Embed Size (px)
Citation preview

INAUGURAL-DISSERTATION
zur Erlangung der Doktorwürde
der Naturwissenschaftlich-Mathematischen Gesamtfakultät
der Ruprecht-Karls-Universität
Heidelberg
vorgelegt von
M.Sc. Neuroscience Laurent-Hervé Perez aus Champigny sur marne (France)
Tag der mündlichen Prüfung.

Dissertation
Submitted to the
Combined Faculties for the Natural Sciences and for Mathematics
of the Rupertus Carolus University of Heidelberg, Germany
for the degree of
Doctor of Natural Sciences
Presented by
M.Sc. Neuroscience. Laurent-Hervé Perez
Born in: Champigny sur Marne (France)
Examiners: Prof. Dr. Eduard C. Hurt
Prof. Dr. Renate Voit

CELL CYCLE REGULATION BY XKID AND
RINGO PROTEINS
Gutachter: Prof. Dr. Eduard C. Hurt Prof. Dr. Renate Voit

ACKNOWLEDGMENTS
To begin with, I would like to thank Angel Nebreda for giving me the opportunity to
carry out my PhD work in his lab and most of all for his support and guidance during my
PhD.
I thank the members of my thesis committee, Guilio Superti-Furga, Isabelle Vernos,
Jochen Wittbrodt and André Picard for their constant advice and support during my PhD.
I would also like to express my appreciation to Eduard Hurt and Renate Voit for
accepting to be my Gutachter.
I would also like to thank the past and the present members of the Nebreda group for
their help, the atmosphere in the lab and discussion. In particular I would like to thank
Philippe Beaufils, Emma Black, Gustavo Gutierrez, Eusebio Perdiguero and Andrea Vögtlin
for the nice environment and their help for this thesis.
Very special thanks go to Gustavo and Stéphane for all the scientific discussions and
party we got.
Pour finir j’aimerais remercier Silvia Palacios pour m’avoir supporté pendant ce travail de thèse et su surmonter avec moi ces années en Allemagne.

A ma Famille : Aurèlie, Olivier, Robert et Suzel.

I
TABLE OF CONTENTS List of figures……………………………………………….………………………………..V
Abbreviations…….……………………………...……………….………………...………..VII
Summary……………………………………………………….………..…………………..XI
1.Introduction............................................................................................................................1
1.1 The mitotic cell cycle........................................................................................................1
1.1.1 Cdks and Cyclins. ......................................................................................................2
1.1.1.1 The Cdk family. ..................................................................................................2
1.1.1.2 The Cyclin family. ..............................................................................................5
1.1.1.3 Mechanism of Cdk regulation.............................................................................8
1.1.1.4 The Cdk inhibitor family. .................................................................................10
1.1.2 The G1/S transition and checkpoints. ......................................................................12
1.1.2.1 The G1/S transition. ..........................................................................................12
1.1.2.2 The G1/S checkpoints. ......................................................................................13
1.1.3 The G2/M transition and checkpoints......................................................................15
1.1.3.1 The G2/M transition..........................................................................................15
1.1.3.2 The G2/M checkpoints and exit from mitosis...................................................17
1.2 The meiotic cell cycle. ....................................................................................................18
1.2.1 Meiotic maturation of Xenopus oocytes as a model system. ...................................18
1.2.2 Signal transduction pathways that trigger meiotic maturation. ...............................19
1.2.3 Activation of the Mos-MAPK pathway and inhibition of DNA replication . .........21
1.2.4 Xkid, a chromokinesin involved in chromosome alignment. ..................................24
1.2.5 RINGO that triggers meiotic maturation in Xenopus oocytes. ................................25
1.3 Aim of the work. .............................................................................................................26
2.Materials and Methods........................................................................................................27
2.1 Materials. ........................................................................................................................27
2.1.1 Solutions. .................................................................................................................27
2.1.2 Antibodies. ...............................................................................................................31
2.1.3 DNA Constructs.......................................................................................................32
2.1.3.1 Vectors. .............................................................................................................32
2.1.3.2 Constructs .........................................................................................................33
2.1.3.3 Primers for mutagenesis....................................................................................34

II
2.1.4 Cells. ........................................................................................................................34
2.1.4.1 Ntera-2. .............................................................................................................34
2.1.4.2 HEK293. ...........................................................................................................35
2.2 Methods...........................................................................................................................35
2.2.1 Molecular Biology. ..................................................................................................35
2.2.1.1 DNA cloning.....................................................................................................35
2.2.1.2 Construction of Cdk1, Ringo1 and Xkid mutants.............................................36
2.2.1.3 Expression of proteins in reticulocyte lysates...................................................36
2.2.1.4 Preparation of total RNA and Northern blotting...............................................37
2.2.1.5 RT-PCR.............................................................................................................38
2.2.2 Biochemistry. ...........................................................................................................38
2.2.2.1 Bacterial expression and purification of recombinant fusion proteins. ............38
2.2.2.2 In vitro Cdk assay with recombinant proteins. .................................................40
2.2.2.3 Baculovirus expression and purification of recombinant His-Cyclin B1. ........40
2.2.2.4 GST pull-down..................................................................................................41
2.2.2.5 Generation and purification of Ringo antibodies..............................................41
2.2.2.6 Covalent coupling of antibodies to protein G-Sepharose. ................................42
2.2.3 The Xenopus laevis oocyte system. .........................................................................42
2.2.3.1 Isolation of stage VI oocytes and induction of meiotic maturation. ................42
2.2.3.2 Preparation of mRNAs for injection into oocytes.............................................43
2.2.3.3 Microinjection of oocytes with mRNAs. ..........................................................43
2.2.3.4 Antisense experiments. .....................................................................................44
2.2.3.5 DNA replication assays.....................................................................................44
2.2.3.6 Preparation of oocyte lysates and immunobloting............................................45
2.2.3.7 Histone H1 kinase assays..................................................................................45
2-2.3.8 In vivo labelling of Xenopus oocyte proteins with 35S-methionine. ................45
2.2.3.9 Immunoprecipitation of Myc-tagged proteins from oocyte lysates..................46
2.2.4 Mammalian Cell Culture..........................................................................................46
2.2.4.1 Conditions of cell culture..................................................................................46
2.2.4.2 Transfection and retroviral infection of cells....................................................46
2.2.4.3 Synchronization of culture cells........................................................................47
2.2.4.4 Mitogenic response. ..........................................................................................47
2.2.4.5 Small Interference RNA. ..................................................................................48
2.2.4.6 Flow cytometry. ................................................................................................48

III
2.2.4.7 Cell growth curves. ...........................................................................................48
2.2.4.8 Cell and tissue extracts and immunoblotting. ...................................................49
2.2.5 Biocomputing...........................................................................................................49
3.Results. ..................................................................................................................................50
3.1 Involvement of the kinesin-like protein Xkid in Xenopus oocyte maturation. ...............50
3.1.1 Xkid synthesis is not required for meiosis I entry of Xenopus oocytes. ..................50
3.1.2 Xkid-depleted oocytes do not re-activate Cdk1-Cyclin B after meiosis I and
undergo DNA replication..................................................................................................54
3.1.3 Xkid is not required for meiosis I but to enter into meiosis II. ................................57
3.1.4 Ectopic expression of Xkid allows Xkid-depleted oocytes to complete meiotic
maturation. ........................................................................................................................60
3.1.5 Ectopic expression of an Xkid mutant lacking the DNA binding domain allows
Xkid-depleted oocytes to complete meiotic
maturation……………………………………………...………………………………..60
3.2 RINGO, a new family of cell cycle regulators. ...............................................................64
3.2.1 RINGO induces Cdk activation independently of T-loop phosphorylation. ...........64
3.2.1.1 RINGO-induced Cdk1 activation is independent of Thr161 phosphorylation. 64
3.2.1.2 Activation of Cdk2 by RINGO is independent of Thr160 phosphorylation.....66
3.2.2 Cloning of RINGO mammalian homologues. .........................................................68
3.2.3 Identification of the RINGO minimal region necessary to bind Cdks.....................71
3.2.4 Biochemical characterization of the mammalian clones. ........................................72
3.2.5 Properties of the mammalian clones in expressed in Xenopus oocytes. ..................75
3.2.5.1 Ringo1...............................................................................................................75
3.2.5.2 Ringo2...............................................................................................................79
3.2.5.3 Ringo3...............................................................................................................79
3.2.5.4 Ringo4...............................................................................................................79
3.2.6 Expression pattern of the mammalian RINGO proteins. .........................................83
3.2.7 RINGO proteins can activate Cdk5. ........................................................................87
3.2.8 Role of the RINGO proteins in the mammalian cell cycle. .....................................89
3.2.8.1 Ringo3 is a cell cycle regulated protein............................................................89
3.2.8.2 Ringo3 interacts with Cdk2 and phosphorylates Rb in vitro. ...........................89
3.2.8.3 Effect of the RINGO proteins on the proliferation rate of Ntera-2 cells. .........92
3.2.8.4 Biochemical and cell cycle properties of the Ntera-2 cell lines overexpressing
RINGO proteins. ...........................................................................................................92

IV
3.2.8.5 Biochemical and cell cycle properties of the Ntera-2 cell lines depleted from
the RINGO proteins. .....................................................................................................96
4.Discussion............................................................................................................................100
4.1 Xkid, more than a molecular motor. .............................................................................100
4.1.1 Xkid is not required for meiosis I entry. ................................................................100
4.1.2 Xkid is required for the meiosis I to meiosis II transition. ....................................100
4.1.3 Putative Xkid functions during the meiosis I to meiosis II transition....................101
4.2 RINGO, a new family of cell cycle regulators. ............................................................104
4.2.1 RINGO bypasses usual Cdk regulatory mechanisms. ...........................................104
4.2.2 Ringo3 activates Cdk5. ..........................................................................................107
4.2.3 RINGO: a novel family of proteins regulating the cell cycle. ...............................108
4.2.4 Role of Ringo1 and Ringo3 in the mammalian cell cycle. ....................................109
4.2.5 Why does the cell cycle need RINGO proteins? ...................................................111
6 Bibliography. ......................................................................................................................113
7 Appendix.............................................................................................................................125
7.1 Nucleotide and deduced amino acid sequence of Human-Ringo1. ..............................125
7.2 Nucleotide and deduced amino acid sequence of Human-Ringo2. ..............................126
7.3 Nucleotide and deduced amino acid sequence of Mouse-Ringo3. ...............................127
7.4 Nucleotide and deduced amino acid sequence of Mouse-Ringo4. ...............................128
7.5 Sequence alignment of Ringo3 alternative splice variants. ..........................................129
7.6 Sequence alignment of Ringo4 alternative splice variants. ..........................................130

V
LIST OF FIGURES Figure 1.1: The phases of the cell cycle......................................................................................3
Figure 1.2: Crystal structure of Cdk2………………………… ………………………………………………………… 5
Figure 1.3: Crystal structure of the Cyclin box...........................................................................7
Figure 1.4: Diversity of cyclins and their putative functions......................................................7
Figure 1.5: Mechanism of Cdk activation...................................................................................9
Figure 1.6 : Mechanisms of Cdk inhibition. .............................................................................11
Figure 1.7: Mechanisms that regulate the G1 to S phase transition..........................................14
Figure 1.8: Model of the G2/M transition.................................................................................16
Figure 1.9: Morphological and biochemical changes during Xenopus oocyte maturation.......20
Figure 1.10: Signalling cascades involved in the Xenopus oocytes meiotic maturation..........23
Figure 3.1: Alignment of the Human and Xenopus kid proteind.Synthesis of Xkid is . ..........53
Figure 3.2: Schematic representation of Xkid protein . ............................................................53
Figure 3.3: Synthesis of Xkid during the Xenopus oocyte meiotic maturation .......................53
Figure 3.4: Synthesis of Xkid is not required for meiosis I entry of Xenopus oocytes. ...........53
Figure 3.5: Biochemical analysis of Xkid-depleted Xenopus oocytes......................................55
Figure 3.6: Xkid-depleted oocytes do not reactivate Cyclin B-Cdk1 after meiosis I. ..............56
Figure 3.7: Xkid-depleted oocytes undergo DNA replication. .................................................58
Figure 3.8: Xkid is required for spindle formation in meiosis II but not meiosis I. .................59
Figure 3.9: Ectopic expression of Xkid allows Xkid-depleted oocytes to enter meiosis II......61
Figure 3.10: Ectopic expression of a Xkid mutant lacking the DNA-binding domain allows
Xkid-depleted oocytes to enter meiosis II. .......................................................................63
Figure 3.11: RINGO activates GST-Cdk1 independently of T161 phosphorylation. ..............63
Figure 3.12: RINGO activates Cdk1 in vitro and in Xenopus oocytes independently of T161
phosphorylation.................................................................................................................67
Figure 3.13: RINGO activates Cdk2 independently of T160 phosphorylation.. ......................63
Figure 3.14: Sequence alignment of the RINGO family of proteins. .......................................69
Figure 3.15: Percentage of identity and phylogenetic tree of the RINGO family of proteins. .70
Figure 3.16: Schematic representation of some of the RINGO proteins . ................................71
Figure 3.17: The core region of RINGO binds and activates Cdk1 in Xenopus oocytes. ........73
Figure 3.18: The RINGO protein family binds and activates Cdks in vitro. ............................74
Figure 3.19: Ringo1 inhibits progesterone induced meiotic maturation...................................76

VI
Figure 3.20: Ringo1 inhibits progesterone induced meiotic maturation but binds and activates
Cdk1..................................................................................................................................77
Figure 3.21: Ringo1 inhibition can be rescued by overexpression of Cdk1.............................78
Figure 3.22: Ringo2 accelerates progesterone induced meiotic maturation and also binds and
activates Cdk1...................................................................................................................80
Figure 3.23: Ringo3 can induce the meiotic maturation of Xenopus oocytes. .........................81
Figure 3.24: Ringo4 does not affect the meiotic maturation in Xenopus oocytes. ...................82
Figure 3.25: Northern blot analysis of the RINGO family . .....................................................83
Figure 3.26: RT-PCR analysis of the expression pattern of RINGO proteins..........................84
Figure 3.27: Schematic representation of the alternative splice variants of Ringo3 and Ringo4.
...........................................................................................................................................85
Figure 3.28: RT-PCR analysis of the expression pattern of the splice variants of the RINGO.
...........................................................................................................................................86
Figure 3.29: GST-pull down from brain and testis extracts......................................................88
Figure 3.30: Ringo3 co-immunoprecipitates with Cdks. ..........................................................90
Figure 3.31: RT-PCR analysis of the expression of the Ringo1 and Ringo3 proteins in the
Ntrera-2 cell line. ..............................................................................................................90
Figure 3.32: Ringo3 is a cell cycle regulated protein. ..............................................................91
Figure 3.33: Effect of RINGO proteins on the proliferation rate of Ntera-2 cells....................93
Figure 3.34: Biochemical properties of the Ntera-2 cell lines expressing Ringo .....................94
Figure 3.35: FACS analysis of RINGO-overexpressing cell lines. ..........................................95
Figure 3.36: Depletion of the RINGO proteins in Ntera-2 cells leads to a cell cycle arrest.....97
Figure 3.37: FACS analysis of Ntera-2 cells depleted of Ringo...............................................98
Figure 3.38: FACS analysis of synchronized cells ...................................................................99
Figure 4.1: Role of Xkid in the meiosis I to meiosis II transition of Xenopus oocytes. .........103
Figure 4.2: Model of the interaction between Cyclin A and Cdk2........................................103
Figure 4.3: The core region of the RINGO proteins contains two hydrophobic stretch of amino
acids ................................................................................................................................106
Figure 4.4: A model for Ringo3 and Ringo1 function during the cell cycle. .........................112

VII
ABBREVIATIONS
32P: phosphor radioisotope35S: sulphur radioisotope
Ab: antibody
ATP: adenosine triphosphate
AEBSF: 4-(2-aminoethyl) benzenesulfonyl fluoride
AP: alkaline phosphatase
bp: base paire
BSA: bovin serum albumine
Ca(NO3)2: calcium nitrate
CaCl2: calcium chloride
CAK: Cdk-activating kinase
Cdc: Cell division cycle
Cdk: Cyclin dependent kinase
cDNA: complementary DNA
CSF: Cytostatic factor
C-terminus: carboxy-end of a protein
Da: dalton
DAPI: 4,6-diamino-2-phenylindole
DEPC: diethyl pyrocarbonate
DMEM: Dulbecco’s modified Eagle’s medium
DMP: dimethyl pimelimidate
DNA: deoxyribonucleic acid
DNAse: deoxyribonuclease
dNTP: deoxy nucleotide triphosphate
DTT: dithiothreitol
E.coli: Escherichia coli
ECL: enhanced chemiluminescence
EDTA: ethylenediaminetetraacetic acid
EGTA: ethylene glycol-bis(b-aminoethyl ether) N,N,N’,N’tetraacetic acid
FA: formaldehyde
FCS: fetal calf serum

VIII
g: gram
GDP: guanosine diphosphate
GST: glutathione-S-transferase
GTP: guanosine triphosphate
GVBD: germinal vesicle breakdown
H1K: histone H1 kinase
HCl: hydrochloric acid
HEPES: N-(2-hydroxyethyl)piperazine-N’-(2-ethanesulfonic acid)
Human: Homo sapiens
hr: hour
HRP: horseradish peroxidase
IgG: immunoglobulin G
IP: immunoprecipitation
IPTG: isopropyl _-D-thiogalactopyranoside
K: kilo
KCl: potassium chloride
KLH: keyhole limpet hemocyanin
LB broth: Luria-Bertani broth
m: mili
M: molar
MAPK: mitogen-activated protein kinase
MEK: MAPK kinase
mBarth: modified Barth
MgCl2 : magnesium chloride
MgSO4: magnesium sulphate
min: minute
ml: milliltre
mm: millimetre
mM: millimolar
MOPS: 3-(N-morpholino) propanesulfonic acid
MPF: M phase promoting factor or maturation-promoting factor
Mouse: Mus musculus
mRNA: messenger RNA
N-terminus: amino-end of a protein

IX
Na2HPO4: disodium hydrogenphosphate
NaAc: sodium acetate
NaCl: sodium chloride
NaF: sodium fluoride
NaHCO3: sodium hydrogencarbonate
NaOH: sodium hydroxide
NaVO3: sodium vanadate
ORF: open reading frame
p: pico
P-: phosphate group
PBS: phosphate buffered saline
PCR: polymerase chain reaction
pfu: plaque forming units
PI: preimmune serum
PIPES: piperazine-N,N’-bis(2-ethanesulfonic acid)
PMSF: phenylmethylsulfonyl fluoride
RINGO: Rapid Inducer of G2/M transition in Oocytes
RNA: ribonucleic acid
s: second
SDS: sodium dodecyl sulfate
SDS-PAGE: SDS-polyacrylamide gel electrophoresis
TBS-T: Tris buffered saline-Tween 20
TCA: trichloroacetic acid
TEMED: N,N,N’,N’-tetramethylethyethyethyethylenediamine
Tris:tris (hydroxymethyl)amniomethane
U: enzyme unit
UTR: untranslated region
UV: ultra violet
v/v: volume per volume
w/v: weight per volume
wt: wild-type
Xenopus: Xenopus laevis
X
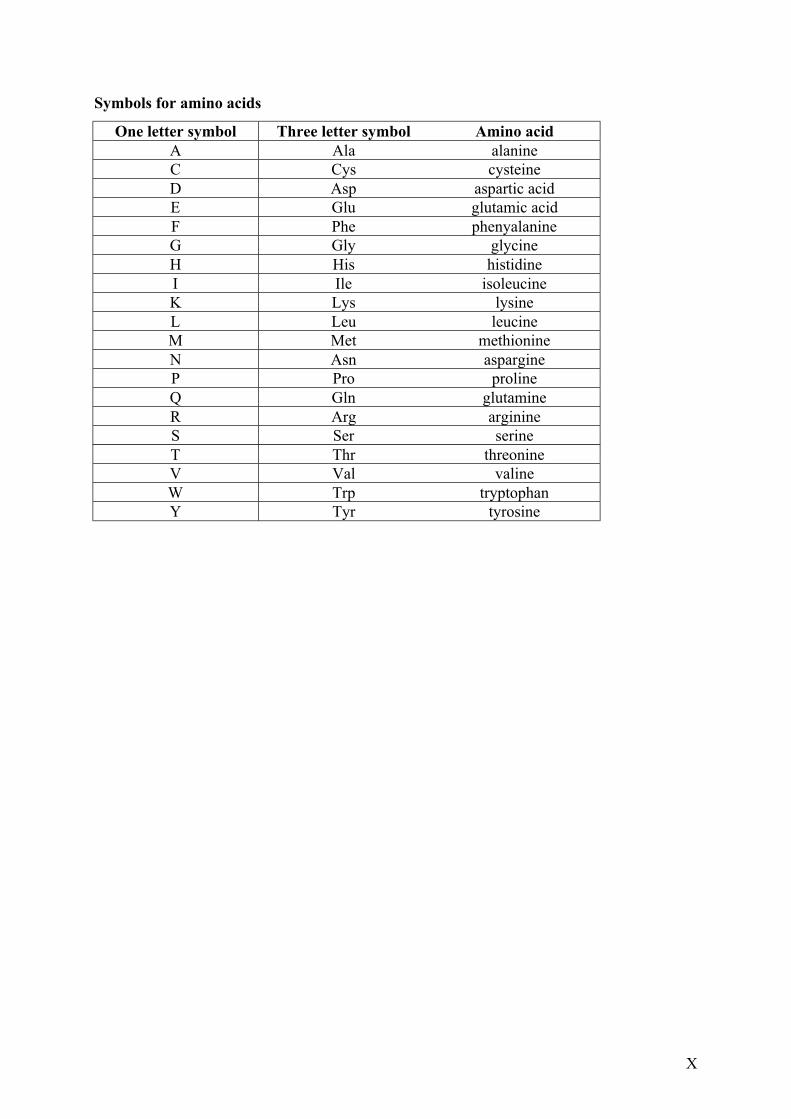
Symbols for amino acids
One letter symbol Three letter symbol Amino acidA Ala alanineC Cys cysteineD Asp aspartic acidE Glu glutamic acidF Phe phenyalanineG Gly glycineH His histidineI Ile isoleucineK Lys lysineL Leu leucineM Met methionineN Asn aspargineP Pro prolineQ Gln glutamineR Arg arginineS Ser serineT Thr threonineV Val valineW Trp tryptophanY Tyr tyrosine

XI
SUMMARY
The chromokinesin Xkid was found in a screen aimed to identify proteins synthesized
de novo during Xenopus oocyte maturation, hence with a potential role in meiotic regulation.
The screen was based on the differential association of mRNAs with polysomes in
progesterone treated versus non treated oocytes. Xkid has previously been shown to play a
crucial role in chromosome alignment at the metaphase plate of the spindle. In progesterone-
treated oocytes, Xkid starts to accumulate at the time of meiosis I and reaches its highest level
at metaphase of meiosis II. We found that spindle assembly at meiosis I can occur normally in
the absence of Xkid. However, Xkid-depleted oocytes cannot reactivate Cyclin B-Cdk1 after
meiosis I and instead of proceeding to meiosis II, they enter an interphase-like state
undergoing DNA replication. Expression of an Xkid mutant that lacks the DNA biding
domain allows Xkid-depleted oocytes to complete meiotic maturation. These results
demonstrate a new role for Xkid in the meiotic cell cycle, which is independent of its role in
metaphase chromosome alignment.
The second part of the work presented here aimed to investigate the mechanism of
Cdk regulation by RINGO. We showed that RINGO does not need the phosphorylation on the
Cdk T-loop that is necessary for Cdk activation by cyclins. Thus, Cdk1 or Cdk2 mutated on
the T-loop display the same histone H1 kinase activity level upon incubation with RINGO as
the wild type Cdk proteins.
We have also identified and characterized mammalian homologues of RINGO, which
were named Ringo1, Ringo2, Ringo3 and Ringo4. Upon injection of these proteins into
oocytes, two members of the family, Ringo2 and Ringo3, are able to stimulate the meiotic
maturation. However, Ringo1 blocks progesterone induced maturation, probably by
sequestering Cdk1 in the oocyte. We have also identified a domain named “core” region,
which is present in all the RINGO proteins and is likely to be involved in binding to Cdks.
We decided to focus on the characterization of Ringo1 and Ringo3 because they displayed
opposite functions in Xenopus oocytes. Overexpression of Ringo3 increases proliferation and
the incorporation of [3-H]-Thymidine in the human Ntera-2 cell line. Moreover, Ringo3
overexpression decreases the overall population of cells in G1 phase of the cell cycle and
increases the population of cell in G2/M. Consistent with this observation, we found that the
Ringo3 protein is only expressed during the G1/S phase of the mammalian cell cycle.

XII
Depletion of Ringo3 slows cell proliferation, suggessting that Ringo3 is required for the G1/S
transition of the mammalian cell cycle.
Ringo1 overexpression decreases proliferation in the human Ntera-2 cell line and
reduces the overall population of cells in G1 and S phase of the cell cycle while increasing the
population of cells in G2/M. Consistent with this observation, we found that depletion of
Ringo1 slows cell growth and increases the population of cell in S phase. These results
suggest that Ringo1 may be involved in S phase exit of the cell cycle.
Part of this work has been published in:
Perez, L.H., Antonio, C., Flament, S., Vernos, I. and Nebreda, A.R. (2002)
Xkid chromokinesin is required for the meiosis I to meiosis II transition in Xenopus laevis
oocytes, Nature Cell Biology 4 (10), 737-742.
Karaiskou, A., Perez, L.H., Ferby, I., Ozon, R., Jessus, C and Nebreda, A.R. (2000)
Differential Regulation of Cdc2 and Cdk2 by RINGO and Cyclins.
The Journal of Biological Chemistry 276 (38), 36028–36034.

Introduction
1
1. Introduction. The capacity of auto-reproduction is an essential property that defines life. The cell
cycle can be defined as the process leading to cell multiplication, which requires both growth
and cell division. During the cell cycle, two daughter cells are formed that are identical to the
mother cell. In unicellular organisms this leads to an increase of the population while in
multicellular organisms this leads to the formation, reparation or regeneration of tissue. In
both cases the decision to enter the cell cycle is regulated, by quantity of food and population
density in unicellular organisms and by growth factors and hormones in multicellular
organisms. Proliferation must be tightly controlled, as a lost of regulation can lead to anarchic
proliferation and oncogenesis. It is therefore crucial to understand the properties of the cell
cycle.
Two major types of cell cycle can be defined: the mitotic cell cycle and the meiotic
cell cycle. The mitotic cell cycle, which aims to produce daughter cells identical to the
mother, is the reproduction mode of unicellular organisms and is also necessary for a total
mass increase in multicellular organisms. However, sexual reproduction requires the fusion of
two germinal cells coming from the two parents. In this case there is a necessity to generate
daughter cells different from the mother (in terms of DNA content). This is the meiotic cell
cycle that results in the reduction of the DNA content of the mother cell by half, leading to the
formation of germinal cells or gametes.
1.1 The mitotic cell cycle. The processes of DNA replication and cell division are separated in time during the
cell cycle. DNA replicates during a phase called S (for Synthesis of DNA) and then the cell
divides in two during a phase called M (for Mitosis). The S and M phases are the two major
phases of the cell cycle. They are separated by two gap phases, called G1 (for Gap phase one
between M and S phases) and G2 (for Gap phase 2 between S and M phases). The two gap
phases provide time for the cell to monitor the internal environment (DNA damage, abnormal
cellular structures) and external environment (presence of growth factors, cell density) to
ensure that the conditions are suitable and the preparations are complete before the cell
commits to S phase or mitosis. For example, if the extracellular conditions are unfavourable,
cells delay progress through G1 and may enter a specialized resting state known as G0, in
which they are quiescent and can remain for years. Indeed, some cells known as post-mitotic

Introduction
2
remain permanently in G0. For example after the nervous system development, most of
neurons remain in G0 until they die.
Thus the cell cycle of most of the somatic cells is divided into four sequential phases:
G1, S, G2 and M. The cell cycle is often represented by a circle, symbolising its cyclic
character (Figure 1.1). G1, S and G2 together are called interphase. In a typical human cell
proliferating in culture, interphase takes about 24 hours (hrs) and only 1 hr is necessary for
mitosis. Although mitosis is short it can be subdivided in five steps. The first step is known as
prophase, where DNA condenses and chromosomes are visible. Prophase ends with the
dissolution of the nuclear envelope which allows mixing between nucleus and cytoplasm.
Microtubules nucleate from the microtubule organizing center, which is also called
centrosome in animal cells. It is duplicated during G1 and organizes the microtubules to form
the bi-polar spindle. After the prophase, metaphase leads to chromosome alignment on the
equatorial plate of the bipolar spindle. During anaphase, the linkage between the sister
chromatids is dissolved and the chromosomes are pulled apart to each pole of the spindle.
Finally in telophase, the chromosomes decondense and the nuclear envelope is reformed.
Cytokinesis then begins, with the formation of a contractile ring that separates the cytoplasms
of the two daughter cells.
Transitions between the different phases of the cell cycle are driven by a family of
serine-threonine protein kinases, the Cyclin dependent kinases (Cdks). Cdks were originally
identified in yeast (Simanis and Nurse, 1986) and are known to be present in all eukaryotic
cells. The essential partner of Cdks was first identified in marine invertebrates, where it was
periodically synthesized and degraded. For this reason they were named cyclins (Evans et al.
1983). Full activation of Cdks requires association with cyclins.
1.1.1 Cdks and Cyclins. 1.1.1.1 The Cdk family.
In mammals, nine Cdks have been identified and they are referred to as Cdk1 to Cdk9,
but only some of them are involved in cell cycle progression (Morgan, 1995; Morgan, 1997;
Simanis and Nurse, 1986).
Cdks display many differences in terms of function but their structure is conserved
(Figure 1.2). The crystal structure of monomeric Cdk2 (De bond et al. 1993) helped to
understand the catalytic activity of Cdks. Cdk2 contains a small N-terminal lobe dominated
by beta sheets and containing a large PSTAIRE helix, and a larger C-terminal lobe that is
Introduction
3
mainly helical. The hydrophobic base of ATP fits into a hydrophobic pocket within the cleft
between the two lobes. When the Cdk is unbound to cyclin, the phosphotransfer reaction is
severely restrained by two mechanisms. First the ATP molecule is incorrectly oriented and
second the activation-loop or T-loop blocks the access of peptide substrates to the active site
(De bond et al. 1993).
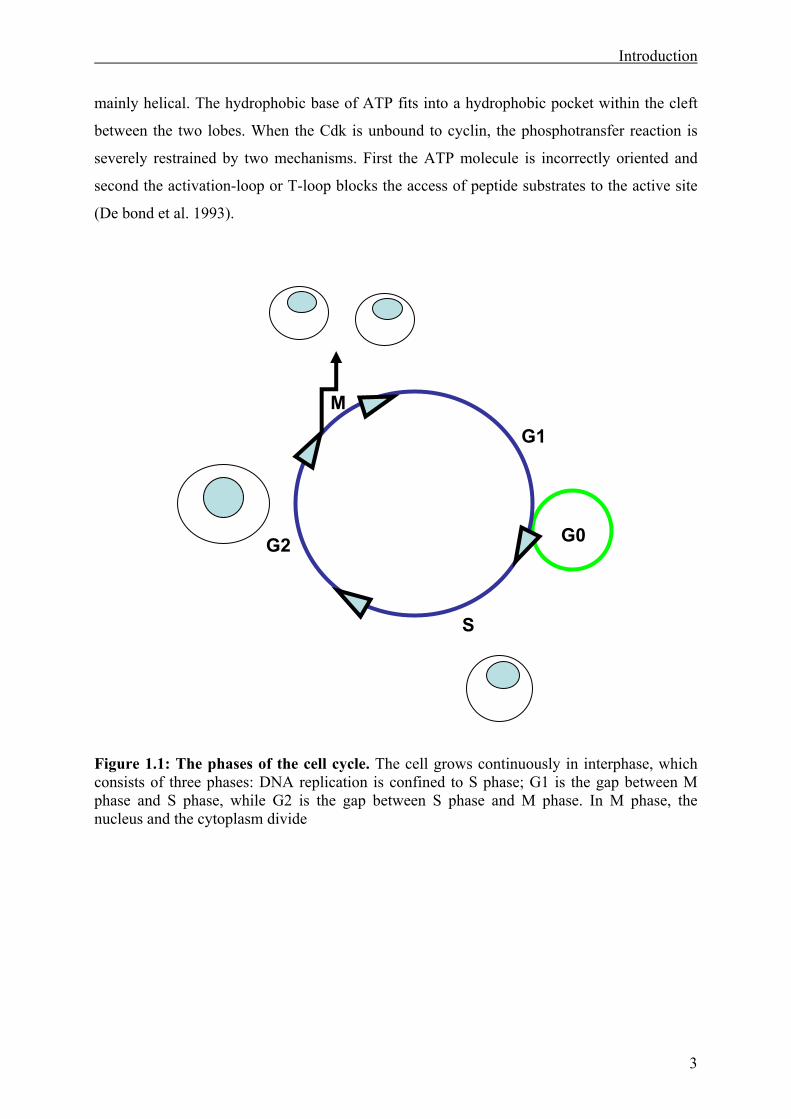
Figure 1.1: The phases of the cell cycle. The cell grows continuously in interphase, which consists of three phases: DNA replication is confined to S phase; G1 is the gap between M phase and S phase, while G2 is the gap between S phase and M phase. In M phase, the nucleus and the cytoplasm divide
M
G1
G2
S
G0

Introduction
4
Cdk1 and Cdk2 are involved in cell cycle progression. Cdk2 interacts with Cyclin E at
the beginning of the S phase to induce the initiation of DNA synthesis and then it binds
Cyclin A throughout the S phase. Mitosis is then initiated by Cdk1, in particular the complex
Cyclin B-Cdk1, also known as M-phase promoting factor (MPF) (Donjerkovic and Scott,
2000; Hunt, 1989; Johnson and Walker, 1999; Takizawa and Morgan, 2000).
Although the structure of Cdk3 is closely related to Cdk1 and Cdk2, its function has
remained relatively obscure. Overexpression of a dominant negative mutant of Cdk3 slows
down the G1 progression. This suggests a putative role at the G1/S transition (Hofmann and
Livingston, 1996).
Cdk4 and Cdk6 are involved in early G1 to S phase transition with their partner Cyclin
D. An interesting property of these two Cdks is their ability to respond to extracellular stimuli.
Indeed the key response to growth factors triggering the G0/G1 transition is the activation of
Cdk4 or Cdk6 (Lucibello et al., 1993; Matsuoka et al., 1994).
Cdk5 has pleiotropic effects from neural differentiation to intracellular transport and
DNA repair (Dhavan and Tsai, 2001; Smith and Tsai, 2002).
Cdk7 can associate with Cyclin H and Mat1 to phosphorylate other Cdks involved in
cell cycle progression. The complex Cdk7-Cyclin H-Mat1 is also called Cdk-Activating
Kinase complex (CAK) (Makela et al., 1994; Tassan et al., 1995). Cdk7 is also involved in
transcriptional regulation.
Cdk8 and Cdk9 are involved in transcriptional regulation and are associated with
Cyclin C and Cyclin T1 or T2, respectively.
Numerous “Cdk-like” proteins have also been identified lately and are named
according to their amino acid sequence in the PSTAIRE region. For example, PISSLRE,
PITALRE and PITSLRE were cloned a decade ago, but their activation mechanisms and
functions are still unknown (Meyerson et al., 1992). Recently PCTAIRE was proposed to
have a role in brain and testis, and it was suggested that it is auto-activated (Graeser et al.,
2002).

Introduction
5
1.1.1.2 The Cyclin family. More than 18 cyclins have been identified in mammals. Cyclins are very diverse, with
a molecular weight from 35 to 90 KDa (Hunt, 1991; Morgan, 1997). However, all the cyclins
contain a common region of homology known as cyclin box, which is the domain used to bind
and activate Cdks (Brown et al., 1995; Jeffrey et al., 1995; Kim et al., 1996). The cyclin box
consists of about one hundred amino acids that are very conserved and fold in a 5-helix
bundle (Figure 1.3).
The most characteristic feature of cyclins is that they are periodically synthesized and
degraded (Evans et al. 1983). Cyclin degradation requires a small sequence motif (the
destruction box) near the N-terminus of the cyclin, this sequence of 9 amino acids is also
very conserved (RXALGXIXN) (Glotzer et al., 1991). Cyclins are polyubiquitinated and
targeted for degradation by the proteasome. Ubiquitination is a complex mechanism that
requires at least three enzymes: the first enzyme (E1) activates the ubiquitin, the second
enzyme (E2) transfers the activated ubiquitin onto the target protein and finally the third
PSTAIREhelix
T-loop
N-terminal lobe
C-terminal lobe
Figure 1.2: Crystal structure of Cdk2. The two lobes of the kinase are easily identifiable, with the PSTAIRE helix in the N-terminal lobe and the T-loop linking the two lobes.

Introduction
6
enzyme (E3) or ubiquitin ligase is required for the transfer of the poly-ubiquitinated cyclin to
the proteasome (Coux et al., 1996).
The E3 ubiquitin ligase for all the mitotic cyclins is a very big protein complex, called
the Anaphase Promoting Complex or Cyclosome (APC/C), because it was identified as being
required for Anaphase and it was also noted that its activity is cyclic (King et al., 1995). The
APC is activated by MPF and its activity remains high until G1 (Peters, 2002).
Figure 1.4 shows the cyclins that have been identified: of the two Cyclin A isoforms,
A1 is required for testis differentiation (Sweeney et al., 1996) and A2 for S phase entry
(Rosenblatt et al., 1992; Strausfeld et al., 1996). Cyclins B1 and B2 have redundant functions
and are both necessary for the G2/M transition (Minshull et al., 1990). Cyclin C associates
with Cdk8 and plays a role in transcriptional regulation and in the G0 to G1 transition.
Cyclins D1, D2, D3 are involved in the G1/S transition and exit from G0 after growth factor
stimulation. They display similar functions but their expression is tissue specific (Lucibello et
al., 1993; Matsuoka et al., 1994). Cyclin E is involved in the G1 to S transition (Geng et al.,
1996; Lucibello et al., 1993). Cyclin F may be involved in the G2/M transition but its Cdk
partner is unknown (Kong et al., 2000). Cyclin G has been involved in apoptosis by regulating
p53 and Mdm2 (Okamoto et al., 2002). Cyclin H contributes to the CAK activity of Cdk7
(Makela et al., 1994; Tassan et al., 1995). Lately the cyclin family has grown exponentially,
with the cloning of the Cyclins I, K, L, T1 and T2, all playing putative roles in transcriptional
regulation.

Figure 1.3: Crystal structure of the cyclin box. The MRAIL motif establishes close contact withCdk2 .The 5 helices that form a bundle are easily recognizable.
Transcriptional regulationCdk9T1, T2
Cdk activation, transcriptional regulation?K
??I
CAK activity, transcriptional regulationCdk7H
DNA damage responseCdk5G1, G2
G2/M transition?F
G1/S transitionCdk2E
G0/G1 transitionCdk4, Cdk6D1, D2, D3
Transcriptional regulationCdk8C
G2/M transitionCdk1B1, B2
S phase entryCdk1, Cdk2A2
testis differentiationCdk1, Cdk2A1
FunctionCdk associatedCyclins
Figure 1.4: Diversity of cyclins and their putative functions. Based on Johnson and walker1999
MRAIL
Cyclin box Helicesbundle
7

Introduction
8
1.1.1.3 Mechanism of Cdk regulation. Cdks are regulated at multiple levels (Figure 1.5). First, by the accumulation of
cyclins, second at the level of cyclin–Cdk complex assembly and third, by specific
phosphorylation and dephosphorylation events (Desai et al., 1995; Morgan, 1995)
(Figure1.6A).
Additional regulation of the Cdks can occur by their association with inhibitory
proteins, the Cyclin dependent Kinase Inhibitors (CKI), that can either physically block
activation or block substrate/ATP access (Figure 1.6B).
The mechanism of Cdk activation was better understood after the co-crystallization of
the Cdk2-cyclin A complex (Brown et al., 1995). Cdk has two lobes. The smaller N-terminal
lobe is dominated by beta sheet and the PSTAIRE helix (major binding site for cyclins)
whereas the C-terminal lobe is mainly helical (Figure 1.5). The active site is located in a cleft
in between the two lobes. ATP binds deeply within the cleft. The protein substrate would
normally interact with the entrance of the active site cleft, but this region is occluded by the
T-loop in the monomeric Cdk2. The binding of Cyclin A induces major structural changes.
Several helices in the cyclin box contact both lobes in a region adjacent to the active site cleft,
and the T-loop is displaced allowing access to the substrate. When the Threonine (Thr) 160 is
phosphorylated by the CAK activity, the T-loop moves closer to the cyclin partner and this
region serves as key part of the binding sites for proteins substrates (Figure1.5).
PSTAIREhelix
Cyclin A
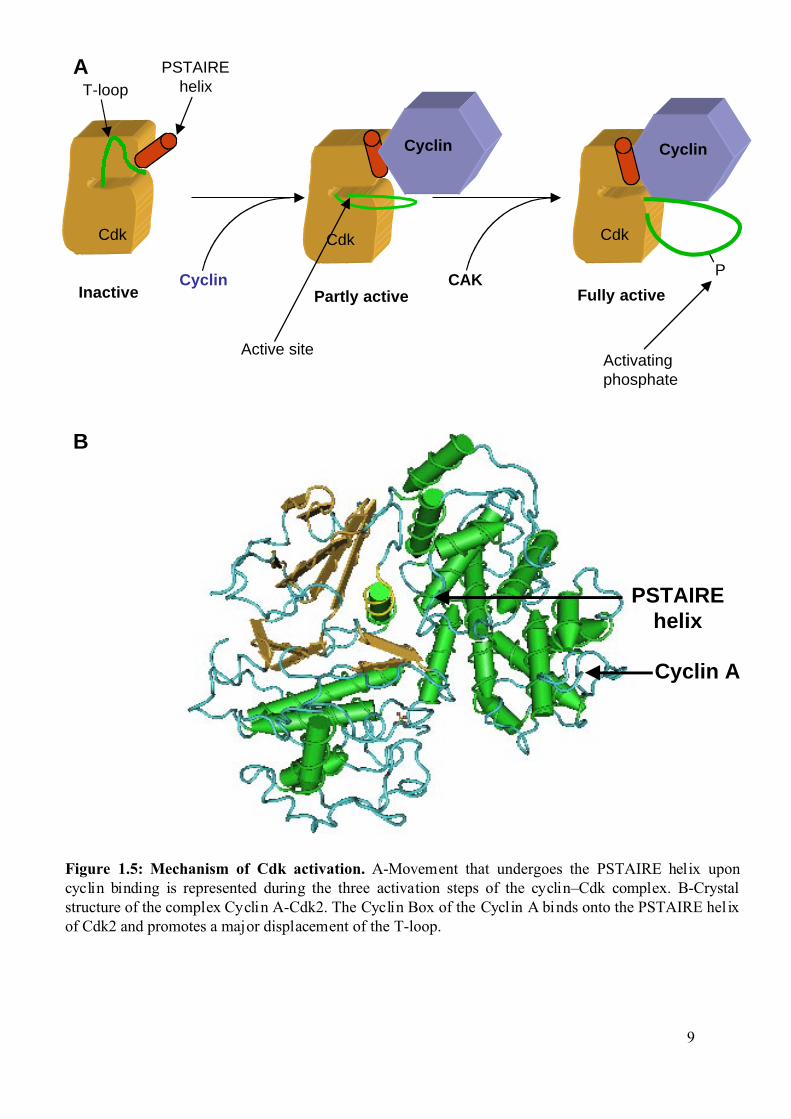
Figure 1.5: Mechanism of Cdk activation. A-Movement that undergoes the PSTAIRE helix uponcyclin binding is represented during the three activation steps of the cyclin–Cdk complex. B-Crystalstructure of the complex Cyclin A-Cdk2. The Cyclin Box of the Cyclin A binds onto the PSTAIRE hel ixof Cdk2 and promotes a major displacement of the T-loop.
9
Cyclin
CdkCdk
PSTAIREhelixT-loop
Inactive Fully active
P
Cyclin
Cdk
Cyclin CAK
Activating phosphate
Active site
Partly active
A
B

Introduction
10
1.1.1.4 The Cdk inhibitor family. CKIs inhibit the Cdk-cyclin complex; they exert their role mainly during the G1 to S
phase transition. There are three categories of CKIs. First, the Ink4 family which includes
Ink4a or p16, Ink4b or p15, Ink4c or p18 and Ink4d or p19. All these proteins share the
presence of ankyrin repeats and compete with cyclin D for the binding to the Cdk. Ink4
proteins are expressed in a cell-type-specific manner and are important regulators of the G1/S
transition. The second category of CKI is the Cip/Kip family, composed of p21/Cip, p27/Kip1
and p57/Kip2. These proteins share a homologous inhibitory domain and act by inhibiting the
Cdk in a stoichiometric manner. They inhibit all G1 Cdks, but especially Cdk2 (Gu et al.,
1992; Morgan, 1995; Zerfass-Thome et al., 1997) (Figure 1.6 B). However these inhibitors
are not as specific as the InK4 family as they also play a role in the G2 to M phase of the cell
cycle. The last family of inhibitors are the members of the Rb pocket protein family. There
are three members: Rb, p107, p130. They bind and modulate the activity of E2F transcription
factors and they can interact with Histone deacetylases, suggesting that they can also regulate
transcription by altering chromatin structure and availability of E2F. They are phosphorylated
by G1 cyclin-Cdk complexes, which impairs their ability to sequester E2F leading to the de-
repression of the E2F transcriptional activity (Brehm et al., 1999; Harbour and Dean, 2000;
Kaye, 2002; Nevins et al., 1997).
Fully active
P
Cyclin
Cdk
Inactive p27-cyclin- CdkComplex
P
Cyclin
Cdk
p27
p27
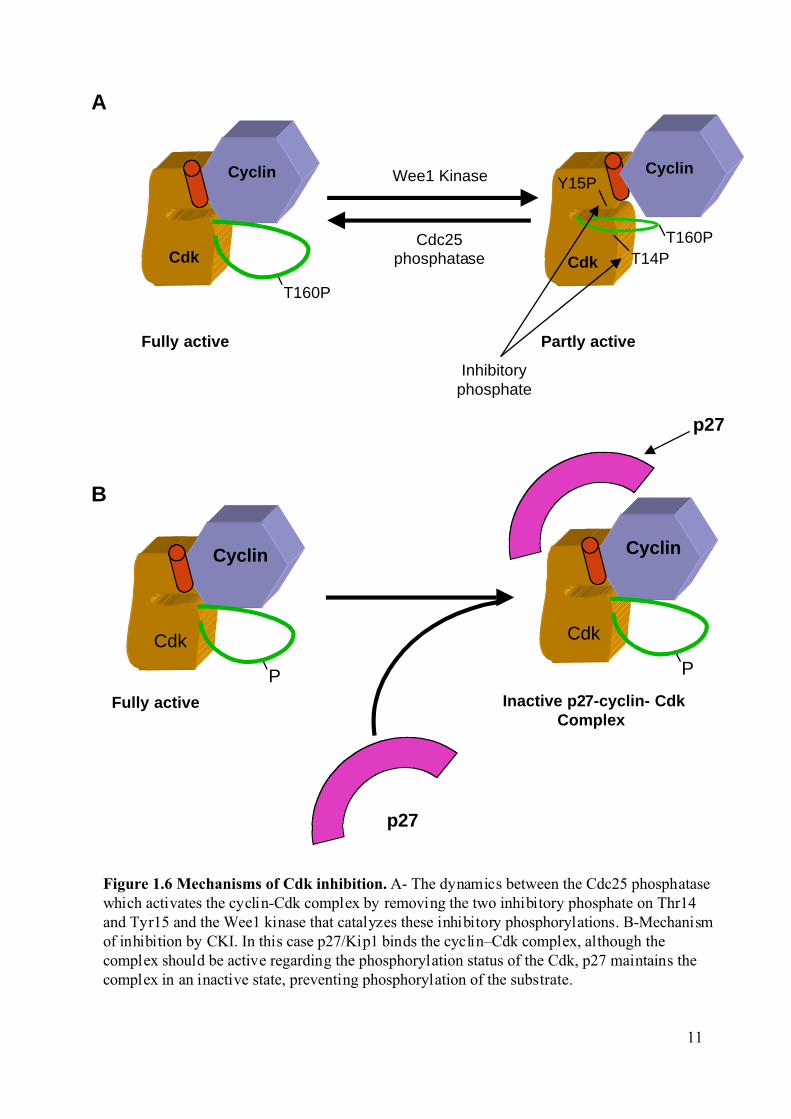
Figure 1.6 Mechanisms of Cdk inhibition. A- The dynamics between the Cdc25 phosphatasewhich activates the cyclin-Cdk complex by removing the two inhibitory phosphate on Thr14and Tyr15 and the Wee1 kinase that catalyzes these inhibitory phosphorylations. B-Mechanismof inhibition by CKI. In this case p27/Kip1 binds the cyclin–Cdk complex, although thecomplex should be active regarding the phosphorylation status of the Cdk, p27 maintains thecomplex in an inactive state, preventing phosphorylation of the substrate.
Cyclin
Cdk
Partly activeFully active
T160P
Cyclin
CdkCdc25
phosphatase
Wee1 Kinase
T14P
Y15P
Inhibitoryphosphate
T160P
A
B
11

Introduction
12
1.1.2 The G1/S transition and checkpoints. 1.1.2.1 The G1/S transition.
Mitogenic signals (growth factors) promote the assembly of active Cyclin D-Cdk4 or
Cyclin D-Cdk6, which results in a partial phosphorylation of Rb. The phosphorylation of Rb
by Cyclin D-Cdk4/6 enable it to sequester E2F and de-repress E2F transcriptional activity
(Brehm et al., 1999; Harbour and Dean, 2000; Kaye, 2002; Nevins et al., 1997). Cyclin E then
starts to be synthesized, as the promoter is under the control of E2F.
The synthesis of Cyclin D promotes cell-cycle progression by two mechanisms, one
direct (Cdk4/6 activation) and one indirect (CKI exchange). Indeed, progression into S phase
depends largely on activation of Cyclin E-Cdk2 that occurs via transcription of Cyclin E after
E2F released from Rb and a mechanism known as CKI exchange. Increased synthesis of D-
type cyclin by c-Myc, leads to the removal of Kip1 from Cyclin E-Cdk2 and the concomitant
activation of the Cyclin E-Cdk2 complexes.
Because Cyclin D genes are transactivated by c-Myc, the resulting accumulation of
Cyclin D leads to a positive feed back-loop and redistribution of Kip from Cyclin E-Cdk2 to
Cyclin D-Cdk4/6. When the level of cyclin E exceeds that of Cip/Kip, Cyclin E-Cdk2
phosphorylates Kip1 on Thr187, and it is then targeted for degradation. After Kip degradation
the activity of Cyclin E-Cdk2 increases again leading to hyper-phosphorylation of Rb and
Cyclin A expression. High levels of Cyclin A lead to competition with Cyclin E for the Cdk2
kinase. In late S phase, Cyclin A will associate with Cdk2 and the free Cyclin E is targeted for
degradation. Although synthesis of Ink4 is poorly understood, it is known that in late S phase
InK4 binds Cyclin D again and the activity of Cyclin D-Cdk4 decreases.
The Cyclin A-Cdk2 complex phosphorylates a subunit of the APC called Cdh1 and
this phosphorylation is inhibitory for APC activity. APC inhibition leads to an increased
expression of Cyclin B which previously was actively degraded by the APC. Cyclin B will
then associate with Cdk1 to promote M phase entry.
The G1/S transition depends largely on one key activator, the phosphatase Cdc25A. In
early S phase Cdk2 is phosphorylated on two inhibitory residues the threonine 14 (Thr14) and
tyrosine 15 (Tyr15) by Wee1 kinase (Booher et al., 1997). When the Cyclin A-Cdk2 complex
starts to be active, it will phosphorylate Cdc25A leading to a second positive feed-back loop
(Bartek and Lukas, 2001a; Bartek and Lukas, 2001b). Interestingly, Cdc25A is positively
regulated by c-Myc at the transcriptional level. The c-Myc transcription factor is also under
the control of E2F.

