Embed Size (px)
Citation preview

Gemeinsames Praktikum
des Fachbereichs Bio- und Chemieingenieurwesen
Der Versuch gehört zum Praktikumsbereich Technische Chemie A
Versuch TC1
Bestimmung des Dampf-Flüssigkeits-Gleichgewichtes und offene Destillation eines realen Zweistoffgemisches
Versuchsinhalt:
• Experimentelle Bestimmung von Dampf-Flüssigkeits-Gleichgewichtsdaten am Beispiel des binären Systems n-Propanol-Wasser
• Darstellung als Siedediagramm (T-x,y-Diagramm)
• Darstellung als Gleichgewichtsdiagramm (x-y-Diagramm)
• Ermittlung der Aktivitätskoeffizienten und der relativen Flüchtigkeit
• Kontrolle der Daten durch den Konsistenztest nach Redlich und Kister
• Modellierung der Messwerte mit Hilfe des Modells von van Laar: Ermittlung der Aktivitätskoeffizienten und der relativen Flüchtigkeit
• aus allen Messwerten
• aus dem azeotropen Punkt
• Vergleich der Ergebnisse aus dem Modell mit den Messdaten
• Destillation eines Gemisches von n-Propanol-Wasser
• experimentelle Bestimmung des Konzentrationsverlaufes im Destillat
• Bilanzierung und Berechnung der offenen Destillation
• Vergleich der gemessenen und berechneten Werte

Universität Dortmund Gemeinsames Praktikum, Praktikumsbereich TCA Zeichen: Fischer/Jörissen Fachbereich Bio- und TC 1 Seite: 2 Chemieingenieurwesen Gleichgewichtsbestimmung und offene Destillation Datum: August 2005
Nur zum persönlichen Gebrauch bestimmt!
Inhaltsverzeichnis Inhaltsverzeichnis .................................................................................................................................. 2
2 Funktionsweise der Versuchsapparaturen................................................................................. 4
2.1 Gleichgewichtsapparatur .................................................................................................................................4 2.2 Offene Destillation ..........................................................................................................................................6
3 Aufgabenstellung .......................................................................................................................... 7
3.1 Bestimmung des Siedegleichgewichtes ...........................................................................................................7 3.2 Offene Destillation ..........................................................................................................................................8
4 Betriebsvorschrift ......................................................................................................................... 8
4.1 Vorbereitungen an den Versuchsapparaturen ..................................................................................................8 4.2 Betrieb der Gleichgewichtsapparatur ..............................................................................................................9 4.3 Betrieb der offenen Destillation ....................................................................................................................10 4.4 Abschalten aller Geräte nach Abschluss beider Versuchsteile ......................................................................11
5 Dichtemessgerät .......................................................................................................................... 11
5.1 Funktionsweise und Bedienung.....................................................................................................................11 5.2 Berechnung des Ergebnisses .........................................................................................................................12
6 Stoffdaten..................................................................................................................................... 12
6.1 Umrechnung von Gewichtsanteilen (ξ) in Mol-Anteile (x)...........................................................................12 6.2 Dampfdruckkurven der reinen Komponenten n-Propanol und Wasser .........................................................12 6.3 Umrechnung der Messwerte auf einen anderen Siededruck Π ......................................................................13
7 Grundlagen.................................................................................................................................. 14
7.1 Ideale Systeme...............................................................................................................................................14 7.2 Die relative Flüchtigkeit (Trennfaktor) .........................................................................................................14 7.3 Die Anwendung der relativen Flüchtigkeit bei idealen Gemischen ..............................................................15 7.4 Darstellung der binären Dampf-Flüssigkeits-Gleichgewichte .......................................................................15 7.5 Nicht-ideale Dampf-Flüssigkeits-Gleichgewichte.........................................................................................16 7.6 Azeotrope Systeme........................................................................................................................................19 7.7 Molekulare Ursachen des nicht-idealen Siedeverhaltens ..............................................................................20 7.8 Systeme mit Mischungslücke ........................................................................................................................21 7.9 Temperatureinfluss auf Siedegleichgewichte, Überwindung des azeotropen Punktes ..................................21
8 Modellgleichungen für Siedegleichgewichte............................................................................. 23
8.1 Die van Laar-Gleichung für binäre Gemische ...............................................................................................23 8.2 Linearisierung der van Laar Gleichung .........................................................................................................24 8.3 Berechnung der Konstanten der van Laar Gleichung aus einem Messpunkt.................................................26 8.4 Berechnung der Aktivitätskoeffizienten und der Gleichgewichtskurve aus der van Laar Gleichung ...........26 8.5 Prüfen der Konsistenz der Messwerte ...........................................................................................................27
9 Verfahren zur Berechnung der offenen Destillation ............................................................... 29
9.1 Berechnung der Blasenkonzentration in Abhängigkeit von der überdestillierten Flüssigkeitsmasse ............29 9.2 Berechnung der Produktkonzentration in Abhängigkeit von der überdestillierten Flüssigkeitsmasse ..........31 9.3 Ermittlung der Blasenkonzentration aus der Konzentration des jeweils kondensierenden Dampfes ............31 9.4 Ermittlung der Produktkonzentration aus der Konzentration des jeweils kondensierten Dampfes ...............31
Formelzeichen...................................................................................................................................... 32
Indizes................................................................................................................................................... 32
Literatur:.............................................................................................................................................. 32

Universität Dortmund Gemeinsames Praktikum, Praktikumsbereich TCA Zeichen: Fischer/Jörissen Fachbereich Bio- und TC 1 Seite: 3 Chemieingenieurwesen Gleichgewichtsbestimmung und offene Destillation Datum: August 2005
Nur zum persönlichen Gebrauch bestimmt!
1 Einführung Aufgabe dieses Praktikumsversuches ist die Darstellung des Siedegleichgewichtes, d.h. des Dampf-Flüssigkeits-Gleichgewichtes und seine Anwendung bei der diskontinuierlichen offenen Destillation für ein reales Zweistoffgemisch (n-Propanol / Wasser).
Bei idealen Gemischen lässt sich zu einer bestimmten Flüssigkeitskonzentration die dazugehörige Gleich-gewichtskonzentration im Dampf allein aus der Kenntnis der Dampfdrücke der reinen Komponenten be-rechnen. Für reale Mischungen sind zur Korrektur Aktivitätskoeffizienten erforderlich, deren Ermittlung normalerweise auf dem Versuchswege erfolgt. Sie können heute auch recht gut aus Molekulardaten abge-schätzt werden [6,7]. Die Ergebnisse ermöglichen die Erstellung des Gleichgewichtsdiagramms, in dem die Dampfkonzentration über der Flüssigkeitskonzentration für den leichter siedenden Stoff im binären System aufgetragen wird. Es gibt eine Reihe von Gleichungssystemen, mit denen diese Gleichgewichts-kurven beschrieben werden können (siehe Abschnitt 8 und [1-7]).
Die Kenntnis des Gleichgewichtsdiagramms eines Systems stellt die Grundlage für alle destillativen Trennverfahren von Flüssigkeitsgemischen dar. Hierzu gehört die in diesem Versuch durchgeführte dis-kontinuierliche offene Destillation als ältestes Destillationsverfahren, das in zahlreichen Produktionspro-zessen nach wie vor zum Einsatz kommt. Die größte industrielle Bedeutung unter den destillativen Trenn-verfahren hat die Rektifikation, d.h. die Destillation mit Rückfluss (siehe auch Praktikumsversuch TV 4).
Aus den Gleichgewichtsdaten kann man zusammen mit der Mengenbilanz der Anlage bei der diskontinu-ierlichen Destillation den Konzentrationsverlauf in Abhängigkeit von der überdestillierten Menge und damit die Auftrennung des Gemisches in Vorlauf, Destillat (ggf. in mehrere Fraktionen) und Rückstand vorausberechnen. Für die Rektifikation können die theoretische Stufenzahl, die entsprechenden Konzen-trationen und das Rücklaufverhältnis ermittelt werden. Anschaulich erfolgt dies mit der graphischen Me-thode nach McCabe-Thiele (siehe Praktikumsversuch Rektifikation TV 4). Üblicherweise werden heute Computerprogramme eingesetzt, die selbstverständlich eines der erwähnten Gleichungssysteme zur mathematischen Beschreibung des Gleichgewichtes erfordern. Diese Gleichungssysteme lassen sich auch auf Mehrstoffgemische übertragen, deren Trennung dann berechnet werden kann. Für einen raschen Über-blick zur Abschätzung der Möglichkeiten destillativer Trennverfahren – z.B. im Rahmen der Verfahrens-entwicklung – sind auch heute weiterhin Methoden mit Nomogrammen sinnvoll. Diese basieren meistens auf der relativen Flüchtigkeit, auch Trennfaktor genannt, dessen Ermittlung ein wesentliches Ziel dieses Praktikumsversuches ist.
Als einfaches Beispiel für die Verwendung eines Siedediagramms soll die offene diskontinuierliche De-stillation berechnet und gemessen werden. In der Technik hat dieser diskontinuierliche Prozess gegenüber kontinuierlichen Verfahren an Bedeutung verloren. Man wendet ihn jedoch häufig vor allem zur Reini-gung von Substanzen an, wenn die anfallenden Mengen so klein sind, dass sich ein kontinuierliches Ver-fahren nicht lohnt.
Die älteste Anwendung der Destillation, die auch heute noch oft als offene Destillation (in meist kleinen Apparaten) durchgeführt wird, ist die Herstellung von Branntwein aus niedrigkonzentrierten Lösungen von Alkohol in Wasser, die durch alkoholische Gärung mit Hilfe von Hefen erhalten wurden.
Auch im chemischen Laboratorium wird die einfache Destillation häufig angewendet. Voraussetzung für eine befriedigende Trennung ist ein relativ hoher Trennfaktor zwischen den Substanzen. Besonders geeig-net ist die einfache Destillation zur Abtrennung des Produktes von schwer oder nicht verdampfbaren Rückständen.
Während des Destillationsvorganges ändern sich die Zusammensetzungen in der Destillierblase, im Dampf und in der Produktvorlage. Im allgemeinen verdampfen zunächst überwiegend leichtflüchtige Nebenpro-dukte und danach das eigentliche Produkt, während sich schwerflüchtige Nebenprodukte in der Destillier-blase anreichern. Wenn man bestimmte Konzentrationen erreichen will, werden deshalb häufig mehrere Produktvorlagen verwendet, die nach einer bestimmten Zeit bzw. nach dem Überdestillieren einer be-stimmten Menge oder auch nach dem Erreichen einer bestimmten Siedetemperatur oder Konzentration

Universität Dortmund Gemeinsames Praktikum, Praktikumsbereich TCA Zeichen: Fischer/Jörissen Fachbereich Bio- und TC 1 Seite: 4 Chemieingenieurwesen Gleichgewichtsbestimmung und offene Destillation Datum: August 2005
Nur zum persönlichen Gebrauch bestimmt!
gewechselt werden, um auf diese Weise verschiedene Fraktionen des Destillats zu sammeln. Damit uner-wünschte Produkte nicht in das Destillat gelangen, wird die Destillation normalerweise auch abgebrochen, bevor der Inhalt der Destillierblase vollständig verdampft ist.
So kommt es bei der Herstellung alkoholischer Getränke darauf an, zunächst unerwünschte leichtflüchtige Produkte (z.B. Ether, Ester) zu entfernen, dann einen möglichst großen Anteil des Alkohols zusammen mit den richtigen Aromastoffen überzudestillieren, während das Wasser und vor allem die unerwünschten Nebenprodukte der Gärung (Fuselöle) möglichst in der Destillierblase zurückbleiben sollen.
Mit Hilfe der in diesem Versuch durchgeführten Bilanzierung unter Anwendung der Gleichgewichtskurve ist die Vorausberechnung des Destillationsverfahrens möglich.
2 Funktionsweise der Versuchsapparaturen 2.1 Gleichgewichtsapparatur
Abb.1 Schematische Darstellung der Gleichgewichtsapparatur Die hier verwendete Gleichgewichtsapparatur erzeugt aus dem siedenden Gemisch Proben der Flüssig-keits- und Gasphase, die dann analysiert werden. Neben dem einfachen Aufbau bietet sie für den Prakti-kumsversuch vor allem den Vorteil, den Vorgang unmittelbar anschaulich zu machen. In vollautomatisch computergesteuerten Anlagen werden häufig andere Messprinzipien eingesetzt, z.B. Ersatz der Analysen durch Präzisions-Dosiereinrichtungen und Messung des Dampfdruckes mit hochgenauen Drucksensoren.

