl, llpenten [l , 1,l~Propellsn Jlethyleiicyclobiiten
A HFSs 175 391 480 3 4 ; 418 380 420 "43
A H:y8 A H;98 123 122
172 166 127 117 33
--.Xi 117
SE") 0
173 329 220 220 336 345 109
") Kingspannungsenergie SE nach 117, 181.
GroBere Abweichurigen sind teils aiif Mangel der Parametrisierung des MlNDO ,ti- Verfahrens, teils anf Unsicherheiten bei der empirischen Approximation der SE-Werte zuriickzrifrihren.
In Tab. 3 Rind die aiif CPD bezogenen Bildriiigsentlialpiediffer~n~eii AAH,, die rnit der MIND0 /3-Methode, dem Irikreinentverfahren sowie durch ab initio-Herechnung rnit zwei versehiedenen Basissatzen [18] fiir die vier l3icyclo[ 2,1, Olpentene erhalten wur- den, zusammengestellt. Fiir die Irikrenieritberechriiing wurden die diirch Vergleich von ah initio-Daten abgeschatzten SE-Werte dieser llingverbindungen [ 171 benutzt. Die Ergebnisse stiniinen gut mit den 4-31 G-Werten und noch besser mit den (i-31 G- Werten [18] iiberein, wahrerid die MINDO/.?-Werte ails ohen bereits genanriten Griinden davon teilmeise bet rachtlich abweichen.

E. GREY u.a., Pyrolyse von ~yclopent~-l ,S-dien 515
Tttbelle 3 (CPD) (in kJ . inol- ' )
Differenz der A Hf-Werte vm Bicyclopentenen (Be[?. l.O]Px(y)en) gegenuber d 11,
Cyclopenta-l,3-BC[2.I.O]P2- RC[2.l.O]Pl- IEC[2.l.O]P1- BC[2.1.O]Pl- dien (3)en (?)en (%>)en (4)cn
Nebeii den bisher betrachteten urid weitereri 8 f im die Diskussion dcs Mechanisrnus der Wandlung von CPD weniger bedcuteriden closed shell-Spezies (s. Abb. I ) sirid zahl- reiche open-shell-Spezies, d. h. Carbene iitid Biradikale vorstellbar, deren elektronischer Grundzustand ein Triplett ist. Hier sollen nur 8 dieser Spezies, d.h. zwei Kiradikale iind sechs Carbene betrachtet werden, die als Jnterrnediate bei der Pyrolyse voii CPD eine Rolle spielen kortnten. Die sowohl firr den Triplettzustarid T, als auch f u r deli Sitignlett- zustand So in MINDO/.?-Nalieriing berechncten AHfg8-Werte sind in Abh. 2 zusammen- gestellt. Analog wie bei Methylen ixnd den Alkylcarberien [29] erhalt inan x. K. fi ir Divinylcsrberi eine Energieaufspaltiing zwischeri den Xustanden T, iind So voii 56 k,J. mol-1. Ahnliche Unterschiede in den AHfqs-Werten der heiden Zirstande erhalt inan auch fur die meisten anderen hier betrachteten C'arbene. Eine Ausriahnie bildet das Penta- I , -I-dienyliden-( l ) , das als Propenyldertvat des Vinylideris ebenso wie diescs im Zustand S,, vorlirgen sollte [30], der genial3 der Kerechnurig iiin 231 ktJ . niol-l stahiler als TI ist.
Ini Falle des Hiradikals Penta-l ,J-dieri-l, ,5-diyl ist T, gegennher S,, urn etwa 45 k J . mol-1 begiinstigt. Wahrerid diese Energiedifferenz etwa gleich grol3 wie bei den Carbenen ist, betragt sie in1 Falle des Penta-1 ,d-dien-1 ,,i-diyIs nahezu 500 k J . mol-l. Die Ursache hierfur hesteht wahrscheinlich darin, da8 die MINUOJ~-SCF-Prozedur fiir den Xustand So eiries Biradikals in riianchen Fallen infolge Vertauschung von besetz- ten urid iinbesetzteri M O nicht die wahre Gesamtenergie liefert. Eirre nachfolgende 2 x 2 CJ- Behandlung hewirkt zwar eine Energiesenkung urn 125 ktJ . rnol-I, iinterhe- wcrtet abet- die Stahilitkt des %ustatides S,, noeh betrachtlich.
