Embed Size (px)
Citation preview

THERAPEUTICSTRATEGIES
DRUG DISCOVERY
TODAY
Drug Discovery Today: Therapeutic Strategies Vol. 1, No. 4 2004
Editors-in-Chief
Raymond Baker – formerly University of Southampton, UK and Merck Sharp & Dohme, UK
Eliot Ohlstein – GlaxoSmithKline, USA
Infectious diseases
Finding the gems using genomicdiscovery: antibacterial drug discoverystrategies – the successes and thechallengesPan F. Chan, David J. Holmes, David J. Payne*Department of Microbiology, Microbial, Musculoskeletal and Proliferative Diseases, Center of Excellence in Drug Discovery, Anti-infectives Research (UP1345),
GlaxoSmithKline, 1250 South Collegeville Road, Collegeville, PA 19426, USA
Since the first bacterial genome was sequenced (1995)
many companies have invested in a variety of antibiotic
discovery strategies but few novel-acting antibacterial
agents have reached human trials. This is not through
lack of trying, as from literature reports alone >125
published studies on high-throughput screens of >60
different novel targets (from 34 companies) have been
identified. Post-genomics should be a new golden era of
antibiotics but there are substantial challenges of deli-
vering much needed, novel-acting antibacterials that
will be discussed.
*Corresponding author. (D.J. Payne) [email protected]
1740-6773/$ � 2004 Elsevier Ltd. All rights reserved. DOI: 10.1016/j.ddstr.2004.11.003
Section Editor:Gary Woodnutt – Diversa Corp., San Diego, CA, USA
The explosion in the bacterial genomics area in the past decade has
been remarkable, but there appears to have been little reward for all of
this effort as judged by the relative lack of new antibacterial agentsentering development. Chan et al. review these developments and
suggest that, as far as target identification is concerned, the use ofgenomics has provided us with a wealth of potentially attractive
opportunities, and failure to consolidate on these reflect on otherdeficits/difficulties in the antibacterial drug discovery process.
Furthermore, the knowledge gained during this period has allowedconsiderable advances in genetic tools that can assist in tracking
structure–activity relationships in the whole cell, and should enable thedissection of compound mode of action. In addition, these advances
have been pivotal in understanding why certain promising candidatesare less viable. Thus, the bacterial genomics era is far from being over
and might actually have just begun to be applied appropriately.
Introduction
In the past 20 years, only two completely novel classes of
antibiotics have reached the market to combat the clinical
threat of multidrug-resistant bacteria (Infectious Disease
Society of America, http://www.idsociety.org/Template.
cfm?Section=AntimicrobialsTemplate=/ContentManagement/
ContentDisplay.cfmContentID=9718). Furthermore, there is
apparently only one novel-acting, systemic agent in the
antibiotic development pipeline [1]. Despite the medical
need for new antibiotics [2] there has been a trend for some
large pharmaceutical companies to downsize or abandon
their antibacterial drug discovery efforts [3]. Reasons for this
withdrawal are complex and multi-factorial but one very
significant factor is that in retrospect, it is significantly more
challenging to deliver novel classes of antibiotics than was
predicted nine years ago when the first bacterial genome was
completed. In this review, we discuss three strategies for
finding new antibiotics – (i) target-based screening (ii) anti-
bacterial whole-cell screening and (iii) structural genomics –
and examine how genomics enable or facilitate each of these
strategies. In addition, we discuss the significant challenges
that compromise the success of each approach.
www.drugdiscoverytoday.com 519

Drug Discovery Today: Therapeutic Strategies | Infectious diseases Vol. 1, No. 4 2004
Glossary
Chemical diversity: different pharmacophores and scaffolds of mole-
cules in a compound-collection. Typically, a compound library bank that is
used in HTS for inhibitors contain a large, chemically diverse range of
compounds, synthesized in combinatorial libraries or gathered from
natural-sources. Lipinski has applied computational and experimental
physical-property measurements to propose the ‘‘drug-like properties’’
of small-molecule inhibitors [66].
High-throughput screens (HTS): automated and rapid method for
identifying compounds that inhibits enzyme activity or bacterial growth.
Assay usually performed in a 96-, 384- or 1536- well-plate format and
screened against a large compound-collection.
Mode-of-action (MOA): mechanism by which a drug exerts its killing
or inhibitory effect on bacteria.
Targets: proteins that are inhibited by antibacterial agents. Microbial
drug targets are typically essential for bacteria to survive, hence, its
inhibition results in bacterial death or reduced ability to cause an
infection.
Target-based screening
To illustrate the impact of genomics on antibiotic drug dis-
covery, a literature search was conducted to identify the
number of published, antibacterial, HIGH-THROUGHPUT SCREENS
(HTS; see Glossary) reported since the release of the first
microbial genome. Fig. 1 summarizes our findings. This graph
shows there has been a clear increase in the number and
diversity of HTS of antibacterial TARGETS (see Glossary) devel-
oped for finding novel-acting antibiotics between 1995 and
2004 that probably reflect the industry’s investment in a
genomics-driven strategy. During this period, 127 antimicro-
bial screens were reportedly run against almost 70 different
bacterial targets, of which about 60 can be considered as
novel targets. However, this survey is clearly a significant
underestimate of the number of antibacterial screens run by
the pharmaceutical industry since much of that information
is proprietary.
Genomics has delivered a large number of diverse and well-
validated, novel drug targets for target-based screening for
novel-acting inhibitors [4–6] (Figs. 1, 2). Although lead anti-
bacterial compounds have been reported to several novel
classes of targets such as those to two-component signal
transduction systems (TCSTS) [4,7], coenzyme metabolism
[8], cell division [9], protein secretion [10] and novel DNA
replication proteins [11,12], only inhibitors to polypeptide
deformylase (PDF) have reached Phase I trials thus far.
Furthermore, to our knowledge, new antibacterial series iden-
tified via this strategy with in vivo efficacy are limited and
have only been reported for four novel targets namely PDF
[13,14], LpxC [15], FabI [16] and aminoacyl-tRNA synthetases
[17,18] and these will be discussed in the following sections.
In addition, rapid target identification in different patho-
genic bacteria has facilitated the development of HTS to well-
validated, biosynthetic pathways that are under-exploited as
antibacterial targets [19]. The impact of genomics on path-
520 www.drugdiscoverytoday.com
way-based screens to identify novel antibacterial agents will
also be discussed.
Polypeptide deformylase (PDF) target
PDF encoded by def (GenBank accession number X77800), is a
well-validated antibacterial target for drug discovery [6,13,
14]. Vircuron Pharmaceuticals (http://www.vicuron.com/)
in collaboration with Novartis (http://www.novartis.com/)
discovered a series of potent broad-spectrum PDF inhibitors,
of which LBM-415 is currently in Phase I clinical trials for oral
treatment of community-acquired pneumoniae [14,20].