Universität Dortmund Gemeinsames Praktikum, Praktikumsbereich TCA Zeichen: Fischer/Jörissen Fachbereich Bio- und TC 1 Seite: 5 Chemieingenieurwesen Gleichgewichtsbestimmung und offene Destillation Datum: August 2005
Nur zum persönlichen Gebrauch bestimmt!
Abb. 1 zeigt schematisch den Aufbau der Gleichgewichtsapparatur. Das Flüssigkeitsgemisch wird im Sie-deraum (2) durch die beiden Quarztauchsieder (1) zum Sieden gebracht. Der Dampf gelangt zusammen mit Flüssigkeit durch die Übersteigrohre (3) in den Dampfraum (5). Dieser von Dampf und Flüssigkeit bei fortlaufender Vermischung gemeinsam zurückgelegte Weg trägt wesentlich dazu bei, den Gleichgewichts-zustand (weitgehend) zu erreichen. Direkt beim Verdampfungsvorgang im Siederaum ist dies nicht mög-lich, zumal dort infolge des durch die Leitung (12) zurückgeführten Kondensates mit einer vom Inhalt des Siederaumes abweichenden Zusammensetzung keine einheitlichen Konzentrationen erwartet werden kön-nen. Die mitgenommene Flüssigkeit scheidet sich beim Eintritt in den Dampfraum (5) ab und wird durch das Rohr (4) bis unter die Flüssigkeitsoberfläche in den unteren Bereich des Siederaums zurückgeführt. Dieser „hydraulische Verschluss“ (4) verhindert, dass der Dampf unter Umgehung der Übersteigrohre (3) unmittelbar in den Dampfraum (5) gelangen kann. Der aufsteigende Dampf hat im Dampfraum (5) durch den großen Querschnitt nur eine geringe Geschwindigkeit, die das Mitreißen von Flüssigkeitströpfchen verhindert. Er wird im Kondensator (10) vollständig kondensiert und gelangt durch die Rückführungs-leitung (12) wieder in den Siederaum zurück. Durch einen ausreichend langen Betrieb im Kreislauf kön-nen stationäre Bedingungen mit konstanten Konzentrationen erreicht werden.
Das Gleichgewicht liegt (annähernd) bei der Trennung von Dampf und Flüssigkeit nach der Durchmi-schung in den Übersteigrohren beim Eintritt in den Dampfraum vor. Deshalb sollten die Gleichgewichts-daten (Siedetemperatur, Konzentration der Flüssigkeit, Konzentration des Dampfes) möglichst auch an dieser Stelle gemessen werden. Dem ist weitgehend Rechnung getragen worden:
• Der Temperaturfühler (8 TIC) des Digitalthermometers (Platin-Widerstandsthermometer) befindet sich an der Trennstelle von Dampf und Flüssigkeit am Eintritt der Übersteigrohre in den Dampfraum.
• Die Proben der flüssigen Phase werden mit Hilfe einer Kapillare gezogen, die nahe an die Trennstelle von Dampf und Flüssigkeit heranreicht (Probehahn 13 a).
• Zur Probenahme des Dampfes wird der Kondensatablauf aus dem Kühler (10) verwendet. Die Dampf- und Kondensat-Konzentration stimmen überein, wenn eine Teilkondensation von Dampf im Dampf-raum verhindert wird. In dem Kondensatsammelraum (11) ist eine Kondensatmenge von ca. 2 ml vor-handen, die nach Erreichen des stationären Zustands durch den Probenahmehahn 13 b abgezogen und analysiert werden kann.
Die Teilkondensation des Dampfes im Dampfraum wird durch die elektrische Heizwendel (6) verhindert. Diese Schutzheizung wird von dem Temperaturdifferenz-Regler gesteuert, der die an der Trennstelle von Dampf und Flüssigkeit gemessene Siedetemperatur (8 TIC) mit der Temperatur im Mantelraum (7 TC) vergleicht und auf einer vorher eingestellten Differenz hält.
Bevor die Proben des kondensierten Dampfes und der flüssigen Phase zur Analyse entnommen werden können, muss die eingefüllte Flüssigkeit ca. 20 Minuten bis zum Erreichen des stationären Zustands im Kreislauf sieden, da eine Konzentrationsverschiebung der flüssigen Phase auftritt, wenn sich die Appa-ratur mit Dampf füllt, das Kondensat (mit der Konzentration des Dampfes) im Raum (11) gesammelt wird und die Flüssigkeit in der Rückführungsleitung (12) nur langsam durch das Kondensat verdrängt wird. Diese Versuchszeit bis zum Erreichen des stationären Zustandes hängt von der Kondensatmenge, d.h. von der Verdampfungsgeschwindigkeit und von der Bauweise der Apparatur, insbesondere von der Güte der Flüssigkeitsdurchmischung im Siederaum (2) ab. Die Erfassung der Siedetemperatur und die Probenahme der beiden Phasen muss möglich gleichzeitig erfolgen, da sich durch die Probenahme die Stoffmengen in der Apparatur und damit die Konzentrationen verschieben, und sich infolgedessen ein neuer Gleich-gewichtszustand einzustellen beginnt.
Die Verdampfungsgeschwindigkeit muss einerseits so hoch gewählt werden, dass in den Übersteigrohren (3) gleichmäßig Flüssigkeit mitgenommen wird und eine gute Durchmischung in der ganzen Apparatur er-folgt, andererseits darf sie nicht zu hoch sein, weil sonst feine Flüssigkeitströpfchen vom Dampf in den Dampfraum (5) mitgerissen werden können, die dann die Konzentrationsmessungen verfälschen.
Die Konzentrationen in den Proben des kondensierten Dampfes und der flüssigen Phase werden durch Messung der Dichte bzw. des Brechungsindex bestimmt.
Universität Dortmund Gemeinsames Praktikum, Praktikumsbereich TCA Zeichen: Fischer/Jörissen Fachbereich Bio- und TC 1 Seite: 6 Chemieingenieurwesen Gleichgewichtsbestimmung und offene Destillation Datum: August 2005
Nur zum persönlichen Gebrauch bestimmt!
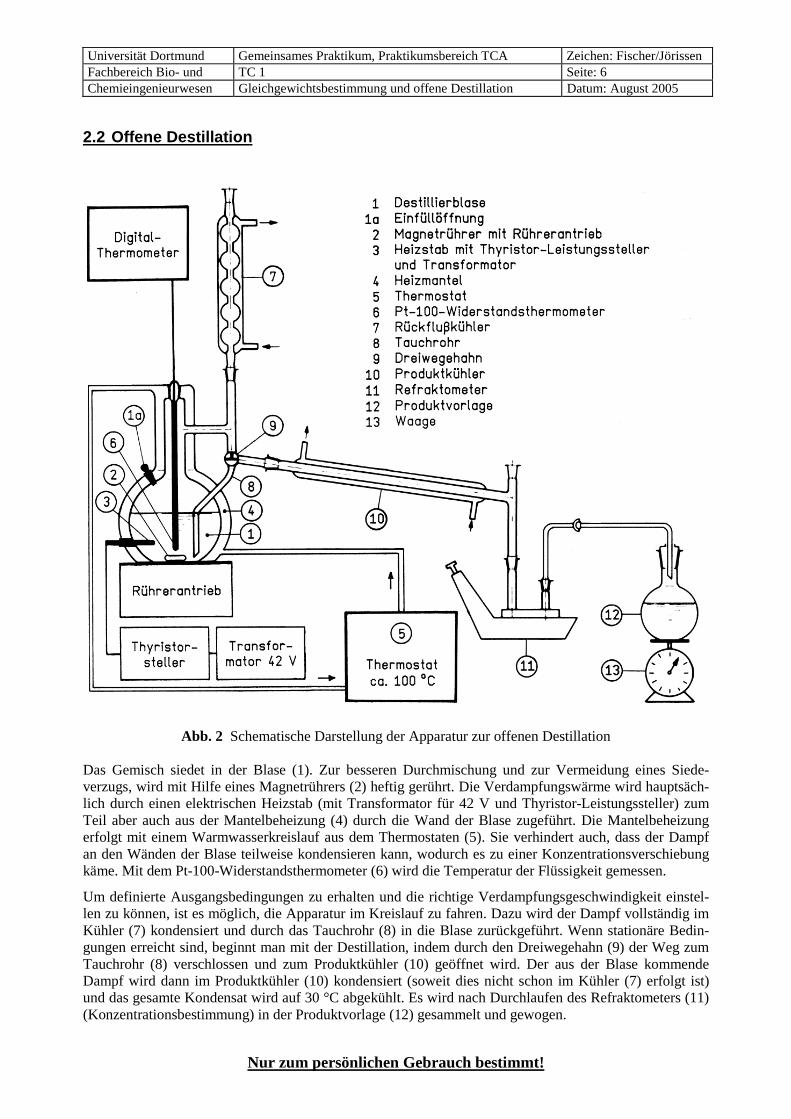
2.2 Offene Destillation
Abb. 2 Schematische Darstellung der Apparatur zur offenen Destillation Das Gemisch siedet in der Blase (1). Zur besseren Durchmischung und zur Vermeidung eines Siede-verzugs, wird mit Hilfe eines Magnetrührers (2) heftig gerührt. Die Verdampfungswärme wird hauptsäch-lich durch einen elektrischen Heizstab (mit Transformator für 42 V und Thyristor-Leistungssteller) zum Teil aber auch aus der Mantelbeheizung (4) durch die Wand der Blase zugeführt. Die Mantelbeheizung erfolgt mit einem Warmwasserkreislauf aus dem Thermostaten (5). Sie verhindert auch, dass der Dampf an den Wänden der Blase teilweise kondensieren kann, wodurch es zu einer Konzentrationsverschiebung käme. Mit dem Pt-100-Widerstandsthermometer (6) wird die Temperatur der Flüssigkeit gemessen.
Um definierte Ausgangsbedingungen zu erhalten und die richtige Verdampfungsgeschwindigkeit einstel-len zu können, ist es möglich, die Apparatur im Kreislauf zu fahren. Dazu wird der Dampf vollständig im Kühler (7) kondensiert und durch das Tauchrohr (8) in die Blase zurückgeführt. Wenn stationäre Bedin-gungen erreicht sind, beginnt man mit der Destillation, indem durch den Dreiwegehahn (9) der Weg zum Tauchrohr (8) verschlossen und zum Produktkühler (10) geöffnet wird. Der aus der Blase kommende Dampf wird dann im Produktkühler (10) kondensiert (soweit dies nicht schon im Kühler (7) erfolgt ist) und das gesamte Kondensat wird auf 30 °C abgekühlt. Es wird nach Durchlaufen des Refraktometers (11) (Konzentrationsbestimmung) in der Produktvorlage (12) gesammelt und gewogen.

Universität Dortmund Gemeinsames Praktikum, Praktikumsbereich TCA Zeichen: Fischer/Jörissen Fachbereich Bio- und TC 1 Seite: 7 Chemieingenieurwesen Gleichgewichtsbestimmung und offene Destillation Datum: August 2005
Nur zum persönlichen Gebrauch bestimmt!
3 Aufgabenstellung
Die Sicherheitsvorschriften für die leichtentzündliche Flüssigkeit n-Propanol sind unbedingt zu beachten (siehe Aushang) !
3.1 Bestimmung des Siedegleichgewichtes
1. Das Siedegleichgewicht des binären Systems n-Propanol / Wasser ist für folgende acht Einsatzgemi-sche zu bestimmen. Die Ergebnisse sollen in ein Versuchsprotokoll eingetragen werden.
Nr. 1 2 3 4 5 6 7 8
Gew-% n-Propanol 100 95 85 70 30 15 5 0
Gew-% Wasser 0 5 15 30 70 85 5 100
Die benötigte Menge beträgt jeweils ca. 100 g. Dabei soll mit reinem n-Propanol begonnen werden, und das nächste wasserreichere Gemisch jeweils aus dem vorhergehenden durch Zuwiegen von Was-ser hergestellt werden. Da die genauen Konzentrationen der Flüssigkeitsproben analysiert werden, müssen die o.g. Einwaage-Konzentrationen nur ungefähr eingehalten werden.
2. Die gemessenen Siedetemperaturen sind auf einen Gesamtdruck von 760 Torr umzurechnen. Der dafür benötigte aktuelle Barometerstand wird vom Assistenten angegeben.
3. Die auf den Gesamtdruck von 760 Torr umgerechneten Werte sind im Siedediagramm (T-x,y-Diagramm) (1 °C = 2 cm, Abszissenlänge 15 cm), und im Gleichgewichtsdiagramm (x,y-Dia-gramm, McCabe-Thiele-Diagramm) (Kantenlänge 15 cm) aufzutragen und zum Vortestat nach Abschluss des Versuches vorzulegen (zur Beurteilung, ob auswertbare Ergebnisse vorliegen).
4. Die Dampfdrücke für n-Propanol (PAo) und Wasser (PBo) sind aus den umgerechneten Siedetempera-turen zu ermitteln (siehe auch Abb. 3).
5. Aus den unter 2. umgerechneten Messwerten sind die Aktivitätskoeffizienten γA und γB sowie die relative Flüchtigkeit αAB (Trennfaktor) zu berechnen.
6. Der Konsistenztest nach Redlich-Kister ist durchzuführen (Abszissenlänge 15 cm, Ordinatenmaßstab 1 = 5 cm).
7. Die Konstanten CA und CB der van Laar-Gleichung sind mit Hilfe des beigefügten Netzes (Abb. 11) aus den unter 5. berechneten Werten zu bestimmen.
8. Berechnung der Aktivitätskoeffizienten γA und γB sowie der relativen Flüchtigkeit αAB aus den Kon-stanten nach 7. für die Konzentrationen der Messpunkte.
9. Die Konstanten der van Laar-Gleichung sind aus den Daten des azeotropen Punktes zu berechnen.
10. Berechnung der Aktivitätskoeffizienten γA und γB sowie der relativen Flüchtigkeit αAB aus den Kon-stanten nach 9. für die Konzentrationen der Messpunkte.
11. Die Logarithmen (Basis 10) aller ermittelten Aktivitätskoeffizienten sind über dem Molenbruch xA aufzutragen (Ordinatenmaßstab 1 = 15 cm, Abszissenlänge 15 cm) und zum Vortestat nach Abschluss des Versuches vorzulegen (zur Beurteilung, ob die Ergebnisse auswertbar sind).
12. Alle ermittelten relativen Flüchtigkeiten αAB sind über dem Molenbruch xA aufzutragen (Ordinatenmaßstab 1 = 2 cm, Abszissenlänge 15 cm).
13. Zur Versuchsdiskussion sind alle gezeichneten Diagramme zu diskutieren: qualitative und quatitative Angaben zu den Kurvenverläufen sind zu machen, besonders für Extrempunkte. Bei der Auftragung von log γA und log γB über xA sind die Achsenabschnitte der Kurven für xA = 0 und xA = 1 anzugeben (Aktivitätskoeffizienten bei unendlicher Verdünnung). Abweichungen zwischen den Ergebnissen aus Messwerten und den Ergebnissen nach van Laar sind zu diskutieren.

Universität Dortmund Gemeinsames Praktikum, Praktikumsbereich TCA Zeichen: Fischer/Jörissen Fachbereich Bio- und TC 1 Seite: 8 Chemieingenieurwesen Gleichgewichtsbestimmung und offene Destillation Datum: August 2005
Nur zum persönlichen Gebrauch bestimmt!
3.2 Offene Destillation
14. Der Versuch ist entsprechend der Betriebsvorschrift (siehe Abschnitt 4.2.16) mit einem Gemisch von 25 Gew-% n-Propanol in Wasser zu starten.
15. Die Siedetemperatur, die Dampfkonzentration ψM und die Produktmenge sind laufend zu messen.
16. Zum Vortestat nach Abschluss des Versuches sind die Konzentrationswerte umgerechnet in Gew.-% vorzulegen (zur Beurteilung, ob die Ergebnisse auswertbar sind).
17. Die im ersten Versuchsteil mit der Gleichgewichtsapparatur gemessenen Gleichgewichtskonzentra-tionen sind auf Massenbrüche umzurechnen und in ein Gleichgewichtsdiagramm (ξ-ψ-Diagramm) mit 15 cm Kantenlänge einzutragen.
18. Auswertung und Bilanzierung der Messwerte: • Ermittlung der Blasenkonzentration ξM aus den Messwerten der Dampfkonzentration ψM • Ermittlung der Produktkonzentration ξPM1 aus den Messwerten der Dampfkonzentration ψM • Ermittlung der Produktkonzentration ξPM2 aus den Konzentrationswerten in der Blase ξM
19. Vergleichswerte für die Bilanzierung während des Destillationsverlaufes aus den Daten der Gleich-gewichtsbestimmung: • Ermittlung des theoretischen Verlaufes der Blasenkonzentration ξV • Ermittlung der dazugehörigen theoretischen Dampfkonzentrationen ψV • Ermittlung der Produktkonzentration ξPV aus den Konzentrationswerten der Blase ξV
20. Tragen Sie alle unter 18) und 19) ermittelten Konzentrationsverläufe sowie die gemessene Dampfzu-sammensetzung ψM als Funktion des überdestillierten Anteils 1 – (mF / mFo) auf und kennzeichnen Sie die Kurven mit den entsprechenden Formelzeichen.
21. Geben Sie unter Verwendung der Messwerte ψM an, welchen Anteil der Anfangsmenge mFo sie über-destillieren müssen: • um ein Destillat mit 50 Gew.-% zu erreichen • um 95% der gesamten Alkoholmenge in das Destillat zu überführen.
22. Zur Versuchsdiskussion sind alle gezeichneten Konzentrationsverläufe als Funktion des überdestil-lierten Anteils 1 – (mF / mFo) zu diskutieren. Anzugeben sind ferner alle theoretischen Anfangs- und Endkonzentrationen bei 1 – (mF / mFo) = 0 und 1 – (mF / mFo) = 1, auch wenn diese nicht gemessen wurden. Abweichungen zwischen gemessenen und berechneten Konzentrationen sind zu diskutieren.
4 Betriebsvorschrift
Die Sicherheitsvorschriften für die leichtentzündliche Flüssigkeit n-Propanol sind unbedingt zu beachten (siehe Aushang) !
4.1 Vorbereitungen an den Versuchsapparaturen
1. Digitales Temperaturmessgerät einschalten (mindestens 30 Minuten vor der ersten Messung), das Gerät wird für beide Versuchsteile genutzt und muss deshalb für alle Messungen jeweils
umgeschaltet werden (Destillation / Gleichgewicht).
2. Thermostat für die Gleichgewichtsapparatur und die offene Destillation einschalten, Heizung: mittlere Stellung, Kühlwasser für den Thermostaten aufdrehen. Wenn auch die offene Destillation läuft, unbedingt Wasser für den zusätzlichen Kühler aufdrehen !
(nicht ausreichende Kühlung macht sich dadurch bemerkbar, dass sich die Heizung des Thermostaten nicht gleichmäßig ein- und ausschaltet). Der Thermostat-Kreislauf versorgt den Kondensator (10) der Gleichgewichtsapparatur sowie den Produktkühler (10) und das Refraktometer (11) der offenen De-stillation, für das eine genaue Einhaltung der Messtemperatur von 30 °C erforderlich ist.