Introduction
13
To summarize, the G1/S transition is complex, requiring positive (phosphorylation of
Rb) and negative (degradation of Kip and repression of the Cyclin D-Cdk4/6 complex) feed-
back loops. In addition, this transition also requires synthesis of positive activators like Cyclin
E, Cyclin A, c-Myc and modulation of positive (Cdc25A) and negative regulators (Wee1)
(Donjerkovic and Scott, 2000; Zarzov et al., 2002). A schematic representation of the G1/S
transition is presented in figure 1.7
1.1.2.2 The G1/S checkpoints. The genetic material of the cell is always very susceptible to damage, for example
agents like reactive oxygen intermediates, UV or inaccurate DNA replication are sources of
trauma for the cell. When damage occurs, it is crucial for the cell to be able either to repair it
or to stop proliferation. These mechanisms of surveillance ensure that the mistakes will not be
transmitted to the daughter cells. They are called checkpoints and are present at G1/S and
G2/M transitions and also operate in M phase (Bartek and Lukas, 2001b; Bulavin et al., 2002;
Hoyt, 2001; Musacchio and Hardwick, 2002; Nurse, 1997; Zarzov et al., 2002).
During the G1/S transition, the first step involves activation of the so called “sensor”
proteins that are still poorly characterised. The current idea is that these proteins probably
scan the DNA and when a defect is found they will activate, by an unknown mechanism, the
phosphoinositide 3-kinase related kinase (PIKK) family of proteins. In mammals, there are
two PIKK members: ATM (atxia-telangiectasia mutated) and ATR (ATM and Rad 3-related)
(Abraham, 2001; Falck et al., 2002).
Activation of ATM or ATR depends on the nature of the DNA damage (Abraham,
2001; Schwab et al., 2001). They act by two mechanisms: a fast one, which is a primary
response after DNA damage which targets Cdc25A for degradation by the checkpoint proteins
Chk1(if ATR is activated) or Chk2 (if ATM is activated) (Bartek et al., 2001; Mailand et al.,
2000). Degradation of Cdc25A leads to an increase of inactive Cdk2 phosphorylated on Thr14
and Tyr15. The second pathway activated by PIKKs involves phosphorylation of p53 (a
transcription factor). The checkpoint proteins Chk1 or Chk2 are phosphorylated and activated
by ATM or ATR. Chk1 or Chk2 will in turn phosphorylate p53 on serine (Ser) 20, which will
increase the stability and therefore increase the level of p53 (Bartek et al., 2001; Winters,
2002). Then p53 activates numerous genes, among them Cip1, which leads to a G1 block by
inhibiting Cdk2.
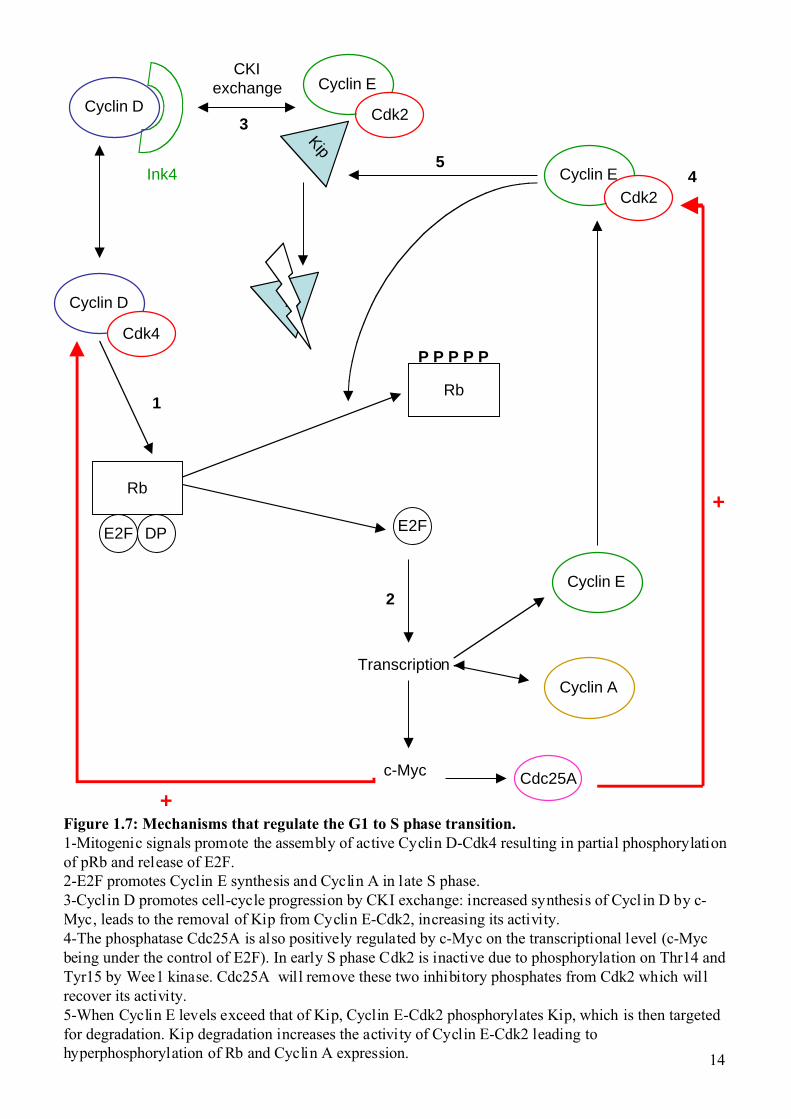
14
Cyclin E
Cdk2
Ink4
Cyclin D
Cyclin D
Cdk4
RbKip
CKIexchange
E2F DP
Rb
E2F
P P P P P
Transcription
Cyclin E
Cyclin A
c-Myc
+
Cyclin E
Cdk2
Cdc25A
+Kip
1
2
3
54
Figure 1.7: Mechanisms that regulate the G1 to S phase transition.1-Mitogenic signals promote the assembly of active Cyclin D-Cdk4 resulting in partial phosphorylationof pRb and release of E2F.2-E2F promotes Cyclin E synthesis and Cyclin A in late S phase.3-Cyclin D promotes cell-cycle progression by CKI exchange: increased synthesis of Cycl in D by c-Myc, leads to the removal of Kip from Cyclin E-Cdk2, increasing its activity.4-The phosphatase Cdc25A is also positively regulated by c-Myc on the transcriptional level (c-Mycbeing under the control of E2F). In early S phase Cdk2 is inactive due to phosphorylation on Thr14 andTyr15 by Wee1 kinase. Cdc25A will remove these two inhibitory phosphates from Cdk2 which willrecover its activity.5-When Cyclin E levels exceed that of Kip, Cyclin E-Cdk2 phosphorylates Kip, which is then targetedfor degradation. Kip degradation increases the activity of Cyclin E-Cdk2 leading tohyperphosphorylation of Rb and Cyclin A expression.

Introduction
15
1.1.3 The G2/M transition and checkpoints. 1.1.3.1 The G2/M transition.
As mentioned previously, at the end of S phase the Cyclin A-Cdk2 complex
phosphorylates Cdh1 (a subunit of the APC). This inhibitory phosphorylation leads to the
accumulation of Cyclin B that was actively degraded by the APC during S phase. Cyclin B
will then associate with Cdk1.
Once the Cyclin B-Cdk1 complex forms, it is immediately inactivated by
phosphorylation on the Thr14 and Tyr15 of Cdk1 by the Wee1 kinase. This complex can be
activated by the Cdc25C phosphatase which removes the two inhibitory phosphates (for
review see Bulavin et al., 2002). In turn the Cyclin B-Cdk1 complex will phosphorylate
Cdc25C and increase its activity (Izumi and Maller, 1993; Kumagai and Dunphy, 1991). A
crucial parameter in this transition is the subcellular localisation of these proteins (Takizawa
and Morgan, 2000). Cyclin A is mainly nuclear from its synthesis to its degradation
(metaphase), but Cyclin B is mainly cytoplasmic, due to active nuclear export. Cyclin B is
translocated to the nucleus at the beginning of mitosis (Jin et al., 1998). Cdc25C also remains
cytoplasmic during interphase, because it is bound to the small acidic 14-3-3 protein
(Kumagai et al., 1998), which inhibits its nuclear import. At the beginning of mitosis Cdc25C
becomes nuclear after dephosphorylation of the 14-3-3 binding site (Kumagai and Dunphy,
1997; Lammer et al., 1998). In a second step, the polo like kinase (Plk1) (Golsteyn et al.,
1996) phosphorylates Cdc25C, enhancing its activity. The upstream activator of Plk1 is still
unknown, but it is known that Cyclin B-Cdk1 can also phosphorylate and enhance the activity
of Cdc25C (Izumi and Maller, 1993; Kumagai and Dunphy, 1991). This network of
phosphorylations is interconnected and is part of a positive feed-back loop, which makes
difficult to know what the initial activatory events are.
When both Cdc25C and Cyclin B are in the nucleus, the dephosphorylation of Thr14
and Tyr15 leads to activation of Cyclin B-Cdk. Cyclin B-Cdk1 is thought to phosphorylate
components of the nuclear envelope to promote nuclear membrane disassembly. A schematic
representation of the G2/M transition is presented in figure 1.8
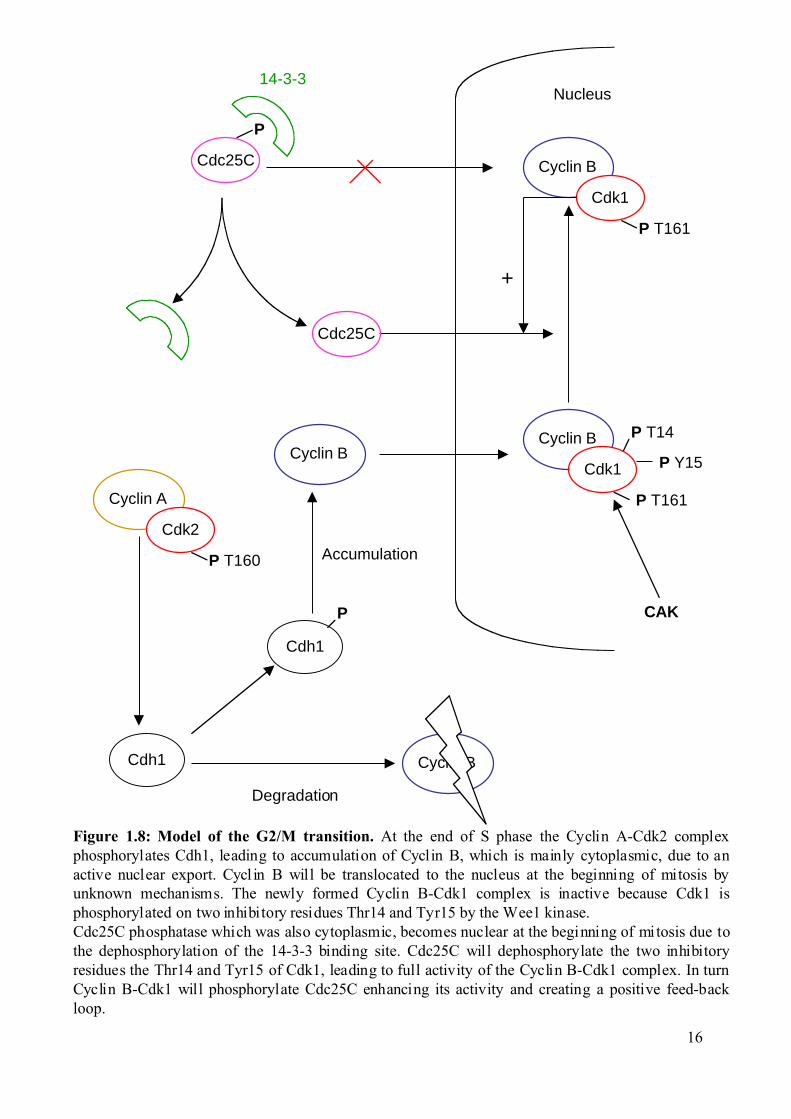
Nucleus
Cyclin B
Cyclin B
Cdk1
Cdk1
P T14
P Y15
P T161
P T161
CAK
Cdc25C
P
14-3-3
Cdc25C
Cyclin A
Cdk2
P T160
Cdh1
Cdh1
P
Cyclin B
Cyclin B
Degradation
Accumulation
+
16
Figure 1.8: Model of the G2/M transition. At the end of S phase the Cyclin A-Cdk2 complexphosphorylates Cdh1, leading to accumulation of Cyclin B, which is mainly cytoplasmic, due to anactive nuclear export. Cycl in B will be translocated to the nucleus at the beginning of mitosis byunknown mechanisms. The newly formed Cyclin B-Cdk1 complex is inactive because Cdk1 isphosphorylated on two inhibitory residues Thr14 and Tyr15 by the Wee1 kinase.Cdc25C phosphatase which was also cytoplasmic, becomes nuclear at the beginning of mitosis due tothe dephosphorylation of the 14-3-3 binding site. Cdc25C will dephosphorylate the two inhibitoryresidues the Thr14 and Tyr15 of Cdk1, leading to full activity of the Cyclin B-Cdk1 complex. In turnCyclin B-Cdk1 will phosphorylate Cdc25C enhancing its activity and creating a positive feed-backloop.

Introduction
17
1.1.3.2 The G2/M checkpoints and exit from mitosis. To maintain integrity of the genome, cells must be able to prevent progression through
the cell cycle when their DNA is not properly replicated; this is achieved by the G2/M
checkpoint (Bulavin et al., 2002; Mailand et al., 2002; O'Connell et al., 2000; Winters, 2002).
The G2/M checkpoint depends on Chk1, if any defect is detected Chk1 will phosphorylate
Cdc25C on Ser216 (Chk2 can also do this depending of the kind of defect). This
phosphorylation provides a binding site for the small acidic 14-3-3 protein (Kumagai et al.,
1998), which inhibits the nuclear import of Cdc25C. Interestingly, this phosphorylation can
also be done by the c-TAK1 kinase and the cAMP dependent protein kinase in vitro, but the
relevance of these kinases for G2/M regulation in vivo is still unknown. Activation of p38
MAP kinase, which inhibits Cdc25B by phosophorylation on Ser309 (Bulavin et al., 2002),
may be also involved in the G2/M block. Indeed Cdc25B is involved in the dephosphorylation
and activation of the cytoplasmic Cdk1.
The spindle checkpoint prevents cells from entering anaphase until all chromosomes
are properly aligned. Kinetochores have been implicated as the source of checkpoint
signalling. The mechanical tension at the kinetochore can determine whether or not the
checkpoint is initiated (Hoyt, 2001; Musacchio and Hardwick, 2002).
Mitotic exit requires sister chromatid separation, spindle disassembly and cytokinesis
(Karsenti and Vernos, 2001; Nasmyth, 2002). First cyclins are targeted for destruction by the
ubiquitin proteasome pathway. To initiate mitosis exit, Securin is targeted for degradation by
the APC/C (Gieffers et al., 2001; King et al., 1995; Peters, 2002; Yang, 2002). The protease
Separase is kept inactive by its interaction with Securin, but when the interaction is broken
Separase will become active and degrade the Cohesin protein complex. Cohesin is “the glue”
of the sister chromatids. The sister chromatids start to separate and anaphase ends when the
two pools of chromosomes go to each pole of the spindle.

Introduction
18
1.2 The meiotic cell cycle. In sexual reproduction, the diploid germ cells divide by meiosis to produce haploid
gametes that upon fertilization will give rise to a new organism with a diploid genome
(Murray and Hunt, 1993). Cells undergo a special division cycle with two subsequent M-
phases without an intervening S-phase. As a consequence, in spermatogenesis, four daughter
cells are generated that are different from the parent cell, although in oogenesis one oocyte
and two polar bodies are produced.
Meiosis is composed of two successive M-phases, which reduce the ploidy level from
4n to n. Both divisions in meiosis are divided into the same phases as mitosis and most of the
differences between the processes occur during meiosis I (MI). At prophase of MI, the
homologue chromosomes pair to form a bivalent and recombination occurs by crossing-over,
leaving several attachment points between the two homologues called the chiasmata. As in
mitotic prophase, the chromosomes condense and the nuclear membrane disassembles. After
assembly of the spindle apparatus in prometaphase I, the paired homologue chromosomes
align at the equator of the spindle in metaphase I. During anaphase I, the chiasmata is
dissolved and the homologue chromosomes are segregated. At telophase I only one set of
chromosomes remains in each daughter cell and the nuclear membrane forms. Meiosis II
(MII) starts with the disassembly of the nuclear membrane formed in telophase I and spindle
is formed.
Meiosis II is very similar to mitosis and undergoes the same series of phases
(prophase, metaphase, anaphase and telophase) and also leads to the segregation of the sister
chromatids. Unlike mitosis, the final products of the meiosis II division are haploid cells.
1.2.1 Meiotic maturation of Xenopus oocytes as a model system. Oocytes of the South African clawed frog Xenopus laevis (Xenopus) provide an
excellent model system for studying the biochemical mechanisms regulating the G2/M
transition (Maller et al., 1989; Maller, 1990; Schmitt and Nebreda, 2002b).These oocytes are
big (about 1,3 mm in diameter) and can easily be maintained in culture and manipulated by
microinjection of mRNA or protein. The fully-grown Xenopus oocytes (stage VI) are easily
identifiable by their specific pigmentation pattern, which define the radial symmetry of the
oocytes. The animal hemisphere is brown, whereas the vegetal hemisphere is weakly
pigmented and appears white (Figure 1.9A).

Introduction
19
The fully-grown Xenopus oocytes are arrested at the G2/M boundary of the first
meiotic division. Progesterone produced by the follicular cells surrounding the oocyte in vivo,
or added to the culture media in vitro, releases the G2 arrest in a process called meiotic
maturation (Baulieu et al., 1978; Godeau et al., 1978; Schorderet-Slatkine et al., 1978).
Approximately 4 to 8 hrs after incubation with progesterone, the nucleus of the G2 arrested
oocytes, called germinal vesicle, disaggregates and the oocyte enters meiosis I. This is called
Germinal Vesicle Break Down (GVBD) (Figure 1.9A). The first meiotic spindle forms, and
one polar body is extruded. After a transient reformation of the nucleus due to a drop in MPF
activity (Daar et al., 1991; Masui and Markert, 1971), the second meiotic spindle forms
perpendicular to the animal pole of the oocyte and the oocyte remains blocked at metaphase II
of the second division, waiting for fertilization. This is called Cytostatic Factor (CSF) arrest
(Erikson and Maller, 1989; Maller et al., 2002; Masui, 2001) (Figure 1.9A).
1.2.2 Signal transduction pathways that trigger meiotic
maturation. Resumption of meiosis is caused by activation of several transduction pathways that
together lead to MPF activation (Schmitt and Nebreda, 2002b). It is important to note that
MPF is activated both in meiosis I and meiosis II, but there is a transient drop of activity
between the two divisions (Figure 1.9B). MPF was first found in the cytoplasm of
progesterone-treated oocytes as an activity able to induce meiotic maturation when injected
into stage VI Xenopus oocytes in the absence of progesterone (Masui, 2001; Shibuya and
Masui, 1989a; Shibuya and Masui, 1989b). MPF is active only in M phase and is a complex
of a catalytic subunit, Cdk1 (also called Cdc2) and a regulatory subunit, a B-type cyclin. In
Xenopus oocytes about 80 to 90% of Cdk1 is found as a monomer. The remaining 10-20 % is
complexed with Cyclin B2 or Cyclin B5 (Hochegger et al., 2001; Kobayashi et al., 1991a;
Kobayashi et al., 1991b). This complexed form is inactive, due to the inhibitory
phosphorylation of Cdk1 on Thr14 and Tyr15. This pre-formed inactive Cdk1-Cyclin B
complex is called pre-MPF. Members of the Wee1 kinase family catalyse the inhibitory Cdk1
phosphorylations. Wee1 is absent from G2 arrested oocytes but Myt1 (a Wee1-related protein
kinase) is likely to be the kinase responsible for the formation of pre-MPF. Two pathways can
activate pre-MPF, Myt1 is inhibited by phosphorylation catalysed by Rsk (Palmer et al.,
1998) and Cdc25C can be activated by the Xenopus polo homologue (Plx1) (Gross et al.,
2001).
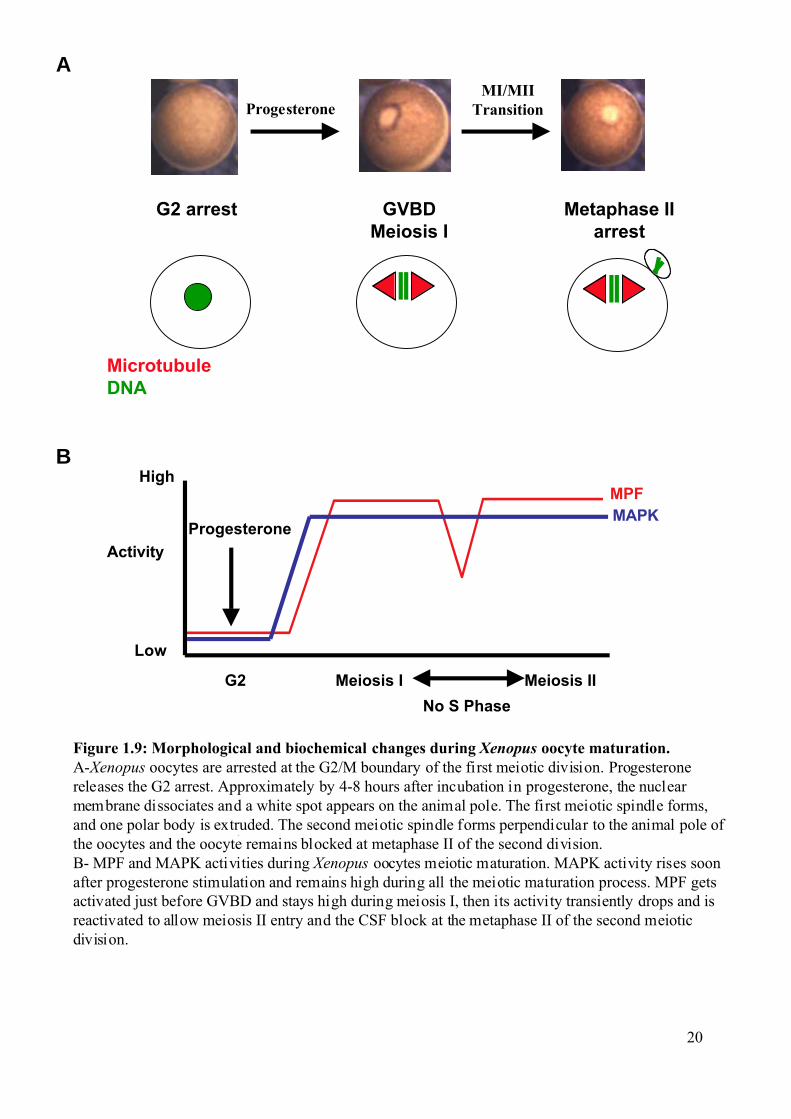
G2 arrest GVBDMeiosis I
Metaphase IIarrest
ProgesteroneMI/MII
Transition
MicrotubuleDNA
Activity
MPFMAPK
High
Low
G2 Meiosis I Meiosis II
No S Phase
Progesterone
Figure 1.9: Morphological and biochemical changes during Xenopus oocyte maturation.A-Xenopus oocytes are arrested at the G2/M boundary of the first meiotic division. Progesteronereleases the G2 arrest. Approximately by 4-8 hours after incubation in progesterone, the nuclearmembrane dissociates and a white spot appears on the animal pole. The first meiotic spindle forms,and one polar body is extruded. The second meiotic spindle forms perpendicular to the animal pole ofthe oocytes and the oocyte remains blocked at metaphase II of the second division.B- MPF and MAPK activities during Xenopus oocytes meiotic maturation. MAPK activity rises soonafter progesterone stimulation and remains high during all the meiotic maturation process. MPF getsactivated just before GVBD and stays high during meiosis I, then its activity transiently drops and isreactivated to allow meiosis II entry and the CSF block at the metaphase II of the second meioticdivision.
A
B
20

Introduction
21
1.2.3 Activation of the Mos-MAPK pathway during and inhibition of DNA
replication . Stimulation of Xenopus oocytes by progesterone initiates maturation probably by
binding to a recently identified 7-transmembrane G-protein coupled receptor (GPCR) (Zhu et
al., 2003a; Zhu et al., 2003b). Progesterone binds to this GPCR, which results in inhibition of
the adenylyl cyclase activity present in the G2 arrested oocytes and therefore a decrease in
the cAMP level and inactivation of PKA (Maller et al., 1979).
PKA blocks oocytes in G2 (Maller and Krebs, 1977) and it has been shown to
phospshorylate the Ser287 of Cdc25C, which is the 14-3-3 binding site for the Xenopus
Cdc25C (Duckworth et al., 2002). PKA can also inhibit oocyte meiotic maturation in a kinase
independent manner (Schmitt and Nebreda, 2002a).
Progesterone leads to the recruitment of specific mRNAs onto polysomes and their
translation is activated (de Moor and Richter, 1999; Groisman et al., 2002; Mendez et al.,
2002; Wu et al., 1998). Indeed, inhibitors of translation like cycloheximide (CHX) completely
block progesterone-induced maturation, showing that translation, unlike transcription is
essential for oocyte meiotic maturation.
Progesterone also leads to the activation of the kinase Eg2. This has been proposed to
be an early event that correlates with translation of the Mos proto-oncogene mRNA. The level
of Mos protein remains high throughout the meiotic maturation process. The injection of Mos
into Xenopus oocytes is sufficient to trigger GVBD (Haccard et al., 1995), but a recent study
proposes that Mos is only essential to maintain the metaphase II block and prevent
parthenogenic activation of the oocytes (Dupre et al., 2002). Mos activates MEK1 by
phosphorylation, which in turn triggers the activation of MAPK. MAPK phosphorylates Rsk
and Rsk phosphorylates and inhibits the Cdk1 inhibitory kinase Myt1.
As previously mentioned, it is crucial that no DNA replication occurs between meiosis
I and meiosis II in order to create haploid cells. However, MPF activity drops during the
meiosis I to meiosis II transition (Figure 1.9B). How can DNA replication be repressed if
MPF is low? One hypothesis is that the Mos-MAPK-Rsk pathway plays an important role in
the S phase omission after meiosis I (Furuno et al., 1994; Gross et al., 2000). However, it is
not clear whether this could be a direct effect on the DNA replication machinery or if it is just
the ability of this signalling pathway to participate in MPF re-activation after meiosis I. MPF
re-activation is driven by synthesis of B-type cyclins (Hochegger et al., 2001). In addition,
the absence of Wee1 inhibition has also been proposed to be important for S phase omission

Introduction
22
(Nakajo et al., 2000; Ohsumi et al., 1994). This suggests that the low level of MPF activity
that remains after meiosis I is still sufficient in combination with the Mos-MAPK pathway to
prevent DNA replication. Consistent with this hypothesis, Xenopus oocytes injected with
cyclin B antisense oligonucleotides or incubated with the MAPK kinase inhibitor U0126, can
enter meiosis I normally, as judged by both biochemical and cytological markers, but then
degenerate and fail to arrest at metaphase II. The collapse of meiotic maturation in these cases
correlates with the inability to re-activate MPF after meiosis I and with the inactivation of
MAPK (Dupre et al., 2002; Furuno et al., 1994; Gross et al., 2000).
The ability of indestructible B-type cyclins to allow oocytes treated with CHX at
meiosis I to progress into meiosis II also supports the idea that MPF is the final target of the
pathway (Hochegger et al., 2001). But since Cyclin B accumulation, MPF re-activation and
spindle assembly are all intimately associated; it is difficult to favour one particular
mechanism. Recent work has also shown that the absence of the pre-replication complex
protein Cdc6 and the cytoplasmic delocalisation of Origin of Replication Complex (ORC)
protein and Cdc7 kinase are responsible for the loss of DNA replication competence in
oocytes (Lemaitre et al., 2002; Whitmire et al., 2002).
Figure 1.10, summarises the interplay between several proteins that participate in
oocyte meiotic maturation.
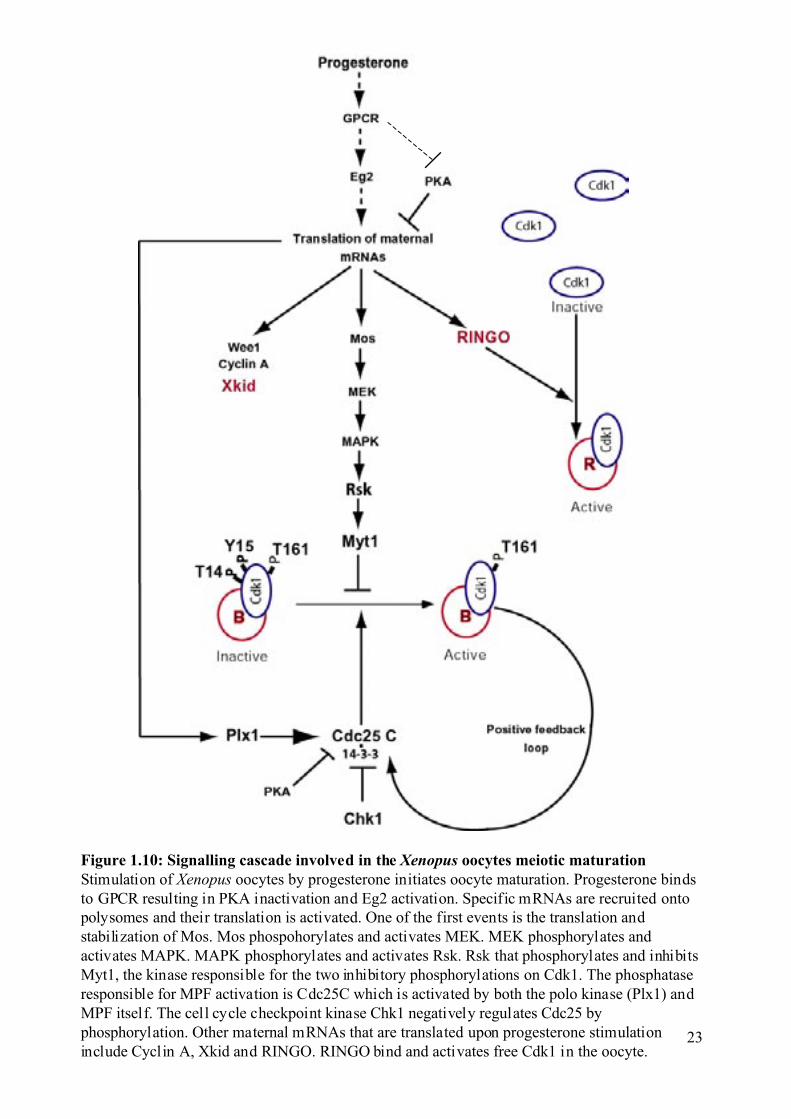
Figure 1.10: Signalling cascade involved in the Xenopus oocytes meiotic maturationStimulation of Xenopus oocytes by progesterone initiates oocyte maturation. Progesterone bindsto GPCR resulting in PKA inactivation and Eg2 activation. Specific mRNAs are recruited ontopolysomes and their translation is activated. One of the first events is the translation andstabilization of Mos. Mos phospohorylates and activates MEK. MEK phosphorylates andactivates MAPK. MAPK phosphorylates and activates Rsk. Rsk that phosphorylates and inhibitsMyt1, the kinase responsible for the two inhibitory phosphorylations on Cdk1. The phosphataseresponsible for MPF activation is Cdc25C which is activated by both the polo kinase (Plx1) andMPF itself. The cell cycle checkpoint kinase Chk1 negatively regulates Cdc25 byphosphorylation. Other maternal mRNAs that are translated upon progesterone stimulationinclude Cyclin A, Xkid and RINGO. RINGO bind and activates free Cdk1 in the oocyte.
23

Introduction
24
1.2.4 Xkid, a chromokinesin involved in chromosome alignment. Xkid was found in a screen performed in our laboratory, the aim of which was to
identify proteins synthesized de novo during Xenopus oocyte maturation and therefore with a
potential role in meiotic regulation. The screen was based on the differential association of
mRNAs with polysomes in progesterone treated versus non treated oocytes. One of the
positive clones was used to screen a Xenopus cDNA oocyte library from which two full-
length cDNAs were isolated, clone 8-2b and clone 8-5, containing ORFs of 650 and 651
amino acids respectively. The proteins encoded by these ORFs are very similar along their
whole length with 92% identity at the amino acid level suggesting that they might correspond
to pseudoalloploid alleles in the tetraploid Xenopus genome (Kobel and Du Pasquier, 1979).
They shared a high sequence identity with a human cDNA encoding the chromokinesin kid
and therefore were named Xkid (Antonio et al., 2000).
Analysis of the amino acid sequence revealed an N-terminus motor domain
characteristic of the kinesin super family. Furthermore, kid was recently proposed to be a
plus-end directed motor (Yajima et al., 2003); therefore, it is likely that Xkid also moves to
the plus-end of the microtubules. Outside the motor domain Xkid is also similar to human kid.
They have a short stretch of 50 amino acids predicted to form coiled coil interactions. Xkid
and kid also share a helix-hairpin-helix (HhH) DNA binding domain at their C-termini. A
construct comprised of the 90 C-terminal amino acids of Xkid containing the two repeats of
the HhH DNA binding domain was shown to be capable of binding DNA (Antonio, 2002).
Xkid localizes to mitotic chromosomes, both in spindles assembled in tissue culture
cells and in Xenopus egg extracts. Analysis of the anti-Xkid staining pattern on chromosomes
revealed an even distribution of Xkid throughout the chromosome arms (Antonio, 2002).
Depletion and antibody addition studies have revealed that Xkid is required for mitotic
chromosome congression in spindles assembled in Xenopus egg extracts. When the activity of
Xkid is inhibited by blocking antibodies or when Xkid is immunodepleted, spindles assemble
normally but the chromosomes fail to align at the metaphase plate and instead are stretched
throughout the spindle. At anaphase, Xkid must disappear to allow sister chromatid
movement to the spindle poles. Funabiki and Murray showed that Xkid is ubiquitinated and
degraded at anaphase in Xenopus egg extracts and they proposed that the APC complex is
responsible for this degradation. Moreover, they showed that a non-degradable form of Xkid
allowed sister chromatid separation but prevented their poleward movement suggesting that

Introduction
25
Xkid degradation/inactivation is required for sister chromatid migration to the poles at
anaphase (Funabiki and Murray, 2000).
1.2.5 RINGO triggers meiotic maturation in Xenopus oocytes. To identify novel proteins involved in G2/M progression during the meiotic
maturation of Xenopus oocytes, our laboratory used an expression cloning strategy. A
Xenopus oocyte cDNA library was subdivided into smaller pools and in vitro transcribed. The
mRNA pools that upon injection in oocytes were able to induce maturation on their own were
subdivided until single positive clones were isolated. Using this approach, two clones named
ls26 and ls27 were isolated, which, were able to induce oocyte maturation in the absence of
progesterone stimulation (Ferby et al., 1999).
Maturation induced by ls26 or ls27 correlated with the activation of both MAPK and
pre-MPF. The ls26 and ls27 ORFs encoded highly related proteins of 300 and 298 amino
acids respectively, which were 88% identical. At that time, searches with the ls26 and ls27
sequences using BLAST against DNA and protein databases did not detect significant
homologies with any other known DNAs or proteins, suggesting that ls26/ls27 belong to a
novel protein family. The close similarity between the sequences of ls26 and ls27 suggests
that they might correspond to pseudoalloploid alleles (Kobel and Du Pasquier, 1979). These
proteins were named RINGO, which stands for Rapid INducer of G2/M progression in
Oocytes.
RINGO induces oocyte maturation in the presence of a protein synthesis inhibitor,
indicating that RINGO act late in the signalling pathways that lead to pre-MPF activation.
When the kinetics of activation for MAPK and pre-MPF were investigated, RINGO was
found to rapidly activate in oocytes Histone H1 kinase activity (H1K) (a marker of MPF
activity) before myelin basic protein (MBP) kinase activity (a marker of MAPK activity).
Thus, the function of RINGO is more likely to be related to the activation of Cdk1 rather than
of MAPK. Moreover, when RINGO antisense oligonucleotides were injected into oocytes a
strong delay in progesterone induced oocyte maturation was observed, meaning that the
protein has a function in the process of oocyte maturation. In the light of these results, the
possibility that RINGO could directly associate with the pre-MPF complex was investigated.
Indeed, RINGO was able to bind and activate Cdk1 and Cdk2 in vitro and in cell free extracts
(Ferby et al., 1999; Karaiskou et al, 2001). Interestingly, a similar protein was independently
cloned by another group in a screen for proteins that complement a rad1 mutant of
Schizosaccharomyces pombe and it was called ‘Speedy’ (Lenormand et al., 1999). The same

Introduction
26
group recently reported the identification and characterization of a novel cell cycle gene name
Human Speedy (Spy1) which is 40% homologous to the Xenopus RINGO.
1.3 Aim of the work. The PhD work has addressed two questions. First, the chromokinesin Xkid has been
shown both to be synthesized upon progesterone stimulation and to play a crucial role in
chromosome alignment (Antonio et al., 2000; Funabiki and Murray, 2000). Therefore, we
decided to investigate the putative role of Xkid during the meiotic maturation of Xenopus
oocytes. This work was done in collaboration with Celia Antonio from the laboratory of
Isabelle Vernos at EMBL. She performed the confocal microscopy analysis of Xkid depleted
oocytes.
The second part of the work presented here aimed to investigate the mechanism of
Cdk regulation by RINGO and the identification and characterization of mammalian
homologues of RINGO. We indeed found that there are at least four RINGO protein
homologues in mammals. We have used in vitro assays and the Xenopus system to investigate
their biochemical properties. We have also characterized some of these RINGO homologues
in mammalian cell lines.
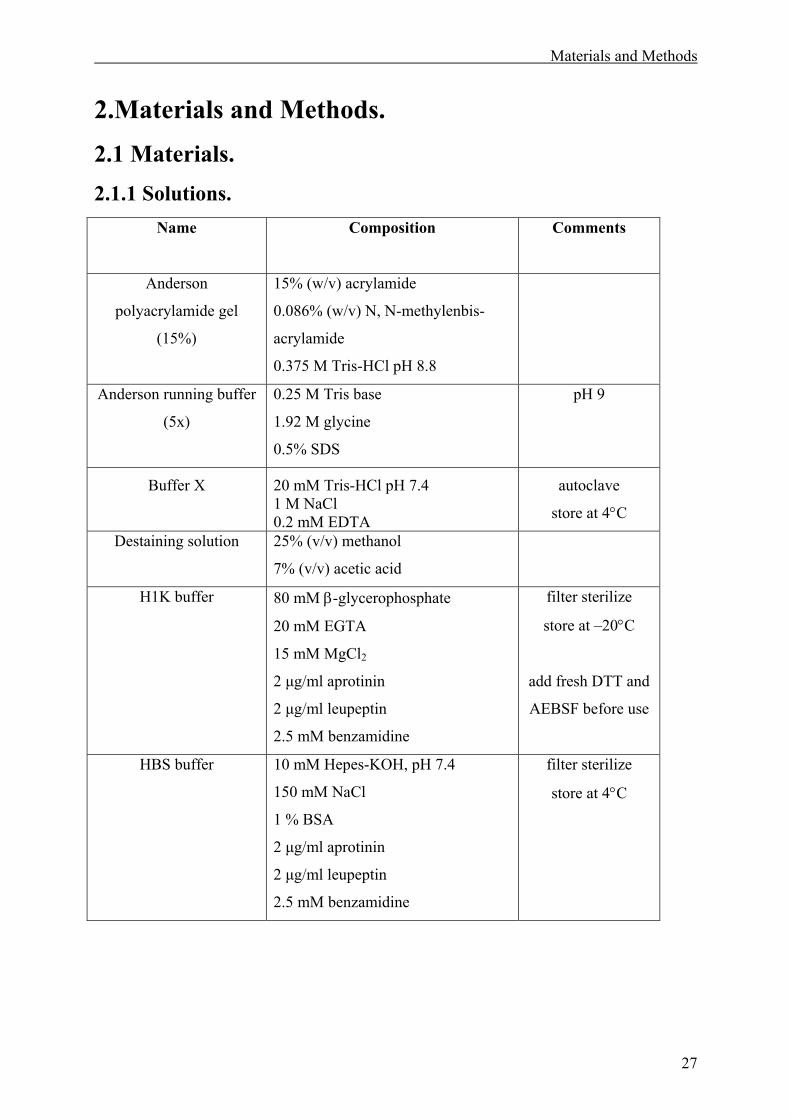
Materials and Methods
27
2.Materials and Methods. 2.1 Materials. 2.1.1 Solutions.
Name
Composition
Comments
Anderson
polyacrylamide gel
(15%)
15% (w/v) acrylamide
0.086% (w/v) N, N-methylenbis-
acrylamide
0.375 M Tris-HCl pH 8.8
Anderson running buffer
(5x)
0.25 M Tris base
1.92 M glycine
0.5% SDS
pH 9
Buffer X 20 mM Tris-HCl pH 7.4 1 M NaCl 0.2 mM EDTA
autoclave
store at 4°C
Destaining solution 25% (v/v) methanol
7% (v/v) acetic acid
H1K buffer 80 mM β-glycerophosphate
20 mM EGTA
15 mM MgCl2
2 µg/ml aprotinin
2 µg/ml leupeptin
2.5 mM benzamidine
filter sterilize
store at –20°C
add fresh DTT and
AEBSF before use
HBS buffer 10 mM Hepes-KOH, pH 7.4
150 mM NaCl
1 % BSA
2 µg/ml aprotinin
2 µg/ml leupeptin
2.5 mM benzamidine
filter sterilize
store at 4°C
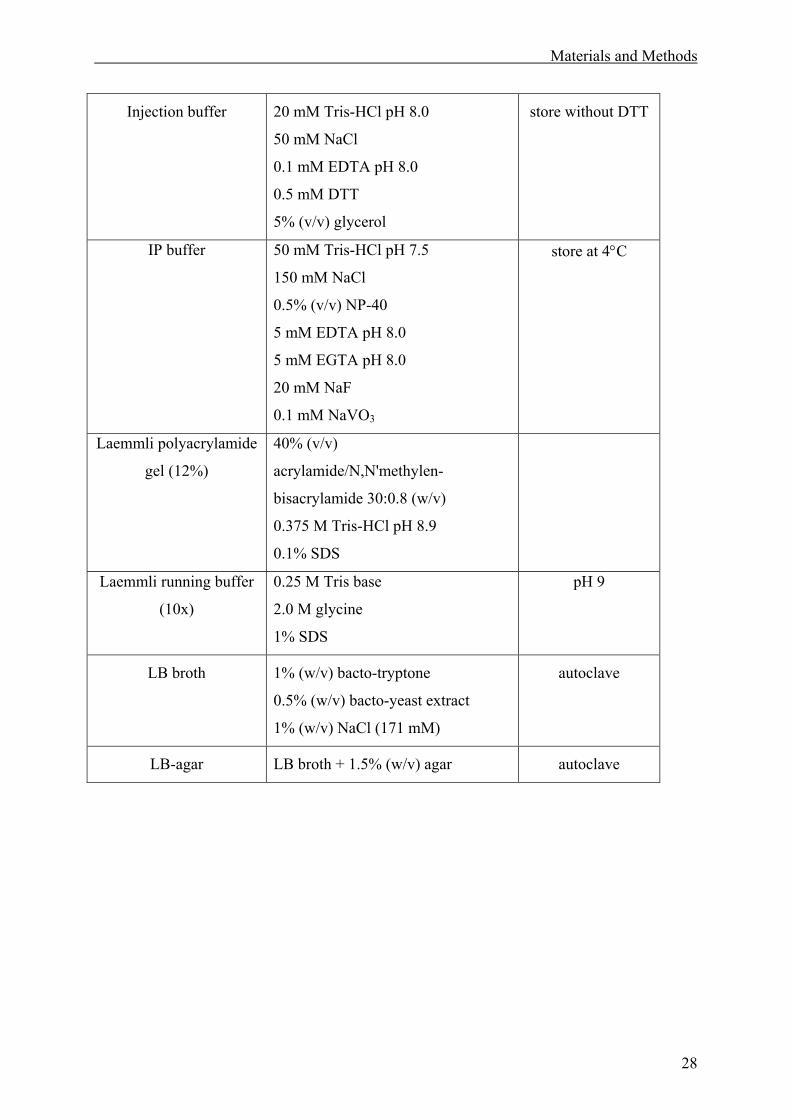
Materials and Methods
28
Injection buffer 20 mM Tris-HCl pH 8.0
50 mM NaCl
0.1 mM EDTA pH 8.0
0.5 mM DTT
5% (v/v) glycerol
store without DTT
IP buffer 50 mM Tris-HCl pH 7.5
150 mM NaCl
0.5% (v/v) NP-40
5 mM EDTA pH 8.0
5 mM EGTA pH 8.0
20 mM NaF
0.1 mM NaVO3
store at 4°C
Laemmli polyacrylamide
gel (12%)
40% (v/v)
acrylamide/N,N'methylen-
bisacrylamide 30:0.8 (w/v)
0.375 M Tris-HCl pH 8.9
0.1% SDS
Laemmli running buffer
(10x)
0.25 M Tris base
2.0 M glycine
1% SDS
pH 9
LB broth 1% (w/v) bacto-tryptone
0.5% (w/v) bacto-yeast extract
1% (w/v) NaCl (171 mM)
autoclave
LB-agar LB broth + 1.5% (w/v) agar autoclave

