Embed Size (px)
Citation preview

Plant Molecular Biology 36: 601–612, 1998. 601c 1998 Kluwer Academic Publishers. Printed in Belgium.
Expression of fission yeast cdc25 alters the frequency of lateral rootformation in transgenic tobacco
Rowan S. McKibbin1;2, Nigel G. Halford1 & Dennis Francis2;�
1IACR – Long Ashton Research Station, Department of Agricultural Sciences, University of Bristol, Long Ashton,Bristol BS18 9AF, UK; 2School of Pure and Applied Biology, University of Wales, PO Box 915, Cardiff CF1 3TL,UK (�author for correspondence)
Received 22 April 1997; accepted in revised form 17 October 1997
Key words: cell division, cell size, lateral roots
Abstract
Lateral root formation was examined following the expression of a fission yeast mitotic regulator gene, cdc25, underthe control of a tetracycline-inducible promoter in cultured roots of tobacco. Over expression of cdc25 in fissionyeast results in premature cell division at a reduced cell size. Our aim was to examine whether cdc25 expressionwould affect cell size in the tobacco roots, and what effect this would have on lateral root morphogenesis. Transgeneintegration was confirmed by Southern blotting; it was inherited as a dominant Mendelian trait. Conditions foroptimal expression, determined using plants transformed with gus under the control of the same promoter, were:addition of tetracycline (5 �g/ml) every 72 h, to cultured roots in Murashige-Skoog liquid medium in darkness at27 �C. After the addition of tetracycline, cdc25 transcripts were detected using RT-PCR, initially after 48 h, andmore strongly after 72 h. Appearance of cdc25 transcripts was followed by major changes in the roots. Comparedwith controls, lateral root primordia were initiated more frequently, were significantly smaller and comprisedsmaller cells at mitosis. However, cdc25 expression did not perturb normal development of the lateral roots. Thedata are consistent with cdc25 expression leading to a greater frequency of lateral root primordium formationand establishing a new threshold size for cell division in the primordia which was then maintained throughoutsubsequent development.
Introduction
Cell division in meristems could be regarded as a mech-anism that partitions protoplasm and as a process thatis separated both spatially and temporally from sub-sequent development. Hence, and according to Bar-low [2], cell division is an optional accompaniment togrowth and development. This is consistent with theview that cells are constituents as opposed to ‘makers’of the organism [22]. Supporting these ideas was thefinding that cell division timing could be uncoupledfrom development in dominant-negative mutants oftobacco [17]. However, since cell size was perturbed inthese mutants, perhaps sizer controls of the cell cyclehave a more important role in development than ratesof division per se.
Newly formed cells resulting from mitotic divisiongrow to a minimum critical size for division in late G1,referred to as START in yeast [34], and the Restrictionpoint in animal cells [35]. In yeasts, passage throughSTART in late G1 commits the cell to the next roundof division [15]. In fission yeast, the cell’s size is alsoregulated during the transition from G2 to M-phase ofthe cell cycle [3, 4] and entry into mitosis is dependentupon the activity of the protein kinase,p34cdc2. In orderto exhibit protein kinase activity in late G2, it mustbe phosphorylated at Thr-167 (Thr-161 in Xenopusand mammalian cells) by a cyclin-dependentactivatingkinase which facilitates the formation of the complex,maturation promoting factor, from p34cdc2 and cyclinB [10, 24, 38, 44]. p34cdc2 is itself regulated by otherprotein kinases and at least one tyrosine phosphatase,p80cdc25,which dephosphorylates p34cdc2 in late G2

602
[23]. Dephosphorylation at Tyr-15 (and Thr-14 in ver-tebrates) is an important prerequisite before p34cdc2
exhibits protein kinase activity [25, 37]. In additionto the positive regulation of p34cdc2 by p80cdc25, anegative regulation is imposed by p107wee1, a proteinkinase that phosphorylates p34cdc2 on the same Tyr-15residue [37]. Mutants of fission yeast with additionalcopies of wee1 exhibit a larger-than-wild-type pheno-type at division [41] while those with additional copiesof cdc25 exhibit a smaller-than-wild-type phenotype atdivision [40]. As noted by Nurse [37], p107wee1 andp80cdc25 lie on antagonistic pathways which regulatecell size at division.
Under optimal conditions, fission yeast grows sothat its mass doubles once per cell cycle [8]. In otherworlds, the cell growth rate and the cell cycle are co-ordinated. However, a plant meristem does not com-prise a uniform population of cells all dividing andgrowing at the same rate. In fact, meristems are het-erogeneous with respect to both cell cycle durationand cell size. For example, in the shoot apical mer-istem basipetal gradients of cell cycle duration and cellsize exist from the summit to the flanks [29, 36]. Inroot meristems, cell size also differs from region toregion. For example, in Vicia faba descendants of thestellar initials are markedly elongate compared withdescendants of the cortical initials [27]. This spatialorganisation of cells suggests that normal co-ordinateddevelopment depends, at least partly, on cells acquiringtissue-specific sizes.
The requirement for a critical cell size for divisionis not obvious in some animal cells (e.g. mammalianneurones), but in plants, the cell cycle stabilises cellsize so that during normal development, size at divisionis altered as meristematic cells are displaced to the peri-phery of the meristem [2, 21]. This suggests stronglythat, on a tissue-specific basis, a minimum size controlregulates cell size at division and that this is one import-ant component of normal development. Testing thishypothesis requires experiments in which cell size con-trol is perturbed and subsequently plant developmentassessed for perturbations or changes. Cell size at divi-sion was reduced substantially in fission yeast mutantsinto which five tandem copies of cdc25 were integratedinto the cdc25 chromosomal locus [40].Although planthomologues of cdc2 have now been identified in a widerange of unrelated plant species [6, 9, 11, 18, 20], planthomologues of cdc25 and wee1 have yet to be dis-covered. However, p34cdc2 from tobacco was respons-ive to phosphoregulationexerted by fission yeast cdc25[46]. Our approach was to transform plants with the
fission yeast cdc25, measure cell size at division andstudy the development of transgenic plants expressingthis gene [4]. Moreover, using yeast cdc25 has theadvantage that problems with co-suppression wouldbe avoided and that the yeast protein might escape reg-ulation by the plant cell, whereas a plant homologuemight not. In tobacco, cdc25 expression under the con-trol of the CaMV 35S gene promoter was correlatedwith precocious flowering, more flowers per floweringhead and the presence of aberrant ‘petalless’ flowersalongside normal ones. These changes in developmentwere accompanied by the appearance of pockets in,and twisting of, the vegetative leaves. In addition, cellsize at division was significantly smaller in secondaryroot meristem cells of the transgenic plants comparedwith the wild type [4].
