Embed Size (px)
Citation preview

Grundkurs Neurobiologie
Wahlpflichtlaborübung für Studierende der Biologie
SS 2019 15.07. - 19.07.2019
Organisation: L. Bakota, R. Brandt Durchführung: N. Abreu, B. Flenker, V. Herkenhoff,
N. Trushina, B. Niewidok, M. Rierola Assistenzmodul: E.M. Flick, J. Winter

Generelle Organisation des Praktikums Bitte bringen Sie einen Laborkittel für das Praktikum mit Wir haben uns große Mühe zur Vorbereitung des Praktikums gegeben. Wir erwarten, daß Sie
ebenfalls vorbereitet kommen und das Praktikumsskript vor dem Praktikumsbeginn durchgearbeitet haben. Wir werden das in einem Kolloquium (lat., "wissenschaftliche Unterhaltung") zu Beginn des Praktikums überprüfen. Beachten Sie dazu bitte die „Leitfragen zum Grundkurs-Praktikum“, die Sie im Lehrmaterialenordner finden.
Die Teilnahme an der Veranstaltung "Grundlagen der Neurobiologie mit Tutorium" ist
Voraussetzung für die Teilnahme am Praktikum. Grundlage für das Bestehen dieses Modulteils ist (1) die Teilnahme am Praktikum, (2) die
Abgabe der bearbeiteten Rechenübungen zu Beginn des Versuchs AChE, und (3) die Abgabe des ausgefüllten Formblattes zum Versuch AChE (enthalten im Praktikumsprotokoll) am Ende des Versuchs.
Das Erarbeiten eines vollständigen Protokolls ist freiwillig und empfehlenswert für alle, die
weitere Praktika oder Abschlussarbeiten in der Abteilung Neurobiologie planen. Bitte geben Sie in diesem Fall ein Protokoll pro Person ab. Beschränken Sie sich dabei auf einen Versuch. Deadline für die Abgabe des Protokolls ist 4 Wochen nach Praktikumsende (Sekretariat der Neurobiologie, Raum 36-312, oder im Postfach vor Raum 36-313 (Fach: R. Brandt)).
Nutzen Sie Leerzeiten während des Praktikums zur Arbeit am Protokoll - es hat sich bewährt,
Protokolle so zeitnah zum Experiment wie irgend möglich zu erarbeiten Bitte befolgen Sie folgende Hinweise zum Aufbau des Protokolls:
Vermerken Sie mögliche Versuchsänderungen, Probleme bei der Versuchsdurchführung etc. Zentraler Teil eines Versuchsprotokolls ist die vollständige Darstellung und Auswertung der Ergebnisse Beantworten Sie alle Fragen in der Versuchsvorschrift; übernehmen Sie für die Beantwortung
die Nummerierung aus der Versuchsvorschrift Keine Wiederholung der Versuchsvorschrift Kein ausführlicher Literaturüberblick
Protokolle können voraussichtlich ab Mitte November aus dem Sekretariat der Neurobiologie
abgeholt werden. PLEASE NOTE: PART OF THE PRACTICAL TRAINING WILL BE IN ENGLISH
LANGUAGE
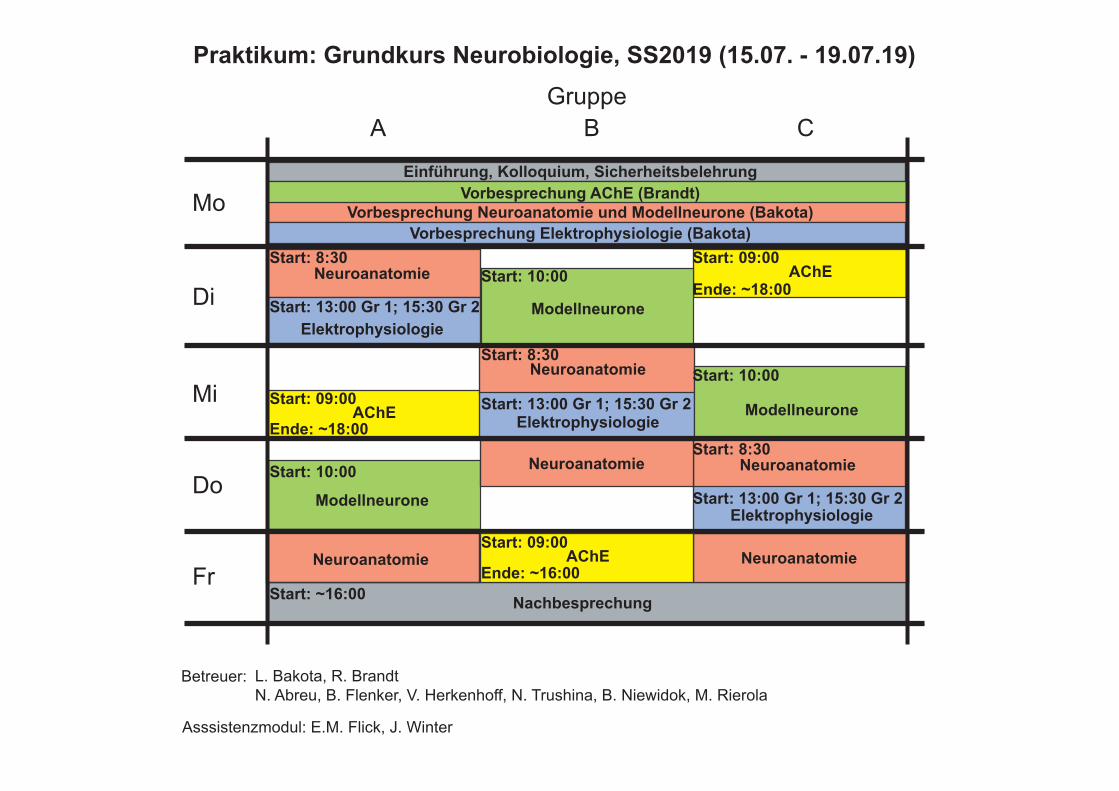
A B CGruppe
Mo
Di
Mi
Do
Fr
Einführung, Kolloquium, Sicherheitsbelehrung
Vorbesprechung Neuroanatomie und Modellneurone (Bakota)Vorbesprechung Elektrophysiologie (Bakota)
Vorbesprechung AChE (Brandt)
Betreuer: L. Bakota, R. BrandtN. Abreu, B. Flenker, V. Herkenhoff, N. Trushina, B. Niewidok, M. Rierola
Neuroanatomie
Neuroanatomie
Neuroanatomie
Neuroanatomie Neuroanatomie
Neuroanatomie
Modellneurone
Modellneurone
Modellneurone
Elektrophysiologie
Elektrophysiologie
Elektrophysiologie
Praktikum: Grundkurs Neurobiologie, SS2019 (15.07. - 19.07.19)
AChE
AChE
AChE
Nachbesprechung
Start: 10:00
Start: 10:00
Start: 10:00
Asssistenzmodul: E.M. Flick, J. Winter
Start: 09:00
Ende: ~18:00
Ende: ~16:00Start: ~16:00
Start: 13:00 Gr 1; 15:30 Gr 2
Start: 8:30
Start: 13:00 Gr 1; 15:30 Gr 2
Start: 8:30
Start: 13:00 Gr 1; 15:30 Gr 2
Start: 8:30
Start: 09:00
Ende: ~18:00
Start: 09:00

Modellneurone - 1 -
Humane Modellneuronen / lichtmikroskopische Immunzytochemie / neuronale Marker (Versuch Modellneurone)
Inhalt:
1. Humane Modellneuronen als Modell terminal differenzierter und polarer Nervenzellen
2. Lichtmikroskopische Verfahren zur Untersuchung dissoziierter Nervenzellen
2.1 Das Mikroskop
2.2 Zellfärbung und Hellfeldmikroskopie
3. Lichtmikroskopische Immunzytochemie zur Identifizierung von Neuronen
4. Protokolle
4.1 Materialien
4.2 Fixierung der Zellen
4.3 Färbung mit Toluidinblau
4.4 Immunzytochemische Färbung
4.5 Einbetten
5. Aufgaben
6. Weiterführende Literatur

Modellneurone - 2 -
1. Humane Modellneuronen als Modell terminal differenzierter und polarer Nervenzellen
Die Entwicklung von Methoden, dissoziierte Zellen aus dem Gehirn zu präparieren
und zu kultivieren, ermöglichte erstmals die Anwendung zellbiologischer Methoden in der
Neurobiologie. Damit wurde es zum Beispiel in den 70er Jahren des vorigen Jahrhunderts
zum erstenmal möglich, die Ausbildung und das Auswachsen neuronaler Zellausläufer zu
beobachten sowie das Verhalten neuronaler Wachstumskegel in einzelnen Nervenzellen zu
analysieren. Interessanterweise entwickeln sich bestimmte Neuronentypen (wie zum Beispiel
Pyramidenzellen, die aus dem Hippocampus der Ratte isoliert werden) in einer stereotypen
Sequenz von Ereignissen (Abbildung 1). In einem ersten Schritt nach dem Ausplattieren
kommt es zur Ausbildung einer dynamischen Membranstruktur (Lamellipodien),
anschließend zum Auswachsen kleiner Zellausläufer, die sich morphologisch und funktional
nicht unterscheiden (“minor processes”). In einem nächsten Schritt wächst dann einer dieser
Zellausläufer schneller als die anderen und entwickelt eine typisch axonale Morphologie. Im
weiteren Verlauf nehmen dann die verbleibenden kleinen Zellausläufer zunehmend eine
dendritische Morphologie mit ihren typischen baumartigen Verzweigungen an.
Abb. 1: Stufen der Entwicklung von Neuronen, die dem Hippocampus der embryonalen Ratte (embryonaler Tag 18) entnommen wurden (nach Dotti et al., 1988).
Seit diesen Pionierarbeiten wurden die Kulturtechniken beständig weiterentwickelt
und verfeinert und es wurden Methoden eingeführt, um zum Beispiel bestimmte Zelltypen
anzureichern, in Kultur zu nehmen und experimentell zu manipulieren (für einen Überblick
über die verschiedenen Kulturmethoden siehe die Bücher von Banker und Goslin (1991) und
Fedoroff und Richardson (2001)). Kulturen, bei denen die Zellen direkt aus dem Tier
entnommen werden, werden dabei als "Primärkulturen" bezeichnet. Aus diesen Kulturen
können Zellen, die sich teilen (nicht also Nervenzellen), mehrmals weiter "passagiert"
werden, bis nach einiger Zeit auch diese Zellen aufhören, sich zu teilen. Im Unterschied zu
"Primärkulturen" können klonale Zelllinien, die zum Beispiel aus einem Tumor gewonnen

