Embed Size (px)
Citation preview

HUMANA GENETIKA
o Opcije prevencije naslednih bolesti: prevencija rađanja obolelih od retkih monogenskih bolesti i poremećaja hromozoma
(prenatalni skrining) otkrivanje fenilketonurije, hipotireoze i galaktozemije kao i sličnih monogenskih bolesti na
samom rađanju što uz pravu terapiju omogućava normalan život (natalni skrining) posmatranjem porodične historije, vrši se posebna pretraga za mutiranim genima
odgovarajućih bolestio Forme genetske terapije:
transplantacija koštane srži transplantacija embrionalnih matičnih ćelija
o Istorijat: pre oko 2000. godina, Talmud – oslobođena obrezivanja muška deca čiji su muški srodnici po
majci pokazivali sklonost krvarenju (hemofilija) druga polovina 17. veka, de Graaf – spoznaja suštine začeća (spermatozoid+jajna ćelija); opis
ispupčenja na jajnicima, danas Grafovi folikuli 18. vek, Maupertuis– proučavanje rodoslovnih stabala; zaključio da albinizam i prekobrojni
prsti imaju različit način nasleđivanja 1865. godina, Mendel – objavljuje rezultate svojih eksperimenata na grašku; njegovo delo
ostaje neprimećeno 1900. godina,de Vries,Correns, Tschermak – ponovo otkrivaju Mendelove eksperimente i
postavljaju Mendelove zakone 1900. godina, Garrod – objašnjava način nasleđivanja alkaptonurije po Mendelovim zakonima
uz prepoznavanje uloge kosngviniteta 1900. godina, Landsteiner – ABO sistem krvnih grupa 1903. godina, Sutton, Boveri – hromozomska teorija nasleđivanja 1903. godina, Otto – recesivno nasleđivanje na X hromozomu 1953. godina, Crick, Watson i Wilkins – molekularna struktura DNK 1956. godina, Tjio i Levan/ Ford i Hamerton – čovek ima 46 hromozoma 1958. godina, Bedle – Nobelova nagrada za otkriće sistema jedan gen-jedan enzim 1959. godina, Lejeune/ Ford i Jacobs – Daunov sindrom uzrokovan prekobrojnim hromozomom
iz grupe G 1961. godina, Jacob i Monodo – koncept operona, genetska regulacija 1988. godina – započet projekat humanog genoma 2003. godina – kompletiran humani genom i genomi pet inicijalnih modela organizama

Ćelijska i molekularna baza nasleđivanja
o Ćelija: ćelijska membrana (plazmalema) – fosfolipidni dvosloj koji se sastoji od hidrofobnih regiona u
unutrašnjiosti i hidrofilnih spolja; selektivno je propustljiva; sadrži proteine koji služe za međućelijsko prepoznavanje, signalizaciju i transport
jedro (nukleus) – ima dvoslojnu membranu; sadrži jedarce (nukleolus) u kojem se sintetiše rRNK za koju se geni nalaze na kratkim kracima akrocentričnih hromozoma (13, 14, 15, 21, 22); sadrži oko 99,5% ćelijske DNK u vidu linearnih hromozoma; nuklearni matriks sadrži proteine i RNK kao i hromozome koje se dodiruju putem MARs sekvenci DNK
citosol – tečni deo citoplazme; u njemu se odvija većina metaboličkih aktivnosti; sadrži citoskelet koji ima ulogu u pokretanju ćelije, daje joj oblik i uključen je u međućelijski transport
endoplazmin retikulum – mreža uvijenih kanalića koja zajedno sa ribozomima čini aparat za biosintezu proteina i lipida; u granuliranom EM sintetišu se proteini i dodaju oligosaharidi a u glatkom ER sintetišu se lipidi
Goldžijev aparat – vrši sekreciju produkata ćelije; funkcionalno i morfološki je povezan sa ER
DNK RNK
DEZOKSIRIBONUKLEOTID RIBONUKLEOTID
DEZOKSIRIBONUKLEOZID
FOSF
ATN
A G
RUPA
RIBONUKLEOZID
FOSF
ATN
A G
RUPAAZOTNA BAZA DEZOKSIRIBOZA AZOTNA BAZA RIBOZA
PURINSKA PIRIMIDINSKA PURINSKA PIRIMIDINSKA
adenin timin adenin uracil
guanin citozinguanin citozin
DIHELIKS (DVOSTRUKI LANAC) HELIKS (JEDNOSTRUKI LANAC)
HROMOZOMI NUKLEOLUS, CITOPLAZMA
mitohondrije – vrše obnavljanje ATPa (ADP---ATP) metodom oksidativne fosforilacije peroksizomi – male vezikule sa jednostrukom membranom koje razlažu vodonik-peroksid,
toksičan za ćeliju lizozomi – digestivne organele, sadrže hidrolitičke enzime ribonukleazu i fosfatazu; vrše
fagocitozu i pinocitozu

o Stabilnost nukleotidnog lanca: fosfodiestarske veze koje nastaju vezivanjem fosfatnih grupa za 3 ugljenikov′ atom i 5 ′
ugljenikov atom jedne pentoze vodonične veze između komplementarnih azotnih baza dva lanca antiparalelnost dva lanca – 3 (-OH) naspram 5 (-P) kraja′ ′
o Replikacija (sinteza) DNK: odvija se u toku S faze ćelijskog ciklusa, pri čemu se količina DNK udvostručuje; od jednog starog lanca (matrice) nastaje novosintetisan koji mu je potpuno identičan:
I. proteini despiralizacije (A) razmotavaju molekul DNKII. DNK heliksaza raskida vodonične veze; nastaju replikacione viljuške (replikacija započinje na
više mesta i u oba smera)III. proteini destabilizacije DNK i DNK topoizomeraze uklanjaju superzavojeIV. RNK polimeraza (primaza) dovodi RNK nukleotide formirajući RNK početnicu (prajmer)V. na jednom starom lancu se uz pomoć DNK polimeraze III na RNK prajmer vezuju DNK
nukleotidi, on se sintetiše ka dnu replikativne viljuške i naziva se vodeći lanacVI. na drugom starom lancu se sintetiše nekoliko RNK prajmera na koje se nastavljaju mali
fragmenti DNK nukleotida koji se nazivaju Okazaki; isecaju se RNK početnice, a uz pomoć DNK polimeraze I praznine nastale tim putem se popunjavaju DNK nukleotidima; DNK ligaza povezuje nukleotide u kontinuirani lanac i tako nastaje zaostajući lanac
VII. rezultat replikacije je jedan hromozom sa dve hromatide i dva molekula DNK u centromerio Sinteza proteina:
geni – delovi DNK (600-1800 bp) koji sadrže informacije za sintezu RNK; tRNK i rRNK se ne prevodi u proteine, iRNK se prevodi u proteine
genetski kod – sekvenca nukleotida u genu (deo molekula DNK) kodon – set od tri nukleotidne baze na iRNK koja odgovara jednoj AK antikodon – set od tri baze na tRNK koja nosi AK kojoj odgovara komplementarni kodon transkripcija – proces stvaranja iRNK translacija – proces prenošenja informacije sa iRNK u polipeptidni lanac
Hromozomi(chroma=boja, soma=telo)
o 15% DNK, 83% proteinio 1 hromozom = 4 cm diheliksa; 46 hromozoma čoveka = 2m DNKo Struktura:
primarna – namotavanje diheliksa sekundarna – namotavanje oko histonskih kuglica; nastaju nukleozomi tercijarna – namotavanje nukleozoma; nastaju hromatinske niti
o Uloga: prenos genetske informacije sa jedne generacije na drugu o Broj hromozoma je stalan za vrstu (do 1956. godine se smatralo da čovek ima 48)

o Nauka o hromozomima – citogenetikao Centromera (primarno suženje) – sastoji se od nekoliko stotina kb repetitivne DNK i uloga joj je kretanje pri ćelijskoj deobi; deli hromozom na dugi (q) i kratki (p) krako Telomera – vrh svakog hromozomskog kraka kome je uloga očuvanje strukturnog itegriteta hromozoma; sastoje se od mnogo ponavljanja TTAGGG sekvence; enzim telomeraza (pri replikaciji) je odgovoran za sprečavanje prebrzog skraćivanja 5′ kraja dugog kraka što omogućava duži ćelijski životo Podela hromozoma prema položaju centromere:
metacentrični – centromera jena sredini; p i q kraci su jednaki submetacentrični – p krak je kraći od q kraka akrocentrični – subterminalna pozicija centromere; p kraci su mnogo manji od q kraka; ponekad
imaju satelite – peteljke koje forimiraju nukleoluse i sadrže gene za rRNK
A B C D E F GVELIKI SREDNJI MALI
MC SMC AC MC SMC MC AC
1,3 2 4,5 6-12, X 13-15 16 17,18
19,20 21,22,Y
o Normalno ćelijsko jedro sadrži 22 para autozoma i jedan par polnih hromozoma – XX kod žene i XY kod muškarca, ovi parovi hromozoma su homolozio Polni hromozomi su odlučujući u determinaciji pola; Y je mnogo manji od X hromozoma i nosi samo nekoliko funkcionalnih gena, najznalajnija je seks determinirajuća regija (SRY) koja se nalazi na distalnom delu p kraka i sadrži gen zaslužan za razvoj testisa (35kbp); dokazi da SRY determiniše muški pol su :
1. ova sekvenca je prisutna i kod XX muškaraca, koji su infertilni fenotipski muškarci sa ženskim kariotipom
2. mutacije ili delecije ove sekvence su pronađene kod mnogih XY žena, to su infertilne fenotipske žene sa muškim kariotipom
o Kariotip – analiza hromozomske konstitucije osobe tj uređena mikrofotografija hromozoma te osobeo Za analizu hromozoma se koriste najčešće limfociti periferne krvi, ali i ćelije kože, koštane srži, horionskih resica, plodove vode; ove ćelije se zaustave u metafazi mitoze jer su tada hromozomi najkondenzovaniji a samim tim i vidljivio Analiza hromozoma sa limfocita periferne krvi:
1. krv se stavjla na hranljivu podlogu kojoj je dodat lecitin(fitohemaglutinin) koji podstiče deobe limfocita i tako se ostavlja 3 dana na 37 stepeni
2. dodaje se kolhicin koji uništava mikrotubulin, glavnu komponentu deobnog vretena čime se sprečava samo formiranje deobnog vretena a ćelija ostaje u prometafazi kada su hromozomi najkondenzovaniji a samim tim i vidljivi
3. tretiranje hipotoničnim rastvorom što dovodi do povećanja ćelije a i uništavanje eritrocita4. ovako fiksirani hromozomi se kaplju na pločice i boje tehnikama traka (bending tehnike):