Even though the various alterations of developmentthat we observed occurred in meristematic regions ofthe plant, we could only make correlations between theeffect of constitutive expression of cdc25 on cell sizein the roots with the alterations of development else-where. For example, the profuse root branching patternof the tobacco plants used in these experiments did notenable us to relate changes in cell size to changes inroot development [4]. Hence, to test directly the rela-tionship between cdc25 expression, the cell cycle andplant development, a system was required in whichcdc25 could be expressed at a specific stage of devel-opment. In this paper, we report on the expression ofcdc25 under the control of an inducible promoter dur-ing a specific developmental event, the initiation anddevelopment of lateral root primordia in transgenictobacco. We show, for the first time, that the expres-sion of this mitotic inducer not only interfered with cellsize regulation, but also caused profound changes inthe pattern of root branching. The lateral root primor-dia were smaller and were initiated at a much greaterfrequency compared with the controls.
Materials and methods
Plant material
Wild-type and transgenic tobacco plants were main-tained in tissue culture under a 16 h light/8 h darkregime at 25 �C on Murashige and Skoog medium[32] containing 3% (w/v) sucrose, 1% (w/v) agar, and200 mg/l cefotaxime. Seed of selfed tetR+ plants [13]was kindly provided by Prof. Christiane Gatz (Insti-tut fur Pflanzenphysiologie, University of Gottingen)

603
and was germinated and propagated under sterile con-ditions. Kanamycin was added to the medium for thetetR+ plants to 100 mg/l, while hygromycin was usedat a concentration of 40 mg/l to select for TX-GUS andTX-CDC25 transformants. Well-rooted transformantswere transferred to compost and maintained in a green-house under a 16 h light/8 h dark regime at a minimumday temperature of 22 �C and minimum night temper-ature of 18 �C.
Construction of plasmids
Plasmids BIN-Hyg-TX-GUS-INT (BTX-GUS) andBIN-HYG-TX (BTX) were provided by ProfessorGatz [12, 13]. Plasmid pCDC25-S9 [39] was kindlyprovided by Professor Paul Nurse FRS (Imperial Can-cer Research Fund, Cell Cycle Control Laboratory,Lincoln’s Inn Fields, London). To construct BTX-CDC25, a 2068 bp DraI/XbaI restriction fragmentwas excised from pCD25-S9 and cloned into plasmidBTX restricted with XbaI and SmaI. The same frag-ment was cloned into the expression vector pGEM-3Zf(�) (Promega) for in vitro transcription/translationstudies. The cdc25 coding region minus flankingsequences was also cloned into pGEM-3Zf(�) afterPCR amplification primed with oligonucleotides 50-GCGGCCCCGGGATGGATTCTCCGCTTTCT and50-GGGCGTCTAGAATTAACGTCTGGGGAAGC,which contain SmaI and XbaI restriction sites respect-ively.
In vitro transcription and translation
The sequences cloned into pGEM-3Zf(�) were tran-scribed using T7 RNA polymerase and the RNA trans-lated in the presence of 35S-methionine. The reactionswere performed using the coupled TNT Wheat GermSystem (Promega), according to the manufacturer’sinstructions.
SDS-PAGE
Proteins were solubilized by adding 2 �l of 5� SDSloading buffer (10% (v/v) glycerol, 2% (w/v) SDS,100 mM Tris-HCl pH 6.8, 20 mM EDTA, 5% v/v2-mercaptoethanol, 0.001% bromophenol BBR250)to 10 �l of the sample and heating to 95 �C for3 min. A 3 �l aliquot was then run on a 10% SDS-polyacrylamide gel in Tris buffer at a constant 40 mA.After electrophoresis, gels were rinsed with distilledwater, fixed in boiling 10% TCA, rinsed again and
soaked in 2 M Tris base for 5–10 min. They werethen dried for 45 min at 80 �C and autoradiographedovernight at room temperature.
Tobacco transformation
Plasmids BTX-GUS and BTX-CDC25 were elec-troporated into Agrobacterium tumefaciens strainLBA4404 using the method of Shen and Forde [42].Agrobacteria which had taken up the plasmid wereselected on medium containing 100 mg/l kanamycinand used to transform the tetR+ tobacco plants throughthe leaf disc transformation method of Horsch [19].Transgenic plants were selected on medium contain-ing 40 mg/l hygromycin and 100 mg/l kanamycin. Theplants were self-pollinated and the T1 generation usedfor subsequent analyses.
Southern blotting
Genomic DNA was extracted from frozen leaves asdescribed by Smith et al. [43]. 50 mg was diges-ted with the appropriate restriction enzyme, electro-phoresed through a 0.8% agarose gel and blottedonto a GeneScreen Plus membrane (NEN ResearchProducts) using the manufacturer’s salt transfer pro-tocol. Radiolabelled probes were synthesised using thePrime-It II Random Primer Labeling Kit (Stratagene),according to the manufacturer’s instructions, and unin-corporated nucleotides removed using NucTrap pushcolumns (Stratagene). Hybridisations were carried outovernight at 65 �C in a solution containing 10% (w/v)1 M dextran sulphate (sodium salt, Mr 500 000), 1%(w/v) SDS. Membranes were washed twice in 2� SSC(0.3 M NaCl, 0.03 M sodium citrate) at 65 �C for30 min, then twice in 0.5� SSC, 1% SDS at 65 �C for60 min, then autoradiographed.
Establishment of primary root cultures
To establish the cultures, 10 mm long primary roottips which were completely devoid of lateral roots andvisible lateral root primordia were excised from trans-genic plants, aseptically, and cultured in a modifiedLinsmaier-Skoog [26] medium supplemented with 3%(v/v) sucrose, 10 �M 2,4-dichlorophenoxyacetic acid,550 �M myoinositol, 150 �M KH2PO4 and 3 �Mthiamine-HCl adjusted to a pH of 5.8 [33]. The rootswere cultured in darkness at 27 �C in 95 ml of medi-um in 300 ml stationary Erlenmeyer flasks. Underthese conditions, the primary roots elongated at 3.5

604
and 3.3 mm per day in the non-induced and inducedcultures, respectively (P > 0:05).