Modellneurone - 3 -
wurden, im Prinzip unbegrenzt passagiert und weiter kultiviert werden. Aus einem Tumor
gewonnene klonale Zelllinien sind z.B die häufig verwendeten Ratten PC12 Zellen und das
humane NT2/NT2-N Zellsystem (Pleasure et al., 1992; Piontek et al., 1999).
Für Untersuchungen zur Pathologie humaner Krankheiten für die kein Tiermodell
vorhanden ist - wie etwa der Alzheimerschen Krankheit - aber auch für mögliche
therapeutische Anwendungen kann es wichtig sein, humane Nervenzellen zu untersuchen.
Aus naheliegenden Gründen ist es jedoch nicht ohne weiteres möglich, humane
Primärneuronen zu präparieren. Ein Ausweg stellt hier die Untersuchung geeigneter humaner
Zelllinien, wie die sogenannten NT2-Zellen, dar. Diese Zellen repräsentieren in ihrem
undifferenzierten Zustand humane neuronale Vorläuferzellen und können, nach einer
zeitaufwendigen Behandlung mit Vitamin A-Säure, in vitro zu postmitotischen und polaren
Nervenzellen (NT2-N Zellen) differenziert werden (Abbildung 2). Die Differenzierung ist
z.B. daran zu erkennen, daß die Nervenzellen typische Markerproteine wie das mikrotubuli-
assoziierte Protein MAP2b exprimieren. Mit Hilfe dieser Modellneurone kann nun z.B. der
Effekt von Faktoren, die bei neurodegenerativen Erkrankungen eine Rolle spielen, auf das
Überleben humaner Nervenzellen untersucht werden (Fath et al., 2002) oder Chemikalien
(„small molecules“) identifiziert werden, die die Differenzierung menschlicher Neurone
beeinflussen (Raasch et al., 2015). Zudem ist es möglich, diese Zellen in Ratten- oder auch
menschliche Hirne zu verpflanzen, in denen sie monatelang überleben, sich in das neuronale
Netzwerk einfügen und zum Beispiel abgestorbene Nervenzellen ersetzen können (Nelson et
al., 2002).
In diesem Versuchsteil sollen Grundfertigkeiten der zellbiologischen Untersuchung
neuraler Zellen am humanen NT2/NT2-N Zellsystem vermittelt werden. Neben den
undifferenzierten Vorläuferzellen (NT2 Zellen) werden Ihnen differenzierte Zellen, die neben
den Neuronen auch nicht-neuronale Zellen (großflächige Zellen mit einem lichtmikroskopisch
größer erscheinenden Zellkern) enthalten und insgesamt 7 Wochen kultviert wurden, zur
immunzytochemischen Behandlung und mikroskopischen Untersuchung übergeben.
2. Lichtmikroskopische Verfahren zur Untersuchung dissoziierter Nervenzellen
Trotz der Möglichkeiten, die heute elektronenmikroskopische Verfahren bieten, um in
hohe Vergrößerungsbereiche vorzustoßen, wird die Lichtmikroskopie eine der wichtigsten
Arbeitsmethoden des (Zell-)Biologen bleiben (Gerlach, 1985; Rieder und Schmidt, 1987).
Das liegt an der häufig einfacheren und schnelleren Vorbereitung der Präparate, die z.B. auch
eine Beobachtung lebender Zellen erlaubt, also mit einem minimalen Eingriff in die Integrität
der Objekte auskommt. Sinn dieses Praktikumsteils ist es, Ihnen wichtige lichtmikroskopische

Modellneurone - 4 -
Techniken, wie sie für die praktische Arbeit relevant sind, nahezubringen und ihnen
exemplarisch an neuronalen Präparaten zu verdeutlichen.
Abb. 2: Schematische Darstellung der Differenzierung von humanen Modellneuronen (NT2-N Neurone).
2.1. Das Mikroskop
Im Mikroskop wird das Bild von zwei getrennten Linsensystemen - dem Objektiv und dem
Okular - erzeugt. Die Gesamtvergrößerung ergibt sich aus der Vergrößerung des Objektivs
multipliziert mit der Vergrößerung des Okulars. Der Informationsgehalt des Bildes wird
hauptsächlich durch die Qualität und Vergrößerung des Objektivs bestimmt, wobei das
Auflösungsvermögen die entscheidende Rolle spielt. Unter "Auflösung" versteht man dabei
das Vermögen eines optischen Systems, zwei nahe beeinanderliegende Strukturen noch
deutlich voneinander getrennt darzustellen. Dabei ist die sogenannte numerische Apertur (A)
die entscheidende Kenngröße für die Qualität eines Objektivs. Aus physikalischen Gründen
bestimmt sich A zu sinD �x n mit D als dem halben Öffnungswinkel unter dem das zur
Bildentstehung wichtige Licht gerade noch in das Objektiv eintritt, und mit n als dem

Modellneurone - 5 -
Brechungsindex des Mediums das sich zwischen Präparat und Frontlinse befindet (Luft: n =
1, Glas oder Öl: n = 1.5). Beim Auflösungsvermögen (d) spielt auch die Wellenlänge des
verwendeten Lichtes nach der Formel d = O/A eine Rolle. Trockenobjektive werden bis zu
einem A = 0.95 gebaut, gute Immersionsobjektive haben ein A von 1.40. Nach einer
einfachen Faustregel sollte die sinnvoll anzuwendende Gesamtvergrößerung zwischen dem
500- bis 1000-fachen der numerischen Apertur des Objektivs liegen. Für das
Auflösungsvermögen des Gesamtsystems ist neben der numerischen Apertur des Objektivs
auch die numerische Apertur des Beleuchtungssystems (des Kondensors) entscheidend (bei
Trockenkondensoren etwa 0.9). Um das System zwischen Objektiv, Präparat und
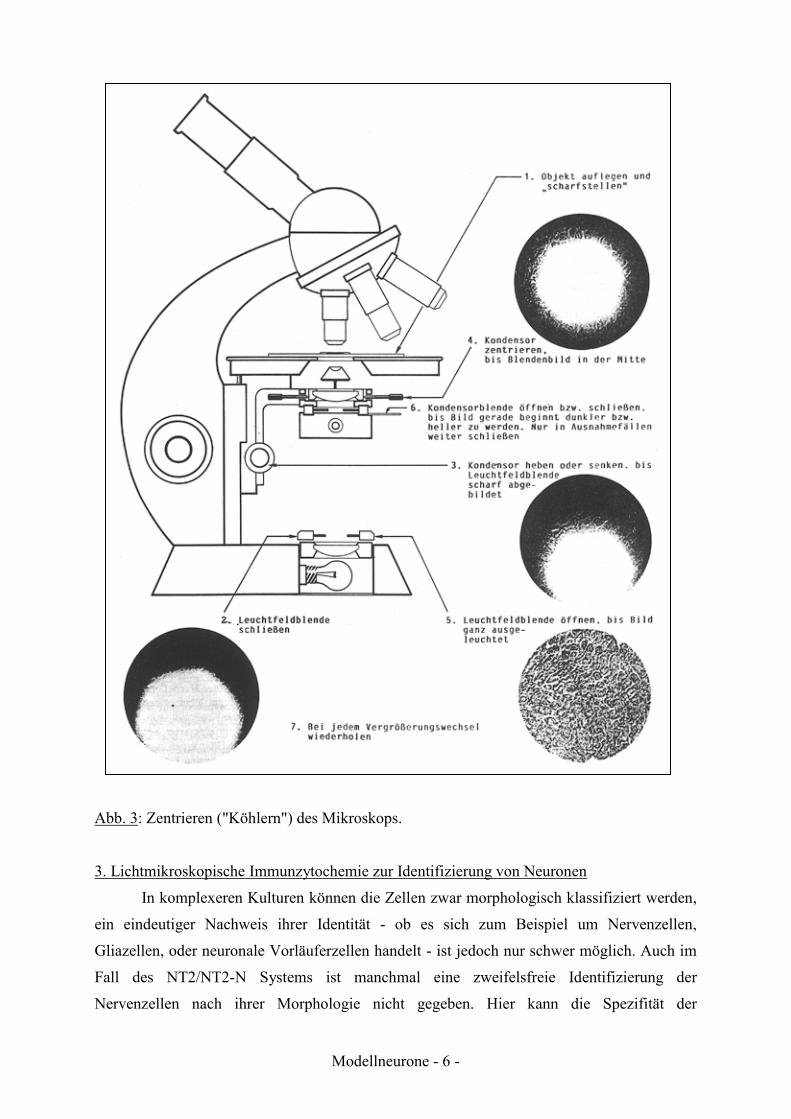
Beleuchtungseinrichtung optimal einzustellen wurde von Köhler (1893) ein entsprechendes
Verfahren entwickelt. Vor dem Beginn des Arbeitens sollte das Mikroskop gemäß diesen
Regeln "geköhlert" werden (Abb. 3). Im Rahmen des Praktikums soll die
Hellfeldmikroskopie gefärbter Proben eingesetzt werden.
2.2 Zellfärbung und Hellfeldmikroskopie
Bei der Hellfeldmikroskopie wird das Präparat vom Licht durchstrahlt, also wie ein
Diapositiv betrachtet. Man sieht deshalb das Objekt an den Stellen dunkler, an denen es einen
Teil der Lichtstrahlen herausfiltert oder abdeckt. Leider sind mit dieser einfachen Technik die
Umrisse einzelner Zellen nur schwer auszumachen und intrazelluläre Details kaum zu
erkennen. Der Kontrast kann jedoch durch geeignete Färbeverfahren erhöht werden. Im
Rahmen dieses Praktikumsteils werden die Zellen mit Toluidinblau gefärbt. Toluidinblau
führt zu einer differenzierten Anfärbung biologischer Bestandteile in unterschiedlichen
Blautönen. Obwohl es - neben solchen unspezifischen Färbeverfahren - auch Methoden gibt,
die mehr oder weniger spezifisch subzelluläre Komponenten anfärben, werden diese
Verfahren heute kaum noch genutzt, da man mit fluoreszenzmikroskopischen Methoden und
den entsprechenden Fluoreszenzfarbstoffen eine wesentlich empfindlichere und spezifischere
Nachweismethode zur Verfügung hat. Die Einführung in die Fluoreszenzmikroskopie wird
einem neurobiologischen Fortgeschrittenenpraktikum vorbehalten sein.
Modellneurone - 6 -
Abb. 3: Zentrieren ("Köhlern") des Mikroskops.
3. Lichtmikroskopische Immunzytochemie zur Identifizierung von Neuronen
In komplexeren Kulturen können die Zellen zwar morphologisch klassifiziert werden,
ein eindeutiger Nachweis ihrer Identität - ob es sich zum Beispiel um Nervenzellen,
Gliazellen, oder neuronale Vorläuferzellen handelt - ist jedoch nur schwer möglich. Auch im
Fall des NT2/NT2-N Systems ist manchmal eine zweifelsfreie Identifizierung der
Nervenzellen nach ihrer Morphologie nicht gegeben. Hier kann die Spezifität der

Modellneurone - 7 -
Antigen/Antikörper Interaktion ausgenutzt werden, um Zellen zu identifizieren, die gewebs-
oder zelltypspezifische Antigene exprimieren. Eine gängige Methode ist dabei die indirekte
Immunperoxidase-Technik (Harlow and Lane, 1988). Dabei wird ein Primärantikörper
eingesetzt, um das Antigen zu erkennen. In einer Art Sandwich-Technik wird die Zelle oder
das Gewebe dann mit einem Sekundärantikörper inkubiert, der die Spezies des
Primärantikörpers erkennt und mit einem Enzym (der Meerrettichperoxidase, oder abgekürzt
"HRP" für engl. "horseradish peroxidase") gekoppelt ist. Die Enzymaktivität kann dann durch
eine Farbreaktion sichtbar gemacht werden bei der das Reaktionsprodukt präzipitiert und das
dann lichtmikroskopisch lokalisiert werden kann. Diese Technik kann noch empfindlicher
gestaltet werden, indem eine Verstärkungstechnik eingesetzt wird, die auf der starken
Bindung von Avidin (aus Hühnereiweiß) an Biotin (Vitamin H) (KD=10-15
) basiert. Hierbei
wird ein biotinylierter Sekundärantikörper eingesetzt, an den dann ein Avidin-Komplex, der
kovalent gekoppeltes HRP enthält, gebunden wird (Abb. 5).
Abb. 5: Methode der Avidin-Biotin Technik zur Verstärkung des immunzytochemischen Nachweises. A: Inkubation mit dem Primärantikörper, B: Inkubation mit dem biotinylierten Sekundärantikörper, C: HRP-konjugiertes Biotin wird mit Avidin inkubiert. Dabei bildet sich ein Komplex von einem Avidin mit mehreren Biotinmolekülen. D: Die Probe wird mit dem Avidin-Biotinkomplex inkubiert der an den biotinylierten Sekundärantikörper bindet. Schraffierte Quadrate: Biotin, karierte Flächen: Avidin.