Independently, Oscient Pharmaceuticals (http://www.os-
cient.com/) are working on another series of PDF inhibitors
that were originally identified by HTS of specific chemical
libraries targeted against metalloproteases [13]. Following
structural optimization, BB-83698 was the first compound
of its class to enter clinical trials [21]. Recent reports, how-
ever, suggest that second generation compounds with
improved Haemophilus influenzae activity may be developed
[22]. Numerous pharmaceutical companies have published
studies on PDF as an antimicrobial target [23-28]. Genomics
has also helped address the issue of development of in vitro
resistance to anti-PDF compounds. In the genome sequences
of some pathogens, the formyltransferase (fmt) (GenBank
accession number X63666) gene has been found that
bypasses the formylation pathway though these resistant
mutants show reduced virulence in vivo [15,28]. Indeed,
PDF inhibitors might prove to be the first novel class of
antibiotic to reach the market as a result of target-screening
approaches in the post-genomics era.
LpxC target
Although inhibitors with in vivo efficacy have been described
that target LpxC (GenBank accession number AAC73207), an
essential metalloenzyme involved in lipid A biosyntheis in
Gram-negative pathogens, these compounds have not pro-
gressed beyond pre-clinical stage [15].
Fatty acid biosyntheis (FAB) targets
Comparative genomics has identified all the enzymes
involved in FAB pathway in bacteria and some of the targets
have been validated with antibacterial compounds [29].
Enoyl-ACP reductase encoded by fabI (GenBank accession
number AF197058) has probably yielded the most promising
leads to date with this target screened by several companies
[16,29,30]. Leads originating from HTS have been described
by GlaxoSmithKline (http://www.gsk.com/) with efficacy in
animal infection models and exquisite antibacterial activity
against multi-resistant strains of Staphylococcus aureus (MIC90
of <0.06 mg/ml) [16]. Furthermore, Genome Therapeutic
Corporation (http://www.oscient.com/) describe a new series
of FabI inhibitors with IC50 of 4 mM and MIC of 2 mg/ml
against S. aureus, adding a new chemical scaffold for devel-
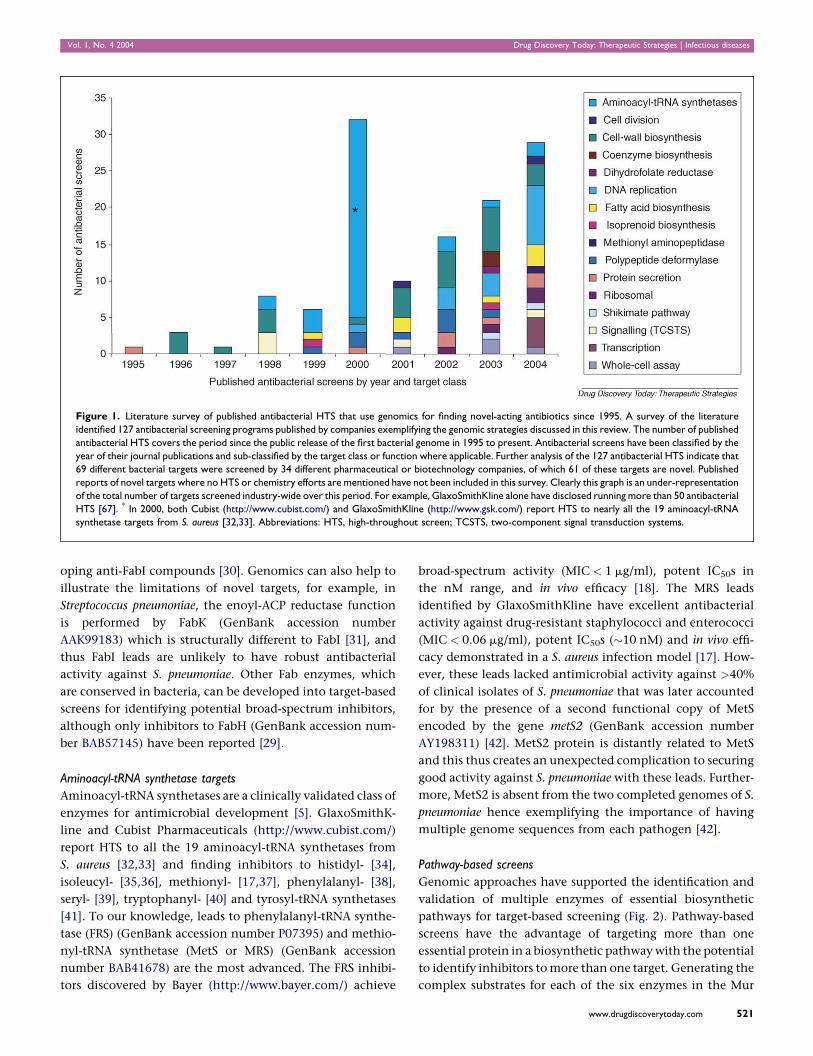
Vol. 1, No. 4 2004 Drug Discovery Today: Therapeutic Strategies | Infectious diseases
Figure 1. Literature survey of published antibacterial HTS that use genomics for finding novel-acting antibiotics since 1995. A survey of the literature
identified 127 antibacterial screening programs published by companies exemplifying the genomic strategies discussed in this review. The number of published
antibacterial HTS covers the period since the public release of the first bacterial genome in 1995 to present. Antibacterial screens have been classified by the
year of their journal publications and sub-classified by the target class or function where applicable. Further analysis of the 127 antibacterial HTS indicate that
69 different bacterial targets were screened by 34 different pharmaceutical or biotechnology companies, of which 61 of these targets are novel. Published
reports of novel targets where no HTS or chemistry efforts are mentioned have not been included in this survey. Clearly this graph is an under-representation
of the total number of targets screened industry-wide over this period. For example, GlaxoSmithKline alone have disclosed running more than 50 antibacterial
HTS [67]. * In 2000, both Cubist (http://www.cubist.com/) and GlaxoSmithKline (http://www.gsk.com/) report HTS to nearly all the 19 aminoacyl-tRNA
synthetase targets from S. aureus [32,33]. Abbreviations: HTS, high-throughout screen; TCSTS, two-component signal transduction systems.
oping anti-FabI compounds [30]. Genomics can also help to
illustrate the limitations of novel targets, for example, in
Streptococcus pneumoniae, the enoyl-ACP reductase function
is performed by FabK (GenBank accession number
AAK99183) which is structurally different to FabI [31], and
thus FabI leads are unlikely to have robust antibacterial
activity against S. pneumoniae. Other Fab enzymes, which
are conserved in bacteria, can be developed into target-based
screens for identifying potential broad-spectrum inhibitors,
although only inhibitors to FabH (GenBank accession num-
ber BAB57145) have been reported [29].