RT-PCR analysis
Total RNA was extracted from fresh plant materi-al using the acid phenol procedure as described byChattopodhyay et al. [5]. First-strand cDNA was syn-thesised by reverse transcription with AMV reversetranscriptase (Promega) of the RNA using oligonuc-leotide dT15 as a primer. The protocol provided bythe supplier was followed with the modification thatthe RNA, primer and RNAse-free water were mixedtogether and heated to 67 �C for three minutes toeliminate secondary structures. The tubes were thencooled to 42 �C and the rest of the reagents addedand left for 20 min. Specific primers were used toamplify cdc25 or gus sequences using 2 �l (from atotal of 20 �l) of the cDNA synthesis reaction as atemplate in a PCR reaction of 30 cycles of 94 �Cfor 1 min, 48–52 �C (depending on primers used)for 1 min, and 72 �C for 1 min, in a Perkin Elmer480 Thermal Cycler. The reactions were incubated fora further 10 min at 72 �C, then stored at �20 �C.The oligonucleotides used as PCR primers were 50-GCGGATCCATGCGGTGACTCATTACG and 50-G-CTCTAGACTGTGACGCACAGTTCA for gus, 50-GCGAATTCGCCCTGCCTTACCGACTCC and 50-GCGAATTCGGCGGGTAGCTTGTCCACC forcdc25, and TGGGAYACNGCNGGNCA and GCNG-ANGTYTCCATAAA (kindly provided by Dr Johnath-an Napier, Long Ashton Research Station) for rab1.The PCR products were electrophoresed, Southernblotted, hybridised with radiolabelled cdc25, gus orrab1 probes and autoradiographed.
Measurements of lateral root formation on culturedprimary roots
At 7 day intervals for 35 days the roots were fixedin 3:1 absolute ethanol/glacial acetic acid (24 h at5 �C), hydrolysed in 5 M HCl for 30 min and thenFeulgen-stained (2 h). The roots were then examinedunder a Nikon stereo dissecting microscope; primor-dia were recognised as deeply-stained structures alongthe length of the primary root (Figure 4). To providecomparability between sampling times, scoring wasdone on 10 mm long segments of primary root whichwere at least 10 mm from the root tip. In this way, wewere able to examine lateral root primordia at com-parable stages of their development in both treatments.
At each sampling time, ten replicate 10 mm lengthsof primary were scored for lateral root primordia. Thesame measurements were also done on roots sampledfrom wild-type, untransformed plants; the roots werecultured, fixed and stained as above.
Primordium volume
At each sampling time the height (length) and diamet-er of 20 primordia were measured at random usinga dissecting microscope. The primordia were taken asconforming to the shape of a cone and,hence, the meas-urements were used to calculate primordium volumein the equation
V = 1=3�r2h:
Subsequently, these lateral root primordia were dis-sected from the primary root tissue. They were placedin droplets of 45% (v/v) acetic acid and dispersedinto a monolayer by overlaying a coverslip. The lat-ter was then detached after placement of the slideonto dry-ice (5 min). The slides were then counter-stained with naphthol yellow S [39] and overlaid withnew coverslips. Mitotic cells were clearly identifiedas purple chromosomes against a yellow backgroundwhich provided an obvious perimeter for each cell.Cellarea was then obtained using image analysis whichcomprised an Olympus photomicroscope interfacedwith a Graf pad graphics tablet [4]. Having establishedthat there were no significant differences in mitoticcell size within treatments, the mitotic cell areas werepooled from all sampling times and presented as acomposite histogram for each treatment.
Results
Transformation of tobacco with gus and fission yeastcdc25 under the control of an inducible promoter
An inducible promoter system was used for expres-sion of cdc25, enabling it to be switched on spe-cifically during root development. The tetracycline-inducible system developed by Gatz et al. [12, 13]was chosen because it has been reported to give upto 500-fold increases in expression of �-glucuronidase(gus) on the application of tetracycline at levels whichcaused no detectable side-effects. The system uses theTriple X (TX) promoter, which is a modified cauli-flower mosaic virus 35S RNA (CaMV 35S) promoterin which three tet operators have been inserted. Theseact as recognition sites for the Tn10-encoded tetra-

605
Figure 1. Structure of chimaeric gene construct TX-CDC25.
cycline repressor molecule (Tet repressor) and bindingof the Tet repressor greatly reduces promoter activity.Expression is induced in the presence of tetracycline,which complexes with the Tet repressor, preventing itfrom binding to the promoter, thus allowing transcrip-tion.
Plasmid BIN-hyg-TX-GUS-INT (BTX-GUS), andseeds from self-pollinated tetR+ transgenic tobaccoplants containing a chimaeric construct of a CaMV35S promoter and the Tn10 gene, were kindly providedby Professor C. Gatz (Institut fur Pflanzenphysiologie,University Gottingen). The TX-GUS chimaeric genecontains the gus coding sequence plus a 189 bp intronwhich increases the efficiency of transcript processing[45], downstream of the TX promoter. The cdc25 con-struct was made by cloning a 2068 bp fragment of fis-sion yeast cdc25 [40], into the plasmid BIN-HYG-TX(BTX) [13]. The resulting chimaeric gene comprisingthe TX promoter and cdc25 sequence was called TX-CDC25 (Figure 1), and the plasmid containing it wascalled BTX-CDC25. The cdc25 fragment includes thecoding region plus 54 bp and 273 bp, respectively, ofthe 50- and 30- untranslated regions.
The TX-CDC25 construct was checked by sequen-cing across the junction of the promoter and cod-ing sequences and by coupled in vitro transcrip-tion/translation. The latter was performed using thecdc25 sequence from BTX-CDC25, with and withoutupstream and downstream sequences, cloned into thetranscription vector pGEM-3Zf(�). RNA was syn-thesised from these templates and translated usingwheat germ extracts in the presence of 35S-methionineand the reaction products were separated by SDS-PAGE and autodiographed (Figure 2). In both cases,a radiolabelled Mr 64 000 polypeptide was synthes-ised, indicating that the cdc25 sequence was tran-scribed and translated correctly. Considerably moreof this polypeptide was produced when the flankingDNA sequences were included, suggesting that theyincreased the efficiency of transcription and/or trans-lation.
Figure 2. SDS-PAGE of products from a coupled in vitro tran-scription/translation reaction using the cdc25 sequence from BTX-CDC25 without flanking sequences (lane 1) and with flankingsequences (lane 2). The position of size markers (kDa) is shownon the left.
BTX-GUS and BTX-CDC25 were used to trans-form tetR+ tobacco plants through A. tumefaciens-mediated leaf disk transformation. Transformed plantswere selected on medium containing 40 mg/l hygro-mycin and 200 mg/l kanamycin. The presence of thetransgene was confirmed by probing Southern blotsof EcoRI/HindIII-restricted genomic DNA with theEcoRI/HindIII fragment from BTX. This showed thepresence in the transformants of a ca. 1.5 kb hybrid-ising band corresponding to the portion of TX-CDC25comprising the promoter and 970 bp of the cdc25sequence up to an internal HindIII site (not shown).Ten independent transformants containing TX-CDC25construct were recovered.