Universität Dortmund Gemeinsames Praktikum, Praktikumsbereich TCA Zeichen: Fischer/Jörissen Fachbereich Bio- und TC 1 Seite: 9 Chemieingenieurwesen Gleichgewichtsbestimmung und offene Destillation Datum: August 2005
Nur zum persönlichen Gebrauch bestimmt!
3. Dichtemessgerät und Thermostaten für das Dichtemessgerät und das separate Refraktometer einschal-ten. Kühlwasser für den Thermostaten aufdrehen. Solltemperatur am Regler so einstellen, dass die Temperatur am Thermometer des Refraktometers 30 ± 0.5 °C beträgt.
4. Transformator und Temperatur-Differenz-Regler für die Gleichgewichtsapparatur einschalten. Darauf achten, dass zunächst alle Heizungen an der Gleichgewichtsapparatur und offenen Destillation
ausgeschaltet sind (die Heizungen dürfen nur bei gefüllten Apparaturen betrieben werden ! ). Der Temperatur-Differenz-Regler muss auf 5 °C und die markierten Schalterstellungen eingestellt sein.
Das Einschalten des Temperatur-Differenz-Reglers darf auf keinen Fall vergessen werden, da sonst die Mantelheizung der Gleichgewichtsapparatur mit voller Leistung (200 Watt) heizt und es zu einer starken Überhitzung kommt. Im gleichen Sinne muss die Funktion des Temperatur-Differenz-Reglers überwacht werden (gleichmäßiges Ein- und Ausschalten).
5. Eichung des Dichtemessgerätes (siehe Abschnitt 5). Dies kann während der Wartezeit auf die Gleich-gewichtseinstellung des ersten Messpunktes der Gleichgewichtskurve erfolgen.
4.2 Betrieb der Gleichgewichtsapparatur
6. Für die Messung mit reinem 1-Propanol muss die Gleichgewichts-Apparatur sauber und trocken sein. Für die Messung mit reinem Wasser (VE-Wasser) genügt gründliches Spülen der Apparatur.
(Trotzdem in beiden Fällen zur Kontrolle Analysen der beiden Phasen vornehmen ! )
Bei allen anderen Messpunkten kommt es nicht darauf an, exakt eine bestimmte Zusammensetzung zu erreichen. Es muss also nur sichergestellt sein, dass die Proben nicht durch Reste von der letzten Messung verunreinigt werden (sorgfältiges Trocknen der Probeentnahmehähne 13 a und 13 b durch Absaugen mit der Wasserstrahlpumpe).
7. Alle Hähne schließen.
Gemisch durch die Einfüllöffnung (9) des Kondensators (10) in die Apparatur bis zur Markierung am Siederaum (obere Kante) einfüllen. Evtl. erforderliche Korrektur des Füllstandes siehe Punkt 9.
8. Widerstandsthermometer nicht herausziehen oder drehen: (Glasbruch-Gefahr !!! )
9. Heizung des Siederaumes und des Temperiermantels mit dem gemeinsamen Schalter einschalten und Thyristorsteller („Thyrotakt TW 42“) so einstellen, dass die Flüssigkeit in den Übersteigrohren (3) gerade vom aufsteigenden Dampf mitgenommen wird.
Zu geringes Sieden führt dazu, dass der Dampf in den Übersteigrohren nicht mehr mit der Flüssigkeit im Gleichgewicht steht. Darüber hinaus wird er von der Heizung des Mantelraumes (6) überhitzt. Das Thermometer am Ende der Übersteigrohre zeigt dann also nicht mehr die Siedetemperatur sondern einen zu hohen Wert an. Durch den Temperatur-Differenz-Regler wird die Manteltemperatur weiter erhöht, die sich dadurch bis auf viel zu hohe Werte aufschaukeln kann.
Zu starkes Sieden ist ebenfalls zu vermeiden, damit Flüssigkeitstropfen nicht durch den Dampf mit-gerissen werden.
Um ein richtiges Arbeiten der Apparatur zu erreichen, muss u.U. eine Korrektur des Flüssigkeitsstan-des erfolgen (teilweises Ablassen am Entleerungs-Hahn 13 c).
10. Gemisch bis zur Gleichgewichtseinstellung (stationärer Zustand) ca. 20 Minuten sieden lassen.
Die folgenden Punkte 11 - 13 müssen möglichst gleichzeitig ausgeführt werden !
Durch die Entnahme der Proben verschieben sich die Stoffmengen und anschließend die Konzentrationen !
11. Temperatur am Digitalthermometer (Schalterstellung Gleichgewicht) ablesen. 30 Minuten Warmlaufzeit beachten !
12. Kondensatprobe am oberen Probehahn (13 b) in trockenes Probegläschen laufen lassen.
13. Probe der flüssigen Phase am unteren Probehahn (13 a) ziehen (trockenes Probegläschen verwenden).
Vor der Probenahme ca. 1 ml Flüssigkeit ablaufen lassen, damit die Kapillare, die zur Flüssigkeits-entnahme dient, gespült wird.

Universität Dortmund Gemeinsames Praktikum, Praktikumsbereich TCA Zeichen: Fischer/Jörissen Fachbereich Bio- und TC 1 Seite: 10 Chemieingenieurwesen Gleichgewichtsbestimmung und offene Destillation Datum: August 2005
Nur zum persönlichen Gebrauch bestimmt!
14. Die Konzentration beider Phasen durch Messung der Dichte (gesamter Konzentrationsbereich) und des Brechungsindex (nur im Konzentrationsbereich 0 – 40 Mol-% n-Propanol) bestimmen.
Eichkurven hängen aus; Temperaturkonstanz 30 °C beachten !
15. Nach Beendigung der Messung Schalter für Siederaum- und Temperiermantel-Heizung ausschalten.
Apparatur etwas abkühlen lassen. Danach Flüssigkeit ablassen und zur Herstellung der nächsten Mischung verwenden (Zuwiegen von Wasser).
Dann unter Beachtung von Punkt 6 erneut bei Punkt 7 beginnen.
16. Nach Abschluss der letzten Messung mit reinem Wasser ist die noch heiße Apparatur durch Absaugen mit der Wasserstrahlpumpe zu trocknen.
Alle Hähne öffnen und geöffnet lassen. 4.3 Betrieb der offenen Destillation 17. Ca. 600 g eines Gemisches mit ca. 25 Gew-% n-Propanol in Wasser ansetzen.
(Ermittlung der genauen Konzentration durch Bestimmung von Dichte und / oder Brechungsindex)
Gemisch genau abwiegen.
Gemisch durch die Einfüllöffnung (1a) in die leere Destillierblase einfüllen. Der Einfluss von Trop-fen, die evtl. im Rückflusskühler (7), im Hahn (9) und im Tauchrohr (8) von der letzten Messung hängengeblieben sind, kann vernachlässigt werden.
18. Rührer der Destillierblase einschalten. (auf keinen Fall vergessen, das Gemisch neigt zu heftigem Siedeverzug ! ).
Thermostat für die Mantelbeheizung einschalten (auf ca. 103 °C).
19. Destillation mit dem Dreiwegehahn (9) auf Kreislaufbetrieb einstellen.
Um den Dreiwegehahn von oben in dem Schutzkasten erreichen zu können, Trittleiter benutzen, auf keinen Fall einen Stuhl !!!
Thyristorsteller („Thyrotakt TW 42“) für den Heizstab einschalten.
Verdampfungsgeschwindigkeit so einstellen, dass ca. 4 Tropfen/sec aus dem Kühler (7) ablaufen.
20. Waage einschalten.
Nullpunkt mit Taravoreinstellung justieren (auf freie Beweglichkeit des Vorlagegefäßes achten).
Beleuchtungseinrichtung am Refraktometer einschalten. (darauf achten, dass die untere Lichtklappe geschlossen ist)
Temperatur kontrollieren (30 ± 0,5 °C) und ggf. am Thermostaten korrigieren.
21. Kontrollieren, wann im Kreislaufbetrieb stationäre Bedingungen (Temperatur, Verdampfungsmenge) erreicht werden.
22. Starten der Produktentnahme durch Umstellen des Dreiwegehahnes (9).
Für jeden Messpunkt sind jetzt gleichzeitig Waage, Refraktometer und Siedetemperatur abzulesen.
Um genau die Konzentration des jeweiligen Dampfes bestimmen zu können, müsste im Kondensator eine exakte Pfropfenströmung herrschen und das Messvolumen des Refraktometers müsste unendlich klein sein. Beides trifft nur näherungsweise zu, die dadurch bedingten Messfehler können aber insge-samt vernachlässigt werden. Am Anfang der Messung befinden sich Reste des Kondensates (über-wiegend Wasser) aus der vorangegangenen Messung im Refraktometer (Totvolumen ca. 2 ml).
Die Anfangswerte sind deshalb verfälscht. Für die Auswertung sollte die Kurve des Dampfkonzen-trations-Verlaufes (Punkt 3.2.20 der Aufgabenstellung) durch Extrapolation auf den Beginn der Mes-sung korrigiert werden.

Universität Dortmund Gemeinsames Praktikum, Praktikumsbereich TCA Zeichen: Fischer/Jörissen Fachbereich Bio- und TC 1 Seite: 11 Chemieingenieurwesen Gleichgewichtsbestimmung und offene Destillation Datum: August 2005
Nur zum persönlichen Gebrauch bestimmt!
Die Konzentration ändert sich zu Beginn der Destillation nur wenig, fällt dann aber immer steiler und zuletzt sehr steil ab. Die Ablesungen müssen daher in ausreichend kleinen Abständen erfolgen, damit sich die Konzentration von Messpunkt zu Messpunkt nicht zu stark ändert, wodurch bei der Aus-wertung Fehler entstehen würden. Im steilen Bereich sollte so oft wie möglich abgelesen werden. Infolge der Konzentrationsänderung bleibt die Verdampfungswärme des Gemisches nicht konstant, sondern steigt in Richtung reinen Wassers an, so dass immer weniger Destillat übergeht. Durch Nach-regulieren des Thyristorstellers ist eine vernünftige Destillationsgeschwindigkeit aufrechtzuerhalten.
22. Wenn die Kondensatkonzentration unter 2 Gew.-% gefallen ist, sollte man noch etwa 50 g abdestil-lieren. Dann kann die Messung beendet werden.
23. Nach dem Abschalten des Heizstabes soll der Restinhalt der Destillationsblase im heißen Zustand mit der Wasserstrahlpumpe (Teflonschlauch) durch die Einfüllöffnung abgesaugt werden, wobei der Dreiwegehahn (9) so einzustellen ist, dass alle drei Wege offen sind.
Solange absaugen, bis die Destillierblase trocken ist! 4.4 Abschalten aller Geräte nach Abschluss beider Versuchsteile
• Gleichgewichtsapparatur: Heizung des Siederaumes und des Temperiermantels (ein Schalter) Temperatur-Differenz-Regler
• offene Destillation: Thyristorsteller des Heizstabes Rührer
• Transformator • 2 Thermostate und das dazugehörende Kühlwasser
(für die Versuchsapparaturen sowie das separate Refraktometer und das Dichtemessgerät) • ggf. Kühlwasser für den zusätzlichen Kühler • alle Waagen (ggf. auch die im Abzug) • Beleuchtungen für die beiden Refraktometer • Digitalthermometer • Dichtemessgerät
5 Dichtemessgerät
5.1 Funktionsweise und Bedienung
Das Dichtemessgerät arbeitet mit einer im Sichtfenster erkennbaren Stimmgabel aus Glasrohr, deren Schwingungsfrequenz von der Dichte des Stoffes im Glasrohr abhängt. Das Gerät misst nach Betätigen der Tasten „RESET“ (Nullstellung) und „START“ die Zeit für eine bestimmte Anzahl von Schwingungen. Die Proben werden an der Seite des Gerätes von unten mit einer Spritze in das Messsystem eingefüllt, wobei sorgfältig auf Blasenfreiheit zu achten ist (nur saubere und trockene Spritzen verwenden!).
Da die Dichte stark von der Temperatur abhängt (Solltemperatur = Bezugstemperatur der ausgehängten Eichkurve = 30 °C), muss unbedingt auf Temperaturkonstanz gewartet werden. Diese darf man annehmen, wenn zwei im Abstand von 15 Sekunden aufeinander folgende Messungen um nicht mehr als ein Digit voneinander abweichen. Um die Wartezeit abzukürzen, sind die Proben unbedingt vorher mit den Probe-gläschen im Thermostatbad (bzw. im Trockenschrank) zu temperieren. Strikt darauf achten, dass der Ther-mostat die Solltemperatur 30 °C ± 0.5 °C (abgelesen am Thermometer des separaten Refraktometers) einhält.
Das Dichtemessgerät enthält eine kleine Luftpumpe zum Trocknen des Messrohres (Schalter an der Frontseite, Ausgang an dem dünnen Silikongummischlauch auf der Seite des Gerätes). Ausreichende Trocknung lässt sich ebenfalls durch Übereinstimmung zweier Messwerte bis auf 1 Digit bei laufendem Luftstrom erkennen. Zur Beschleunigung der Trocknung sollten Flüssigkeitsreste im Messrohr zunächst mit der Wasserstrahlpumpe abgesaugt werden.

Universität Dortmund Gemeinsames Praktikum, Praktikumsbereich TCA Zeichen: Fischer/Jörissen Fachbereich Bio- und TC 1 Seite: 12 Chemieingenieurwesen Gleichgewichtsbestimmung und offene Destillation Datum: August 2005
Nur zum persönlichen Gebrauch bestimmt!
5.2 Berechnung des Ergebnisses
Dichte und Anzeigewerte hängen gemäß folgender Gleichung zusammen:
d dA
t t1 21
2
2
21− = −( ) (1)
di = Dichte des Stoffes i dWasser = 0.9957 bei 30 °C dLuft = 0.0012 bei 30 °C ti = Schwingungszeit = Anzeigewert für den Stoff i A = Gerätekonstante (beinhaltet Federkonstante und Volumen des Schwin-
gungssystems), die durch Eichmessungen ermittelt werden muss:
At t
d d
Wasser Luft
Wasser Luft=
−−
2 2
(2)
Zur Bestimmung der Konstanten A müssen also zunächst Eichmessungen (auf Temperaturkonstanz achten ! ) mit Luft (sorgfältig trocknen und für die Messung Luftstrom abschalten ! ) und mit Wasser durchgeführt werden.
Die Dichte eines Stoffes i kann dann ermittelt werden nach:
d dA
t ti Wasser i Wasser= + −1 2 2( ) (3)
6 Stoffdaten
6.1 Umrechnung von Gewichtsanteilen (ξ) in Mol-Anteile (x)
Für ein Zweistoffgemisch gelten die folgenden Formeln:
xM
M
AA
B
A
A
=+
−
1
11 ξ
ξ
(4)
oder umgekehrt:
ξAB
A
A
A
M
M
x
x
=+
−
1
11
(5)
Die Molmassen von n-Propanol (A) bzw. Wasser (B) betragen: MA = 60,10 g/mol ; MB = 18,02 g/mol
6.2 Dampfdruckkurven der reinen Komponenten n-Propanol und Wasser Alle Modellgleichungen zur Beschreibung von Dampf-Flüssigkeits-Gleichgewichten basieren auf den Dampfdrücken Pi o der reinen Komponenten bei der betreffenden Siedetemperatur T [°C].
Diese lassen sich z.B. mit Hilfe der Antoine-Gleichung darstellen: log P AB
T Co = −
+ (6)
A B C Die Koeffizienten lauten für T in [°C] n-Propanol 7,74416 1437,686 198,463 (Daten aus [5]) Wasser 8,07131 1730,630 233,426
Universität Dortmund Gemeinsames Praktikum, Praktikumsbereich TCA Zeichen: Fischer/Jörissen Fachbereich Bio- und TC 1 Seite: 13 Chemieingenieurwesen Gleichgewichtsbestimmung und offene Destillation Datum: August 2005
Nur zum persönlichen Gebrauch bestimmt!
400
500
600
700
800
900
1000
85 90 95 100 105
Temperatur [°C]
Dam
pfd
ruck
[T
orr
]
n-Propanol
Wasser
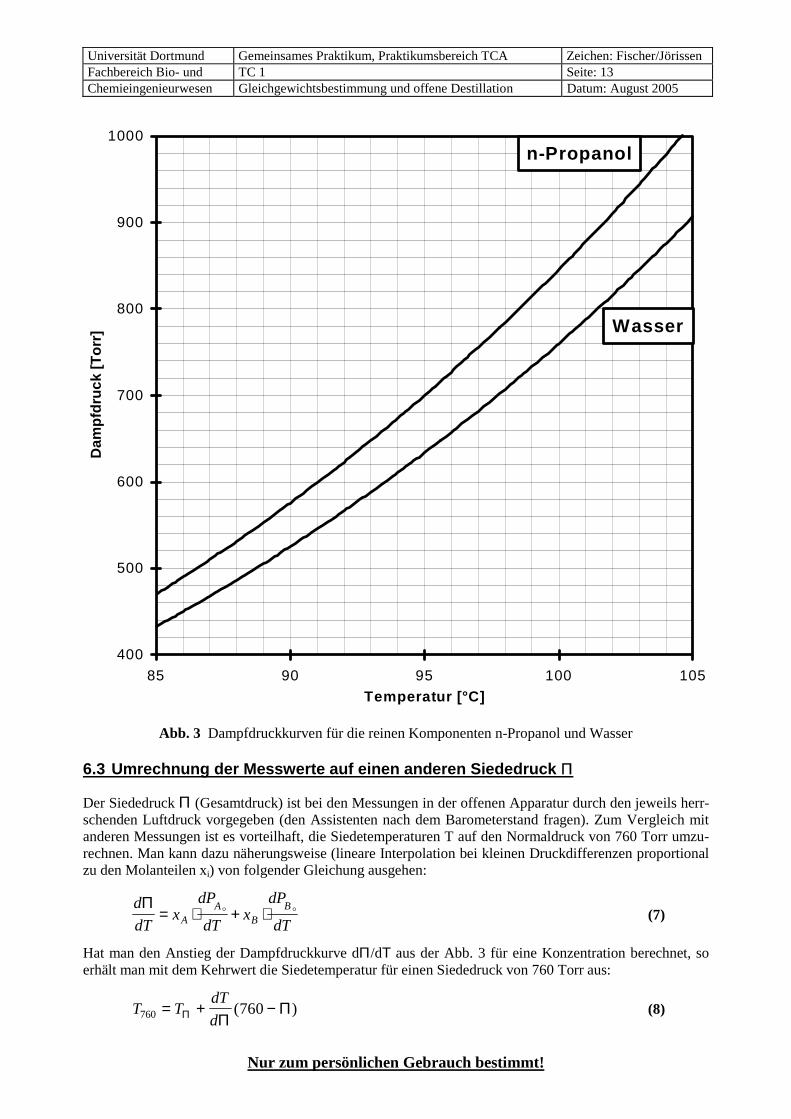
Abb. 3 Dampfdruckkurven für die reinen Komponenten n-Propanol und Wasser 6.3 Umrechnung der Messwerte auf einen anderen Siededruck Π
Der Siededruck Π (Gesamtdruck) ist bei den Messungen in der offenen Apparatur durch den jeweils herr-schenden Luftdruck vorgegeben (den Assistenten nach dem Barometerstand fragen). Zum Vergleich mit anderen Messungen ist es vorteilhaft, die Siedetemperaturen T auf den Normaldruck von 760 Torr umzu-rechnen. Man kann dazu näherungsweise (lineare Interpolation bei kleinen Druckdifferenzen proportional zu den Molanteilen xi) von folgender Gleichung ausgehen:
d
dTx
dP
dTx
dP
dTAA
BBΠ
= ⋅ + ⋅o o (7)
Hat man den Anstieg der Dampfdruckkurve dΠ/dΤ aus der Abb. 3 für eine Konzentration berechnet, so erhält man mit dem Kehrwert die Siedetemperatur für einen Siededruck von 760 Torr aus:
T TdT
d760 760= + −Π ΠΠ( ) (8)