Gimza (G) bending – hromozomi se tretiraju tripsinom koji denaturiše proteine a potom gimza bojom koja vezuje DNK i daje svetle i tamne trake
Quinacrin (Q) bending – bojenje kvinakrin mastardom koji daje fluorescentne trake koje se mogu posmatrati samo pod fluorescentnim mikroskopom
Reverzni (R) bending – daje šaru suprotnu G bendingu a dobija se denaturacijom toplotom ili akridin oranžom pre gimza bojenja; vrsta R-bendinga je T-bending koji pokazuje telomere
C-bending –dobija se kada se pre tretiranja gimzom dodaju kiseline a zatim i baze čime se boje centromere i druge heterohromatinske regije sa visokorepetitivnom DNK
5. fotografišu se, isečene mikrofotografije se poređaju u homologe i daju definitivan kariotipo Ideogram – šematski prikaz bending šare idealnog kariotipa o Fluorescentna in situ hibridizacija (FISH) – metoda podrazumeva spavjanje dela lanca DNK tj probe sa komplementarnom ciljnom sekvencom bilo gde na metafaznom (ili interfaznom) hromozomu; mesto gde se proba vezala posmatra se pod fluorescentnim mikroskopom; postoje razne vrste ove metode a najznačajnije su centromerična proba koja je korisna za brzu prenatalnu dijagnozu trizomija 13, 18 i 21 i telomerična proba koja je korisna za pronalaženja delecija i translokacija koje su uzrok nerazjašnjene mentalne retardacijeo Kvantitativna fluorescentna PCR tehnika (QF-PCR) – metoda koja služi za brzo otkrivanje monogenskih oboljenja, determinaciju pola i hromozomske aneuploidijeo Normalna ženska osoba 46,XXo Normalna muška osoba 46,XYo Žena sa Daunovim sindromom 47,XX+21o Sindrom mačijeg plača 46,XXdel(5p) ili 46,XX5p-o Translokacija 46,XYt(7q;8q)+t(7p;8p)o Mozaicizam 47,XY+21/64,XY
Ćelijski ciklus
o INTERFAZA (16-24h): G1 – PREDSINTETSKI PERIOD – sinteza proteina neophodnih za rast ćelije, izgradnju organela i regulaciju aktivnosti G0 – PERIOD MIROVANJA –neke ćelije kao što su nervne, pop.mišićne napuštaju ćelijski ciklus i ne dele se, limfociti se mogu po potrebi vratiti u ćelijski ciklus S – SINTETSKI PERIOD – replikacija DNK; rezultat su dve hromatide koje imaju dva molekula DNK u centromeri ali su još uvek samo jedan hromozom G2 – POSTSINTETSKI PERIOD – sinteza proteina potrebnih za niti deobnog vretena, enzima za reparaciju DNK, histona, energije
o DEOBA:A. MITOZA (1-2h)– deoba somatskih ćelija:
PROFAZA – kondenzacija hromozoma; resorpcija nukleolusa i jedrove membrane; deoba centriola i njihovo udaljavanje; počinje obrazovanje niti deobnog vretena (kontinuirane, hromozomske i aster)

METAFAZA – centrioli na suprotnim polovima; hromozomi u ekvatorijalnoj ravni; formirano deobno vreteno ANAFAZA – podela centromera; skraćivanje niti deobnog vretena; odvajanje sestrinskih hromatida i njihovo kretanje ka suprotnim polovima; počinje citokineza TELOFAZA – grupisanje hromozoma na polovima i njihova despiralizacija; formiranje jedrove membrane i nukleolusa; završetak citokineze obrazovanjem deobne brazde
B. MEJOZA – deoba gametskih ćelija u ovarijumima i testisima; mejoza 1 (redukciona deoba ) + mejoza 2 (ekvaciona deoba) *TOKOM MEJOZE I SE RAZDVAJAJU HOMOLOZI, A TOKOM MEJOZE II SESTRINSKE HROMATIDE* PROFAZA I – traje i do nekoliko godina; hromozomi se spiralizuju; jedrova membrana i jedarce resorbuju; centriole se dele
LEPTOTEN – hromozomi su dugi, tanki i isprepletani ZIGOTEN – spiralizacija i sparivanje homologa (sinapsis) PAHITEN – homolozi spojeni čitavom dužinom formiraju bivalente; može doći do crossingovera; resporpcija jedrove membrane i jedarceta DIPLOTEN – bivalenti ostaju spojeni samo u mestima koja se nazivaju hijazme; kod žena su oocite zaustavljene u ovoj fazi pri čemu jedna nastavlja deobu svakog meseca od puberteta DIJAKINEZA – bivalenti spojeni samo u terminalnim hijazmama; počinje obrazovanje niti deobnog vretena; centriole na suprotnim polovima
METAFAZA I – bivalenti u ekvatorijalnoj ravni; maksimalna spiralizacija ANAFAZA I – skraćivanje niti deobnog vretena; homolozi kreću ka polovima TELOFAZA I – haploidan broj hromozoma na polovima ćelije; formiranje jedrove membrane i jedarceta; despiralizacija hromozoma; citokineza; rezultat su dve diade sa haploidnim brojem hromozoma koji imaju dve hromatide i 2 molekula DNK G1,G2 PROFAZA II - kondenzacija hromozoma; resorpcija nukleolusa i jedrove membrane; deoba centriola i njihovo udaljavanje; počinje obrazovanje niti deobnog vretena METAFAZA II– centrioli na suprotnim polovima; hromozomi u ekvatorijalnoj ravni; počinje deoba centromera ANAFAZA II – hromatide kreću ka suprotnim polovima skraćivanjem niti deobnog vretena TELOFAZA II – formiranje jedrove membrane i jedarceta; citokineza; rezultat su četiri tetrade sa haploidnim brojem hromozoma sa jednom hromatidom i jednim molekulom DNK
o Gametogeneza predstavlja sazrevanje polnih ćelija – gametogeneza ženskih je oogeneza a muških spermatogenezao Razlike između oogeneze i spermatogeneze:
oogeneza počine u 7. mesecu razvica fetusa, a spermatogeneza u pubertetu oogeneza traje 10-50 godina, a spermatogeneza 60-65 dana broj mitoza u oogenezi je 20-30, a u spermatogenezi 30-500

rezultat oogeneze je jedna jajna ćelija i tri polarna telašca, a spermatogeneze četiri spermatozoida produkcija gameta je jedna jajna ćelija po menstrualnom ciklusu, a 100-200 miliona spermatozoida po ejakulatu
Poremećaj broja hromozoma
ime uzrok incidenca klinička slikaDaunov sindrom (trizomija 21)
trizomija 21 (95%); kariotip 47, XX+21 ili 47, XY+21; poreklo viška hromozoma je u 90% slučajeva iz majčine mejoze I Robertsonova translokacija (4%); kariotip 46,XX-14+t(14q;21q) ili 46,XY-14+t(14q;21q); u preko 30% je jedan od roditelja nosilac balansirane translokacije (rekurentni rizik za muškarce 1-3% a za žene 10-15%) mozaicizam (1%); kariotip 47,XY+21/46,XY; ove osobe imaju blažu mentalnu retardaciju parcijalna trizomija 21; kritična regija za Daunov sindrom je na distalnom kraju dugog kraka (21q22)
1:600-700 živorođene dece
hipotonija (mlitavost), sanjivost, višak kože na vratu novorođenčeta i odojčeta mentalna retardacija kod deteta i starije osobe, IQ 25-75 nizak rast, koso postavljene oči posle treće decenije života pojavljuju se simptomi Alchajmerove bolesti brahicefalija, hipertelorizam, unutrašnji epikantus, Brushfeildove pege(u 80% slučajeva), nizak koren nosa usna duplja mala, jezik prominira, zubi kariozni srčane mane (30-40%) suženje ili neprolaznost anusa i duodenuma (1-2,5%) linija četiri prsta(50%) i kratke šake povećan prostor između palca i drugog prsta – sandalski prostor produžen u sandalsku brazdu imunološki eoplas 10-20x veći rizik od akutne leukemije, sklonost tireodnoj disfunkciji kod muških osoba sterilnost
Patau sindrom (trizomija 13)
čista trizomija 13 (90%) Robertsonova translokacija, mozaicizam (10%)
1:5000 živorođene dece
većina novorođenčadi umire u prvim nedeljama života teška mentalna zaostalost defekt kože glave, rascepi srednje linije lica od rascepa usne, vilice i nepca kiklopija, mikroftalmija, kolobom irisa anomalije mozga – agnezija, fuzija bazalnih ganglija, hidrocefalus, holoprozenkefalija srčane mane (u 90% slučajeva) anomalije bubrega i digestivnog trakta
Edvardsov sindrom (trizomija 18)
čista trizomija 18 (hromozom submetacentričan)
1:5000-6000 živorođene dece
mala težina i dužina na rođenju prominentan potiljačni deo glave mala brada povučena nazad nisko postavljene i malformisane uši fleksione kontrakture prstiju urođene srčane mane, anomalije bubrega i digestivnog trata teška mentalna retardacija i velika smrtnost u prvoj godini života