Segregation analysis
The 10 independent TX-CDC25 transformants wereself-pollinated and seed was collected, surface-sterilised and germinated on medium containing100 mg/l hygromycin. A total of 792 seedlings weregerminated, of which 207 showed symptoms of hygro-mycin sensitivity (small size, bleached leaves and poorroot growth) 12 days after sowing. For each line, about100 seedlings were generated and, in each case, theratio of resistant to non-resistant seedlings did not dif-fer significantly from 3:1 (�2
= 0:545; r = 0:5).

606
Optimisation of induction conditions in root cultures
To determine the optimum induction parameters foruse with this system, tetracycline titration and timecourse experiments were carried out using TX-GUSplants. The plants were grown aseptically and primaryroot tips devoid of lateral roots and visible lateral rootprimordia were excised when they had reached 10 mmin length. These were cultured in a modified Murashigeand Skoog [32] medium, in the dark (to prevent light-degradation of the tetracycline) at 27 �C. Each culturewas divided into six subcultures and expression of theTX-GUS construct was induced by the addition of tet-racycline to the medium to final concentrations of 0,2, 4, 6, 8 or 10 mg/l, applied every 72 h. RNA wasextracted from the roots after one week of the treat-ment and the relative levels of gus expression weredetermined by reverse-transcription polymerase chainreaction (RT-PCR) to amplify a 500 bp sequence fromthe gus transcript. This method was used in preferenceto assays of gus activity to enable direct comparisonwith experiments performed on the TX-CDC25 plants.A 200 bp fragment of the transcript of the rab1 gene,which is constitutively expressed in tobacco roots [13],was also amplified as a control. The PCR products wereSouthern-blotted, hybridised with a radiolabelled gusor rab1 probe and autoradiographed (Figure 3a). Theamount of rab1 product remained approximately con-stant, confirming that addition of the tetracycline didnot affect rab1 gene expression and that the RT-PCRwas giving a quantitative result. In contrast, the quant-ity of gus amplified fragments was directly related tothe concentration of tetracycline in the culture medi-um, indicating that expression of the gene was beinginduced by the addition of tetracycline.
The results showed that significant induction of gusexpression could be achieved by applying tetracyclineat a concentration of ca. 5 mg/l. This concentration wasthen used in a second experiment to determine howrapidly expression of the gene was induced. RNA wasextracted 0, 24, 48 and 72 h after induction and it wasfound that transcripts were not detectable using RT-PCR until after 48 h and further accumulated after 72 h(Figure 3b). Some gus product was also amplified fromRNA extracted from uninduced roots after 72 hours inculture in this experiment. This indicates incompleterepression of the promoter even when uninduced, asreported previously [12, 13].
Figure 3. Autoradiographs of Southern-blotted RT-PCR productsamplified using gus, rab1 and cdc25-specific primers from RNAprepared from transgenic root cultures. a. TX-GUS roots, treatedwith 0–10 mg/l tetracycline for 1 week. Top panel shows gus PCRproducts detected by a gus probe, bottom panel rab1 products detec-ted by a rab1 probe. b. TX-GUS roots treated (t) with 5 mg/l tet-racycline and sampled every 24 h, compared with untreated roots(u). Top panel shows gus PCR products detected by a gus probe,bottom panel rab1 products detected by a rab1 probe. c. TX-GUSand TX-CDC25 roots treated (t) with 5 mg/l tetracycline for 1 week,compared with untreated roots (u). Top panel shows cdc25 PCRproducts detected by a cdc25 probe, middle panel gus PCR productsdetected by a gus probe, bottom panel rab1 products detected by arab1 probe.
Inducible expression of cdc25
Root cultures were set up using plants grown fromseed of selfed TX-CDC25 plants as for the TX-GUS

607
plants. Each culture was divided into two subculturesand tetracycline was added to the medium of one ofthe subcultures to a final concentration of 5 mg/l andreapplied every 72 h. RNA was extracted from theroots after 1 week and used as the template for RT-PCRusing primers which amplify a 429 bp sequence fromthe cdc25 transcript. A rab1 sequence was amplified asa control as before. The PCR products were Southern-blotted, hybridised with cdc25 or rab1 probes and auto-radiographed. Cdc25 transcripts were detectable in thisway in RNA from all of the TX-CDC25 cultures afteraddition of tetracycline. However, small amounts ofproduct were also produced from RNA from unin-duced cultures, again indicating that the promoter wasnot fully repressed. No change was observed in thelevel of rab1 transcripts throughout. Autoradiographsof PCR products from two TX-GUS cultures and twoTX-CDC25 cultures are shown in Figure 3c and showthat the cdc25 transcript accumulates to levels similarto that of the gus transcript.
The levels of induction obtained here are consid-erably lower than the 500-fold increase in transcriptlevels reported by Gatz et al. [13]. The length of timerequired for full induction, and the concentration of tet-racycline required, are also much greater. The reasonfor this relatively poor induction is unknown, but sincethe achieved levels of induction of the TX-CDC25 con-struct were causing significant changes in the develop-ment of the roots, it was not investigated further.
All subsequent observations and data were pooledfrom the same independent transgenic lines, TX-CDC25.1 and TX-CDC25.2, RT-PCR analyses ofwhich are shown in Figure 3c.
Frequency of lateral root primordia and primordiumvolume
At each sampling time, there was a substantial increasein the number of primordia in the induced comparedwith the uninduced cultures (Figures 4 and 5; day 7, 2-fold; day 14, 3-fold; days 21–35, 3.5-fold); the meanswere significantly different (P < 0:001). As above,10 mm long primary roots tips, devoid of lateral rootprimordia, were excised from TX-CDC25 plants andcultured in the presence (induced cultures) or absence(uninduced cultures) of 5 mg/l tetracycline at 27 �Cin darkness (tetracycline was added once every 72 h).Over the 35 day interval of these experiments, therate of elongation of primary TX-CDC25 roots in theuninduced and induced cultures was 3.5 and 3.3 mmper day, respectively (P > 0:05).
Figure 4. Feulgen-stained TX-CD25 primary roots exhibiting lateralroot primordia in (a) uninduced and (b) induced 21-day old cultures.Bar scale (for both): 2 mm. Uninduced (c) and (d) induced (d) 35-dayold cultures (overlying scale).