Universität Dortmund Gemeinsames Praktikum, Praktikumsbereich TCA Zeichen: Fischer/Jörissen Fachbereich Bio- und TC 1 Seite: 14 Chemieingenieurwesen Gleichgewichtsbestimmung und offene Destillation Datum: August 2005
Nur zum persönlichen Gebrauch bestimmt!
7 Grundlagen 7.1 Ideale Systeme Bei einem idealen System geht man davon aus, dass sich in der Flüssigkeit die einzelnen Komponenten vollständig miteinander mischen und gegenseitig keinen Effekt aufeinander ausüben (Mischungswärme ∆H misch = 0). Die Wechselwirkungen zwischen allen Molekülen in der flüssigen Phase sind in diesem Fall also identisch, für das hier betrachtete binäre Gemisch sowohl zwischen den gleichen Molekülen A und A bzw. B und B als auch zwischen den ungleichen Molekülen A und B. Jede Komponente i verhält sich dann unabhängig und zeigt eine Verdampfungstendenz – ausgedrückt durch den Partialdruck p i im Dampf –, die nur eine Funktion der Konzentration (Molenbruch x i) in der flüssigen Phase und des Dampfdruckes der reinen Komponente P i o bei der Systemtemperatur ist.
Das Raoult’sche Gesetz beschreibt diese Verhältnisse des idealen Systems wie folgt (siehe Abb. 4)
pA = PAo · xA pB = PBo · xB (9)
Die Dampfphase kann man bis zu mäßigen Drücken im allgemeinen näherungsweise als ideales Gas an-nehmen, so dass das Dalton’sche Gesetz gilt (Π = Gesamtdruck):
pii∑ = Π (10)
Daraus folgt (y i = Molenbruch der Komponente i in der Dampfphase):
pA = yA · ∏ pB = yB · ∏ (11)
Aus Gleichung (9) und (11) kombiniert erhält man als Beschreibung idealer Siedegleichgewichte:
yP x
yP x
A
A A
B
B B=
⋅=
⋅o o
Π Πbzw. (12)
0
20
40
60
80
100
120
140
160
180
200
0 0,1 0,2 0,3 0,4 0,5 0,6 0,7 0,8 0,9 1
Molenbruch des Leichtersieders A
Dam
pfd
ruck
[T
orr
]
AB
Dalton´sches Gesetz
Raoult´sches Gesetz
System:A = DibrommethanB = Dibrompropan
bei konstanterTemperatur 85 °C
Abb. 4 Gültigkeit des Raoult’schen Gesetzes bei einem idealen System
7.2 Die relative Flüchtigkeit (Trennfaktor)
Wenn man ein Gemisch zweier Flüssigkeiten erhitzt, dann tendiert die flüchtigere Komponente dazu, in größerem Ausmaß zu verdampfen als die weniger flüchtige Substanz. Demzufolge enthält der mit der flüssigen Mischung im Gleichgewicht stehende Dampf einen höheren Anteil der flüchtigeren Komponente als die Flüssigkeit. Das Ausmaß dieser Anreicherung im Dampf lässt sich bequem mit Hilfe des Begriffes der relativen Flüchtigkeit, dem Verhältnis aus den Flüchtigkeiten zweier Substanzen A und B in einer Lösung beschreiben:

Universität Dortmund Gemeinsames Praktikum, Praktikumsbereich TCA Zeichen: Fischer/Jörissen Fachbereich Bio- und TC 1 Seite: 15 Chemieingenieurwesen Gleichgewichtsbestimmung und offene Destillation Datum: August 2005
Nur zum persönlichen Gebrauch bestimmt!
α AB
A
A
B
B
A
A
B
B
yx
yx
y
x
x
y= = ⋅ (13)
Vereinbarungsgemäß bezieht sich der Index A auf die leichter siedende und der Index B auf die schwerer siedende Substanz. Dabei muss die leichter siedende Substanz nicht auch die leichter flüchtige Substanz sein (siehe azeotrope Systeme, Abschnitt 8.3). Häufig wird statt der Bezeichnung „relative Flüchtigkeit“ der Ausdruck Trennfaktor für αAB gebraucht. 7.3 Die Anwendung der relativen Flüchtigkeit bei idealen Gemischen
Mit pA = xA · PAo (9) pA = yA · ∏ (10) und
pB = xB · PBo (9) pB = yB · ∏ (10)
erhält man für ideale Systeme als relative Flüchtigkeit αAB das Verhältnis der Dampfdrücke P i o der beiden reinen Komponenten aus Gleichung 13:
α AB
A
B
P
P= o
o
(14)
Hierbei ist zu beachten, dass das Verhältnis der Dampfdrücke im allgemeinen nur bei konstanter Tem-peratur konstant bleibt (die Dampfdrücke ändern sich nicht linear und wegen der unterschiedlichen Ver-dampfungswärme nicht in gleicher Form für verschiedene Stoffe mit der Temperatur, siehe Abb. 3).
Eine Destillation wird aber normalerweise nicht bei konstanter Temperatur, sondern bei (annähernd) kon-stantem Druck betrieben, so dass sich die Siedetemperatur mit der Zusammensetzung ändert. Deshalb kann der Trennfaktor αAB bei idealen Gemischen zwar aus Gleichung 14 für jede Temperatur bestimmt werden, er ist aber nicht für den ganzen Konzentrationsbereich konstant. Bei kleinen Siedepunktsdifferen-zen, also kleinem Trennfaktor αAB , sind dessen Veränderungen im allgemeinen praktisch vernachlässig-bar. Bei großer Siedepunktsdifferenz ändert sich der Trennfaktor aber deutlich mit der Siedetemperatur (siehe System Benzol / p-Xylol, Abb. 5). 7.4 Darstellung der binären Dampf-Flüssigkeits-Gleichgewichte Die Abb. 5 a zeigt das Siedediagramm mit den Koordinaten Temperatur und Zusammensetzung (xA für die Flüssigkeit, yA für den Dampf; A bezieht sich auf die Komponente mit dem niedrigeren Siedepunkt). Die obere Kurve (Taulinie) ist die Dampfzusammensetzung , die untere Kurve (Siedelinie) entspricht der Flüssigkeitszusammensetzung. Wenn man beide Kurven durch eine horizontale Linie verbindet – entspre-chend einer konstanten Temperatur – dann repräsentieren die Schnittpunkte die Flüssigkeits- und Dampf-zusammensetzungen im Gleichgewicht zueinander bei dieser Temperatur. Der Dampf ist hier stets reicher an der flüchtigeren Komponente A als die Flüssigkeit.
Die Abb. 5 b stellt das gleiche System dar, wobei aber als Koordinaten die Zusammensetzungen (Molen-brüche) von Flüssigkeit und Dampf gewählt wurden (Gleichgewichtsdiagramm, McCabe-Thiele-Dia-gramm). Das McCabe-Thiele-Diagramm ist besonders übersichtlich und gestattet die graphische Ausle-gung der Rektifikation (siehe Versuch TV4), es enthält aber im Gegensatz zum Siedediagramm nicht mehr die Information der Siedetemperatur. Da die Dampfzusammensetzung yA bei diesem System stets größer als die Flüssigkeitszusammensetzung xA ist, tritt die flüchtigere Komponente immer in höherer Konzen-tration in der Dampfphase als in der flüssigen Phase auf, d.h. die Kurve verläuft oberhalb der Diagonalen.
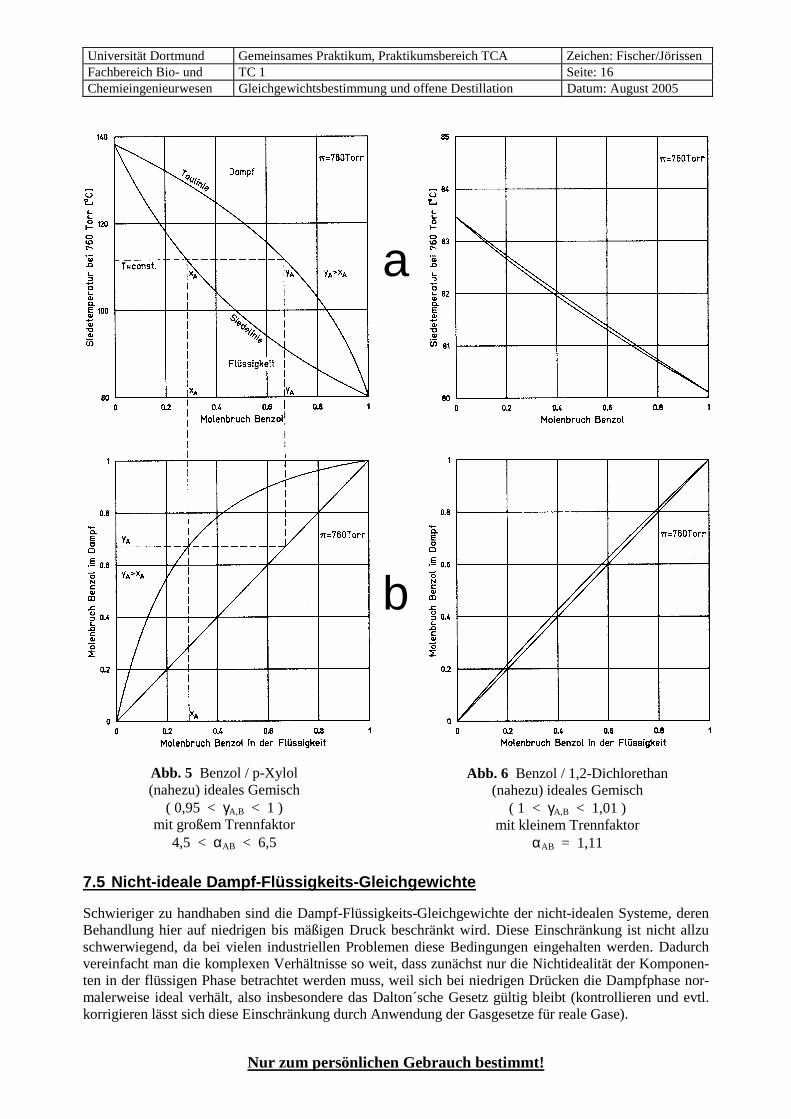
Bei konstantem Trennfaktor αAB wird das Gleichgewicht im McCabe-Thiele-Diagramm durch eine sym-metrische Kurve dargestellt, die sich in der Mitte um so weiter von der Diagonalen entfernt, je größer der Trennfaktor ist (siehe Abb. 5 b und 6 b). Wie oben erläutert, ist der Trennfaktor αAB bei konstantem Druck, also wechselnder Temperatur, nicht konstant, so dass die Gleichgewichtskurve auch bei idealen Systemen etwas verzerrt ist. Bei kleiner Siedepunktdifferenz (Abb. 6) sind die Abweichungen meistens vernach-lässigbar klein, bei einer großen Siedepunktdifferenz sind sie aber erkennbar (Abb. 5).
Universität Dortmund Gemeinsames Praktikum, Praktikumsbereich TCA Zeichen: Fischer/Jörissen Fachbereich Bio- und TC 1 Seite: 16 Chemieingenieurwesen Gleichgewichtsbestimmung und offene Destillation Datum: August 2005
Nur zum persönlichen Gebrauch bestimmt!
Abb. 5 Benzol / p-Xylol (nahezu) ideales Gemisch
( 0,95 < γA,B < 1 ) mit großem Trennfaktor
4,5 < αAB < 6,5
a
b
Abb. 6 Benzol / 1,2-Dichlorethan (nahezu) ideales Gemisch
( 1 < γA,B < 1,01 ) mit kleinem Trennfaktor
αAB = 1,11
7.5 Nicht-ideale Dampf-Flüssigkeits-Gleichgewichte Schwieriger zu handhaben sind die Dampf-Flüssigkeits-Gleichgewichte der nicht-idealen Systeme, deren Behandlung hier auf niedrigen bis mäßigen Druck beschränkt wird. Diese Einschränkung ist nicht allzu schwerwiegend, da bei vielen industriellen Problemen diese Bedingungen eingehalten werden. Dadurch vereinfacht man die komplexen Verhältnisse so weit, dass zunächst nur die Nichtidealität der Komponen-ten in der flüssigen Phase betrachtet werden muss, weil sich bei niedrigen Drücken die Dampfphase nor-malerweise ideal verhält, also insbesondere das Dalton´sche Gesetz gültig bleibt (kontrollieren und evtl. korrigieren lässt sich diese Einschränkung durch Anwendung der Gasgesetze für reale Gase).