Re a k t ion se t i t h a l p i en A H ," vori CPD
f u I E le me n t a r s c h r i t t e d e r 1' y r o 1 y se
Reaktionsenthalpien A H f g 8 kbnnen nach den1 HeBschen Satz aus den AH;"-Wertcii berechriet werden. Fur sigmatrope Umlagerungen [ 11 sind dicse AH,-Werte glcich null; fiir ,,walk rearrangements" [2] wird gefnnden, dab unter der Voraussctzung gleichhlei- bender entropischer Einfliisse a u f das Reaktionsgeschehen die Bildung voii 3-Tinyl- cyclopropen als Intermediat (AH:9* etwa 170 k J . mol-l) gegeniiber der von Bicyclo- [a, 1,0]-pent-2(3)-en (AH;qX etwa 255 k,J . niol-l) bevorzugt sein sollte. Einige Isomeri- sieriingsreaktionen wie z. B. 22 12 + Methylericyclobuteii, Vinylidencyclopropan -+ y2 13 irnd 2 113 bzw. Diinethylencyclopropan sowie Cyclopentin + Vinylidencyclopro- pan (x. 11) solltcn entweder exotherm oder zumindest ohne Energiezufiihr ahlaufen.
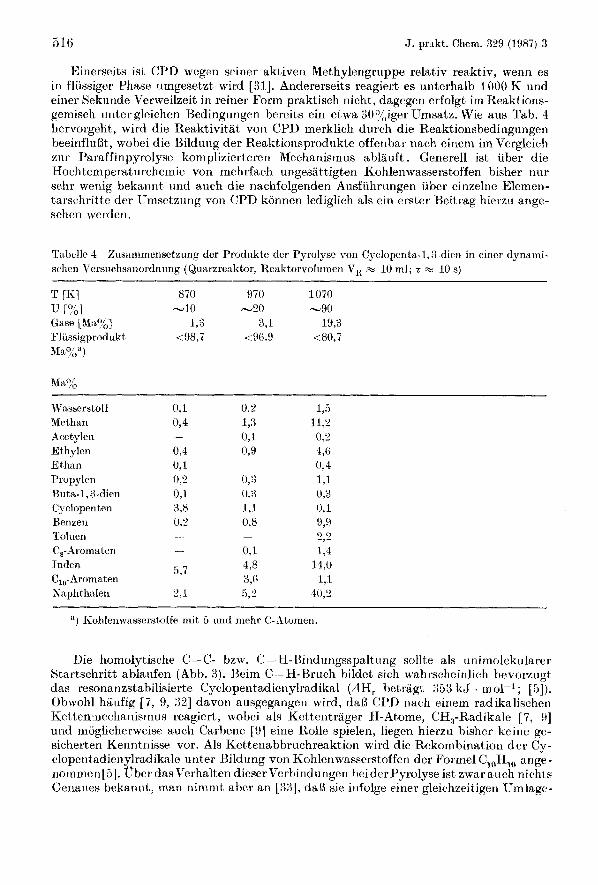
516 J. prakt. Chem. 329 (1987) 3
Einerseits is1 CPD wegen seiner aktiven Methylerigruppe relativ reaktiv, wenn es in fliissiger Phase umgesetzt wird 1311. Andererseits reagiert es unterhalb 1000 K und einer Sekuride Verweilzeit in reirier Form praktisch nicht, dagegen erfolgt im Reaktions- geinisch linter gleichen Hedingringen bereits ein etwa 30%,iger Umsatz. Wie aus Tab. 4 hervorgeht, wird die Reaktivitat von CPD merklich durch die Reaktiorisbedingungeri beeinflullt, wohei die Bildung der Reaktionsprodrikte offenbar riach eineni im Vergleich zrir Paraffinpyrolyse komplizierteren Mechanismus ablauft . Generell ist iiber die Hochtemperaturchernie von rriehrfach ungesattigten Kohlenwasserstofferi hisher nur sehr wenig hekannt m d auch die nachfolgenden Ausfuhrringen uber einzelrie Elemeri- tarschritte der ITmsetzrmg von CPD konnen lediglich als eiri erster Keitrag hierzu ange- sehen werderi.