Aminoacyl-tRNA synthetase targets
Aminoacyl-tRNA synthetases are a clinically validated class of
enzymes for antimicrobial development [5]. GlaxoSmithK-
line and Cubist Pharmaceuticals (http://www.cubist.com/)
report HTS to all the 19 aminoacyl-tRNA synthetases from
S. aureus [32,33] and finding inhibitors to histidyl- [34],
isoleucyl- [35,36], methionyl- [17,37], phenylalanyl- [38],
seryl- [39], tryptophanyl- [40] and tyrosyl-tRNA synthetases
[41]. To our knowledge, leads to phenylalanyl-tRNA synthe-
tase (FRS) (GenBank accession number P07395) and methio-
nyl-tRNA synthetase (MetS or MRS) (GenBank accession
number BAB41678) are the most advanced. The FRS inhibi-
tors discovered by Bayer (http://www.bayer.com/) achieve
broad-spectrum activity (MIC < 1 mg/ml), potent IC50s in
the nM range, and in vivo efficacy [18]. The MRS leads
identified by GlaxoSmithKline have excellent antibacterial
activity against drug-resistant staphylococci and enterococci
(MIC < 0.06 mg/ml), potent IC50s (�10 nM) and in vivo effi-
cacy demonstrated in a S. aureus infection model [17]. How-
ever, these leads lacked antimicrobial activity against >40%
of clinical isolates of S. pneumoniae that was later accounted
for by the presence of a second functional copy of MetS
encoded by the gene metS2 (GenBank accession number
AY198311) [42]. MetS2 protein is distantly related to MetS
and this thus creates an unexpected complication to securing
good activity against S. pneumoniae with these leads. Further-
more, MetS2 is absent from the two completed genomes of S.
pneumoniae hence exemplifying the importance of having
multiple genome sequences from each pathogen [42].
Pathway-based screens
Genomic approaches have supported the identification and
validation of multiple enzymes of essential biosynthetic
pathways for target-based screening (Fig. 2). Pathway-based
screens have the advantage of targeting more than one
essential protein in a biosynthetic pathway with the potential
to identify inhibitors to more than one target. Generating the
complex substrates for each of the six enzymes in the Mur
www.drugdiscoverytoday.com 521

Drug Discovery Today: Therapeutic Strategies | Infectious diseases Vol. 1, No. 4 2004
Figure 2. Integrated strategies for antibiotic drug discovery using genomics. Antibacterial drug discovery strategies that use genomics for finding novel-acting
antibiotics can be divided into three areas: target-based screening, antibacterial whole-cell screening and structural genomics/screening. Once a hit molecule is
identified from screening, the target of the molecule is determined (whole-cell screen) or confirmed (target-based screen) using several MOA assays/tools. A
chemistry lead-optimization program is initiated to synthesise chemical derivatives of the hit compound with the aim of incorporating improved antibacterial
activity and other desirable clinical properties to the molecule. This flowchart only shows genomic tools used for MOA/SAR tracking, and does not include
biochemical and genetic assays reviewed elsewhere [5].
Abbreviations: HT, high-throughput; HTS, high-throughout screen; MOA, mode-of-action; OE, overexpression; ORF, open reading frame; SAR, structure–
activity relationship.
pathway involved in cell wall biosynthesis (MurA-F) (Gen-
Bank accession numbers AAC76221, AAC76950, AAC73202,
AAC73199, AAC73196, AAC73197) has likely led to the
underexploitation of the Mur enzymes as targets, while a
pathway assay circumvents the need to generate these sub-
strates [19]. Similarly, high-throughput pathway screens for
inhibitors of multiple membrane-associated proteins of pep-
tidoglycan biosynthesis that include MraY (GenBank acces-
sion number AAC73198), MurG (GenBank accession number
AAC73201) and penicillin-binding proteins (PBPs) have been
reported [43–45].
Another example of a pathway-based antibacterial screen is
an assay that targets the shikimate (Aro) pathway. The Aro
pathway is a multi-enzyme, essential process in the biosynth-
esis of chorismate, ubiquinone and folate in diverse numbers
of bacteria. An optimized, homogeneous assay of all four
essential enzymes, AroB, AroD, AroE and AroK (GenBank
accession numbers AAL00037, AAL00039, AAL00038,
AAL00032) has been developed for HTS (Goryanin I. et al,
Glaxo SmithKline, submitted for publication).
Although the essential proteins of the peptidoglycan bio-
synthesis pathway have been widely screened in pathway
assays by several groups, to date there have been no reports of
522 www.drugdiscoverytoday.com
sustainable leads from these types of screens progressing to
clinical development.
In summary, a large number and variety of novel geno-
mics-derived targets have been screened by many companies
for finding new antibiotics but the approach has delivered
relatively few new chemical series with in vivo efficacy. It is
likely that the main causes of this are the lower than expected
hit rate at HTS and that the chemical optimization of HTS hits
is far more challenging than making new derivatives of
established classes of antibiotics. In addition, progressing
entirely novel antibacterial targets can be further compli-
cated by ‘‘novel target surprises’’, such as the discovery of
a second MRS in S. pneumoniae.
Antibacterial whole-cell screening
A second strategy for identifying potential novel-acting anti-
bacterials is the HTS of large combinatorial chemical libraries
and new natural products for their ability to inhibit growth of
whole bacterial cells. Most currently marketed antibiotics
were originally discovered by random screening of com-
pounds from natural sources. Once hits have been identified,
there follows the complex task of identifying the molecular
target of the antibacterial hit as well as ensuring that the

Vol. 1, No. 4 2004 Drug Discovery Today: Therapeutic Strategies | Infectious diseases
activity is selective using biochemical and genetic tools [5].
Recent developments in expression technologies that have
facilitated MODE-OF-ACTION MOA; see Glossary) studies of com-
pounds of unknown mechanism are discussed below (Fig. 2).
Regulated strains as MOA tools
Overexpression-gene and antisense-RNA libraries have been
constructed that cover almost every single open reading
frame (ORF) in the S. aureus genome [46,47]. In theory, these
libraries enable hundreds of targets to be rapidly screened as
the potential molecular targets of whole-cell assay hits [47].
This approach has been validated for established antibiotics
of known inhibitory mechanisms but there are no reports
where this approach has identified the MOA of an unchar-
acterized antibacterial.