o Aneuploidija – gubitak ili višak jednog ili više hromozomao Poliploidija – dodatak jednog ili više haploidnih setova hromozoma; vrste poliploidije su triploidija (3n), tetraploidija (4n); triploidija može biti greška u mejozi muškarca ili žene ili rezultat fertilizacije sa dva spermatozoida(dispermija); triploidija je čest nalaz kod spontanih pobačaja, pošto dolazi do intrauterine retardacije rasta sa zaostajanjem rasta trupa u odnosu na glavu i sindaktilijama trećeg i četvrtog i li drugog i trećeg prsta na nogamao Monozomija – gubitak jednog hromozoma; kod autozomnih hromozoma dovodi skoro uvek do pobačaja a kod polnih hromozoma rezultira stanjem koje se naziva Turnerov sindrom; rezultat je nerazdvajanja u mejozi ili gubitkom hromozoma tokom anafaze pri kretanju ka polu ćelije (anafazno zaostajanje)o Trizomija – dodatak jednog homologog hromozoma (najčešće se rode bolesnici sa trizomijama 13,18 i 21 koji su mali hromozomi, ostale se završavaju spontanim pobačajem, a najčešće trizomija 16)o Tetrazomija – dodatak dva homologa hromozomao Višak autozomnog hromozoma dovodi ili do pobačaja ili do retardacije ploda a višak polnog hromozoma ima blage fenotipske efekteo Trizomija 21 je uglavnom posledica nerazdvajanja homologa u anafazi i kod majke ili ređe u mejozi II kod nerazdvajanja hromatidao Mozaicizam – pojava uslovljena nerazdvajanjem u ranim mitotskim deobama ploda; nastajanje dve ili više ćelijskih linijao Himerizam – pojava da kod jedne osobe postoje dve ili više različitih ćelijskih linija različitog genetskog porekla; dispermične himere su rezultat dvostruke fertilizacije(mogu dovesti do pojave pravog hermafrodita sa kariotipom XX/XY), a krvne himere su rezultat izmene ćelija između neidentičnih blizanaca putem placenteo Povećana učestalost trizomija 13, 18 i 21 pojavljuje se sa starošću majke, a ostali faktori uticaja na nerazdvajanje hromozoma su odsustvo rekombinacija u profazi I fetalnih ovarijuma, abnormalnost u deobnom vretenu, radijacija i odložena fertilizacijao Postoji prirodna poliplodija, npr kod ćelija jetre (4n), mekariocita koštane srži(i do 16n), mišićne ćelije, itd.
Poremećaj strukture hromozoma
o Rezultat lomova hromozoma sa ponovnim spajanjem i mogu biti balansirane (bez gubitka i dodatka genetskog materijala, fenotipski normalne osobe, sa rizikom od dobijanja bolesne dece) i nebalansirane(štetan klinički efekat)o Translokacija – prenos genetskog materijala sa jednog hromozoma na drugi
recipročna (1/500) – lom na oba hromozoma; segmenti se razmene i fomiraju se dva nova derivata hromozoma; nosioci ovih translokacija nose 1-10% rizik za bolesnu bebu; rezultat deobe može biti normalan hromozom, balansirana translokacija i nebalansirana translokacija Robertsonova (1/1000) – dešava se kada je tačka prekida u ili blizu centromera akrocentričnih hromozoma uz fuziju njihovih q krakova; naziva se i centrična fuzija; dolazi do gubitka p krakova što nema klinički značaj jer se na njima nalaze geni samo rRNK; ukupan broj hromozoma je

redukovan do 45 ali ne dolazi do gubitka ni viška genetskog materijala; najčešće kod hromozoma 13 i 14
o Delecija – gubitak dela hromozoma što uzrokuje monozomiju za taj deo hromozoma; velike delecije (više od 2% ukupnog haploidnnog genoma) dovode do pobačaja
mikroskopska – velike delecije vidljive pod svetlosnim mikroskopom; izazivaju Wolf-Hirschhorn sindrom (delecija dela kratkog kraka hromozoma 4) i Cri du chat tj sindrom mačjeg plača (delecija dela kratkog kraka hromozoma 5) submikroskopska – vidljive uz pomoć FISH metoda; izazivaju Prader-Willi i Angelman sindrom; može obuhvatati samo nekoliko susednih gena i u tom slučaju se naziva sindrom susednih gena
o Insercija – segment jednog hromozoma se umetne u drugi hromozomo Inverzija – dva loma na jednom hromozomu; ukoliko uključuje centromeru onda je pericentrična, a ako je na kraku ona je paracentrična; retko uzrokuju problema kod nosioca, osim ako je tačka prekida oštetila bitan gen; mogu biti štetne za potomstvoo Ring hromozom – kada se lomovi dese na oba kraka ostavljajući dva lepljiva kraja koja se spoje u prsten; kod autozomnih hromozoma su letalneo Izohromozom – gubitak jednog kraka i duplikacija drugog; 15% uzroka Tarnerovog sindroma je izohromozom sa dva duga kraka X hromozomao Genetički imprinting je pojava da ne dolazi do ispoljavanja oba roditeljska alela kod deteta; ukoliko su oba ispoljena može doći do poremećaja kao što su Angelman, Prader-Willi, Wilms tumor, Bekwith Wiedmann i Silver Russel sindrom
ime uzrok incidenca klinička slikaWolf-Hirschhorn sindrom
delecija kratkog kraka 4. hromozoma; u toku gametogeneze dolazi do recipročne translokacije otcepljenog dela što daljom segregacijom dovodi do nastajanja gameta sa deletiranim p krakom 4. hromozoma; kariotip 46,XX,4p- ili 46,XY,4p-
1:50000 živorođene dece
teška mentalna retardacija mala težina i zaostajanje u rastu glava mikrocefalična, linija nosa spojena sa linijom obrva, širok i visok koren nosa, hipertelorizam i epikantusi “kaciga grčkog ratnika” umiru u toku prve decenije života
Cri du chat sindrom (sindrom mačijeg plača)
delecija kratkog kraka 5. hromozoma; kariotip 46,XX,5p- ili 46,XY,5p-
1:50000 živorođene dece
plač nalik mjauku usled nerazvijenosti larinksa mala težina mentalna retardacija mikrocefalična glava, okruglo lice, kasnije trouglasto hipertelorizam, epikantusi, antimongoloidno postavljene oči
Retinoblastom (maligni tumor retine)
mikrodelecija hromozoma 13q14; autozomno dominantno se nasleđuje
retko može biti na obe strane, pa i na više mesta u oku (multifokalno) što ne važi za nenasledni oblik može ići uz mentalnu zaostalost i druge anomalije
WAGR sindrom
mikrodelecija hromozoma 11p13; u okviru ove delecije nalazi se gen PAX 6 koji je
retko retka embrionalna bubrežna neoplazma Wilms tumor, aniridia, genitourinarne anomalije, retardacija rasta i razvoja
odgovoran za aniridiju, WAT1 uzrokuje Wilms tumor
Angelmanov i Prader-Willi sindromi
mikrodelecija proksimalnog dela dugog kraka 15. hromozoma; 15q(15q11-12); ukoliko anomalija potiče od oca dete će imati Prader-Willi sindrom, a ukoliko potiče od majke Angelman sindrom uniparentalna dizomija; slučaj kada je par hromozoma nasleđen samo od jednog roditelja; kada su oba od oca dete će imati Angelman sindrom, a kada su oba od majke Prater-Willi sindrom
retko deca sa Angelmanovim sindromom se bezrazložno smeju, imaju konvulzije, mentalnu retardaciju deca sa Prater- Willi sindromom su ekstremno mlitava u ranom uzrastu, kasnije postaju pojasno debela sa blagom do srednjom mentalnom retardacijom
DiGeorge sindrom
mikrodelecija proksimalnog dugog kraka hromozoma 22
retko srčane mane, hipoplazija timusa i paratireoidnih žlezda
Poremećaji polnih hromozoma
ime uzrok incidenca klinička slikaTarnerov sindrom
osobe sa ovim sindromom nemaju Barovo telašce; inaktivni X hromozom koji se u ranom embrionalnom životu inaktivira kariotip je 45,X u 55% slučajeva uzrok je gubitak X hromozoma u mejozi majke ili X ili Z hromozoma u mejozi oca 45,X/46,XX 46,X,I(Xq) 46,X,r(X) 46,Xdel(Xp)
1:2000 živorođene dece (devojčica) tj 1-2%od ukupno začetih Tarnerovih sindroma
zadebljan vrat otoci šaka i stopala mali rast,gojaznost i nedostatak sekundarne seksualne karakteristike loše formirane, odstojeće uši, uska gornja vilica i nepce, mala donja vilica, kratak i zadebljan vrat, niska linija kose udubljen grudni koš, piroko postavljene bradavice srčane mane, potkovičast bubreg ovarijalna dizgeneza sa hipoplazijom sterilitet višak mladeža na koži normalna inteligencija, perceptivne slušne tegobe u adolescenciji terapija estrogenom radi razvijanja sekundarnih polnih karakteristika i sprečavanja osteoporoze; poznati su slučajevi rađanja veštačkom oplodnjom
XXX žene višak X hromozoma je u 95% slučajeva majčinog porekla i vezan je za
1:1000 živorođenih devojčica
normalna inteligencija fenotipski potpuno normalne manja porođajna težina i dužina