Modellneurone - 8 -
4. Protokolle
4.1 Materialien
Zur Verfügung gestellt werden:
Undifferenzierte neuronale Vorläuferzellen (NT2 Zellen) kultiviert auf mikroskopischen
Deckgläschen (je Gruppe: 1 x NT2)
Differenzierte humane Modellneurone (NT2-N), kultiviert auf mikroskopischen
Deckgläschen (je Gruppe: 3 x NT2-N)
Fixierungs-Lösung: 4% (w/v) Paraformaldehyd, 4% (w/v) Saccharose in PBS. Lagerung bei -
20°C in Aliquots.
Phosphatgepufferte Saline (PBS): 10 mM Phosphatpuffer pH 7.4, 2.7 mM KCl, 137 mM
NaCl. Lagerung bei 4°C.
Toluidinblau-Lösung: 0.5%(w/v) Toluidinblau in 2.5% (w/v) NaHCO3
Glycin-Lösung: 0.1 M Glycin in PBS.
Triton X-100 Lösung: 0.2%(v/v) Triton X-100 in PBS.
3% Wasserstoffperoxid (vorsicht ätzend)
BSA/PBS-Tween: 1%(w/v) BSA, 0.1%(v/v) Tween in PBS. Enthält 0.02% Natriumazid.
Gelagert bei 4oC.
Primärantikörper: Monoklonaler MAP2b Antikörper aus Maus (AP20; Boehringer
Mannheim)
Sekundärantikörper: Biotinylierter Ziegen anti-Maus Antikörper (Sigma, ExtrAvidin-Kit).
Avidin Peroxidase (Sigma, ExtrAvidin-Kit).
AEC (3-Amino-9-Äthylcarbazol)-Stammlösung: 20 mg AEC in 2.5 ml Dimethylformamid
auflösen (vorsicht giftig). Gelagert bei 4°C.
Acetatpuffer: 50 mM Na-acetat, pH 5.0
Einbettmedium: 90%(v/v) Glycerin/10%(v/v) PBS. Gelagert bei 4°C.
Nagellack
4.2 Fixierung der Zellen (alle 4 Deckgläschen)
ACHTUNG: Die Deckgläschen dürfen während der gesamten Präparation keinesfalls
austrocknen - bei Immunfärbungen würde dies den Hintergrund erhöhen! - Entnehmen Sie die Deckgläschen aus dem Kulturgefäß mit einer Pinzette (Sterilität ist nicht
mehr nötig) und legen Sie diese in derselben Orientierung (Zellen nach oben) auf ein
Stück Parafilm (Kennzeichnen Sie die Position). Addieren Sie einen Tropfen PBS zu
jedem Deckgläschen und saugen Sie es danach wieder ab.

Modellneurone - 9 -
- Pipettieren Sie 50 µl der Fixierungslösung auf die Deckgläschen und inkubieren Sie für 20
Minuten.
4.3 Färbung mit Toluidinblau (1 x NT2 und 1 x NT2-N)
- Saugen Sie die Lösung ab. Waschen Sie die Deckgläschen 5×2 Minuten mit PBS.
- Bedecken Sie das Deckgläschen mit 50 µl Toluidinblau-Lösung. Färben Sie die Zellen 20
Minuten lang in einer "Feuchtkammer" ein. Befeuchten Sie dazu etwas Papier, legen Sie
es neben die Deckgläschen und decken Sie alles ab. Waschen Sie die Zellen 5×2 Minuten
mit PBS und betten diese entsprechend Punkt 4.5 ein.
4.4 Immunzytochemische Färbung (2 x NT2-N)
- Waschen Sie die Deckgläschen 5×2 Minuten mit PBS.
- Bedecken Sie die Deckgläschen mit 50 µl Glycin-Lösung (zum Blockieren verbliebener
Aldehydgruppen). Inkubieren Sie 20 Minuten.
- Saugen Sie die Glycin-Lösung ab. Waschen Sie einmal mit PBS.
- Saugen Sie die PBS-Lösung ab und bedecken Sie die Deckgläschen mit 50 µl Triton X-100-
Lösung (Permeabilisieren der Zellen).
- Saugen Sie nach 5 Minuten die Triton-Lösung ab, waschen Sie einmal mit PBS und
bedecken Sie die Deckgläschen mit 50 µl 3% Wasserstoffperoxid.
- Saugen Sie nach 5 Minuten die Lösung ab und waschen Sie einmal mit PBS.
- Waschen Sie die Deckgläschen 5×2 Minuten mit jeweils einigen Tropfen BSA/PBS-Tween.
- Bereiten Sie die Primärantikörperlösung vor: Pipettieren Sie 200 µl BSA/PBS-Tween in ein
Eppendorf-Reaktionsgefäß und addieren Sie den Primärantikörper (anti MAP2b) in einer
Verdünnung von 1:200. Mischen Sie gut und addieren Sie jeweils 48 µl zu einem der
beiden Deckgläschen. Inkubieren Sie 1 Stunde in der "Feuchtkammer" (Mittagspause ?).
- Saugen Sie die Lösung ab und waschen Sie die Deckgläschen 5×2 Minuten in BSA/PBS-
Tween.
- Bereiten Sie die Sekundärantikörperlösung vor, indem Sie eine Verdünnung (1:50) des
biotinylierten anti-Maus Antikörpers in BSA/PBS-Tween in einem Eppendorf-
Reaktionsgefäß ansetzen. Mischen Sie gut und addieren Sie jeweils 48 µl zu den
Deckgläschen. Inkubieren Sie 30 Minuten in der "Feuchtkammer".
- Saugen Sie die Lösung ab und waschen Sie die Deckgläschen 5×2 Minuten mit BSA/PBS-
Tween.
- Setzen Sie eine Verdünnung (1:20) der Avidin-Peroxidase Lösung in BSA/PBS-Tween in
einem Eppendorf-Reaktionsgefäß an und addieren Sie jeweils 50 µl zu den Deckgläschen.

Modellneurone - 10 -
- Inkubieren Sie 30 Minuten in der "Feuchtkammer".
- Saugen Sie die Lösung ab und waschen Sie die Deckgläschen 5×2 Minuten in PBS. Bereiten
Sie in dieser Zeit die Substratmischung vor: Mischen sie 100 µl AEC Stammlösung mit
1.9 ml Acetat Puffer. Fügen Sie 10 µl 3% Wasserstoffperoxid zu.
- Addieren Sie jeweils 50 µl der Substratmischung zu den Deckgläschen. Inkubieren Sie 5-10
Minuten.
- Beenden Sie die Reaktion, indem Sie die Deckgläschen 3x mit Wasser waschen. Betten Sie
die Deckgläschen ein (Punkt 4.5).
4.5 Einbetten
Plazieren Sie für jedes Deckgläschen ein kleines Tröpfchen der Einbettlösung auf einen
Objekttäger (maximal 3 Deckgläschen können auf einen Objektträger montiert werden).
Saugen Sie die Deckgläschen völlig trocken, und plazieren Sie sie jeweils kopfüber mit
Hilfe einer Pinzette möglichst frei von Luftblasen auf dem Tropfen der Einbettlösung.
Versiegeln Sie die Deckgläschen am Rand mit klarem Nagellack. Sobald der Nagellack
trocken ist (das dauert wenige Minuten), können die Deckgläschen mikroskopisch
betrachtet werden.
5. Aufgaben
5.1 Betrachten Sie die Toluidinblau- und immunzytochemisch gefärbten Präparate in der
Hellfeldmikroskopie. Verwenden Sie die dabei das 40x (Trocken)Objektiv. Fertigen Sie
gruppenweise von jedem Präparat repräsentative Bilder an.
5.2 Beschreiben und diskutieren Sie die Bilder aus 5.1 im Hinblick auf die abgebildeten
Zelltypen und mikroskopischen Techniken.
5.3 Wie unterscheiden sich morphologisch die Stammzellen von Nervenzellen? Beschreiben
Sie die morphologischen Kriterien für Nervenzellen anhand ihrer Bilder.
5.4 Welche Typen von Neuronen können Sie anhand morphologischer Kriterien in ihren
Bildern unterscheiden? Wo finden Sie diese Nervenzellen in einem realen Nervensystem?
5.5 Diskutieren Sie Möglichkeiten und Grenzen der rein morphologischen Beschreibung und
stellen Sie diese Technik der immunzytochemischen Untersuchung gegenüber.
5.6 Als Kontrolle zur Spezifität der Färbung haben Sie parallel eine Färbung ohne den
Primärantikörper durchgeführt. Welche weiteren Kontrollen für die Spezifität einer
Immunfärbung können Sie vorschlagen?
5.7 Sie können dieselbe Gesamtvergrößerung eines Objekts mit einer Objektivvergrößerung
von 100 und einer Okularvergrößerung von 10 oder einer Objektivvergrößerung von 50

Modellneurone - 11 -
und einer Okularvergrößerung von 20 erzielen. Ist der Informationsgehalt des erhaltenen
Bildes gleich? Diskutieren sie an diesem Beispiel das Konzept der "förderlichen" bzw.
"leeren" Vergrößerung.
6. Weiterführende Literatur
Banker G, and Goslin K (1991) Culturing Nerve Cells. Cambridge: MIT Press (Einführung in die neuronale Zellkultur)
Dotti CG, Sullivan CA, and Banker GA (1988) The establishment of polarity by hippocampal neurons in culture. J. Neurosci. 8:1454-1468.
Fath T, Eidenmüller J, and Brandt R (2002) Tau-mediated cytotoxicity in a pseudohyperphosphorylation model of Alzheimer's disease. J. Neurosci. 22:9733-9741
Fedoroff, S., and Richardson, A. (2001) Protocols for Neural Cell Culture. Totowa: Humana Press (Praxisnahe Einführung in die Kultur neuraler Zellen)
Gerlach D (1985) Das Lichtmikroskop. Stuttgart: Thieme (ausführlich, auch in der Beschreibung der optischen Grundlagen; für mikroskopisch Interessierte)
Harlow E, and Lane D (1988) Antibodies. Cold Spring Harbor, CSH ("Kochbuch" für immunologische Techniken; sehr praxisorientiert)
Nelson PT, Kondziolka D, Wechsler L, Goldstein S, Gebel J, DeCesare S, Elder EM, Zhang PJ, Jacobs A, McGrogan M, Lee VM, and Trojanowski JQ (2002) Clonal human (hNT) neuron grafts for stroke therapy: neuropathology in a patient 27 months after implantation. Am. J. Pathol.160:1201-12066 (therapeutische Anwendung von humanen Modellneuronen)
Piontek J, Chen CC, Kempf M, and Brandt R (1999) Neurotrophins differentially regulate the survival and morphological complexity of human CNS model neurons. J. Neurochem. 73:139-146 (zur Kultur der NT2-N Zellen)
Pleasure SJ, Page C, and Lee VM (1992) Pure, postmitotic, polarized human neurons derived from NTera 2 cells provide a system for expressing exogenous proteins in terminally differentiated neurons. J. Neurosci. 12:1802-1815 (zur Einführung in das NT2/NT2-N System)
Raasch K, Malecki E, Siemann M, Montilla Martinez M, Heinisch JJ, Müller J, Bakota L, Kaltschmidt C, Kaltschmidt B, Rosemeyer H, and Brandt R (2015) Identification of nucleoside analogues as inducers of neuronal differentiation in a human reporter cell line and adult stem cells. Chem. Biol. Drug Des. 86:129-143
Rieder N, and Schmidt K (1987) Morphologische Arbeitsmethoden in der Biologie. Weinheim: VCH, 50-65 (kompakte Einführung; hilfreich für den schnellen Einstieg in die Mikroskopierpraxis)