Proteome- and genome-profiling as MOA tools
Expression profiling can also be potentially powerful tools for
antibacterial MOA studies [48]. One successful application of
gene arrays has been the confirmation of a compound, ori-
ginally identified by Abbott (http://www.abbott.com/) from a
coupled transcription–translation HTS, as a ribosomal inhi-
bitor by comparative profiling [49]. Recently, Bayer has used
expression profiling to elucidate the MOA of two whole-cell
hits as acetyl-CoA carboxylase, AccA (GenBank accession
number BAB57862) and peptidyltransfer inhibitors, respec-
tively. [50,51]. These are some of the firsts examples of
proteome- and genome-profiling being used to identify the
MOA of an antimicrobial agent of unknown mechanism [48].
Whole-cell screening strategies targeting known MOAs
Whole-cell screens specific for targets have also been devel-
oped [52]. An advantage of this strategy is that a particular
enzyme is screened in situ so that the characteristics of an
antibacterial (for example, cell penetration) are already a
prerequisite to obtaining a hit. Screening of an Escherichia
coli strain with downregulated MurA (GenBank accession
number AAC76221) successfully identified a series of MurA
inhibitors that exemplifies the potential of the approach [53].
Another method exploits whole-cell pathway reporter strains
containing antibiotic-responsive promoters often of stress-
inducible genes, fused to a sensitive reporter gene [54,55]. A
Bacillus subtilis strain carrying a FabH reporter screened
against a chemical bank, successfully identified hits by induc-
tion of the reporter that showed weak antibacterial activity
against S. aureus, and specific inhibition of fatty acid bio-
synthesis [54].
Overall, whole-cell screening has some key advantages over
enzyme-based screening for finding novel antibacterial
agents (Table 1). However, success at defining MOA of hits
to enable rational SAR programs has been low, which might
be a function of the diversity of the compound-collection
screened. Using a whole-cell screening approach, GlaxoS-
mithKline have discovered a class of benzylidenethiazoli-
diones with Gram-positive activity though the molecular
target has yet to be reported [56], while Pharmacia (http://
www.pfizer.com/) identified a translation elongation factor-
Tu inhibitor whose MOA was elucidated using ligand-affinity
fishing and macromolecular synthesis [57]. Bayer has demon-
strated the use of expression analysis to identify novel inhi-
bitors of acetyl-CoA carboxylase and peptidyltransferase
activities [48,50,51]. Many genomic tools are now available
to study MOA of novel agents. In addition to identifying
antibacterial targets, these MOA tools are also valuable for
confirming the activity of target-based screening hits and
allowing rapid monitoring of SAR to ensure that the target
does not shift during a lead-optimization program (Fig. 2).
Structural genomics
To find novel-acting antibacterial agents, structure-based
drug design is a key strategy widely employed to assist che-
mical optimization of leads identified from antibacterial
target-based screens (Table 1). Genomics has accelerated
the identification of targets and enabled high-throughput
cloning, expression and purification of novel target proteins
for crystallisation studies. 3D structures can be exploited by
virtual screening of compound databases using docking tech-
niques that provide hits in addition to those predicted by in
silico modelling approaches [58]. Finally, structural genomics
can facilitate studies to identify the function of essential
proteins that have no significant homology with previously
characterized proteins and termed ‘‘proteins of unknown
function’’ (Fig. 2).
Structural genomics of novel targets
Extensive genomic and structural information on novel tar-
gets help characterize the active sites of targets across differ-
ent pathogens, and enable robust and rational, lead-
optimization programs to improve the inhibitory potency,
and potentially expand the antibacterial spectrum of lead-
chemical series. PDF is a good example to illustrate the impact
of structural genomics on a lead-optimization program. Sev-
eral co-crystal structures of the PDF target and anti-PDF lead
compounds have been described that have helped guide
design of potent inhibitors [13,14,24,25,28]. Furthermore,
at the 44th Interscience Conference on Antimicrobial Agents
and Chemotherapy (ICAAC) a new class of broad-spectrum
inhibitor that dual targets topoisomerase IV ParE (GenBank
accession number AAC76066) and DNA gyrase GyrB (Gen-
Bank accession number AAT48201) subunits was reported by
Vertex Pharmaceuticals (http://www.vrtx.com/) that also
achieves in vivo efficacy in animal models [59].
Structural genomics of targets of unknown function
Structural genomics is particularly useful for identifying the
function of novel protein of unknown function [60]. At
www.drugdiscoverytoday.com 523
Dru
gD
iscovery
Today:
Therap
eutic
Strategies|
Infectio
us
diseases
Vol.
1,N
o.4
2004
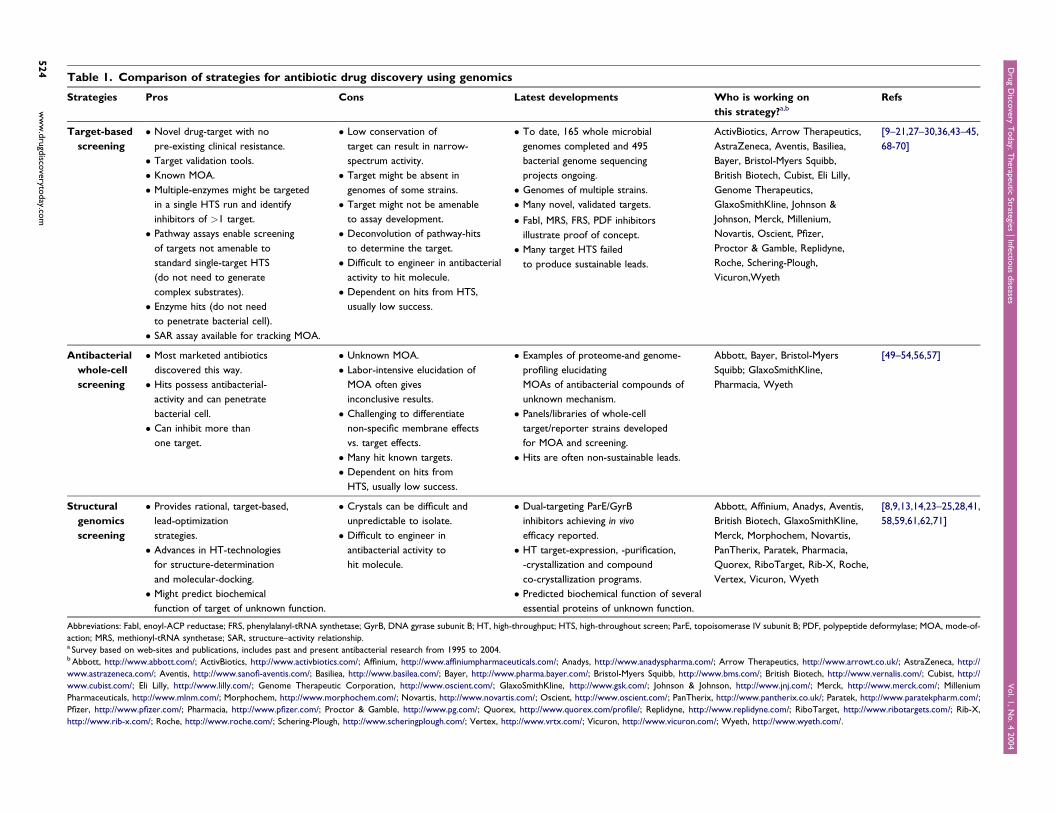
Table 1. Comparison of strategies for antibiotic drug discovery using genomics
Strategies Pros Cons Latest developments Who is working on
this strategy?a,b
Refs
Target-based
screening
� Novel drug-target with no
pre-existing clinical resistance.