Materials and Methods
29
mBarth (1x) 1.0 mM KCl
8.8 mM NaCl
2.4 mM NaHCO3
15 mM Hepes pH 7.6
0.3 mM Ca(NO3)2.4H2O
0.4 mM CaCl2.6H2O
0.8 mM MgSO4.7H2O
10 µg/ml penicillin G/Streptomycin
autoclave and store
as 25x buffer A
and 25x buffer B
add before use
MBP elution buffer 100 mM Hepes pH 7.6
12 mM maltose
0.1 mM EDTA
1 mM DTT
filter sterilize
MOPS buffer (1x)
20 mM MOPS
5 mM NaAc
1 mM EDTA
pH to 6.5 - 7.0 with NaOH
do not autoclave
store at 4°C
PBS (1x) 2.7 mM KCl
1.75 mM KH2PO4
137 mM NaCl
10 mM Na2HPO4
1.75 mM KH2PO4
pH 7.4
autoclave
Ponceau staining solution 0.2% Ponceau-S
3.0% TCA
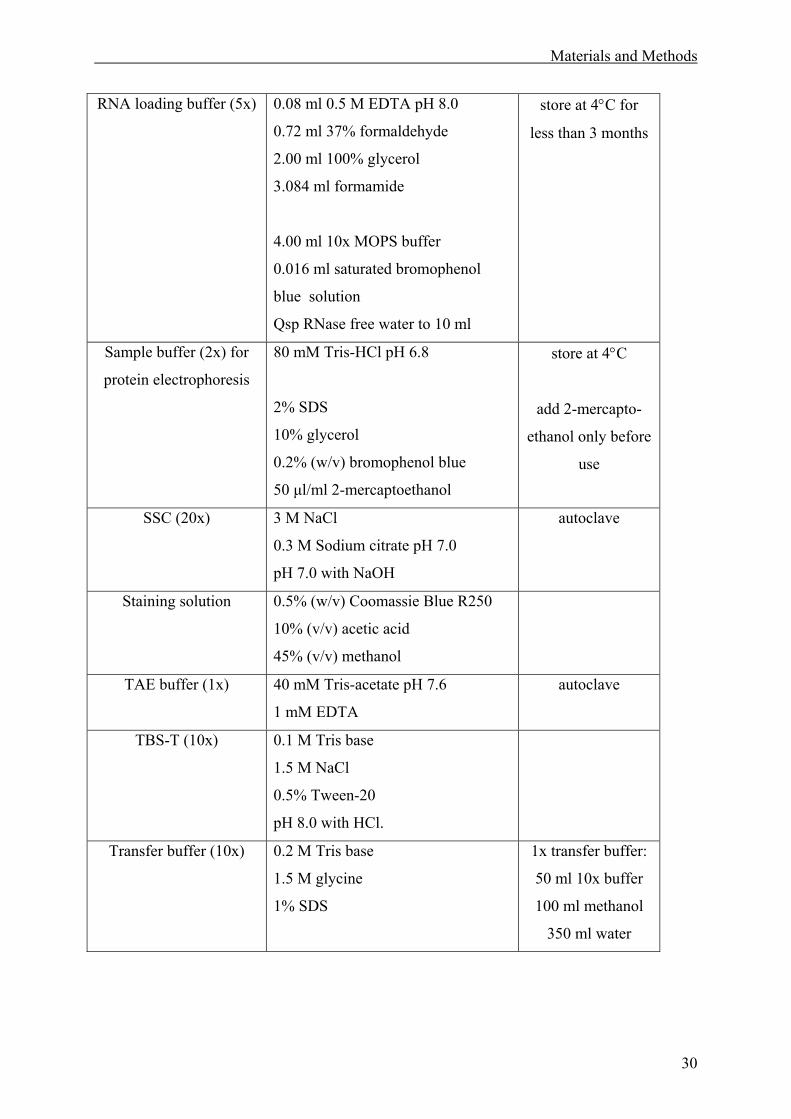
Materials and Methods
30
RNA loading buffer (5x) 0.08 ml 0.5 M EDTA pH 8.0
0.72 ml 37% formaldehyde
2.00 ml 100% glycerol
3.084 ml formamide
4.00 ml 10x MOPS buffer
0.016 ml saturated bromophenol
blue solution
Qsp RNase free water to 10 ml
store at 4°C for
less than 3 months
Sample buffer (2x) for
protein electrophoresis
80 mM Tris-HCl pH 6.8
2% SDS
10% glycerol
0.2% (w/v) bromophenol blue
50 µl/ml 2-mercaptoethanol
store at 4°C
add 2-mercapto-
ethanol only before
use
SSC (20x) 3 M NaCl
0.3 M Sodium citrate pH 7.0
pH 7.0 with NaOH
autoclave
Staining solution
0.5% (w/v) Coomassie Blue R250
10% (v/v) acetic acid
45% (v/v) methanol
TAE buffer (1x) 40 mM Tris-acetate pH 7.6
1 mM EDTA
autoclave
TBS-T (10x) 0.1 M Tris base
1.5 M NaCl
0.5% Tween-20
pH 8.0 with HCl.
Transfer buffer (10x)
0.2 M Tris base
1.5 M glycine
1% SDS
1x transfer buffer:
50 ml 10x buffer
100 ml methanol
350 ml water
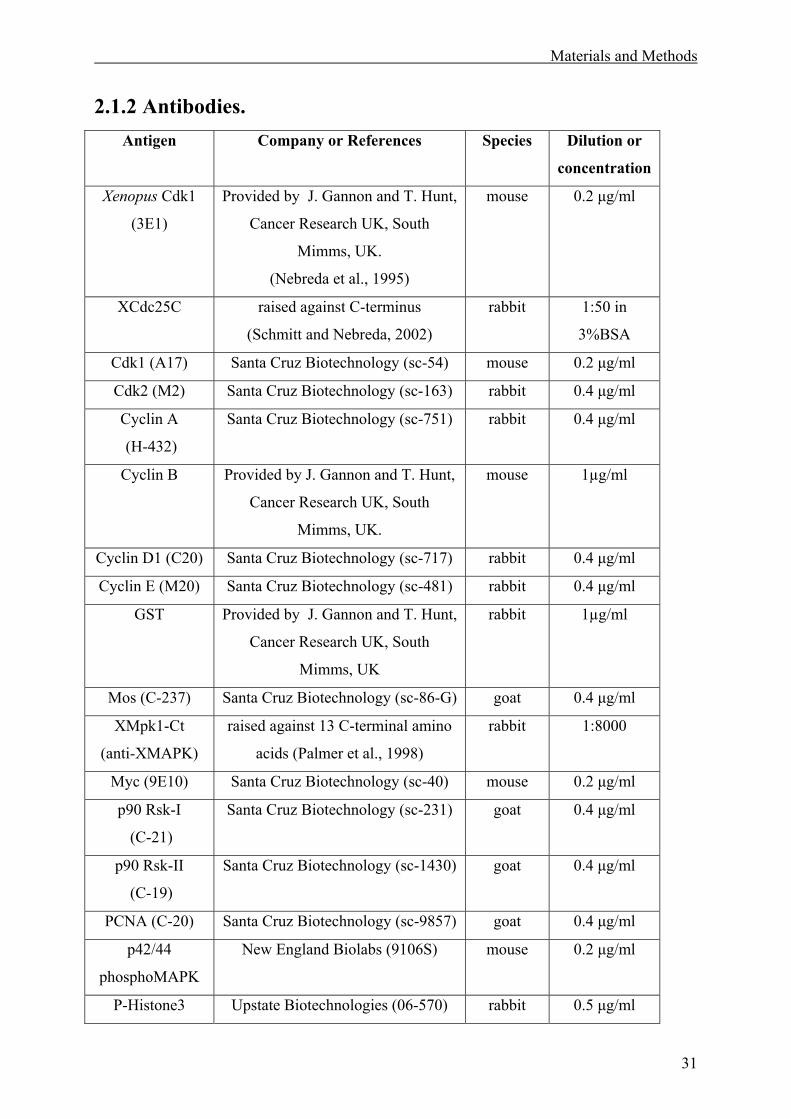
Materials and Methods
31
2.1.2 Antibodies. Antigen Company or References Species Dilution or
concentration
Xenopus Cdk1
(3E1)
Provided by J. Gannon and T. Hunt,
Cancer Research UK, South
Mimms, UK.
(Nebreda et al., 1995)
mouse 0.2 µg/ml
XCdc25C raised against C-terminus
(Schmitt and Nebreda, 2002)
rabbit 1:50 in
3%BSA
Cdk1 (A17) Santa Cruz Biotechnology (sc-54) mouse 0.2 µg/ml
Cdk2 (M2) Santa Cruz Biotechnology (sc-163) rabbit 0.4 µg/ml
Cyclin A
(H-432)
Santa Cruz Biotechnology (sc-751) rabbit 0.4 µg/ml
Cyclin B Provided by J. Gannon and T. Hunt,
Cancer Research UK, South
Mimms, UK.
mouse 1µg/ml
Cyclin D1 (C20) Santa Cruz Biotechnology (sc-717) rabbit 0.4 µg/ml
Cyclin E (M20) Santa Cruz Biotechnology (sc-481) rabbit 0.4 µg/ml
GST Provided by J. Gannon and T. Hunt,
Cancer Research UK, South
Mimms, UK
rabbit 1µg/ml
Mos (C-237) Santa Cruz Biotechnology (sc-86-G) goat 0.4 µg/ml
XMpk1-Ct
(anti-XMAPK)
raised against 13 C-terminal amino
acids (Palmer et al., 1998)
rabbit 1:8000
Myc (9E10) Santa Cruz Biotechnology (sc-40) mouse 0.2 µg/ml
p90 Rsk-I
(C-21)
Santa Cruz Biotechnology (sc-231) goat 0.4 µg/ml
p90 Rsk-II
(C-19)
Santa Cruz Biotechnology (sc-1430) goat 0.4 µg/ml
PCNA (C-20) Santa Cruz Biotechnology (sc-9857) goat 0.4 µg/ml
p42/44
phosphoMAPK
New England Biolabs (9106S) mouse 0.2 µg/ml
P-Histone3 Upstate Biotechnologies (06-570) rabbit 0.5 µg/ml
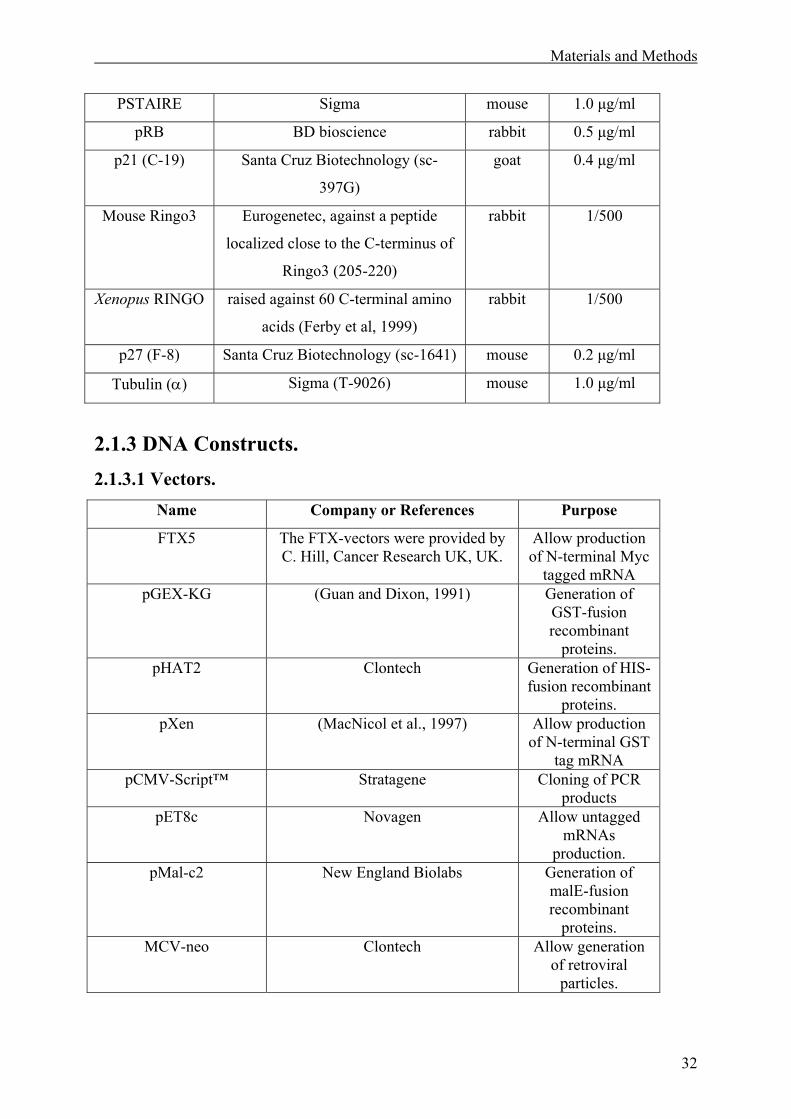
Materials and Methods
32
PSTAIRE Sigma mouse 1.0 µg/ml
pRB BD bioscience rabbit 0.5 µg/ml
p21 (C-19) Santa Cruz Biotechnology (sc-
397G)
goat 0.4 µg/ml
Mouse Ringo3 Eurogenetec, against a peptide
localized close to the C-terminus of
Ringo3 (205-220)
rabbit 1/500
Xenopus RINGO raised against 60 C-terminal amino
acids (Ferby et al, 1999)
rabbit 1/500
p27 (F-8) Santa Cruz Biotechnology (sc-1641) mouse 0.2 µg/ml
Tubulin (α) Sigma (T-9026) mouse 1.0 µg/ml
2.1.3 DNA Constructs. 2.1.3.1 Vectors.
Name Company or References Purpose
FTX5 The FTX-vectors were provided by C. Hill, Cancer Research UK, UK.
Allow production of N-terminal Myc
tagged mRNA pGEX-KG (Guan and Dixon, 1991) Generation of
GST-fusion recombinant
proteins. pHAT2 Clontech Generation of HIS-
fusion recombinant proteins.
pXen (MacNicol et al., 1997)
Allow production of N-terminal GST
tag mRNA pCMV-Script™ Stratagene
Cloning of PCR
products pET8c
Novagen
Allow untagged
mRNAs production.
pMal-c2 New England Biolabs Generation of malE-fusion recombinant
proteins. MCV-neo Clontech Allow generation
of retroviral particles.
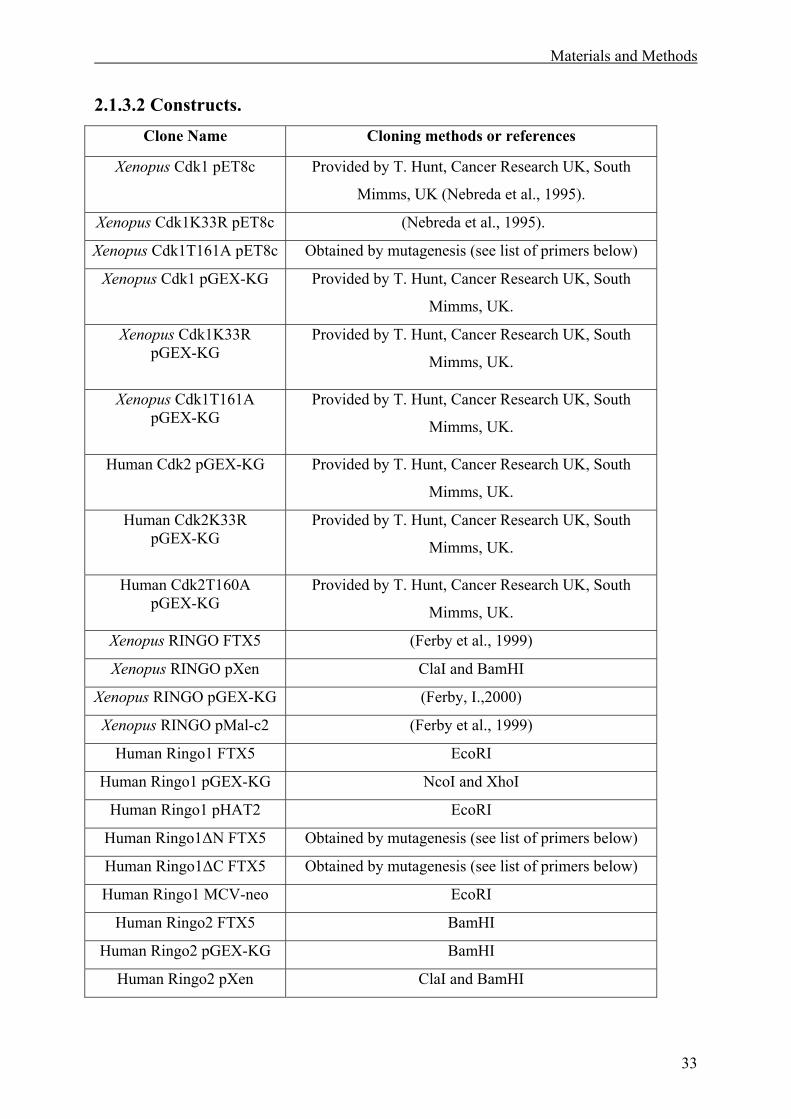
Materials and Methods
33
2.1.3.2 Constructs. Clone Name Cloning methods or references
Xenopus Cdk1 pET8c
Provided by T. Hunt, Cancer Research UK, South
Mimms, UK (Nebreda et al., 1995).
Xenopus Cdk1K33R pET8c (Nebreda et al., 1995).
Xenopus Cdk1T161A pET8c Obtained by mutagenesis (see list of primers below)
Xenopus Cdk1 pGEX-KG
Provided by T. Hunt, Cancer Research UK, South
Mimms, UK.
Xenopus Cdk1K33R pGEX-KG
Provided by T. Hunt, Cancer Research UK, South
Mimms, UK.
Xenopus Cdk1T161A pGEX-KG
Provided by T. Hunt, Cancer Research UK, South
Mimms, UK.
Human Cdk2 pGEX-KG
Provided by T. Hunt, Cancer Research UK, South
Mimms, UK.
Human Cdk2K33R pGEX-KG
Provided by T. Hunt, Cancer Research UK, South
Mimms, UK.
Human Cdk2T160A pGEX-KG
Provided by T. Hunt, Cancer Research UK, South
Mimms, UK.
Xenopus RINGO FTX5 (Ferby et al., 1999)
Xenopus RINGO pXen ClaI and BamHI
Xenopus RINGO pGEX-KG (Ferby, I.,2000)
Xenopus RINGO pMal-c2 (Ferby et al., 1999)
Human Ringo1 FTX5 EcoRI
Human Ringo1 pGEX-KG NcoI and XhoI
Human Ringo1 pHAT2 EcoRI
Human Ringo1∆N FTX5 Obtained by mutagenesis (see list of primers below)
Human Ringo1∆C FTX5 Obtained by mutagenesis (see list of primers below)
Human Ringo1 MCV-neo EcoRI
Human Ringo2 FTX5 BamHI
Human Ringo2 pGEX-KG BamHI
Human Ringo2 pXen ClaI and BamHI

Materials and Methods
34
Mouse Ringo3 FTX5 EcoRI
Mouse Ringo3 pGEX-KG NcoI and XhoI
Mouse Ringo3 pHAT2 EcoRI
Mouse Ringo3 MCV-neo EcoRI
Mouse Ringo4 FTX5 EcoRI
Xenopus CORE pXen ClaI and BamHI
Xkid FTX5 NcoI and XhoI
Xkid∆DNA FTX5 Obtained by mutagenesis (see list of primers below)
2.1.3.3 Primers for mutagenesis.
Clone Name Primer sequence
Xenopus Cdk1T161A pET8c
5' -TGTGCCTGTGAGAACCTTCgcgCATGAGGTTGTCACT -3'
5'-AGTGACAACCTCATGcgcGAAGGTTCTCACAGGCACA -3'
Human Ringo1∆N FTX5 5'-gaattcATGGAGTGGTGGGACAAATCTG-3'
5'-gaattcATGGAGGAGGACGACGAGGAC-3'
Human Ringo1∆C FTX5 5'-ATGAACCCGAGGGCCAGGAtgaACCGCTCTCACATA-3'
5-'TATGTGAGAGCGGTTcatCCTGGCCCTCGGGTTCAT-3'
Xkid∆DNA FTX5 5'- GAGTTACAAAGCGTAtaatagGGGATCCCCGTTTTT-3'
5'- AAAAAACGGGGATCCCCTAttatacTACGCTTTGTAACTC-3'
Small and blue letters indicates the modified sequences.
2.1.4 Cells. 2.1.4.1 Ntera-2.
Ntera-2 is a human teratocarcinoma cell line (American Type Culture Collection,
CRL-1973). The parental NTERA-2 lines were established in 1980 from a nude mouse
xenograft of the Tera-2 cell line. The cells exhibit high expression levels of the N-myc
oncogene.

Materials and Methods
35
2.1.4.2 HEK293. The 293 cell line is a permanent line of primary human embryonal kidney transformed
by sheared human adenovirus type 5 (Ad 5) DNA.
2.2 Methods. 2.2.1 Molecular Biology. 2.2.1.1 DNA cloning.
For subcloning of cDNAs into different plasmid vectors, Polymerase Chain Reaction
(PCR) was used in many cases to provide the cDNAs with appropriate restriction sites at their
5'- and/or 3'-ends. All primers were obtained from Sigma-ARK. For the PCR, 100 ng template
DNAs were mixed with 200 nM of each primer, 20 µM dNTPs, 1x Pfu reaction buffer and 2.5
U cloned Pfu polymerase (Stratagene). The PCR consisted of 20 cycles (1 min 94°C
denaturation, 1 min 50°C annealing, 2 min/kb 68°C extension) followed by a final 10 min
extension at 68°C. The PCR products were cloned directly into the pCMV-Script vector
following the instructions of the manufacturer (pCMV-ScriptTM PCR Cloning Kit,
Stratagene). The PCR was purified with the StrataPrep PCR purification kit, ligated for 1 h at
room temperature with 10 ng SrfI-predigested pCMV Script vector in the presence of 5 U SrfI
restriction enzyme and transformed into E. coli XL10-Gold Cam ultracompetent cells
provided in the kit.
Positive colonies were selected on LB-kanamycin (50 µg/ml) agar plates. DNA
sequencing analysis controlled the fidelity of the DNA sequence. Then 4 µg of plasmid DNA
was digested with the respective restriction enzymes used for cloning in a total volume of 20
µl for at least 2 hrs at 37°C. After heat-inactivation of the restriction enzymes, 2 µg of the
digestion reaction was directly used for ligation with 0.5 µg of linearized vector in a total
volume of 10 µl containing 1x of ligase buffer and 1 U T4-DNA-ligase (Roche) overnight at
16°C. E. coli TG-1 cells, transformed with the new plasmid construct, were selected on LB-
antibiotic (50 µg/ml for ampicilin and kanamycin, 30 µg/ml for chloramphenicol) agar plates.
For plasmid preparation (Qiaprep Spin Miniprep Kit, Qiagen) was used. Individual colonies
were examined by restriction endonuclease analysis.

Materials and Methods
36
2.2.1.2 Construction of Cdk1, Ringo1 and Xkid mutants. Mutants were constructed using the QuikChangeTM site-directed mutagenesis kit
(Stratagene). Complementary oligonucleotide primer pairs (40-45 bp long) were designed to
contain the desired point mutations in the middle of the primer sequence. The primers were
obtained from Sigma-ARK GmbH, Darmstadt, and are listed in section 2.1.3.3. The primers
were extended by PCR using the reagents provided in the kit. A mix of 50 µl final volume
containing 10-50 ng template plasmid DNA, 1x reaction buffer (10 mM KCl, 10 mM
(NH4)2SO4, 20 mM Tris-HCl pH 8.8, 2 mM MgSO4, 0.1% Triton X®-100, 10% (w/v) BSA),
250 nM of each primer, 1 µl dNTP mixture and 2.5 U of PfuTurboTM DNA polymerase was
subjected to 12-15 cycles (30 sec denaturing at 95°C, 1 min annealing at 55°C and 12 min
extension at 68°C). The PCR reactions were incubated with 10 U DpnI restriction enzyme for
2 hrs at 37°C to digest parental DNA, directly transformed into 50 µl of E.coli XL1-Blue
supercompetent cells by heat-shock and plated onto LB-ampicillin (50 µg/ml) agar plates.
Resulting colonies were grown in LB broth-ampicilin (10 µg/ml) for plasmid preparation
(Qiaprep Spin Miniprep Kit, Qiagen). Plasmids were subjected to DNA sequencing to verify
the mutation and the fidelity of the rest of the cDNA sequence.
To generate a Xkid mutant lacking the C-terminal 109 amino acids including the
DNA-binding region two consecutive stop codons were introduced at amino acid positions
542 and 543, using the primers listed above. To generate a Cdk1 mutant for the Thr160 in the
T-loop, this amino acid was replaced by alanine, using the primers listed above. To create
deletion at the 5’end of the Human Ringo1 protein, EcoRI sites were created by PCR and the
amplified fragment was directly cloned into FTX5. The ∆C mutant was obtained by creating a
stop codon at the position of amino acid 200 in Human Ringo1 FTX5, using the primers listed
above.
2.2.1.3 Expression of proteins in reticulocyte lysates.
In order to verify expression of cDNAs cloned into FTX5 or other vectors containing
the T7 promoter, the TNT® T7 coupled reticulocyte lysate system (Promega) was used. For
testing a plasmid construct, 1 µl of a standard Qiagen plasmid mini preparation (about 0,2 µg)
was incubated in a total volume of 10 µl with 5 µl TNT reticulocyte lysate, 0.4 µl TNT
reaction buffer, 0.2 µl amino acid mixture minus methionine (1 mM), 0.4 µl RedivueTM Pro-
mixTM L-(35S) in vitro cell labelling mix (10 mCi/ml; Amersham), 0.2 µl T7 RNA polymerase
and 8 U RNasin® ribonuclease inhibitor (40 U/µl, Promega) for 90 min at 30°C. 1 µl of this

Materials and Methods
37
reaction was subjected to SDS-PAGE followed by autoradiography of the Coomassie-stained
dried gel with Biomax films (Kodak, Sigma).
2.2.1.4 Preparation of total RNA and Northern blotting.
Total RNAs were extracted from tissue or cells using the Trizol RNA isolation reagent
(Gibco BRL) following the manufacturer's protocol. 0,3 mg of material was homogenized in 1
ml Trizol reagent using a 5 ml syringe with a 21G needle (Becton Dickinson) and incubated at
30°C for 15 min. 160 µl chloroform was added, the solution was mixed and incubated for 3
min at 30°C. After centrifugation at 10000 xg for 15 min at 4°C, the upper phase was
recovered and supplemented with 0.4 ml isopropanol for RNA precipitation. The mixture was
incubated for 15 min at room temperature and centrifuged at 10000 xg for 30 min at 4°C.
After one wash with ice cold 70% ethanol, the RNA pellet was dissolved in 50 µl 5 mM Tris-
HCl pH 7.5 and the RNA concentration determined in a UV spectrophotometer, measuring
the absorbance at 260 nm. About 20 µg of total RNA was mixed with 5x RNA loading buffer
and heated to 65°C for 5 min before being resolved on a 1.2% MOPS/FA agarose gel (1x
MOPS buffer, 2% formaldehyde) at 10 V/cm. The RNA was transferred from the gel to a
nylon membrane (GeneScreen Plus® Hybridization Transfer Membrane, NEN Life Science,
USA) by capillary-blotting overnight with 10x SSC and UV cross-linked to the membrane the
following day. Successful blotting was verified by staining the RNA on the membrane with
0.04% methylene blue (Sigma) in 0.5 M NaAc pH 5.2. The membrane was destained with
30% ethanol and then extensively washed with water.
In some cases First ChoiceTM Northern blot (Ambion) membrane was used.
Membranes were prehybridized in ExpressHyb Hybridization Solution (Clontech) for 1 hour
at 68°C. For the hybridization, the respective cDNA probes were labelled with 5 µCi [α-32P]dCTP (3000 Ci/mmol, Amersham) using the MegaprimeTM labelling kit (Amersham
Pharmacia Biotech) and purified using Chroma Spin +STE-30 columns (Clontech). The
radiolabelled, denatured probes were incubated with the Northern blots membrane in 5 ml
ExpressHyb at 68°C for 1 hr. After extensive washes (twice for 20 min at room temperature
in 2x SSC, 0.05% SDS, three times for 30 min at 50°C in 0.1x SSC, 0.1% SDS), the Northern
blot membrane was exposed to Kodak X-OMAT film (Sigma) at -70°C with two intensifier
screens (Kisker, Mühlhausen, Germany). For all the Northern blots a probe corresponding to
the Ubiquitin ORF was used as a loading control.

Materials and Methods
38
2.2.1.5 RT-PCR. RNAs were isolated from Ntera-2 cells and tissues (embryonic brain, adult brain and
testis) as described in section 2.2.1.4. For some of the expression pattern analyse First Choice
TM Total RNAs (Ambion) were used. Total RNA (1µg) was treated with DNase I (Life
Technology) and used for RT-PCR, with the Superscript kit (Life Technology). PCR was
performed using the oligonucleotides indicated below.
Ringo3 and Ringo4 cloning.
Ringo3-5’: (5'-ATGCGGCATAATCAGATGTATTGTG-3')
Ringo3-3’: (5'-CCCACACAGTGAGCAATCCACTGGT-3')
Ringo4-5’: (5'-CTAGGTCAGGTTGGTGCGATCTCGT-3')
Ringo4-3’: (5'-CTAGGTCAGGTTGGTGCGATCTCGT-3')
Expression analysis.
Ringo1-5’: (5'-TCTACAGTGAGGCCTGAACACCACAAGGTC-3')
Ringo1-3’: (5'-GCGCAGTTTGTGGAACATGGGGCGCTGGGC-3')
Ringo3-5’: (5'-CTAATCATACAGCGCCAGGAAATGACTGCT-3')
Ringo3-3’: (5'-TAACTTTAAGAAATTAGGGAACAGTTTTCT-3')
Ringo4-5’: (5'-CTAGGTCAGGTTGGTGCGATCTCGT-3')
Ringo4-3’: (5'-CTAGGTCAGGTTGGTGCGATCTCGT-3')
GAPDH5’: (5'-GAGGGGCCATCCACAGTCTTC-3')
GAPDH3’: (5'- CAGTATGACTCCACTCACGGC -3')
Alternative splicing analysis.
Ringo3-5’: (5'-ATGCGGCATAATCAGATGTATTGTG-3')
Ringo3-B: (5'-TTAAGCCATACCTTGAGAATAGGTAT-3')
Ringo3-C/D: (5'-TCATTCTTCACTCTCTGCAAACCAT-3')
Ringo4-A: (5'-CTAGGTCAGGTTGGTGCGATCTCGT-3')
Ringo4-B: (5'-GTCATTGGCCAGGTAGAGAAGCAAA-3')
2.2.2 Biochemistry. 2.2.2.1 Bacterial expression and purification of recombinant fusion
proteins. For expression and purification of all recombinant GST-fusion proteins, pGEX-KG
constructs were transformed into E.coli BL21-CodonPlus (DE-3) competent cells (Stratagene)

Materials and Methods
39
(Guan and Dixon, 1991). For induction of GST-proteins, fresh overnight cultures grown in a
gyratory shaker (200 rpm) at 37°C in LB-ampicillin (10 µg/ml) were diluted 10-fold into 500
ml LB-ampicillin and further incubated at 37°C until OD600 was 0.8. The cultures were
diluted 1:1 with fresh LB-ampicillin and induced with 0.1 mM β-D-isopropyl-thiogalactoside
(IPTG) for 6 hrs at 23°C. Cells from 1 litre of induced bacteria culture were harvested,
washed with cold PBS and lysed in 13.5 ml PBS containing 1 mM PMSF, 10 µg/ml aprotinin,
10 µg/ml leupeptin and 1 mg/ml lysozyme. After incubation on ice for 10 min, the cells were
homogenized by sonication on ice and supplemented with 1% Triton X-100 (1.5 ml 10%
TritonX-100 in PBS). The lysate was then centrifuged at 10000 xgfor 20 min and the
supernatant incubated with 1 ml washed glutathione Sepharose 4B beads (Amersham
Pharmacia) for 2 hrs at 4°C on a rotating wheel. The beads were washed three times with cold
PBS and once with cold 50 mM Tris-HCl pH 8.0. GST-tagged proteins were eluted three
times from the beads with 0.5 ml 10 mM glutathione in 50 mM Tris-HCl pH 8.0 for each
elution, dialyzed against injection buffer (see materials) and stored at -70°C in small aliquots.
Recombinant His-Ringo1 and His-Ringo3 were induced as described above, and the
bacterial pellet was washed once in PBS prior to lysis in ice cold lysis buffer (50 mM
NaH2PO4 pH8. 0, 0.1 mM NaCl, 10 mM Tris-HCl pH 8.0). The cells lysate was obtained by
sonication, supplemented with 0.1% NP-40 and centrifuged at 10000xgfor 30 min. The lysate
was incubated with 1 ml washed TALON metal affinity beads (Clontech) for two hrs at 4°C.
The beads were then washed four times in lysis buffer supplemented with 50 mM imidazole.
Elution of His-Ringo1 and His-Ringo3 from the beads was done with 1M imidazole, 0.1 M
NaCl, 20 mM Tris-HCl pH 6.0. Purified proteins were dialyzed against injection buffer (see
materials) and stored at -70°C in small aliquots.
Xenopus RINGO-pMal-c2 was transformed into E.coli BL 21-CodonPlus (DE-3)
competent cells (Stratagene). For induction of MalE-proteins, fresh overnight cultures grown
in LB-ampicillin (100 µg/ml) were diluted 10-fold into 500 ml LB-ampicillin and further
incubated at 37°C until OD600 was 0.8. The cultures were diluted 1:1 with fresh LB-ampicillin
and induced with 0.1 mM β-D-isopropyl-thiogalactoside (IPTG) for 6 hrs at 23°C. Cells were
collected by centrifugation (4000 rpm, Sorvall 34 rotor), washed with cold PBS and
resuspended in 20 ml buffer X supplemented with 1 mM dithiothreitol (DTT), 1mM PMSF
(Sigma), 1 mg/ml lysozyme (Sigma) and 2 tablets of CompleteTM Mini Protease Inhibitor
Cocktail (Roche). After incubation on ice for 10 min, the cells were broken with a French cell
press and centrifuged at 12000 xg for 30 min. The supernatant was incubated with 1 ml
washed amylose resin (New England Biolabs) for 1 to 2 hrs at 4°C on a rotating wheel. The

Materials and Methods
40
resin was washed twice with 10 ml buffer X supplemented with 1 mM DTT and 1 tablet of
CompleteTM Mini Protease Inhibitor Cocktail per 50 ml buffer before elution of the bound
protein with 5 times 600 µl MBP elution buffer. The different elutions were analysed by SDS-
PAGE and elutions with high concentrations were dialyzed against injection buffer. The
proteins were stored in aliquots at -70°C.
2.2.2.2 In vitro Cdk assay with recombinant proteins.
Recombinant protein substrates were incubated with different Cdks in the presence of
recombinant RINGO proteins in a final volume of 12 µl 50 mM Tris-HCl pH 7.5 containing
10-50 µM cold ATP, 10 mM MgCl2, 2 µM microcystin, 2 µM DTT and 2 µCi [γ-32P]ATP
(3000 Ci/mmol, Amersham) for 15 min at 30°C. Typically 1µg of each protein was used. The
reactions were stopped by adding 4 µl of 4x sample buffer and the samples were boiled for 5
min at 95°C prior to SDS-PAGE. Autoradiography was performed on the coomassie-stained,
dried gel with Kodak X-OMAT film (Sigma).
2.2.2.3 Baculovirus expression and purification of recombinant His-Cyclin
B1. For purification, seven 245×45 mm Nunc tissue culture dishes of High five™ cells
(Invitrogen) were grown at 27°C in Express Five ® SFM medium (Invitrogen) and infected
for 48 hrs with the His-Cyclin B1 Baculovirus (obtained from W. Dunphy). Cells were
scraped off and washed in cold PBS. The cell pellet was resuspended in 25 ml of 50 mM NaPi
(pH 8), 10 mM KCl, 1.5 mM MgCl2, 5 mM β-mercaptoethanol, and protease inhibitor mix (1
mg/ml of leupeptin, pepstatin, aprotinin, and 1 mM PMSF) and incubated for 15 min on ice.
The suspension was homogenized with 25 strokes in a B-type dounce and centrifuged at 700
×g for 10 min at 4°C. The pellet was reextracted in 7 ml of 50 mM NaPi (pH 8), 500 mM
KCl, 5 mM MgCl2, 10% glycerol, 0.1% Tween20, 5 mM β-mercaptoethanol, and protease
inhibitor mix for 30 min on ice, homogenized as described above, and centrifuged for 10 min
at 10000 xg. The two supernatants were combined and mixed in batch with 3 ml of TALON
metal affinity beads (Clontech) for 2 hrs at 4°C, beads. The beads were previously
preequilibrated in 50 mM NaPi (pH 8), 150 mM KCl, 1.5 mM MgCl2, 10% glycerol, 2.5 mM
β-mercaptoethanol, and protease inhibitor mix at 4°C for 1 hr. The beads were then washed
four times with the equilibration buffer. Elution was performed in five steps of with 2 ml of
wash buffer suplemented with 100 mM imidazole (pH 7.2).

Materials and Methods
41
2.2.2.4 GST pull-down. Extracts from Sf9 insect cells expressing Cdk1 were prepared as described by (Ferby
et al., 1999). The extracts (usually 20 µl) were diluted 1:5 in HBS and incubated for 20 min at
room temperature with different concentrations of either GST or GST-RINGO pre-bound to
10 µl of GSH beads. After centrifugation, the beads were washed four times in IP buffer and
once in HBS buffer. The samples were boiled for 5 min at 95°C prior to SDS-PAGE and
analyzed by immunobloting.
2.2.2.5 Generation and purification of Ringo antibodies.
Polyclonal anti-Ringo1 and anti-Ringo3 antibodies were produced against
recombinant His-tagged proteins or against a peptide localized close to the C-terminus of
Ringo3 (amino acids 205-220). Polyclonal anti-His-Ringo1 and anti-His-Ringo3 were
generated by the Laboratory Animal Resources facility of EMBL. For immunization, New
Zealand White (NZW) rabbits were injected with 0.25 mg recombinant protein in 1 ml
PBS containing 0.25 mg purified microbial components in 1% squalene oil as adjuvant
(Ribi R-730 adjuvant; Ribi ImmunoChem Research, Hamilton, MT, USA). Immunized
rabbits were boosted five times every four weeks with 0.25 mg protein in adjuvant. The final
bleed (used in subsequent experiments) was taken 22 weeks after the initial antigen injection.
Synthesis of a peptide corresponding to residues near the C-terminus of Ringo3 and
immunization with the keyhole limpet hemocyanin (KLH)-coupled peptide was done by
Eurogenetec (Belgium) using a three-month protocol including two boosts every four weeks
and the final bleed 14 weeks after the initial antigen injection. All antibodies prepared were
affinity purified against recombinant GST-Ringo3 bound to a nitrocellulose strip. 2 mg of
GST-Ringo3 was separated on a 12% Laemmli gel (Laemmli and Quittner, 1974). The band
corresponding to the GST-Ringo3 protein was visualized by Ponceau staining, cut out and
blocked for 2 hrs with 5% non-fat dry milk in PBS prior to incubation with 5 ml anti-RINGO
antiserum diluted 1:1 in PBS overnight at 4°C with rocking. The strip was washed three times
for 10 min with TBS-T, and bound antibodies were eluted 3 times with 900 µl of 100 mM
glycine, pH 2.5. The eluates were immediately neutralized with 1/10 volume 1 M Tris-HCl
pH 9.0. Affinity purified antibodies were dialyzed against PBS and stored at -70°C in small
aliquots.

Materials and Methods
42
2.2.2.6 Covalent coupling of antibodies to protein G-Sepharose. The protocol used was a modified version of the protocol of Harlow and Lane (Harlow
and Lane, 1999). Purified antiserum (usually 10 µg of IgG) was incubated on a rotary wheel
at Room Temperature (RT) with 5 µl beads in a total volume of 500 µl PBS. The beads with
bound IgG were washed three times with 5 to 10 volumes of 0.2 M Na borate pH 9.0 and
resuspended in 5 to 10 volumes of the same borate buffer. Fresh dimethyl pimelimidate
(DMP, Sigma) was added to a final concentration of 20 mM (5.2 mg/ml) and incubated with
the beads for 30 min at RT. This step was repeated once by adding more DMP to the same
sample. The reaction was stopped by washing the beads once with 5 to 10 volumes of 0.2 M
ethanolamine pH 8.0 followed by incubation in 5 to 10 volumes of 0.2 M ethanolamine pH
8.0 for 30 min on a rotary wheel at RT. After this blocking reaction, the beads were washed
twice with 5 to10 volumes 0.1 M glycine pH 2.5 for 5 min, followed by two washes with
PBS. The beads with covalently coupled IgG were resuspented in 3 volumes of PBS
containing 0.1% NaN3 as antibacterial agent and stored at 4°C.
2.2.3 The Xenopus laevis system.
2.2.3.1 Isolation of stage VI oocytes and induction of meiotic maturation.
Female Xenopus laevis frogs (African Reptile Park, South Africa) were
anaesthetized with 0.4% 3-aminobenzoic acid ethyl ester (Sigma) for 30 min at room
temperature. The ovaries were removed surgically and stored in mBarth medium. Small
pieces of ovary were treated with 0.5 mg/ml collagenase B (Roche Diagnostics GmbH,
Mannheim) in mBarth medium for 60 min to release oocytes from connective tissue and
facilitate sorting of stage VI oocytes. After washes with 0.1 M NaCl, the collagenase-treated
oocytes were left at 18°C in mBarth medium for at least 2 hrs before injection.
For the in vitro induction of meiotic maturation, stage VI oocytes were incubated with
5 µg/ml (15.9 µM) progesterone (Sigma; a 5 mg/ml stock solution was prepared in ethanol).
Maturation was scored by the appearance of a white spot on the animal pole of the oocytes.
Germinal Vesicle BreakDown (GVBD) was confirmed by dissection of oocytes fixed in 3%
trichloroacetic acid (TCA) for 10 min.