Universität Dortmund Gemeinsames Praktikum, Praktikumsbereich TCA Zeichen: Fischer/Jörissen Fachbereich Bio- und TC 1 Seite: 17 Chemieingenieurwesen Gleichgewichtsbestimmung und offene Destillation Datum: August 2005
Nur zum persönlichen Gebrauch bestimmt!
Zurückgehend auf die Thermodynamik gebraucht man das von Lewis eingeführte Konzept der „Aktivität“, wonach die Konzentration xA durch die Aktivität aA – als „wirksame“ Konzentration – ersetzt wird, indem man die Konzentration xA mit einem Korrekturfaktor, dem Aktivitätskoeffizienten γA multipliziert:
aA = γA · xA (15)
Wie besprochen beschreibt für ideale Systeme das Raoult’sche Gesetz den Partialdruck pA der Kompo-nente A in der Dampfphase über einer idealen Lösung:
pA = PAo · xA (9)
In nicht-idealen Lösungen stellt jedoch xA nicht die tatsächlich wirksame Konzentration dar, die die Flüchtigkeitstendenz der Komponente A aus der flüssigen Phase bestimmt. In Wirklichkeit ist die Aktivi-tät aA das wirksame Maß dieser Konzentration. Demzufolge wird geschrieben:
ideal x xA B( , ) real a x a xA A A B B B( , )= ⋅ = ⋅γ γ (15)
Raoult: p x PA A A= ⋅
o (9) p x PA A A A
= ⋅ ⋅γo
(16)
Dalton: p yA A= ⋅ Π (11) p yA A= ⋅ Π (11)
yA
x PA A=
⋅o
Π (12) yA
x PA A A=
⋅ ⋅γo
Π (17)
α AB
A
A
B
B
A
B
yx
yx
P
P= =
o
o
(13 / 14) αγ
γAB
A
A
B
B
A A
B B
yx
yx
P
P= =
⋅
⋅o
o
(13 / 18)
Tabelle 1 Gegenüberstellung der Gleichungen für ideale und reale Flüssigkeitsgemische
Die realen Gleichungen unterscheiden sich von der Beschreibung idealer Systeme nur durch die Ein-führung der Aktivitätskoeffizienten γA und γB als Korrekturfaktoren, die für ideale Systeme und bei Rein-stoffen den Wert γ = 1 annehmen. Zu Abweichungen von der Idealität führen Änderungen der molekularen Wechselwirkungen in der Flüssigkeit bei Zugabe der zweiten Komponente. Die Wechselwirkungen zwi-schen den ungleichen Molekülen A und B können entweder
• vermindert oder
• verstärkt
sein gegenüber denjenigen zwischen den gleichen Molekülen A und A bzw. B und B. Im ersten Fall erhö-hen die gegenseitigen molekularen Beeinflussungen die Flüchtigkeit einer Komponente verglichen mit dem idealen Zustand p i = Pi o · x.i (positive Abweichung vom Raoult’schen Gesetz) und γ i wird größer als 1 sein. Im zweiten Fall verkleinert die gegenseitige molekulare Beeinflussung die Flüchtigkeit auf einen Wert unter p i = Pi o · x i (negative Abweichung), dann wird γ i niedriger als 1 liegen.
Üblicherweise wird die Abweichung von der Idealität durch den Logarithmus der Aktivitätskoeffizienten beschrieben. Falls γ > 1 ist, dann wird log γ > 0 sein, es liegt also eine positive Abweichung vor. Ist γ < 1, dann wird der log γ < 0 sein und entsprechend handelt es sich um eine negative Abweichung.
Die Abweichung vom idealen Verhalten und damit die Aktivitätskoeffizienten – sowie die relative Flüch-tigkeit αAB – hängen von der Zusammensetzung der Mischung ab. Die Gleichgewichtskurve im McCabe-Thiele-Diagramm weicht deshalb mehr oder weniger deutlich von der symmetrischen Kurvenform ab (siehe Abb. 7 b bis 9 b). Am stärksten unterscheidet sich der Aktivitätskoeffizient γ i von 1, wenn der be-treffende Stoff nur in kleiner Konzentration gegenüber einem großen Überschuss der anderen Komponen-te vorliegt (Aktivitätskoeffizient bei unendlicher Verdünnung, siehe Abb. 7 c bis 9 c). Gemäß Gleichung (16) macht sich dies im Flüssig-Dampf-Gleichgewicht aber nur wenig bemerkbar, weil γ i mit der kleinen Konzentration x i multipliziert wird.
Universität Dortmund Gemeinsames Praktikum, Praktikumsbereich TCA Zeichen: Fischer/Jörissen Fachbereich Bio- und TC 1 Seite: 18 Chemieingenieurwesen Gleichgewichtsbestimmung und offene Destillation Datum: August 2005
Nur zum persönlichen Gebrauch bestimmt!
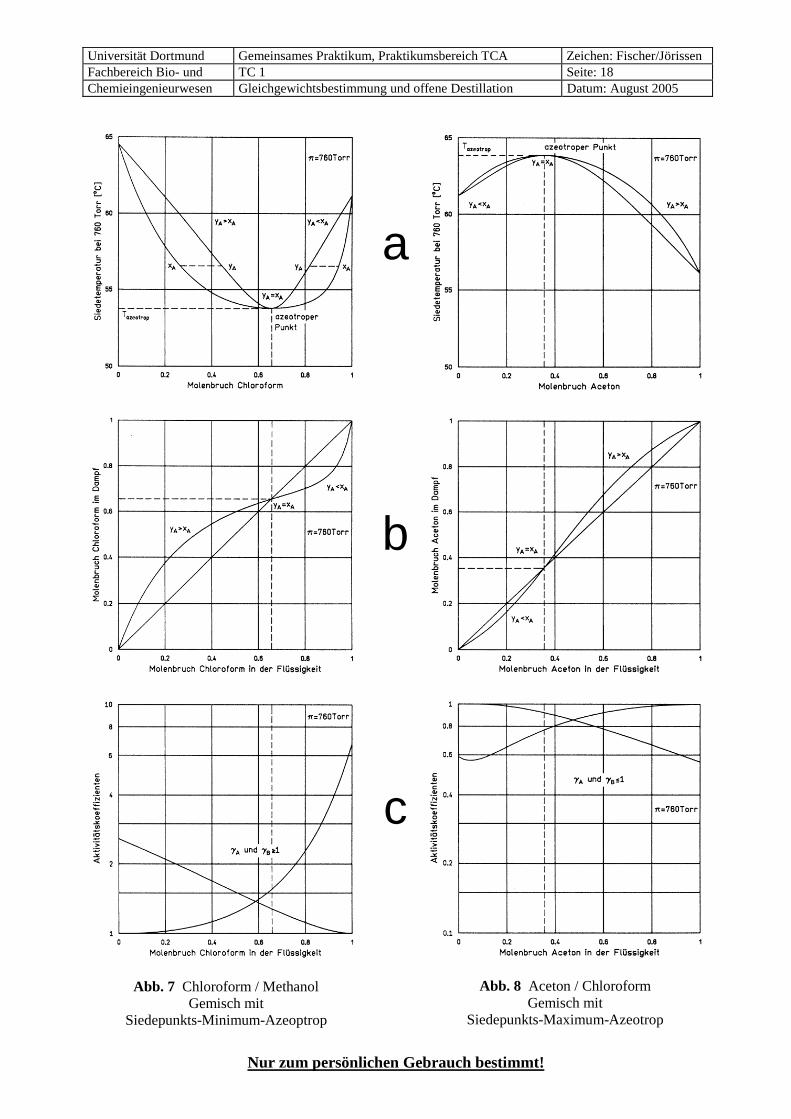
Abb. 7 Chloroform / Methanol Gemisch mit
Siedepunkts-Minimum-Azeoptrop
a
b
c
Abb. 8 Aceton / Chloroform Gemisch mit
Siedepunkts-Maximum-Azeotrop

Universität Dortmund Gemeinsames Praktikum, Praktikumsbereich TCA Zeichen: Fischer/Jörissen Fachbereich Bio- und TC 1 Seite: 19 Chemieingenieurwesen Gleichgewichtsbestimmung und offene Destillation Datum: August 2005
Nur zum persönlichen Gebrauch bestimmt!
7.6 Azeotrope Systeme Die Abweichungen vom idealen Verhalten können so groß werden, dass die Gleichgewichtskurve die Diagonale im sogenannten „azeotropen Punkt“ schneidet (Abb. 7b und 8b), d.h. an diesem Punkt haben Dampf und Flüssigkeit die gleiche Zusammensetzung (xA = yA, xB = yB, Trennfaktor αAB = 1). Eine Kom-ponente ist links vom azeotropen Punkt die leichter flüchtige und rechts davon die schwerer flüchtige *).
Am azeotropen Punkt ist eine destillative Trennung des Gemisches nicht möglich, und er kann bei einem destillativen Trennverfahren ohne weitere Maßnahmen nicht überschritten werden. Einige Möglichkeiten zur Überwindung des azeotropen Punktes werden im Abschnitt 7.9 genannt.
Es kann also vorkommen, dass die Komponente B eine höhere Flüchtigkeit als die Komponente A besitzt, obwohl letztere als reiner Stoff den höheren Dampfdruck hat. Anhand von Gleichung (19) (siehe auch Gleichung (18) in Tabelle 1) lässt sich dieser Zusammenhang erläutern. Am azeotropen Punkt gilt:
αγ
γAB
A A
B B
P
P=
⋅
⋅=o
o
1 (19)
und daraus folgt:
γB / γA = PAo / PBo (20)
Für den azeotropen Punkt (αAB = 1) ist die Abweichung beider Substanzen von der Idealität also gerade ausreichend, um die Wirkung des Unterschiedes in den Dampfdrücken der reinen Stoffe auf die relative Flüchtigkeit αAB genau aufzuheben.
Wenn das Verhältnis der Aktivitätskoeffizienten γB / γA größer als das Verhältnis der Dampfdrücke der reinen Stoffe PAo / PBo wird, dann wird αAB < 1, d.h. A – die leichter siedende Komponente – wird zur weniger flüchtigen und B zur leichter flüchtigen Komponente.
Es gibt zwei Arten von azeotropen Systemen:
Beim Siedepunkts-Minimum-Azeotrop (Abb. 7, meistens einfach „Minimum-Azeotrop“ genannt) erfolgt bei Zugabe der leichter siedenden Komponente A zur schwerer siedenden Komponente B zunächst eine stärkere Erniedrigung des Siedepunktes, als dem Raoult´schen Gesetz entspricht. Die leichter siedende Komponente A ist auch die leichter flüchtige, die im Dampf eine höhere Konzentration hat als in der Flüssigkeit. Dies entspricht einem normalen, nicht-azeotropen System, wie in Abb. 5, d.h. die Gleich-gewichtskurve in Abb. 7 b liegt oberhalb der Diagonalen. Am azeotropen Punkt, der den niedrigsten Sie-depunkt im ganzen System aufweist, drehen sich die Verhältnisse um. Bei weiterer Zugabe von A steigt der Siedepunkt wieder an, der Dampf enthält weniger A als die Flüssigkeit, A ist also zur schwerer flüchtigen Komponente geworden, und die Gleichgewichtskurve liegt unterhalb der Diagonalen.
Beim Siedepunkts-Maximum-Azeotrop (Abb. 8, „Maximum-Azeotrop“) liegen die Verhältnisse genau umgekehrt. Damit bei den Abb. 7 b und 8 b keine Verwechslungen auftreten können, verwendet man im Siede- und Gleichgewichts-Diagramm grundsätzlich die Konzentrationen xA und yA der leichter siedenden Komponente A, also der Komponente mit dem niedrigeren Siedepunkt, die ja nicht immer die leichter flüchtige Komponente ist. Nur so ist es möglich, im Mc-Cabe-Thiele-Diagramm eindeutig zu erkennen, ob es sich um ein Minimum- (Abb. 7 b) oder ein Maximum-Azeotrop (Abb. 8 b) handelt.
Vereinfachend kann man sich vorstellen, dass es sich bei dem Stoffgemisch mit der azeotropen Zusam-mensetzung um einen zusätzlichen dritten Stoff handelt. Im Falle des Siedepunkts-Minimum-Azeotrops hat er den niedrigsten Siedepunkt der drei Stoffe. Der linke Teil des Gleichgewichtsdiagramms (Abb. 7 b) bis zum azeotropen Punkt stellt demnach ein Siedegleichgewicht dar zwischen der schwerer siedenden _____________________________________________________________________________________ *) Hinweis: Bei einer nur kleinen Differenz der Siedepunkte können u.U. schon geringe Abweichungen
von der Idealität oder sogar nur der unterschiedliche Verlauf der Dampfdruckkurven (siehe Abb. 3) bei (nahezu) idealem Verhalten zu einem Azeotrop führen.

Universität Dortmund Gemeinsames Praktikum, Praktikumsbereich TCA Zeichen: Fischer/Jörissen Fachbereich Bio- und TC 1 Seite: 20 Chemieingenieurwesen Gleichgewichtsbestimmung und offene Destillation Datum: August 2005
Nur zum persönlichen Gebrauch bestimmt!
Komponente B und dem azeotropen Gemisch in der üblichen Auftragungsart, d.h. die Gleichgewichtskur-ve liegt in Abb. 7 b oberhalb der Diagonalen. Der rechte Teil von Abb. 7 b stellt das Siedegleichgewicht zwischen dem azeotropen Gemisch und der leichter siedenden Komponente A dar, aber in der nicht übli-chen Auftragungsart, denn die Komponente A ist nur gegenüber der Komponente B die leichter siedende Substanz, während A gegenüber der azeotropen Zusammensetzung den höheren Siedepunkt hat. Die Gleichgewichtskurve liegt deshalb in diesem Bereich der Abb. 7 b unterhalb der Diagonalen. Für Systeme mit einem Siedepunkts-Maximum-Azeotrop sind die Verhältnisse genau umgekehrt (Abb. 8). 7.7 Molekulare Ursachen des nicht-idealen Siedeverhaltens
Während man für ein ideales Gas keine Wechselwirkungen zwischen den Molekülen annimmt, wird eine Flüssigkeit durch Wechselwirkungen zusammengehalten, für deren Überwindung beim Verdampfungs-vorgang die Verdampfungswärme aufgebracht werden muss. Wie bereits erwähnt sind diese Wechselwir-kungen bei einem idealen Flüssigkeitsgemisch von den Konzentrationen unabhängig. In einem realen Ge-misch können die Wechselwirkungen entweder gelockert oder verstärkt werden, dementsprechend nimmt die Verdampfungstendenz zu oder ab (positive oder negative Abweichung vom Raoult´schen Gesetz).
Einen besonders großen Einfluss auf die Siedegleichgewichte haben die sogenannten Wasserstoffbrücken (häufig auch Wasserstoffbrücken-Bindung genannt), die sehr starke Wechselwirkungskräfte zwischen den Molekülen bewirken können. Am deutlichsten sind sie im Wasser ausgeprägt: in dem gewinkelten Was-sermolekül tritt eine starke Polarisation mit einer negativen Teilladung am Sauerstoff und einer positiven Teilladung am Wasserstoff auf. Zwischen diesen Teilladungen benachbarter Moleküle existieren starke Anziehungskräfte. Sie führen zu einem dreidimensionalen Netzwerk, in dem man Bindungen zwischen allen Sauerstoff- und Wassersoff-Atomen annehmen kann. Damit erklärt sich auch der – gemessen am Molekulargewicht – ungewöhnlich hohe Siedepunkt des Wassers.
Wasserstoffbrücken können gebildet werden, wenn:
a) im Molekül ein oder mehrere Wasserstoffatome infolge der Molekülaufbaus positiv polarisiert sind,
b) im Molekül ein oder mehrere Atome negativ polarisiert sind (meistens Sauerstoff, aber auch Stickstoff u.a.).
Man kann die Stoffe dementsprechend in fünf Gruppen einteilen:
I Verbindungen, die die Bedingungen a) und b) erfüllen, und dabei dreidimensionale Netzwerke von starken Wasserstoffbrücken zwischen OH- oder NH-Gruppen ausbilden,
typisches Beispiel ist Wasser, aber auch mehrwertige Alkohole gehören dazu,
II andere Verbindungen, die die Bedingungen a) und b) erfüllen, z.B. Alkohole, Säuren,
III Verbindungen, die nur Bedingung b) erfüllen, z.B. Ketone, Ether,
IV Verbindungen, die nur Bedingung a) erfüllen, z.B. Chloroform,
V Verbindungen, die weder Bedingung a) noch b) erfüllen, z.B. Kohlenwasserstoffe.
Mit Hilfe dieser Einteilung lassen sich Voraussagen über das Siedegleichgewicht machen:
• positive Abweichungen vom Raoult’schen Gesetz treten auf, wenn die Wasserstoffbrücken, die in den reinen Stoffen der Gruppen I und II vorliegen, gelockert werden. Sicher findet dies bei Mischung mit Stoffen der Gruppe V statt, die ja keine neuen Wasserstoffbrücken bilden können. Auch bei Mischun-gen mit Stoffen aus Gruppe IV wirkt sich der Abbau bestehender Wasserstoffbrücken meistens stärker aus als der Aufbau von neuen. Das System Chloroform / Methanol ist ein Beispiel dafür (Abb. 7).
• im allgemeinen ergeben sich bei Mischungen aus Stoffen der Gruppen I und II mit Stoffen der Gruppe III auch positive Abweichungen vom Raoult’schen Gesetz. Da hier aber relativ starke neue Wasser-stoffbrücken möglich sind, kann es auch zu einem Ausgleich oder sogar negativen Abweichungen kommen. Das gleiche gilt für Mischungen innerhalb der Gruppen I und II. Wegen der sehr starken Wasserstoffbrücken im Wasser zeigen sich bei Systemen organischer Stoffe zusammen mit Wasser fast immer positive Abweichungen.