� Target validation tools.
� Known MOA.
� Multiple-enzymes might be targeted
in a single HTS run and identify
inhibitors of >1 target.
� Pathway assays enable screening
of targets not amenable to
standard single-target HTS
(do not need to generate
complex substrates).
� Enzyme hits (do not need
to penetrate bacterial cell).
� SAR assay available for tracking MOA.
� Low conservation of
target can result in narrow-
spectrum activity.
� To date, 165 whole microbial
genomes completed and 495
bacterial genome sequencing
projects ongoing.
ActivBiotics, Arrow Therapeutics,
AstraZeneca, Aventis, Basiliea,
Bayer, Bristol-Myers Squibb,
British Biotech, Cubist, Eli Lilly,
Genome Therapeutics,
GlaxoSmithKline, Johnson &
Johnson, Merck, Millenium,
Novartis, Oscient, Pfizer,
Proctor & Gamble, Replidyne,
Roche, Schering-Plough,
Vicuron,Wyeth
[9–21,27–30,36,43–45,
68-70]
� Target might be absent in
genomes of some strains. � Genomes of multiple strains.
� Target might not be amenable
to assay development.
� Many novel, validated targets.
� Deconvolution of pathway-hits
to determine the target.
� FabI, MRS, FRS, PDF inhibitors
illustrate proof of concept.
� Difficult to engineer in antibacterial
activity to hit molecule.
� Many target HTS failed
to produce sustainable leads.
� Dependent on hits from HTS,
usually low success.
Antibacterial
whole-cell
screening
� Most marketed antibiotics
discovered this way.
� Hits possess antibacterial-
activity and can penetrate
bacterial cell.
� Can inhibit more than
one target.
� Unknown MOA.
� Labor-intensive elucidation of
MOA often gives
inconclusive results.
� Challenging to differentiate
non-specific membrane effects
vs. target effects.
� Many hit known targets.
� Dependent on hits from
HTS, usually low success.
� Examples of proteome-and genome-
profiling elucidating
MOAs of antibacterial compounds of
unknown mechanism.
Abbott, Bayer, Bristol-Myers
Squibb; GlaxoSmithKline,
Pharmacia, Wyeth
[49–54,56,57]
� Panels/libraries of whole-cell
target/reporter strains developed
for MOA and screening.
� Hits are often non-sustainable leads.
Structural
genomics
screening
� Provides rational, target-based,
lead-optimization
strategies.
� Crystals can be difficult and
unpredictable to isolate.
� Dual-targeting ParE/GyrB
inhibitors achieving in vivo
efficacy reported.
Abbott, Affinium, Anadys, Aventis,
British Biotech, GlaxoSmithKline,
Merck, Morphochem, Novartis,
PanTherix, Paratek, Pharmacia,
Quorex, RiboTarget, Rib-X, Roche,
Vertex, Vicuron, Wyeth
[8,9,13,14,23–25,28,41,
58,59,61,62,71]
� Advances in HT-technologies
for structure-determination
and molecular-docking.
� Difficult to engineer in
antibacterial activity to
hit molecule.
� HT target-expression, -purification,
-crystallization and compound
co-crystallization programs.
� Might predict biochemical
function of target of unknown function.
� Predicted biochemical function of several
essential proteins of unknown function.
Abbreviations: FabI, enoyl-ACP reductase; FRS, phenylalanyl-tRNA synthetase; GyrB, DNA gyrase subunit B; HT, high-throughput; HTS, high-throughout screen; ParE, topoisomerase IV subunit B; PDF, polypeptide deformylase; MOA, mode-of-
action; MRS, methionyl-tRNA synthetase; SAR, structure–activity relationship.a Survey based on web-sites and publications, includes past and present antibacterial research from 1995 to 2004.b Abbott, http://www.abbott.com/; ActivBiotics, http://www.activbiotics.com/; Affinium, http://www.affiniumpharmaceuticals.com/; Anadys, http://www.anadyspharma.com/; Arrow Therapeutics, http://www.arrowt.co.uk/; AstraZeneca, http://
www.astrazeneca.com/; Aventis, http://www.sanofi-aventis.com/; Basiliea, http://www.basilea.com/; Bayer, http://www.pharma.bayer.com/; Bristol-Myers Squibb, http://www.bms.com/; British Biotech, http://www.vernalis.com/; Cubist, http://
www.cubist.com/; Eli Lilly, http://www.lilly.com/; Genome Therapeutic Corporation, http://www.oscient.com/; GlaxoSmithKline, http://www.gsk.com/; Johnson & Johnson, http://www.jnj.com/; Merck, http://www.merck.com/; Millenium
Pharmaceuticals, http://www.mlnm.com/; Morphochem, http://www.morphochem.com/; Novartis, http://www.novartis.com/; Oscient, http://www.oscient.com/; PanTherix, http://www.pantherix.co.uk/; Paratek, http://www.paratekpharm.com/;
Pfizer, http://www.pfizer.com/; Pharmacia, http://www.pfizer.com/; Proctor & Gamble, http://www.pg.com/; Quorex, http://www.quorex.com/profile/; Replidyne, http://www.replidyne.com/; RiboTarget, http://www.ribotargets.com/; Rib-X,
http://www.rib-x.com/; Roche, http://www.roche.com/; Schering-Plough, http://www.scheringplough.com/; Vertex, http://www.vrtx.com/; Vicuron, http://www.vicuron.com/; Wyeth, http://www.wyeth.com/.