Materials and Methods
43
2.2.3.2 Preparation of mRNAs for injection into oocytes.
To prepare mRNAs for injection into oocytes, cDNAs of interest were cloned into the
vectors FTX5 (N-terminal Myc tag).The FTX5 constructs were linearized with either XbaI or
XmnI (depending on the presence of internal restriction sites), which cut the FTX-vector
downstream of its poly (A) sequence. The pXen constructs were linearized with either PstI or
XbaI (depending on the presence of internal restriction sites), which cut the pXen-vector
downstream of its poly(A) sequence. Usually, 2 µg DNA was digested for 2 hrs at 37°C in a
total volume of 20 µl using 20 units (U) of the respective enzymes. The linearized DNA was
directly used to prepare mRNA with the MEGAscriptTM in vitro transcription kit for large-
scale synthesis of RNAs (Ambion). In a total volume of 20 µl, 3 µl linearized DNA was
incubated for 3 hrs at 37°C with 1x reaction buffer, 3 mM RNA Cap Structure Analog
m7G(5')ppp(5')G (New England Biolabs), 7.5 mM ATP, CTP and UTP, 1.875 mM GTP and 2
µl T7 enzyme mixture (Ambion). After RNA synthesis, the plasmid DNA was removed by
digestion with 2 U RNase free DNase (Ambion) for 15 min at 37°C.
The reaction was stopped with 10 µl 10% SDS, diluted to 200 µl with RNase free
water, and 18 µl ammonium acetate stop solution was added. The mixture was vigorously
vortexed prior to phenol/chloroform extraction and precipitation of the mRNA with 1 volume
(200µl) isopropanol at -20°C for 30 min. The RNA pellet was washed twice with 70% ethanol
and dissolved in 50 µl RNase-free water. 1 µl of this mRNA preparation was analysed on a
1% TAE-agarose gel and the stock was frozen in aliquots at -70°C. The standard protocol of
mRNA preparation yields mRNA concentrations between 0.1 and 2 mg/ml.
2.2.3.3 Microinjection of oocytes with mRNAs.
Xenopus oocytes were microinjected with 50 nl of mRNA solution. Control injections
were done with water. Injections were done with an IM 300 microinjector (Narishige). The
oocytes were put onto a small plastic grid to prevent them from moving during the
micromanipulation and injected preferentially at the border between the animal and the
vegetal hemispheres, as this ensures both expression of the mRNAs and optimal survival of
the oocytes. After mRNA injection, the oocytes were transferred into mBarth medium
incubated for 8 to 16 hrs at 18°C to allow protein expression.

Materials and Methods
44
2.2.3.4 Antisense experiments. Xenopus oocytes were microinjected with 50 nl of either antisense or sense (control)
oligonucleotides in solution. 3′-propanediol oligonucleotides were used, which were
synthesized and purified as described in (Ferby et al., 1999). The antisense oligonucleotides
were based on nucleotides 54 to 78 of the Xkid open reading frame. Xenopus oocytes were
microinjected with 100 ng of each oligonucleotide and incubated for four hours, before
progesterone stimulation. For the rescue experiments, we used the antisense morpholino
oligonucleotides and control morpholinos that contain four mismatches provided by Genetool.
Xenopus oocytes were microinjected with 50 nl of each morpholino oligonucleotide (0.5 mM
stock). Sequences of the primers are listed bellow.
Antisense oligonucleotide: (5′-CTTCTTGTGCTGATCCAACATGCTC-3′)
Sense oligonucelotide: (5’-CGCTGGAGATTGTCAACTGGAGAAA-3’)
Antisense morpholino oligonucleotide: (5’-GCCCAGTAAGAACCATTCCCGCCTC-3’)
Control morpholino oligonucleotide: (5’-GCCgAGTtAGAACCATTCgCGCgTC-3’)
Small and blue letters indicate the four mismatches.
2.2.3.5 DNA replication assays. Xenopus oocytes were microinjected with 50 nl of a 1 µCi of [α-32P]dCTP (3,000 Ci
mmol) about 2 hrs before GVBD. Oocytes were lysed (30 µl per oocyte) in 1% SDS, 10 mM
Tris (pH 7) and 10 mM EDTA. The lysed mixture was then incubated with 10 µg/ml of
proteinase K (Roche) for 60 min at 37 °C. Then an equivalent volume of phenol-chloroform
was added to the mixture. After centrifugation at 10000 xg for 1 min at 4°C, the upper phase
was recovered and supplemented with the equivalent volume of chloroform. After
centrifugation at 10000 xg for 1 min at 4°C, the upper phase was recovered and supplemented
with three volumes of ice cold 100% ethanol supplemented with 10% of Sodium acetate 3M,
Ph 5.5 for DNA precipitation. The mixture was incubated for 10 min at -70°C and 30 min -
20°C and centrifuged at 10000 xg for 30 min at 4°C. After one wash with ice cold 70%
ethanol, the DNA pellet was dissolved in 10 µl of 5 mM Tris-HCl pH 7.5 and 1mM EDTA
per oocyte. The equivalent of three oocytes (genomic DNA) was analysed on a 1% TAE-
agarose gel, followed by autoradiography of the dried gel with Kodak X-OMAT film. The
results were also quantified on the gels using a Fuji phosphorimaging device.

Materials and Methods
45
2.2.3.6 Preparation of oocyte lysates and immunobloting.
Oocyte lysates were prepared by crushing fresh or frozen oocytes in 10 µl H1K buffer
per oocyte followed by centrifugation for 15 min at 4°C (at 10000 xg). The cytosolic lysate,
separated from lipids and insoluble debris, was transferred to fresh ice precooled 1.5 ml
reaction tubes.
For immunoblotting, oocyte lysates were separated by SDS-PAGE (sodium dodecyl
sulphate-polyacrylamide gel electrophoresis) on a 15% Anderson gel (Anderson et al.,
1973), applying the equivalent of 1 oocyte per lane. After electrophoresis, the proteins were
transferred from the polyacrylamide gels to nitrocellulose membranes (Protran 0.22 µm) using
a semi-dry blotting apparatus (Hoeffer). The membranes were blocked for 1 hr at room
temperature in a TBS-T buffer, supplemented with 5% non-fat dry milk. Primary antibodies
were usually diluted in TBS-T supplemented with 1% milk and incubated with the
membranes overnight at 4°C or for several hrs at room temperature.
After incubation with the first antibody, the blots were washed three times for 10 min
in TBS-T followed by incubation with the secondary antibodies for 1 hr at room temperature.
The secondary antibodies were horseradish peroxidase (HRP)-coupled (Dako) or Alexa 680-
coupled anti-mouse, rabbit or goat antibodies, diluted 1:4000 or 1:8000 in TBS-T
supplemented with 1% milk. After extensive washing with TBS-T, HRP-coupled antibodies
were detected using the enhanced chemiluminescence (ECL) Western blotting analysis kit
(Amersham Pharmacia Biotech) and Alexa 680-coupled antibodies were detected using the
Odissey® infrared imaging system (Li-Cor).
2.2.3.7 Histone H1 kinase assays.
Oocytes were lysed as described above. 4 µl oocyte lysate were incubated in a total
volume of 12 µl H1K buffer containing 4 µg histone H1 (Sigma), 2 µCi [γ-32P] ATP (3000
Ci/mmol, Amersham) and 50 µM cold ATP for 15 min at 30°C. The kinase reactions were
analysed by SDS-PAGE followed by autoradiography. In some experiments, the results were
quantified on the gels using a Fuji phosphorimaging device.
2-2.3.8 In vivo labelling of Xenopus oocyte proteins with 35S methionine. For in vivo 35S-labelling of Xenopus oocyte proteins, oocytes that had been injected
with Xkid antisense, were incubated for 16 hrs at 18°C , in 150 µl mBarth buffer containing 1
mCi/ml of RedivueTM Pro-mixTM L-(35S) in vitro cell labelling mix (Amersham). Labelled

Materials and Methods
46
oocytes were transferred to a fresh tube, washed 4 times with mBarth buffer and lysed in H1k
buffer (10 µl/oocyte). The equivalent of one oocyte was subjected to SDS-PAGE followed by
autoradiography of the Coomassie-stained dried gel with Kodak Biomax films.
2.2.3.9 Immunoprecipitation of Myc-tagged proteins from oocyte lysates.
G2-arrested or mature oocytes expressing Myc-RINGO, Myc-Ringo1, Myc-Ringo2 or
Myc-Ringo3 or water-injected control oocytes were lysed in IP buffer containing protease
inhibitors. 10 µg of agarose-conjugated Myc antibodies were added to the cleared oocyte
lysates and they were incubated at 4°C on a rotating wheel for 2 hrs. The beads were washed
twice with IP buffer and twice with H1K buffer. After buffer removal the beads were
incubated for 30 min at 30°C with 15 µl of H1K buffer containing 50 µM cold ATP, 2 µCi [γ-32P]ATP (3000 Ci/mmol, Amersham), 2 µM microcystin and 1 µg of Histone H1 as substrate.
The reactions were stopped by addition of 5 µl 4x sample buffer and boiling at 95°C for 5 min
prior to analysis by SDS-PAGE on a 15% Anderson polyacrylamide gel. The gels were
stained with Coomassie Blue, dried and exposed to Kodak X-OMAT film for 4 to 24 hrs at
room temperature. In some cases, Myc immunoprecipitates were analysed by immunobloting
to detect associated proteins.
2.2.4 Mammalian Cell Culture. 2.2.4.1 Conditions of cell culture.
Ntera-2 cells were grown on tissue culture plates (Falcon) containing Dulbecco
minimal essential medium plus 10% fetal calf serum (Gibco-BRL), 2 mM glutamine (Gibco-
BRL), 100 U of penicillin per ml and 100 mg of streptomycin per ml (Gibco-BRL), at 37°C
and 10% CO2.
The AmphoPack-293 cells (Clontech) are HEK293 cells which were grown in the
same medium as the Ntera-2 but in 5% CO2.
2.2.4.2 Transfection and retroviral infection of cells. The viruses were generated by transfection of the plasmidic form of the retroviral
vectors (MSCV-neo plasmid) in the AmphoPack-293 cell line (Clontech) by calcium
phosphate precipitation. The day before the transfection, the AmphoPack-293 cells were
seeded at 70% confluence. For a single transfection of a 10 cm diameter plate, the following
mix was prepared: 525 µl CaCl2 (1M), 10 µg of DNA of interest and sterile water to get a

Materials and Methods
47
final volume of 2.1 ml. Then 2.6 ml of 2x HBS buffer was added drop by drop, while
continuously bubbling air into the solution through a pipette. The following mix was left to
precipitate for 5 min at room temperature. 1 ml of the solution was added to the plate, drop by
drop, while gently shaking it. The following day, the precipitates were removed and fresh
medium was added. This medium, containing the viral particles, was recovered 24 hrs later,
centrifuged at 3,000 xg for 10 min, and filtered through a 0.45-µm-pore size filter (Millipore).
Sub-confluent cultures of Ntera-2 were infected by adding 12 ml of medium
containing the viral particles and 4 µg of polybrene (Sigma) per ml to each 10 cm diameter
plate. A second round of infection was performed 12 hrs later. The medium was changed 30
hrs later, and the cells were selected for 15 days in the presence of 250 µg of G418 per ml.
2.2.4.3 Synchronization of culture cells. Exponentially growing Ntera-2 cells were synchronized by three different techniques:
* Serum starvation: Ntera-2 cells were starved in DMEM + 0.25% foetal calf
serum (FCS, Gibco-BRL) for 48 hrs to arrest the cells in G0. FCS was then added to stimulate
cell cycle re-entry. After various time periods in the presence of FCS cells were collected and
their cell cycle profiles were analysed.
* Nocodazole block (Nocodazole is a tubulin depolymerising drug
preventing the formation of the mitotic spindle) to arrest the cells at the G2/M boundary.
Nocodazole was added at 100 ng/ml for 16 hrs. The release was done by extensive washing,
at least three times with DMEM.
* Hydroxy-Urea (HU, a nucleotide analogue that prevents DNA synthesis) to
arrest the cells at the G1/S boundary. HU was added at 100 mM for 16 hrs. The release was
done by extensive washing, at least three times with DMEM.
2.2.4.4 Mitogenic response. Mitogenic response of Ntera-2 cells either overexpressing or depleted of Ringo3 or
Ringo1 was performed as previously described (Yasumoto et al., 2001). Cells were seeded at
105 cells per well (24-well plate) and labelled with [3H]Thymidine (6.7 Ci/mM, 2 µCi/ml) for
48 hrs. After labelling, cells were washed three times with 5% trichloroacetic acid at 0°C and
resuspended in 0.1 M NaOH. The incorporated radioactivity was measured by using a
scintillation counter.

Materials and Methods
48
2.2.4.5 Small Interference RNA. RNA interference assays for Ringo1 and Ringo3 were conducted through the
construction of siRNAs (McManus and Sharp, 2002). The siRingo3 oligonucleotides and
siRingo1 oligonucleotides were synthesized, purified, and duplexed using the siSilencer Kit
(Ambion). As a negative control, a luciferase siRNA was used. For siRNA transfection,
subconfluent Ntera-2 cells (105 cells/ml) were seeded in 10 cm dishes.
Cells were transfected using si-PORT Lipid (Ambion), with 0.6 nM of siRingo 3
oligonucleotides, siRingo1 oligonucleotides or siLuc oligonucleotides per plate. Cells were
incubated for 72 hrs before being collected and analysed by immunobloting or FACS.
The sequence targeted by the siRNA is listed below.
siRingo3: (5’-GTACGAAATTTTTTCCATGG-‘3)
siRingo1: (5’-CCGCTCTCGCATACCCTTG-‘3)
siLuc: (5’-CGTACGCGGAATACTTGGA-‘3)
2.2.4.6 Flow cytometry. Ntera-2 cells were trypsinized, pelleted by centrifugation at 1000 xg and washed twice
with 4 ml of PBS. After another centrifugation at 1000 xg, the cells were fixed with 4 ml of
ice cold ethanol (70%) for 2 hrs at -20°C, cells were then spun and washed with 4 ml of PBS,
to finally be resuspended in 1 ml of PBS containing RNAse A (25 µg/106 cells, stock at 10
mg/ml, Roche), 0,05% propidium iodide (stock at 10 mg/ml, Boehringer) and 0,1% of Triton
X-100 (Sigma) and incubated in the dark for 30 min at 37°C. Cells were analysed in the
cytometer, data acquisition and statistical analyses were done using the FACScan (Beckson &
Dickinson) using the software Cell Quest Pro.
2.2.4.7 Cell growth curves. Ntera-2 cells were seeded at a density of 106 cells/plate (10 cm plates) and infected
with either the retrovirus expressing the empty vector, Ringo1 or Ringo3. Every 24 hrs after
transfection, cells were trypsinized and counted by trypan blue exclusion. The average values
of three plates are represented (±SEM). Remaining cells for the last time point were pooled,
lysed, subjected to SDS-PAGE, and immunoblotted.

Materials and Methods
49
2.2.4.8 Cell and tissue extracts and immunoblotting. Cell extracts were prepared as reported previously (Ambrosino et al., 2003). Cells
were washed in cold PBS and lysed in 100 µl IP buffer for a 10 cm plate. After 10 min of
incubation on ice, lysates were centrifuged for 20 min at 10,000 x g. Mouse tissues were
homogenized in 5 volumes of IP buffer, incubated on ice for 10 min, sonicated three times for
10 sec each, and centrifuged for 15 min at 10,000 xg. For immunoblotting, about 75 µg of
total protein was separated by SDS-PAGE gel electrophoresis and transferred to nitrocellulose
using a semidry blotting apparatus as described in section 2.2.3.6.
2.2.5 Biocomputing. DNA sequence analysis and comparisons were done with the DNASTAR program
(London). Protein sequences were obtained from the NCBI (National Center for
Biotechnology Information) and the EMBL databases. Secondary structure predictions were
done using the program PHD (Rost, 1996). The SMART program was used to find domains
(http://smart.embl-heidelberg.de). Protein sequence alignments were made using the clustal X
program (Jeanmougin et al., 1998).
The mammalian homologues of RINGO were found using the BLAST (Altschul et al.,
1997) search of Ensembl (http://www.ensembl.org).

Results
50
3. Results. 3.1 Involvement of the kinesin-like protein Xkid in
Xenopus oocyte maturation. Xkid was found in a screen aimed at identifying proteins synthesized de novo during
Xenopus oocyte maturation and hence have a potential role in meiotic regulation. The screen
was based on the differential association of mRNA with polysomes in progesterone treated
versus non treated oocytes. Using this approach, two full-length cDNAs were isolated, clone
8-2b and clone 8-5, containing two ORFs of 650 and 651 amino acids respectively. These
proteins were 92% identical at the amino acid level suggesting that they might correspond to
pseudoalloploid alleles in the tetraploid Xenopus genome (Kobel and Du Pasquier, 1979).
Database searches showed that the predicted proteins were very similar to the human kinesin-
like DNA binding protein kid and were named Xkid (Figure 3.1 and 3.2). All the subsequent
studies were made using the Xkid 8-5 clone. The Xkid protein has been shown to be present
in meiosis II, but not in G2-arrested Xenopus oocytes (Antonio et al., 2000), suggesting that
the Xkid maternal mRNA becomes translated during oocyte maturation. The aim of this
project was to investigate the importance of Xkid for the Xenopus oocyte maturation.
3.1.1 Xkid synthesis is not required for meiosis I entry of Xenopus
oocytes. We performed a time-course analysis of Xkid synthesis upon progesterone stimulation
of the oocytes. The Xkid protein started to be detected at GVBD and continued accumulating
until the metaphase II arrest, where it reached its highest levels (Figure 3.3).
We decided to assay the stability of Xkid during the meiotic maturation. Oocytes that
underwent GVBD where treated with CHX, at different times after GVBD. Interestingly, we
discovered that Xkid was very unstable during meiosis I, but its stability dramatically
increased in meiosis II-arrested oocytes (Figure 3.4A). These observations suggested a
putative role for Xkid during meiotic maturation.

1 11410 20 30 40 50 60 70 80 90 100MAAGGSTQQRRREMAAASAAAISGAGRCRLSKIGATRRPPPARVRVAVRLRPFVDG-TAGASDPPCVRGMDSCSLEIANWRNHQETLKYQFDAFYGERSTQQDIYAGSVQPILRKid (1)MVLTGPLQ---------RE--S-VSMAKRVSMLDQHKKSSCARVRVAVRLRPYMDEKDEAKATTVCVRGLDSQSLEIVNWRNQLETMQYQFDAFYGDSASQREIYMGSVCYILPXkid (1)
114 227120 130 140 150 160 170 180 190 200 210RHLLEGQNAKVVLAYGPTGAGKT-THAGQPRATWGDPAGSHGPPAAHKGGGCR--GPAMGLSVTMSYLEIYQEKVLDLLDPASGDLVIREDCRGNILIPGLSQKPISSFADFERKid (113)PHLLIGQNAS-VFAYGPTGAGKTHTMLGNPNQPGVIPRAVRDLLQMSRTAASAPENENWTYTINMSYVEIYQEKVMDLLEPKNKDLPIREDKDHNILIPGVTQKMINSFADFDEXkid (102)
227 340240 250 260 270 280 290 300 310 320 330RHFLPASRNRTVGATRLNQRSSRSHAVLLVKVDQRERLAPFRQREGKLYLIDLAGSEDNRRTGNKGLRLKESGAINTSLFALGKVVDALNQGLPRVPYRDSKLTRLLQDSLGGSKid (223)EHFIPASQNRTVASTKLNDRSSRSHAVLLIKVQKSQQVVPFRQLTGKLYLIDLAGSEDNRRTGNQGIRLKESGAINSSLFTLSKVVDALNQGLPRIPYRDSKLTRLLQDSLGGSXkid (214)
340 453350 360 370 380 390 400 410 420 430 440SAHSILIANIAPERRFYLDTVSALNFAARSKEVINRPFTNESLQPHALGPVKLSQKELLGPPEAKRARGPEEEEIGSPEPMASSSLCLPETQPPTEAKAAWTRPCGAPPQLGPSKid (336)SAHSVMITNIAPEQTYYFDTLTALNFAAKSKQIINKPFSQETTQTVVQPAMKRPRE------ETGHIAGSQKRKKSKNDSTESSPNSSMDTAGKQKLNLATLDPAVVERLLKLDXkid (327)
453 566460 470 480 490 500 510 520 530 540 550SACLP--GEPGAPLLSTPKRERMVLMKTVEEKDLEIERLKTKQKELEAKMLAQKAEEKR----------TIVPQCSGPLSHRTVTGAKPLKKAVVMPLQLIQEQAASPNAEIHIKid (449)DKILTEKGKKKAQLLSTPKRERMALLKKWEESQMEIERLKEKQKELEQKAMEAEARLEKSNNSDLSDSSVSENTFRAPLRGRNTSTAKVKKVLRVLPMQGNSQLQSTVEEGIPVXkid (434)
555 668560 570 580 590 600 610 620 630 640 650EQAASPNAEIHILKNKGRKRKLESLDALEPEEKAEDCWELQISPELLAHGRQKILDLLNEGSARDLRSLQRIGPKKAQLIVGWRELHGPFSQVEDLERVEGITGKQMESFLKANKid (539)QLQSTVEEGIPVFEKK-KKKKQVTCEGLENQP----TWEMNMRTDLLESGKERILKLLNTGSVKELKSLQRIGDKKAKLIIGWREVNGPFKNVEELACLEGISAKQVSSFIKANXkid (536)
Figure 3.1: Alignment of the Human and Xenopus kid proteins. The alignment was done using the Clustal X program. The consensussequences are shown in red
51
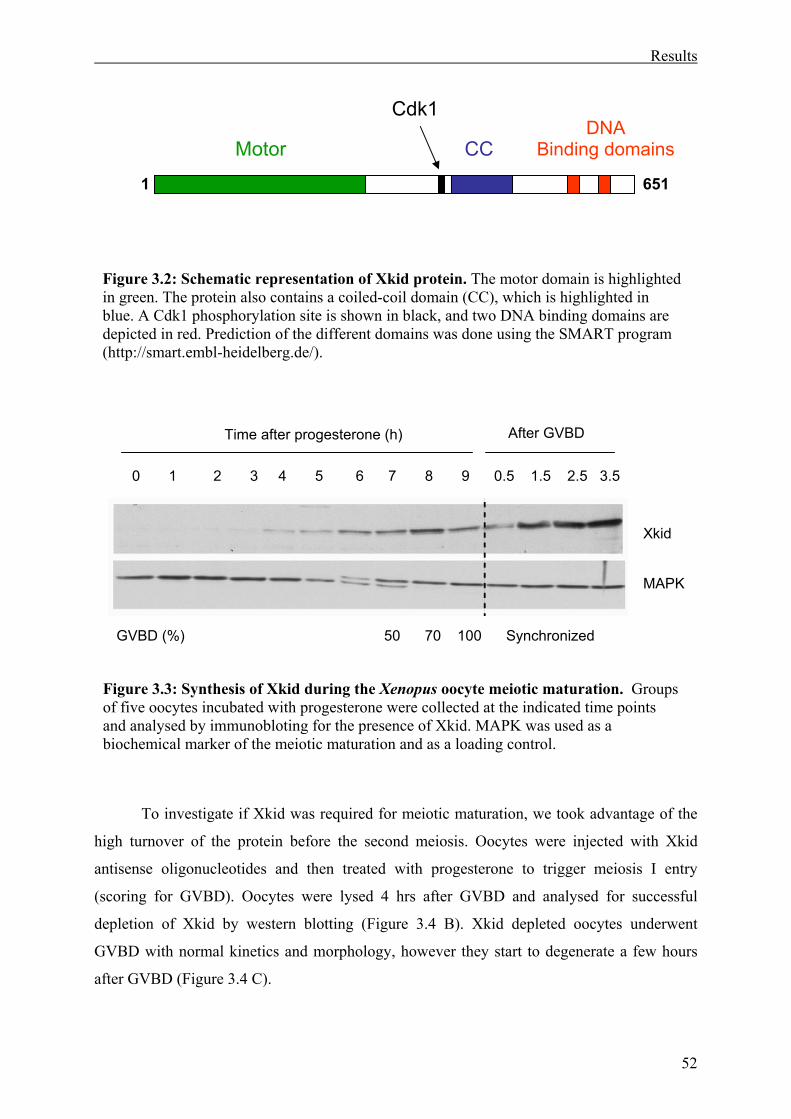
Results
52
To investigate if Xkid was required for meiotic maturation, we took advantage of the
high turnover of the protein before the second meiosis. Oocytes were injected with Xkid
antisense oligonucleotides and then treated with progesterone to trigger meiosis I entry
(scoring for GVBD). Oocytes were lysed 4 hrs after GVBD and analysed for successful
depletion of Xkid by western blotting (Figure 3.4 B). Xkid depleted oocytes underwent
GVBD with normal kinetics and morphology, however they start to degenerate a few hours
after GVBD (Figure 3.4 C).
Figure 3.3: Synthesis of Xkid during the Xenopus oocyte meiotic maturation. Groups of five oocytes incubated with progesterone were collected at the indicated time points and analysed by immunobloting for the presence of Xkid. MAPK was used as a biochemical marker of the meiotic maturation and as a loading control.
0 1 2 3 4 5 6 7 8 9 0.5 1.5 2.5 3.5
MAPK
After GVBD
GVBD (%) 50 70 100 Synchronized
Time after progesterone (h)
Xkid
1 651
CCMotor
Cdk1DNA
Binding domains
Figure 3.2: Schematic representation of Xkid protein. The motor domain is highlightedin green. The protein also contains a coiled-coil domain (CC), which is highlighted in blue. A Cdk1 phosphorylation site is shown in black, and two DNA binding domains are depicted in red. Prediction of the different domains was done using the SMART program (http://smart.embl-heidelberg.de/).
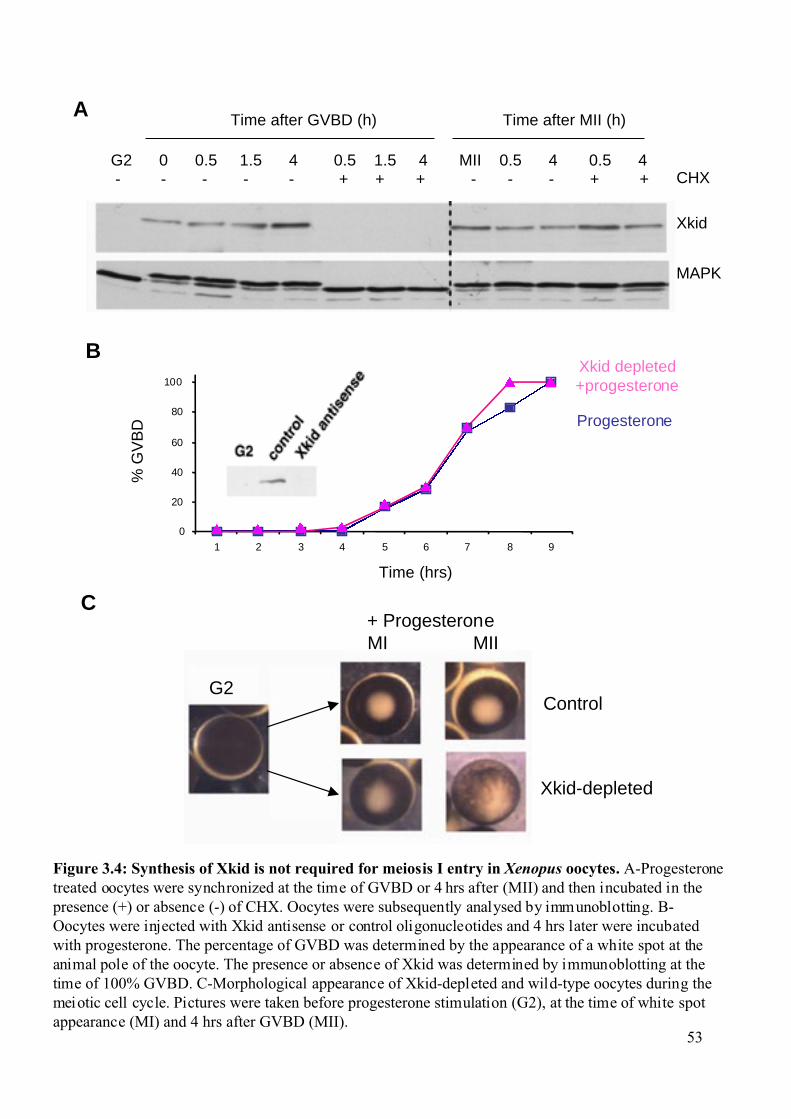
Figure 3.4: Synthesis of Xkid is not required for meiosis I entry in Xenopus oocytes. A-Progesteronetreated oocytes were synchronized at the time of GVBD or 4 hrs after (MII) and then incubated in thepresence (+) or absence (-) of CHX. Oocytes were subsequently analysed by immunoblotting. B-Oocytes were injected with Xkid antisense or control oligonucleotides and 4 hrs later were incubatedwith progesterone. The percentage of GVBD was determined by the appearance of a white spot at theanimal pole of the oocyte. The presence or absence of Xkid was determined by immunoblotting at thetime of 100% GVBD. C-Morphological appearance of Xkid-depleted and wild-type oocytes during themeiotic cell cycle. Pictures were taken before progesterone stimulation (G2), at the time of white spotappearance (MI) and 4 hrs after GVBD (MII).
Control
Xkid-depleted
+ Progesterone
Time (hrs)
BXkid depleted+progesterone
0
20
40
60
80
100
1 2 3 4 5 6 7 8 9
Progesterone
% G
VB
D
C
G2 0 0.5 1.5 4 0.5 1.5 4 MII 0.5 4 0.5 4 - - - - - + + + - - - + +
Time after GVBD (h) Time after MII (h)
MAPK
Xkid
CHX
MI MII
G2
A
53

Results
54
This degeneration of the oocytes correlates with apoptotic death as shown by the
cleavage of the cytoskeletal protein Fodrin (a marker of apoptosis in Xenopus oocytes)
(Figure 3.5). These results indicate that Xkid-depleted oocytes are able to enter meiosis I
normally, but are probably unable to arrest at metaphase II.
To investigate whether Xkid depleted oocytes arrest or not at metaphase II, some
markers of oocyte maturation were analysed. As shown in figure 3.5, Xkid depleted oocytes
underwent pre-MPF activation normally as indicated by the disappearance of the anti-
phospho-Cdk1/Tyr15 signal present in G2 arrested oocytes and by the disappearance of the
slower migrating Cdk1 band. We could also detect activation of the MAPK pathway, by
phosphorylation of both MAPK and Rsk, which are easily identifiable by the retardation in
the electrophoretic mobility of these two proteins. Finally, activation of the Plx1/Cdc25C
pathway occurred normally as shown by the hyper-phosphorylation of the Cdc25C.
3.1.2 Xkid-depleted oocytes do not re-activate Cdk1-Cyclin B after
meiosis I and undergo DNA replication. We investigated the levels of H1K activity during oocyte maturation. As expected, the
H1K activity was high at GVBD and transiently inactivated in wild type oocytes during the
meiosis I to meiosis II transition. In contrast, the H1K activity decreased after GVBD in Xkid-
depleted oocytes at about the same rate as in wild type oocytes but then stayed low,
suggesting that these oocytes did not enter into meiosis II (Figure 3.6). As the
dephosphorylation of Cdk1 happens normally, we investigated the level of B-type cyclins. As
shown in figure 3.6, the inability to re-activate MPF after GVBD correlated with significantly
reduced levels of B-type cyclins in oocytes that undergo maturation in the absence of Xkid.
However, bulk protein synthesis, as determined by 35S-methionine incorporation, was
unchanged in control and Xkid-depleted oocytes (Figure 3.6).
Previous work has shown that when the meiotic cell cycle is blocked after GVBD, the
oocytes proceed from meiosis I to an interphase-like state in which DNA replication is de-
repressed (Furuno et al., 1994). To address whether that Xkid depleted oocytes enter this
interphase-like state, we assayed the ability of Xkid depleted oocytes to undergo DNA
replication after GVBD by investigating their ability to incorporate radiolabelled dCTP.
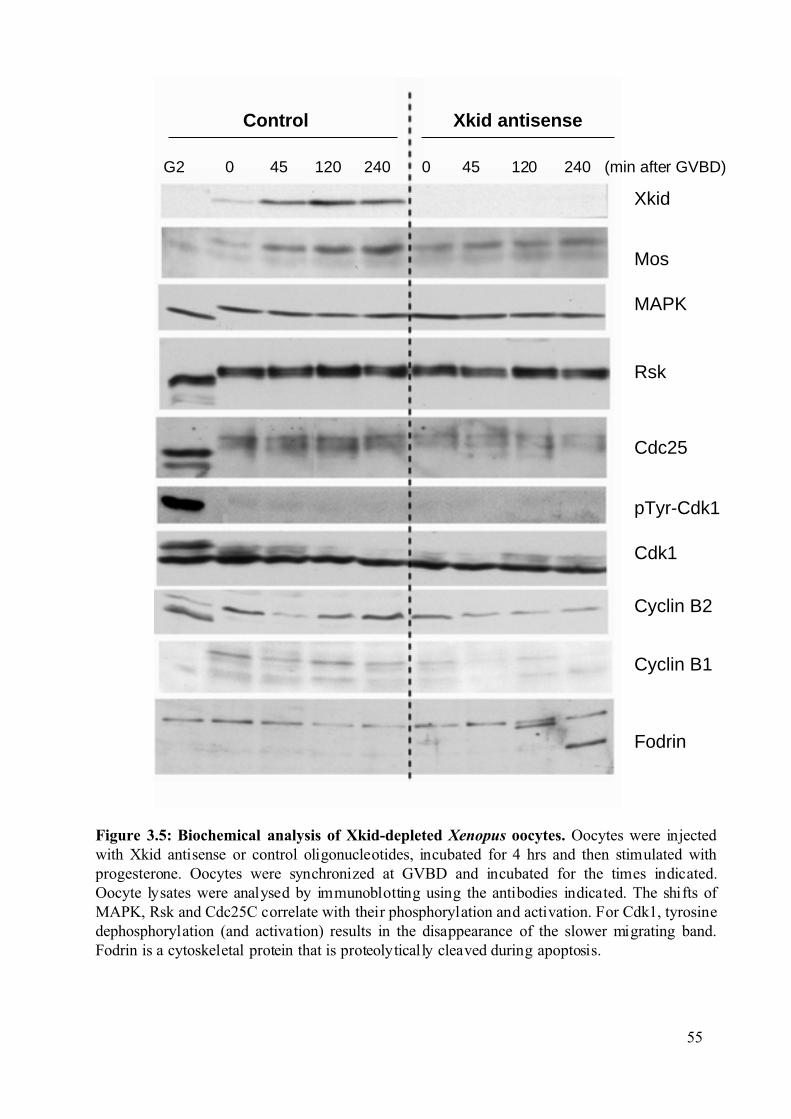
Control Xkid antisense
G2 0 45 120 240 0 45 120 240 (min after GVBD)
Figure 3.5: Biochemical analysis of Xkid-depleted Xenopus oocytes. Oocytes were injectedwith Xkid antisense or control oligonucleotides, incubated for 4 hrs and then stimulated withprogesterone. Oocytes were synchronized at GVBD and incubated for the times indicated.Oocyte lysates were analysed by immunoblotting using the antibodies indicated. The shifts ofMAPK, Rsk and Cdc25C correlate with their phosphorylation and activation. For Cdk1, tyrosinedephosphorylation (and activation) results in the disappearance of the slower migrating band.Fodrin is a cytoskeletal protein that is proteolytically cleaved during apoptosis.
pTyr-Cdk1
Xkid
Mos
MAPK
Rsk
Fodrin
Cyclin B1
Cyclin B2
Cdk1
Cdc25
55
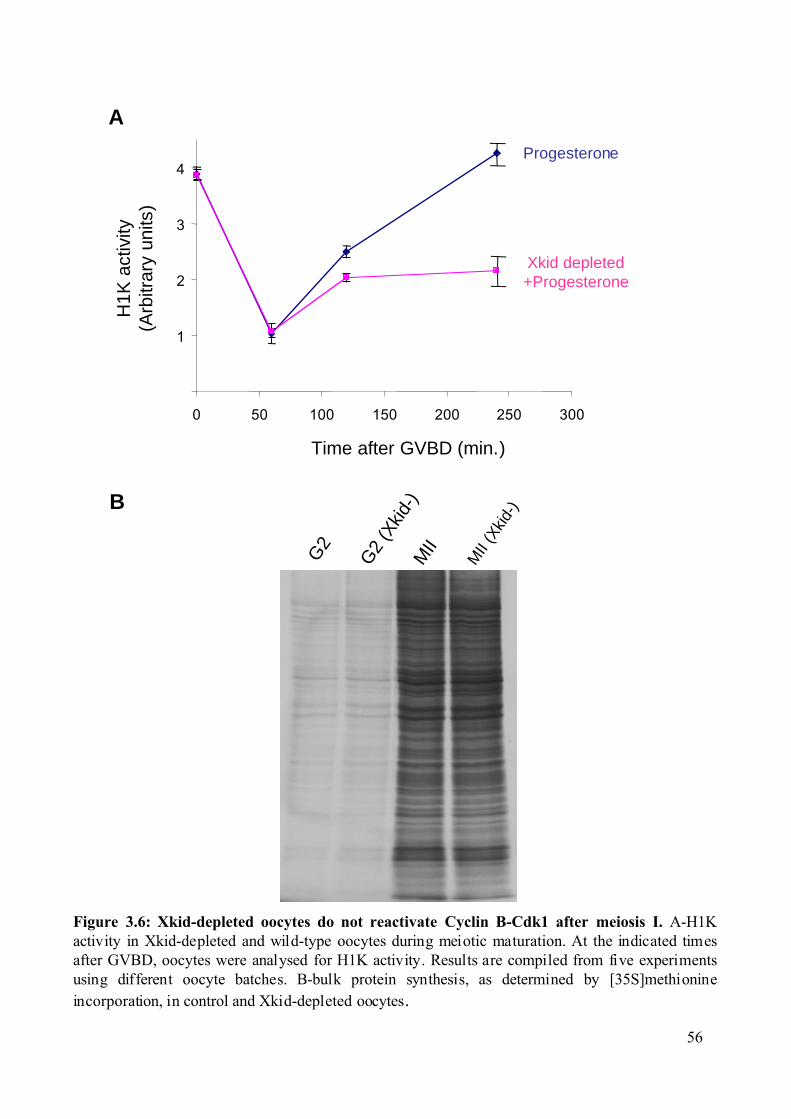
1
2
3
4
0 50 100 150 200 250 300
Time after GVBD (min.)
H1K
act
ivity
(Arb
itrar
y un
its)
A
Figure 3.6: Xkid-depleted oocytes do not reactivate Cyclin B-Cdk1 after meiosis I. A-H1Kactivity in Xkid-depleted and wild-type oocytes during meiotic maturation. At the indicated timesafter GVBD, oocytes were analysed for H1K activity. Results are compiled from five experimentsusing different oocyte batches. B-bulk protein synthesis, as determined by [35S]methionineincorporation, in control and Xkid-depleted oocytes.
B
MII
(Xki
d-)
G2
G2
(Xki
d-)
MII
Progesterone
Xkid depleted+Progesterone
56

Results
57
We could detect DNA replication in the Xkid-depleted oocytes, suggesting that these
oocytes entered an interphase-like state (Figure 3.7A). In order to know if this level of DNA
replication was significant, we compared it to the effect of CHX, which is known to induce
DNA replication, if added 10-30 minutes after GVBD. CHX induces DNA replication
through the inhibition of re-synthesis of B-type cyclins, which repress DNA replication
machinery after meiosis I (Hochegger et al., 2001).
The level of DNA replication in the Xkid-depleted maturing oocytes was similar to
that observed upon addition of CHX to wild type oocytes 30 minutes after GVBD.
Furthermore the incorporation of radiolabelled dCTP was sensitive to aphidicolin (an inhibitor
of the DNA polymerase α and hence of the DNA replication machinery), indicating that DNA
replication rather than DNA repair was occurring (Figure 3.7B).
3.1.3 Xkid is not required for meiosis I but to enter into meiosis II. To complement the biochemical analysis of Xkid depletion, we decided to investigate
the putative morphological effect by confocal microscopy. Oocytes were fixed in methanol at
GVBD, 30, 60, 90, 120 and 240 min after GVBD and processed for immunofluorescence.
Confocal microscopy analysis of control and Xkid depleted oocytes showed that both in the
presence and absence of Xkid, the meiosis I spindle assembled and chromosomes condensed
and aligned at the metaphase plate (Figure 3.8). The process of meiosis I spindle formation
observed in both samples was similar to the one already described (Furuno et al., 1994). Just
after the appearance of the white spot which is characteristic of GVBD, a large MTOC
assembled at the basal side of the germinal vesicle irradiating microtubules that capture the
condensed chromosomes. In both Xkid depleted and control oocytes, the spindle rotated and
during this process and the chromosomes that were initially scattered through the spindle
aligned at the metaphase plate. Around 60 min after GVBD, the meiosis I spindle reached a
final position, perpendicular to the cortex and the chromosomes aligned in the equatorial
plate. The control spindles proceeded to anaphase and formed a meiosis II spindle following a
similar pathway as for meiosis I. Four hours after GVBD most of the oocytes were in
metaphase II
In contrast, at the time of meiosis II, oocytes that did not synthesise Xkid were clearly
different from wild type oocytes. When analysed 4 hrs after GVBD, most of the Xkid-
depleted oocytes contained no spindle, although the chromosomes were partially condensed
they lay scattered under the oocyte cortex with no associated microtubules.
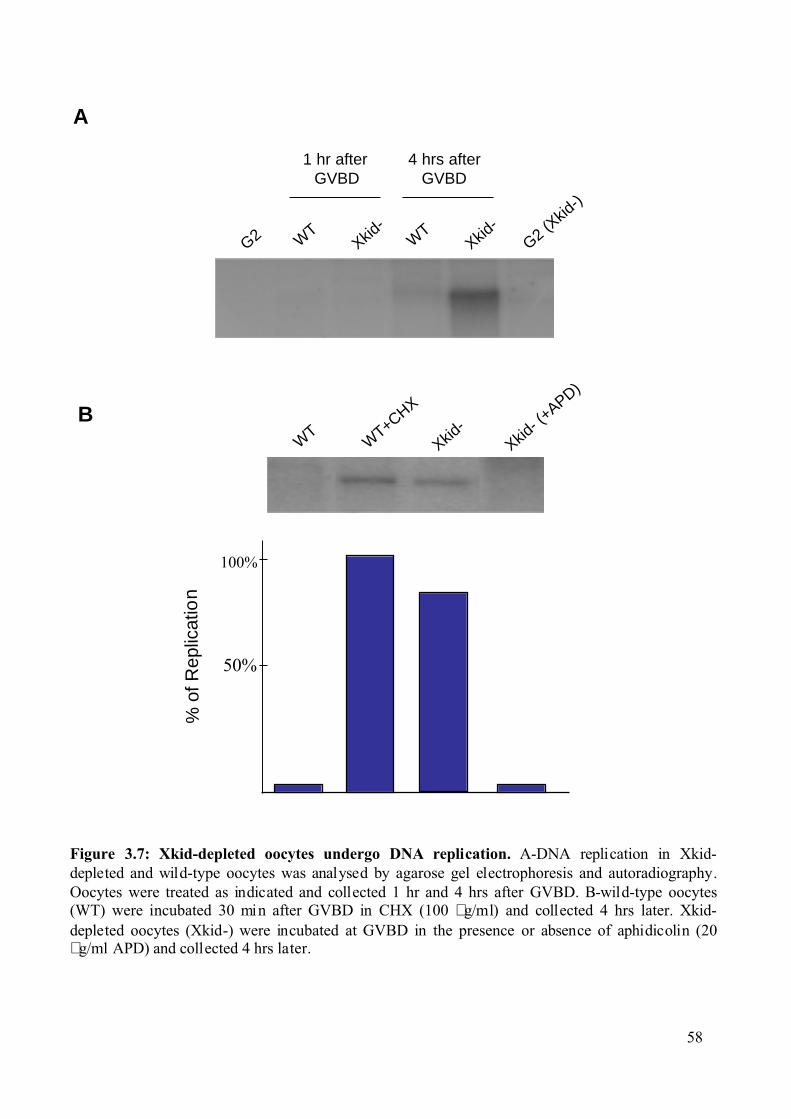
G2
Figure 3.7: Xkid-depleted oocytes undergo DNA replication. A-DNA replication in Xkid-depleted and wild-type oocytes was analysed by agarose gel electrophoresis and autoradiography.Oocytes were treated as indicated and collected 1 hr and 4 hrs after GVBD. B-wild-type oocytes(WT) were incubated 30 min after GVBD in CHX (100 µg/ml) and collected 4 hrs later. Xkid-depleted oocytes (Xkid-) were incubated at GVBD in the presence or absence of aphidicolin (20µg/ml APD) and collected 4 hrs later.
B
1 hr after GVBD
4 hrs afterGVBD
% o
f Rep
licat
ion
100%
50%
WT
Xkid-
G2 (Xkid
-)
WT
Xkid-
WT
WT+CHX
Xkid- (
+APD)
Xkid-
A
58
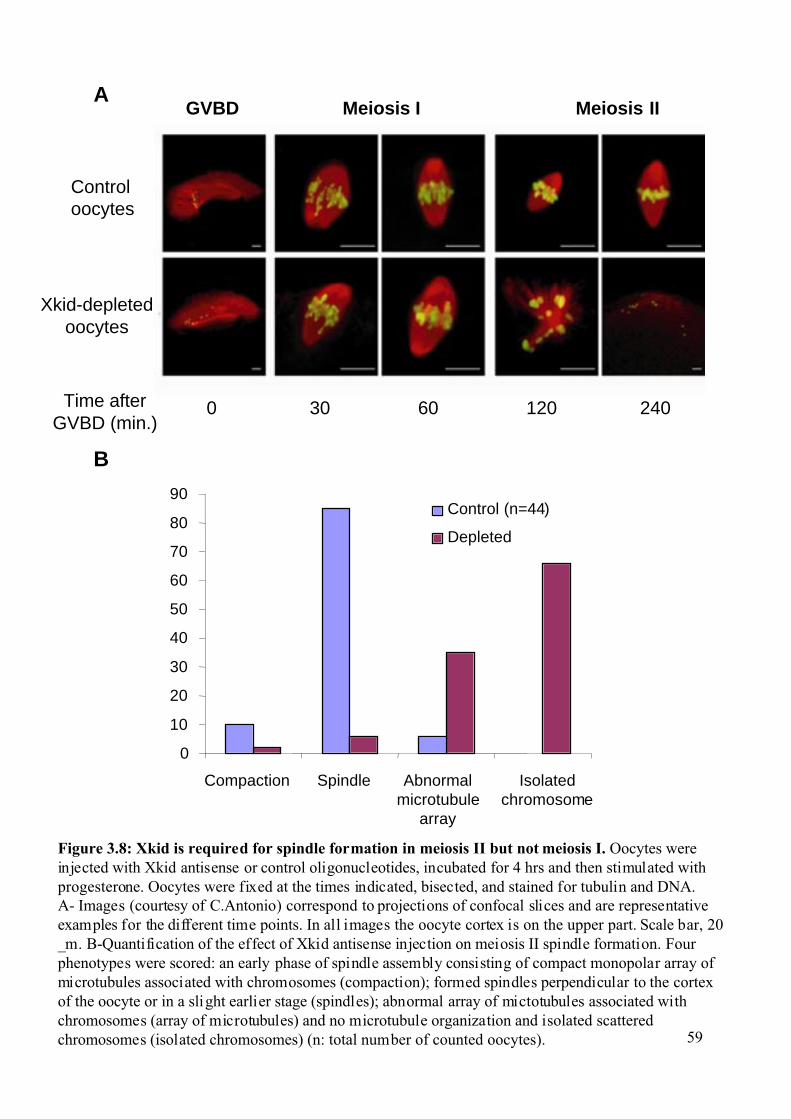
GVBD Meiosis I Meiosis II
Xkid-depletedoocytes
Controloocytes
Time afterGVBD (min.)
0 30 60 120 240
Figure 3.8: Xkid is required for spindle formation in meiosis II but not meiosis I. Oocytes wereinjected with Xkid antisense or control oligonucleotides, incubated for 4 hrs and then stimulated withprogesterone. Oocytes were fixed at the times indicated, bisected, and stained for tubulin and DNA.A- Images (courtesy of C.Antonio) correspond to projections of confocal slices and are representativeexamples for the different time points. In all images the oocyte cortex is on the upper part. Scale bar, 20_m. B-Quantification of the effect of Xkid antisense injection on meiosis II spindle formation. Fourphenotypes were scored: an early phase of spindle assembly consisting of compact monopolar array ofmicrotubules associated with chromosomes (compaction); formed spindles perpendicular to the cortexof the oocyte or in a slight earlier stage (spindles); abnormal array of mictotubules associated withchromosomes (array of microtubules) and no microtubule organization and isolated scatteredchromosomes (isolated chromosomes) (n: total number of counted oocytes).
B
A
0
10
20
30
40
50
60
70
80
90
Compaction Spindle Abnormalmicrotubule
array
Isolatedchromosome
Control (n=44)
Depleted
59