majline godine kasne u progovaranju i motorici kao odrasle natprosečno visoke i sa govornim poteškoćama i neregularnim menstruacijama nezgrapne, trapave, loša koordinacija, spore i nezrelog ponašanja blaga redukcija inteligencije blaga depresija, problem socijalizacije, stidljivost, povučenost normalno fertilne, deca im imaju normalne kariotipove
Klinefelterov sindrom
ovo su muškarci koji imaju Barovo telo za razliku od normalnih muškaraca; kariotip 47,XXY; višak X hromozoma može podjednako poticati i od majke i od oca; u majčinom slučaju velik uticaj imaju godine postoje i slučajevi mozaicizma 47,XXY /46,XY
1:1000 živorođenih dečaka
nespretnost i blagi problema sa učenjem, posebno verbalne prirode inteligencija blago niža natprosečno visoki sa dugim nogama ginekomastija, mali testisi u kojima nema spermatogeneze retka pubična maljavost i brada 4-6% muškog steriliteta u slučajevima mozaicizma posebnim tehnikama mogu dobiti potomstvo od puberteta neophodan tretman testosteronom radi razvijanja sekundarnih polnih karakteristika i sprečavanja osteoporoze kao i sprečavanje daljeg porasta ekstremiteta
Jacobs sindrom
rezultat greške u mejozi oca ili postzigotični događaj, višak Y hromozoma; kariotip 47,XYY
1:1000 živorođenih dečaka
2-3% čjudi u institucijama za mentalno zaostale i zatvorima uglavnom normalnog ponašanja, ali mogu biti emocionalno nezreli, impulsivni malo veća visina od prosečne, veliki zubi uglavnom fertilni
Fragilno X sindrom (Martin – Bellov sindrom)
X hromozom pokazuje fragilno mesto blizu telomere dugog kraka; to je prostor u tački u kojoj se hromozom lomi Fragilno X mesto A (FRAXA) sadrži dugu CGG ponovljenu sekvencu čije ponavljanje na 50-200 sekvenci ga čini nestabilnim a naziva se premutacija, a na 200-2000 mutacija postaje potpuna Fragilno X mesto E (FRAXE) ima lokaciju na Xq28 i sadrži ponovljenu sekvencu CCG, a nestabilno ponavljanje je iznad 200
1:2000 živorođenih dečaka (žene su nosioci)
4-8% muških mentalno zaostalih osoba imaju ovu bolest visoko čelo, velike uši, dugo lice, prominentna vilica posle puberteta veliki testisi hiperekstenzibilni zglobovi, strije na koži, prolaps mitralne valvule srednje do teška mentalna retardacija, autistično i/ili hiperaktivno ponašanje govor sa odmorima i ponavljanjem ženski prenosioci mogu pokazati karakteristične crte lice i blago su ili srednje mentalno zaostale