524
ww
w.d
rugd
iscoveryto
day.co
m

Vol. 1, No. 4 2004 Drug Discovery Today: Therapeutic Strategies | Infectious diseases
Links
� Infectious Diseases Society of America (IDSA) 2004 report on ‘Bad
Bugs, No Drugs’, http://www.idsociety.org/
� IDSA/PhRma/FDA Working Group Meeting, 19th November
2002 on drug development for resistant-pathogens sponsored by
the Center for Drug Evaluation and Research, Food and Drug
Administration (FDA), http://www.fda.gov/cder/present/idsaphrma/
default.htm
� The Institute for Genomic Research (TIGR) Pathogen Functional
Genomics Resource sponsored by the National Institute of Allergy
and Infectious Diseases (NIAID), http://pfgrc.tigr.org/
� Genomes Online Database. An updated, comprehensive resource of
completed and ongoing microbial genomes projects, http://www.
genomesonline.org/
� Mycobacterium Tuberculosis Structural Consortium, http://www.doe-
mbi.ucla.edu/TB/
� Structure 2 Function Project is a structural genomics project that
aims to solve structures of poorly characterised Haemophilus influ-
enzae proteins by using X-ray crystallography and protein NMR
techniques, http://s2f.carb.nist.gov/.
� The ExPASy (Expert Protein Analysis System) server of the Swiss
Institute of Bioinformatics dedicated to the analysis of protein
sequences and structures, http://us.expasy.org/
Abbott, crystallization programs have solved the structure
and determined the functions of several essential genes of
unknown function from S. pneumoniae [61]. The approach by
Aventis (http://www.sanofi-aventis.com/) involves the high-
throughput crystallization and X-ray elucidation of over 200
conserved microbial targets of unknown function that so far
has inferred the function of seven of these proteins [62]. In
addition, several consortiums are applying structural geno-
mics to systematically solve the structures of proteins of
uncharacterised function in pathogens H. influenzae and
Mycobacterium tuberculosis [63,64]. Assignments of the bio-
chemical function of these proteins by these approaches
certainly help our understanding of the genome. However,
progression of such targets as well as essential, broad-spec-
trum targets of unknown functions [65], are dependent on
identifying assayable functions that can be employed in HTS,
which adds an additional complexity to exploitation of the
unknowns.
Conclusion
The application of genomics has led to an unprecedented
number of novel, validated targets for screening of com-
pound collections to identify new classes of antibiotics. A
survey of the literature has identified more than 125 pub-
lished studies of antibacterial HTS in the nine years since the
completion of the first bacterial genome. These HTS have
targeted more than 60 different novel protein targets
screened by 34 different companies (Fig. 1). However, tar-
get-based screening has been significantly less successful in
finding novel-acting antibiotics than expected. From our
experience and others, antibacterial targets appear to have
very low hit rate and hit-to-lead success rate which compro-
mise the approach at a very early and fundamental step.
Furthermore, this strategy is frustrated by the fact that fre-
quently hits lack whole-cell antibacterial activity and no
rational SAR processes exist to improve this property. Never-
theless, there are successes such as FabI, FRS and MRS inhi-
bitors achieving promising in vivo efficacy [16–18]. However,
MRS illustrates one of the limitations of the novel target-
based strategy with the presence of a second MRS severely
compromising the broader clinical utility of these leads [42].
Target screens for PDF inhibitors by Vircuron/Novartis and
Oscient have also led to promising lead molecules, two
progressing to Phase I trials [20,21]. In contrast, there are
many more examples of enzyme-based screens of antibacter-
ial targets in the industry that have failed to deliver high-
quality antibacterial leads.
Antibacterial whole-cell screening for novel antibiotics has
the major benefit that antibacterial activity has already been
incorporated into the hit molecule. Genomics has facilitated
this approach in providing new technologies for MOA hunt-
ing studies. However, despite the availability of these power-
ful tools such studies are challenging and rarely definitive.
However, there are at least four literature examples of leads
from this approach [50,51,56,57].
Structural genomics plays a significant role in facilitating
the other antibacterial discovery strategies and there is a
promising report of a novel, dual-target inhibitor to GyrB
and ParE, predominately derived from a structural genomics
strategy that achieve broad-spectrum and in vivo activities
[59].
At present, the investment in genomics for antibacterial
research has resulted in no new genomics-derived drugs on
the market from either target-based or whole-cell screens.
However, genomics has provided numerous new antibacter-
ial target strategies and provided tools to track and identify
the MOA of antibacterial hits. One of the reasons for the low
success rate of antibacterial target screening probably lies
with the CHEMICAL DIVERSITY (see Glossary) of the compounds
being screened against these targets. There is some evidence
that chemical diversity that favors antibiotic screening can be
unique and different from the ‘‘drug-like’’ diversity defined
by Lipinski [66] that is more suited to other therapeutic areas
[4]. Consequently, one approach to increase success at anti-
bacterial HTS would be to generate new ‘‘antibacterial tar-
geted’’ diversity and instigate broader screening of natural
products.
Many companies have invested in genomics-based anti-
bacterial discovery strategies to find new classes of antibio-
tics. However, the output from this investment in terms of
new antibacterial agents in development has been disap-
pointingly low. This does not appear to be through a lack
of trying but more of a function of the unexpected complex-
ities and challenges of delivering new antibiotics targeting
www.drugdiscoverytoday.com 525

Drug Discovery Today: Therapeutic Strategies | Infectious diseases Vol. 1, No. 4 2004
novel targets. This factor might have played a contributing
role to the withdrawal of some companies from antibacterial
research just when the clinical need for new antibiotics is
beginning to peak. Although the tools of antibacterial
research have now reached a level of sophistication unim-
aginable several years ago, it is clear that new research stra-
tegies and continued investment to overcome the challenges
in antibacterial research are required.
Outstanding issues
� When will the antibacterial development pipeline give us confidence
that we have the drugs to effectively treat bacterial infections in the
10–20 year time frame?
� How can the success rate of target- and whole-cell antibacterial
screening approaches be increased?
� Do the chemical libraries assayed in HTS by pharmaceutical compa-
nies contain adequate diversity to include compounds with the
complex requirements of an antibacterial agent? If not, can effective
‘‘antibacterial-targeted’’ diversity be created?
� Will compound penetration into bacteria ever be understood to the
extent that rational SAR processes will exist to improve antibacterial
activity?