Results
60
These results confirmed the biochemical analysis indicating that synthesis of Xkid was
required for oocytes to complete the meiotic cell cycle and arrest at metaphase II.
3.1.4 Ectopic expression of Xkid allows Xkid-depleted oocytes to
complete meiotic maturation. To demonstrate the specificity of the antisense phenotype, a rescue experiment was
performed. Oocytes were injected with Xkid morpholino oligonucleotides and then injected
again with an Xkid mRNA that contained a Myc-tag N-terminal to the Xkid ORF. The
mopholino oligonucleotides target a region upstream of the start codon of the protein and act
by preventing the ribosome from a haching to the mRNA. The Myc-tagged Xkid mRNA
injected into these oocytes does not contain Xkid untranslated regions and is not targeted by
the morpholino oligonucleotides.
Oocytes injected only with morpholino displayed the same phenotype as the oocytes
depleted of Xkid by ‘standard’ antisense oligos: no MPF reactivation and low level of B type
cyclin (Figure 3.9). When morpholino oligos and Myc-Xkid mRNA were co-injected, we
found that the expression of myc-Xkid allowed the accumulation of B-type cyclins to normal
levels and the re-activation of MPF after GVBD in Xkid-depleted oocytes, suggesting that
these oocytes were now able to enter meiosis II. Our results indicate that the absence of Xkid
may interfere with the re-activation of MPF during the meiosis I to meiosis II transition.
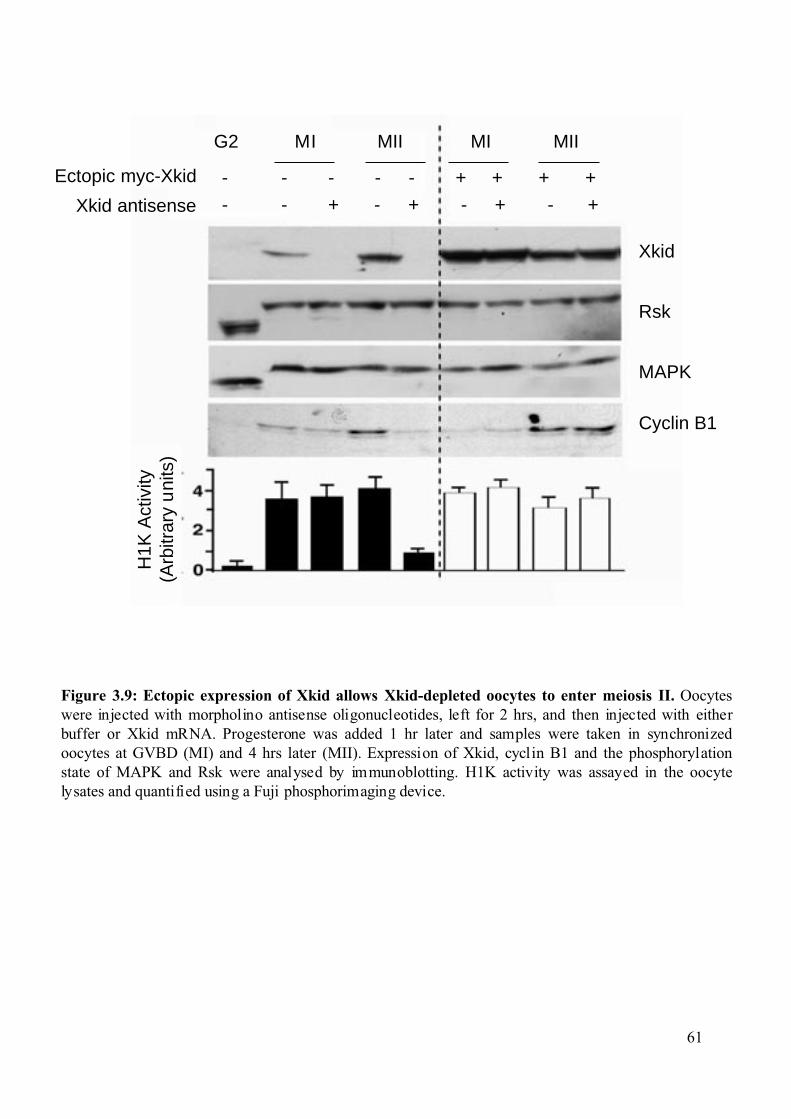
Figure 3.9: Ectopic expression of Xkid allows Xkid-depleted oocytes to enter meiosis II. Oocyteswere injected with morpholino antisense oligonucleotides, left for 2 hrs, and then injected with eitherbuffer or Xkid mRNA. Progesterone was added 1 hr later and samples were taken in synchronizedoocytes at GVBD (MI) and 4 hrs later (MII). Expression of Xkid, cyclin B1 and the phosphorylationstate of MAPK and Rsk were analysed by immunoblotting. H1K activity was assayed in the oocytelysates and quantified using a Fuji phosphorimaging device.
G2 MI MII MI MII
Ectopic myc-Xkid
Xkid antisense
Xkid
Rsk
MAPK
Cyclin B1
H1K
Act
ivity
(Arb
itrar
y u
nits
)- - - - - + + + +
- - + - + - + - +
61

Results
62
3.1.5 Ectopic expression of an Xkid mutant lacking the DNA
binding domain allows Xkid-depleted oocytes to complete meiotic
maturation. Xkid has been shown to be required for chromosome alignment at the metaphase plate
of the mitotic spindle (Antonio et al., 2000; Funabiki and Murray, 2000).
We investigated whether the inability to reactivate MPF after meiosis I, in Xkid
depleted oocytes was a direct consequence of Xkid absence or a consequence of DNA
misalignment. To address this question and discover whether DNA binding of Xkid was
required for meiosis II entry, we created a mutant that lacks the C-terminal DNA binding
domain. This Xkid protein is unable to bind DNA and to align the chromosome. Interestingly,
the Xkid mutant lacking the C-terminal DNA binding domain could also rescue the Xkid
antisense phenotype this was confirmed by the normal morphological appearance of the
oocytes and their biochemical markers, such as the accumulation of normal levels of B type-
cyclins (Figure 3.10).
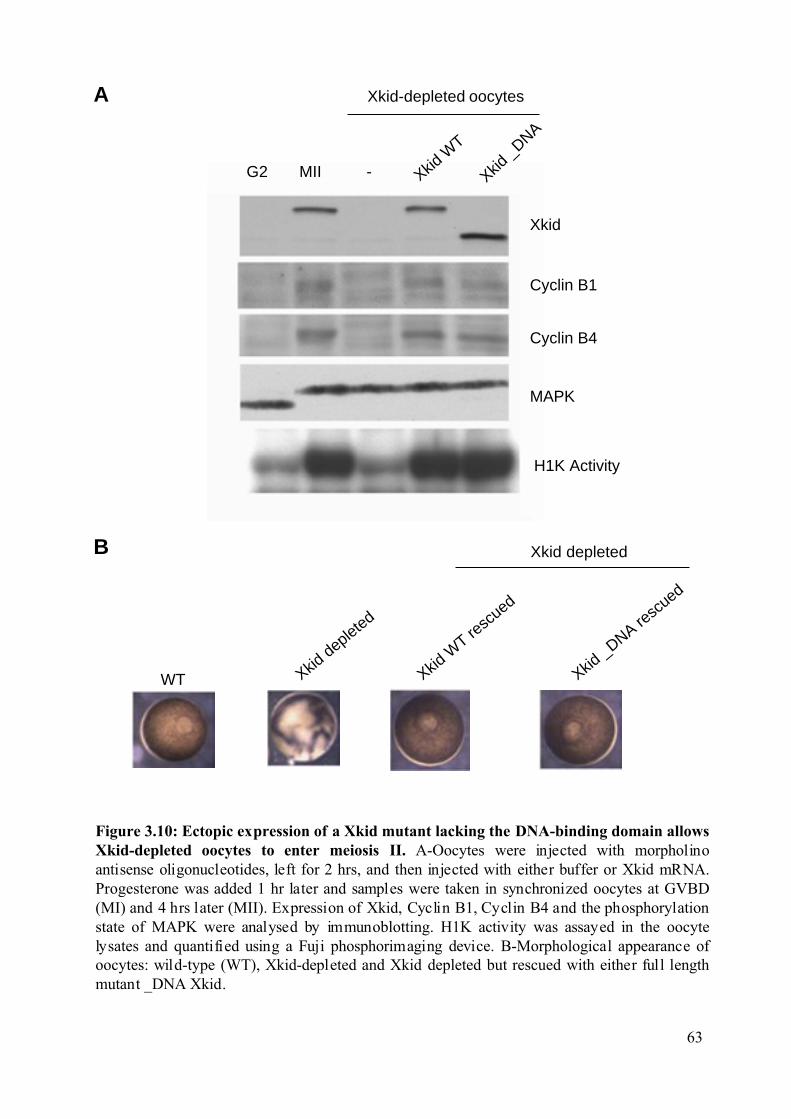
Figure 3.10: Ectopic expression of a Xkid mutant lacking the DNA-binding domain allowsXkid-depleted oocytes to enter meiosis II. A-Oocytes were injected with morpholinoantisense oligonucleotides, left for 2 hrs, and then injected with either buffer or Xkid mRNA.Progesterone was added 1 hr later and samples were taken in synchronized oocytes at GVBD(MI) and 4 hrs later (MII). Expression of Xkid, Cyclin B1, Cyclin B4 and the phosphorylationstate of MAPK were analysed by immunoblotting. H1K activity was assayed in the oocytelysates and quantified using a Fuji phosphorimaging device. B-Morphological appearance ofoocytes: wild-type (WT), Xkid-depleted and Xkid depleted but rescued with either full lengthmutant _DNA Xkid.
G2 MII -
Xkid-depleted oocytes
Xkid
Cyclin B1
Cyclin B4
MAPK
H1K Activity
A
B
Xkid
WT
Xkid
_DNA
Xkid depleted
WT Xkid depleted
Xkid W
T resc
ued
Xkid _DNA re
scued
63

Results
64
3.2 RINGO, a new family of cell cycle regulators. RINGO was found in a screen performed to identify proteins that can trigger Xenopus
oocyte maturation. The screen was based on the identification of mRNAs that when injected
in oocytes were able to trigger maturation in the absence of progesterone stimulation. Two
clones named ls26 and ls27 were identified, which were predicted to encode proteins 88%
identical to each other. They were called RINGO.
RINGO was able to induce MPF activation in Xenopus oocytes independently of
protein synthesis. RINGO was also shown to be required for Xenopus oocyte maturation,
based on the inhibitory effect of RINGO antisense. Biochemically RINGO has been shown to
bind and activate the protein kinases Cdk1 and Cdk2 (Ferby et al., 1999; Lenormand et al.,
1999; Karaiskou et al., 2001).
Cdk1 and Cdk2 should be bound to regulatory subunits named cyclins as well as
phosphorylated on a conserved thr located in the T-loop for full enzymatic activity. Cdk1 and
Cdk2-cyclin complexes can be inactivated by phosphorylation on the catalytic cleft-located
Thr14 and Tyr15 residues or by association with inhibitory subunits such as Cip1.
We decided to investigate the biochemical properties of Cdk activation by RINGO, in
terms of requirement for the phosphorylation of the Cdk and regulation by CKIs. These
studies were all made using the RINGO ls26 clone.
3.2.1 RINGO induces Cdk activation independently of T-loop
phosphorylation. 3.2.1.1 RINGO-induced Cdk1 activation is independent of Thr161
phosphorylation. It has previously been shown that purified recombinant RINGO can stimulate the
kinase activity of Cdk1 expressed in insect cells or in Xenopus oocytes as well as bacterially
produced Cdk1 (Ferby et al., 1999). For full activation of the Cdk1-Cyclin B complex the
Thr161 should be phosphorylated (Morgan, 1997). We investigated whether this
phosphorylation was also required for activation of Cdk1 by RINGO using a mutant in which
the Thr161 of Cdk1 was replaced by an alanine (Ala) (see Materials and Methods).
Purified RINGO or Cyclin B proteins were incubated with purified recombinant Cdk1
pre-incubated in concentrated Xenopus oocyte extracts (Figure 3.11). As expected, Cyclin B
was able to activate the H1K activity of Cdk1 but was a poor activator of Cdk1 T161A.

Results
65
Cyclin B was also unable to activate the kinase-dead Cdk1K33R (this mutation is located in
the ATP binding pocket and interferes with phosphate transfer onto the substrate). In contrast,
RINGO was able to stimulate the H1K activity of wild type Cdk1 and Cdk1T161A to similar
extents, but not in the case of an inactive Cdk1K33R mutant protein which was processed in
parallel. The inability to activate the Cdk1K33R confirms that the observed increased in H1K
activity was not an artefact due to the pre-incubation of Cdk1 in oocyte extracts.
These results indicate that the activation of purified recombinant Cdk1 by purified
RINGO can occur in the absence of Thr161 phosphorylation.
Consistent with these in vitro results, we also found that overexpression of either wild
type Cdk1 or the Cdk1T161A mutant in Xenopus oocytes resulted in similar levels of RINGO
triggered H1K activity (Figure 3.12A). In contrast, the level of H1K activity triggered by
ectopic Cyclin B1 was strongly reduced in Cdk1T161A-expressing oocytes.
These results indicate that RINGO is also able to induce Cdk1 activation
independently of T161-phosphorylation in oocytes. Moreover, RINGO binds Cdk1 under
Figure 3.11: RINGO activates GST-Cdc2 independently of T161 phosphorylation. Bacterially produced GST-Cdk1 wild type (WT), K33R or T161A mutants were pre-incubated in concentrated Xenopus oocyte extracts and recovered on Glutathione-Sepharose beads. After washing, the beads were incubated in the presence of MalE-RINGO (2 µM) or His-cyclin B1 (1 µM) at 30 °C for 15 min prior to H1K assay. .
H1K activity
WT T161A K33R WT T161A K33R
RINGO Cyclin B
Cyclin B GST-Cdk1
Coomassiestaining
Autoradiography
MalE-RINGO
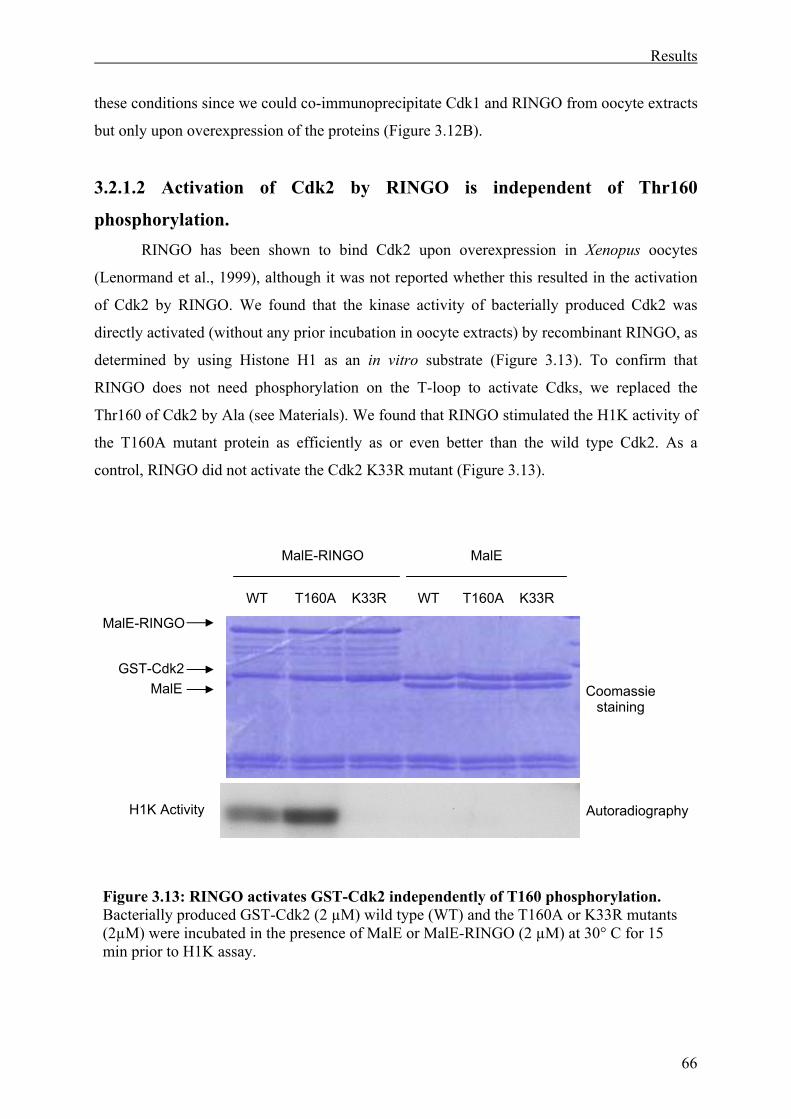
Results
66
these conditions since we could co-immunoprecipitate Cdk1 and RINGO from oocyte extracts
but only upon overexpression of the proteins (Figure 3.12B).
3.2.1.2 Activation of Cdk2 by RINGO is independent of Thr160
phosphorylation. RINGO has been shown to bind Cdk2 upon overexpression in Xenopus oocytes
(Lenormand et al., 1999), although it was not reported whether this resulted in the activation
of Cdk2 by RINGO. We found that the kinase activity of bacterially produced Cdk2 was
directly activated (without any prior incubation in oocyte extracts) by recombinant RINGO, as
determined by using Histone H1 as an in vitro substrate (Figure 3.13). To confirm that
RINGO does not need phosphorylation on the T-loop to activate Cdks, we replaced the
Thr160 of Cdk2 by Ala (see Materials). We found that RINGO stimulated the H1K activity of
the T160A mutant protein as efficiently as or even better than the wild type Cdk2. As a
control, RINGO did not activate the Cdk2 K33R mutant (Figure 3.13).
Figure 3.13: RINGO activates GST-Cdk2 independently of T160 phosphorylation. Bacterially produced GST-Cdk2 (2 µM) wild type (WT) and the T160A or K33R mutants (2µM) were incubated in the presence of MalE or MalE-RINGO (2 µM) at 30° C for 15 min prior to H1K assay.
H1K Activity
MalE-RINGO MalE
MalE-RINGO
MalE GST-Cdk2
Coomassiestaining
Autoradiography
WT T160A K33R WT T160A K33R
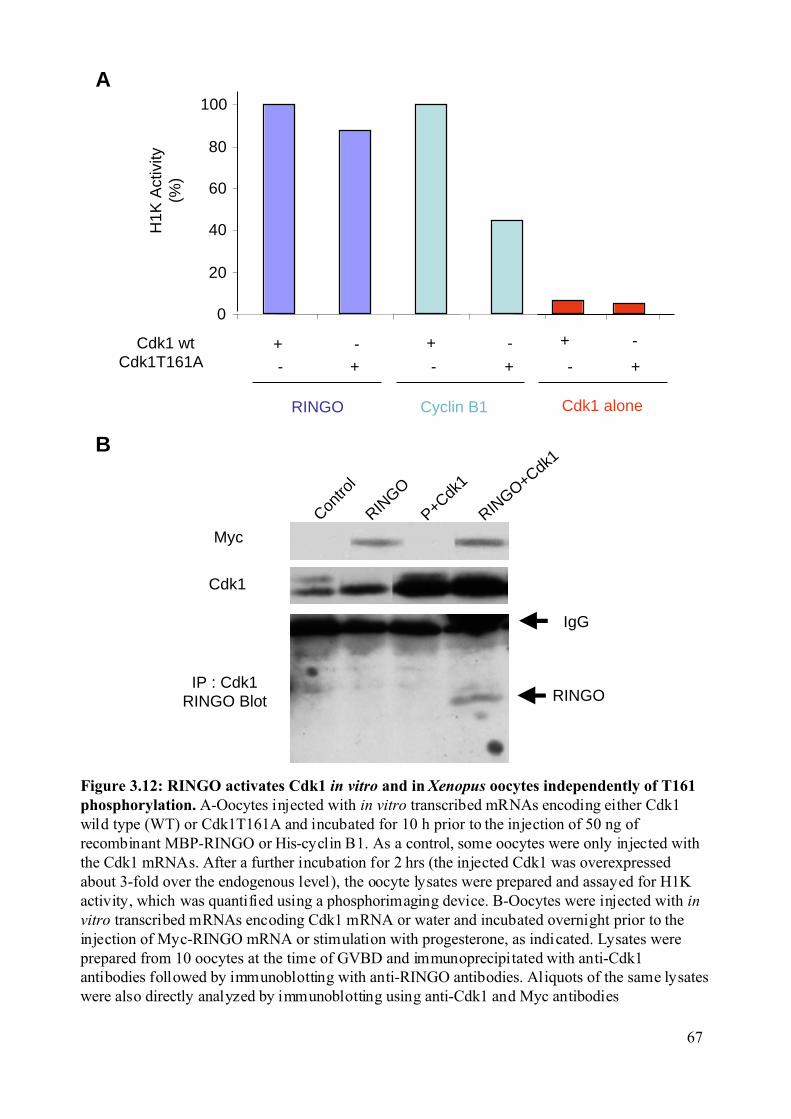
Figure 3.12: RINGO activates Cdk1 in vitro and in Xenopus oocytes independently of T161phosphorylation. A-Oocytes injected with in vitro transcribed mRNAs encoding either Cdk1wild type (WT) or Cdk1T161A and incubated for 10 h prior to the injection of 50 ng ofrecombinant MBP-RINGO or His-cyclin B1. As a control, some oocytes were only injected withthe Cdk1 mRNAs. After a further incubation for 2 hrs (the injected Cdk1 was overexpressedabout 3-fold over the endogenous level), the oocyte lysates were prepared and assayed for H1Kactivity, which was quantified using a phosphorimaging device. B-Oocytes were injected with invitro transcribed mRNAs encoding Cdk1 mRNA or water and incubated overnight prior to theinjection of Myc-RINGO mRNA or stimulation with progesterone, as indicated. Lysates wereprepared from 10 oocytes at the time of GVBD and immunoprecipitated with anti-Cdk1antibodies followed by immunoblotting with anti-RINGO antibodies. Aliquots of the same lysateswere also directly analyzed by immunoblotting using anti-Cdk1 and Myc antibodies
A
B
RINGO
Contro
l
P+Cdk1
RINGO
RINGO+Cdk1
Myc
IP : Cdk1RINGO Blot
IgG
H1K
Act
ivity
(%)
0
20
40
60
80
100
Cdk1 wtCdk1T161A
RINGO Cyclin B1
+ - + -
- + - + - +
+ -
Cdk1 alone
Cdk1
67

Results
68
3.2.2 Cloning of RINGO mammalian homologues. We searched databases for putative homologues of Xenopus RINGO in mammals. We
found a partial match with several Expressed Sequence Tag (ESTs) clones from humans, mice
and fish. However, no related sequence could be found in invertebrates.
In total, we identified two human, two mouse and one Zebrafish homologue of
Xenopus RINGO (Figure 3.14).
The two human clones were available as commercial ESTs. The two mouse
homologues were cloned by RT-PCR from mouse testis mRNA.
We use the following nomenclature to describe the different proteins: Ringo1 and
Ringo2 were initially identified as human proteins and Ringo3 and Ringo4 were initially
identified as mouse proteins.
Interestingly, we noticed that these four proteins were as different to the Xenopus
protein as they were to each other (Figure 3.15A). This suggested that they form a new family
of proteins, as shown by the phylogenetic tree (Figure 3.15B). As shown in the sequence
alignment, the different proteins display a low level of homology, but a central stretch of 100
amino acids appeared to be highly conserved in all members of the family (Figure 3.16).
We also identified the mouse homologue of the Human-Ringo1 clone, which displays
95% of identity at the amino acid level and the human homologue of mouse-Ringo3 and
mouse-Ringo4 that are also very similar to the mouse clone. However, no mouse homologue
has been found for Human-Ringo2. As the mouse and human proteins were almost identical
we decided to work with the original clones.
Despite numerous efforts, we did not manage to clone the full length Ringo4 by RT-
PCR. We only obtained a truncated version of this protein, with the last 80 amino acids
missing. A possible explanation could be that the 3’ end of the putative mRNA is not
correctly annotated in the database.
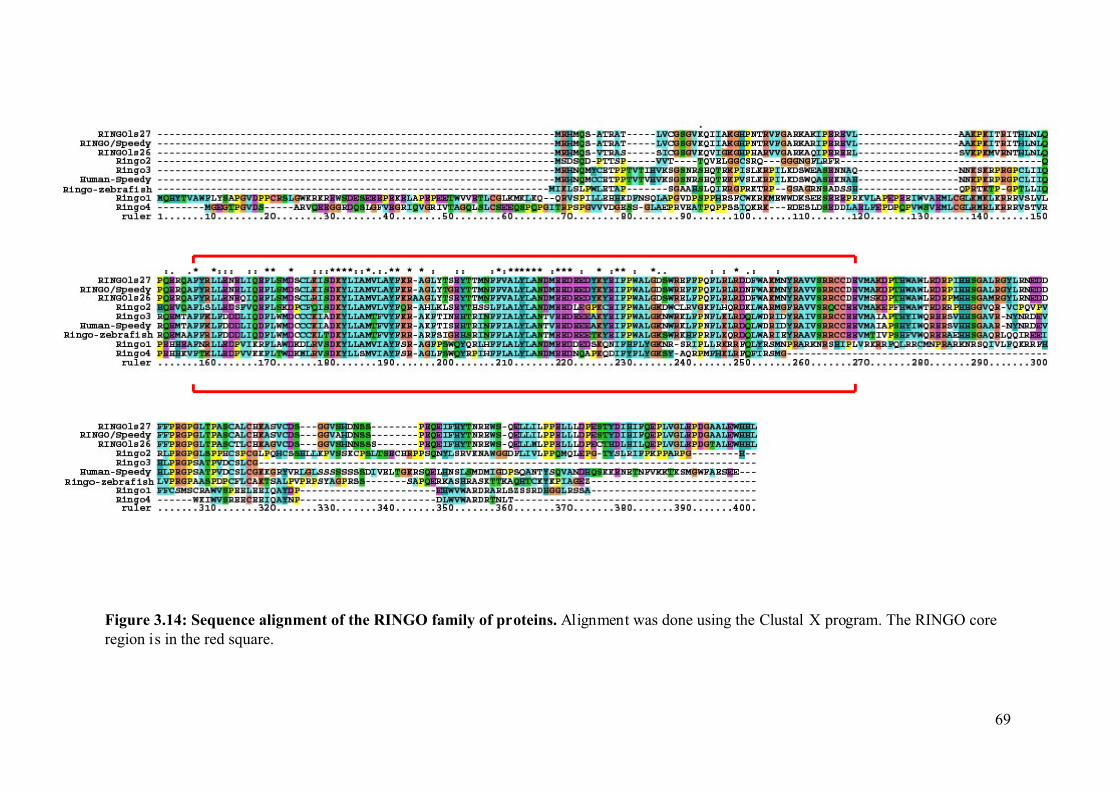
Figure 3.14: Sequence alignment of the RINGO family of proteins. Alignment was done using the Clustal X program. The RINGO coreregion is in the red square.
69
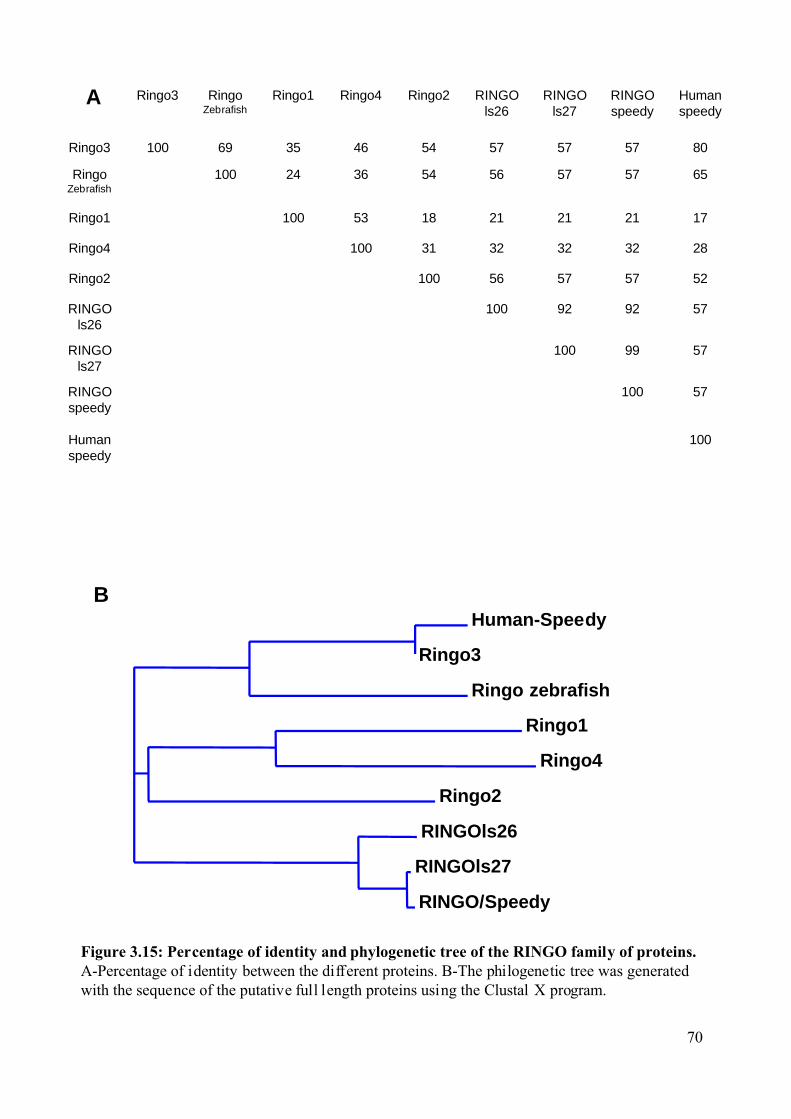
Figure 3.15: Percentage of identity and phylogenetic tree of the RINGO family of proteins.A-Percentage of identity between the different proteins. B-The philogenetic tree was generatedwith the sequence of the putative full length proteins using the Clustal X program.
Human-Speedy
Ringo3
Ringo zebrafish
Ringo1
Ringo4
Ringo2
RINGOls26
RINGOls27
RINGO/Speedy
B
A
100Humanspeedy
57100RINGOspeedy
5799100RINGOls27
579292100RINGOls26
52575756100Ringo2
2832323231100Ringo4
172121211853100Ringo1
65575756543624100RingoZebrafish
8057575754463569100Ringo3
Humanspeedy
RINGOspeedy
RINGOls27
RINGOls26
Ringo2Ringo4Ringo1RingoZebrafish
Ringo3
70
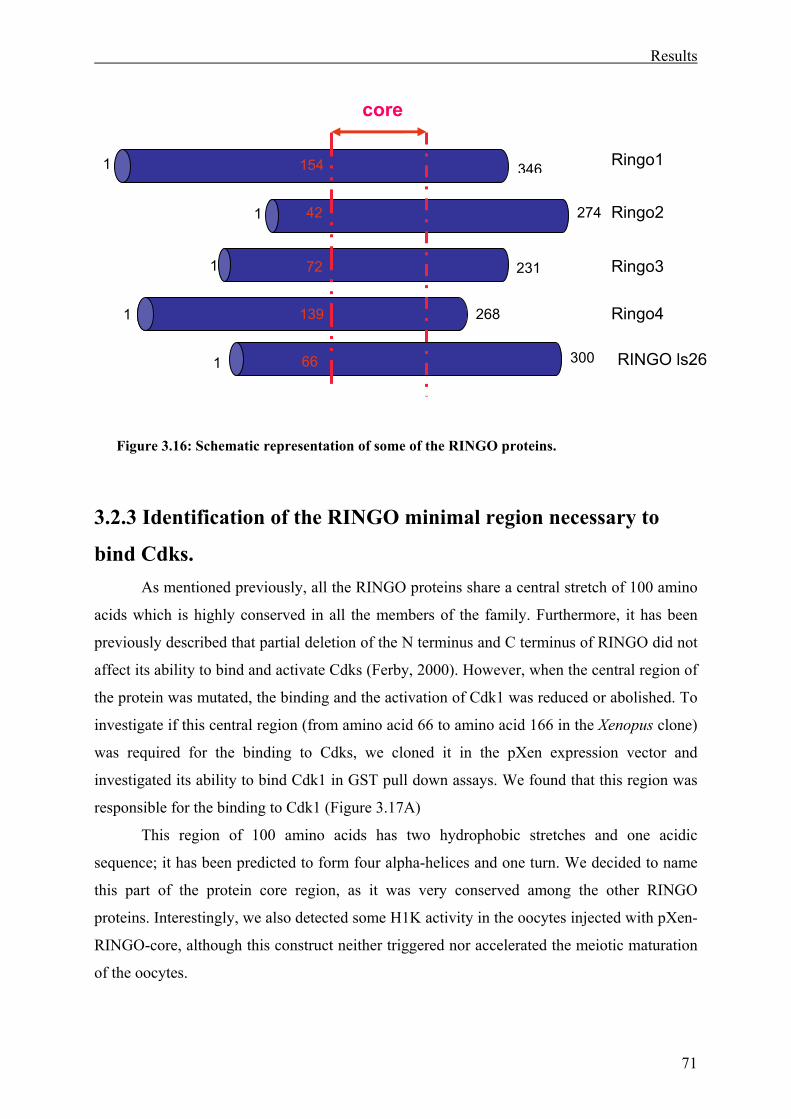
Results
71
3.2.3 Identification of the RINGO minimal region necessary to
bind Cdks. As mentioned previously, all the RINGO proteins share a central stretch of 100 amino
acids which is highly conserved in all the members of the family. Furthermore, it has been
previously described that partial deletion of the N terminus and C terminus of RINGO did not
affect its ability to bind and activate Cdks (Ferby, 2000). However, when the central region of
the protein was mutated, the binding and the activation of Cdk1 was reduced or abolished. To
investigate if this central region (from amino acid 66 to amino acid 166 in the Xenopus clone)
was required for the binding to Cdks, we cloned it in the pXen expression vector and
investigated its ability to bind Cdk1 in GST pull down assays. We found that this region was
responsible for the binding to Cdk1 (Figure 3.17A)
This region of 100 amino acids has two hydrophobic stretches and one acidic
sequence; it has been predicted to form four alpha-helices and one turn. We decided to name
this part of the protein core region, as it was very conserved among the other RINGO
proteins. Interestingly, we also detected some H1K activity in the oocytes injected with pXen-
RINGO-core, although this construct neither triggered nor accelerated the meiotic maturation
of the oocytes.
Figure 3.16: Schematic representation of some of the RINGO proteins.
1 346
274
231
268
1
1
1
core
300 1
154
42
72
139
66
Ringo1
Ringo2
Ringo3
Ringo4
RINGO ls26

Results
72
To confirm that the core region was responsible for binding to Cdks we decided to
generate GST-core recombinant protein. When incubated in oocytes extract this region was
able to bind Cdk1 as shown by GST pull down, but not MAPK, indicating that the binding
was specific for Cdks (Figure 3.17B).
3.2.4 Biochemical characterization of the mammalian clones. To confirm that the identified mammalian clones were part of a new family of
proteins, we investigated if these proteins display the same biochemical properties as Xenopus
RINGO. Indeed, they are all able to bind Cdk1, apparently with similar affinity. Furthermore
all these proteins are able to activate the kinase activity of Cdk2 in vitro (Figure 3.18).
It is interesting to note that Ringo1 and Ringo2 were poor activators of Cdk2 against
histone H1, in comparison to the Xenopus RINGO and Ringo3. However, we do not know if
this observation might be the consequence of improper folding of the proteins when they are
bacterially produced.
To summarize, we found that all the new members of the RINGO family display
similar biochemical properties. We therefore decided to investigate their effect in the Xenopus
oocyte system.
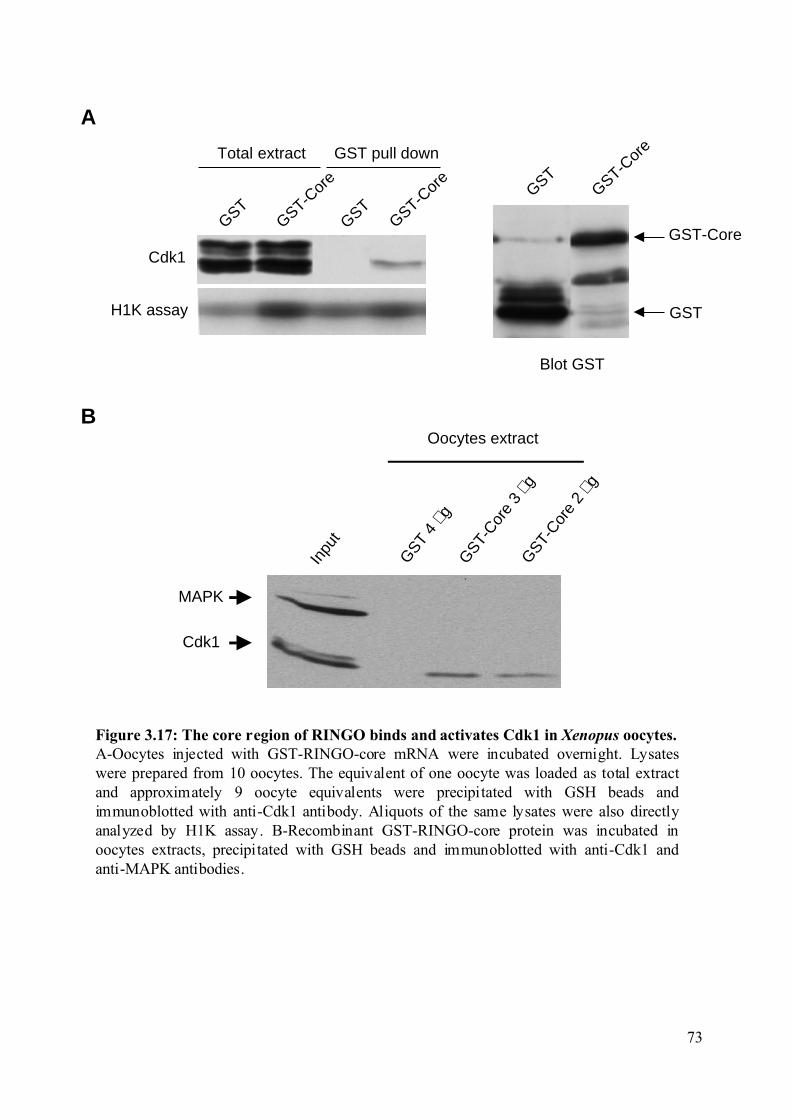
Figure 3.17: The core region of RINGO binds and activates Cdk1 in Xenopus oocytes.A-Oocytes injected with GST-RINGO-core mRNA were incubated overnight. Lysateswere prepared from 10 oocytes. The equivalent of one oocyte was loaded as total extractand approximately 9 oocyte equivalents were precipitated with GSH beads andimmunoblotted with anti-Cdk1 antibody. Aliquots of the same lysates were also directlyanalyzed by H1K assay. B-Recombinant GST-RINGO-core protein was incubated inoocytes extracts, precipitated with GSH beads and immunoblotted with anti-Cdk1 andanti-MAPK antibodies.
Blot GST
GSTGST-C
ore
GST
GST-Core
H1K assay
GSTGST-C
ore
GST-Cor
e
GST
Total extract GST pull down
Cdk1
Inpu
t
GST
4 µg
GST-
Core
3 µg
GST-
Core
2 µg
Oocytes extract
Cdk1
MAPK
A
B
73
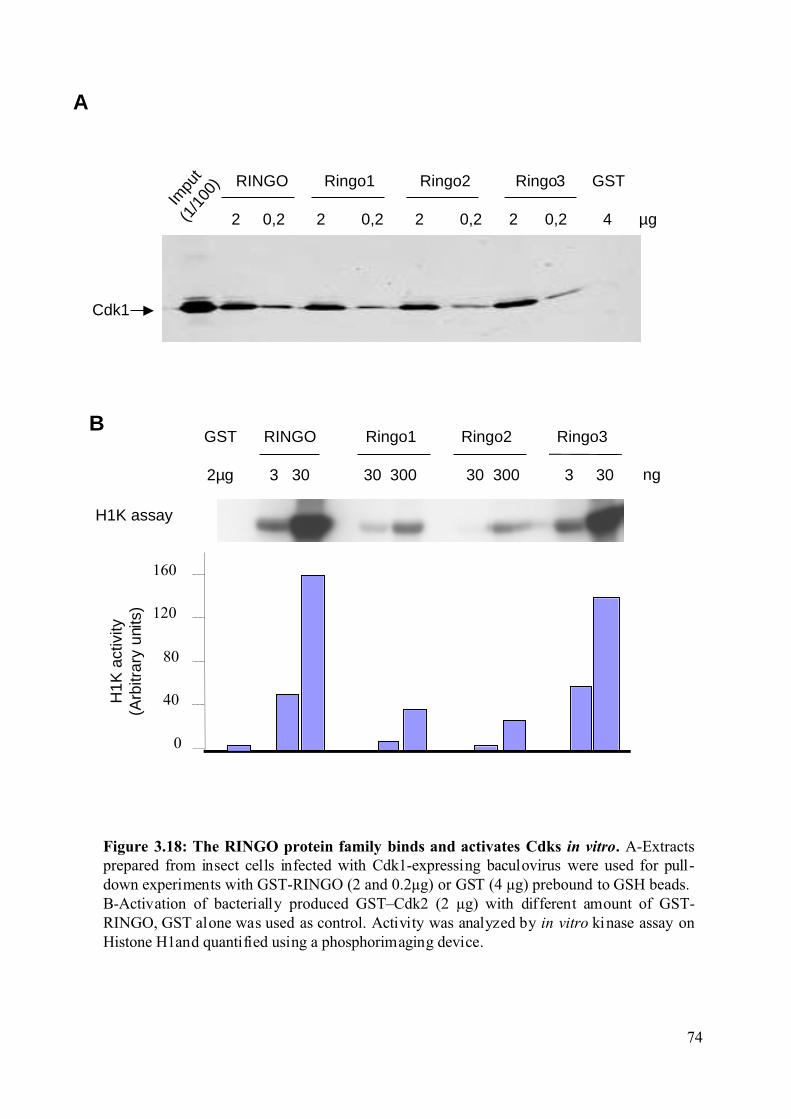
A
Figure 3.18: The RINGO protein family binds and activates Cdks in vitro. A-Extractsprepared from insect cells infected with Cdk1-expressing baculovirus were used for pull-down experiments with GST-RINGO (2 and 0.2µg) or GST (4 µg) prebound to GSH beads.B-Activation of bacterially produced GST–Cdk2 (2 µg) with different amount of GST-RINGO, GST alone was used as control. Activity was analyzed by in vitro kinase assay onHistone H1and quantified using a phosphorimaging device.
RINGO Ringo1 Ringo2 Ringo3 GST
2 0,2 2 0,2 2 0,2 2 0,2 4 µg
Impu
t(1
/100
)
2µg 3 30 30 300 30 300 3 30 ng
GST RINGO Ringo1 Ringo2 Ringo3
0
40
80
120
160
B
Cdk1
H1K assay
H1K
act
ivity
(Arb
itrar
y un
its)
74