Monogensko (Mendelijansko nasleđivanje)
o Alel – gen na jednom lokusu svakog hromozoma jednog homologog parao Genotip lokusa – dva alela datog lokusa; ako su aleli isti radi se o homozigotu za dati lokus, a ako su različiti u pitanju je heterozigoto Za vreme mejoze aleli se dele i svaki ide u posebnu gametuo Fenotip – spoljašni izgled uslovljen genotipom i uticajem sredine; različiti genotipovi mogu imati isti fenotipo Rodoslovno stablo (pedigre) slži za praćenje monogenskih karakteristika; autozomno dominatno nasleđivanje se prepoznaje po vertikalnoj pojavi bolesti i jednakoj zastupljenosti kod oba pola; kod recesivnog nasleđivanja je pojava bolesti horizontalna – zahvaćena je četvrtina potomaka, polovina su nosioci a četvrtina ne nosi patološki gen; X vezano recesivno nasleđivanje se prepoznaje po kosoj pojavi, pri čemu su zahvaćeni muški članovi porodice; X vezano dominanto nasleđivanj se razlikuje od autozomno dominantog jer se ne prenosi sa oca na sina pošto otac sinu daje samo Y hromozom o Pravila rodoslovnog stabla:
zdrava žena obeležava se simbolom praznog kruga, a obolela punim krugom žena koja je heterozigotni prenosilac krugom sa tačkom u sredini ili krugom u kome je polovina
zatamnjena zdrav muškarac obeležava se praznim kvadratićem, a ostalo je isto kao za ženu samo što je
simbol kvadrat, a ne krug; roditeljski par povezan je jednom horizontalnom (bračnom) linijom; brak u srodstvu označen je
sa dve linije Ispod roditeljske generacije unose se potomci F1 generacije i obeležavaju se rednim brojevima
prema redosledu rađanja i povezani su međusobno horizontalnom linijom; jedan horizontalan niz osoba čini jednu generaciju i obeležen je rimskim brojem, pri čemu najstarija generacija (roditeljska) nosi broj I
Pri konstrukciji rodoslovnog stabla uvek se polazi od osobe koja se javila na pregled (proband ili propozitus)
monozigotni i dizigotni blizanci se obeležavaju povezanim simbolima za pol umrle osobe se obeležavaju precrtanim simbolom za pol, a mrtvorođeno dete simbolom za pol
u kome ja upisan krst
Autozomno dominantne nasledne bolesti
o Aficirane su osobe heterozigotni nosioci patološkog gena; homozigostnost uslovljava težu kliničku sliku ili češće letalnost pre rođenja; za njih se vezuje efekat očevih godinao Ove bolesti karakteriše ekspresivnost tj. izražajnost (primer – višak prstiju može biti izražen i kao kvržica a i kao ceo prst) i penetrantnost tj. probojnost (osoba izgleda kao da nije aficirana a ima patološki gen što znači da on nije penetrantan)
o Ekspresivnost se može razlikovati kod muških i ženskih osoba (npr. ćelavost koja je kod žena ispoljena samo u homozigotnom stanju tog gena jer ekspresiju gena zahtevaju androgeni kojih ima više kod muškaraca)o Fenomen anticipacije podrazumeva da se u svakoj novoj generaciji boelst javlja ranije i u težem obliku (Hantingtonova horea)o Kodominantnost je pojava kada se oba alela kod heterozigota ispoljavaju (krvne grupe)o Plejotropija je pojava da se jedan mutirani gen ispoljava na više načina u različitim organima i tkivimao U ove bolesti spadaju: Opitzov sindrom, Peutz-Jeghersov sindrom, Marfanov sindrom, ahondroplazija, familijarna hiperholesterolemija, miotonična distrofija, Hantingtonova horea, hepatična porfirija, neurofibromatoza, tuberozna skleroza, osteogenesis imperfekta, policistična bolest bubrega, porodični retinoblastom, Von Willebrandova bolest, hereditarna sferocitoza
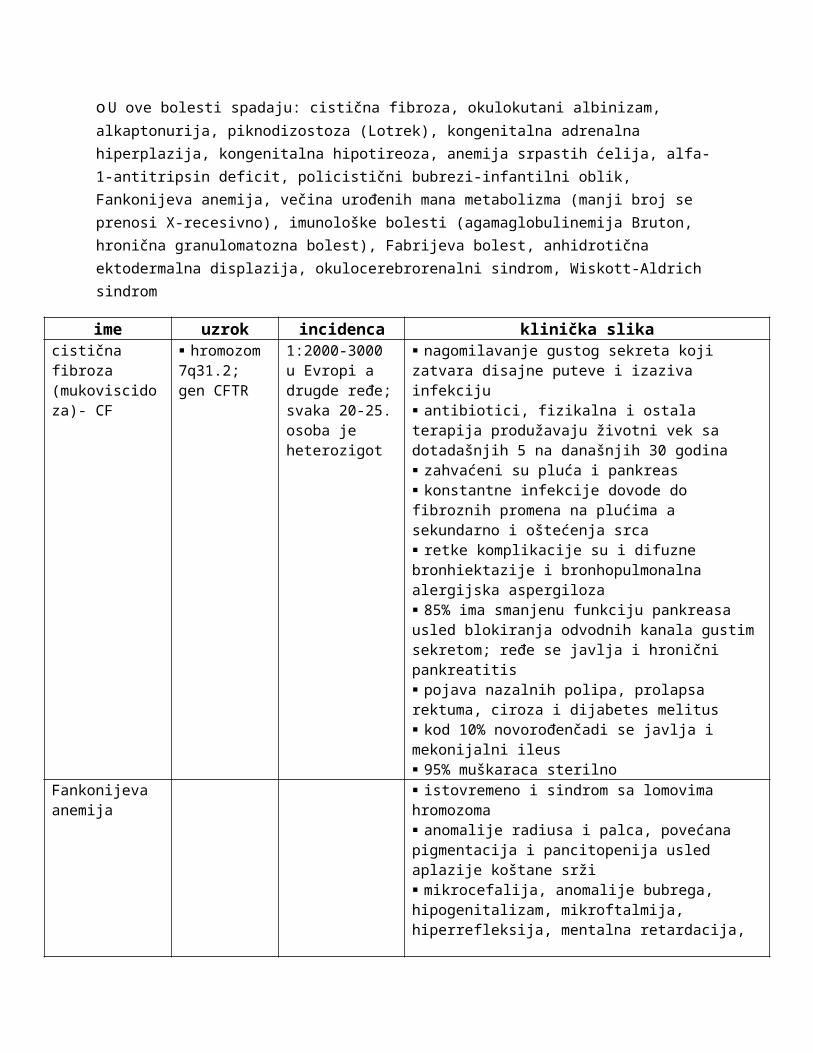
Autozomno recesivne nasledne bolesti
oAficirana su samo homozihotna deca heterozigotnih roditelja (zdravih), u rodoslovnom stablu se ispoljava horizontalno, a bolest se pojavlajuje podjednako kod oba pola; kod potomaka se očekuje odnos 1:2:1 oboleli:heterozigoti:zdavi homozigotio Za recesivne gene veća je mogućnost sretanja kod konsangvinitetao Kod ovih bolesti dolazi do mutacije gena koji vodi do promene ili nedostatka nekog proteinao Pseudodominantno nasleđivanje je pojava kada su roditelji homozigotna osoba za autozomno- recesivnu bolest i heterozigot za istu bolest; deca u tom slučaju imaju 50% šanse da oboleoU ove bolesti spadaju: cistična fibroza, okulokutani albinizam, alkaptonurija, piknodizostoza (Lotrek), kongenitalna adrenalna hiperplazija, kongenitalna hipotireoza, anemija srpastih ćelija, alfa-1-antitripsin deficit, policistični bubrezi-infantilni oblik, Fankonijeva anemija, večina urođenih mana metabolizma (manji broj se prenosi X-recesivno), imunološke bolesti (agamaglobulinemija Bruton, hronična granulomatozna bolest), Fabrijeva bolest, anhidrotična ektodermalna displazija, okulocerebrorenalni sindrom, Wiskott-Aldrich sindrom
ime uzrok incidenca klinička slikacistična fibroza (mukoviscidoza)- CF
hromozom 7q31.2; gen CFTR
1:2000-3000 u Evropi a drugde ređe; svaka 20-25. osoba je heterozigot
nagomilavanje gustog sekreta koji zatvara disajne puteve i izaziva infekciju antibiotici, fizikalna i ostala terapija produžavaju životni vek sa dotadašnjih 5 na današnjih 30 godina zahvaćeni su pluća i pankreas konstantne infekcije dovode do fibroznih promena na plućima a sekundarno i oštećenja srca retke komplikacije su i difuzne bronhiektazije i bronhopulmonalna alergijska aspergiloza 85% ima smanjenu funkciju pankreasa usled blokiranja odvodnih kanala gustim sekretom; ređe se javlja i hronični pankreatitis pojava nazalnih polipa, prolapsa rektuma, ciroza i dijabetes melitus
kod 10% novorođenčadi se javlja i mekonijalni ileus 95% muškaraca sterilno
Fankonijeva anemija
istovremeno i sindrom sa lomovima hromozoma anomalije radiusa i palca, povećana pigmentacija i pancitopenija usled aplazije koštane srži mikrocefalija, anomalije bubrega, hipogenitalizam, mikroftalmija, hiperrefleksija, mentalna retardacija, gluvoća dve trećine obolelih ima anomalije skeleta – odsutan, hipoplastičan ili palac pripojen preko mekog tkiva, aplazija i hipoplazija metakarpalne kosti, hipoplazija tenara, sindaktilija palca na stopalu, udubljen grudni koš, urođeno iščašenje kuka, abnormalnosti donje vilice, mikrodoncija, skolioza, kratak vrat, sprengelov deformitet, Klippel-Feil sindrom, spina bífida, osteoporoza, pojačane prstaste impresije na lobanji u kasnijem razvoju javlja se mrka pigmentacija kože a posebno vrata, tela i intertriginoznih mesta mrlje boje bele kafe i bele mrlje opšta hemosideroza
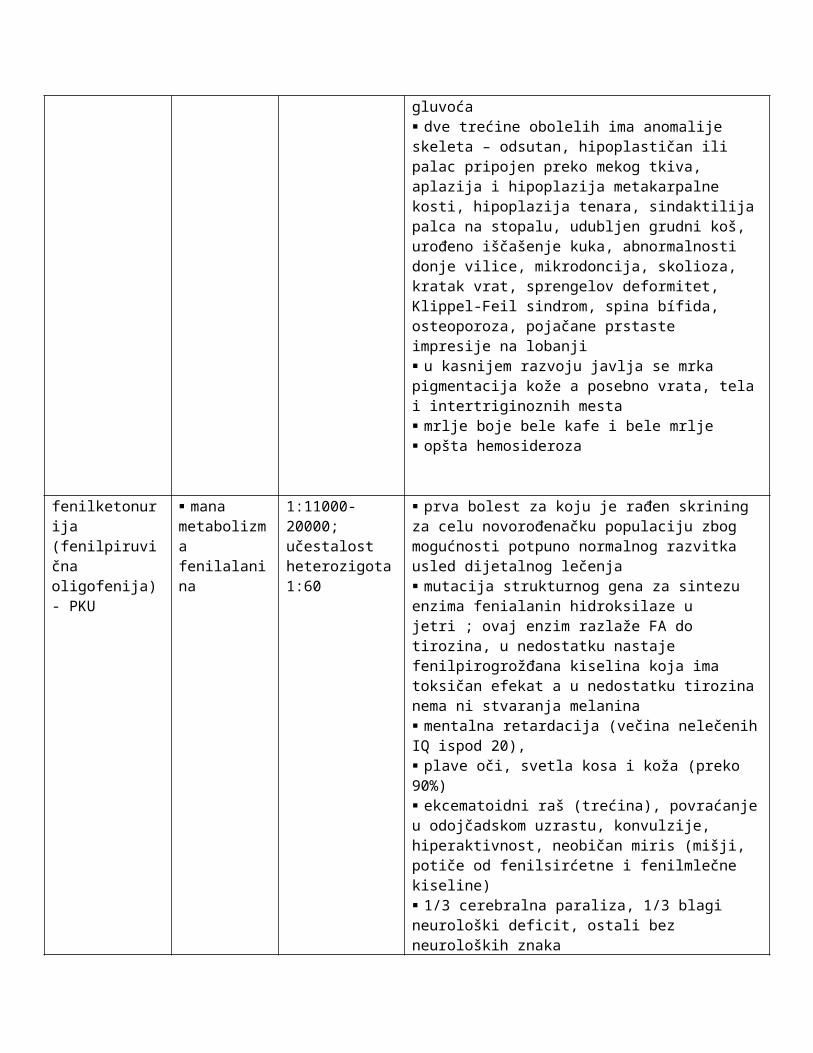
fenilketonurija (fenilpiruvična oligofenija) - PKU
mana metabolizma fenilalanina
1:11000-20000; učestalost heterozigota 1:60
prva bolest za koju je rađen skrining za celu novorođenačku populaciju zbog mogućnosti potpuno normalnog razvitka usled dijetalnog lečenja mutacija strukturnog gena za sintezu enzima fenialanin hidroksilaze u jetri ; ovaj enzim razlaže FA do tirozina, u nedostatku nastaje fenilpirogrožđana kiselina koja ima toksičan efekat a u nedostatku tirozina nema ni stvaranja melanina mentalna retardacija (večina nelečenih IQ ispod 20), plave oči, svetla kosa i koža (preko 90%) ekcematoidni raš (trećina), povraćanje u odojčadskom uzrastu, konvulzije, hiperaktivnost, neobičan miris (mišji, potiče od fenilsirćetne i fenilmlečne kiseline) 1/3 cerebralna paraliza, 1/3 blagi neurološki deficit, ostali bez neuroloških znaka psihomotorni nemir i promene ponašanja epileptični napadi (25%), EEG abnormalnosti (80%) mikrocefalija, zaostajanje u telesnom razvoju
X-vezano recesivno nasledne bolesti
o Određene su genom na X hromozomu i uglavnom se manifestuju samo kod muškog potomstva koje se naziva hemizigotnim za taj mutantni alelo Prenose ih žene nosioci na bolesne muškarce čije su sve ćerke nosioci a nijedan sin oboleo