References1 Bush, K. et al. (2004) Taking inventory: antibacterial agents currently at or
beyond Phase 1. Curr. Opin. Microbiol. 7, 466–476
2 Walsh, F.M. et al. (2004) Microbiology and drug resistance mechanisms of
fully resistant pathogens. Curr. Opin. Microbiol. 7, 439–444
3 Projan, S.J. (2003) Why is big Pharma getting out of antibacterial drug
discovery? Curr. Opin. Microbiol. 6, 427–430
4 Chan, P.F. et al. (2002) Novel antibacterials: a genomics approach to drug
discovery. Curr. Drug Targets Infect. Disord. 2, 291–308
5 Payne, D.J. et al. (2004) Genomic approaches to antibacterial discovery.
Methods Mol. Biol. 266, 231–259
6 Miesel, L. et al. (2003) Genetic strategies for antibacterial drug discovery.
Nat. Rev. Genet. 4, 442–456
7 Stephenson, K. et al. (2004) Developing inhibitors to selectively target
two-component and phosphorelay signal transduction systems of patho-
genic microorganisms. Curr. Med. Chem. 11, 765–773
8 Zhao, L. et al. (2003) Inhibitors of phosphopantetheine adenylyltransfer-
ase. Eur. J. Med. Chem. 38, 345–349
9 Jennings, L.D. et al. (2004) Design and synthesis of indolo[2,3-a]quino-
lizin-7-one inhibitors of the ZipA–FtsZ interaction. Bioorg. Med. Chem.
Lett. 14, 1427–1431
10 Kulanthaivel, P. et al. (2004) Novel lipoglycopeptides as inhibitors of
bacterial signal peptidase I. J. Biol. Chem. 279, 36250–36258
11 Zhang, Y. et al. (2002) Homogenous assays for Escherichia coli DnaB-
stimulated DnaG primase and DnaB helicase and their use in screening for
chemical inhibitors. Anal. Biochem. 304, 174–179
12 Brotz-Oesterhelt, H. et al. (2003) Specific and potent inhibition of NAD+-
dependent DNA ligase by pyridochromanones. J. Biol. Chem. 278, 39435–
39442
13 Clements, J.M. et al. (2001) Antibiotic activity and characterization of BB-
3497, a novel peptide deformylase inhibitor. Antimicrob. Agents Che-
mother. 45, 563–570
14 Hackbarth, C.J. et al. (2002) N-alkyl urea hydroxamic acids as a new class
of peptide deformylase inhibitors with antibacterial activity. Antimicrob.
Agents Chemother. 46, 2752–2764
15 White, R.J. et al. (2003) Targeting metalloenzymes: a strategy that works.
Curr. Opin. Pharmacol. 3, 502–507
16 Payne, D.J. et al. (2002) Discovery of a novel and potent class of FabI-
directed antibacterial agents. Antimicrob. Agents Chemother. 46, 3118–3124
526 www.drugdiscoverytoday.com
17 Jarvest, R.L. et al. (2000) Nanomolar inhibitors of Staphylococcus aureus
methionyl tRNA synthetase with potent antibacterial activity against
Gram-positive pathogens. J. Med. Chem. 45, 1959–1962
18 Beyer, D. et al. (2004) New class of bacterial phenylalanyl-tRNA synthe-
tase inhibitors with high potency and broad-spectrum activity. Antimicrob.
Agents Chemother. 48, 525–532
19 Trias, J. et al. (1999) Mining bacterial cell wall biosynthesis with new
tools: multitarget screens. Drug Resist. Updat. 2, 358–362
20 Jones, R.N. et al. (2004) Antimicrobial spectrum and activity of
NVP PDF-713, a novel peptide deformylase inhibitor, tested against
1,837 recent Gram-positive clinical isolates. Diagn. Microbiol. Infect.
Dis. 49, 63–65
21 Lofland, D. et al. (2004) In vitro antibacterial activity of the peptide
deformylase inhibitor BB-83698. J. Antimicrob. Chemother. 53, 664–
668
22 Barrett, J.F. et al. (2004) Antibacterial drug discovery & development
summit. Expert Opin. Investig. Drugs 13, 715–721
23 Guilloteau, J.P. et al. (2002) The crystal structures of four peptide
deformylases bound to the antibiotic actinonin reveal two distinct types:
a platform for the structure-based design of antibacterial agents. J. Mol.
Biol. 320, 951–962
24 Smith, K.J. et al. (2003) Structural variation and inhibitor binding in
polypeptide deformylase from four different bacterial species. Protein Sci.
12, 349–360
25 Harris, M.S. et al. (2002) Co-crystallization of Staphylococcus aureus
peptide deformylase (PDF) with potent inhibitors. Acta Crystallogr. D:
Biol. Crystallogr. 58, 2153–2156
26 Green, B.G. et al. (2000) Inhibition of bacterial peptide deformylase by
biaryl acid analogs. Arch. Biochem. Biophys. 375, 355–358
27 Wabnitz, P.A. et al. (2002) Drug screening of pharmaceutical discovery
compounds by micro-size exclusion chromatography/mass spectrometry.
Rapid Commun. Mass Spectrom. 16, 85–91
28 Apfel, C.M. et al. (2001) Peptide deformylase as an antibacterial drug
target: target validation and resistance development. Antimicrob. Agents
Chemother. 45, 1058–1064
29 Payne, D.J. (2004) The potential of bacterial fatty acid biosynthetic
enzymes as a source of novel antibacterial agents. Drug News Perspect.
17, 187–194
30 Ling, L.L. et al. (2004) Identification and characterization of inhibitors of
bacterial enoyl-acyl carrier protein reductase. Antimicrob. Agents Che-
mother. 48, 1541–1547
31 Marrakchi, H. et al. (2003) Characterization of Streptococcus pneumoniae
enoyl-(acyl-carrier protein) reductase (FabK). Biochem. J. 370, 1055–1062
32 Macarron, R. et al. (2000) A homogeneous method to measure aminoacyl-
tRNA synthetase aminoacylation activity using scintillation proximity
assay technology. Anal. Biochem. 284, 183–190
33 Gallant, P. et al. (2000) The identification of quality antibacterial drug
discovery targets: a case study with aminoacyl-tRNA synthetases. Emerg.
Ther. Targets 4, 1–9
34 Qiu, X. et al. (1999) Cooperative structural dynamics and a novel fidelity
mechanism in histidyl-tRNA synthetases. Biochemistry 38, 12296–
12304
35 Stefanska, A.L. et al. (2000) SB-203207 and SB-203208, two novel
isoleucyl tRNA synthetase inhibitors from a Streptomyces sp. I. Fermenta-
tion, isolation and properties. J. Antibiot. (Tokyo) 53, 357–363
36 Tao, J. et al. (2000) Inhibitors of aminoacyl-tRNA synthetases as novel
anti-infectives. Expert Opin. Investig. Drugs 9, 1767–1775
37 Finn, J. et al. (2003) Discovery of a potent and selective series of pyrazole
bacterial methionyl-tRNA synthetase inhibitors. Bioorg. Med. Chem. Lett.