AChE - 1 -
Acetylcholinesterase aus Mäusehirnen (Versuch AChE)
Inhalt:
1. Cholinerge Neurotransmission
2. Protokolle
2.1 Materialien
2.2 Präparation der Homogenate aus Mäusehirnen
2.3 Proteinbestimmung nach Bradford
2.4 Bestimmung der Acetylcholinesterase-Aktivitäten mit Hilfe des Ellman-Tests
3. Aufgaben (Übersicht)
4. weiterführende Literatur
5. Rechenübung

AChE - 2 -
1. Cholinerge Neurotransmission
Acetylcholin wirkt als Neurotransmitter im autonomen Nervensystem, bei der Innervation der
Skelettmuskulatur durch Motorneurone und in einigen Regionen des Zentralnervensystems,
wie zum Beispiel des zentralen Aufmerksamkeitssystems. Acetylcholin wirkt auf der
postsynaptischen Seite auf zwei Typen von Rezeptoren, das sind zum einen die
muskarinischen und zum anderen die nikotinischen Acetylcholinrezeptoren. Bei
verschiedenen Erkrankungen ist die cholinerge Neurotransmission beeinträchtigt. Dazu
gehören die Alzheimersche Erkrankung und die Autoimmunkrankheit Myasthenia gravis. Bei
der Alzheimerschen Erkrankung degenerieren vor allem cholinerge Neurone, die den
Hippocampus innervieren, der bei Gedächtnisvorgängen beteiligt ist. Bei der Myasthenia
gravis produziert das Immunsystem fälschlicherweise Antikörper gegen den nikotinischen
Acetylcholinrezeptor, was den Abbau der Rezeptoren induziert und damit eine
Muskelschwäche bewirkt.
Wenn Acetylcholin von seinem Rezeptor abdissoziiert, wird es durch die im synaptischen
Spalt lokalisierte Acetylcholinesterase (AChE) gespalten und damit inaktiviert (Abb 1). Die
Wechselzahl (kcat) der AchE ist mit 7,4 × 105 min-1 für ein Enzym außergewöhnlich hoch
(Wilson and Harrison, 1961).
Abb.1: Reaktion der Acetylcholinesterase (AChE)
Die Hemmung der AChE bewirkt dadurch eine Erhöhung der verfügbaren Menge von
Acetylcholin. Dies wird zum Beispiel zur symptomatischen Behandlung der Alzheimerschen
Erkrankung genutzt bei der die AChE-Inhibitoren Aricept® (Donepezil) und Exelon®
(Rivastigmin) eingesetzt werden. Bei der Behandlung von Myasthenia gravis wird der AchE -
Hemmer Neostigmin eingesetzt. Ein weiterer AChE-inhibitor, Physostigmin (Eserin), findet
AChE - 3 -
als Gegenmittel bei Vergiftungen zum Beispiel mit Atropin (dem Gift der Tollkirsche), das
den Acetylcholinrezeptor blockiert, Verwendung.
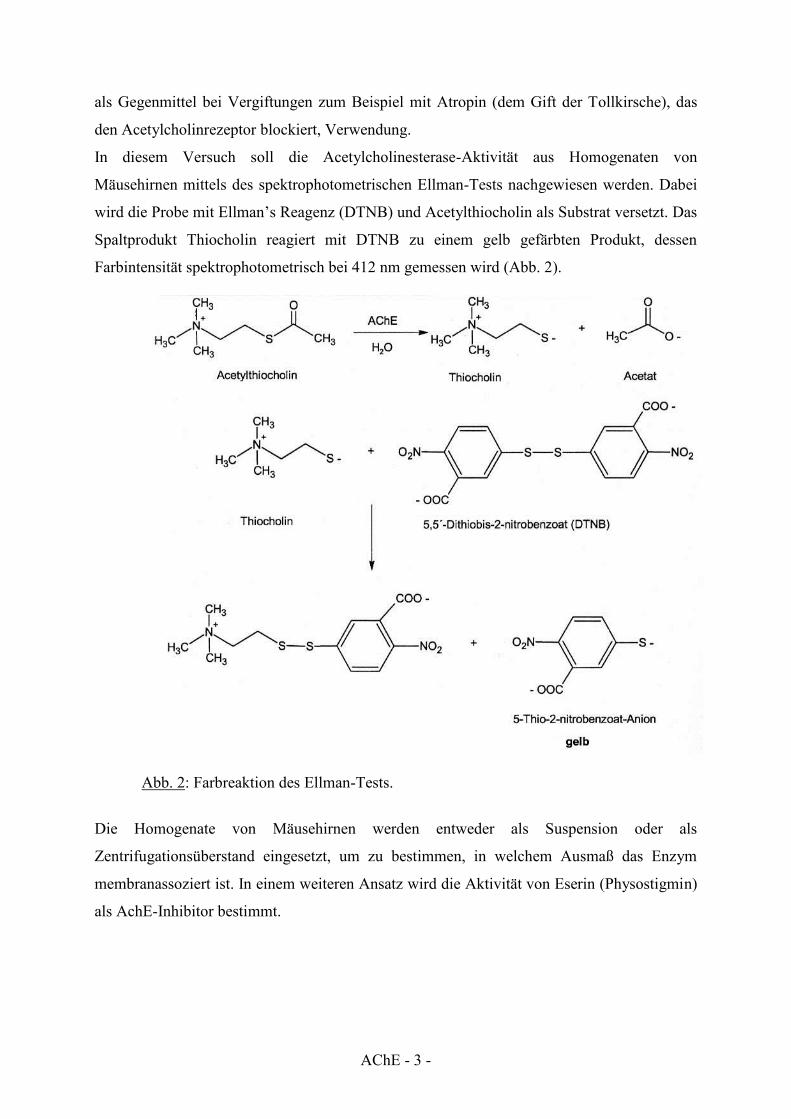
In diesem Versuch soll die Acetylcholinesterase-Aktivität aus Homogenaten von
Mäusehirnen mittels des spektrophotometrischen Ellman-Tests nachgewiesen werden. Dabei
wird die Probe mit Ellman’s Reagenz (DTNB) und Acetylthiocholin als Substrat versetzt. Das
Spaltprodukt Thiocholin reagiert mit DTNB zu einem gelb gefärbten Produkt, dessen
Farbintensität spektrophotometrisch bei 412 nm gemessen wird (Abb. 2).
Abb. 2: Farbreaktion des Ellman-Tests.
Die Homogenate von Mäusehirnen werden entweder als Suspension oder als
Zentrifugationsüberstand eingesetzt, um zu bestimmen, in welchem Ausmaß das Enzym
membranassoziert ist. In einem weiteren Ansatz wird die Aktivität von Eserin (Physostigmin)
als AchE-Inhibitor bestimmt.