o Rodoslovno stablo je koso, dijagonalno ili kao pokret konja u šahu; bolesni su muški hemizigoti i ženski homozigoti za taj alel; žene nosioci sa zdravim partnerom imaju 50% šanse da imaju sina koji je oboleo i ćeku nosiocao Bolesne mogu biti i žene sa samo jednim X hromozomom
ime uzrok incidenca klinička slikaHemofilija A deficit faktora
VIII, plazma proteina koji je odgovoran za stvaranje krvnog ugruška na mestu oštećenja delecije (5%), frameshift, nonsence mutacije, insercije i flip inverzije (5%) težih slučajeva, trećini slučajeva spontana mutacija
1:5000-10000 muškaraca
epizode krvarenja u velike zglobove posebno kolena, gležnja, lakta koje su u početku bolne a kasnije dovode do nepokretnosti i deformiteta krvarenja mogu biti spontana nakon povreda u igri, nakon vađenja ili ispadanja zuba, gastrointestinalna, hematurija, u centralni nervni sistem simptomi na rođenju mogu biti veliki kefalhematom ili intrakranijalno krvarenje, ali mogu se primetiti i tek pri razvoju motornih funkcija pri pojavi hematoma posle minimalne traume najlakši slučajevi imaju 6-30% normalne aktivnosti faktora VIII, srednji 2-5%, a najteži 0-2% aktivnosti
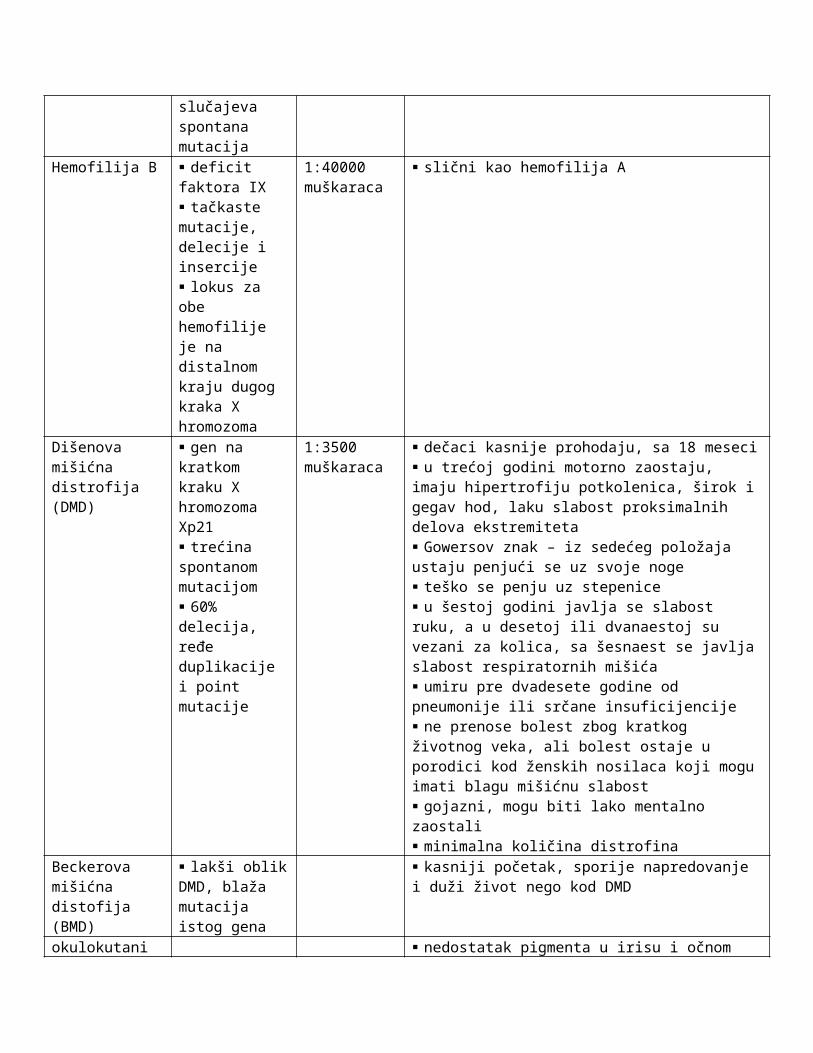
Hemofilija B deficit faktora IX tačkaste mutacije, delecije i insercije lokus za obe hemofilije je na distalnom kraju dugog kraka X hromozoma
1:40000 muškaraca
slični kao hemofilija A
Dišenova mišićna distrofija (DMD)
gen na kratkom kraku X hromozoma Xp21 trećina spontanom mutacijom 60% delecija, ređe duplikacije i point mutacije
1:3500 muškaraca
dečaci kasnije prohodaju, sa 18 meseci u trećoj godini motorno zaostaju, imaju hipertrofiju potkolenica, širok i gegav hod, laku slabost proksimalnih delova ekstremiteta Gowersov znak – iz sedećeg položaja ustaju penjući se uz svoje noge teško se penju uz stepenice u šestoj godini javlja se slabost ruku, a u desetoj ili dvanaestoj su vezani za kolica, sa šesnaest se javlja slabost respiratornih mišića umiru pre dvadesete godine od pneumonije ili srčane insuficijencije ne prenose bolest zbog kratkog životnog veka, ali bolest ostaje u porodici kod ženskih nosilaca koji mogu imati blagu mišićnu slabost gojazni, mogu biti lako mentalno zaostali minimalna količina distrofina
Beckerova mišićna distofija (BMD)
lakši oblik DMD, blaža mutacija istog gena
kasniji početak, sporije napredovanje i duži život nego kod DMD
okulokutani albinizam
nedostatak pigmenta u irisu i očnom dnu kod muškarca mozaična šara pigmentacije kod žene nosioca
Daltonizam 8% muškaraca 1:150 žena
nesposobnost razlikovanja crvene i zelene boje može da se ispolji i kod žene nosioca u osnovi čega je nebalansirana lajonizacija
testikularna feminizacija
odsustvo androgenih receptora u ćelijskim membranama koji vezuju testosteron
osoba sa normalnim muškim kariotipom ima spoljne ženske genitalije; vagina se završava slepo a uterus i Falopijeve tube ne postoje, testisi su u abdomenu ili ingvinalnom kanalu
X-vezano dominantno nasledne bolesti
o Vertikalna pojava u rodoslovu i zahvaćenost oba pola; oboleli su hemizigotni muškarci i heterozigotne ženeo Obolela heterozigotna žena prenosi bolest polovini svojih potomaka, homozigotna svom potomstvu, a oboleli muškarac svim ćerkama ali ne i sinovima pa je zahvaćenost žena duplo veća nego muškaracao Ovim putem se nasleđuju: porodični hipofosfatemilni rahitis (vitamin D rezistentni rahitis), Alportov sindrom (nasledni hefritis i gluvoća), inkontinencija pigmenti, fokalna dermalna hipoplazija
ime uzrok incidenca klinička slikaVitamin D rezistentni rahitis (fosfatni dijabetes)
urođena mana transporta fosfata u bubrezima
hiperfosfaturija i hipofosfatemija manifestuje se u drugoj godini života deformacijom u obliku O nogu i drugim kostnim znacima rahitisa usporen rast u visinu, otežan hod teža je kod muškaraca nego kod žena heterozigota terapija su fosfati i D vitamin
Y-vezano nasleđivanje (holandrično)
o Prenošenje sa oca na sina; do sada je jedino pokazano da se snop dlaka u ušima prenosi na ovaj načino Na ovom hromozomu se nalazi H-Y histokompatibilni antigen i gen uključen u spermatogenezu; usled njegovog nedostatka nema spermatozoida u spermio Ako postoji normalan Y hromozom oko 10. nedelje trudnoće formira se rani embrionalni testis usled TDF; ako ga nema razvijaju se jajnicio Volfovi kanali, preteča vas deferens, seminalniih vezikula i prostate razvijaju se pod uticajem testosterona koji se produkuje u fetalnim testisima

Poligenski (multifaktorski) uslovljene bolesti
o Na njih se ne mogu primeniti Mendelovi zakoni; incidencija kod bliskih rođaka je 2-4% što je znatno manje neko kod monogenskih bolestio Nastaju interakcijom genetskih i spoljnosredinskih faktora; ekspresija fenotipa determinisana je sa mnogo gena na različitim lokusima sa mali aditivnim efektom svakog gena pri tom nijedan od gena nije dominantan ni recesivano Ovim putem se nasleđuju: kongenitalne malformacije (rascerp usne/nepsca, kongenitalna dislokacija kukam urođene srčane mane, defekti neuralne tube, pilorična stenoza, krivo stopalo) i stečene bolesti u dečijem i odraslom doobu (astma, autizam, dijabetes melitus, epilepsija, glaukom, hipertenzija, ishemična bolest srcam peptični ulkus, manična depresija, šizofrenija, ankilozirajući spondilitis)o Procena rekurentnog rizika:
1. incidencija stanja je najveća među rođacima najteže aficiranih pacijenata2. rizik je najveći među bliskim rođacima; incidencija opada sa udaljenošću srodstva3. ako je veći broj aficirane braće i sestara veći je rizik za ponovnu pojavu4. ako je stanje mnogo češće kod jednog pola, veća je verovatnoća da obolela osoba bude
suprotnog pola5. rekurentni rizik za rođake prvog stepena (braću, sestre, potomka) približno je jednak
kvadratnom korenu incidencije u opštoj populacijio Heritabilnost je stepen prosečne naslednosti neke osobine jedinki određene populacije pod specifičnim uslovima sredine tj stepen do kojeg se jedno svojstvo prenosi sa roditelja na potomstvo; izražava se kao odnos genetičke varijanse i ukupne fenotipske varijanse tj koeficijent heritabilnostio Identifikacija gena uzročnika:
1. analizom asocijacije dve osobine ili asocijacije markera i osobine2. analizom vezanosti gena (linkage)
ime uzrok incidenca klinička slikaAutizam nije jasan, ali
genetski faktori dosta utiču; udružen je sa 15 ili više lokusa od kojih svaki doprinosi na opšti fenotip bolesti
1:2500 dece teška neurorazvojna bolest, javlja se tokom prve tri godine života osustvo verbalne komunikacije, nedostatak socijalnog odgovora i stereotipno ponašanje autistično ponašanje (javlja se i kod dece sa tuberoznom sklerozom, Rettovim sindromom i fragilnim X sindromom)
Insulin zavisni dijabetes melitus
TIP 1: udružen sa HLA lokusima DR3i DR4 TIP2: nije udružen sa HLA lokusima TIP 3: udružen sa HLA lokusima; razmatra se
TIP 1: 0,4% populacije, TIP 2: 5-6% populacije, TIP 3:
bolest metabolizma delimični ili potupni nedostatak insulina, hormona koji se stvara u pankreasu a služi za metabolizam ugljenih hidrata polidipsija (pojačana žeđ), polofagija (prekomerna glad), poliurija (pojačano mokrenje), gubitak na težini TIP 1: redak juvenilni , insulin zavisan; renalni, retinalni, vaskularni problemi; nije udružen sa gojaznošću TIP 2: početak u zrelom dobu, nije insulin zavisan;