Results
75
3.2.5 Properties of the mammalian clones in expressed in Xenopus
oocytes. As indicated in the Materials and Methods section the different cDNAs were cloned in
FTX5 or pXen expression vectors to produce mRNAs which were injected into Xenopus
oocytes.
3.2.5.1 Ringo1. Ringo1, the more distant member of the family compared to Xenopus RINGO, does
not induce oocyte maturation but inhibits progesterone-induced meiotic maturation. In
oocytes injected with Ringo1 and treated with progesterone, no accumulation of Mos was
detected and no phosphorylated MAPK or dephosphorylated Cdk1 were observed (Figure
3.19).
Surprisingly, we found that although Ringo1 is not able to induce meiotic maturation,
it is still able to bind and activate Cdk1 (Figure 3.20A). However, the H1K activity observed
with Ringo1 did not come from Cdk1 alone (Figure 3.20A), indicating that other kinases
might also associated with Ringo1.
To investigate if the inhibition by Ringo1 was a consequence of the sequestration of
endogenous Cdk1, which should normally be available for newly synthesized proteins upon
progesterone stimulation (Nebreda et al., 1995), we overexpressed either Cdk1 or Cdk1K33R
in oocytes expressing Ringo1. The inhibition could be rescued by overexpression of Cdk1.
Interestingly, we found that co-overexpression of Cdk1 and Ringo1 was not only able to
rescue the meiotic maturation, but even accelerate it when oocytes were treated with
progesterone. However, overexpression of the Cdk1K33R kinase dead mutant was not able to
rescue the inhibition by Ringo1 (Figure 3.21).
Mutants of Ringo1 lacking either the N-terminus or the C-terminus region of this
protein were generated (Figure 3.20B) and assayed for their ability to bind and activate Cdk1
in oocytes. Deletion of 100 amino acids at the N-terminus of the protein did not impair its
ability to bind and activate Cdk1, when injected in Xenopus oocytes. However, a deletion at
the C-terminus of 70 amino acids (that include the core region) rendered the protein unable to
bind and activate Cdk1 in Xenopus oocytes.
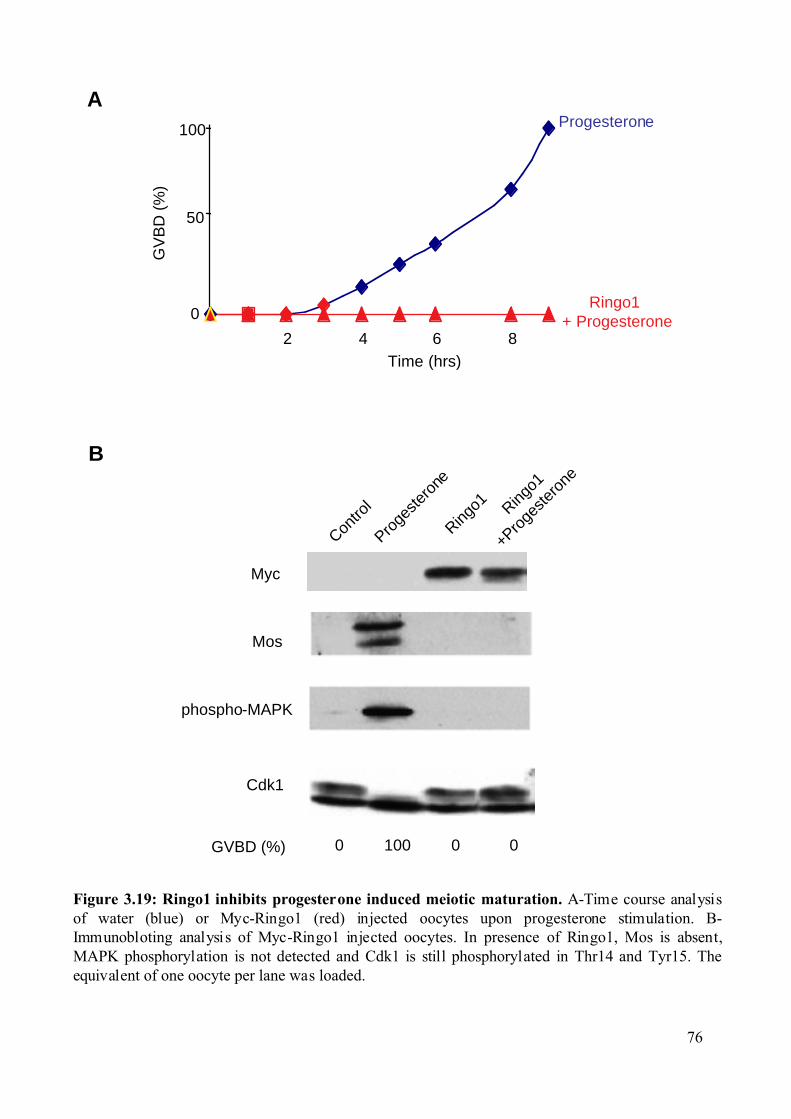
0
50
100
2 4 6 8Time (hrs)
GV
BD
(%
)
Progesterone
Ringo1 + Progesterone
Figure 3.19: Ringo1 inhibits progesterone induced meiotic maturation. A-Time course analysisof water (blue) or Myc-Ringo1 (red) injected oocytes upon progesterone stimulation. B-Immunobloting analysis of Myc-Ringo1 injected oocytes. In presence of Ringo1, Mos is absent,MAPK phosphorylation is not detected and Cdk1 is still phosphorylated in Thr14 and Tyr15. Theequivalent of one oocyte per lane was loaded.
Contro
l
Proge
stero
ne
Ringo1
+Proge
stero
ne
Ringo1
Mos
phospho-MAPK
Cdk1
GVBD (%) 0 100 0 0
Myc
A
B
76
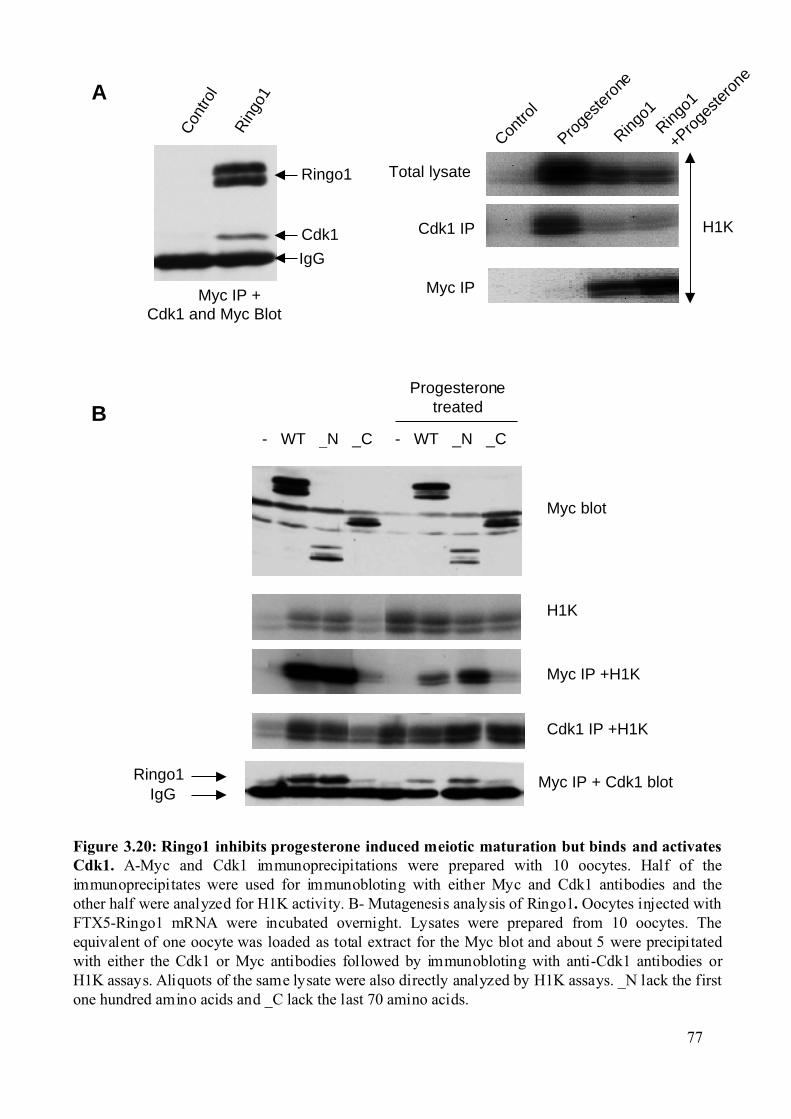
Con
trol
Rin
go1
Figure 3.20: Ringo1 inhibits progesterone induced meiotic maturation but binds and activatesCdk1. A-Myc and Cdk1 immunoprecipitations were prepared with 10 oocytes. Half of theimmunoprecipitates were used for immunobloting with either Myc and Cdk1 antibodies and theother half were analyzed for H1K activity. B- Mutagenesis analysis of Ringo1. Oocytes injected withFTX5-Ringo1 mRNA were incubated overnight. Lysates were prepared from 10 oocytes. Theequivalent of one oocyte was loaded as total extract for the Myc blot and about 5 were precipitatedwith either the Cdk1 or Myc antibodies followed by immunobloting with anti-Cdk1 antibodies orH1K assays. Aliquots of the same lysate were also directly analyzed by H1K assays. _N lack the firstone hundred amino acids and _C lack the last 70 amino acids.
Contro
l
Proge
stero
ne
Ringo1
+Proge
stero
ne
Ringo1
Total lysate
Cdk1 IP
Myc IP
H1K
A
Ringo1 Myc IP + Cdk1 blot
- WT _N _C - WT _N _C
Myc blot
Cdk1 IP +H1K
Myc IP +H1K
H1K
IgG
ProgesteronetreatedB
Myc IP + Cdk1 and Myc Blot
Cdk1
IgG
Ringo1
77
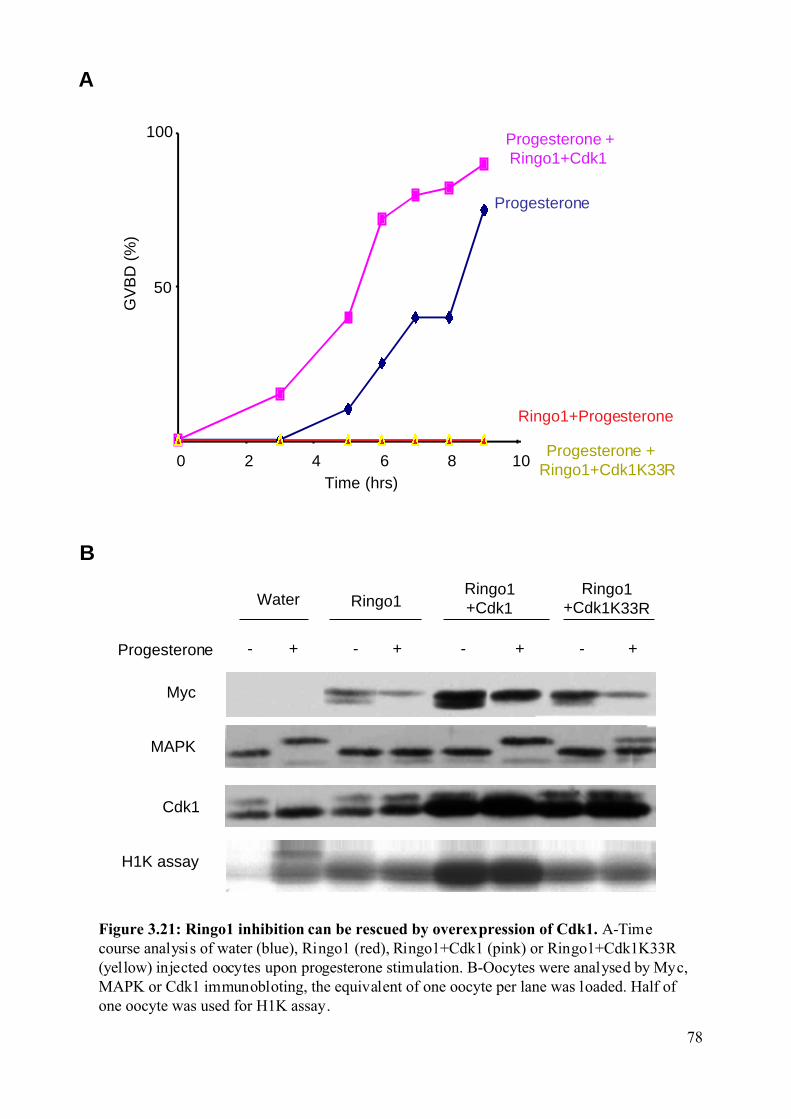
Time (hrs)
Progesterone + Ringo1+Cdk1
Progesterone
Ringo1+Progesterone
GV
BD
(%
)
50
100
0 2 4 6 8 10 Progesterone +
Ringo1+Cdk1K33R
Figure 3.21: Ringo1 inhibition can be rescued by overexpression of Cdk1. A-Timecourse analysis of water (blue), Ringo1 (red), Ringo1+Cdk1 (pink) or Ringo1+Cdk1K33R(yellow) injected oocytes upon progesterone stimulation. B-Oocytes were analysed by Myc,MAPK or Cdk1 immunobloting, the equivalent of one oocyte per lane was loaded. Half ofone oocyte was used for H1K assay.
A
B
H1K assay
Ringo1Ringo1+Cdk1
Progesterone - + - + - +
Water
Myc
MAPK
Cdk1
Ringo1+Cdk1K33R
- +
78

Results
79
3.2.5.2 Ringo2. Ringo2 displays 56% identity to Xenopus RINGO. When FTX5-Ringo2 was injected
in Xenopus oocytes Ringo2 was expressed at very low level and did not trigger oocyte
maturation, although it was able to significantly accelerate progesterone-induced meiotic
maturation (Figure 3.22A). To try to achieve higher levels of expression, Ringo2 was cloned
in the pXen vector. In this case, the level of expression in Xenopus oocytes was higher (Figure
3.22B), but we were still unable to observe oocyte maturation in the absence of progesterone.
However, as with Xenopus RINGO and Ringo1, Ringo2 was also able to bind and activate
Cdk1 in Xenopus oocytes (Figure 3.22C).
3.2.5.3 Ringo3. Ringo3 like Ringo2 is structurally very close to Xenopus RINGO. As expected this
clone was able to induce meiotic maturation (Figure 3.23A). The speed of the process was
slower than for Xenopus RINGO or progesterone induced meiotic maturation. But as shown
in figure 3.23B, the biochemical markers of Ringo3-induced maturation were identical to
those in oocytes induced to mature by progesterone.
3.2.5.4 Ringo4. We were not able to clone the full length Ringo4. However we cloned a partial cDNA
of Ringo4 which does not include the last 80 amino acids (this clone lacks part of the core
domain). Expression of this truncated Ringo4 in oocytes was neither able to trigger meiotic
maturation nor to accelerate it upon progesterone stimulation (Figure 3.24A). No differences
in H1K activity could be detected compared to G2 arrested oocytes (Figure 3.24B). These
results probably reflect that we did not look at the full length protein.
In the light of these results, we decided to further characterize in mammalian cells the
Ringo1 and Ringo3 proteins, which displayed opposite function in Xenopus oocytes.
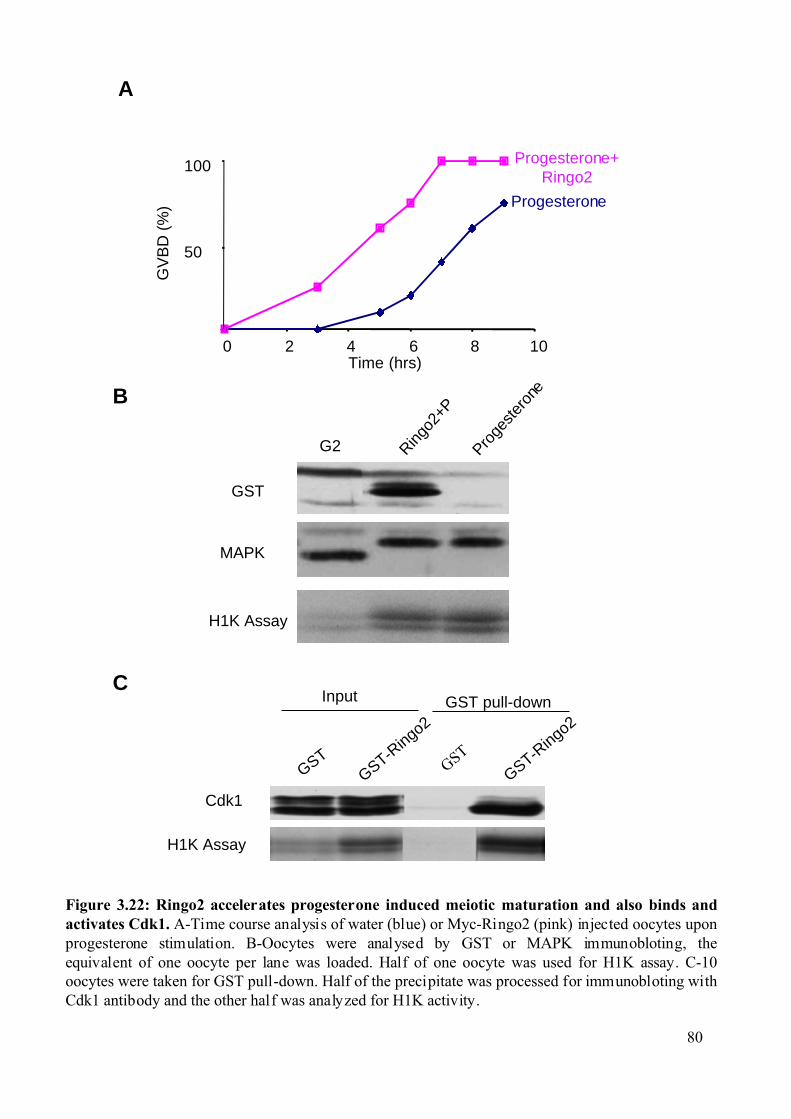
B
A
GST
Time (hrs)
Progesterone+Ringo2
100
0 2 4 6 8 10
50
Progesterone
GV
BD
(%
)
Figure 3.22: Ringo2 accelerates progesterone induced meiotic maturation and also binds andactivates Cdk1. A-Time course analysis of water (blue) or Myc-Ringo2 (pink) injected oocytes uponprogesterone stimulation. B-Oocytes were analysed by GST or MAPK immunobloting, theequivalent of one oocyte per lane was loaded. Half of one oocyte was used for H1K assay. C-10oocytes were taken for GST pull-down. Half of the precipitate was processed for immunobloting withCdk1 antibody and the other half was analyzed for H1K activity.
C
Cdk1
GST pull-down
H1K Assay
GST-Ringo2
GST-Ringo2
Input
Proge
stero
ne
G2
GST
MAPK
Ringo2
+P
H1K Assay
GST
80
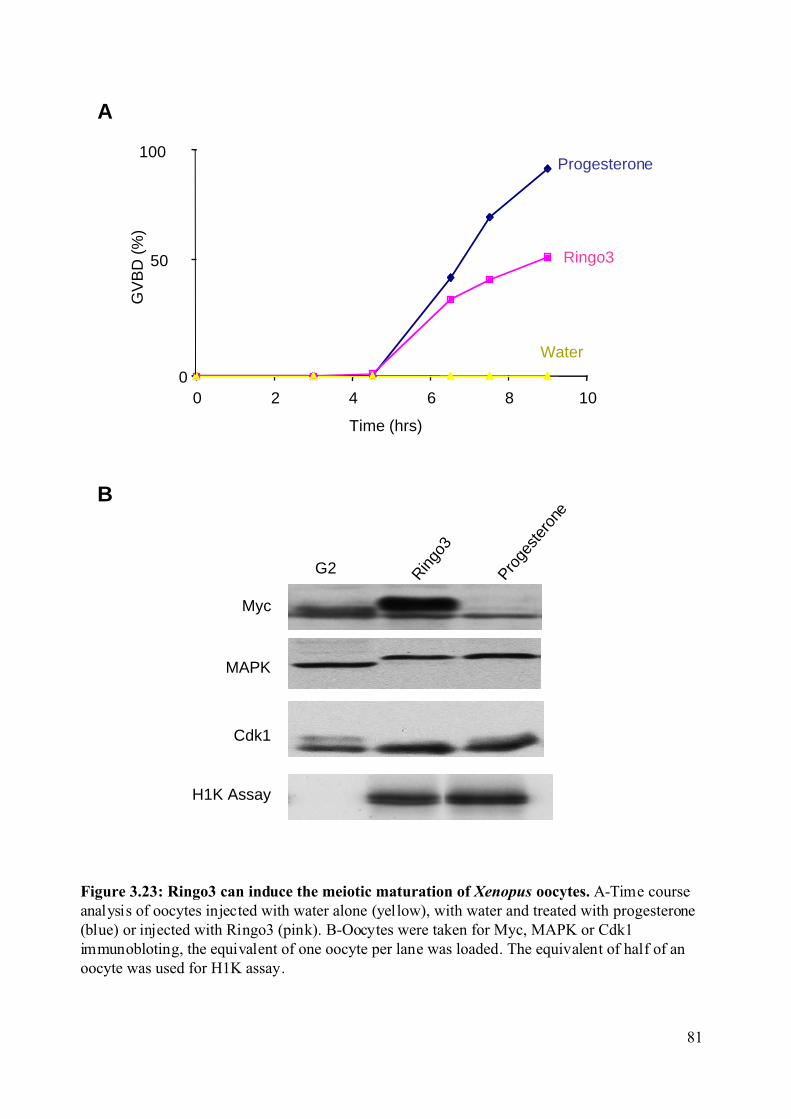
Ringo3
GV
BD
(%
)
0
50
100
0 2 4 6 8 10
Time (hrs)
Progesterone
Water
B
A
Figure 3.23: Ringo3 can induce the meiotic maturation of Xenopus oocytes. A-Time courseanalysis of oocytes injected with water alone (yellow), with water and treated with progesterone(blue) or injected with Ringo3 (pink). B-Oocytes were taken for Myc, MAPK or Cdk1immunobloting, the equivalent of one oocyte per lane was loaded. The equivalent of half of anoocyte was used for H1K assay.
G2
Myc
MAPK
Cdk1
Ringo
3
Proge
ster
one
H1K Assay
81
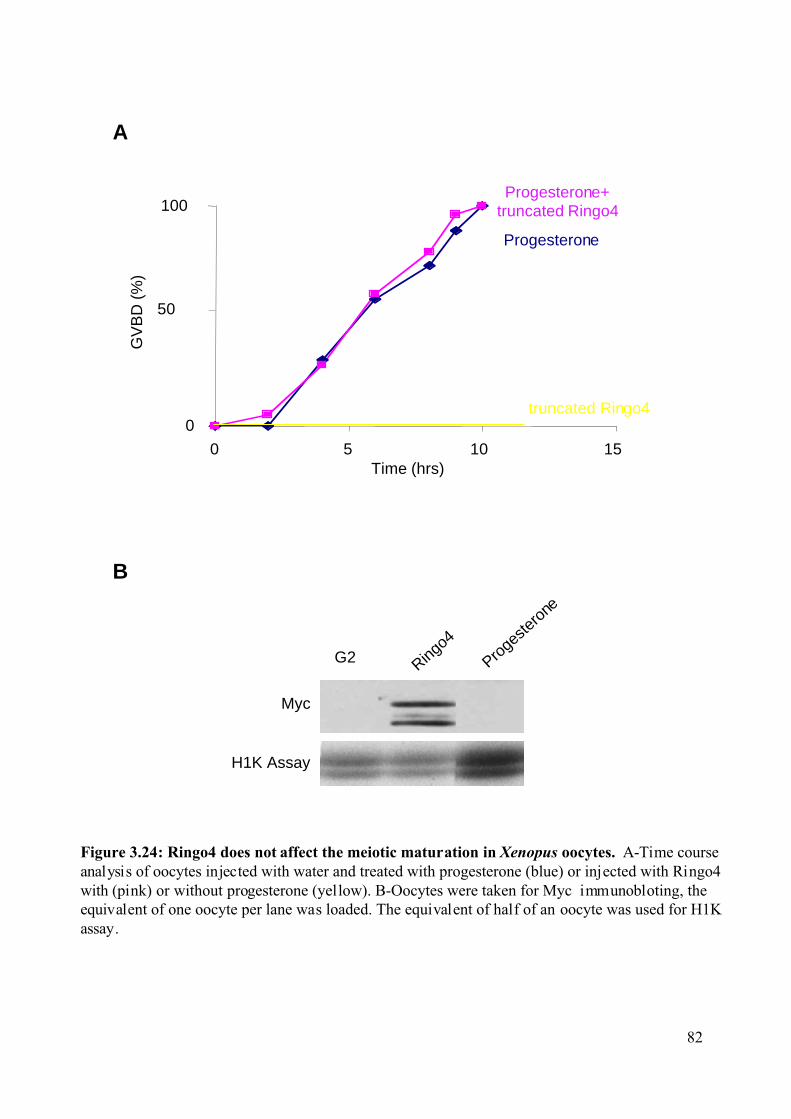
B
Figure 3.24: Ringo4 does not affect the meiotic maturation in Xenopus oocytes. A-Time courseanalysis of oocytes injected with water and treated with progesterone (blue) or injected with Ringo4with (pink) or without progesterone (yellow). B-Oocytes were taken for Myc immunobloting, theequivalent of one oocyte per lane was loaded. The equivalent of half of an oocyte was used for H1Kassay.
G2
Myc
Ringo4
Proge
stero
ne
A
0
100
0 5 10 15
50
GV
BD
(%
)
Time (hrs)
Progesterone+truncated Ringo4
Progesterone
H1K Assay
truncated Ringo4
82

Results
83
3.2.6 Expression pattern of the mammalian RINGO proteins. To determine the expression of RINGO mRNAs in mammalian tissues, we performed
Northern blot analysis (Figure 3.25). Ringo1 appears as a unique band of 1,5 Kilo bases (Kb)
expressed only in testis. Ringo3 appears as a doublet of 1 to 2 Kb, which could indicate the
presence of alternative splicing variant forms. This approach did not allow us to detect tissues
expressing Ringo2 or Ringo4. We therefore decided to use a more sensitive technique. RT-
PCR was performed on mRNA isolated from ten different mouse tissues and three embryonic
brains. The primers used were specific for the core region of the RINGO proteins. We found
that Ringo1 and Ringo4 are testis specific. Ringo3 is more ubiquitously expressed, with high
expression levels found in foetal brain and testis, significant amounts of Ringo3 mRNAs were
also detected in liver, heart, thymus and weak expression was also detected in ovary (Figure
3.26). Interestingly, expression of Ringo3 appeared to be “switched off” during brain
development, Ringo3 is highly expressed in foetal brain and its expression decreased,
becoming almost absent in new born mouse brain (Figure 3.26).
By searching databases (Ensembl software, http:\\www.ensembl.org), we identified
multiple spliced forms for Ringo3 and Ringo4 (Figure 3.27). Ringo3 was predicted to have
four putative alternative splice variants, but we could only detect three of them by RT-PCR.
The expressions of these alternative splice forms were restricted to testis (Figure 3.28). By
RT-PCR, we could only detect one of two putative Ringo4 alternative splice variants (Figure
3.28).
Ringo1
Ringo3
Ubiquitin
Blan
kla
neHe
art
Brai
nLi
ver
Sple
enKi
dney
Emnb
ryo
Lung
Thym
usTe
stes
Ova
ry
Figure 3.25: Northern blot analysis of the RINGO family. The expression analysis was done using the First Choice™ Northern blot (Ambion) membrane. No expression was detected for Ringo2 and Ringo4 A probe corresponding to the Ubiquitin Open Reading Frame (ORF) was used as a loading control.
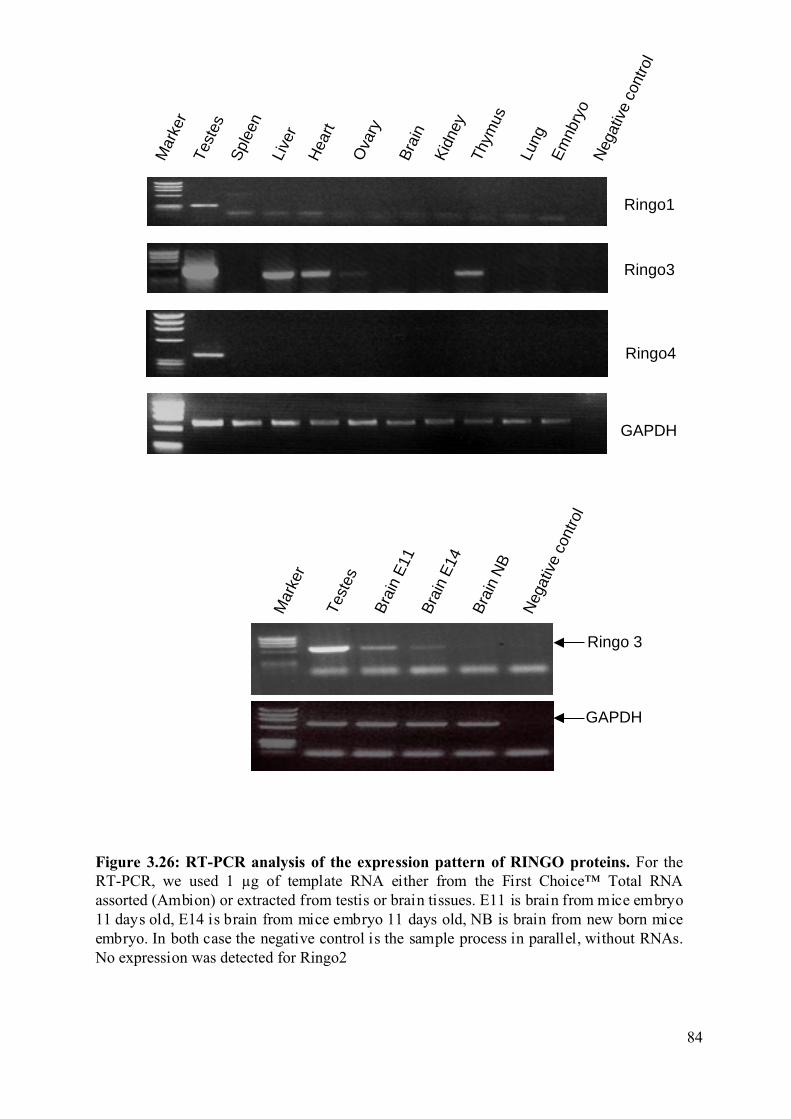
Figure 3.26: RT-PCR analysis of the expression pattern of RINGO proteins. For theRT-PCR, we used 1 µg of template RNA either from the First Choice™ Total RNAassorted (Ambion) or extracted from testis or brain tissues. E11 is brain from mice embryo11 days old, E14 is brain from mice embryo 11 days old, NB is brain from new born miceembryo. In both case the negative control is the sample process in parallel, without RNAs.No expression was detected for Ringo2
Neg
ativ
e co
ntro
l
Ringo1
Ringo3
GAPDH
Ringo4
Hea
rt
Bra
in
Live
r
Spl
een
Kid
ney
Em
nbry
o
Lung
Thym
us
Test
es
Ova
ry
Mar
ker
Neg
ativ
e co
ntro
l
Ringo 3
GAPDH
Bra
in E
11
Mar
ker
Test
es
Bra
in E
14
Bra
in N
B
84
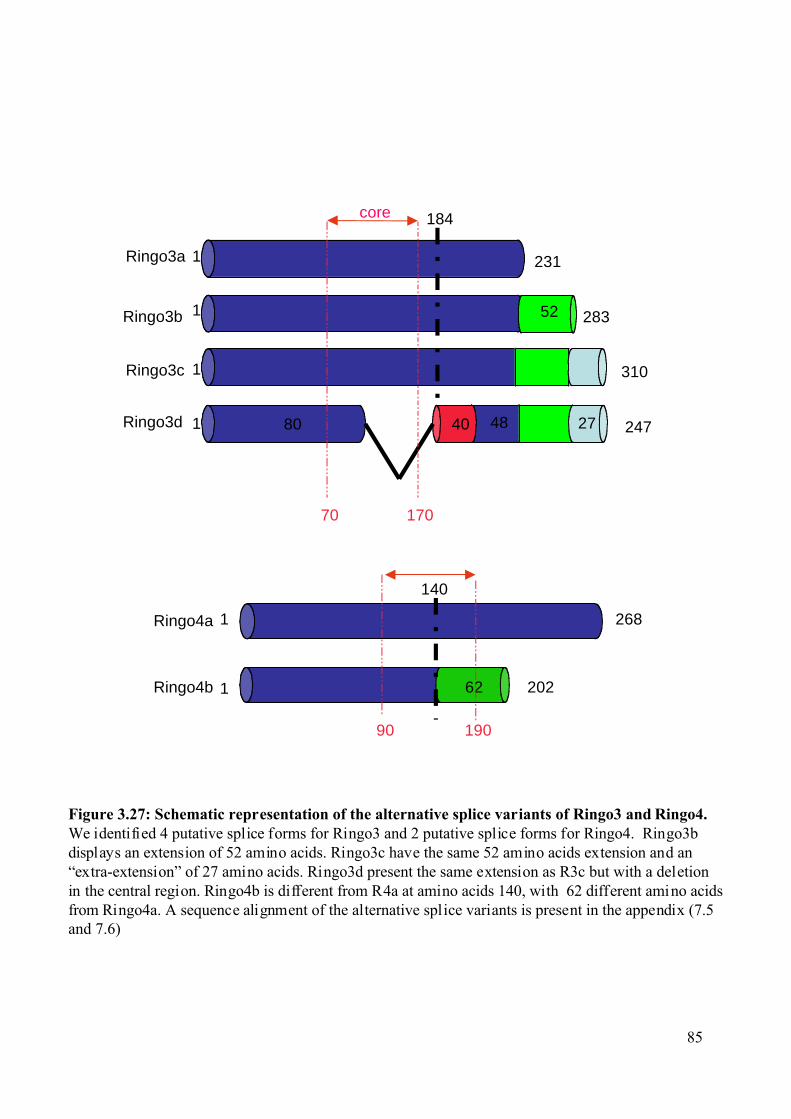
Figure 3.27: Schematic representation of the alternative splice variants of Ringo3 and Ringo4.We identified 4 putative splice forms for Ringo3 and 2 putative splice forms for Ringo4. Ringo3bdisplays an extension of 52 amino acids. Ringo3c have the same 52 amino acids extension and an“extra-extension” of 27 amino acids. Ringo3d present the same extension as R3c but with a deletionin the central region. Ringo4b is different from R4a at amino acids 140, with 62 different amino acidsfrom Ringo4a. A sequence alignment of the alternative splice variants is present in the appendix (7.5and 7.6)
1 231
283
310
40 4880 247
184
1
1
1
70 170
Ringo3a
Ringo3b
Ringo3c
Ringo3d
core
52
27
140
1
1
268
62 202
Ringo4a
Ringo4b
90 190
85
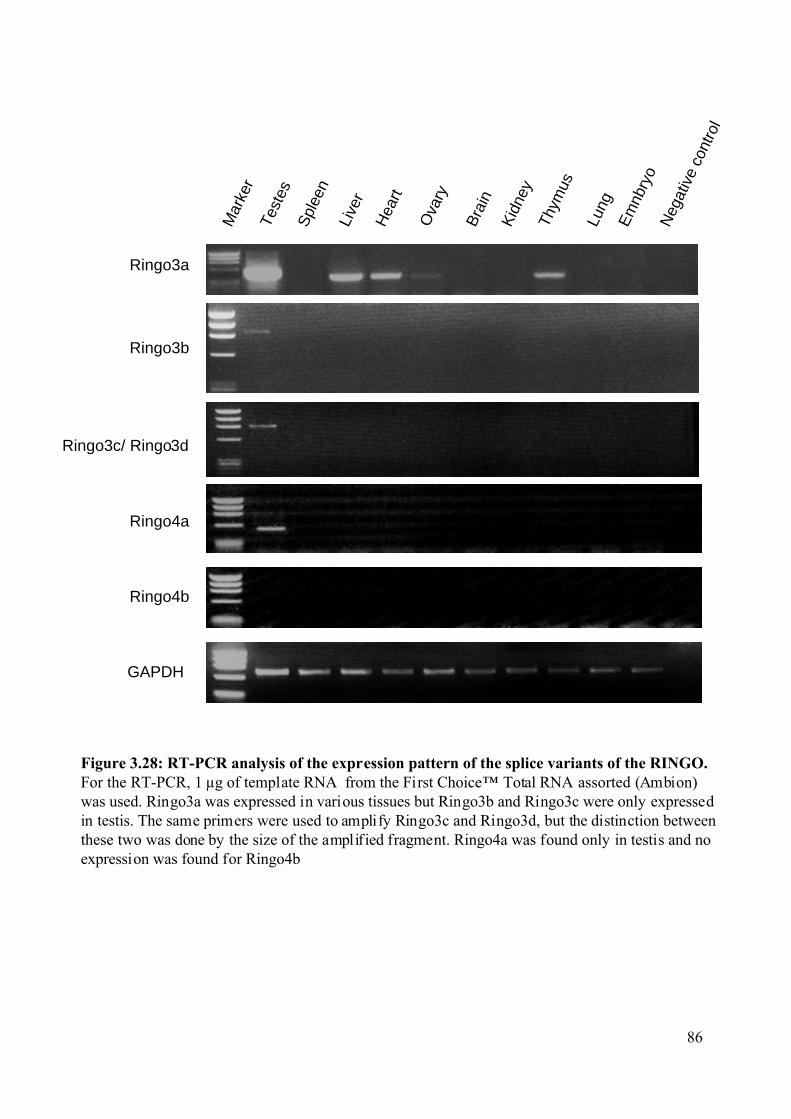
Figure 3.28: RT-PCR analysis of the expression pattern of the splice variants of the RINGO.For the RT-PCR, 1 µg of template RNA from the First Choice™ Total RNA assorted (Ambion)was used. Ringo3a was expressed in various tissues but Ringo3b and Ringo3c were only expressedin testis. The same primers were used to amplify Ringo3c and Ringo3d, but the distinction betweenthese two was done by the size of the amplified fragment. Ringo4a was found only in testis and noexpression was found for Ringo4b
GAPDH
Ringo3a
Ringo3b
Ringo3c/ Ringo3d
Ringo4a
Ringo4b
Neg
ativ
e co
ntro
l
Hea
rt
Bra
in
Live
r
Spl
een
Kid
ney
Em
nbry
o
Lung
Thym
us
Test
es
Ova
ry
Mar
ker
86

Results
87
3.2.7 RINGO proteins can activate Cdk5. Cdk5 is involved in numerous processes such as neurite outgrowth, brain
development, neurodegenerative disease and drug addiction. Interestingly, Cdk5 activity has
also been found in testis, where Cdk5 is believed to play a role in spermatogenesis (Ino et al.,
1994; Musa et al., 1998; Session et al., 2001). However, no Cdk5 activator has been found in
testis.
Although the p35 protein has no sequence homology with cyclins, it is able to activate
Cdk5 (Lew et al., 1994; Tsai et al., 1994). Moreover, it has been shown that p35 allows Cdk5
to escape inhibition by p27Kip1 in neurons (Lee et al., 1996). A crystallisation study of the
p25-Cdk5 complex (p25 is a proteolitic fragment of p35 generated by the protease calpain
(Lee et al., 2000)), showed that the p35/p25 proteins indeed fold like cyclins. Moreover, they
are able to activate Cdk5 by a mechanism independent of T-loop phosphorylation (Tarricone
et al., 2001). This resemble to the RINGO proteins that also do not have homology with
cyclins and do not need T-loop phosphorylation for activation of Cdks (Karaiskou et al.,
2001).
We investigated if Ringo3 protein (which we know to be present in foetal brain and
testis) could activate Cdk5. We incubated GST-Ringo3 in brain or testis extract and after GST
pull-down, we could detect by immunobloting specific binding of Cdk1 and Cdk2 in testis
extract and Cdk5 in brain extract (Figure 3.29A). When the GST-Ringo3 recovered after
incubation in testis or brain extract was subsequently used for in vitro kinase assay, we could
detect H1K activity, indicating that indeed Ringo3 can bind and activate Cdk5 (Figure 3.29B).
Furthermore, we found that the H1K activity of Cdk5 was similar when incubated with
recombinant GST-Ringo3 or recombinant GST-p25 (Figure 3.29C).
As Ringo3 was well expressed in some tissues we immunoprecipitated Ringo3 to try
to detect co-immunoprecipitation of Cdks and the associated kinase activity. We could detect
interaction between Ringo3 and Cdks (probably Cdk1 and Cdk2), especially in testis, heart,
liver and ovary (Figure 3.30).
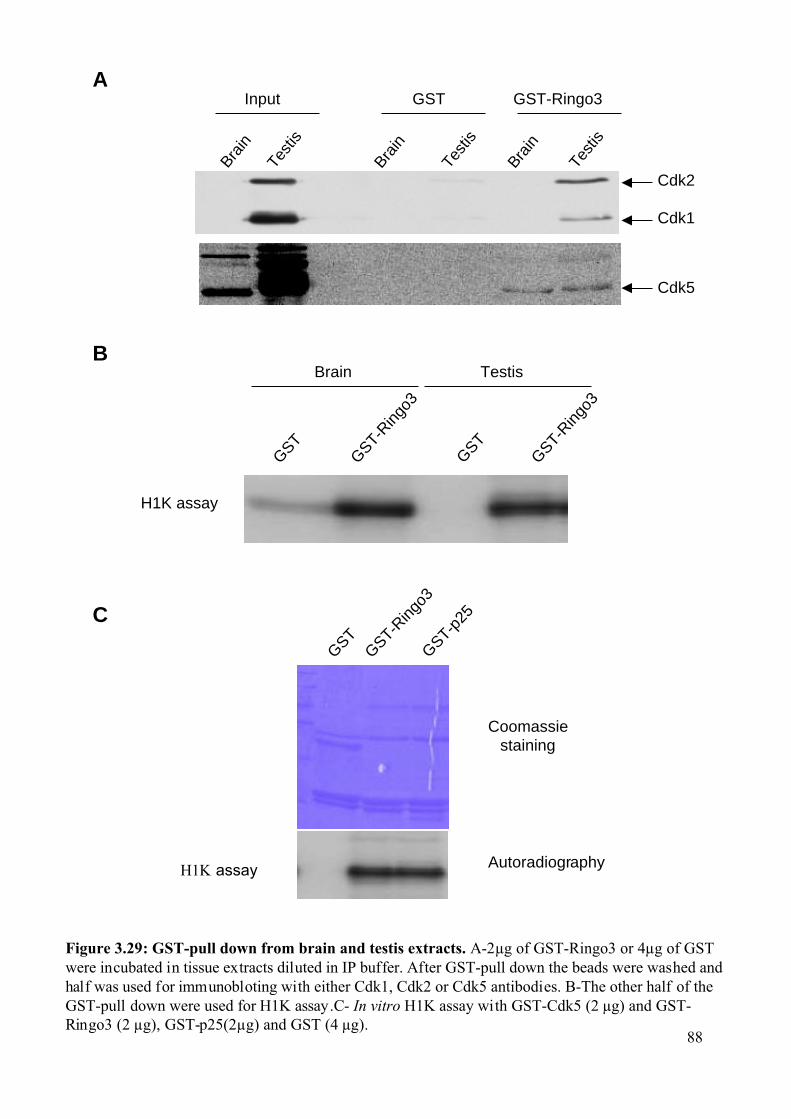
Figure 3.29: GST-pull down from brain and testis extracts. A-2µg of GST-Ringo3 or 4µg of GSTwere incubated in tissue extracts diluted in IP buffer. After GST-pull down the beads were washed andhalf was used for immunobloting with either Cdk1, Cdk2 or Cdk5 antibodies. B-The other half of theGST-pull down were used for H1K assay.C- In vitro H1K assay with GST-Cdk5 (2 µg) and GST-Ringo3 (2 µg), GST-p25(2µg) and GST (4 µg).
Brain
Test
is
GST GST-Ringo3
Brain
Test
is
Brain
Test
is
Input
Cdk2
Cdk1
Cdk5
A
B
C
H1K assay
GSTGST-R
ingo3
GST-p25
Coomassiestaining
Autoradiography
Brain Testis
GST
GST-Ring
o3
GST
GST-Ring
o3
H1K assay
88

Results
89
3.2.8 Role of the RINGO proteins in the mammalian cell cycle. Since All the Ringo proteins are expressed in testis, we decided to investigate if they
were also present in a testis cell line. The human teratocarcinoma cell line (Ntera-2) expressed
both Ringo1 and Ringo3, as determined by RT-PCR (Figure 3.31).
3.2.8.1 Ringo3 is a cell cycle regulated protein. To determine if Ringo3 was cell cycle regulated, Ntera-2 cells were synchronized by
serum starvation. After 48 hrs without serum, cells were released from the G0 block by
addition of serum. Progression through the cell cycle was monitored by using cyclin synthesis
as a biochemical marker.
We found that Ringo3 started to accumulate in early G1 before Cyclin A, around the
time of Cyclin D synthesis, and it was then degraded in late S phase, when Cyclin B starts to
be synthesized (Figure 3.32A). The pattern of expression of Ringo3 overlaps with that of
G1/S cyclins.
We were not able to detect expression of the endogenous Ringo1 protein in Ntera-2
cells. However, overexpression and depletion data indicate that Ringo1 is more likely to be
involved in the S phase exit (see paragraph 3.2.8.4 and 3.2.8.5).
3.2.8.2 Ringo3 interacts with Cdk2 and phosphorylates Rb in vitro. It has been reported that Xenopus RINGO can bind and activate Cdk2 (Karaiskou et
al., 2001; Lenormand et al., 1999). We decided to verify these observations in mammalian
cells. Immunoblotting with Ringo3 antibody of Cdk1 and Cdk2 immunoprecipitated
asynchronous Ntera-2 cell extracts revealed an interaction between Ringo3 and Cdk2, but no
interaction with Cdk1 was detected (Figure 3.32B).
The correlation between the G1/S pattern of expression and the ability of Ringo3 to
bind Cdk2 suggests a possible role for Ringo3 in S phase entry.
An obvious candidate substrate for Ringo3-Cdk2 is the retinoblastoma protein (Rb).
Rb is the first target of the G1/S Cyclin-Cdk complexes. When we performed a kinase assay
with the RINGO proteins and Cdk2, we could detect that the incubation of Cdk2 with Ringo3
could phosphorylate Rb in vitro (Figure 3.32C). Interestingly, the other RINGO family
members can also induce Rb phosphorylation in vitro (Figure 3.32C).
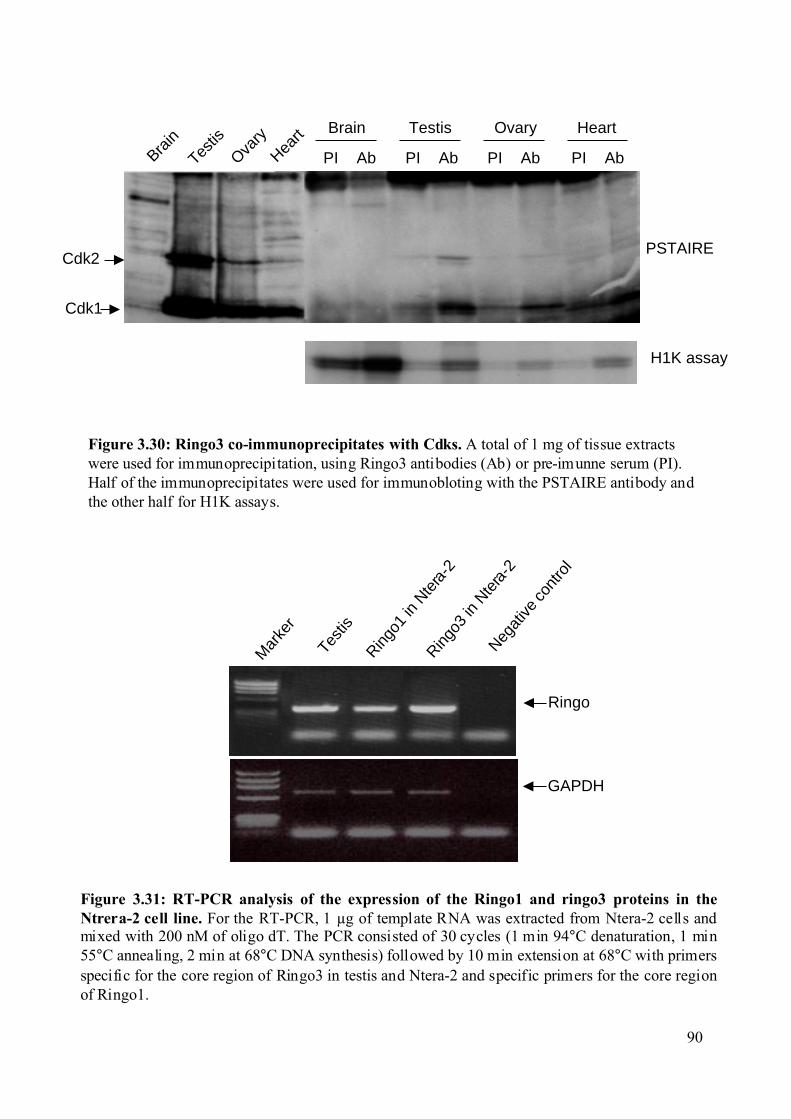
Cdk1
Cdk2
H1K assay
PSTAIRE
Brain
Testis
Ovary
Heart
PI Ab PI Ab
Brain Testis Ovary Heart
PI Ab PI Ab
Figure 3.30: Ringo3 co-immunoprecipitates with Cdks. A total of 1 mg of tissue extractswere used for immunoprecipitation, using Ringo3 antibodies (Ab) or pre-imunne serum (PI).Half of the immunoprecipitates were used for immunobloting with the PSTAIRE antibody andthe other half for H1K assays.
Figure 3.31: RT-PCR analysis of the expression of the Ringo1 and ringo3 proteins in theNtrera-2 cell line. For the RT-PCR, 1 µg of template RNA was extracted from Ntera-2 cells andmixed with 200 nM of oligo dT. The PCR consisted of 30 cycles (1 min 94°C denaturation, 1 min55°C annealing, 2 min at 68°C DNA synthesis) followed by 10 min extension at 68°C with primersspecific for the core region of Ringo3 in testis and Ntera-2 and specific primers for the core regionof Ringo1.
GAPDH
Mar
ker
Test
is
Ringo
1 in
Nte
ra-2
Ringo
3 in
Nte
ra-2
Negat
ive c
ontro
l
Ringo
90
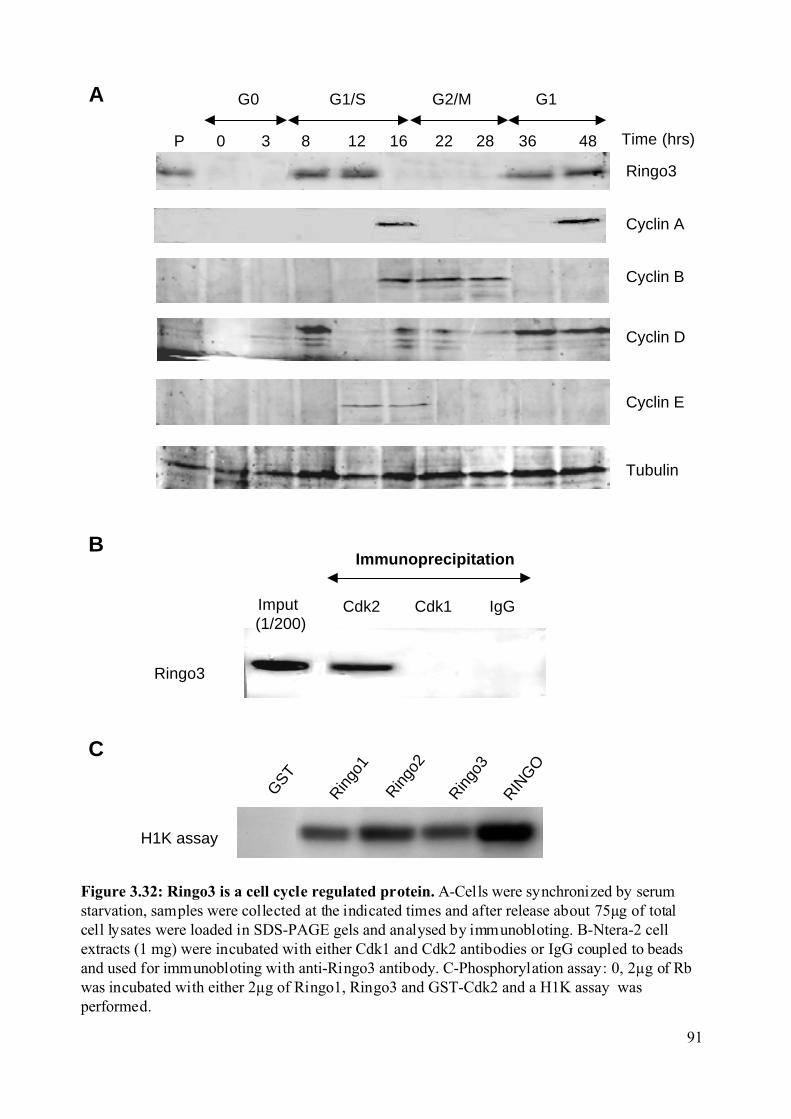
Figure 3.32: Ringo3 is a cell cycle regulated protein. A-Cells were synchronized by serumstarvation, samples were collected at the indicated times and after release about 75µg of totalcell lysates were loaded in SDS-PAGE gels and analysed by immunobloting. B-Ntera-2 cellextracts (1 mg) were incubated with either Cdk1 and Cdk2 antibodies or IgG coupled to beadsand used for immunobloting with anti-Ringo3 antibody. C-Phosphorylation assay: 0, 2µg of Rbwas incubated with either 2µg of Ringo1, Ringo3 and GST-Cdk2 and a H1K assay wasperformed.
A
B
Ringo3
Imput (1/200)
Cdk2 Cdk1 IgG
Immunoprecipitation
C
Rin
go1
Rin
go3
GST
Rin
go2
RIN
GO
Ringo3
Cyclin B
Cyclin D
Cyclin A
G1/S G2/M G1
36 482816 2212830P
G0
Cyclin E
Tubulin
Time (hrs)
H1K assay
91