'I'abellc 4 schen Versuchsanordnung (Quarzreaktor, Reaktorvolumeri V,, w 10 ml; t R+ 10 s)
Zrisamniensetzung der Produkte der Pyrolyse von Cyclopenta-1,:I-dien in einer dynami-
AT'LO,,
Wssscrstoff 0,l 0,2 1,5 Methan 074 I,:$ l l , ? Acetylen - 0,1 0;i Ethylen (44 0,9 4,ci Ethan 0,l - 0.4 1'1 opylcn 0,t' 0,:; 1,1 Buta-l,:Ldien 0,1. u,:\ 0,3 Cy clopenten 3,s 1,1 0, l Benzen 0.2 0,s 9,9 Toluen __ -
0,1 L4 C,-Aromaten - 4 3 14.0 5,7 Indrn
C,,-Aromaten 3,G 1,1 Napht halen t',l 5,2 40,2
L> -) -,-
") Kohlenwasserstoffe mit 6 n r i d niehr C'-Atonien.
Die homolytische C- C- bzw. C-H-l~indurigsspaltririg sollte als unimolekrrlarer Startschritt ablaufen (Abh. 3). h i m C- H-Hruch bildet sich wahrscheinlich bevorzugt das resonarizstabilisierte Cyclopentadicnylradikal (AH, betragt 353 kJ . mol-1; [>I). Obwohl haufig [7, 9, 321 davon ausgegarigen wird, daB CPD nach einem radikalischen Kettenmechanismus reagiert, wobei als Kettentrager H-Atome, CH,-Radikale [ 7, ! I ] und mciglicherweise auch Carbene [O] eirie ltolle spielen, liegen hierzu bisher keine ge- sicherten Kemitriisse vor. Als Kettenabbruchreaktion wird die Rekombination der Cy- cloperitadienylradikalc linter Rildung vori Kohleriwasserstoffen der Formel C,H,, ange- nonimeri[5]. u b e r das Verhalten dieser Verhiridungeri 1)eiderPyrolyse ist zwar auch nichts Genaues bekannt, man riirnmt aher an [%I, daB sie infolge einer gleichzeitigeri Uinlage-

R. GKEY u.R., Pyrolyse v o ~ i Cyclopent:L-l,R-dien 51 7
riiiig und Dehydrierung entweder in mehrkcrnige Aromaten, insbesondere Kaphthalen CluH8 oder in ruBartige Substanzen iimgewandelt werden. Gem6B Formelschema 1 (vgl. [MI) eritsteht Naphthalen iiber das lntermediat Fulvalen in Analogie zri der Um-
lagerling Fulven + Berizvalen -+ Benzen [35]. Man nimint ferner an, daS die Cyclo- pentadienyldimeren somie die Mjschdiineren des Cyclopentadienyl- und des Cyclo- pentenylradikals signiatropen H-Umlagerungen und Isomerisierungsreaktionen, die zii Doppelhindungsverschiebring~n fiihren, unterliegen [35] (Formelschema 2) :
Die Bulvalerie lagern sich in Dihydronaphthalerie iind Tetralin um ; eine nachfol- gende Dehydrierung fiihrt entweder zii Naphthalen oder im Palle des Tetralins zrir Hil- diing von l nden, Methan und RTX-Aroinaten [Xi]. Eiii solcher Mechanismus der Pyro- lys3 von CPD unter Aromatenbildung erscheint plausibler als die Vorstellung, wonach infolge [4 + 21-Cycloaddition und anschlieBender Isomerisierung ein Dihydroinden mit einer Methylgruppe am Sechsring entsteht (Formelschema 3), das unter Kildung von Methylinden, Inden rind Methari weiterreagiert [ 81.