mogućnost autozomno dominantnog nasleđivanja
benigan; gojaznost TIP 3(Mody): juvenilni početak; redak je;ne zahteva insulin, nema gojaznosti;
Spoljašnji činioci koji mogu da oštete plod
o Teratogeni – činioci iz spoljašnje sredine koje mogu da oštete embron ili fetus koji se razvija normalno; u njih spadaju hemikalije, fizički i biološki agensio Infekcije(biološki agensi) koje utiču su: rubella, citomegalovirus (CMV), varicela zoster virus (VZV), a retko herpes simpleks virus (HSV), humaniparvovirus (HPV), virus humane imunodeficijencije (HIV), toksoplazmoza i sifiliso Virus rubellae (rubeole, crvenke) – RNK virus iz toga viridae porodice virusa; opasan je po plod u prva tri meseca trudnoće (posebno u prvom), ako virus prodre kroz placentu dovodi do smrti, spontanog pobačaja embriona ili teškog oštećenja srca, oka, uha, mentalne retardacije ploda; uzrok teratogenog uticaja je u tome što virus dopušta deobu ćelije a menja je u osnovi, što u periodu od prva 3 meseca trudnoće kada još nije završena organogeneza, a od jedne ćelije nastaje ceo organ dovodi do fatalnih posledica; takođe, ćelije embriona ne mogu da produkucju interferon iako su inficirane virusom; posle infekcije imunitet je solidan, dugotrajan i verovatno doživotan; majka koja poseduje imunitet transplacentarno prenosi antitela pa je dete nekoliko meseci nakon porođaja zaštićenoo Kod nas se tokom poslednjih decenija vrši imunizacija Mo-Ru-Par vakcinom (morbilli, rubella, parotitis)o Virus morbilla – iz paramyxoviridae porodice virusa; može biti uzročnik spontanih pobačaja, anomalija embriona i prevremenih porođajao Virus mumpsa - iz paramyxoviridae porodice virusa;može uzrokovati endokardijalnu fibroelastozu plodao Citomegalovirus (CMV) – DNK virus iz porodice herpes viridae;retko se ispoljava kod odraslih ljudi, ali može da dugo postoji latentno, najčešće u pljuvačnim žlezdama, bubrezima, pankreasu, jetri, endokrinim žlezdama, mozgu; nalazi se unutar karakterističnih džinovkih ćelija po kojima je i dobio ime; kod novorođenčeta se javlja transplacentarnom, kongeitalnom infekcijom u toku viremije majke nastale tokom primarne infekcije u trudnoći; dospeva u sve organe ploda a najviše su zahvaćene epitelne ćelije bubrega, jetre, pankreasa, pljuvačne žlezde; rizik se povećava ukoliko je do infekcije došlo u prvom trimestre; oštećuje 5% inficiranih trudnoća; citomegalna inkluzivna bolest se javlja kod ploda a njeni siptomi su generalizovane infekcije sa pojavom hepatosplenomegalije, trombocitopenične purpure, hemolitičke anemije, žutice, horioretinitisa uz oštećenje vida, oštećenje sluha, infekcije CNSa, motorna i fizička retardacija, periventrikularne kalcifikacije; u ovom obliku je smrtnost visoka, ugavnom u prve dve godine života; antitela protiv CMV ima 50-80% osobao Herpes simpleks virus (HSV) - DNK virus iz porodice herpes viridae; retko daje mikrocefaliju, aplaziju pojedinih delova mozga, mikroftalmiju, horioretinitis, mentalnu retardaciju, gluvoću i srčane maneo Varicella zoster virus (VZV) - DNK virus iz porodice herpes viridae; pošto je ovo česta dečija bolest, 95% žena ima prethodnu infekciju; rizik od intrauterine infekcije zbog varičele majke je 24%, a kongenitalne malformacije se pojavljuju kod 5% i karakterišu se kožnim lezijama, muskuloskeletnim
deficitom, mikrocefalijom, kortikalnom i cerebralnom atrofijom, konvulzijama, kalcifikacijama mozga, mentalnom retardacijom, horioretinitisom, mikroftalmijom, kataraktom, atrofijom mišićao Variolla virus – DNK virus iz porodice poxviridae; izaiva smrt fetusa ili multiorganska oštećenjao Humani parvovirus (HPV) – RNK virus iz porodice parvoviridae; infekcija se najčešće javlja između 5. i 15. godine tako da više od polovine odraslih ima antitela; žene koje su izbegle tu infekciju osetljivije su na nju u trudnoći, a ukoliko dođe do infekcije u trudnoći imaju blage simptome dok se u 1-10% slučajeva događa spontani pobačaj, u 35% može da nastane oštećenje ploda – hidrops kao rezultat anemije i miokardita, srčane mane i teška oštećenja raznih organao Virus humane imunodeficijencije (HIV) – RNK virus iz porodice retrovirae; intrauterina infekcija je ređa u odnosu na intrapartalnu ili postpartalnu; intrauterino se najčešće infekcija dešava manje od 2 meseca pre porođaja putem plodove vode ili transplacentarno iz majčine krvi; kod HIVa je broj spontanih pobačaja 67%, telesna masa je smanjena, povećan prematuritet, pojavljuje se mentalna zaostalost, mikrocefalija, kranijalne malformacije i hronično nosilaštvo HIV virusao Echo virusi – pripadaju porodici picornaviridae, izazivaju simptome slične rubelli, aseptični meningitis ili fatalnu diseminovanu virusni infekciju sa nekritičnim hepatitisomo Respiratnorni virusi influence, parainfluence, rhinovirue RSV, virusi hepatitisa HVC i HVB nemaju dokazan teratogeni efekato Toksoplazmoza – antropozoonoza široko rasprostranjena a izaziva je parazit tokspolazma gondii; fetalna infekcija se događa samo kad je majka incijalno inficirana tokom trudnoće jer majčina ranije stečena antitela potpuno štite plod; majčina infekcija nosi rizik od 20% da embrion bude zaražen a oštećenja ploda podrazumevaju mikrocefaliju, hidrocefalus, mikroftalmiju, kataraktu, horioretinitis, gluvoću; kombinacije ovih oštećenja mogu da izazovu smrt plodao SIfilis – izazivač ove bolesti je bakterija Treponema pallidum; posledice su hidrocefalus, mikrocefalus, osteitis i rinitis; rizik od infekcije ploda zavisi od stadijuma majčine bolesti – najveća verovatnoća je da se fetus zarazi od strane majke koja je u prvom ili drugom stadijumu a nije lečena; takođe verovatnoća zavisi i od starosti cfetusa, najveća je u kasnom stadijumu trudnoće; intrauterino inficirani plodovi se rađaju na vreme u 15% slučajeva, u 60% je letalan ishod; plod ima povećanu slezinu i jetru, osteohondritis sifilitka uglavnom u distalnim delovima femura; može se roditi zdravo na oko ali da kasnije obolio Lekovi i hemikalije (hemijski agensi) – izazivaju oko 2% kongenitalnih anomalija; uticaj lekova je shvaćen posle tragedije sa Talidomidom, lekom protiv muke u trudnoći; majke koje su uimale ovaj lek u prvom trimestre trudnoće rodile decu sa teškim anomalijama, nedostatkom jedne ili svih dugih kostiju, sa prstima, što detetu daje izgled foke, a prisutne su bile i anomalije uha, oka, rasepi usana, vilice i nepca, 40% beba umrlo u odojčadskom uzrastu; rođeno je 10000 što je tek četvrtina od svih trudnica koje su uzimale lek, ovaj podatak pokazuje da štetnost lekova zavisi i od genetske konstitucije same majke; ovaj slučaj je doveo do stroge provere svih lekova pre davanja trudnicamao Teratogeni efekat imaju: ACE inhibitori, hlorokvin, DES, litijum, benzodiazepini, fenitoin, retinoidi, streptomici, tetraciklini, talidomid, valproilna kiselina, Warfarin, Aminopterin, metotreksat, antitireoidni lekovi kao i mnogi drugi lekovi; potpuno suprotan efekat ima folna kiselina koja je poželjna u trudnoći jer njen nedostatak dovodi do poremećaja neuralne cevi
o Takođe, odvojeni slučajevi neposrednog trovanja organskom živom u Japanu i Argentini su doveli do ploda rođenog sa simptomima cerebralne paralizeo Fetalni alkoholni sindrom (FAS) – majke koje stalno uzimaju veće količine alkohola tokom trudnoće dobijaju decu karakteristično male glave, sa zaostajanjem u rastu, kratkim očnim otvorinam dugim prostorom između nosa i usta, uskom gornjom usnom, malom bradom i urođenim srčanim manama, u kasnijem uzrastu hiperaktivni su i nespretni; u slučajevima kada je majka konzumirala više od 60ml etanola dnevno dolazi i do mentalne retardacije ploda; čak su i male količine alkohola štetne pa je on zabranjen u trudnoćio Udisanje olovnog benzina – dovodi do rađanja dece sa teškom mentalnom retardacijom, veveričjim licem i splastičnom diplegijomo Fizički agensi – u njih spadaju jonizujuće zračenje i prolongirana hipotermijao Doze zračenja veće od 250 rada koje se koriste u terapiji mogu uzrokovati mikrocefaliju, mentalnu i retardaciju rasta, anomalije oka; najosetljiviji je plod u 2-5. nedelji od začeća; zračenje ispod 5 rada ne ostavljaju posledice; ovo zračenje ima mutageni i karcinogeni efekat pa se izbegava u trudnoćio Prolongirana hipertermija u ranoj trudnoći izaziva mikrocefaliju, mikroftalmiju i defekte migracije neurona; zbog toga se izbegavaju vruće kupke i saune u prvom trimestru o Bolesti majke koje utiču na plod su insulin zavisni dijabetes melitus koji izaziva 2-3x veću pojavu urođenih anomalija ploda (srčana mana, defekt neuralne tube, sakralna ageneza, hipoplazija femura, holoprozencefalija i sirenomelija; ostali tipovi dijabetesa ne utiču na plod) i fenilketonurija (ukoliko nije na adekvatnoj dijeti majka rađa nefenilketonurično, mentalno zaostalno dete koje je i mikrocefalično, sa urođenom srčanom manom) kao i nedostatak folne kiseline
Genetsko savetovanje
o Jedan od najznačajnijih vidova prevencije u medicinskoj geneticio Termin genetsko savetovanje ili konsultacija uveo je Harper 1981. ali taj termin nije adekvatan jer stručno lice ne daje savet, na roditeljima je da donesu odluku, ono samo pruža genetsku informacijuo Radi genetske informacije u savetovalište se javljaju:
osobe u čijim porodicama ima naslednih bolesti osobe koje su nosioci mirnih translokacija osobe koje imaju ili su imale u rodoslovu poremećaje morfogeneze, odstupanje u rastu i razvoju, nedovoljnu mentalnu razvijenost, poremećaje pola, metaboličke i endokrine bolesti porodice sa brakom u srodstvu žene kod kojih postoji ponavljanje spontanih pobačaja ili mrtvorođenja, sterilitet ili parovi zbog pripreme za veštačku oplodnju trudnice sa 33 i više godina kao i njihovi partneri stariji od 45 trudnice koje su tokom trudnoće bile izložene dejstvu lekova, hemikalija, infekcija ili zračenja
o Faze genetičkog savetovanja:1.postavljane tačne dijagnoze probanda (na osnovu anamneze sa porodičnom istorijom bolesti, rodoslovnog stabla, kliničkog i laboratorijskog ispitivanja; anamneza podrazumeva saznavanja godina supružnika, zanimanja, informacija o prethodnim trudnoćama kao i sadašnjoj)