When wild-type roots were cultured in the presenceor absence of tetracycline there were no significant dif-ferences between the cultures in four out of the fivesampling times (P > 0:05) but there was a marginallysignificant reduction in the number of primordia inthe tetracycline-treated culture after 28 days (Table 1).Overall, the numbers forming per unit time were notappreciably different from those scored on the unin-duced TX-CDC25 roots. In other words, tetracyclineper se had no apparent effect on the frequency of lateralroot primordium formation over this interval.However,between days 7 and 21, there was a 3-fold increase inthe number of lateral root primordia in wild-type roots(Table 1) compared with only a 0.2-fold increase inuninduced transgenic roots over the same period (Fig-ure 5) whereas there were no significant differencesbetween days 21 and 35. If, due to experimental vari-ation, the wild-type cultures lagged slightly behind theothers, then some primordia would have been too smallto resolve (primordia of<800 cells cannot be resolvedwith a dissecting microscope). On day 7, the wild type
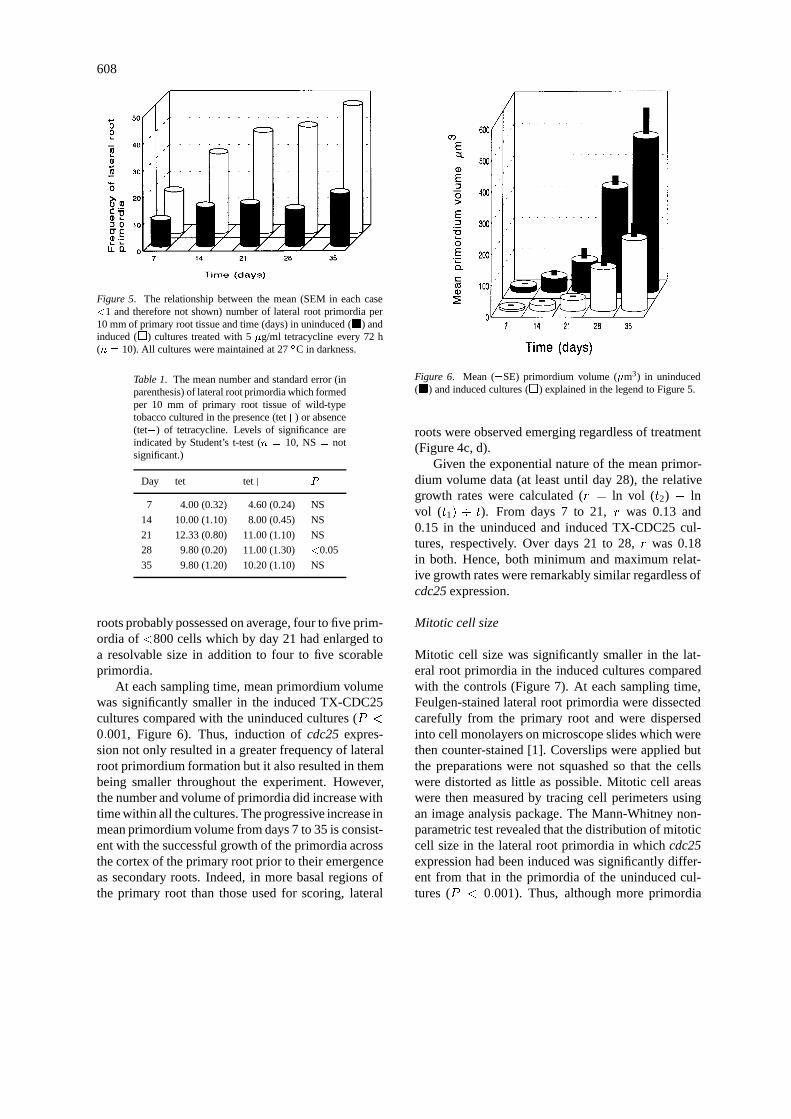
608
Figure 5. The relationship between the mean (SEM in each case<1 and therefore not shown) number of lateral root primordia per10 mm of primary root tissue and time (days) in uninduced (�) andinduced (�) cultures treated with 5 �g/ml tetracycline every 72 h(n = 10). All cultures were maintained at 27 �C in darkness.
Table 1. The mean number and standard error (inparenthesis) of lateral root primordia which formedper 10 mm of primary root tissue of wild-typetobacco cultured in the presence (tet+) or absence(tet�) of tetracycline. Levels of significance areindicated by Student’s t-test (n = 10, NS = notsignificant.)
Day tet� tet+ P
7 4.00 (0.32) 4.60 (0.24) NS
14 10.00 (1.10) 8.00 (0.45) NS
21 12.33 (0.80) 11.00 (1.10) NS
28 9.80 (0.20) 11.00 (1.30) <0.05
35 9.80 (1.20) 10.20 (1.10) NS
roots probably possessed on average, four to five prim-ordia of <800 cells which by day 21 had enlarged toa resolvable size in addition to four to five scorableprimordia.
At each sampling time, mean primordium volumewas significantly smaller in the induced TX-CDC25cultures compared with the uninduced cultures (P <
0:001, Figure 6). Thus, induction of cdc25 expres-sion not only resulted in a greater frequency of lateralroot primordium formation but it also resulted in thembeing smaller throughout the experiment. However,the number and volume of primordia did increase withtime within all the cultures. The progressive increase inmean primordium volume from days 7 to 35 is consist-ent with the successful growth of the primordia acrossthe cortex of the primary root prior to their emergenceas secondary roots. Indeed, in more basal regions ofthe primary root than those used for scoring, lateral
Figure 6. Mean (�SE) primordium volume (�m3) in uninduced(�) and induced cultures (�) explained in the legend to Figure 5.
roots were observed emerging regardless of treatment(Figure 4c, d).
Given the exponential nature of the mean primor-dium volume data (at least until day 28), the relativegrowth rates were calculated (r = ln vol (t2) � lnvol (t1) � t). From days 7 to 21, r was 0.13 and0.15 in the uninduced and induced TX-CDC25 cul-tures, respectively. Over days 21 to 28, r was 0.18in both. Hence, both minimum and maximum relat-ive growth rates were remarkably similar regardless ofcdc25 expression.