AChE - 4 -
2. Protokolle
2.1 Materialien
Zur Verfügung gestellt werden:
Homogenat aus Mäusehirn (Suspension (S) oder Zentrifugationsüberstand (Z)) Bradford Reagenz 10 mg/ml Stammlösung BSA 0,1 M Na,K Phosphatpuffer, pH 7,9 0,1 M Na,K Phosphatpuffer, pH 7,2 (Zur Verdünnung des Homogenats) DTNB (5,5’-Dithiobis-(2-nitrobenzoesäure) 0,3 mM Acetylthiocholinjodid 7,5 mM Eserin (Physostigmin) 2 mM 2.2 Präparation von Homogenat aus Mäusehirnen (wurde von den Betreuern übernommen) Das Mäusehirn wird mechanisch zerkleinert und durch Pottern (ca 30x) homogenisiert, dabei
wird ein 10%iges Homogenat hergestellt (d.h. pro g Gewebe wird 9 ml Na,K-Phosphatpuffer
0,1 M pH 7,2 zugesetzt).
Eine Hälfte des Homegenats wird 10 min bei 13000 rpm und 4°C in einer Mikrozentrifuge
abzentrifugiert, der Überstand abpipettiert und in ein neues Eppendorf-Cup überführt
(„Zentrifugationsüberstand“), die andere Hälfte wird auf Eis gestellt, um proteolytische
Aktivität möglichst gering zu halten („Suspension“).
Danach erfolgt jeweils eine Bestimmung des Proteingehaltes der Homogenate nach Bradford.
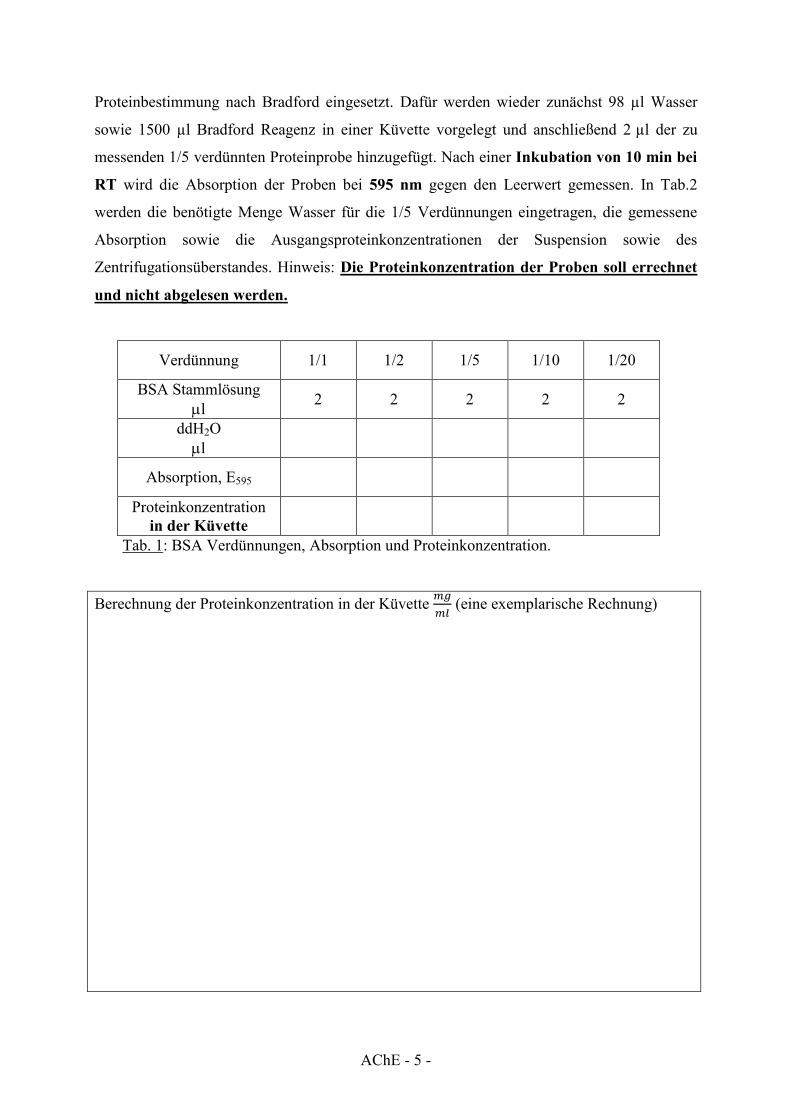
2.3 Proteinbestimmungen nach Bradford
Für die Proteinbestimmung nach Bradford werden zuerst definierte BSA Verdünnungen
hergestellt. Dazu werden ausgehend von der BSA Stammlösung (10 mg/ml) folgende
Verdünnungen (in Wasser) hergestellt: 1/1 1/2 1/5 1/10 1/20. Anschließend werden von
den Verdünnungen jeweils 2 µl für die Proteinbestimmung nach Bradford eingesetzt. Dafür
werden zunächst 98 µl Wasser sowie 1500 µl Bradford Reagenz in einer Küvette vorgelegt
und anschließend 2 µl des jeweiligen BSA-Standards hinzugefügt. Nach einer Inkubation
von 10 min bei RT wird die Absorption der BSA-Standards bei 595 nm gegen einen
Leerwert (100 µl Wasser + 1500 µl Bradford Reagenz) gemessen. In Tab.1 werden die
benötigten Mengen an Wasser für Verdünnungen eingetragen, die gemessene Absorption
sowie die Proteinkonzentration der BSA-Standards in der Küvette. Die gemessene Absorption
wird anschließend gegen die Proteinkonzentration aufgetragen (Abb. 3.).
Für die Proteinbestimmung der Suspension und des Zentrifugationsüberstandes wird jeweils
eine 1/5 Verdünnung (in Wasser) angesetzt. Anschließend werden davon jeweils 2 µl für die
AChE - 5 -
Proteinbestimmung nach Bradford eingesetzt. Dafür werden wieder zunächst 98 µl Wasser
sowie 1500 µl Bradford Reagenz in einer Küvette vorgelegt und anschließend 2 µl der zu
messenden 1/5 verdünnten Proteinprobe hinzugefügt. Nach einer Inkubation von 10 min bei
RT wird die Absorption der Proben bei 595 nm gegen den Leerwert gemessen. In Tab.2
werden die benötigte Menge Wasser für die 1/5 Verdünnungen eingetragen, die gemessene
Absorption sowie die Ausgangsproteinkonzentrationen der Suspension sowie des
Zentrifugationsüberstandes. Hinweis: Die Proteinkonzentration der Proben soll errechnet
und nicht abgelesen werden.
Verdünnung 1/1 1/2 1/5 1/10 1/20
BSA Stammlösung Pl 2 2 2 2 2
ddH2O Pl
Absorption, E595
Proteinkonzentration in der Küvette
Tab. 1: BSA Verdünnungen, Absorption und Proteinkonzentration.
Berechnung der Proteinkonzentration in der Küvette 𝑚𝑔𝑚𝑙
(eine exemplarische Rechnung)
AChE - 6 -
Suspension Zentrifugationsüberstand
Verdünnung (1/5) 2 µl 2 µl
ddH2O für die Verdünung _____µl _____µl
Absorption, E595
Proteinkonzentration der Ausgangslösung
𝑚𝑔𝑚𝑙
𝑚𝑔𝑚𝑙
Tab. 2: Verdünnungen, Absorption und Proteinkonzentration.
E 59
5
Proteinkonzentration Abb. 3: Auftragung der Absorption pro Proteinkonzentration.

AChE - 7 -
Berechnung der Proteinkonzentration in der Küvette (Suspension + Zentrifugationsüberstand) 𝑚𝑔𝑚𝑙
Berechnung der Ausgangsproteinkonzentration (Suspension + Zentrifugationsüberstand) 𝑚𝑔𝑚𝑙
(dieses Ergebnis in die Tabelle 2 eintragen)
2.4 Bestimmung der Acetylcholinesterase-Aktivitäten mit Hilfe des Ellman-Tests
Die Aktivität der Acetylcholinesterase in der Suspension sowie im Zentrifugationsüberstand
wird mit Hilfe des spektrophotometrischen Ellman-Tests bestimmt. Für diesen werden die
Ansätze I-V wie in Tab. 3 beschrieben vorbereitet.
AChE - 8 -
Tab. 4: Änderung der Absorption von DNTB bei 412 nm pro Zeit.
Zeit [min] Ansatz
I (Leerwert) II III IV V
Suspension 0 µl 22,5 µl 0 µl 22,5 µl 0 µl
Zentrifugationsüberstand 0 µl 0 µl 22,5 µl 0 µl 22,5 µl
Na,K Phosphatpuffer, 0,1M, pH 7,2 25 µl 2,5 µl 2,5 µl 0 µl 0 µl
Eserin, 2 mM 0 µl 0 µl 0 µl 2,5 µl 2,5 µl
Tab. 3: Pipettierschema für die Ansätze I-V.
In fünf Küvetten werden anschließend vorgelegt:
x 1460 µl Na,K Phosphatpuffer 0,1M pH 7,9
x 50 µl DTNB (Dithiobisnitrobenzoesäure 0,3 mM)
x 10 µl Acethylthiocholinjodid ( 2 mM)
Danach werden die Ansätze I-V zur jeweiligen Küvette zugegeben (siehe Hinweis unten!).
Dabei ist zu beachten, dass I den Leerwert repräsentiert, gegen den bei 412 nm gemessen
werden soll. Ebenfalls soll jeweils direkt nach Zugabe gemischt (Parafilm) und gemessen
werden. In Folge wird die Absorption aller Ansätze zum Zeitpunkt 0 min, 1 min, 2 min,
3 min, 4 min, 5 min,10 min sowie 15 min bestimmt und in Tab. 4 vermerkt sowie in Abb. 4
aufgetragen. (Messen Sie in den jeweils ersten 5 min die Proben bitte nacheinander und nicht
gleichzeitig! Wenn Sie also mit II starten, bitte erst nach 5 min die Probe III in die Küvette
pipettieren, mischen und messen, nach ablaufen der 5-minütigen Messung von Probe III dann
den Wert für 10 min von Probe II messen usw.)
0 1 2 3 4 5 10 15
I (Leerwert) - - - - - - -
II (Suspension)
III (Zentrifugationsüberstand)
IV (Suspension + Eserin)
V (Zentrifugationsüberstand +Eserin)
AChE - 9 -
Der Substratumsatz der AchE wird indirekt durch die Änderung der Absorption von DTNB
gemessen. Aus der Auftragung von 'E als Funktion der Zeit t wird die anfängliche
Extinktionsänderung pro Zeiteinheit ('E/min) ausgerechnet. Die Änderung der Extinktion pro
Zeit entspricht einer Änderung der Konzentration nach dem Lambert-Beerschen Gesetz. Mit
Hilfe des spezifischen Extinktionskoeffizienten für DTNB (H412=14150 M-1cm-1) läßt sich aus
der Änderung der Extinktion die Änderung der Konzentration pro Zeit ermitteln. Die
spezifische Enzymaktivität ist als Unit (µmol Substrat / (min*mg Protein)) definiert.
Errechnen sie die spezifische Enzymaktivität der Acetylcholinesterase!
E 41
2
Zeit, min
Abb. 4: Auftragung der Absorption pro Zeit.

AChE - 10 -
Beispiel für die Berechnung der Änderung der Proteinkonzentration pro Zeit [ 𝑚𝑜𝑙𝑙∗𝑚𝑖𝑛
]
Ergebnisse für die übrigen Proteinlösungen
Proteinlösung (röm. Ziffer + Name) Änderung der Proteinkonzentration pro Zeit
Berechnung der Proteinkonzentration in der Küvette [mg/l]
Suspension:
Zentrifugationsüberstand:

AChE - 11 -
Beispiel für die Berechnung der spezifischen Enzymaktivität [ µ𝑚𝑜𝑙 𝑆𝑢𝑏𝑠𝑡𝑟𝑎𝑡𝑚𝑖𝑛∗𝑚𝑔 𝑃𝑟𝑜𝑡𝑒𝑖𝑛
]
Ergebnisse für die übrigen Proteinlösungen
Proteinlösung (röm. Ziffer + Name) Spezifische Enzymaktivität
Ermitteln sie den prozentualen Anteil an Enzymhemmung bei den mit Eserin inkubierten
Ansätzen. Diskutieren sie den möglichen Mechanismus der Enzyminaktivierung.
Prozentualer Anteil an Enzymhemmung
Suspension:
Zentrifugationsüberstand:

AChE - 12 -
Möglicher Mechanismus der Enzyminaktivierung
Vergleichen Sie nun die beiden Homogenat-Proben (Suspension bzw.
Zentrifugationsüberstand). Wie unterscheiden sich die beiden Probe in der Enzymaktivität?
Versuchen Sie, eine Erklärung zu geben.
Vergleich beider Homogenat-Proben

AChE - 13 -
Zur symptomatischen Behandlung der Alzheimer Erkrankung, zur Behandlung der
Myasthenia gravis und als Antidot gegen Atropin werden unterschiedliche AchE-
Inhibitoren verwendet. Versuchen Sie, dafür eine Erklärung zu geben.
3. Auswertung und Aufgaben die sie während des Praktikums durchführen (Übersicht)
3.1 Berechnen sie die Proteinkonzentration der Homogenate in mg/ml anhand der
Geradengleichung der Eichgerade bei der Bradford Bestimmung
3.2 Der Substratumsatz der AchE wird indirekt durch die Änderung der Absorption von
DTNB gemessen. Aus der Auftragung von 'E als Funktion der Zeit t wird die anfängliche
Extinktionsänderung pro Zeiteinheit ('E/min) ausgerechnet. Die Änderung der Extinktion
pro Zeit entspricht einer Änderung der Konzentration nach dem Lambert-Beerschen
Gesetz. Die spezifische Enzymaktivität ist als Unit (µmol Substrat / (min·mg Protein))
definiert. Mit Hilfe des spezifischen Extinktionskoeffizienten für DTNB (H412=14150 M-

AChE - 14 -
1cm-1) läßt sich aus der Änderung der Extinktion die Änderung der Konzentration pro Zeit
ermitteln. Errechnen sie die spezifische Enzymaktivität der Acetylcholinesterase!
3.3 Ermitteln sie den prozentualen Anteil an Enzymhemmung bei den mit Eserin inkubierten
Ansätzen. Diskutieren sie den möglichen Mechanismus der Enzyminaktivierung.
3.4 Vergleichen Sie die beiden Homogenat-Proben (Suspension bzw.
Zentrifugationsüberstand). Wie unterscheiden sich die beiden Probe in der
Enzymaktivität? Versuchen Sie, eine Erklärung zu geben.
3.5 Zur symptomatischen Behandlung der Alzheimer Erkrankung, zur Behandlung der
Myasthenia gravis und als Antidot gegen Atropin werden unterschiedliche AchE-
Inhibitoren verwendet. Versuchen Sie, dafür eine Erklärung zu geben.
4. Weiterführende Literatur Lang, B., Vincent, A. (2009) Autoimmune disorders of the neuromuscular junction. Curr Opin
Pharmacol. 9: 336-40. Rafii, M.S., Aisen, P.S. (2009) Recent developments in Alzheimer’s disease therapeutics. BMC Med.
Feb 19;7:7 Riener, C.K., Kada, G., Gruber, H.J. (2002). Quick measurement of protein sulfhydryls with Ellman's
reagent and with 4,4'-dithiodipyridine. Anal Bioanal Chem 373: 266–276. Wilson, I.B., Harrison M.A. (1961) Turnover number of acetylcholinesterase. J. Biol. Chem. 236:
2292-2295.