Universität Dortmund Gemeinsames Praktikum, Praktikumsbereich TCA Zeichen: Fischer/Jörissen Fachbereich Bio- und TC 1 Seite: 21 Chemieingenieurwesen Gleichgewichtsbestimmung und offene Destillation Datum: August 2005
Nur zum persönlichen Gebrauch bestimmt!
• Stets negative Abweichungen treten bei Gemischen der Gruppen III mit IV auf, da dann ja nur neue Wasserstoffbrücken gebildet werden. Ein Beispiel dafür ist das System Aceton / Chloroform (Abb. 8).
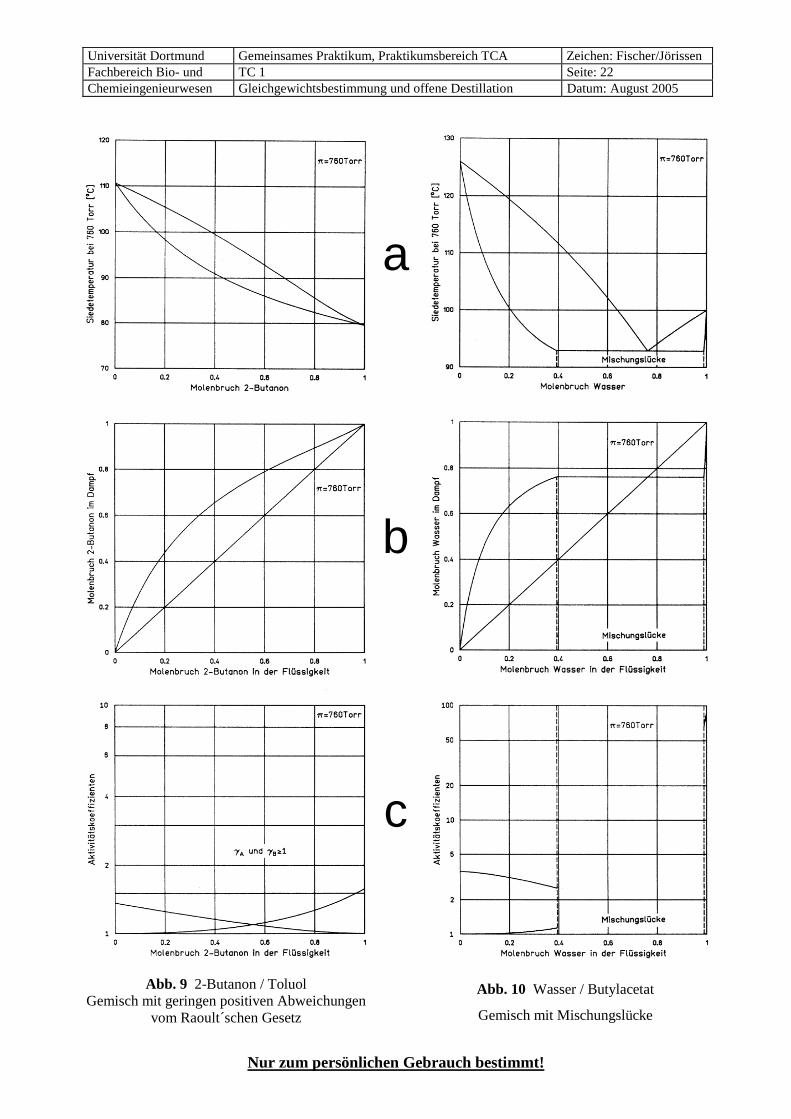
• Bei allen übrigen Mischungen sind keine oder nur schwache Wasserstoffbrücken möglich und andere Wechselwirkungen haben meistens nur einen geringen Effekt auf das Dampf-Flüssigkeits-Gleich-gewicht. Diese Mischungen verhalten sich deshalb im allgemeinen weitgehend ideal, wie das Beispiel 2-Butanon / Toluol (Abb. 9) zeigt.
7.8 Systeme mit Mischungslücke Alles bisher Gesagte galt stets für vollständig mischbare Systeme. Systeme mit einer Mischungslücke müssen gesondert betrachtet werden. Diese treten besonders bei der Kombination von Gruppe I mit V oder IV häufig auf, also wenn starke Wasserstoffbrücken in dem reinen Stoff der Gruppe I durch die Mischung mit der anderen Komponente abgebaut werden müssten, ohne dass neue gebildet werden können (typische Beispiele: Wasser / Kohlenwasserstoffe, Wasser / Chlorkohlenwasserstoffe).
Im Idealfall von zwei völlig unmischbaren Flüssigkeiten (der natürlich real nicht existiert) beeinflussen sich die beiden Komponenten in ihrem Siedeverhalten gegenseitig nicht, so dass sich die Dampfdrücke der beiden Phasen einfach addieren. Das bedeutet, dass der Siedepunkt des Gemisches immer niedriger liegt als derjenige der beiden Einzelkomponenten (ähnlich wie bei einem Siedepunkts-Minimum-Azeotrop).
Diesen Effekt, der im Bereich der Mischungslücke qualitativ auch für die realen, teilweise mischbaren Systeme gilt, macht man sich bei der Wasserdampf-Destillation zunutze. Dabei kann ein mit Wasser nicht mischbarer Stoff zusammen mit Wasserdampf unterhalb seiner Siedetemperatur abdestilliert und in einem Phasenabscheider wieder vom Wasser getrennt werden.
Im Bereich der Mischungslücke haben die beiden flüssigen Phasen die gleiche Dampfphase und die glei-che Siedetemperatur, wobei nur wichtig ist, dass sie vorliegen, aber nicht, in welcher Menge sie vorhan-den sind. Es gilt also nicht das Raoult’sche Gesetz (mit Korrektur durch die Aktivitätskoeffizienten nach Gleichung 16), sondern Dampfzusammensetzung und Siedetemperatur bleiben im gesamten Bereich der Mischungslücke konstant (siehe Abb. 10). 7.9 Temperatureinfluss auf Siedegleichgewichte,
Überwindung des azeotropen Punktes Bei dem Auf- und Abbau der beschriebenen Wechselwirkungen zwischen den Molekülen handelt es sich im Prinzip um chemische Reaktionen, deren Ablauf bekanntlich stark von der Temperatur abhängig ist. Vor allem bei den azeotropen Systemen, bei denen sich das nicht-ideale Verhalten besonders deutlich be-merkbar macht, kann sich das Siedegleichgewicht mit der Siedetemperatur und damit dem Druck ändern.
Wenn sich ein azeotropes Stoffsystem so verhält, kann man dies zur Überwindung des azeotropen Punktes bei der Destillation nach dem „Zweidruck-Verfahren“ nutzen. Dabei wird die destillative Trennung in einer ersten Stufe bis in die Nähe des azeotropen Punktes durchgeführt. Für die Fortsetzung der Destil-lation wählt man den Druck (und damit die Temperatur) so, dass die Endkonzentration der ersten Stufe als Anfangskonzentration der zweiten Stufe auf der anderen Seite des azeotropen Punktes liegt.
Andere Methoden zur Überwindung eines azeotropen Punktes verwenden einen Zusatzstoff, der danach wieder (destillativ) abgetrennt werden muss. Bei der „Azeotrop-Rektifikation“ setzt man ein „Schlepp-mittel“ zu, das mit den beiden Komponenten ein ternäres Azeotrop bildet, das den niedrigsten Siedepunkt aller Komponenten aufweist und abdestilliert werden kann. Besonders günstig ist es, wenn sich dieses Gemisch nach der Kondensation in zwei flüssige Phasen trennt („Heteroazeotrop-Rektifikation“), die man dann destillativ aufarbeiten kann (z.B. Trennung von Ethanol und Wasser mit Cyclohexan [1-4]). Bei der „Extraktiv-Rektifikation“ [1-4] wählt man einen Zusatzstoff (z.B. Ethylengycol), der die Flüchtigkeit einer Komponente erniedrigt (z.B. Wasser), so dass sich die andere rein abdestillieren lässt (z.B. Ethanol).
Universität Dortmund Gemeinsames Praktikum, Praktikumsbereich TCA Zeichen: Fischer/Jörissen Fachbereich Bio- und TC 1 Seite: 22 Chemieingenieurwesen Gleichgewichtsbestimmung und offene Destillation Datum: August 2005
Nur zum persönlichen Gebrauch bestimmt!
Abb. 9 2-Butanon / Toluol Gemisch mit geringen positiven Abweichungen
vom Raoult´schen Gesetz
a
b
c
Abb. 10 Wasser / Butylacetat
Gemisch mit Mischungslücke

Universität Dortmund Gemeinsames Praktikum, Praktikumsbereich TCA Zeichen: Fischer/Jörissen Fachbereich Bio- und TC 1 Seite: 23 Chemieingenieurwesen Gleichgewichtsbestimmung und offene Destillation Datum: August 2005
Nur zum persönlichen Gebrauch bestimmt!
8 Modellgleichungen für Siedegleichgewichte
Eine Beziehung zwischen den Aktivitätskoeffizienten γA , γB und der Zusammensetzung lässt sich mit der aus der physikalischen Chemie bekannten Gibbs-Duhem-Gleichung bei konstanter Temperatur und kon-stantem Druck aufstellen:
xd
dxx
d
dxAA
A T PB
B
A T P
=
= −
ln ln
, ,
γ γ (21)
Diese hier für binäre Systeme formulierte Gleichung gilt ganz allgemein und beinhaltet keinerlei Annah-men. Zur Auswertung müssen jedoch einige Annahmen gemacht werden.
Um den in den Abb. 7 c - 10 c dargestellten Zusammenhang zwischen den Aktivitätskoeffizienten und der Zusammensetzung einer Mischung formelmäßig zu erfassen, wurden eine Reihe von Ansätzen vorge-schlagen, die meist von der Gibbs-Duhem-Gleichung ausgehen. Da nicht sowohl die Temperatur als auch der Druck in einem zweiphasigen Gleichgewicht über den gesamten Mischungsbereich konstant gehalten werden können, hängen die vorgeschlagenen Lösungen davon ab, welche Annahmen oder Vereinfachun-gen eingeführt werden.
Die bekanntesten Ansätze sind: 1. Margules 2. van Laar 3. Wilson 4. NRTL-Gleichung (Non-Random Two-Liquid Equation) 5. UNIQUAC-Gleichung (Universal Quasi-Chemical Equation)
Die Ansätze zur Beschreibung von Dampf-Flüssigkeitsgleichgewichten werden in [1-6] näher behandelt (mit ergänzenden Literaturangaben). Je nach Stoffsystem kann jeder dieser Ansätze die beste Lösung lie-fern. In der „Dortmunder Datenbank“, deren Aufbau am Lehrstuhl für Technische Chemie B der Univer-sität Dortmund begonnen wurde, sind die in der Literatur veröffentlichten Gleichgewichtsdaten und je-weils die Koeffizienten der o.g. Modellgleichungen aufgeführt [5]. Ausgehend von dem UNIQUAC-Ansatz, der chemische Strukturelemente berücksichtigt, wurde die UNIFAC-Methode (Universal Func-tional Group Activity Coefficients) entwickelt, die aus der Kenntnis der chemischen Struktur der Stoffe eine Vorhersage ihres Dampf-Flüssigkeits-Gleichgewichtes gestattet [6,7]. Sowohl die Dortmunder Daten-bank als auch die UNIFAC-Methode werden fortlaufend aktualisiert und weiterentwickelt [5-7]. 8.1 Die van Laar-Gleichung für binäre Gemische
Als relativ leicht nachvollziehbares Beispiel sollen hier die van Laar-Gleichung behandelt werden, ein Zwei-Konstanten-Gleichungssystem mit den stoffspezifischen Konstanten CA und CB:
log γA = CA / [1 + (CAxA / CBxB)]2 (22)
log γB = CB / [ 1 + (CBxB / CAxA)]2 (23)
bzw. umgeformt (Ableitung siehe folgende Seite):
CA = log γA [1 + (xB log γB / xA log γA)]2 (24)
CB = log γB [1 + (xA log γA / xB log γB)]2 (25)
mit den Randbedingungen: xA = 0 → log γA = CA xA = 1 → log γA = 0 und γA = 1 xB = 0 → log γB = CB xB = 1 → log γB = 0 und γB = 1
CA und CB sind also die Logarithmen der Aktivitätskoeffizienten γA und γB bei unendlicher Verdünnung. Umformung von Gleichung 22 in 24 (23 in 25 analog) :