Hinsichtlich der miiglichen Hildung vori Henien ails CPD ist sicher, dafi es iiicht als Primarprod ukt eirier uniinolekularen Reaktion entsteht, sondern bei Vorhandensein von Methylradikalen als Kettentrager wahrscheinlich iiber eirie Arizahl von Zwischen- st rifen geinafi Fornielschenia 4 (vgl. [9]).
Um die Vorstellungen zum Mechanismus der Yyrolyse von CPD experimentell zii iiberpriifen, miifiten geeignete Experimente durchgefiihrt werden, die es gestatten, den Reaktionsverlauf zu verfolgen, gegebenenfalls gekoppelt mit einer R;latrixisolations- technik. Es bleibt allerdings offen, ob die in Abb. 3 zusammenfassend dargestellteri Startschritte fiir die Charakterisierung des Ablaufes der Reaktion ausreichend sind. Mit an Sicherheit grenzender Wahrscheinlichkeit kann bisher lediglich der molekril- assistierte Wasserstoff-Transfer, der zur Hildung von Cyclopentadienyl- und Cyclopen- teiiylradikalen fiihrt (AH, betragt 119 kJ . mol-l), als mafigeblieher Startschrit t fi ir die Hochtemperaturwandlung von CPD angesehen werden.

51.8 J. prakt. Chem. 329 (1987) 3
p- C-H A Hr Ck J - mop4 Q - - - Q + t f 353
c-c -0 . . 696 __ 339
-n 234 469 Retro - insertion .. __
288 && R e insertion
Abh. 3 Blternative Startschritte der Py-olyse von CPD. Anmerkung: Die A H,-Werte werden aus dcri niit der 3IIXD0/3-81ethode erhaltenen A Hf298-Werten nach dem HeSschen Satz berechnet.
Bcsrhreibung der Versuche Cyclopenta-l,3-dien (CPD) wurde :bus einer Dicyclopentadien-Fraktion (VEB Leund-Werke
,,\Valter Ulbricht") gewonnm nnd durch Kolben-zu-Kolben-Destillation m e h r f d i gereinigt (GC- Reinheit 3 99%).
Die Analysc der Reaktionsproditkte und die Auswcrtung drr Pyrolyseversuche wurde bereits frChcr beschrieben [Xi].
Literatiirverzeichnis 111 GAJEWSKI, J. J . : Hydrocarbon Thermal Isomerization, New York: Acad. Press, 1981,s . $1. p2] KLARNEK, F. G . : Top. Stereochem. 15 (198-2) 1. [:$I Mc Coxaca~ , J. S., ~VOORE, K. N.: Erdol Kohle 2G (1973) 27.3. [41 ZTEGLER, L'. : iinvcroffcntlichte VLPP-Ergebnisse, Leipzig 1983. [c51 HEDAYA, E.: Aw. Chem. Res. 2 (1969) 367. [(il KOPISKIF, P. D. ; ONDRIJWHKA, B. : ZIBIIIIEKK~NN, (4. : Manuskript in Vorbereitung. ['i] SZWAKC, JI.: Chem. Rev. 47 (1950) 135. [8] S~WCLMAWS, It.; C a a n m ~ s , C. A.: Chrom:itographia 3 (1972) 2%. 191 MEL~XSII)E, M. M.; 3 v ~ ~ ~ r ~ ~ < ~ ~ 7 , I<. 31.: Axcrb. Khim. Zh. 1981 (6) G O .