AChE - 15 -
5. Rechenübung
Die Rechenübung muss am Versuchstag des Praktikums vor Beginn der Lehrveranstaltung bei den durchführenden Dozenten vollständig bearbeitet abgegeben werden. Eine nicht-Abgabe führt zum Ausschluss vom Praktikum.
Aufgabe 1 Ausgehend von einer 10 mg/ml konzentrierten BSA Stammlösung soll jeweils eine der folgenden Verdünnungen mit 2 µl BSA angesetzt werden.
Endkonzentration [mg/ml] 1/1 1/2 1/5 1/10 1/20 BSA Stammlösung [µl] 2 2 2 2 2
ddH2O [Pl] Beispielrechnung:
Aufgabe 2
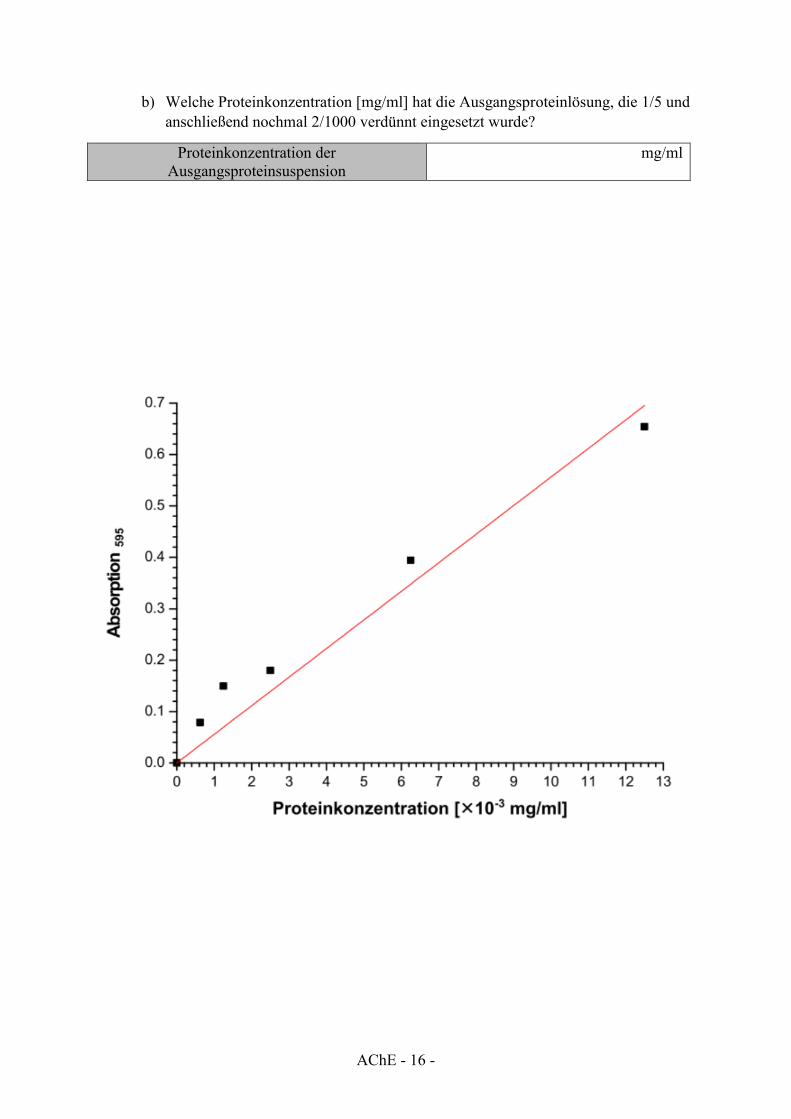
In der untenstehenden Grafik ist eine exemplarische BSA Eichgerade zur Bestimmung der Proteinkonzentration gezeigt. Die Absorption einer gemessenen Proteinprobe beträgt 0.122.
a) Welche Proteinkonzentration [mg/ml] befindet sich in der entsprechenden Küvette?
Proteinkonzentration in der Küvette mg/ml
AChE - 16 -
b) Welche Proteinkonzentration [mg/ml] hat die Ausgangsproteinlösung, die 1/5 und anschließend nochmal 2/1000 verdünnt eingesetzt wurde?
Proteinkonzentration der Ausgangsproteinsuspension
mg/ml
AChE - 17 -
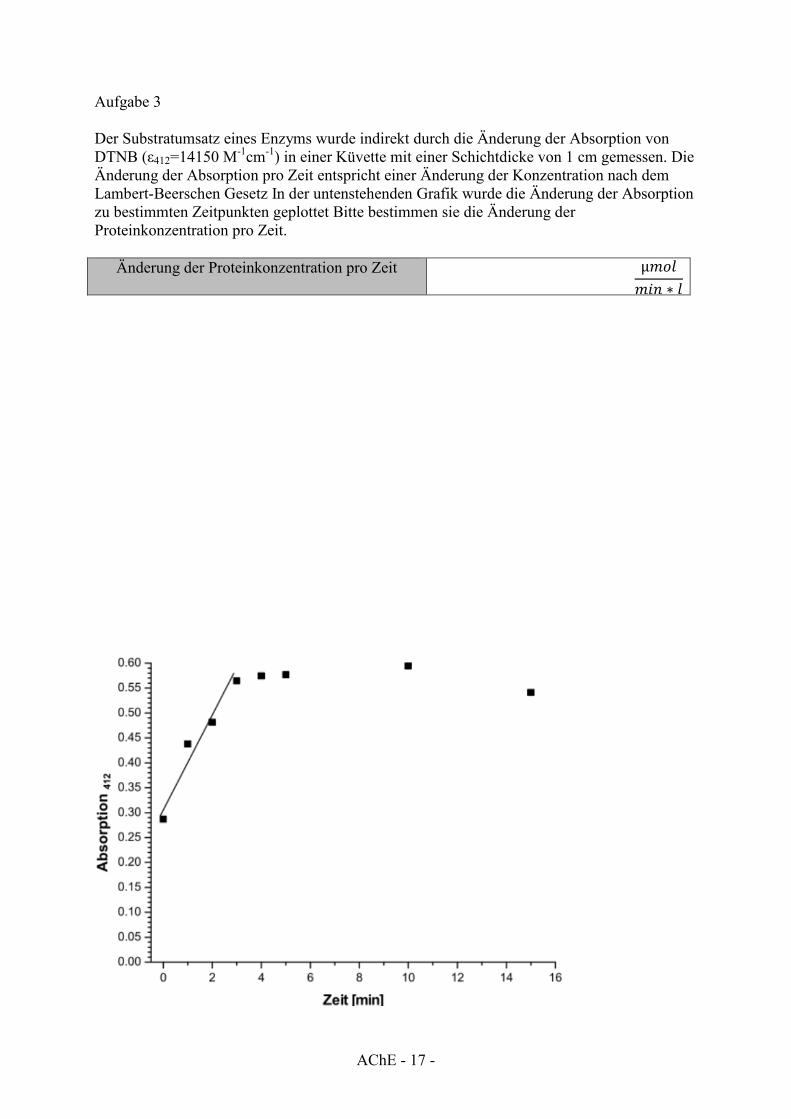
Aufgabe 3 Der Substratumsatz eines Enzyms wurde indirekt durch die Änderung der Absorption von DTNB (ε412=14150 M-1cm-1) in einer Küvette mit einer Schichtdicke von 1 cm gemessen. Die Änderung der Absorption pro Zeit entspricht einer Änderung der Konzentration nach dem Lambert-Beerschen Gesetz In der untenstehenden Grafik wurde die Änderung der Absorption zu bestimmten Zeitpunkten geplottet Bitte bestimmen sie die Änderung der Proteinkonzentration pro Zeit.
Änderung der Proteinkonzentration pro Zeit µ𝑚𝑜𝑙𝑚𝑖𝑛 ∗ 𝑙

AChE - 18 -
Aufgabe 4 Das Enzym aus Aufgabe 3 sei die AchE, deren Substratumsatz in einer Proteinprobe mit einer Konzentration von 0.127 mg/ml in der Küvette gemessen wurde. Die spezifische Enzymaktivität ist als Unit[ µ𝑚𝑜𝑙 𝑆𝑢𝑏𝑠𝑡𝑟𝑎𝑡
𝑚𝑖𝑛∗𝑚𝑔 𝑃𝑟𝑜𝑡𝑒𝑖𝑛 ] definiert. Errechnen sie die spezifische
Enzymaktivität der Acetylcholinesterase.
Spezifische Aktivität µ𝑚𝑜𝑙𝑚𝑖𝑛 ∗ 𝑚𝑔

Elektrophysiologie - 1 -
Elektrophysiologie / Computersimulation "Virtuelles Labor"
(Versuch Elektrophysiologie)
Inhalt:
1. Simulation elektrophysiologischer Ableitungen am Ischiasnerv des Frosches: Ableitung
von Summenaktionspotentialen („SimNerv“)
2. Durchführung eines Experiments
3. Aufgaben

Elektrophysiologie - 2 -
1. Simulation elektrophysiologischer Ableitungen am Ischiasnerv des Frosches:
Ableitung von Summenaktionspotentialen („SimNerv“)
Die Experimente werden in den Cip-Pool-Räumen am Computer durchgeführt.
Einleitung: Die Informationsverarbeitung im Nervensystem beruht auf der Generierung und
Fortleitung bioelektrischer Signale, die als Aktionspotentiale bezeichnet werden. Aktions-
potentiale sind kurzfristige Änderungen der Membranspannung (Depolarisation), die im
Bereich des Axonhügels initiiert und entlang der Axone zu den Nervenendigung
weitergeleitet werden. Im Verlauf eines Aktionspotentials kommt es zu schlagartigen
Permeabilitätsänderungen der Membran vor allem für Natrium- und Kaliumionen. In der
Depolarisationsphase werden zuerst spannungsabhängige Natrium-Kanäle geöffnet. Aufgrund
des hohen Konzentrationsgefälles strömen Natrium-Ionen in die Zelle ein (außen hohe Na+-
Ionenkonzentration gegenüber dem Zellinnern), wodurch eine Ladungsumkehr der
Zellmembran von negativ zu positiv bis zu einem Spitzenwert, dem Peak, erfolgt. Bereits in
der ansteigenden Phase des Aktionspotentials kommt es zu einer spontanen Inaktivierung der
Natriumkanäle, während spannungsabhängige Kalium-kanäle geöffnet werden; es erfolgt ein
Ausstrom von Kalium-Ionen (außen niedrigere K+-Ionenkonzentration gegenüber dem
Zellinnern), bis das Ruhepotential der Zelle wieder hergestellt ist. Dieser Vorgang läuft
immer selbsttätig ab, sobald ein bestimmtes Schwellenpotential überschritten wird, dessen
absoluter Betrag keinen Einfluß auf die Amplitudenhöhe des Aktionspotentials hat. Diese
Tatsache wird als Alles-oder Nichts-Gesetz bezeichnet. Durch die Aktivität der Natrium-
Kalium-ATPase wird die ursprüngliche Ionenverteilung wieder hergestellt.
Direkt nach dem Aktionspotential ist die Membran nicht sofort wieder erregbar, da die
spannungsabhängigen Natriumkanäle noch inaktiviert sind; man spricht von der Refraktärzeit.
Die Refraktärzeit ist ein wichtiger Aspekt für die Erregungsleitung durch Aktionspotentiale,
da sie gewährleistet, dass der Reiz nur in eine Richtung geleitet wird und nicht wieder zum
Ursprungsort "zurückläuft" (unidirektionale Leitung). Gelangt ein Aktionspotential an eine
Synapse, so wird durch die Depolarisation die Exocytose einiger mit Neurotransmitter
gefüllter Vesikel initiiert, wodurch das Signal an die nachfolgende postsynaptische Zelle
weitergegeben wird.
Am Beispiel des großen Beinnerven des Frosches (Nervus ischiadicus) sollen einige wichtige
Grundlagen der Nervenerregung erarbeitet werden. Da die Experimente an einem kompletten
Nerven und nicht an einzelnen Axonen durchgeführt werden, treten nach überschwelliger