13, 2231–2234
38 Yu, X.Y. et al. (2004) A series of heterocyclic inhibitors of phenylalanyl-
tRNA synthetases with antibacterial activity. Bioorg. Med. Chem. Lett. 14,
1343–1346
39 Stefanska, A.L. et al. (2000) A potent seryl tRNA synthetase inhibitor SB-
217452 isolated from a Streptomyces species. J. Antibiot. (Tokyo) 53,
1346–1353
40 Brown, M.J. et al. (2002) The antimicrobial natural product chuangxin-
mycin and some synthetic analogues are potent and selective inhibitors of

Vol. 1, No. 4 2004 Drug Discovery Today: Therapeutic Strategies | Infectious diseases
bacterial tryptophanyl tRNA synthetase. Bioorg. Med. Chem. Lett. 12,
3171–3174
41 Qiu, X. et al. (2001) Crystal structure of Staphylococcus aureus tyrosyl-
tRNA synthetase in complex with a class of potent and specific inhibitors.
Protein Sci. 10, 2008–2016
42 Gentry, D.R. et al. (2003) Variable sensitivity to bacterial methionyl-tRNA
synthetase inhibitors reveals subpopulations of Streptococcus pneumoniae
with two distinct methionyl-tRNA synthetase genes. Antimicrob. Agents
Chemother. 47, 1784–1789
43 Chandrakala, B. et al. (2004) High-throughput screen for inhibitors of
transglycosylase and/or transpeptidase activities of Escherichia coli peni-
cillin binding protein 1b. Antimicrob. Agents Chemother. 48, 30–40
44 Barbosa, M.D. et al. (2002) A multitarget assay for inhibitors of mem-
brane-associated steps of peptidoglycan biosynthesis. Anal. Biochem. 306,
17–22
45 Zawadzke, L.E. et al. (2003) Targeting the MraY and MurG bacterial
enzymes for antimicrobial therapeutic intervention. Anal. Biochem. 314,
243–252
46 Huang, J. et al. (2004) Novel chromosomally encoded multidrug efflux
transporter MdeA in Staphylococcus aureus. Antimicrob. Agents Che-
mother. 48, 909–917
47 Yin, D. et al. (2004) Identification of antimicrobial targets using a
comprehensive genomic approach. Pharmacogenomics 5, 101–113
48 Freiberg, C. et al. (2004) The impact of transcriptome and proteome
analyses on antibiotic drug discovery. Curr. Opin. Microbiol. 7, 451–459
49 Dandliker, P.J. et al. (2003) Novel antibacterial class. Antimicrob. Agents
Chemother. 47, 3831–3839
50 Bandow, J.E. et al. (2003) Proteomic approach to understanding antibiotic
action. Antimicrob. Agents Chemother. 47, 948–955
51 Freiberg, C. et al. (2004) Identification and characterization of the first
class of potent bacterial acetyl-CoA carboxylase inhibitors with antibac-
terial activity. J. Biol. Chem. 279, 26066–26073
52 Alksne, L.E. et al. (2000) Identification and analysis of bacterial protein
secretion inhibitors utilizing a SecA–LacZ reporter fusion system. Anti-
microb. Agents Chemother. 44, 1418–1427
53 DeVito, J.A. et al. (2002) An array of target-specific screening strains for
antibacterial discovery. Nat. Biotechnol. 20, 478–483
54 Fischer, H.P. et al. (2004) Identification of antibiotic stress-inducible
promoters: a systematic approach to novel pathway-specific reporter assays
for antibacterial drug discovery. Genome Res. 14, 90–98
55 Hutter, B. et al. (2004) Panel of Bacillus subtilis reporter strains indicative
of various modes of action. Antimicrob. Agents Chemother. 48, 2588–2594
56 Heerding, D.A. et al. (2003) New benzylidenethiazolidinediones as anti-
bacterial agents. Bioorg. Med. Chem. Lett. 13, 3771–3773
57 Deibel, M.R. Jr. et al. (2004) Immobilization of a novel antibacterial agent
on solid phase and subsequent isolation of EF-Tu. Bioconjug. Chem. 15,
333–343
58 Knowles, D.J. et al. (2002) The bacterial ribosome, a promising focus for
structure-based drug design. Curr. Opin. Pharmacol. 2, 501–506
59 Grillot, A. et al. (2004) A new class of dual targeting inhibitors of GyrB and
ParE. (Abstract F-1951)Proceedings of the 44th Interscience Conference
on Antimicrobial Agents and Chemotherapy, 30 October–2 November
2004, Washington D.C., USA.
60 Buchanan, S.G. et al. (2002) The promise of structural genomics in the
discovery of new antimicrobial agents. Curr. Pharm. Des. 8, 1173–1188
61 Yu, L. et al. (2003) Solution structure and function of an essential CMP
kinase of Streptococcus pneumoniae. Protein Sci. 12, 2613–2621
62 Abergel, C. et al. (2003) Structural genomics of highly conserved micro-
bial genes of unknown function in search of new antibacterial targets. J.
Struct. Funct. Genomics 4, 141–157
63 Terwilliger, T.C. et al. (2003) The TB structural genomics consortium: a
resource for Mycobacterium tuberculosis biology. Tuberculosis (Edinb.)
83, 223–249
64 Gilliland, G.L. et al. (2002) Assisting functional assignment for hypothe-
tical Haemophilus influenzae gene products through structural genomics.
Curr. Drug Targets Infect. Disord. 2, 339–353
65 Zalacain, M. et al. (2003) A global approach to identify novel broad-
spectrum antibacterial targets among proteins of unknown function. J. Mol.
Microbiol. Biotechnol. 6, 109–126
66 Lipinski, C.A. (2000) Drug-like properties and the causes of poor solubility
and poor permeability. J. Pharmacol. Toxicol. Methods 44, 235–249
67 Payne, D.J. (2004) Antimicrobials – where next? Microbiol Today 31
68 Stachyra, T. et al. (2004) Fluorescence detection-based functional assay for
high-throughput screening for MraY. Antimicrob. Agents Chemother. 48,
897–902
69 Baum, E.Z. et al. (2001) Identification and characterization of new
inhibitors of the Escherichia coli MurA enzyme. Antimicrob. Agents
Chemother. 45, 3182–3188
70 Chu, M. et al. (2003) Structure elucidation of Sch 538415, a novel acyl
carrier protein synthase inhibitor from a microorganism. Bioorg. Med.
Chem. Lett. 13, 3827–3829
71 Oefner, C. et al. (2003) The 1.15A crystal structure of the Staphylococcus
aureus methionyl-aminopeptidase and complexes with triazole based
inhibitors. J. Mol. Biol. 332, 13–21
www.drugdiscoverytoday.com 527





