Results
92
3.2.8.3 Effect of the RINGO proteins on the proliferation rate of Ntera-2
cells. To investigate the function of Ringo1 and Ringo3 proteins during the cell cycle, we
addressed the effects of both overexpression and loss of function of the RINGO proteins in
the Ntera-2 cell line. We generated an Ntera-2 cell line overexpressing Ringo1 or Ringo3
proteins by retroviral infection. Interestingly, overexpression of either Ringo1 or Ringo3
displayed different effects.
Ringo3 increased the speed of proliferation (growth curve, figure 3.33A), which
correlated with an increased incorporation of [3H]-Thymidine (Figure 3.33B). This indicates
that Ringo3 promotes a faster proliferation rate.
Ringo1 overexpression reduces the speed of proliferation (growth curve,figure 3.33A).
However, we did not observe any significant decrease in the level of [3H]-Thymidine
incorporation (Figure 3.33B).
3.2.8.4 Biochemical and cell cycle properties of the Ntera-2 cell lines
expressing RINGO proteins. As shown by the growth curves in figure 3.33A, the proliferation rate of Ntera-2 cells
overexpressing RINGO proteins changed. We used both biochemical and FACS analysis to
further characterize RINGO-overexpressing cell lines. High levels of Cyclin A proteins were
detected in the Ringo3-overexpressing cell line and this also correlated with higher levels of
Cyclin A mRNA as detected by Northern blotting (Figure 3.34B). In the Ringo1-
overexpressing cell line we detected a reduced amount of Ringo3 protein (Figure 3.34A).
Using FACS analysis on an asynchronously growing Ntera-2 cell population, we found a
subtle, but reproducible difference between wild type, Ringo1 and Ringo3-overexpressing cell
lines (Figure 3.35). Ringo3-overexpressing cells had a shorter G1 phase and a higher rate of
proliferation. This is consistent with the hypothesis that Ringo3 behaves like a G1/S cyclin
and its overexpression shortens the G1 phase of the cell cycle leading to a premature entry of
the cell into S phase (Figure 3.35). However, in spite of their high rate of proliferation, these
cells undergo a normal cell cycle and proliferate normally but faster. Ringo1-overexpressing
cells also had a shorter G1 phase and a short S phase. Surprisingly, the G2/M phase (normally
considered as one of the indicators of the proliferation rate) was significantly increased in the
Ringo1-overexpressing cells (Figure 3.35).
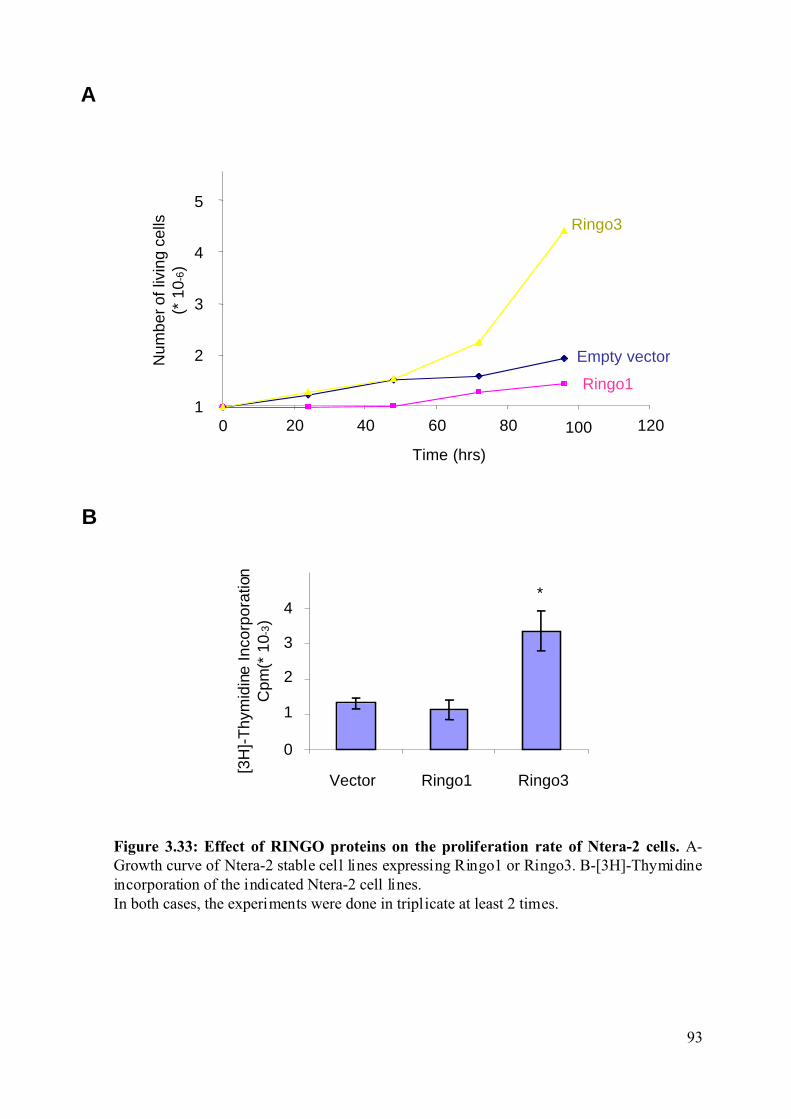
Figure 3.33: Effect of RINGO proteins on the proliferation rate of Ntera-2 cells. A-Growth curve of Ntera-2 stable cell lines expressing Ringo1 or Ringo3. B-[3H]-Thymidineincorporation of the indicated Ntera-2 cell lines.In both cases, the experiments were done in triplicate at least 2 times.
A
B
2
3
4
Vector Ringo1 Ringo3
1
0
[3H
]-T
hym
idin
e In
corp
orat
ion
Cpm
(* 1
0-3)
*
0 20 40 60 80 100 120
Num
ber o
f liv
ing
cells
(* 1
0 -6)
Empty vector
Ringo3
Ringo11
2
4
3
Time (hrs)
5
93
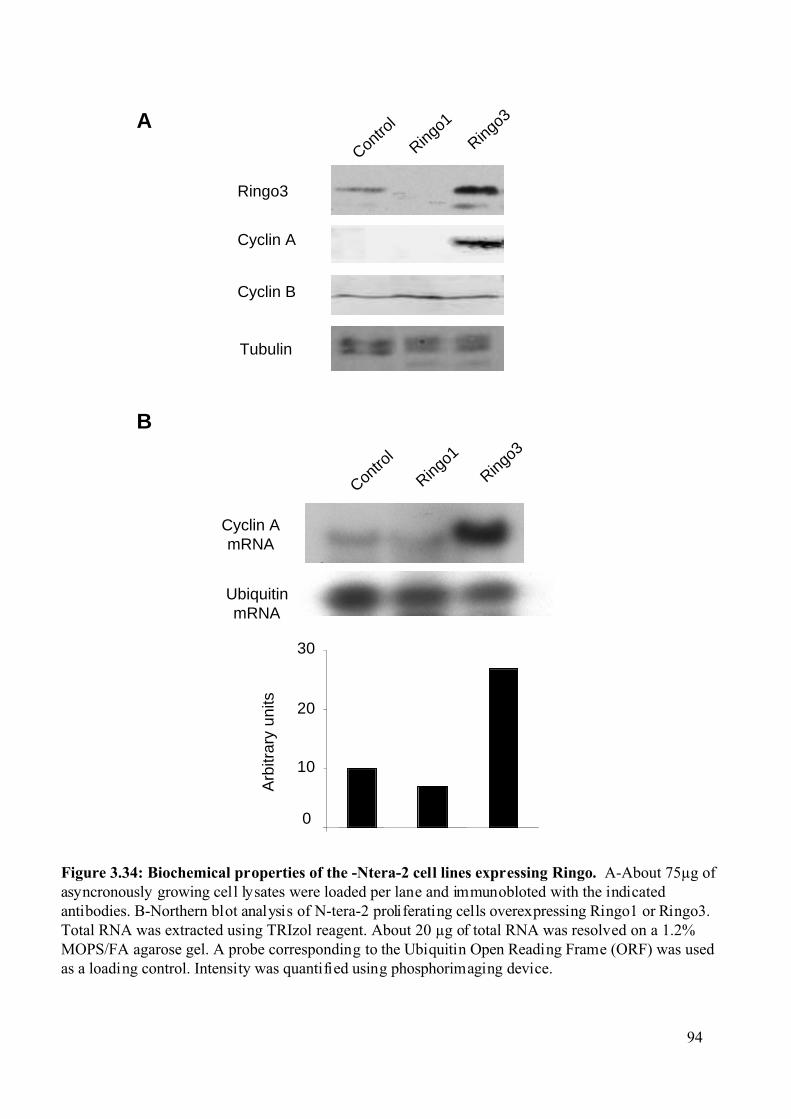
0
10
20
30
Arb
itrar
y un
its
UbiquitinmRNA
Cyclin AmRNA
Figure 3.34: Biochemical properties of the -Ntera-2 cell lines expressing Ringo. A-About 75µg ofasyncronously growing cell lysates were loaded per lane and immunobloted with the indicatedantibodies. B-Northern blot analysis of N-tera-2 proliferating cells overexpressing Ringo1 or Ringo3.Total RNA was extracted using TRIzol reagent. About 20 µg of total RNA was resolved on a 1.2%MOPS/FA agarose gel. A probe corresponding to the Ubiquitin Open Reading Frame (ORF) was usedas a loading control. Intensity was quantified using phosphorimaging device.
A
B
Tubulin
Cyclin B
Cyclin A
Ringo3
Con
trol
Ringo1
Ringo3
Con
trol
Ringo1
Ringo3
94
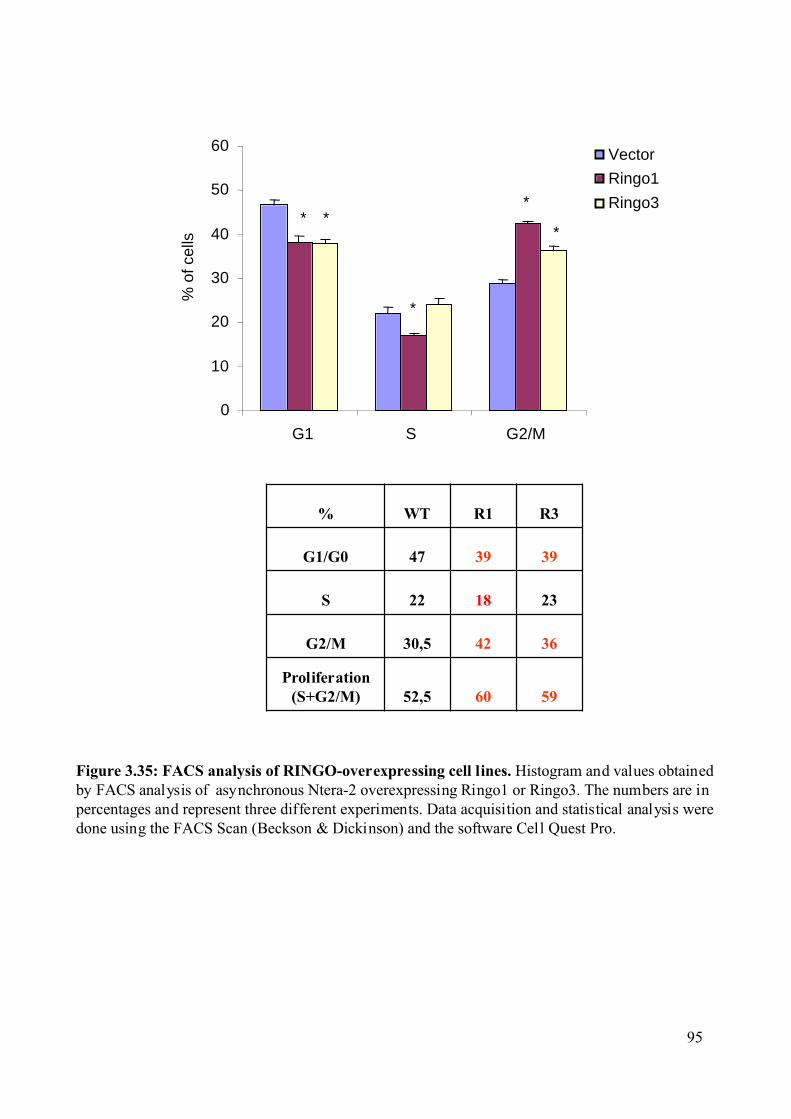
Figure 3.35: FACS analysis of RINGO-overexpressing cell lines. Histogram and values obtainedby FACS analysis of asynchronous Ntera-2 overexpressing Ringo1 or Ringo3. The numbers are inpercentages and represent three different experiments. Data acquisition and statistical analysis weredone using the FACS Scan (Beckson & Dickinson) and the software Cell Quest Pro.
Vector
Ringo1
Ringo3
G1 S G2/M
596052,5Proliferation
(S+G2/M)
364230,5G2/M
231822S
393947G1/G0
R3R1WT%
* *
*
*%
of c
ells
0
10
20
30
40
50
60
*
95

Results
96
3.2.8.5 Biochemical and cell cycle properties of the Ntera-2 cell lines
depleted from the RINGO proteins. We used small interference RNA (siRNA) (McManus and Sharp, 2002) to deplete
Ntera-2 cells from either Ringo1 or Ringo3. We combined both biochemical and FACS
analysis to investigate the role of the RINGO proteins during the cell cycle. As expected,
depletion of either Ringo1 or Ringo3 led to cell cycle arrest. This is shown by the dramatic
loss of [3H]-Thymidine incorporation (Figure 3.36A). We also detected reduced amounts of
the Proliferation Cellular Nuclear Antigen (PCNA), which is a marker of cell proliferation. In
Ringo3-depleted cells, we were able to detect accumulation of p21/Cip1 protein, characteristic
of a G1 arrest, indicating that these cells are probably blocked in G1 (Figure 3.36B). In
Ringo1-depleted cells, high amounts of Cyclin A were detected, indicating that these cells are
not able to enter M phase, since Cyclin A is degraded in pro-metaphase (Geley et al., 2001)
(Figure 3.36B). Interestingly, Ringo3 expression was not detected in these cells (Figure
3.36B).
Ringo1 or Ringo3-depleted cells were also processed for FACS analysis (Figure 3.37).
In Ringo3-depleted cells, we observed an increase of the G1/S population. However in
Ringo1-depleted cells, we observed an increase of cells present in the S/G2 phases of the cell
cycle. To confirm the hypothesis that Ringo3 is involved in the G1/S transition of the cell
cycle and to investigate whether Ringo1 is involved in S phase exit or M phase entry, we
synchronized Ntera-2 cells depleted for Ringo1 or Ringo3 at either the G1/S or G2/M
boundary using Hydroxyurea or Nocodazole respectively. The control and Ringo1-depleted
cells underwent an impressive arrest in G2/M when treated with Nocodazole (Figure 3.38A).
However, if the Ringo3-depleted cells were mainly arrested in G2/M but we could also detect
more cells in G1 than in the control and Ringo1-depleted cells. Hydroxyurea treatment
blocked all the cells in G1 (Figure 3.38A). 24 hrs after release from the Nocodazole or
Hydroxyurea blocks, the control cells entered a normal cell cycle. However, Ringo3 depleted
cells displayed a higher percentage of cells in G1 compared to the control cells (Figure
3.38B). This could indicate that the cells are partially blocked at the G1/S boundary,
suggesting that Ringo3 may be necessary for the G1/S transition of the cell cycle. Ringo1-
depleted cells re-entered the cell cycle after Nocodazole block normally, suggesting that
Ringo1 is probably not required for the G2/M transition (Figure 3.38B). However, there was
an important increase of the percentage of cells in S phase after the Hydroxyurea release,
suggesting that Ringo1 may be required for the S/G2 transition.
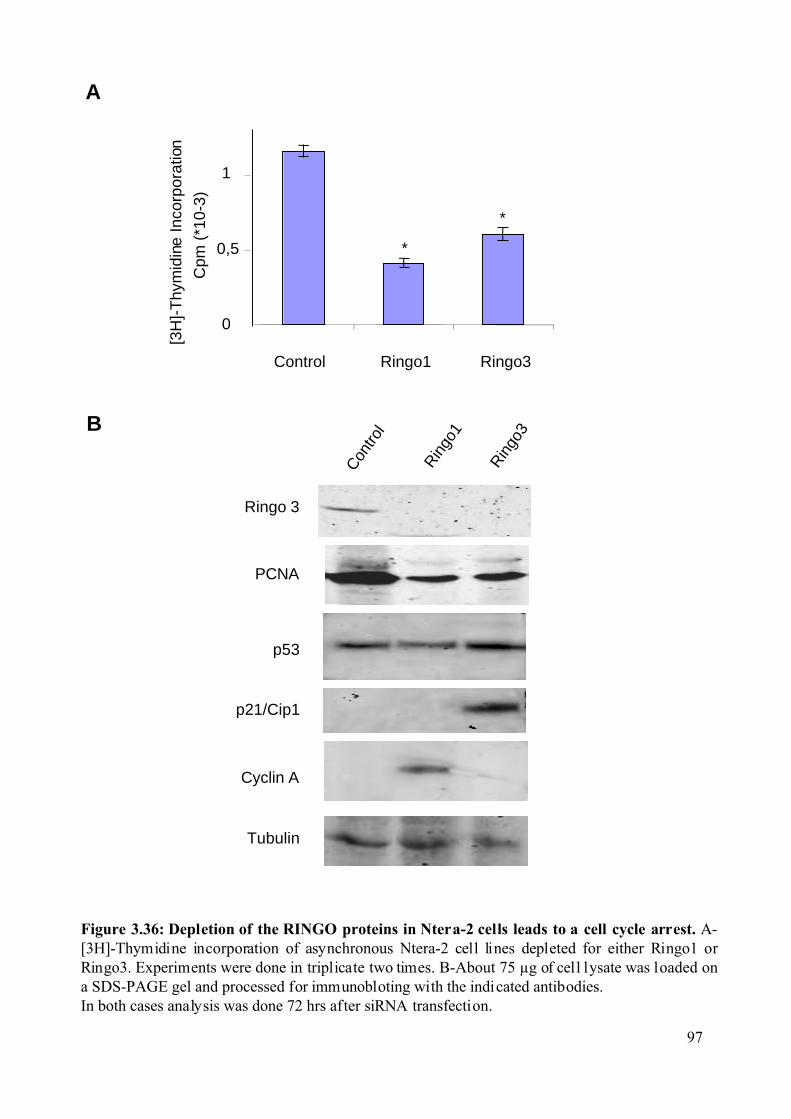
Figure 3.36: Depletion of the RINGO proteins in Ntera-2 cells leads to a cell cycle arrest. A-[3H]-Thymidine incorporation of asynchronous Ntera-2 cell lines depleted for either Ringo1 orRingo3. Experiments were done in triplicate two times. B-About 75 µg of cell lysate was loaded ona SDS-PAGE gel and processed for immunobloting with the indicated antibodies.In both cases analysis was done 72 hrs after siRNA transfection.
A
B
1
0,5
Control Ringo1 Ringo3
0
*
*
[3H
]-T
hym
idin
e In
corp
orat
ion
Cpm
(*1
0-3)
Con
trol
Ringo
3Ringo 3
PCNA
Tubulin
p21/Cip1
p53
Cyclin A
Ringo
1
97
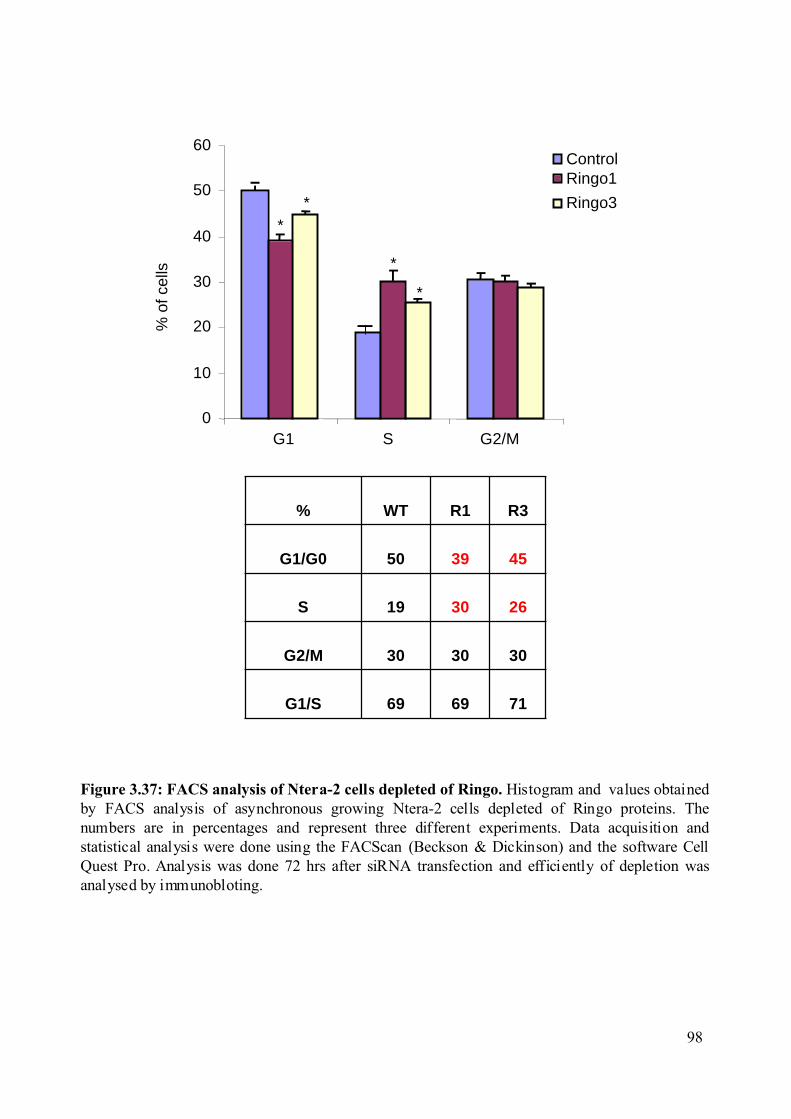
0
10
20
30
40
50
60
G1 S G2/M
716969G1/S
303030G2/M
263019S
453950G1/G0
R3R1WT%
Figure 3.37: FACS analysis of Ntera-2 cells depleted of Ringo. Histogram and values obtainedby FACS analysis of asynchronous growing Ntera-2 cells depleted of Ringo proteins. Thenumbers are in percentages and represent three different experiments. Data acquisition andstatistical analysis were done using the FACScan (Beckson & Dickinson) and the software CellQuest Pro. Analysis was done 72 hrs after siRNA transfection and efficiently of depletion wasanalysed by immunobloting.
*
*
*
*
% o
f cel
lsRingo1
Ringo3
Control
98
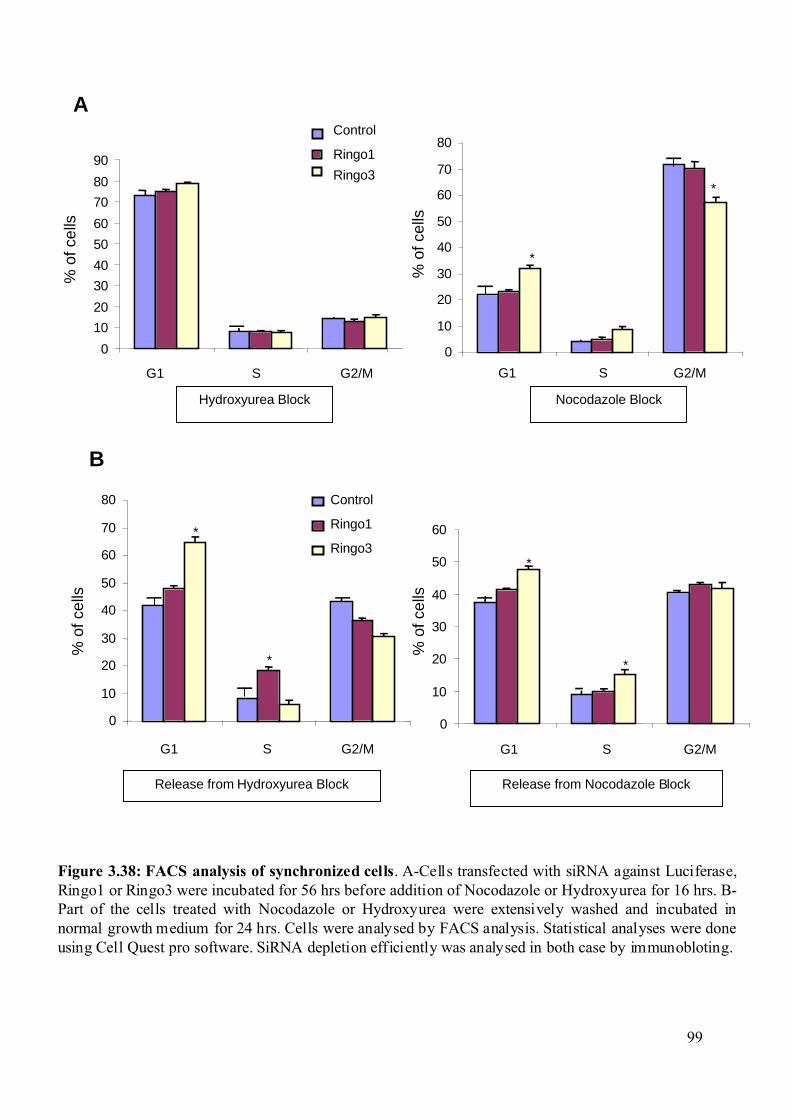
Figure 3.38: FACS analysis of synchronized cells. A-Cells transfected with siRNA against Luciferase,Ringo1 or Ringo3 were incubated for 56 hrs before addition of Nocodazole or Hydroxyurea for 16 hrs. B-Part of the cells treated with Nocodazole or Hydroxyurea were extensively washed and incubated innormal growth medium for 24 hrs. Cells were analysed by FACS analysis. Statistical analyses were doneusing Cell Quest pro software. SiRNA depletion efficiently was analysed in both case by immunobloting.
Release from Hydroxyurea Block
G1 G2/M
Control
Ringo1
Ringo3
0
10
20
30
40
50
60
70
80
S
*
*
Release from Nocodazole Block
G2/MG1
0
10
20
30
40
50
60
S
*
*
G2/M
0
10
20
30
40
50
60
70
80
90
G1 S
Ringo1
Ringo3
Control
Hydroxyurea Block
A
B
% o
f cel
ls
Nocodazole Block
G1 G2/M
0
10
20
30
40
50
60
70
80
S
*
*
% o
f cel
ls
% o
f cel
ls
% o
f cel
ls
99

Discussion
100
4.Discussion. 4.1 Xkid, more than a molecular motor. 4.1.1 Xkid is not required for meiosis I entry.
Xkid was discovered as a protein synthesised during the meiotic maturation of
Xenopus oocytes (Antonio et al., 2000). It was first described that Xkid has a role in
chromosome alignment (Antonio et al., 2000; Funabiki and Murray, 2000) and recently it has
been characterized as a plus end directed motor (Yajima et al., 2003).
Progesterone-treated oocytes can undergo GVBD and assemble the first meiotic
spindle normally in the absence of Xkid. However, these oocytes are unable to form the
second meiotic spindle and appear to enter an interphase-like state.
Xkid-depleted oocytes can enter meiosis I normally, as judged both by biochemical
markers of oocyte maturation (MPF dynamics, activation of the MAPK and Plx1 pathways)
and the cytological markers (the first meiotic spindle forms normally at metaphase I in Xkid-
depleted oocytes).
These observations are in agreement with earlier work showing that spindles assemble
normally around DNA coated beads or sperm nuclei in Xkid-depleted egg extracts (Antonio et
al., 2000). However, while in egg extracts Xkid is required for chromosome alignment
(Antonio et al., 2000), we did not observe any obvious defect in chromosome alignment
during metaphase I in maturing oocytes. The reason for this apparent discrepancy may be that
in meiosis I homologue chromosomes are physically connected by chiasmata and the
mechanisms involved in chromosome congression are probably different (Murray and Hunt,
1993). In addition, at this stage the levels of Xkid are still very low and the protein turnover is
high indicating that Xkid may indeed not be required for meiosis I.
4.1.2 Xkid is required for meiosis I to meiosis II transition. Xkid is not required for meiosis I entry but is essential to reach the metaphase II arrest.
We could observe, about four hours after GVBD, that Xkid depleted oocytes display low level
of H1K activity and start to replicate their DNA, while the second meiotic spindle was absent
and only scattered DNA was present. Eventually, Xkid–depleted oocytes died as a result of
apoptosis.
Interestingly, a similar phenotype of aborted meiotic progression occurs when
Xenopus oocytes are injected with Cyclin B antisense oligonucleotides or incubated with

Discussion
101
either the MAPK kinase inhibitor U0126 or CHX at the time of GVBD (Furuno et al., 1994;
Gross et al., 2000; Hochegger et al., 2001; Kanki and Donoghue, 1991). These oocytes can
enter meiosis I normally, as judged by both biochemical and cytological markers, but then
degenerate and fail to arrest at metaphase II. The collapse of meiotic maturation in these cases
correlates with the inability to re-activate MPF after meiosis I and with the inactivation of
MAPK. In Xkid-depleted oocytes, we do observe low levels of MPF activity, although the
MAPK pathway remains active. The Mos-MAPK-Rsk pathway is thought to play an
important role in the inhibition of S phase after meiosis I. However, it is not clear if this effect
is a direct inhibition on the DNA replication machinery or just reflects the involvement of this
signalling pathway in MPF re-activation after meiosis I, which is thought to be driven by
synthesis of B-type cyclins (Hochegger et al., 2001).
Our work indicates that the absence of Xkid interferes with the re-activation of MPF
during the meiosis I to meiosis II transition, even though MAPK and Rsk are active.
Consistent with this idea, we observed reduced levels of B-type cyclins in maturing Xkid-
depleted oocytes. Moreover, ectopic expression of Xkid was able to fully rescue this effect
and restored cyclin B1 and B4 expression to the normal levels of mature wild type oocytes.
This suggests that Xkid somehow regulates the accumulation of Cyclin B during the meiosis I
to meiosis II transition. These results indicate that the chromokinesin Xkid plays a
fundamental role in the meiotic cell cycle of oocytes, which seems to be independent of its
role in chromosome positioning.
4.1.3 Putative Xkid functions during the meiosis I to meiosis II
transition. What is the DNA-independent function of Xkid in the meiotic cell cycle? Several
possibilities can be considered.
First Xkid could be directly involved in the inhibition of the DNA replication
machinery, but this possibility is unlikely. Indeed, it is difficult to imagine how Xkid could
interfere with the DNA replication machinery. Furthermore, it has recently been found that
Xenopus oocytes temporarily lose their ability to replicate their chromosomes due to the
absence of the essential prereplication complex component Cdc6 and the cytoplasmic
delocalization of Orc proteins and Cdc7 kinase (Lemaitre et al., 2002; Whitmire et al., 2002).
The more likely possibility is that Xkid regulates the meiotic progression, by
regulating either the degradation or synthesis of B type cyclins, which is turn would indirectly

Discussion
102
inhibit the DNA replication machinery, it is known that local translation can occur at the
spindle (Groisman et al., 2000; Richter, 2001). One hypothesis could be that Xkid binds
proteins that participate in translation of B type cyclin mRNAs at the spindle (Cao and
Richter, 2002; de Moor and Richter, 1999; Kobayashi et al., 1991b; Stebbins-Boaz et al.,
1996) ensuring rapid synthesis of cyclins when it is needed, for example after the drop of
MPF activity. Alternatively, Xkid may bind proteins that inhibit Cyclin B synthesis, such as
Maskin (Cao and Richter, 2002; de Moor and Richter, 1999; Groisman et al., 2000; Groisman
et al., 2002). It is also possible that Xkid directly binds Cyclin B mRNAs as it has the ability
to bind nucleic acids (Antonio et al., 2000; Funabiki and Murray, 2000), therefore directly
regulating its translation.
Finally, another possibility could be that Xkid affects the proteins degradation
machinery. It is conceivable that Xkid could interfere with cyclin degradation. Indeed, cyclin
levels decrease during meiosis I to meiosis II transition but they are not completely degraded.
It has been shown that a total degradation of cyclins or the inhibition of Cdk1 by Wee1 results
in a phenotype similar to the one observed in Xkid depleted oocytes (Furuno et al., 1994;
Hochegger et al., 2001; Nakajo et al., 2000).
We can imagine a model where Xkid regulates the meiosis I to meiosis II transition
acting on MPF reactivation (Figure 4.1). Xkid could control either the synthesis or
degradation of B-type cyclins between the meiosis I to meiosis II, leading to MPF reactivation
after meiosis I and preventing the oocytes entering an interphase like state (where DNA
replication would be de-repressed).
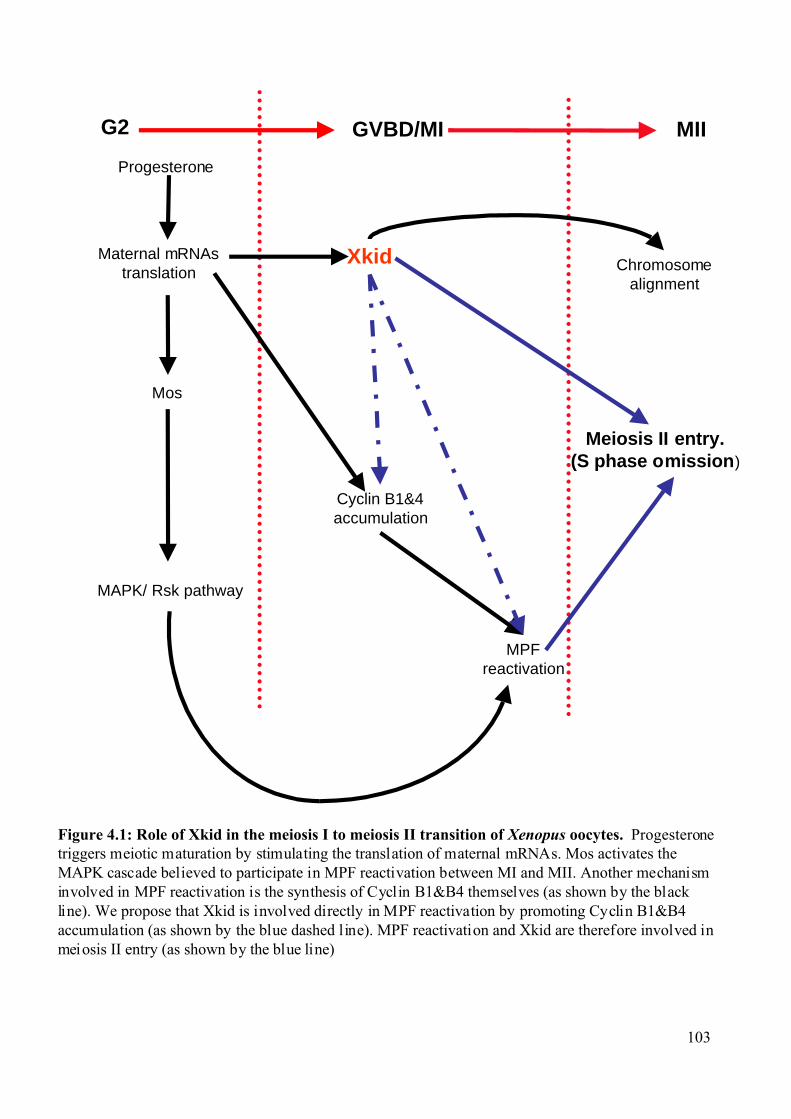
MAPK/ Rsk pathway
Meiosis II entry.(S phase omission)
Xkid Chromosomealignment
G2 GVBD/MI MII
Maternal mRNAstranslation
Cyclin B1&4accumulation
MPFreactivation
Mos
Progesterone
Figure 4.1: Role of Xkid in the meiosis I to meiosis II transition of Xenopus oocytes. Progesteronetriggers meiotic maturation by stimulating the translation of maternal mRNAs. Mos activates theMAPK cascade believed to participate in MPF reactivation between MI and MII. Another mechanisminvolved in MPF reactivation is the synthesis of Cyclin B1&B4 themselves (as shown by the blackline). We propose that Xkid is involved directly in MPF reactivation by promoting Cyclin B1&B4accumulation (as shown by the blue dashed line). MPF reactivation and Xkid are therefore involved inmeiosis II entry (as shown by the blue line)
103

Discussion
104
4.2 RINGO, a new family of cell cycle regulators. 4.2.1 RINGO bypasses usual Cdk regulatory mechanisms.
Cdks are catalytically inactive as monomers and should be bound to cyclins for full
kinase activity. Cdks should be phosphorylated on a threonine located on the T-loop, whereas
Thr14 and Tyr15 residues located in the catalytic cleft should be dephosphorylated for full
activity (Morgan, 1997). We found that Cdks can be activated by RINGO, which has no
amino acid sequence homology to cyclins. RINGO also does not respond to the same
regulatory mechanisms as the Cdk-cyclin complexes. RINGO induced kinase activity of Cdk1
and Cdk2 can bypass the requirement for phosphorylation of the threonine located on the T-
loop, both in vitro and in Xenopus oocytes. This phosphorylation is essential for full
activation of these Cdks by cyclins.
Crystallographic studies have revealed that phosphorylation of this threonine (Thr161
in Cdk1 and Thr160 in Cdk2) induces important conformational changes (Brown et al., 1995;
Cavalli et al., 2001; De Bondt et al., 1993; Jeffrey et al., 1995). The phosphate acts as an
organizing centre, stabilizing the T-loop region (which probably participates in substrate
recognition) as well as the Cdk-Cyclin complex (Brown et al., 1995; Kim et al., 1996;
Morgan, 1997; Tarricone et al., 2001). One of the functions of the T-loop phosphorylation
may be to improve the binding of protein substrates independently of the catalytic activity.
A possibility consistent with the finding that RINGO-induced activation of Cdk1 and
Cdk2 is independent of the T-loop phosphorylation, would be that RINGO itself induces the
conformational changes in the T-loop region that are normally induced upon threonine
phosphorylation. It is interesting to note that, in contrast to Cdk1 and Cdk2, other Cdks have
been reported to be active in the absence of phosphorylation in the T-loop, for example, Cdk7
when associated with the Cyclin H-Mat1 complex (Kim et al., 1996), and Cdk5, when
associated with and activated by the neuronal p39 and p35 proteins (Lee et al., 1996; Lew et
al., 1994). A recent crystallographic study of the neuronal Cdk5-p25 complex reveals that the
interaction between the p25 activator and the T-loop of Cdk5 results in a T-loop
conformational change. This closely resembles to the conformational change observed in
Thr160-phosphorylated Cdk2 associated with Cyclin A (Tarricone et al., 2001).
These data suggest a common mechanism by which non-cyclin Cdk activators could
bypass the requirement for T- loop phosphorylation.
Interestingly, the similarity between RINGO and other non-cyclin Cdk activators also
extends to the negative regulators. Indeed, p25 allows Cdk5 to escape inhibition by the Tyr15-

Discussion
105
specific kinase Wee1 and by CKIs such as Kip1 (Lee et al., 1996). RINGO-bound Cdk1 and
Cdk2 have also been shown to be less susceptible to the CKI inhibitory mechanisms that
negatively regulate the activity of Cdk-cyclin complexes (Karaiskou et al., 2001). The
RINGO-Cdk complexes also display a weaker interaction with Cip1 and are less efficiently
phosphorylated by Myt1 (Karaiskou et al., 2001). This could be explained because RINGO
proteins lack the MRAIL hydrophobic motif typical of cyclins (Figure 4.2) (Brown et al.,
1995; Morgan, 1997). The MRAIL alpha-helix is crucial for the binding of the cyclins to
Cdks, substrates recognition and it is also the target of the Cip/Kip proteins (Davies et al.,
2001; Moskowitz et al., 1996). Interestingly, when RINGO-Cdk complexes were tested with
Rb as a substrate, which also requires the MRAIL motif, we detected phosphorylation of Rb
in vitro. It is therefore conceivable that RINGO proteins may provide Cdks with a substrate
recognition and recruitment patch, perhaps also of a hydrophobic nature as the MRAIL motif
of cyclins (Brown et al., 1995; Jeffrey et al., 1995; Kim et al., 1996).
T-loop
Cdk2 Cyclin A
From Morgan,1997
MRAIL
Figure 4.2: Model of the interaction between Cyclin A and Cdk2. The two lobes of Cdk2 are easily identified, with the PSTAIRE helix in the N-terminal part and the T-loop linking the two lobes. Cyclin A binds with its cyclin box (the MRAIL helix participle in this binding) to the PSTAIRE helix of Cdk2. Cyclin A promotes a major displacement of the T-loop, allowing substrate access to the catalytic cleft located between the two lobes of Cdk2.
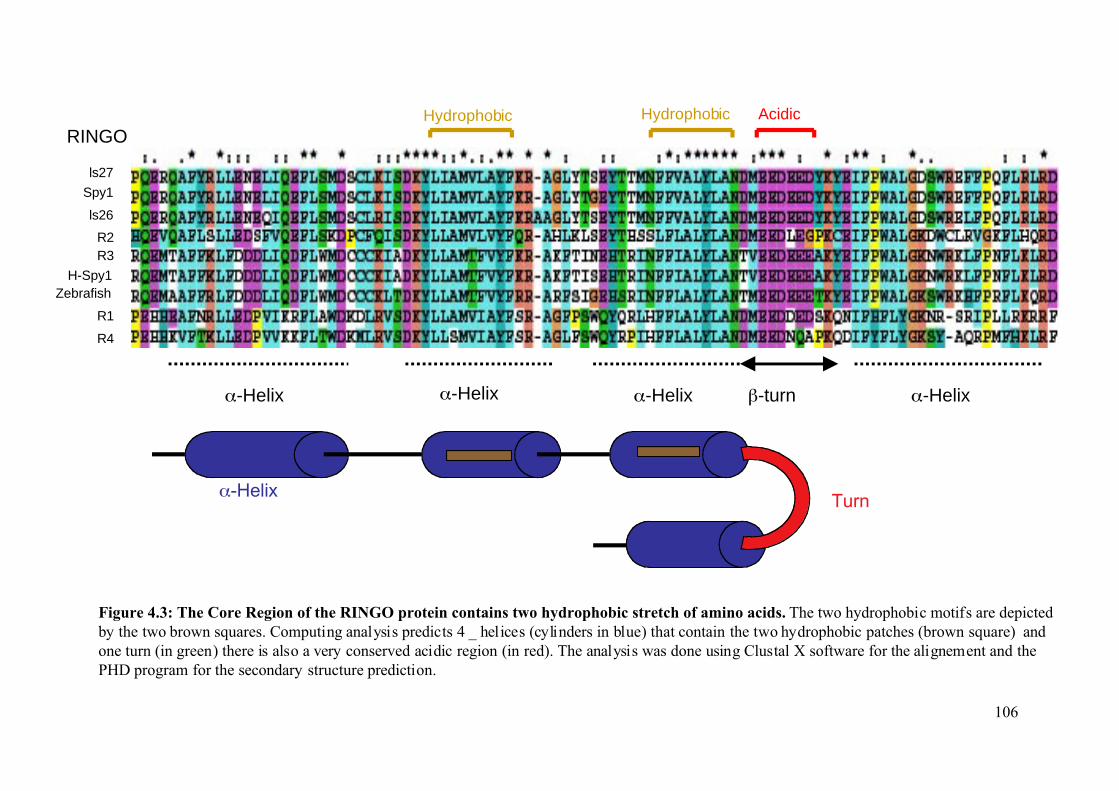
Figure 4.3: The Core Region of the RINGO protein contains two hydrophobic stretch of amino acids. The two hydrophobic motifs are depictedby the two brown squares. Computing analysis predicts 4 _ helices (cylinders in blue) that contain the two hydrophobic patches (brown square) andone turn (in green) there is also a very conserved acidic region (in red). The analysis was done using Clustal X software for the alignement and thePHD program for the secondary structure prediction.
ls27
Spy1
ls26
R2R3
H-Spy1Zebrafish
R1
R4
RINGO
α-Helix α-Helix α-Helixβ-turn
Hydrophobic Hydrophobic Acidic
α-Helix
α-HelixTurn
106