Elektrophysiologie - 3 -
Reizung stets Summenaktionspotentiale (SAP) auf. Der N. ischiadicus ist ein gemischter
Nerv, d.h. er enthält sowohl myelinisierte als auch unmyelinisierte Nervenfasern.
Auf dem Bildschirm erscheint das virtuelle Labor. Folgende Geräte und Utensilien werden für
die Experimente benötigt:
x Präparierschale mit zwei präparierten Nerven
x Experimentierkammer
x Stimulator
x Oszilloskop
2. Durchführung eines Experiments
In der Präparierschale befinden sich zwei aus den Beinen eines Frosches heraus
präparierte Nerven. Beide Nerven haben ein unterschiedliches Eigenschaftsprofil.
Zuerst wird die weiße Experimentierkammer durch Anklicken mit dem Mauscursor
geöffnet.
Anschließend müssen Sie einen der beiden frei präparierten Nerven aus der Präparierschale
heraus nehmen (Anklicken mit dem Mauscursor) und durch Ziehen mit der Maus in die
geöffnete Experimentierkammer bringen.
Sobald Sie die Maustaste loslassen, legt sich der Nerv in die Kammer. Durch erneutes
Anklicken schließt sich die Kammer wieder.
1. Der Stimulator dient der Applikation elektrischer Reize. Er wird durch Mausklick an
der On/Off Taste eingeschaltet, die grüne Leuchtdiode wird sichtbar. Über drei Schieberegler
können drei verschiedene Reizparameter stufenlos eingestellt werden:
Amplitude : Intensität der Reizung (Rechteckimpuls, mV)
Duration : Dauer der Reizung in Millisekunden
Delay: Zeitabstand zwischen zwei Reizungen (in Millisekunden)
(Mode muss dabei auf Twin eingestellt sein!)
Jedem Schieberegler ist ein Multiplier zugeordnet, durch den der Arbeitsbereich um das
Zehn bis Hundertfache erweitert werden kann.

Elektrophysiologie - 4 -
2. Das Oszilloskop dient der Aufzeichnung und Registrierung der Nervenaktivität
(Summenaktionspotentiale). Das Gerät wird durch Anklicken am Power Schalter (Off-On)
eingeschaltet, die grüne Leuchtdiode wird sichtbar.
x Auf Kanal 1 können die elektrischen Reize, auf Kanal 2 die Reizantworten
sichtbar gemacht werden. Über die Marker CH1 und CH2 am linken Rand des
Schirmes kann die Position der beiden Kanalspuren nach oben oder nach unten
verschoben werden. Die aufgezeichneten Meßspuren können nach einem
Experiment sofort wieder gelöscht (clear screen) oder auf dem Schirm gespeichert
werden (store).
x Durch die beiden Drehregler (Channel 1, Channel 2) kann die
Kanalempfindlichkeit eingestellt werden.
x Der Drehregler „Timebase“ (Zeitskala) erlaubt es, die zeitliche Auflösung der
Meßspuren einzustellen.
All drei Regler lassen sich bei gedrückter Maustaste in beide Richtungen bewegen, ein roter
Punkt zeigt die aktuelle Einstellung an.
Vor Beginn der Experimente müssen die Regler am Stimulator und am Oszilloskop auf
passende Werte eingestellt werden, damit die Nervenaktivität am Schirm des Oszilloskops gut
abgelesen werden kann.
x Dazu werden am Stimulator zuerst „Amplitude“ auf 100 mV und „Duration“
auf 1 ms vorgewählt. Durch Anklicken der Taste „Stimulus On“ wird stimuliert.
x Nach jeder Einzelstimulation werden der Channel 2- und der Timebase-Regler des
Oszilloskops so lange stufenweise hochgedreht, bis auf dem Schirm ein
befriedigendes Resultat zu sehen ist (z.B. 1ms/Div für die Zeitskala und 2 mV/Div
für die Höhe des Aktionspotenials).
x Anschließend wird auch Channel 1 entsprechend eingeregelt. Die Zeit (ms) und
die Spannungsstufen (mV) können am Gitter auf dem Schirm des Oszilloskops
abgelesen werden.
Entsprechend der Stellung der Reglerknöpfe entspricht 1 Kästchen jeweils einer
Zeit – bzw. Spannungseinheit.

Elektrophysiologie - 5 -
Amplitude (mV)
Zeit (ms)
3. Aufgaben:
Erstellen Sie eine Skizze über den Aufbau der Apparatur einschließlich der Verschaltung!
Stellen Sie eine Reizdauer von 1ms sowie eine Reizamplitude von 100 mV ein und
kalibrieren Sie nun das Oszilloskop, so dass Sie Reiz- und Reizantwort auf dem Schirm
gut ablesen können.
Stellen Sie eine Reizdauer von 1ms ein und reizen Sie den Nerven – beginnend mit 20 mV-
mit langsam ansteigenden Reizamplituden. Stellen Sie fest, ab welcher Reizintensität die
erste Reaktion sichtbar wird (minimale Reizschwelle) bzw. ab wann keine Steigerung
mehr zu erzielen ist (maximale Reizschwelle, Sättigung)!
Stellen Sie die Abhängigkeit zwischen Reizintensität und Amplitude (Höhe) der
Aktionspotentiale in einem Diagramm grafisch dar (mindestens acht Wertepaare, die
Höhenwerte der Aktionspotentiale sind am Oszilloskop abzulesen)! Weshalb kommt es
bei diesem Experiment zu einem Anstieg der Amplitude (im Widerspruch zum Alles-
oder-Nichts Prinzip) ?
Zur Bestimmung der Refraktärzeit stellen Sie die Reizzeit auf 1ms und schalten Sie den
Stimulator auf TWIN-Mode. Er gibt dann an seinem Ausgang Doppelimpulse aus.
x Den zeitlichen Abstand zwischen beiden Reizimpulsen kontrollieren Sie mittels des
Delay-Schiebereglers. Stellen Sie ihn zunächst auf 10 ms ein.
x Stellen Sie die Reizamplitude auf einen Wert etwas oberhalb des zuvor (Punkt 2)
ermittelten Sättigungswertes ein und stimulieren Sie den Nerv. Achten Sie darauf,
dass beide SAPs gleiche, maximale Amplituden zeigen. Stellen Sie die Zeitauflösung
(time base) so ein, dass auf dem Schirm des Oszilloskops beide APs zu sehen sind.

Elektrophysiologie - 6 -
Reduzieren Sie nun „Delay“ und reizen sie den Nerv erneut. Ab einem bestimmten
Delay-Wert wird die Amplitude des zweiten SAP kleiner und verschwindet schließlich
ganz. Erstellen Sie ein Diagramm, aus dem die Abhängigkeit der SAP-Amplitude vom
Delay deutlich wird und bestimmen Sie aus ihrer Zeichnung die absolute und die
relative Refraktärzeit. Worauf ist die Refraktärzeit zurückzuführen? Welche
physiologische Bedeutung hat die Refraktärzeit?
Bestimmung der Leitungsgeschwindigkeit öffnen Sie die Reizkammer erneut und schieben
Sie die beiden Reizelektroden (gelb und blau) und die beiden Ableitelektroden (grün und
rot) möglichst weit auseinander: (z.B. gelb und blau auf 0 bzw. 1cm, grün und rot auf 9
bzw. 10 cm). Notieren Sie sich die Elektrodenpositionen.
x Stellen Sie den Stimulator wieder von Twin auf Single zurück.
x Stellen Sie „Duration“ auf 2ms, die Reizstärke auf 200 mV ein und lösen Sie ein
Summenaktionspotenzial aus.
x Lesen sie am Oszilloskop die zeitliche Verzögerung (Latenz) vom Reizbeginn bis
zum Beginn des Aktionspotenzials ab. (Reizbeginn ist, wie aus Kanal 1 ersichtlich,
unmittelbar am linken Rand des grünen Leuchtschirmes). Um eine gute Zeitauflösung
zu erhalten sollte Timebase passend eingestellt werden.
Die Leitungsgeschwindigkeit lässt sich aus dem Quotienten von Elektrodenabstand
( = Laufstrecke des Signals) und Latenzzeit in cm/ms ausrechnen. Rechnen Sie in
m/sec um und vergleichen Sie mit Literaturwerten von Säugern. Welche Faktoren
beeinflussen die Leitungsgeschwindigkeit eines Nerven?

Neuroanatomie - 1 -
Neuroanatomie / Neurohistologie
(Versuch Neuroanatomie)
Inhalt:
1. Einleitung
2. Struktur und Lage des Kleinhirns (Cerebellum) der Labormaus
3. Goßhirnrinde (Cortex cerebri) des Hausschweins
4. Struktur der Netzhaut (Retina) der Regenbogenforelle

Neuroanatomie - 2 -
1. Einleitung
In diesem Teil sollen Sie durch vergleichende Untersuchung gefärbter Schnittpräparate
einen Eindruck gewinnen, welche Methoden zur gezielten Darstellung der unterschiedlichen
zellulären Komponenten des Nervengewebes angewendet werden können. Es sollen
Zeichnungen ausgewählter Hirnregionen anfertigt werden, in denen charakteristische
Gewebestrukturen - in leicht schematisierter Form - zeichnerisch wider gegeben werden. Bei
gefärbten Präparaten bieten vor allem Größe und Verteilungsmuster von Zellkernen wichtige
Anhaltspunkte zum Verständnis der zellulären Struktur einer Hirnregion.
Für diese Untersuchungen stehen verschiedene Hirnschnittserien folgender Spezies zur
Verfügung, die mikroskopisch untersucht werden sollen.
1) Labormaus (Längsschnitte durch das Gesamthirn)
2) Hausschwein (Silberimprägnierte Schnitte durch das Vorderhirn)
3) Regenbogenforelle (Querschnitte durch das Auge)
Die Schnittserien wurden nach einer der folgenden Techniken gefärbt sind (Rezepte siehe
Romeis, B.: Mikroskopische Technik)
a) Azan (Azokarmin/Anilinblau)-Färbung: kombinierte Mehrfachfärbung zur
Darstellung sämtlicher Nerven- und Gliazellen (Schnittdicke 10 Mikrometer).
b) Silberimprägnierung nach Bubenaite bzw.nach Golgi: selektive Darstellung einiger
weniger Nerven-bzw. Gliazellen auf ungefärbtem Hintergrund. Auf den 20 bis zu 40
Mikrometer dicken Schnitten können die Zellfortsätze dieser Zellen über weite Strecken
verfolgt werden
2. Struktur und Lage des Kleinhirns (Cerebellum) der Labormaus
Das Kleinhirn (Cerebellum) spielt eine wesentliche Rolle bei der neuronalen Kontrolle der
Körperhaltung und bei der Koordination von Bewegungsabläufen im dreidimensionalen Raum.
Insbesondere ist es zuständig für
x die Steuerung und Korrektur der stützmotorischen Anteile von Haltung und Bewegungen
x für die Kurskorrektur langsamer zielmotorischer Bewegungen
x für die reibungslose Durchführung der im Großhirn entworfenen schnellen Zielmotorik
Das Cerebellum ist außerdem beim Erlernen motorischer Fertigkeiten beteiligt.