I101 HEHKE. IY. J. ; POPLE. J. A.; LATH , E.: J . Am. ("hem.
1111 FITZGER~LD, (+.; SCHAEFER, 111, H. F.: Isr. J. ('hem. 23 (1983) 93; SIXE, R. ; SCHUWER 111,
I I ? ] Hov.ror-. S.: PACAUWY, $J.; YOSHIJILNE, M.: J. Am. Chcm. Suc. 106 (1981) 5361. 1131 A F I ) R ~ I ) ~ , J. G . : C H A N I ) R ~ S F . K I I ~ R , J . ; SCHLICYLR, P. v. I t . : J. Comput. Cliern. 'J (1981) "07. 1141 KOLLMIR, H.: CARRION, F.: DEUAR, M. J . S.; BEWHIM, R. C.: ,J. Am. C'hern. Sorb. 103 (1981)
. \V. 8.; Rruonr, I,.; WaSsERnl
Soc. 98 (1976) 4378.
H. F.: .T. Am. ('hem. SOP. 102 (1980) 3239.
j.2 "1.

E. GREY 11. a., Pyrolyse yon Cyclopentn- 1 , M i e n 519
1151 FITZGERbLI) . G.; SAXE, P.; SCHAEFER 111, H. F.: J. Am. Chern. Soc. 103 (1983) (80. [lti] ANGUS, R. 0.; JOHNSON, R. P.: J . Org. Chem. 49 (1981) 2880. [17] WIBERC, K.; BONNEVILLE, G. ; DENIPSEY, R. : Isr. J. Chem. 23 (1983) 85. [18] WIBERG, K.: J. Comput. Chem. 3 (1984) 197. [19] VAN DEN BERG, A. B. A.; DEN BOER, D. H. W.: Rec. Trav. Chim. Pays-Bas 9s (1979) 432. [20] WIBERG, K.: J. Am. Chem. Soc. 106 (1983) 1227. [21] SIDBERG, S. P.; YOFFE, A. I.; NEFEDOV, 0.11.: I L V . Akad. N<tuk SSYR, Sor. Khim. 1981,
[22] OLIVELLA, S.; PERICAS, M. A.; RIERA, 8 . ; SOLE, A.: J. Chem. Res. (S) 1985, 328; J. Chem.
[23] SCHOELLER, W. W.; BRINKER, U. H.: J. Am. Chem. Soc. 100 (1978) 6012; BRINKER, U. H.;
[24] ANGUS, R. 0.; SCHMIDT, M. W.; JOHNSON, R. P.: J. Am. Chern. SOC. 107 (1988) 532. [25] K ~ H N E L , W.; GEY, E.; ONDRUSGHKA, B.: Z. phys. Chem. (Leipzig), im Druck. [26] DEWAR, 31. J. S.; BINGHAM, D. A.; LO, D. H.: J. Am. Chem. SOC. 97 (1975) 1283. [27] DEWAR, M. J. S.; HASHMALL, J. A.; VENIER, C. G.: J. Am. Chem. Soc. 90 (1968) 1953. [28] LEROY, G.: Adv. Quantum Chem. 17 (1985) 1. ["9] GEY, E.; KUHNEL, W.; ONDRUSCHKA, B.: Z. Chem. 21 (1981) 2-25. (301 GEY, E. : unveroffentlichte MIND0/3-Berechnungen an Vmyliden, vgl. DYKSTRA, C. E. ; SCHAE-
FER 111, H. F.: J. Am. Chem. SOC. 100 (1978) 1378. [31] BEYER, H. : Lehrbuch der organischen Chemie, 11./13. Aufl. Leipzig: S. Hirzel, 1966, S. 364,365. [32] CULLIS, C. F.; NORRIS, A. C.: Carbon 10 (1972) 525. [33] SCOTT, L. T.; JONES, J. JR.: Chem. Rev. 72 (1972) 181. [34] SCHULTZ, J. C.; BEAUCHAMP, J. L.: J. Phys. Chem. 87 (1983) 3587. [35] REMMLER, M. ; ZIMMERMANN, G. ; ONDRUSCHKA, B. : unveroffentlichte Xrbeitsergebnisse, Leip-
L36] GUNSCHEL, H.; ZIMMERMANN, G.; LORENZ, R.; ONDRUSCHKA, B.; NOWAK, S.: J. priikt. Chem.