Mitotic cell size
Mitotic cell size was significantly smaller in the lat-eral root primordia in the induced cultures comparedwith the controls (Figure 7). At each sampling time,Feulgen-stained lateral root primordia were dissectedcarefully from the primary root and were dispersedinto cell monolayers on microscope slides which werethen counter-stained [1]. Coverslips were applied butthe preparations were not squashed so that the cellswere distorted as little as possible. Mitotic cell areaswere then measured by tracing cell perimeters usingan image analysis package. The Mann-Whitney non-parametric test revealed that the distribution of mitoticcell size in the lateral root primordia in which cdc25expression had been induced was significantly differ-ent from that in the primordia of the uninduced cul-tures (P < 0:001). Thus, although more primordia
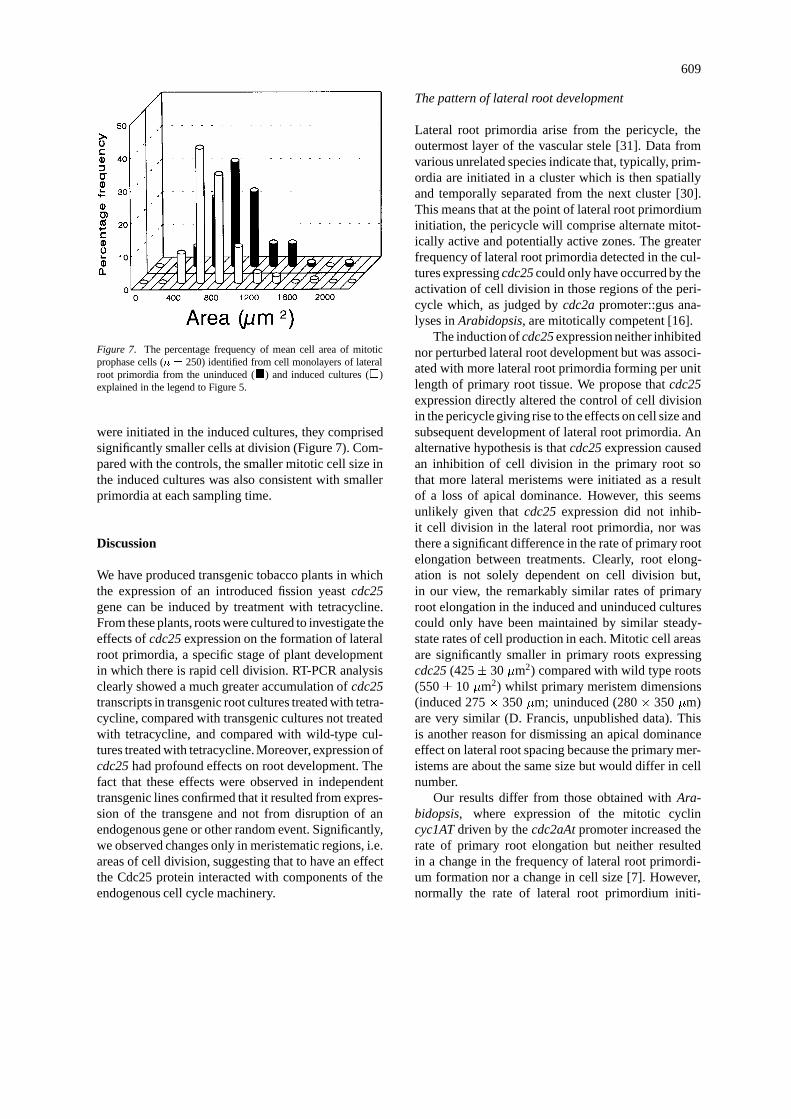
609
Figure 7. The percentage frequency of mean cell area of mitoticprophase cells (n = 250) identified from cell monolayers of lateralroot primordia from the uninduced (�) and induced cultures (�)explained in the legend to Figure 5.
were initiated in the induced cultures, they comprisedsignificantly smaller cells at division (Figure 7). Com-pared with the controls, the smaller mitotic cell size inthe induced cultures was also consistent with smallerprimordia at each sampling time.
Discussion
We have produced transgenic tobacco plants in whichthe expression of an introduced fission yeast cdc25gene can be induced by treatment with tetracycline.From these plants, roots were cultured to investigate theeffects of cdc25 expression on the formation of lateralroot primordia, a specific stage of plant developmentin which there is rapid cell division. RT-PCR analysisclearly showed a much greater accumulation of cdc25transcripts in transgenic root cultures treated with tetra-cycline, compared with transgenic cultures not treatedwith tetracycline, and compared with wild-type cul-tures treated with tetracycline.Moreover, expression ofcdc25 had profound effects on root development. Thefact that these effects were observed in independenttransgenic lines confirmed that it resulted from expres-sion of the transgene and not from disruption of anendogenous gene or other random event. Significantly,we observed changes only in meristematic regions, i.e.areas of cell division, suggesting that to have an effectthe Cdc25 protein interacted with components of theendogenous cell cycle machinery.
The pattern of lateral root development
Lateral root primordia arise from the pericycle, theoutermost layer of the vascular stele [31]. Data fromvarious unrelated species indicate that, typically, prim-ordia are initiated in a cluster which is then spatiallyand temporally separated from the next cluster [30].This means that at the point of lateral root primordiuminitiation, the pericycle will comprise alternate mitot-ically active and potentially active zones. The greaterfrequency of lateral root primordia detected in the cul-tures expressing cdc25 could only have occurred by theactivation of cell division in those regions of the peri-cycle which, as judged by cdc2a promoter::gus ana-lyses in Arabidopsis, are mitotically competent [16].
The induction of cdc25 expression neither inhibitednor perturbed lateral root development but was associ-ated with more lateral root primordia forming per unitlength of primary root tissue. We propose that cdc25expression directly altered the control of cell divisionin the pericycle giving rise to the effects on cell size andsubsequent development of lateral root primordia. Analternative hypothesis is that cdc25 expression causedan inhibition of cell division in the primary root sothat more lateral meristems were initiated as a resultof a loss of apical dominance. However, this seemsunlikely given that cdc25 expression did not inhib-it cell division in the lateral root primordia, nor wasthere a significant difference in the rate of primary rootelongation between treatments. Clearly, root elong-ation is not solely dependent on cell division but,in our view, the remarkably similar rates of primaryroot elongation in the induced and uninduced culturescould only have been maintained by similar steady-state rates of cell production in each. Mitotic cell areasare significantly smaller in primary roots expressingcdc25 (425� 30 �m2) compared with wild type roots(550� 10 �m2) whilst primary meristem dimensions(induced 275 � 350 �m; uninduced (280 � 350 �m)are very similar (D. Francis, unpublished data). Thisis another reason for dismissing an apical dominanceeffect on lateral root spacing because the primary mer-istems are about the same size but would differ in cellnumber.
Our results differ from those obtained with Ara-bidopsis, where expression of the mitotic cyclincyc1AT driven by the cdc2aAt promoter increased therate of primary root elongation but neither resultedin a change in the frequency of lateral root primordi-um formation nor a change in cell size [7]. However,normally the rate of lateral root primordium initi-

610
ation shows a strong positive relationship with rate ofprimary root elongation [29]. Hence, the Arabidopsiswork is consistent in showing that lateral root initiationkept pace with overall primary elongation rates. Ourdata, show an increased frequency of lateral root prim-ordium formation in the absence of a greater increasein primary root elongation.