Universität Dortmund Gemeinsames Praktikum, Praktikumsbereich TCA Zeichen: Fischer/Jörissen Fachbereich Bio- und TC 1 Seite: 24 Chemieingenieurwesen Gleichgewichtsbestimmung und offene Destillation Datum: August 2005
Nur zum persönlichen Gebrauch bestimmt!
[ ]logγ A
A
A A
B B
A B B
B B A A
C
C x
C x
C C x
C x C x=
+⋅⋅
=⋅
⋅ + ⋅1
2
2 2
2 (26)
[ ]logγ B
B
B B
A A
B A A
A A B B
C
C x
C x
C C x
C x C x=
+⋅⋅
=⋅
⋅ + ⋅1
2
2 2
2 (27)
Daraus folgt: log
log
γγ
A
B
A B B
A B A
C C x
C C x=
⋅ ⋅⋅ ⋅
2 2
2 2 und C
C
x
xB
A
A A
B B
=⋅⋅
2
2
log
log
γγ
eingesetzt in (27):
loglog
log
γγγ
BB
A A
B B
B
A
C
x
x
x
x
=
+⋅⋅
⋅
12
2
2
ergibt: Cx
xB BA A
B B
= ⋅ +⋅⋅
log
log
logγ
γγ
12
(25) analog: Cx
xA AB B
A A
= ⋅ +⋅⋅
log
log
logγ
γγ
12
(24)
8.2 Linearisierung der van Laar Gleichung Nach der vorangehenden Ableitung gilt:
log
log
log
log
,γγ
γγ
A
B
A B B
A B A
A A
B B
A
B
B
A
C C x
C C x
C x
C x
C
C=
⋅ ⋅⋅ ⋅
⋅⋅
= ⋅
2 2
2 2
0 5
und umgeformt
Setzt man diesen Ausdruck in die van Laar´sche Gleichung (22) ein:
logγ AA
A A
B B
C
C x
C x
=
+⋅⋅
1
2 (22)
ergibt sich:
loglog
log
,γ
γγ
AA
A B
B A
C
C
C
=
+⋅⋅
10 5 2
Umgeformt:
( )log log,
,,γ γA
A
BB A
C
CC
0 50 5
0 5+ ⋅
=
und weiter umgeformt:
( ) ( )log log, ,
,,γ γA A
A
BBC
C
C0 5 0 5
0 50 5= −
⋅ (28)
analog: ( ) ( )log log, ,
,,
γ γB BB
AAC
C
C
0 5 0 50 5
0 5= −
⋅ (29)
Universität Dortmund Gemeinsames Praktikum, Praktikumsbereich TCA Zeichen: Fischer/Jörissen Fachbereich Bio- und TC 1 Seite: 25 Chemieingenieurwesen Gleichgewichtsbestimmung und offene Destillation Datum: August 2005
Nur zum persönlichen Gebrauch bestimmt!
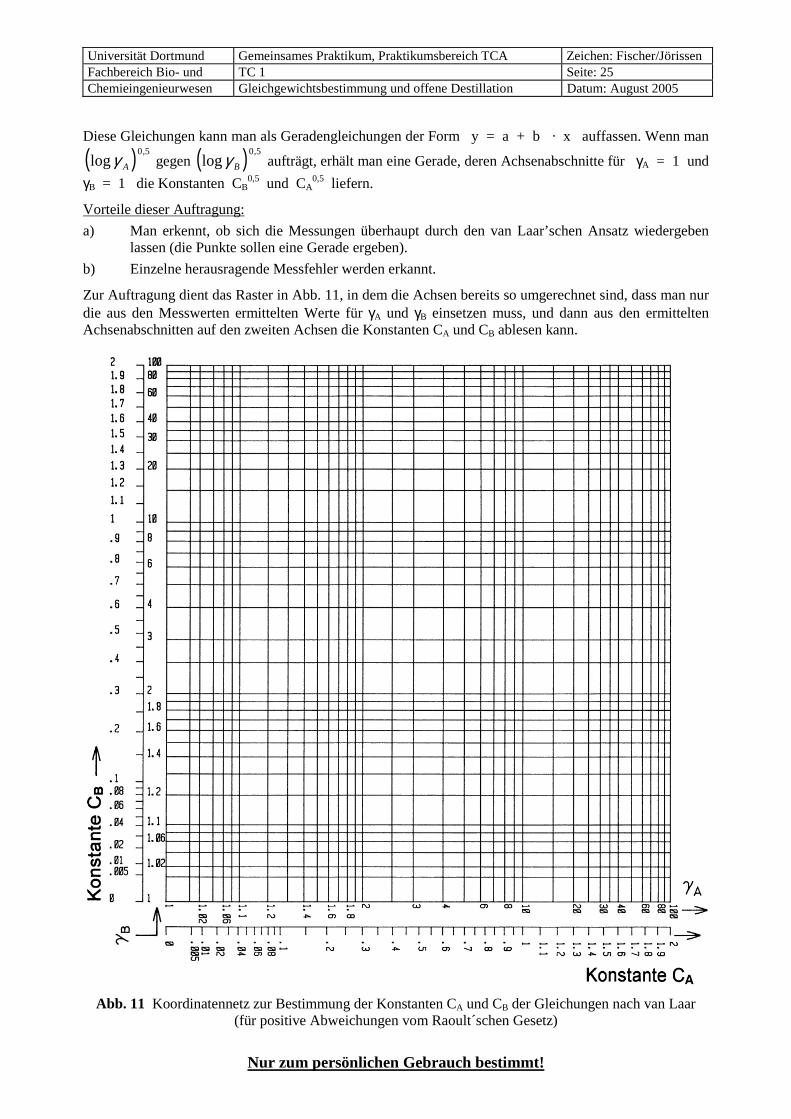
Diese Gleichungen kann man als Geradengleichungen der Form y = a + b · x auffassen. Wenn man
( )log,
γ A
0 5 gegen ( )log
,γ B
0 5 aufträgt, erhält man eine Gerade, deren Achsenabschnitte für γA = 1 und
γB = 1 die Konstanten CB0,5 und CA
0,5 liefern.
Vorteile dieser Auftragung:
a) Man erkennt, ob sich die Messungen überhaupt durch den van Laar’schen Ansatz wiedergeben lassen (die Punkte sollen eine Gerade ergeben).
b) Einzelne herausragende Messfehler werden erkannt.
Zur Auftragung dient das Raster in Abb. 11, in dem die Achsen bereits so umgerechnet sind, dass man nur die aus den Messwerten ermittelten Werte für γA und γB einsetzen muss, und dann aus den ermittelten Achsenabschnitten auf den zweiten Achsen die Konstanten CA und CB ablesen kann.
Abb. 11 Koordinatennetz zur Bestimmung der Konstanten CA und CB der Gleichungen nach van Laar (für positive Abweichungen vom Raoult´schen Gesetz)

Universität Dortmund Gemeinsames Praktikum, Praktikumsbereich TCA Zeichen: Fischer/Jörissen Fachbereich Bio- und TC 1 Seite: 26 Chemieingenieurwesen Gleichgewichtsbestimmung und offene Destillation Datum: August 2005
Nur zum persönlichen Gebrauch bestimmt!
8.3 Berechnung der Konstanten der van Laar Gleichung aus einem Messpunkt 8.3.1 Berechnung aus dem azeotropen Punkt Wenn der Siedepunkt und die Zusammensetzung eines azeotropen Gemisches für einen gegebenen Gesamtdruck Π bekannt sind, lässt sich das gesamte Siedegleichgewicht einfach berechnen.
Am azeotropen Punkt haben Dampf und Flüssigkeit die gleiche Zusammensetzung. Dadurch lassen sich aus der (umgeformten) Gleichung (17):
y
x
PA
A
A A=⋅γ
o
Π (17)
die Aktivitätskoeffizienten einfach für das Azeotrop berechnen. Aus xA = yA am azeotropen Punkt folgt:
γ γAA
BBP P
= =Π Π
o o
am azeotropen Punkt (30)
Setzt man die Logarithmen dieser beiden Aktivitätskoeffizienten in die Gleichungen 24 und 25 ein
CA = log γA [1 + (xB log γB / xA log γA)]2 (24)
CB = log γB [1 + (xA log γA / xB log γB)]2 (25)
dann lassen sich die beiden Konstanten der van Laar’schen Gleichung CA und CB unmittelbar berechnen. 8.3.2 Berechnung aus einem beliebigen Messpunkt Die gemessenen Konzentrationen für Dampf (yA) und Flüssigkeit (xA) werden in die aus der umgeformten Gleichung (17) erhaltene Gleichung (31) eingesetzt:
γ γAA
A AB
B
B B
y
x P
y
x P=
⋅⋅
=⋅
⋅Π Π
o o
(31)
Mit den erhaltenen Aktivitätskoeffizienten verfährt man weiter wie unter 8.3.1.
Hinweis:
Die Berechnung von Siedegleichgewichten aus nur einem Messpunkt (Azeotrop oder ein anderer Punkt) liefert im allgemeinen gute Ergebnisse, wenn dieser im Konzentrationsbereich 0,25 ≤ xA ≤ 0,75 liegt; sie kann aber auch außerhalb dieses Bereiches angewandt werden. Man muss sich selbstverständlich dar-über im klaren sein, dass das Ergebnis nur auf einem einzigen Messpunkt beruht, dass also – im Gegensatz zu der in 8.2 behandelten Methode der linearen Auftragung mehrerer Messpunkte – Fehler nicht auffallen, und dass nicht beurteilt werden kann, ob die van Laar´sche Gleichung für das betrachtete System geeignet ist.
8.4 Berechnung der Aktivitätskoeffizienten und der Gleichgewichtskurve aus der van Laar Gleichung
8.4.1 Berechnung der Aktivitätskoeffizienten ( γA , γB / x - Diagramm ) Mit Hilfe der van Laar’schen Gleichungen (22) und (23) kann man die Aktivitätskoeffizienten für jede Zusammensetzung berechnen. Es ist sinnvoll, sie in einem γA , γB / x -Diagramm aufzutragen (siehe Abb. 7 – 10 c), aus dem dann die Aktivitätskoeffizienten γA und γB für beliebige Konzentrationen abgelesen werden können und ihr Verlauf in Abhängigkeit von den Konzentrationen erkennbar wird.
Universität Dortmund Gemeinsames Praktikum, Praktikumsbereich TCA Zeichen: Fischer/Jörissen Fachbereich Bio- und TC 1 Seite: 27 Chemieingenieurwesen Gleichgewichtsbestimmung und offene Destillation Datum: August 2005
Nur zum persönlichen Gebrauch bestimmt!
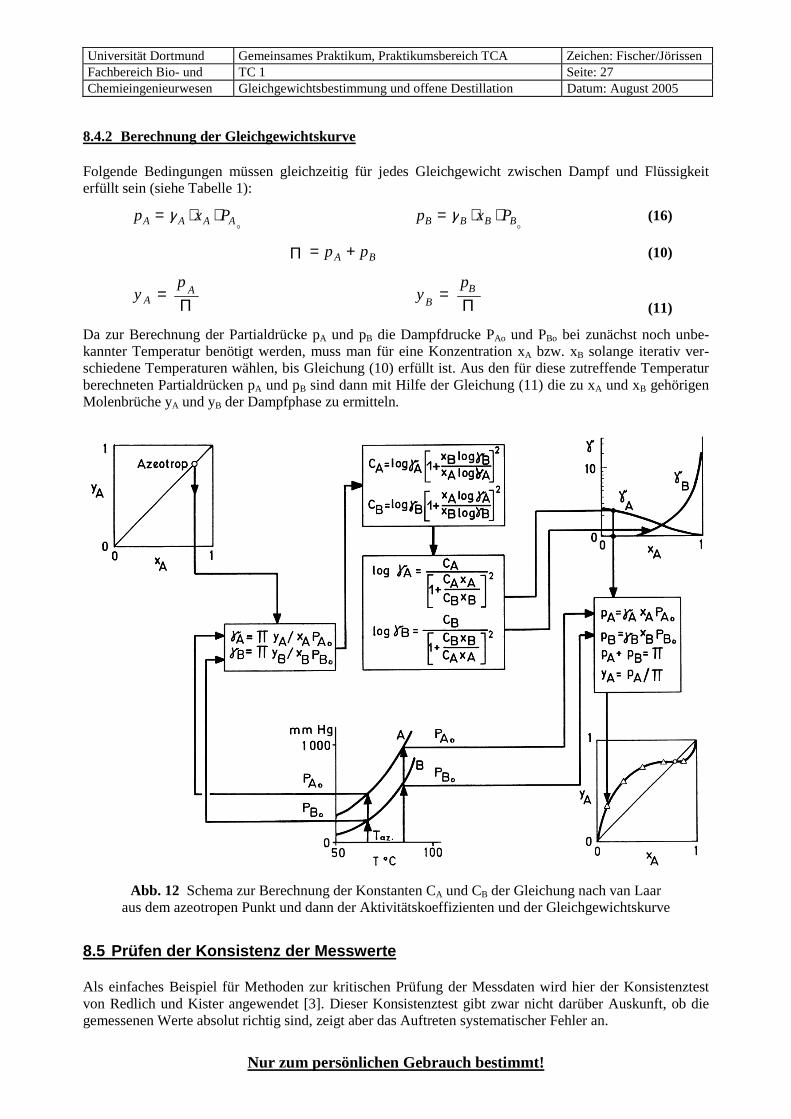
8.4.2 Berechnung der Gleichgewichtskurve Folgende Bedingungen müssen gleichzeitig für jedes Gleichgewicht zwischen Dampf und Flüssigkeit erfüllt sein (siehe Tabelle 1):
p x PA A A A= ⋅ ⋅γo p x PB B B B= ⋅ ⋅γ
o (16)
Π = +p pA B (10)
yp
AA=
Π y
pB
B=Π (11)
Da zur Berechnung der Partialdrücke pA und pB die Dampfdrucke PAo und PBo bei zunächst noch unbe-kannter Temperatur benötigt werden, muss man für eine Konzentration xA bzw. xB solange iterativ ver-schiedene Temperaturen wählen, bis Gleichung (10) erfüllt ist. Aus den für diese zutreffende Temperatur berechneten Partialdrücken pA und pB sind dann mit Hilfe der Gleichung (11) die zu xA und xB gehörigen Molenbrüche yA und yB der Dampfphase zu ermitteln.
Abb. 12 Schema zur Berechnung der Konstanten CA und CB der Gleichung nach van Laar aus dem azeotropen Punkt und dann der Aktivitätskoeffizienten und der Gleichgewichtskurve
8.5 Prüfen der Konsistenz der Messwerte Als einfaches Beispiel für Methoden zur kritischen Prüfung der Messdaten wird hier der Konsistenztest von Redlich und Kister angewendet [3]. Dieser Konsistenztest gibt zwar nicht darüber Auskunft, ob die gemessenen Werte absolut richtig sind, zeigt aber das Auftreten systematischer Fehler an.
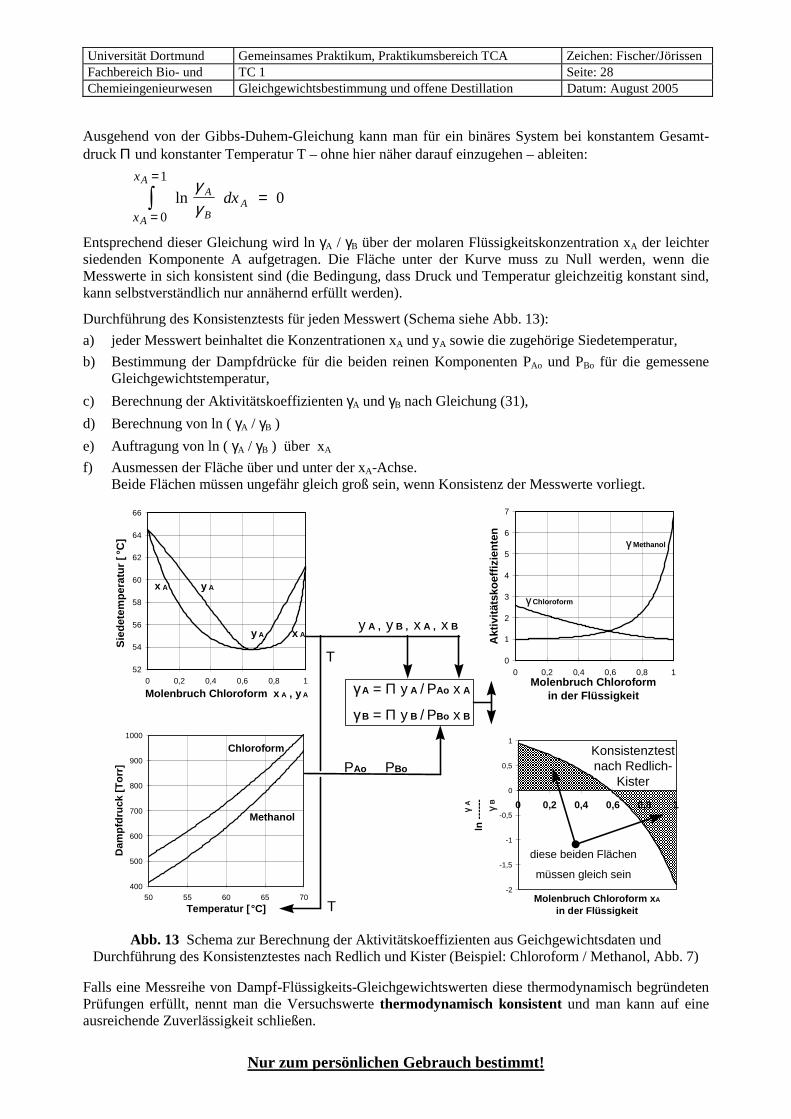
Universität Dortmund Gemeinsames Praktikum, Praktikumsbereich TCA Zeichen: Fischer/Jörissen Fachbereich Bio- und TC 1 Seite: 28 Chemieingenieurwesen Gleichgewichtsbestimmung und offene Destillation Datum: August 2005
Nur zum persönlichen Gebrauch bestimmt!
Ausgehend von der Gibbs-Duhem-Gleichung kann man für ein binäres System bei konstantem Gesamt-druck Π und konstanter Temperatur T – ohne hier näher darauf einzugehen – ableiten:
lnγγ
A
BA
A
Ax
x
dx=
=
∫ =0
1
0
Entsprechend dieser Gleichung wird ln γA / γB über der molaren Flüssigkeitskonzentration xA der leichter siedenden Komponente A aufgetragen. Die Fläche unter der Kurve muss zu Null werden, wenn die Messwerte in sich konsistent sind (die Bedingung, dass Druck und Temperatur gleichzeitig konstant sind, kann selbstverständlich nur annähernd erfüllt werden).
Durchführung des Konsistenztests für jeden Messwert (Schema siehe Abb. 13):
a) jeder Messwert beinhaltet die Konzentrationen xA und yA sowie die zugehörige Siedetemperatur,
b) Bestimmung der Dampfdrücke für die beiden reinen Komponenten PAo und PBo für die gemessene Gleichgewichtstemperatur,
c) Berechnung der Aktivitätskoeffizienten γA und γB nach Gleichung (31),
d) Berechnung von ln ( γA / γB )
e) Auftragung von ln ( γA / γB ) über xA
f) Ausmessen der Fläche über und unter der xA-Achse. Beide Flächen müssen ungefähr gleich groß sein, wenn Konsistenz der Messwerte vorliegt.
52
54
56
58
60
62
64
66
0 0,2 0,4 0,6 0,8 1
Molenbruch Chloroform x A , y A
Sie
det
emp
erat
ur
[ °C
]
x A y A
x Ay A
0
1
2
3
4
5
6
7
0 0,2 0,4 0,6 0,8 1Molenbruch Chloroform
in der Flüssigkeit
Akt
ivit
ätsk
oef
fizi
ente
n
γ Chloroform
γ Methanol
400
500
600
700
800
900
1000
50 55 60 65 70
Temperatur [ °C]
Dam
pfd
ruck
[T
orr
]
Chloroform
Methanol
Konsistenztestnach Redlich-
Kister
diese beiden Flächen
müssen gleich sein
γ A = Π y A / PAo x A
γ B = Π y B / PBo x B
PBoPAo
y A , y B , x A , x B
T
T-2
-1,5
-1
-0,5
0
0,5
1
0 0,2 0,4 0,6 0,8 1
Molenbruch Chloroform xA
in der Flüssigkeit
γ A
ln -
----
-
γ B
Abb. 13 Schema zur Berechnung der Aktivitätskoeffizienten aus Geichgewichtsdaten und Durchführung des Konsistenztestes nach Redlich und Kister (Beispiel: Chloroform / Methanol, Abb. 7)
Falls eine Messreihe von Dampf-Flüssigkeits-Gleichgewichtswerten diese thermodynamisch begründeten Prüfungen erfüllt, nennt man die Versuchswerte thermodynamisch konsistent und man kann auf eine ausreichende Zuverlässigkeit schließen.