398.
SOC., Perkin Trans. 2 1986, 613.
FLEISCHHAUER, I. : Chem. Bcr. 119 (1986) 1244.
zig 1986-1986.
323 (1981) 0 7 .
Bei der Redaktion eingegangen am 3. November bzw. 22. Dezember 1986.
Anschr. d. Verf.: Ilr. E. GEY, Zentralinstitut fur physikalische Chemie der Akitdemie der Wissen- schaften der DDR, Rudower Chatissee 5, Berlin, DDR-1199.
Dr. B. ONDRUSCHKA, Prof. Dr. G . ZIMMERXANN, Zentralinstitut fiir Organische Chemie der Akademie der M'issenschaften der DDR, Brreich Organische Grrind- stoffe, PermoserstraBe 15, Leipzig, DDR-7050
![Membrány a membránový transport - ulbld.lf1.cuni.cz · STEROL LIPIDS STEROL LIPIDS = lipid molecules with backbone derived from cyclopenta[a]phenanthrene (?) Division according](https://img.pdfslide.net/doc/110x75/5e14db8b3fcccd648c5ac62a/membrny-a-membrnov-transport-ulbldlf1cunicz-sterol-lipids-sterol-lipids.jpg)



![Synthetic Routes towards 2-thia-7,8-diaza-cyclopenta[l ...277519/FULLTEXT01.pdf · 1-thia-7,8-diaza-cyclopenta[l]phenanthrene for Molecular Electronics Applications ... Kemin som](https://img.pdfslide.net/doc/110x75/5d1b94de88c993dc468d14cb/synthetic-routes-towards-2-thia-78-diaza-cyclopental-277519fulltext01pdf.jpg)


![Photoreactive low-bandgap 4H-cyclopenta[2,1-b:3,4 …phome.postech.ac.kr/user/pnel/publication/87. U. R. Lee...Photoreactive low-bandgap 4H-cyclopenta[2,1-b:3,4-b0]dithiophene and](https://img.pdfslide.net/doc/110x75/5f049ea57e708231d40edef6/photoreactive-low-bandgap-4h-cyclopenta21-b34-phome-u-r-lee-photoreactive.jpg)


![Gold(I)-Catalyzed Dearomative Rautenstrauch Rearrangement ...Gold(I)-Catalyzed Dearomative Rautenstrauch Rearrangement: Enantioselective Access to Cyclopenta[b]indoles Weiwei Zi,†](https://img.pdfslide.net/doc/110x75/5e448a31894df825a95b8ad3/goldi-catalyzed-dearomative-rautenstrauch-rearrangement-goldi-catalyzed.jpg)




![NOMENCLATURE OF STEROIDSpublications.iupac.org/pac/1989/pdf/6110x1783.pdf · Steroids are compounds possessing the skeleton of cyclopenta[a]phenanthrene or a skeleton derived therefrom](https://img.pdfslide.net/doc/110x75/5eca6f92f0330a21ce79ff19/nomenclature-of-steroids-are-compounds-possessing-the-skeleton-of-cyclopentaaphenanthrene.jpg)


![Sequential Five-Component Construction of the …sites.northwestern.edu/scheidt/files/2011/11/9_13_2010...Sequential Five-Component Construction of the Cyclopenta[e]-[1,3]oxazine Skeleton](https://img.pdfslide.net/doc/110x75/5b0761747f8b9ac33f8e30a0/sequential-five-component-construction-of-the-sites-five-component-construction.jpg)