Molecular mechanisms governing the change inmitotic cell size and development
The simplest molecular model to explain the resultspresented here is that expression of the fission yeastcdc25 maintained the native p34cdc2 in a dephos-phorylated, active state. This resulted in premature celldivision in a manner analogous to that observed in themutants of fission yeast which over-expressed cdc25in late G2 [40]. This model implies that regulation ofp34cdc2 activity in plants is regulated in part by dephos-phorylation through the action of a p80cdc25-like phos-phatase at the G2/M transition, as it is in animals andyeast. In tobacco, p34cdc2 kinase activity was shownto be regulated by an inhibitory tyrosine phosphoryla-tion consistent with the inactivation of purified plantenzyme from pith and suspension cells held in lateG2 [46]. However, fission yeast Cdc25 phosphatasecaused extensive activation of purified plant enzyme inthe same cells [46]. This is an important observationbecause it demonstrates that the late G2 form of plantp34cdc2 can be phosphoregulated. In our experiments,it is possible that tyrosine15 is not the only target forp80cdc25 in plant p34cdc2 in vivo. For example, maybethe ectopic expression of cdc25 activated Cdks thatfunction at the G1/S transition which in turn activatedpericycle cells. However, we favour the G2/M modelgiven the small mitotic cell size phenotype reportedhere, before [4], and in fission yeast [40]; all of thesecell phenotypes were associated with high levels ofcdc25 transcripts.
Cell size and development
Circumstantial evidence exists in the literature that cellsizer controls are an inherent part of normal develop-mental patterns that emanate from the meristems (seeIntroduction). Our data are entirely consistent with thishypothesis. Induction of cdc25 expression in culturedprimary roots was strongly associated not only witha reduced mitotic cell size, confirming earlier work[4], but also with a much greater frequency of lateralroot initiation per unit time. In other words, establish-
ment of a reduced cell size at division was linked to anincrease in the frequency of lateral root formation.
In an experiment involving the transformation oftobacco with mutant cdc2 genes, Hemerly et al. [17]produced plants showing a reduced frequency of celldivision. Seedlings of these plants contained fewer,but larger, cells than the wild type and, remarkably,exhibited shorter roots and a reduced number of lat-eral roots, the converse of the characteristics shownby the induced TX-CDC25 cultures. We accept this asfurther evidence for a cell sizer control as an integralcomponent of a developmental programme.
In our study, the question remains as to why cellsexpressing cdc25 did not become progressively smallerduring each successive cell cycle, as would be expec-ted if cell size at division became progressively reducedat each successive division resulting ultimately in celldeath. What we observed was a significantly smal-ler mitotic cell size which persisted throughout thedevelopment of the lateral root primordia. Notably, therelative growth rates of the induced and uninduced TX-CDC25 lateral root primordia were remarkable similarwhich suggests that the duration of each cell cycle wasnot affected. We suggest that cdc25 expression resultsin an excess of tyrosine dephosphorylation of p34cdc2
in the pericycle (and in other mitotically competentregions) resulting in the induction a new threshold sizefor division in cells of the pericycle which, once estab-lished, persisted throughout subsequent developmentof the lateral root primordia. Consequently, the cellswould be smaller from beginning to end of the cellcycle, but would grow at the normal rate. This is sim-ilar to Hemerly et al.’s [17] view that the controlsof development can act independently of cell divisionrates.
Acknowledgements
IACR receives grant-aided support from the Biotech-nology and Biological Sciences Research Council ofthe United Kingdom. Rowan McKibbin thanks theBBSRC for a CASE studentship.
References
1. Armstrong SW, Francis D: Differences in cell cycle durationof sister cells in secondary root meristems of Cocos muciferaL. Ann Bot 56: 803–813 (1985).
2. Barlow PW: Properties of cells in the root apex. Rev Fac AgronUniv Nac La Plata 47: 275–301 (1971).

611
3. Barlow PW: Cell division in meristems and their contribution toorganogenesis and plant form. In: Ingram DS, Hudson A (eds)Shape and Form in Plants and Fungi, pp. 169–193. AcademicPress, London (1994).
4. Bell MH, Halford NG, Ormrod JC, Francis D: Tobacco plantstransformed with cdc25, a mitotic inducer gene from fissionyeast. Plant Mol Biol 23: 445–451 (1993).
5. Chattopodhyay N, Kher R, Godbole M: Inexpensive SDS phen-ol method for RNA extraction from tissues. Biotechniques 15:24 (1993).
6. Colasanti J, Tyers M, Sunderesan V: Isolation and character-isation of cDNA clones encoding a functional p34cdc2 homo-logue from Zea mays. Proc Natl Acad Sci USA 88: 3377–3381(1991).
7. Doerner P, Jorgensen JE, Yu R, Steppuhn J, Lamb C: Controlof root growth and development by cyclin expression. Nature380: 520–523 (1996).
8. Fantes PA, Nurse P: Division timing: controls models andmechanisms. In: John PCL (ed) The Cell Cycle, pp. 11–33.Cambridge University Press, Cambridge, UK (1981).
9. Feiler HS, Jacobs T: Cell division in higher plants: A cdc2gene, its 34-dKa product, and histone activity in pea. Proc.Nat. Acad. Sci. 87: 5397–5401 (1990).
10. Fesquet D, Labbe J-C, Derancourt J, Capony J-C, Galas S,Girand F, Lorca T, Shuttleworth J, Doree M, Cavadore J-C:The MO15 gene encodes the catalytic subunit of a proteinkinase that activates cdc2 and other cyclin-dependent kinases(CDKs) through phosphorylation of Thr161 and its homo-logues. EMBO J 12: 3111–3121 (1993).
11. Francis D, Halford NG: The plant cell cycle. Physiol Plant 93:365–374 (1995).
12. Gatz C, Kaiser A, Wendenburg R: Regulation of a modifiedCaMV 35S promoter by the Tn10-encoded Tet repressor intransgenic tobacco. Mol Gen Genet 227: 229–237 (1991).
13. Gatz C, Frohberg C, Wendenburg R: Stringent repression andhomogenous derepression by tetracycline of a modified CaMV35S promoter in intact transgenic tobacco plants. Plant J 2:397–404 (1992).
14. Grimwade B, Tatham AS, Freedman RB, Shewry PR, Napi-er JA: Comparison of the expression patterns of wheat glu-tein proteins and proteins involved in the secretory pathwayin developing caryopses of wheat. Plant Mol Biol, in press(1997).
