Embed Size (px)
Citation preview

212
Table ronde
© 2014 Elsevier Masson SAS. Tous droits réservés.Archives de Pédiatrie 2014;21:212-214
Hyperlaxités syndromiques de l’enfant et de l’adolescentB. Chevalliera,b,c,e,*, G. Benoista, M. Jouneauxa,c, C. Stheneura,c, D. Germaina,b,d
aPôle de pédiatrie-génétique des hôpitaux Ile-de-France-Ouest, AP-HPbUniversité de Versailles-Saint-Quentin-en-Yvelines, 55, avenue de Paris, 78000 Versailles, FrancecCentre de référence pour le syndrome de Marfan, AP-HP, hôpital Bichat, 46, rue Henri-Huchard, 75877 Paris Cedex 18, FrancedCentre de référence, syndrome d’Ehlers-Danlos, hôpital Raymond-Poincaré, 104, boulevard Raymond-Poincaré, 92380 Garches, FranceeService de pédiatrie, AP-HP, hôpital Ambroise-Paré, 9, avenue Charles-de-Gaulle, 92100 Boulogne-Billancourt, France
*Auteur correspondant e-mail : [email protected]
Hyperlaxité de l’enfant et de l’adolescent (GPG, SOFOP)
La base de données française des maladies rares (Orphanet) indique 139 maladies ou syndromes incluant la présence d’une hypermobilité. Il existe une hyperlaxité articulaire
physiologique chez 4 à 40 % des enfants. Dans la majorité des cas, elle est asymptomatique et régresse avec l’âge, ne nécessitant pas de prise en charge médicale. Elle est un des signes habituels de certains tableaux cliniques comme dans la trisomie 21, l’ostéogenèse imparfaite, la cutis-laxa, le syndrome de Stickler, etc. Si les maladies héréditaires du tissu conjonctif sont rares, il importe néanmoins de ne pas méconnaître l’hypo-thèse d’un syndrome d’Ehlers-Danlos (SED) ou d’un syndrome de Marfan (SM) devant une hyperlaxité articulaire découverte chez un enfant. Nous présentons ici la démarche visant à confirmer ces deux hypothèses diagnostiques.
1. L’hyperlaxité est-elle cliniquement confirmée ?Le premier temps de la démarche diagnostique vise à confirmer l’hyperlaxité qui peut varier selon l’ethnie, l’âge et le sexe (plus fréquente chez les filles). Le score de Beighton permet de confir-mer ou d’écarter l’hyperlaxité. Chez l’enfant, il est difficile de parler d’hyperlaxité avant 6 ans ; compte tenu de la plus grande souplesse des enfants, il est habituel de retenir un score de 6, voire 7 points pour l’évoquer.
2. Cette hyperlaxité est-elle isolée ou accompagnée de signes associés ?A) Sur le plan locomoteur, une luxation congénitale de hanche, un pied-bot en varus équin, des chutes et/ou entorses fré-quentes, un genu valgum, des luxations à répétition, un affais-
sement des voûtes plantaires sont recherchés. On interrogera sur l’existence de douleurs, plus ou moins systématisées, de nature nociceptive ou neuropathique, en faisant préciser leur topographie.B) Sur le plan dermatologique, l’hyperextensibilité cutanée s’apprécie par une extension de la peau au-delà des limites de la normale, notamment au niveau de la face antérieure (en position anatomique) des avant-bras et des faces latérales du cou. La texture de la peau est veloutée et douce, surtout au niveau des paumes de mains. On peut rencontrer des ecchy-moses, des contusions, une fragilité cutanée, un retard de cicatrisation, des cicatrices dystrophiques, un réseau veineux sous-cutané trop visible.C) L’examen neurologique pourra aussi rechercher des troubles de la proprioception et de l’équilibre, un syndrome dysautono-mique.