2.procena rizika (na osnovu principa mendelijanskog i multifaktorskog načina nasleđivanja; potom saopštavanje svih informacija i rizika dok budući roditelji ne budu potpuno svesni situacije)3.komunikacija (savetodavac daje informaciju, a prima želje i strahove bračnog para; najbitnija je istina)4.diskusija o mogućnostima (neće savetodavac već onaj koji prima informaciju živeti sa posledicama; mogućnost prenatalne dijagnostike)5.dugotrajni kontakt i podrška
Prenatalna dijagnostika
o Prenatalno dijagnostičke metode se dele na invazivne i neinvazivne(ultrazvučne i bihemijske); metode prvog i metode drugog trimesterao Savetuje se i analiza titrova na teratogene viruse, bakterije i parazite (‘’TORCH’’ toksoplazma, rubelam, citomegalovirus, herpes simpleks) a po potrebi i drugeo Invazivne metode se preporučuju parovima koji su pod većim rizikom da imaju oštećenje ploda, a najčešće su uzrok:
godine majke (računa se rizik samo u vreme porođaja jer se veliki deo trudnoća sa hromozomskim aberacijama izgubi spontanim pobačajima u prvom i drugom trimestre);
rizik za par koji je već dobio dete sa hromozomskom aberacijom (bez obzira na godine) odgovara riziku za godine uvećanim za 0,5%;
ukoliko jedan od supružnika nosi balansiranu promenu hromozoma,a prvo dete je rođeno sa balansiranom promenom, teško bolesno, rekurentni rizik je 15-20%;
kod bračnog para koji u široj porodici ima hromozomske anomalije rizik nije veći od ostatka populacije;
bračni par koji ima dete sa nekom od monogenskih bolesti koje se dijagnostifikuju biohemijskim i DNK analizama šalje se na invazivnu dijagnostiku
o Vrste invazivnih metoda: 1. amniocenteza – radi se od 16. nedelje trudnoće; metoda podrazumeva uzimanje plodove vode
(koja sadrži ćelije amniona, fetalne kože i urinarnog epitela) kroz trbušni zid pod ultrazvučnom kontrolom i kasniju analizu uzoraka; ova metoda nosi rizik od 0,5% za spontani pobačaj
2. rana amniocenteza – radi se 12-14. nedelje; rizik od spontanog pobačaja je 1%3. uzimanje uzoraka resica horiona – prednost je postavljanje dijagnoze u prvom trimestru, tačnije
11-12. nedelje; izvodi se pod kontrolom ultrazvuka kroz trbuh ili grlić materice uzimanjem resica horiona koje vode poreklo od blastociste; rezultati se mogu dobiti posle 24h ili kasnije, ako se vrši kultivacija ćelija a tako su rezultati pouzdaniji; ova metoda se u kasnijim stadijumima trudnoće naziva placentalna biopsija; postoji rizik od spontanog pobačaja 2-3% a ukoliko metodu vrši iskusni stručnjak rizik se smanjuje na 0,5%; materijal se koristi za biohemijske i DNK analize monogenskih bolesti
4. kordiocenteza – potkožno uzimanje uzoraka fetalne krvi kroz stomak; koristi se za razjašnjavanje mozaicizma dobijenih drugim metodama i za dijagnostiku hematoloških bolesti
5. fetoskopija – vizualizacija fetusa endoskopom u drugom trimestru; služi za otkrivanje strukturnih anomalija ploda i uzimanje tkiva fetusa u dijagnostici teških, retkih naslednih poremećaja; rizik od spontanog pobačaja kod ove metode je 3-5%
o Rizici na dobijanje rezultata: kod amniocenteze može da se dogodi da ne dođe do porasta ćelijske kulture kod uzimanja resica horiona može doći do pojave mozaicizma u ćelijskim kulturama; uzrok je
kontaminacija majčinim ćelijama, zato se pravi više kultura u više posuda; u kultivaciji ćelija plodove vode kod amniocenteze rade se dve odvojene kulture; u slučaju da
se u jednoj nađe samo jedna nenormalna ćelija onda je to pseudomozaicizam, smatra se lažno pozitivnim rezultatom i naziva se nivo 1 mozaicizma; nivo 2 je kada se mozaicizam nađe u 2 ili više ćelija jedne kulture i težak je za interpretaciju; nivo 3 je pojava mozaicizma u obe kulturei tad se sumnja na pravi mozaicizam koji se proverava kordocentezom
ukoliko se nađe balansirana provera kod amniocenteze proveravaju se roditeljski kariotipovi, pa ukoliko neki od roditelja ima istu promenu plod se smatra zdravim
o Neinvazivne metode : Metode koje se zasnivaju na izolaciji i pregledu fetalnih ćelija (fetalni limfociti, granulociti,
eritroblasti) u cirkulaciji majke; one se FISH metodom ili upotrebom DNK proba testiraju na hromozomske aberacije; nije dovoljno precizna metoda pa se više koristi za procenu rizika
Biohemijski markeri hromozomskih aberacija: double test (prvi trimestar), triple test (drugi trimestar, stopa detekcije poremećaja 63-67%), quadruple test (stopa detekcije 70% uz 5% lažno pozitivnih nalaza)
Ultrazvučni markeri se mogu primetiti pomoću ultrazvuka i to u prvom trimestre; najznačajnija je povećana nuhana translucencija (NT) koja predstavlja ultrazvučni prikaz potkožne nakupine tečnosti iza fetalnog vrata; NT kombinovana sa starošću majke i biohemijskim skriningom detektuje hromozomopatije u 85-90% slučajeva;
o Preimplantaciona prenatalna dijagnostika – koristi se kod porodica u kojima je nasledna bolest evidentno prisutna; tri metode:
analiza prvog i drugog polarnog tela (PB1 i PB2) biopsija blastomere biopsija blastociste























![Molekularna Genetika Skripta by Jernej[1]](https://img.pdfslide.net/doc/110x75/547ed9e8b4af9fc9158b5770/molekularna-genetika-skripta-by-jernej1.jpg)