15. Hayles J, Nurse P: The controls acting at mitosis in Schizosac-charomyces pombe. In: Ormrod JC, Francis D (eds) Molecularand Cell Biology of the Plant Cell Cycle, pp. 1–7. KluwerAcademic Publishers, Dordrecht, Netherlands (1993).
16. Hemerly A, Ferreira P, Engler JD, Van Montague M, Inze D:Cdc2a expression in Arabidopsis is linked with competencefor cell division. Plant Cell 5: 1711–1723 (1993).
17. Hemerley A, de Almeida Engler J, Bergounioux C, VanMontague M, Engler G, Inze D, Ferreira P: Dominant neg-ative mutants of the Cdc2 kinase uncouple cell division fromiterative development. EMBO J. 14: 3925–3926 (1995).
18. Hirt H, Pay A, Gyorgyey J, Bako L, Nemeth K, Bogre L,Schweyen RJ, Heberle-Bors E, Dudits D: Complementation ofa yeast cell cycle mutant by an alfalfa cDNA encoding a proteinkinase homologous to p34cdc2. Proc Natl Acad Sci USA 82:820–823 (1991).
19. Horsch RB, Fry JF, Hoffman NL, Eichholtz D, Rogers SG,Frayley RT: A simple and general method for transferring genesinto plants. Science 227: 1229–1231 (1985).
20. John PCL, Sek FJ, Lee MG: A homologue of the cell cyclecontrol protein p34cdc2 participates in the division cycle of
Chlamydomonas, and a similar protein is detectable in higherplants and remote taxa. Plant Cell 1: 1185–1193 (1989).
21. John PCL, Zhang K, Dong C: A p34cdc2-based cell cycle: itssignificance in monocotyledonous, dicotyledonous and unicel-lular plants. In: Ormrod JC, Francis, D (eds) Molecular andCell Biology of the Plant Cell Cycle, pp. 9–34. Kluwer Aca-demic Publishers, Dordrecht, Netherlands (1993).
22. Kaplan DR, Hagemann W: The relationship of cell and organ-ism in vascular plants. Bioscience 41: 693–703 (1991).
23. Kumagai A, Dunphy WG: The cdc25 protein controls tyrosinedephosphorylation of the cdc2 protein in a cell-free system.Cell 64: 904–914 (1991).
24. Labbe J-C, Capony J-P, Caput D, Cavadore J-C, Derrancourt J,Kaghad M, Lelias J-M, Picard A, Doree M: MPF from starfishoocytes at first meiotic metaphase is a heterodimer containingone molecule of cdc2 and a molecule of cyclin B. EMBO J 8:3053–3058 (1989).
25. Lee MG, Nurse P: Complementation used to clone a humanhomologue of the fission yeast cell cycle control gene cdc2.Nature 327: 31–35 (1987).
26. Linsmaier E, Skoog F: Organic growth factor requirements oftobacco tissue. Physiol Plant 18: 100–127 (1965).
27. Luxova M, Murin A: The extent and differences in mitoticactivity in the root top of Vicia faba L. Biol. Plant. 15: 37–43(1973).
28. Lyndon RF: The cell cycle in the shoot apex. In: Balls M, BillettFS (eds) The Cell Cycle in Development and Differentiation,pp. 167–183. Cambridge University Press, Cambridge, UK(1973).
29. MacLeod RD: Growth of lateral root primordia. New Phytol76: 143–151 (1976).
30. Mallory TE, Chiang S, Cutter EG, Gifford EM: Sequence andpattern of lateral root formation in five selected species. Am JBot 57: 800–809 (1970).
31. McCully ME: The developmental of lateral roots. In: Tor-rey JG, Clarkson DT (eds) The Development and Function ofRoots, pp. 105–124. Academic Press, London (1975).
32. Murashige T, Skoog F: A revised medium for rapid growthand bioassays with tobacco tissue cultures. Physiol Plant 15:473–497 (1962).
33. Nagata TN, Nemoto Y, Hasezawa S: Tobacco BY-2 cell line asthe ‘Hela’ cell line in the cell biology of higher plants. Int RevCytol 132: 1–30 (1992).
34. Nasmyth K: Control of the yeast cell cycle by the Cdc28 proteinkinase. Curr Opin Cell Biol 5: 166–179 (1993).
35. Norbury C, Nurse P: Animal cell cycles and their control. AnnuRev Biochem 61: 441–470 (1992).
36. Nougarede A: Experimental cytology of the shoot apical cellsduring growth and flowering. Int Rev Cytol 21: 203–351(1967).
37. Nurse P: Universal control mechanism regulating onset of M-phase. Nature 344: 503–508 (1990).
38. Poon RYC, Yamashita K, Adamcczewski JP, Hunt T, Shuttle-worth J: The cdc2-related protein p40MO15 is the catalytic sub-unit of a protein kinase that can activate p33cdk2 and p34cdc2.EMBO J 12: 3113–3118 (1993).
39. Powell MJ, Davies MS, Francis D: Effects of zinc on cell,nuclear and nucleolar size and on RNA and protein content inthe root meristem of a zinc-tolerant and non-tolerant cultivarof Festuca rubra L. New Phytol 104: 671–679 (1986).
40. Russell P, Nurse P: cdc25 functions as an inducer in the mitoticcontrol of fission yeast. Cell 5: 145–153 (1986).

612
41. Russell P, Nurse P: Negative regulation of mitosis by wee1+, agene encoding a protein kinase homologue. Cell 49: 559–567(1987).
42. Shen W, Forde BG: Efficient transformation of Agrobacteriumspp. by high voltage electroporation. Nucl Acids Res 17: 8385(1989).
43. Smith MA, Stobart AK, Shewry PR, Napier JA: Tobacco cyto-chrome b5:cDNA isolation, expression analysis and in vitroprotein targeting. Plant Mol Biol 25: 527–537 (1994).
44. Solomon MJ, Harper JW, Shuttleworth J: CAK, the p34cdc2
activating kinase, contains a protein identical or closely relatedto p40MO15. EMBO J 12: 3133–3142 (1993).
45. Vancanneyt G, Schmidt R, O’Connor-Sanchez A, Willmitzer L,Rocha-Sosa M: Construction of an intron-containing markergene: splicing of the intron in transgenic plants and its usein monitoring early events in Agrobacterium-mediated planttransformation. Mol Gen Genet 220: 245–250 (1990).
46. Zhang K, Letham DS, John PCL: Cytokinin controls the cellcycle at mitosis by stimulating the tyrosine dephosphorylationand activation of p34cdc2-like H1 histone kinase. Planta 200:2–12 (1996).





