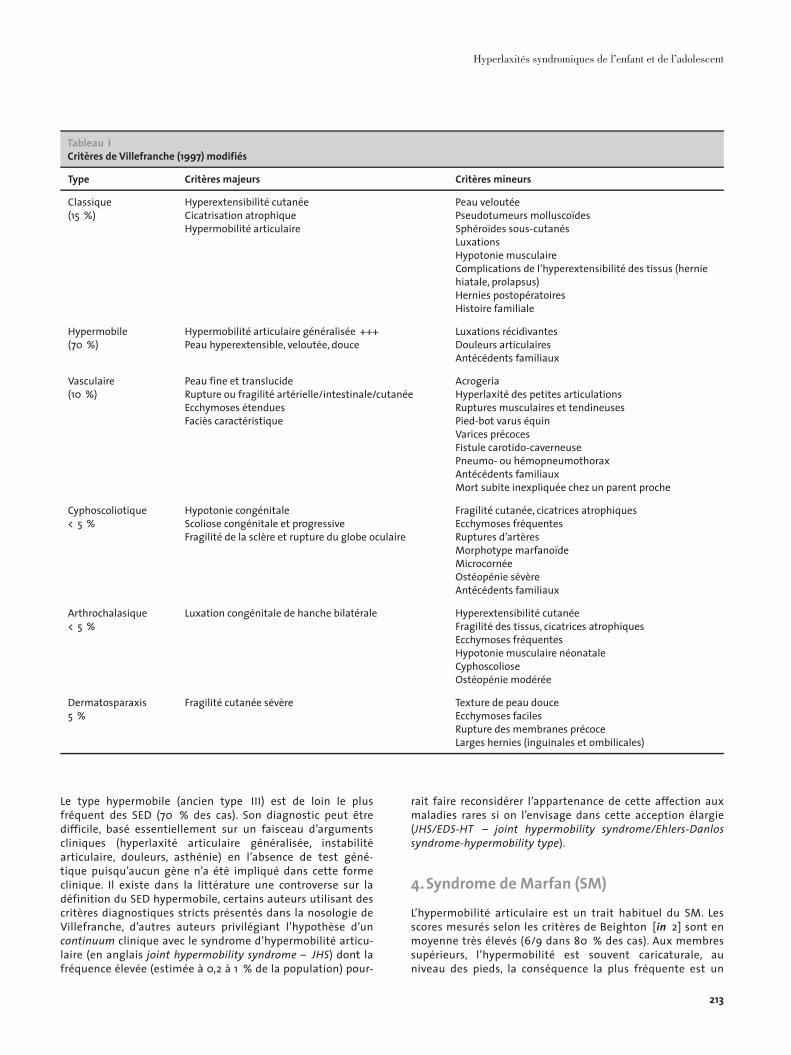
3. L’hyperlaxité est-elle évocatrice d’un SED ?Les SED sont des maladies héréditaires du tissu conjonctif affectant le collagène. Il existe différents types cliniques encore actuellement classés selon la nosologie de Villefranche (1997). Les signes les plus caractéristiques des 6 principaux types de SED sont présentés dans le cadre de la nosologie de Villefranche, en insistant sur la prévalence élevée du SED hypermobile (ancien type III), qui rend compte d’au moins 70 % des observations (Tableau 1).Les SED sont le plus souvent de transmission autosomique dominante, mais 50 % des cas sont dus à des mutations de novo. La pénétrance est incomplète et l’expressivité de la maladie est très variable. Le diagnostic de SED classique (anciens types I et II) est le plus aisé, basé sur une triade associant hyperlaxité articulaire, hyperélasticité cutanée et cicatrisation dystrophique. Le diagnostic est donc essentiellement clinique, ne nécessitant pas forcément le génotypage des gènes impliqués dans cette forme clinique (COL5A1, COL5A2).
213
Hyperlaxités syndromiques de l’enfant et de l’adolescent
rait faire reconsidérer l’appartenance de cette affection aux maladies rares si on l’envisage dans cette acception élargie (JHS/EDS-HT – joint hypermobility syndrome/Ehlers-Danlos syndrome-hypermobility type).