Universität Dortmund Gemeinsames Praktikum, Praktikumsbereich TCA Zeichen: Fischer/Jörissen Fachbereich Bio- und TC 1 Seite: 29 Chemieingenieurwesen Gleichgewichtsbestimmung und offene Destillation Datum: August 2005
Nur zum persönlichen Gebrauch bestimmt!
Erfüllen die Messwerte nicht diesen Test, dann können verschiedene Gründe vorliegen:
a) die Gleichgewichtswerte sind falsch,
b) die x-y-Daten sind zwar richtig, es liegen aber Fehler in der Temperaturmessung vor,
c) die zur Errechnung der Aktivitätskoeffizienten herangezogenen Dampfdrucke sind falsch,
d) die Dampfphase verhält sich nicht ideal (z.B. bei der Assoziation von Essigsäuremolekülen im Dampf),
e) zu große Abweichungen von der Voraussetzung, dass Temperatur und Druck konstant sind, treten auf.
Es ist jedoch nicht möglich, bei Nichterfüllung des thermodynamischen Konsistenztests die Richtigkeit der Messwerte grundsätzlich abzulehnen. Es kann lediglich keine Bestätigung für die Zuverlässigkeit er-bracht werden. 9 Verfahren zur Berechnung der offenen Destillation
Beim Betrieb einer offenen Destillation interessieren die Konzentration des Inhalts von Blase und Pro-duktvorlage, da die Destillation häufig beim Erreichen einer bestimmten Konzentration abgebrochen wer-den soll. Hier sollen Methoden zur Vorausberechnung dieser Konzentration gezeigt und im Experiment nachgeprüft werden. 9.1 Berechnung der Blasenkonzentration in Abhängigkeit von der
überdestillierten Flüssigkeitsmasse
In den folgenden Ableitungen bezeichnet ξ den Massenbruch der leichter flüchtigen Komponente in der flüssigen Phase und ψ den Massenbruch der leichter flüchtigen Komponente im Dampf. Durch die ver-stärkte Verdampfung der leichter flüchtigen Komponente nimmt deren Konzentration sowohl in der Blase (ξ) als auch im Dampf (ψ) stetig ab. Der Dampf wird vollständig kondensiert und ergibt also auch eine Flüssigkeit mit der kontinuierlich abnehmenden Konzentration ψ, die sich in der Produktvorlage zu der Konzentration ξ.P mischt.
Man sollte sich für ein azeotropes Stoffsystem klarmachen, welche Konsequenzen es hat: • ob es sich um ein Minimum- oder Maximum-Azeotrop handelt, • ob man rechts oder links vom azeotropen Punkt anfängt.
Abb. 14 Definition der Konzentrationen bei der offenen Destillation Es werden folgende Voraussetzungen gemacht (siehe Abb. 14):
a) Die Flüssigkeiten sind ideal durchmischt, d.h. in der Blase herrscht die einheitliche Konzentration ξ und in der Vorlage die einheitliche Produktionskonzentration ξ p.

Universität Dortmund Gemeinsames Praktikum, Praktikumsbereich TCA Zeichen: Fischer/Jörissen Fachbereich Bio- und TC 1 Seite: 30 Chemieingenieurwesen Gleichgewichtsbestimmung und offene Destillation Datum: August 2005
Nur zum persönlichen Gebrauch bestimmt!
b) Der aus der Blase aufsteigende Dampf (ψ) steht im Gleichgewicht mit der Flüssigkeit (ξ). In einem unendlich kurzen Zeitintervall verdampft aus der Flüssigkeit in der Blase (Masse m F , Konzen-tration ξ) eine differentielle Dampfmenge d m D mit der Konzentration ψ. Dabei ändert sich die Konzen-tration der Flüssigkeit um d ξ.
Dieser Vorgang wird durch die Bilanzgleichung beschrieben:
( ) ( )m dm m dm dF D F D⋅ = ⋅ + − ⋅ −ξ ψ ξ ξ (32)
Vernachlässigt man bei der Auflösung das differentielle Glied zweiter Ordnung (d m D · d ξ) und berück-sichtigt, dass die Masse des aufsteigenden Dampfes d m D gleich der Massenverringerung der flüssigen Phase d m F ist, so bekommt man die Differentialgleichung:
ψ ξ ξ⋅ − ⋅ − ⋅ =dm dm d mD F F 0 (33)
Nach der Trennung der Variablen erhält man:
dm
m
dF
F
=−ξ
ψ ξ (34)
Die Integrationsgrenzen sind der Anfangszustand in der Blase mit der Masse m F o und der Konzentration ξo sowie ein späterer Zustand mit der Masse m F (< m F o) und der Konzentration ξ (< ξ o).
dm
mm
mdF
FFo
F
o
∫ ∫=−ξ
ψ ξξ
ξ (35)
Daraus ergibt sich:
lnm
m
dF
Fo o
=−∫ξ
ψ ξξ
ξ (36)
Diese Gleichung liefert den Zusammenhang zwischen der Blasen-konzentration ξ und dem in der Blase verbliebenen Restanteil der anfänglichen Flüssigkeitsmenge (m F / m F o).
Eine analytische Lösung des Integrals auf der rechten Seite ist mög-lich, wenn man den Zusammenhang zwischen der Flüssigkeitskon-zentration ξ und der Dampfkonzentration ψ durch einen ausreichend konstanten Wert der relativen Flüchtigkeit αAB beschreiben kann (auf die erforderlichen Formeln soll hier nicht eingegangen werden). Da in dem untersuchten azeotropen System n-Propanol / Wasser keine konstante relative Flüchtigkeit αAB angenommen werden kann, soll hier das in der Abb. 15 anschaulich gemachte graphische Verfahren vorgestellt werden (es ist analog auch iterativ auf einem Computer z.B. mit einem Tabellenkalkulations-Programm zu lösen).
Um das Integral in Gleichung (36) zu lösen, wird zunächst 1 / (ψ – ξ) über ξ aufgetragen (Abb. 15 b). Die Differenz ψ – ξ kann direkt aus dem Gleichgewichtsdiagramm Abb. 15 a (dargestellt in Massen-brüchen ξ und ψ und nicht wie beim McCabe-Thiele-Diagramm in Molenbrüchen x und y ! ) abgegriffen werden. Ausgehend von der bekannten Anfangskonzentration ξ o bestimmt man in Abb. 15 b für verschiedene Endkonzentrationen jeweils die Fläche unter der Kurve. Man kann nun außerdem diese Beträge über den Konzentrationen ξ auftragen und erhält dann die Integralkurve in Abb. 15 c. Aus dieser Kurve kann man für beliebige Restanteile der Flüssigkeit in der Bla-se, ausgedrückt durch das Verhältnis m F / m F o , die Blasenzusam-mensetzung abgreifen.
Abb. 15 Graphische Lösung für die offene Destillation: Abhängigkeit der Blasen- konzentration ξ vom Rest- anteil m F / m F o in der Blase

Universität Dortmund Gemeinsames Praktikum, Praktikumsbereich TCA Zeichen: Fischer/Jörissen Fachbereich Bio- und TC 1 Seite: 31 Chemieingenieurwesen Gleichgewichtsbestimmung und offene Destillation Datum: August 2005
Nur zum persönlichen Gebrauch bestimmt!
9.2 Berechnung der Produktkonzentration in Abhängigkeit von der überdestillierten Flüssigkeitsmasse
Eine offene Destillation kann auf verschiedene Weisen betrieben werden: Man kann das Destillat in einer Vorlage sammeln und bricht die Destillation ab, wenn die Konzentration in der Vorlage den gewünschten Wert erreicht hat bzw. die Blasenkonzentration auf den gewünschten Wert abgefallen ist. Man kann sich auch mehrere Fraktionen erzeugen, indem man die Vorlage nach gewissen Zeiten auswechselt.
Zunächst soll der erste Fall – das gesamte Destillat wird in einer Vorlage gesammelt – behandelt werden. Die Masse m.P des Produktes in der Vorlage ergibt sich aus der Anfangsmasse in der Blase m F o und der zu einem beliebigen späteren Zeitpunkt darin befindlichen Menge m F :
m m mP Fo F= − (37)
Für die Menge der leichter flüchtigen Komponente, die dabei in die Vorlage übergetrieben wurde, gilt entsprechend:
ξ ξ ξP P Fo o Fm m m⋅ = ⋅ − ⋅ (38)
Daraus ergibt sich die Konzentration in der Vorlage:
ξ
ξ ξ
P
oF
Fo
F
Fo
m
m
m
m
=
− ⋅
−1
=⋅ − ⋅
−
m m
m m
Fo o F
Fo F
ξ ξ (39)
Für ein beliebig wählbares Verhältnis m F / m F o kann nach dieser Gleichung die Konzentration in der Produktvorlage berechnet werden. Die dazu erforderliche Konzentration ξ in der Blase kann nach dem im Abschnitt 9.1 beschriebenen Verfahren bestimmt werden.
9.3 Ermittlung der Blasenkonzentration aus der Konzentration des jeweils kondensierenden Dampfes
Im vorliegenden Versuch wird die Produktmenge m P sowie die Konzentration des Dampfkondensates gemessen. Da vorausgesetzt wurde, dass der Dampf und die siedende Flüssigkeit in der Blase miteinander im Gleichgewicht stehen, kann man zu jeder Dampfkonzentration die zugehörige Blasenkonzentration aus dem Gleichgewichtsdiagramm ablesen.
9.4 Ermittlung der Produktkonzentration aus der Konzentration des jeweils kondensierten Dampfes
Die Produktkonzentration kann man sich ebenfalls aus der Dampfkonzentration ermitteln. Das Produkt in der Vorlage ist eine Mischung infinitesimal kleiner Kondensattröpfchen der Masse d m D = d m P mit der sich dauernd ändernden Konzentration ψ. Die Mischung aller dieser Tröpfchen ergibt die Produktmenge m P und die Konzentration ξ P. Mathematisch lässt sich das darstellen als:
ψ ξ⋅ = ⋅∫ dm mP P P
mP
0
beziehungsweise
ψξ
⋅=
∫ dm
m
P
mP
PP
0 (40)
Zur Ermittlung der Produktkonzentration ξ P trägt man zunächst die Dampfkonzentration ψ über der Masse des Destillats m P auf. Die Fläche unter dieser Kurve für verschiedene Werte von m P ist die Masse der leichter flüchtigen Komponente, die bis dahin in das Destillat übergegangen ist. Wenn man jeweils durch m P dividiert, erhält man die Produktkonzentration ξ P.
Zweckmäßigerweise wird man ψ direkt über 1 – m
mF
Fo
, was nichts anderes als m
mP
Fo
ist, auftragen.

Universität Dortmund Gemeinsames Praktikum, Praktikumsbereich TCA Zeichen: Fischer/Jörissen Fachbereich Bio- und TC 1 Seite: 32 Chemieingenieurwesen Gleichgewichtsbestimmung und offene Destillation Datum: August 2005
Nur zum persönlichen Gebrauch bestimmt!
Formelzeichen
a i Aktivität der Komponente i [-] C i Konstante der van Laar-Gleichung [-] m Masse [g] M i Molmasse der Komponente i [g/mol] p i Partialdruck der Komponente i [Torr] Pi o Dampfdruck der reinen Komponente i [Torr] T Temperatur [°C] x i Molenbruch der Komponente i in der flüssigen Phase [-] (mol/mol) y i Molenbruch der Komponente i in der Dampfphase [-] (mol/mol) αAB relative Flüchtigkeit (Trennfaktor)
der Komponente A gegenüber der Komponente B [-]
γ i Aktivitätskoeffizient der Komponente i [-] Π Gesamtdruck [Torr] ξ i Massenbruch der Komponente i in der flüssigen Phase [-] (g/g) ψ i Massenbruch der Komponente i in der Dampfphase [-] (g/g)
Indizes
A leichter siedende Komponente B schwerer siedende Komponente o Anfangszustand der offenen Destillation F Flüssigkeit in der Blase P Produkt M Messwert V Vergleichswert
Literatur:
[1] Ullmann's Encyclopedia of Industrial Chemistry, Chapter: Distillation and Rectification, aktuelle elektronische Ausgabe über die Homepage der Universitätsbibliothek zugänglich.
letzte deutsche Ausgabe in Buchform: Ullmanns Encyklopädie der technischen Chemie, 4. Auflage, Band 2, Kapitel Destillation und Rektifikation, Verlag Chemie, Weinheim, 1972
[2] Gmehling, J.; Brehm, A.: Lehrbuch der Technischen Chemie, Band 2, Grundoperationen, Georg Thieme Verlag, Stuttgart, 1996
[3] Sattler, K.: Thermische Trennverfahren, VCH Verlagsgesellschaft mbH, Weinheim, 1995
[4] Schlünder, E., Thurner, F., Destillation, Absorption, Extraktion, Georg Thieme Verlag, Stuttgart, 1986
[5] Gmehling, J.; Onken, U. u.a., Vapor-Liquid Equilibrium Data Collection, DECHEMA Chemistry Data Series, Frankfurt am Main, ab 1977 „Dortmunder Datenbank“, bis auf eine Einführung zum Gebrauch der Werte reines Daten- verzeichnis, Tabellen, McCabe-Thiele-Diagramme, umfassende Sammlung der Literaturdaten (laufend ergänzt)
[6] Fredenslund, A.; Gmehling, J.; Rasmussen, P.: Vapor-Liquid Equilibria Using UNIFAC, Elsevier, Amsterdam, 1977 Tabellen werden zusätzlich mit Text erläutert, teilweise Rechenbeispiele und Formeln angegeben
[7] Lohmann, J.; Joh, R.; Nienhaus, B,; Gmehling, J.: „Verbesserung der Vorhersagequalität und Erwei-terung der Anwendungsgebiete der Gruppenbeitragsmethode Modified UNIFAC (Dortmund)“,
Chem. Ing. Tech. 70 (1998) 138-142
[8] Welzenbacher, U., Neue Datenblätter für gefährliche Arbeitsstoffe nach der Gefahrstoffverordnung, WEKA, Augsburg 1994





