Neuroanatomie - 3 -
Die Rinde des Kleinhirns ist im Gegensatz zu der des Großhirns durch einen
dreischichtigen histologischen Aufbau charakterisiert (vgl. Abbildung 1): eine tief liegende
kleinzellige Körnerzellschicht wird durch eine großzellige Purkinjezellschicht von einer
außen liegenden Molekularschicht abgegrenzt. Darunter liegt das Marklager, das aus
myelinisierten Axonen besteht.
Material: Azan-gefärbte Längsschnitte durch das Gehirn der Hausmaus
Aufgaben:
x Fertigen Sie an Hand eines sagittalen Längsschnittes eine Detailskizze der
Kleinhirnrinde an und bezeichnen Sie die drei Hauptschichten (40fach Objektiv).
Skizzieren Sie insbesondere ein Purkinje-Neuron. Zur besseren Darstellung des
stark verzweigten Dendritenbaums ist ein leichtes Schließen der Kondensorblende
vorteilhaft.
x Welche Funktion haben Purkinje- Zellen?
x Welche beiden Fasersysteme führen ihnen Signale zu (Lehrbuch)?
Abb. 1 :Ausschnitt aus der Kleinhirnrinde eines Säugers

Neuroanatomie - 4 -
3. Goßhirnrinde (Cortex cerebri) des Hausschweins
Die Großhirnrinde der Säuger (graue Substanz) zeigt einen sechsschichtigen
histologischen Aufbau. Der dominierende Nervenzelltyp sind Pyramidenzellen, deren
Zellkörper einen stark verzweigten Dendriten aufweist, der stets senkrecht zu Hirnoberfläche
ausgerichtet ist. Außer Nervenzellen findet man in der grauen Substanz vor allem die zu den
Gliazellen gehörenden protoplasmatischen Astrozyten. In der darunter liegenden weißen
Substanz, die überwiegend aus myelinisierten Nervenfasern besteht, treten vor allem faserige
Astrocyten und Oligodendrocyten, die myelinbildenden Gliazellen des Zentralnervensystems,
auf.
Darstellung von Nerven-und Gliazellen durch Silberimprägnierung von Nervengewebe:
Im Jahre 1883 fand der Italiener Camillo Golgi eine Methode, mit der man Nervenzellen
bis in ihre feinsten Ausläufer mit Silber imprägnieren kann. Es gibt inzwischen zahlreiche
Modifikationen dieser Methode, bei der aus noch ungeklärter Ursache stets nur ein Bruchteil der
tatsächlich vorhandenen Zellen geschwärzt wird. Auf diese Weise erhält man sehr klare und
übersichtliche mikroskopische Bilder. Die Modifikation nach Bubenaite kann auch an Formol-
fixierten Gewebeproben durchgeführt werden.
Material:
Es stehen bereits fertige Präparate silberimprägnierter Hirnschnitte (Cortex) des Hausschweines
zur Verfügung. Auf den Schnitten sind neben den großen Nervenzellen (vor allem
Pyramidenzellen) auch die deutlich kleineren Gliazellen (Astrocyten und Oligodendrocyten),
sowie Blutgefäße zu erkennen.
Beschreibung der Methode
x ca. 1-2 cm lange Gewebefragmente werden für zwei Tage bei 34°C (Wärmeschrank) in
2,5% ige Kaliumdichromatlösung inkubiert.
x Danach werden die Gewebeblöcke kurz mit Filterpapier abgetupft, in ein frisches
Glasgefäß gebracht und einmal mit 2% iger Silbernitratlösung kurz, aber gründlich
abgespült. Anschließend werden sie in frische Silbernitratlösung gebracht und dort für
zwei Tage bei 34°C (Wärmeschrank) belassen.

Neuroanatomie - 5 -
x Nach Abgießen der Silbernitratlösung werden die Gewebeblöcke zur Härtung in 90%
Äthylakohol überführt.
x Sobald das Gewebe etwas durchgehärtet ist (nach ca. 30 min.), können mit einem
Tischmikrotom Schnitte angefertigt und anschließend mikroskopiert werden.
Aufgaben:
1. Suchen Sie bei schwacher Vergrößerung (10- bzw. 20-faches Objektiv) nach großen,
möglichst einzeln liegenden Nervenzellen mit gut imprägnierten Zellfortsätzen.
Wechseln Sie zum 40-fach Objektiv und verfolgen Sie die Zellausläufer durch
Drehen an der Feinscharfstellung bis zu deren Ende. Fertigen Sie eine möglichst
genaue Skizze von einer reich verzweigten Nervenzelle mit sämtlichen Zellfortsätzen
an.
2. Untersuchen Sie die Feinstruktur der Dendritenverzweigungen bei 40-facher
Objektivvergrößerung. Wie ist die Oberfläche der Dendriten strukturiert, welche
Bedeutung könnten die beobachteten Strukturen haben?
3. Suchen Sie in den tieferen, myelinisierten Schichten nach Oligodendrocyten (kleine
Zellen mit kurzen Zellfortsätzen). Skizzieren Sie eine solche Zelle und
berücksichtigen Sie den Größenunterschied zu Nervenzellen..
4. Versuchen Sie die kleinen, sternförmig verzweigten Astrocyten zu identifizieren, und
fertigen Sie eine Zeichnen an, die die Größenverhältnisse zu Nervenzellen und
Oligodendrocyten berücksichtigt.
Astrocyten stehen häufig in direktem Kontakt mit Blutgefäßen. Welche funktionelle
Bedeutung der Astrocyten kann man daraus ableiten?

Neuroanatomie - 6 -
4. Struktur der Netzhaut (Retina) der Regenbogenforelle
Das menschliche Auge wird auf Grund seines Aufbaues häufig mit einem Fotoapparat
verglichen (Abb.1). Wie bei einer Kamera ist das „Objektiv“ des Auges ein zusammengesetztes
optisches System, das aus Cornea (Hornhaut), vorderer Augenkammer und Linse besteht. Die
Hauptbrechkraft geht von der Cornea aus, während die Linse durch Veränderung ihrer
Krümmung das Auge an Nah- bzw. Fernsicht anpasst (Akkommodation). Mitten im
Strahlengang sitzt beim Auge – wie bei der Kamera – eine Blende mit verstellbarem
Durchmesser, die Iris, die den Lichteinfall ins Auge steuert. Die Achse des optischen Systems
trifft auf der Netzhaut (Retina) auf eine Stelle mit einer kleinen Einbuchtung, die als Sehgrube
(Fovea) bezeichnet wird. Sie ist der Ort des schärfsten Sehens. Wenn man ein Objekt genau
betrachten will („fixeren“), richtet man den Blick automatisch so ein, dass sein Abbild auf die
Sehgrube beider Augen fällt. Eine weitere Auffälligkeit ist der Netzhautbereich, wo der Sehnerv
ins Auge eintritt. Da dort keine lichtempfindlichen Sensoren (Photorezeptoren) auftreten, wird
diese Stelle als blinder Fleck bezeichnet. Durch die ständigen Augenbewegungen, wird das
Sehzentrum jedoch fortlaufend über die optischen Eigenschaften des gesamten Sehfeldes
informiert, so dass dieser blinde Fleck normalerweise nicht wahrgenommen wird.
Die Netzhaut entsteht während der Embryonalentwicklung aus einer Ausstülpung des
Zwischenhirnbodens, ist also genau genommen ein Teil des Gehirns. Daher ist es nicht
überraschend, dass sie ein komplexes neuronales Netzwerk besitzt, das bereits eine erhebliche
Vorverarbeitung visueller Informationen betreibt. Die wichtigsten zellulären Elemente und ihre
Verknüpfungen sind in Abb. 2 dargestellt. Grundsätzlich ist zwischen einem radialen und einem
horizontalen Signalweg innerhalb der Retina zu unterscheiden. Die radialeVerknüpfung wird
durch Bipolarzellen vermittelt, welche die Photorezeptoren (Stäbchen, Zapfen) mit den
Dendriten der Retinaganglienzellen verknüpfen. Die horizontaleVerschaltung erfolgt in der
äußeren Retina über Horizontalzellen, während dies in der inneren Retina durch amakrine
Zellen bewerkstelligt wird. Die äußerst regelmäßige Anordnung dieser Zellen in klar definierten
Schichten erlaubt es, jeden der beteiligten Zelltypen aufhistologischen Schnitten schon auf
Grund ihrer Position zu identifizieren.

Neuroanatomie - 7 -
Material: Azan-gefärbte Querschnitte durch das Auge der Regenbogenforelle
Aufgaben:
1. Untersuchen Sie eines der Retinapräparate aus dem Auge der Forelle und versuchen
sie die o.g. Zellen und Zellschichten zu identifizieren.
2. Erstellen Sie eine einfache Skizze, auf der die Proportionen der einzelnen
Retinaschichten und Zell(kern)größen richtig wiedergegeben sind.
3. Vor den Retinaganglienzellen befindet sich eine faserige Schicht. Vergleichen Sie
deren Breite im Randbereich bzw. in der Mitte der Retina. Wodurch sind die zu
beobachtenden Unterschiede zu erklären?
4. Bestimmen Sie die Anzahl von Retinaganglienzellen in einem ausgewählten Bildfeld.
Vergleichen Sie die Anzahl von Photorezeptoren bzw. Bipolarzellen im gleichen
Bildfeld (Schätzung). Welche Schlussfolgerung ziehen sie daraus bezüglich der
Zellverschaltung (1:1 Verhältnis oder Konvergenz bzw. Divergenzschaltung)? Welche
Auswirkungen auf die Sehschärfe bzw. Lichtempfindlichkeit ergeben sich aus einer
solchen Verschaltung?

- Sicherheit 1 -
Sicherheitsbelehrung Teil I
Leisten Sie – auch zu Ihrer eigenen Sicherheit - den Anweisungen der Betreuerinnen und Betreuer des Versuchs unbedingt Folge. Die Betreuerinnen und Betreuer sind auch die ersten Ansprechpartner, falls Sie weitere Fragen zur Laborsicherheit haben.

- Sicherheit 2 -
Sicherheitsbelehrung Teil II





