4. Syndrome de Marfan (SM)
L’hypermobilité articulaire est un trait habituel du SM. Les scores mesurés selon les critères de Beighton [in 2] sont en moyenne très élevés (6/9 dans 80 % des cas). Aux membres supérieurs, l’hypermobilité est souvent caricaturale, au niveau des pieds, la conséquence la plus fréquente est un
Tableau ICritères de Villefranche (1997) modifiés
Type Critères majeurs Critères mineurs
Classique(15 %)
Hyperextensibilité cutanéeCicatrisation atrophiqueHypermobilité articulaire
Peau veloutéePseudotumeurs molluscoïdesSphéroïdes sous-cutanésLuxationsHypotonie musculaireComplications de l’hyperextensibilité des tissus (hernie hiatale, prolapsus)Hernies postopératoiresHistoire familiale
Hypermobile(70 %)
Hypermobilité articulaire généralisée +++Peau hyperextensible, veloutée, douce
Luxations récidivantesDouleurs articulairesAntécédents familiaux
Vasculaire(10 %)
Peau fine et translucideRupture ou fragilité artérielle/intestinale/cutanéeEcchymoses étenduesFaciès caractéristique
AcrogeriaHyperlaxité des petites articulationsRuptures musculaires et tendineuses Pied-bot varus équinVarices précocesFistule carotido-caverneusePneumo- ou hémopneumothoraxAntécédents familiauxMort subite inexpliquée chez un parent proche
Cyphoscoliotique< 5 %
Hypotonie congénitaleScoliose congénitale et progressiveFragilité de la sclère et rupture du globe oculaire
Fragilité cutanée, cicatrices atrophiquesEcchymoses fréquentesRuptures d’artèresMorphotype marfanoïdeMicrocornéeOstéopénie sévèreAntécédents familiaux
Arthrochalasique< 5 %
Luxation congénitale de hanche bilatérale Hyperextensibilité cutanéeFragilité des tissus, cicatrices atrophiquesEcchymoses fréquentesHypotonie musculaire néonataleCyphoscolioseOstéopénie modérée
Dermatosparaxis5 %
Fragilité cutanée sévère Texture de peau douce Ecchymoses facilesRupture des membranes précoceLarges hernies (inguinales et ombilicales)
Le type hypermobile (ancien type III) est de loin le plus fréquent des SED (70 % des cas). Son diagnostic peut être difficile, basé essentiellement sur un faisceau d’arguments cliniques (hyperlaxité articulaire généralisée, instabilité articulaire, douleurs, asthénie) en l’absence de test géné-tique puisqu’aucun gène n’a été impliqué dans cette forme clinique. Il existe dans la littérature une controverse sur la définition du SED hypermobile, certains auteurs utilisant des critères diagnostiques stricts présentés dans la nosologie de Villefranche, d’autres auteurs privilégiant l’hypothèse d’un continuum clinique avec le syndrome d’hypermobilité articu-laire (en anglais joint hypermobility syndrome – JHS) dont la fréquence élevée (estimée à 0,2 à 1 % de la population) pour-

214
B. Chevallier et al. Archives de Pédiatrie 2014;21:212-214
pied plat valgus, évolutif, retentissant sur les articulations tibiotarsienne et sous-astragalienne. Les symptômes asso-ciés faisant évoquer le diagnostic de SM à l’âge pédiatrique sont squelettiques (grande taille associée à une scoliose et/ou une déformation thoracique) ou oculaires. La difficulté du diagnostic est renforcée par le fait que le tableau clinique se constitue de façon progressive avec l’âge. Ainsi les signes squelettiques ne sont souvent évidents qu’à partir de l’âge pubertaire. L’intérêt de dépister précocement les cas pédia-triques de SM est de pouvoir mettre en place une série de mesures préventives permettant d’allonger significativement l’espérance de vie de ces patients et de prévenir la survenue de handicaps divers. Le diagnostic repose sur les critères de Gand modifiés.Le SM est une maladie rare (1/5 000 – 1/10 000), autosomique dominante, secondaire le plus souvent à une mutation du gène de la fibrilline de type 1. L’histoire familiale n’est pas toujours contributive étant donné qu’une néomutation est en cause dans près de 25 % des cas. Sa pénétrance est complète, mais son expressivité est variable, d’une forme gravissime, le SM néonatal, à une forme minime passant inaperçue sans un screening systé-matique. L’extrême variabilité d’expression du SM, même au sein d’une famille, rend le diagnostic parfois difficile. La fibrilline-1 est codée par un acide ribonucléique messager ARNm provenant d’un grand gène de 235 kb, appelé FNB1, localisé en 15q21. À ce jour, plus de 2 000 mutations ont été identifiées sur le gène
FBN1 : les mutations sont essentiellement privées, c’est-à-dire spécifiques à une famille.
5. Conclusion
La distinction entre hypermobilité articulaire bénigne généralisée et hyperlaxité articulaire syndromique n’est pas toujours aisée et nombreux sont les patients qui passent d’une entité à l’autre au cours de leur vie, induisant une perte de chance en raison des retards au diagnostic précis. Seule une enquête anamnestique et clinique approfondie peut faire évoquer un syndrome avec hypermobilité. Chez l’enfant, une scoliose, une grande taille, des anomalies cardio-vasculaires ou oculaires (sclérotiques bleues, ectopie du cristallin), des anomalies du pectus, des fractures multiples, un palais ogival et des anomalies cutanées doivent faire évoquer la possibilité d’une cause syndromique. L’hyperlordose, les atteintes musculaires, les troubles digestifs et urinaires associés, l’atteinte des articulations temporo-mandibulaires orientent plutôt vers un JHS. Le continuum SED/JHS rend souvent la tâche ardue pour le clinicien.
Références
Les références complètes peuvent être obtenues sur demande auprès de l’auteur.





