Discussion
107
Indeed, we identified two highly hydrophobic motifs in all the RINGO proteins. These
hydrophobic motifs are located in the core region. Using the PHD programme (Rost, 1996)
we predicted that these two hydrophobic motifs should be part of two alpha-helices up to the
four alpha-helices located in the core region (Figure 4.3). These observations suggest that
RINGO is a new class of non-cyclin Cdk activator that bypasses the usual biochemical
properties expected for the Cdks. Crystallization of the RINGO-Cdk complex should help to
elucidate if it follows the same activation mode as Cdk5 and Cdk7 or a different mechanism.
4.2.2 Ringo3 activates Cdk5. The common general function of the Cdk family members is to ensure the normal
progression through the cell cycle. As previously mentioned, Cdks are tightly regulated by the
sequential expression of cyclins. Cdk5 is a Cdk family member which is neither activated by
cyclins nor involved in cell cycle regulation. Like other Cdks, monomeric Cdk5 shows no
enzymatic activity and requires association with a regulatory partner for activation.
Interestingly, although Cdk5 is ubiquitously expressed, the only known activators of Cdk5 are
the p35 and p39 proteins, the expression of which is restricted to the nervous system. The
highest expression of Cdk5 and associated kinase activity can be detected in the nervous
system (Ino et al., 1994; Lew et al., 1994; Tsai et al., 1994; Tsai et al., 1993). Cdk5 activity
has been linked to the regulation of the cytoskeleton, axon guidance, membrane transport,
synaptic function, dopamine signalling and drug addiction (Dhavan and Tsai, 2001; Smith and
Tsai, 2002).
Several groups have since demonstrated the presence of active Cdk5 in non-neuronal
tissues. For example, low level of Cdk5 kinase activity is present in adult mouse testis and
muscles (Dhavan and Tsai, 2001; Musa et al., 1998; Session et al., 2001).
Immunohistochemical analysis showed that Cdk5 is expressed in Leydig cells, Sertoli cells,
spermatogonia and peritubular cells of the developing rat testis. Cdk5 kinase activity
coincides with Cdk5 protein expression in these cells, suggesting a role in spermatogenesis
(Musa et al., 1998; Session et al., 2001). Although Cdk5 function in non-neuronal tissues is
only just beginning to be explored, the studies described above indicate that the role of this
kinase extends far beyond what was originally suspected (Dhavan and Tsai, 2001).
We have found that Ringo3 protein is able to activate Cdk5 to similar extent as p25,
which is one of the physiological activators of Cdk5. This is a surprising result and it is the
first time that a protein that does not belong to the p35 family has been shown to be able to

Discussion
108
directly activate Cdk5. Moreover, Cdk5 has been implicated in multiple processes as
described above, but not in cell cycle regulation.
Since Ringo3 is present in foetal brain but not in adult brain, it is possible that Ringo3
could be involved in brain cytoarchitecture during development, suggesting that Ringo3 may
also be involved in cell differentiation. Ringo3 is mostly present in highly proliferative tissues
and in combination with Cdk5 it might regulate the cell cycle in specific tissues, for example
foetal brain or testis, where both proteins are present. A careful examination of the expression
pattern of Ringo3 and Cdk5 in testis, by in situ hybridization will be useful to determine if
these proteins co-localize in dividing or post mitotic cells. This information could help to
elucidate whether Ringo3-Cdk5 complexes are involved in proliferation or differentiation.
4.2.3 RINGO: a novel family of proteins regulating the cell cycle. In this thesis, we have characterized a novel family of cell cycle regulators. The first
member of this family was discovered in Xenopus, for its ability to induce the G2/M transition
in Xenopus oocytes (Ferby et al., 1999; Lenormand et al., 1999).
We have identified and cloned four mammalian family members, which we have
named Ringo1 to Ringo4. We have also identified several others which are only present in
vertebrates. The absence of RINGO in invertebrates could be due to two possibilities. First,
RINGO could be a new addition to the vertebrate cell cycle, which is more elaborated and
complex than the invertebrate one. The second possibility is that a functional homologue with
low sequence homology exists in invertebrate organisms, but we could not identify it by
BLAST searches.
Despite their low level of homology, the RINGO proteins share common features and
they are all able to bind and activate Cdks. Expression of the mammalian homologues in
Xenopus oocytes showed some surprising results. While Ringo2 and Ringo3 were able to
accelerate or trigger the meiotic maturation, Ringo1 blocked progesterone induced
maturation. It is important to note that Ringo2 is very close to the Xenopus RINGO, but did
not induce the maturation. In the case of the Myc tagged version of this protein, we could only
detect an acceleration of the meiotic maturation process, although the protein was expressed
at very low level. Interestingly, the same result was observed when Xenopus RINGO was
expressed at low levels (Ferby, 2000). Taken together, these results suggest that higher level
of expression of Myc-Ringo2 could trigger meiotic maturation. Higher levels of expression
were achieved when GST-Ringo2 was injected into oocytes, but the GST tag seems to affect

Discussion
109
the properties of RINGO proteins, as the Xenopus RINGO fused to GST is also unable to
trigger the meiotic maturation.
The fact that Ringo1inhibits oocyte maturation but is still able to bind and activate
Cdk1 is puzzling. This could be explained by the inability of Ringo1 to target Cdk1 to the
proper substrates, either because the kinase is mislocalized or because Ringo1 does not
contain the necessary information for a proper recognition of the right substrates. It is also
important to mention that although part of Ringo1 is associated to Cdk1, another part of the
Ringo1 pool is likely to be associated with another kinase, as we could not recover all the
H1K activity associated with Ringo1 by Cdk1 immunoprecipitation. The obvious candidates
are Cdk2 and Cdk5, although neither are considered essential for the meiotic maturation
process (Furuno et al., 1997; Gervasi and Szaro, 1995). The ability of overexpressed Cdk1 to
rescue inhibition by Ringo1 is an important point in favour of an inhibitory mechanism by
Ringo1 due to sequestration of the endogenous Cdk1. One explanation could be that Ringo1
binds the free Cdk1 in the oocyte and makes it inaccessible to other proteins, such as
endogenous RINGO that may need to bind to the free Cdk1 for progesterone-induced
maturation (Nebreda et al., 1995).
Finally, we have identified the RINGO-core domain as a stretch of 100 amino acids
that is highly conserved in all the family members. It is predicted to fold with four alpha-
helices and one turn (PHD programme (Rost, 1996)). The core domain is likely to be involved
in binding the Cdks, as we could detect interaction with Cdk1 in vitro and in Xenopus oocytes
using only this RINGO region.
4.2.4 Role of Ringo1 and Ringo3 in the mammalian cell cycle. We have focused our attention on two members of the family: Ringo1 and Ringo3,
because they display opposite effects when expressed in Xenopus oocytes.
We demonstrate that the RINGO proteins are regulators of the cell cycle. We found
that the Ringo3 protein is cell cycle regulated and present during the G1/S phase of the
mammalian cell cycle. Ringo3 mRNA is found at different levels in specific tissues, but it is
more abundant in tissues with a high proliferation rate such as testis, thymus and foetal brain.
We were also able to identify alternative spliced variants of this protein, but in this case their
expression appears to be restricted to testis, suggesting a putative role for these proteins in
testis function. It is also possible that the proteins are expressed at very low levels in other
tissues and we could only detect them in testis.

Discussion
110
The identification and characterization of a novel cell cycle gene name Human Speedy
(Spy1) has been recently reported (Porter et al., 2002). Spy1 is 40% homologous to the
Xenopus RINGO and is likely to be the human homologue of Ringo3. The Spy1 mRNA is
only expressed during the G1/S phase of the cell cycle and overexpressed Myc-tagged Spy1 is
mainly localized in the nucleus. Overexpression of Spy1 enhances cell proliferation in a Cdk2
dependent manner. Human Speedy is proposed to be able of promoting cell proliferation
through the premature activation of Cdk2 at the G1/S phase transition (Porter et al., 2002).
Although, this study overlaps with part of the work presented in this thesis, it should be noted
that the endogenous mammalian Spy1 was not characterised and most of the experimental
work was done by overexpression of the protein in HeLa or HEK293 cells, which themselves
do not express Spy1 (Porter et al., 2002). Moreover no molecular insight was provided into
how Spy1 could enhance cell proliferation.
We have worked with a cell line expressing both the endogenous Ringo3 and Ringo1.
We found that overexpression of Ringo3 increases the proliferation rate in human Ntera-2
cells, as indicated by cell growth curves and increased level of [3H]-Thymidine incorporation.
One possibility could be that Ringo3 binds to and prematurely activates Cdk2, as we could
detect binding between the endogenous Ringo3 and Cdk2. Indeed, flow cytometry studies
demonstrate that Ringo3 overexpression decreases the overall population of cells in the G1
phase of the cell cycle and increase the population of cells in G2/M. Consistent with this
observation cells undergo a G1/S arrest as indicated by both biochemical and FACS analysis,
upon depletion of Ringo3. The most well-established pathway leading to Cdk2 activation and
entry into S phase is the Rb pathway, comprising Rb protein along with its upstream
regulators cyclin D and Cdk4/6 and Rb-regulated E2F transcription factors (Bartek et al.,
1996). We have shown that Ringo3-Cdk2 can phosphorylate Rb in vitro. This activity could
be responsible for premature Cyclin A synthesis and therefore S phase entry. Another
attractive posibility is that the Ringo3-Cdk2 complex forms at early G1 and phosphorylates
Kip1, which in return would be targeted for degradation. This would allow the activation of
the Cyclin E-Cdk2 complex to promote earlier S phase entry (Figure 4.5).
Ringo1 mRNAs are only expressed in testis. Although we generated several anti-sera
we could not detect expression of the endogenous Ringo1 protein, perhaps because it is
expressed at very low levels. Overexpression of Ringo1 decreases proliferation in the human
Ntera-2 cell line. One hypothesis could be that Ringo1 is involved in S phase exit. This could
explain the inability of Ringo1 overexpressing cells to compensate defects accumulated
during a premature S phase exit and a block at the G2/M boundary. Flow cytometry studies

Discussion
111
demonstrate that Ringo1 decreases the overall population of cells in G1/S phase of the cell
cycle and increases the population of cells in G2/M. Consistent with this observation we have
found that depletion of Ringo1 slows cell growth considerably and increases the population of
cells in S phase. These results suggest that Ringo1 is likely to be involved in the S phase exit
of the cell cycle.
All the data presented in this thesis demonstrate that the RINGO proteins are a new
type of cyclin-like proteins, which are necessary for cell cycle progression. They can bind and
activate Cdks, and at least Ringo3 is transiently expressed during the cell cycle. Our results
indicate that Ringo3 is involved in the G1/S transition of the cell cycle and Ringo1 is more
likely to be involved in S phase exit.
4.2.5 Why does the cell cycle need the RINGO proteins? Cdk activity must be tightly controlled, as defects in this process may result in the gain
or loss of chromosomes and can lead to cell death or contribute to oncogenic transformation
(Smits and Medema, 2001). One obvious question when looking at the cell cycle regulation is
why does the cell need RINGO proteins, if there are already cyclins? RINGO is a new class of
non-cyclin Cdk activator that can bypass the usual biochemical mechanisms regulating Cdk
activation by cyclins.
This implies that RINGO can activate Cdks via a unique mechanism. This kind of
“super-cyclin” property would be very useful in conditions where the organism needs fast cell
proliferation. Consistent with this idea we found that Ringo3 is expressed in highly
proliferative tissues. We could imagine that RINGO is present when the organisms need to
increase its cell numbers, in a short period of time, for example during brain development.
An interesting idea would be that the RINGO proteins can bypass the cell cycle
checkpoints and act under conditions where the Cdk-cyclins complex is normally inactive.
What would be the interest for the cell to be able to bypass checkpoint? This remains to be
investigated, but one possibility could that the RINGO proteins are necessary to “liberate” the
cell from the checkpoint arrest. Indeed if the mechanisms of checkpoint establishment are
well known, exit from them is still poorly understood. A further characterization of the
RINGO proteins in cell cycle regulation and especially the mechanisms of checkpoint
maintenance will be of a great interest.
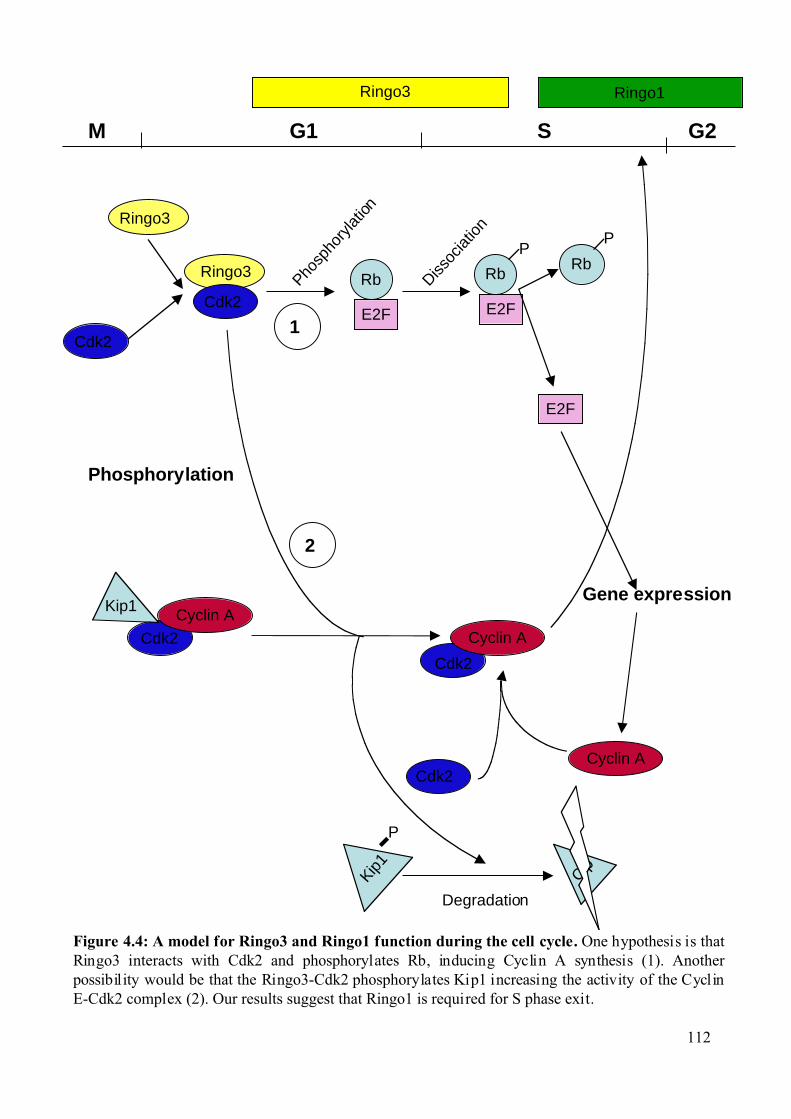
Figure 4.4: A model for Ringo3 and Ringo1 function during the cell cycle. One hypothesis is thatRingo3 interacts with Cdk2 and phosphorylates Rb, inducing Cyclin A synthesis (1). Anotherpossibility would be that the Ringo3-Cdk2 phosphorylates Kip1 increasing the activity of the CyclinE-Cdk2 complex (2). Our results suggest that Ringo1 is required for S phase exit.
G1 S
Ringo3
Cdk2
Ringo3
Cdk2
Rb
E2F
PRb
P
E2F
Gene expression
Cyclin A
G2M
Cdk2
Cyclin A
Cdk2
Cyclin A
Kip
1
P
Cip
Degradation
Ringo3 Ringo1
Rb
E2FPho
spho
rylat
ion
Dissoc
iation
Kip1
Phosphorylation
Cdk2
1
2
112

Bibliography
113
6 Bibliography.
Abraham, R. T. (2001). Cell cycle checkpoint signaling through the ATM and ATR
kinases. Genes Dev 15, 2177-2196.
Altschul, S. F., Madden, T. L., Schaffer, A. A., Zhang, J., Zhang, Z., Miller, W., and
Lipman, D. J. (1997). Gapped BLAST and PSI-BLAST: a new generation of protein database
search programs. Nucleic Acids Res 25, 3389-3402.
Ambrosino, C., Mace, G., Galban, S., Fritsch, C., Vintersten, K., Black, E., Gorospe, M.,
and Nebreda, A. R. (2003). Negative feedback regulation of MKK6 mRNA stability by
p38alpha mitogen-activated protein kinase. Mol Cell Biol 23, 370-381.
Anderson, C. W., Baum, P. R., and Gesteland, R. F. (1973). Processing of adenovirus 2-
induced proteins. J Virol 12, 241-252.
Antonio, C. (2002). Heidelberg Univesity. PhD thesis.
Antonio, C., Ferby, I., Wilhelm, H., Jones, M., Karsenti, E., Nebreda, A. R., and Vernos,
I. (2000). Xkid, a chromokinesin required for chromosome alignment on the metaphase plate.
Cell 102, 425-435.
Bartek, J., Falck, J., and Lukas, J. (2001). CHK2 kinase--a busy messenger. Nat Rev
Mol Cell Biol 2, 877-886.
Bartek, J., and Lukas, J. (2001a). Mammalian G1- and S-phase checkpoints in response
to DNA damage. Curr Opin Cell Biol 13, 738-747.
Bartek, J., and Lukas, J. (2001b). Pathways governing G1/S transition and their response
to DNA damage. FEBS Lett 490, 117-122.
Baulieu, E. E., Godeau, F., Schorderet, M., and Schorderet-Slatkine, S. (1978). Steroid-
induced meiotic division in Xenopus laevis oocytes: surface and calcium. Nature 275, 593-
598.
Booher, R. N., Holman, P. S., and Fattaey, A. (1997). Human Myt1 is a cell cycle-
regulated kinase that inhibits Cdc2 but not Cdk2 activity. J Biol Chem 272, 22300-22306.
Brehm, A., Miska, E., Reid, J., Bannister, A., and Kouzarides, T. (1999). The cell cycle-
regulating transcription factors E2F-RB. Br J Cancer 80 Suppl 1, 38-41.
Brown, N. R., Noble, M. E., Endicott, J. A., Garman, E. F., Wakatsuki, S., Mitchell, E.,
Rasmussen, B., Hunt, T., and Johnson, L. N. (1995). The crystal structure of cyclin A.
Structure 3, 1235-1247.
Bulavin, D. V., Amundson, S. A., and Fornace, A. J. (2002). p38 and Chk1 kinases:
different conductors for the G(2)/M checkpoint symphony. Curr Opin Genet Dev 12, 92-97.

Bibliography
114
Cao, Q., and Richter, J. D. (2002). Dissolution of the maskin-eIF4E complex by
cytoplasmic polyadenylation and poly(A)-binding protein controls cyclin B1 mRNA
translation and oocyte maturation. Embo J 21, 3852-3862.
Cavalli, A., Dezi, C., Folkers, G., Scapozza, L., and Recanatini, M. (2001). Three-
dimensional model of the cyclin-dependent kinase 1 (CDK1): Ab initio active site parameters
for molecular dynamics studies of CDKS. Proteins 45, 478-485.
Coux, O., Tanaka, K., and Goldberg, A. L. (1996). Structure and functions of the 20S
and 26S proteasomes. Annu Rev Biochem 65, 801-847.
Daar, I., Paules, R. S., and Vande Woude, G. F. (1991). A characterization of cytostatic
factor activity from Xenopus eggs and c-mos-transformed cells. J Cell Biol 114, 329-335.
Davies, T. G., Tunnah, P., Meijer, L., Marko, D., Eisenbrand, G., Endicott, J. A., and
Noble, M. E. (2001). Inhibitor binding to active and inactive CDK2: the crystal structure of
CDK2-cyclin A/indirubin-5-sulphonate. Structure (Camb) 9, 389-397.
De Bondt, H. L., Rosenblatt, J., Jancarik, J., Jones, H. D., Morgan, D. O., and Kim, S. H.
(1993). Crystal structure of cyclin-dependent kinase 2. Nature 363, 595-602.
De Moor, C. H., and Richter, J. D. (1999). Cytoplasmic polyadenylation elements
mediate masking and unmasking of cyclin B1 mRNA. Embo J 18, 2294-2303.
Desai, D., Wessling, H. C., Fisher, R. P., and Morgan, D. O. (1995). Effects of
phosphorylation by CAK on cyclin binding by CDC2 and CDK2. Mol Cell Biol 15, 345-350.
Dhavan, R., and Tsai, L. H. (2001). A decade of CDK5. Nat Rev Mol Cell Biol 2, 749-
759.
Donjerkovic, D., and Scott, D. W. (2000). Regulation of the G1 phase of the mammalian
cell cycle. Cell Res 10, 1-16.
Duckworth, B. C., Weaver, J. S., and Ruderman, J. V. (2002). G2 arrest in Xenopus
oocytes depends on phosphorylation of cdc25 by protein kinase A. Proc Natl Acad Sci U S A
99, 16794-16799.
Dupre, A., Jessus, C., Ozon, R., and Haccard, O. (2002). Mos is not required for the
initiation of meiotic maturation in Xenopus oocytes. Embo J 21, 4026-4036.
Erikson, E., and Maller, J. L. (1989). Biochemical characterization of the p34cdc2
protein kinase component of purified maturation-promoting factor from Xenopus eggs. J Biol
Chem 264, 19577-19582.
Evans, T., Rosenthal, E. T., Youngblom, J., Distel, D., and Hunt, T. (1983). Cyclin: a
protein specified by maternal mRNA in sea urchin eggs that is destroyed at each cleavage
division. Cell 33, 389-396.

Bibliography
115
Falck, J., Petrini, J. H., Williams, B. R., Lukas, J., and Bartek, J. (2002). The DNA
damage-dependent intra-S phase checkpoint is regulated by parallel pathways. Nat Genet 30,
290-294.
Ferby, I. (2000). Heidelberg Univesity. PhD thesis.
Ferby, I., Blazquez, M., Palmer, A., Eritja, R., and Nebreda, A. R. (1999). A novel
p34(cdc2)-binding and activating protein that is necessary and sufficient to trigger G(2)/M
progression in Xenopus oocytes. Genes Dev 13, 2177-2189.
Funabiki, H., and Murray, A. W. (2000). The Xenopus chromokinesin Xkid is essential
for metaphase chromosome alignment and must be degraded to allow anaphase chromosome
movement. Cell 102, 411-424.
Furuno, N., Nishizawa, M., Okazaki, K., Tanaka, H., Iwashita, J., Nakajo, N., Ogawa,
Y., and Sagata, N. (1994). Suppression of DNA replication via Mos function during meiotic
divisions in Xenopus oocytes. Embo J 13, 2399-2410.
Furuno, N., Ogawa, Y., Iwashita, J., Nakajo, N., and Sagata, N. (1997). Meiotic cell
cycle in Xenopus oocytes is independent of cdk2 kinase. Embo J 16, 3860-3865.
Geley, S., Kramer, E., Gieffers, C., Gannon, J., Peters, J. M., and Hunt, T. (2001).
Anaphase-promoting complex/cyclosome-dependent proteolysis of human cyclin A starts at
the beginning of mitosis and is not subject to the spindle assembly checkpoint. J Cell Biol 153,
137-148.
Geng, Y., Eaton, E. N., Picon, M., Roberts, J. M., Lundberg, A. S., Gifford, A., Sardet,
C., and Weinberg, R. A. (1996). Regulation of cyclin E transcription by E2Fs and
retinoblastoma protein. Oncogene 12, 1173-1180.
Gervasi, C., and Szaro, B. G. (1995). The Xenopus laevis homologue to the neuronal
cyclin-dependent kinase (cdk5) is expressed in embryos by gastrulation. Brain Res Mol Brain
Res 33, 192-200.
Gieffers, C., Dube, P., Harris, J. R., Stark, H., and Peters, J. M. (2001). Three-
dimensional structure of the anaphase-promoting complex. Mol Cell 7, 907-913.
Glotzer, M., Murray, A. W., and Kirschner, M. W. (1991). Cyclin is degraded by the
ubiquitin pathway. Nature 349, 132-138.
Godeau, J. F., Schorderet-Slatkine, S., Hubert, P., and Baulieu, E. E. (1978). Induction
of maturation in Xenopus laevis oocytes by a steroid linked to a polymer. Proc Natl Acad Sci
U S A 75, 2353-2357.
Golsteyn RM, Lane HA, Mundt KE, Arnaud L, Nigg EA.(1996) The family of polo-like
kinases.Prog Cell Cycle Res. 1996;2:107-14.

Bibliography
116
Graeser, R., Gannon, J., Poon, R. Y., Dubois, T., Aitken, A., and Hunt, T. (2002).
Regulation of the CDK-related protein kinase PCTAIRE-1 and its possible role in neurite
outgrowth in Neuro-2A cells. J Cell Sci 115, 3479-3490.
Groisman, I., Huang, Y. S., Mendez, R., Cao, Q., Theurkauf, W., and Richter, J. D.
(2000). CPEB, maskin, and cyclin B1 mRNA at the mitotic apparatus: implications for local
translational control of cell division. Cell 103, 435-447.
Groisman, I., Jung, M. Y., Sarkissian, M., Cao, Q., and Richter, J. D. (2002).
Translational control of the embryonic cell cycle. Cell 109, 473-483.
Gross, S. D., Lewellyn, A. L., and Maller, J. L. (2001). A constitutively active form of
the protein kinase p90Rsk1 is sufficient to trigger the G2/M transition in Xenopus oocytes. J
Biol Chem 276, 46099-46103.
Gross, S. D., Schwab, M. S., Taieb, F. E., Lewellyn, A. L., Qian, Y. W., and Maller, J.
L. (2000). The critical role of the MAP kinase pathway in meiosis II in Xenopus oocytes is
mediated by p90(Rsk). Curr Biol 10, 430-438.
Gu, Y., Rosenblatt, J., and Morgan, D. O. (1992). Cell cycle regulation of CDK2 activity
by phosphorylation of Thr160 and Tyr15. Embo J 11, 3995-4005.
Guan, K. L., and Dixon, J. E. (1991). Eukaryotic proteins expressed in Escherichia coli:
an improved thrombin cleavage and purification procedure of fusion proteins with glutathione
S-transferase. Anal Biochem 192, 262-267.
Haccard, O., Lewellyn, A., Hartley, R. S., Erikson, E., and Maller, J. L. (1995).
Induction of Xenopus oocyte meiotic maturation by MAP kinase. Dev Biol 168, 677-682.
Harbour, J. W., and Dean, D. C. (2000). The Rb/E2F pathway: expanding roles and
emerging paradigms. Genes Dev 14, 2393-2409.
Harlow, E., and Lane, D. (1999). Using antibodies : a laboratory manual (Cold Spring
Harbor, N.Y., Cold Spring Harbor Laboratory Press).
Hochegger, H., Klotzbucher, A., Kirk, J., Howell, M., le Guellec, K., Fletcher, K.,
Duncan, T., Sohail, M., and Hunt, T. (2001). New B-type cyclin synthesis is required between
meiosis I and II during Xenopus oocyte maturation. Development 128, 3795-3807.
Hofmann, F., and Livingston, D. M. (1996). Differential effects of cdk2 and cdk3 on the
control of pRb and E2F function during G1 exit. Genes Dev 10, 851-861.
Hoyt, M. A. (2001). A new view of the spindle checkpoint. J Cell Biol 154, 909-911.
Huang, D., Patrick, G., Moffat, J., Tsai, L. H., and Andrews, B. (1999). Mammalian
Cdk5 is a functional homologue of the budding yeast Pho85 cyclin-dependent protein kinase.
Proc Natl Acad Sci U S A 96, 14445-14450.

Bibliography
117
Hunt, T. (1989). Maturation promoting factor, cyclin and the control of M-phase. Curr
Opin Cell Biol 1, 268-274.
Hunt, T. (1991). Cell biology. Cell cycle gets more cyclins. Nature 350, 462-463.
Ino, H., Ishizuka, T., Chiba, T., and Tatibana, M. (1994). Expression of CDK5
(PSSALRE kinase), a neural cdc2-related protein kinase, in the mature and developing mouse
central and peripheral nervous systems. Brain Res 661, 196-206.
Irniger, S. (2002). Cyclin destruction in mitosis: a crucial task of Cdc20. FEBS Lett 532,
7-11.
Izumi, T., and Maller, J. L. (1993). Elimination of cdc2 phosphorylation sites in the
cdc25 phosphatase blocks initiation of M-phase. Mol Biol Cell 4, 1337-1350.
Jeanmougin, F., Thompson, J. D., Gouy, M., Higgins, D. G., and Gibson, T. J. (1998).
Multiple sequence alignment with Clustal X. Trends Biochem Sci 23, 403-405.
Jeffrey, P. D., Russo, A. A., Polyak, K., Gibbs, E., Hurwitz, J., Massague, J., and
Pavletich, N. P. (1995). Mechanism of CDK activation revealed by the structure of a cyclinA-
CDK2 complex. Nature 376, 313-320.
Jin, P., Hardy, S., and Morgan, D. O. (1998). Nuclear localization of cyclin B1 controls
mitotic entry after DNA damage. J Cell Biol 141, 875-885.
Johnson, D. G., and Walker, C. L. (1999). Cyclins and cell cycle checkpoints. Annu Rev
Pharmacol Toxicol 39, 295-312.
Kanki, J. P., and Donoghue, D. J. (1991). Progression from meiosis I to meiosis II in
Xenopus oocytes requires de novo translation of the mosxe protooncogene. Proc Natl Acad
Sci U S A 88, 5794-5798.
Karaiskou, A., Perez, L. H., Ferby, I., Ozon, R., Jessus, C., and Nebreda, A. R. (2001).
Differential regulation of Cdc2 and Cdk2 by RINGO and cyclins. J Biol Chem 276, 36028-
36034.
Karsenti, E., and Vernos, I. (2001). The mitotic spindle: a self-made machine. Science
294, 543-547.
Kaye, F. J. (2002). RB and cyclin dependent kinase pathways: defining a distinction
between RB and p16 loss in lung cancer. Oncogene 21, 6908-6914.
Kim, K. K., Chamberlin, H. M., Morgan, D. O., and Kim, S. H. (1996). Three-
dimensional structure of human cyclin H, a positive regulator of the CDK-activating kinase.
Nat Struct Biol 3, 849-855.

Bibliography
118
King, R. W., Peters, J. M., Tugendreich, S., Rolfe, M., Hieter, P., and Kirschner, M. W.
(1995). A 20S complex containing CDC27 and CDC16 catalyzes the mitosis-specific
conjugation of ubiquitin to cyclin B. Cell 81, 279-288.
Kobayashi, H., Golsteyn, R., Poon, R., Stewart, E., Gannon, J., Minshull, J., Smith, R.,
and Hunt, T. (1991a). Cyclins and their partners during Xenopus oocyte maturation. Cold
Spring Harb Symp Quant Biol 56, 437-447.
Kobayashi, H., Minshull, J., Ford, C., Golsteyn, R., Poon, R., and Hunt, T. (1991b). On
the synthesis and destruction of A- and B-type cyclins during oogenesis and meiotic
maturation in Xenopus laevis. J Cell Biol 114, 755-765.
Kobel, H. R., and Du Pasquier, L. (1979). Hyperdiploid species hybrids for gene
mapping in Xenopus. Nature 279, 157-158.
Kong, M., Barnes, E. A., Ollendorff, V., and Donoghue, D. J. (2000). Cyclin F regulates
the nuclear localization of cyclin B1 through a cyclin-cyclin interaction. Embo J 19, 1378-
1388.
Kumagai, A., and Dunphy, W. G. (1991). The cdc25 protein controls tyrosine
dephosphorylation of the cdc2 protein in a cell-free system. Cell 64, 903-914.
Kumagai, A., and Dunphy, W. G. (1997). Regulation of Xenopus Cdc25 protein.
Methods Enzymol 283, 564-571.
Kumagai, A., Yakowec, P. S., and Dunphy, W. G. (1998). 14-3-3 proteins act as
negative regulators of the mitotic inducer Cdc25 in Xenopus egg extracts. Mol Biol Cell 9,
345-354.
Laemmli, U. K., and Quittner, S. F. (1974). Maturation of the head of bacteriophage T4.
IV. The proteins of the core of the tubular polyheads and in vitro cleavage of the head
proteins. Virology 62, 483-499.
Lammer, C., Wagerer, S., Saffrich, R., Mertens, D., Ansorge, W., and Hoffmann, I.
(1998). The cdc25B phosphatase is essential for the G2/M phase transition in human cells. J
Cell Sci 111, 2445-2453.
Lee, M. H., Nikolic, M., Baptista, C. A., Lai, E., Tsai, L. H., and Massague, J. (1996).
The brain-specific activator p35 allows Cdk5 to escape inhibition by p27Kip1 in neurons. Proc
Natl Acad Sci U S A 93, 3259-3263.
Lee, M. S., Kwon, Y. T., Li, M., Peng, J., Friedlander, R. M., and Tsai, L. H. (2000).
Neurotoxicity induces cleavage of p35 to p25 by calpain. Nature 405, 360-364.
Lemaitre, J. M., Bocquet, S., and Mechali, M. (2002). Competence to replicate in the
unfertilized egg is conferred by Cdc6 during meiotic maturation. Nature 419, 718-722.

Bibliography
119
Lenormand, J. L., Dellinger, R. W., Knudsen, K. E., Subramani, S., and Donoghue, D. J.
(1999). Speedy: a novel cell cycle regulator of the G2/M transition. Embo J 18, 1869-1877.
Lew, J., Huang, Q. Q., Qi, Z., Winkfein, R. J., Aebersold, R., Hunt, T., and Wang, J. H.
(1994). A brain-specific activator of cyclin-dependent kinase 5. Nature 371, 423-426.
Lucibello, F. C., Sewing, A., Brusselbach, S., Burger, C., and Muller, R. (1993).
Deregulation of cyclins D1 and E and suppression of cdk2 and cdk4 in senescent human
fibroblasts. J Cell Sci 105 ( Pt 1), 123-133.
MacNicol, M. C., Pot, D., and MacNicol, A. M. (1997). pXen, a utility vector for the
expression of GST-fusion proteins in Xenopus laevis oocytes and embryos. Gene 196, 25-29.
Mailand, N., Falck, J., Lukas, C., Syljuasen, R. G., Welcker, M., Bartek, J., and Lukas, J.
(2000). Rapid destruction of human Cdc25A in response to DNA damage. Science 288, 1425-
1429.
Mailand, N., Podtelejnikov, A. V., Groth, A., Mann, M., Bartek, J., and Lukas, J. (2002).
Regulation of G(2)/M events by Cdc25A through phosphorylation-dependent modulation of
its stability. Embo J 21, 5911-5920.
Makela, T. P., Tassan, J. P., Nigg, E. A., Frutiger, S., Hughes, G. J., and Weinberg, R.
A. (1994). A cyclin associated with the CDK-activating kinase MO15. Nature 371, 254-257.
Maller, J., Gautier, J., Langan, T. A., Lohka, M. J., Shenoy, S., Shalloway, D., and
Nurse, P. (1989). Maturation-promoting factor and the regulation of the cell cycle. J Cell Sci
Suppl 12, 53-63.
Maller, J. L. (1990). MPF and cell cycle control. Adv Second Messenger
Phosphoprotein Res 24, 323-328.
Maller, J. L., Butcher, F. R., and Krebs, E. G. (1979). Early effect of progesterone on
levels of cyclic adenosine 3':5'-monophosphate in Xenopus oocytes. J Biol Chem 254, 579-
582.
Maller, J. L., and Krebs, E. G. (1977). Progesterone-stimulated meiotic cell division in
Xenopus oocytes. Induction by regulatory subunit and inhibition by catalytic subunit of
adenosine 3':5'-monophosphate-dependent protein kinase. J Biol Chem 252, 1712-1718.
Maller, J. L., Schwab, M. S., Gross, S. D., Taieb, F. E., Roberts, B. T., and Tunquist, B.
J. (2002). The mechanism of CSF arrest in vertebrate oocytes. Mol Cell Endocrinol 187, 173-
178.
Masui, Y. (2001). From oocyte maturation to the in vitro cell cycle: the history of
discoveries of Maturation-Promoting Factor (MPF) and Cytostatic Factor (CSF).
Differentiation 69, 1-17.

Bibliography
120
Masui, Y., and Markert, C. L. (1971). Cytoplasmic control of nuclear behavior during
meiotic maturation of frog oocytes. J Exp Zool 177, 129-145.
Matsuoka, M., Kato, J. Y., Fisher, R. P., Morgan, D. O., and Sherr, C. J. (1994).
Activation of cyclin-dependent kinase 4 (cdk4) by mouse MO15-associated kinase. Mol Cell
Biol 14, 7265-7275.
McManus, M. T., and Sharp, P. A. (2002). Gene silencing in mammals by small
interfering RNAs. Nat Rev Genet 3, 737-747.
Mendez, R., Barnard, D., and Richter, J. D. (2002). Differential mRNA translation and
meiotic progression require Cdc2-mediated CPEB destruction. Embo J 21, 1833-1844.
Meyerson, M., Enders, G. H., Wu, C. L., Su, L. K., Gorka, C., Nelson, C., Harlow, E.,
and Tsai, L. H. (1992). A family of human cdc2-related protein kinases. Embo J 11, 2909-
2917.
Minshull, J., Golsteyn, R., Hill, C. S., and Hunt, T. (1990). The A- and B-type cyclin
associated cdc2 kinases in Xenopus turn on and off at different times in the cell cycle. Embo J
9, 2865-2875.
Morgan, D. O. (1995). Principles of CDK regulation. Nature 374, 131-134.
Morgan, D. O. (1997). Cyclin-dependent kinases: engines, clocks, and microprocessors.
Annu Rev Cell Dev Biol 13, 261-291.
Murray, A and Hunt, T. (1993). The cell cycle, an introduction. Oxford university Press.
Moskowitz, N. K., Borao, F. J., Dardashti, O., Cohen, H. D., and Germino, F. J. (1996). The
amino terminus of Cdk2 binds p21. Oncol Res 8, 343-352.
Musa, F. R., Tokuda, M., Kuwata, Y., Ogawa, T., Tomizawa, K., Konishi, R., Takenaka,
I., and Hatase, O. (1998). Expression of cyclin-dependent kinase 5 and associated cyclins in
Leydig and Sertoli cells of the testis. J Androl 19, 657-666.
Musacchio, A., and Hardwick, K. G. (2002). The spindle checkpoint: structural insights
into dynamic signalling. Nat Rev Mol Cell Biol 3, 731-741.
Nakajo, N., Yoshitome, S., Iwashita, J., Iida, M., Uto, K., Ueno, S., Okamoto, K., and
Sagata, N. (2000). Absence of Wee1 ensures the meiotic cell cycle in Xenopus oocytes. Genes
Dev 14, 328-338.
Nasmyth, K. (2002). Segregating sister genomes: the molecular biology of chromosome
separation. Science 297, 559-565.
Nebreda, A. R., and Ferby, I. (2000). Regulation of the meiotic cell cycle in oocytes.
Curr Opin Cell Biol 12, 666-675.

Bibliography
121
Nebreda, A. R., Gannon, J. V., and Hunt, T. (1995). Newly synthesized protein(s) must
associate with p34cdc2 to activate MAP kinase and MPF during progesterone-induced
maturation of Xenopus oocytes. Embo J 14, 5597-5607.
Nevins, J. R., Leone, G., DeGregori, J., and Jakoi, L. (1997). Role of the Rb/E2F
pathway in cell growth control. J Cell Physiol 173, 233-236.
Nishizawa, M., Furuno, N., Okazaki, K., Tanaka, H., Ogawa, Y., and Sagata, N. (1993).
Degradation of Mos by the N-terminal proline (Pro2)-dependent ubiquitin pathway on
fertilization of Xenopus eggs: possible significance of natural selection for Pro2 in Mos. Embo
J 12, 4021-4027.
Nishizawa, M., Okazaki, K., Furuno, N., Watanabe, N., and Sagata, N. (1992). The
'second-codon rule' and autophosphorylation govern the stability and activity of Mos during
the meiotic cell cycle in Xenopus oocytes. Embo J 11, 2433-2446.
Nurse, P. (1997). Checkpoint pathways come of age. Cell 91, 865-867.
O'Connell, M. J., Walworth, N. C., and Carr, A. M. (2000). The G2-phase DNA-damage
checkpoint. Trends Cell Biol 10, 296-303.
Ohsumi, K., Sawada, W., and Kishimoto, T. (1994). Meiosis-specific cell cycle
regulation in maturing Xenopus oocytes. J Cell Sci 107 ( Pt 11), 3005-3013.
Okamoto K, Li H, Jensen MR, Zhang T, Taya Y, Thorgeirsson SS, Prives C. (2002)
Cyclin G recruits PP2A to dephosphorylate Mdm2..Mol Cell. 9(4):761-71.
Palmer, A., Gavin, A. C., and Nebreda, A. R. (1998). A link between MAP kinase and
p34(cdc2)/cyclin B during oocyte maturation: p90(rsk) phosphorylates and inactivates the
p34(cdc2) inhibitory kinase Myt1. Embo J 17, 5037-5047.
Peters, J. M. (2002). The anaphase-promoting complex: proteolysis in mitosis and
beyond. Mol Cell 9, 931-943.
Porter, L. A., Dellinger, R. W., Tynan, J. A., Barnes, E. A., Kong, M., Lenormand, J. L.,
and Donoghue, D. J. (2002). Human Speedy: a novel cell cycle regulator that enhances
proliferation through activation of Cdk2. J Cell Biol 157, 357-366.
Richter, J. D. (2001). Think globally, translate locally: what mitotic spindles and
neuronal synapses have in common. Proc Natl Acad Sci U S A 98, 7069-7071.
Rosenblatt, J., Gu, Y., and Morgan, D. O. (1992). Human cyclin-dependent kinase 2 is
activated during the S and G2 phases of the cell cycle and associates with cyclin A. Proc Natl
Acad Sci U S A 89, 2824-2828.
Rost, B. (1996). PHD: predicting one-dimensional protein structure by profile-based
neural networks. Methods Enzymol 266, 525-539.

Bibliography
122
Schmitt, A., and Nebreda, A. R. (2002a). Inhibition of Xenopus oocyte meiotic
maturation by catalytically inactive protein kinase A. Proc Natl Acad Sci U S A 99, 4361-
4366.
Schmitt, A., and Nebreda, A. R. (2002b). Signalling pathways in oocyte meiotic
maturation. J Cell Sci 115, 2457-2459.
Schorderet-Slatkine, S., Schorderet, M., Boquet, P., Godeau, F., and Baulieu, E. E.
(1978). Progesterone-induced meiosis in Xenopus laevis oocytes: a role for cAMP at the
"maturation-promoting factor" level. Cell 15, 1269-1275.
Schwab, M. S., Roberts, B. T., Gross, S. D., Tunquist, B. J., Taieb, F. E., Lewellyn, A.
L., and Maller, J. L. (2001). Bub1 is activated by the protein kinase p90(Rsk) during Xenopus
oocyte maturation. Curr Biol 11, 141-150.
Session, D. R., Fautsch, M. P., Avula, R., Jones, W. R., Nehra, A., and Wieben, E. D.
(2001). Cyclin-dependent kinase 5 is expressed in both Sertoli cells and metaphase
spermatocytes. Fertil Steril 75, 669-673.
Shibuya, E. K., and Masui, Y. (1989a). Fractionation of cytostatic factors from cytosols
of amphibian eggs. Dev Biol 135, 212-219.
Shibuya, E. K., and Masui, Y. (1989b). Molecular characteristics of cytostatic factors in
amphibian egg cytosols. Development 106, 799-808.
Silver, E. T., Gwozd, T. J., Ptak, C., Goebl, M., and Ellison, M. J. (1992). A chimeric
ubiquitin conjugating enzyme that combines the cell cycle properties of CDC34 (UBC3) and
the DNA repair properties of RAD6 (UBC2): implications for the structure, function and
evolution of the E2s. Embo J 11, 3091-3098.
Simanis, V., and Nurse, P. (1986). The cell cycle control gene cdc2+ of fission yeast
encodes a protein kinase potentially regulated by phosphorylation. Cell 45, 261-268.
Smith, D. S., and Tsai, L. H. (2002). Cdk5 behind the wheel: a role in trafficking and
transport? Trends Cell Biol 12, 28-36.
Smits, V. A., and Medema, R. H. (2001). Checking out the G(2)/M transition. Biochim
Biophys Acta 1519, 1-12.
Stebbins-Boaz, B., Hake, L. E., and Richter, J. D. (1996). CPEB controls the
cytoplasmic polyadenylation of cyclin, Cdk2 and c-mos mRNAs and is necessary for oocyte
maturation in Xenopus. Embo J 15, 2582-2592.
Strausfeld, U. P., Howell, M., Descombes, P., Chevalier, S., Rempel, R. E.,
Adamczewski, J., Maller, J. L., Hunt, T., and Blow, J. J. (1996). Both cyclin A and cyclin E

Bibliography
123
have S-phase promoting (SPF) activity in Xenopus egg extracts. J Cell Sci 109 ( Pt 6), 1555-
1563.
Sweeney, C., Murphy, M., Kubelka, M., Ravnik, S. E., Hawkins, C. F., Wolgemuth, D.
J., and Carrington, M. (1996). A distinct cyclin A is expressed in germ cells in the mouse.
Development 122, 53-64.
Takizawa, C. G., and Morgan, D. O. (2000). Control of mitosis by changes in the
subcellular location of cyclin-B1-Cdk1 and Cdc25C. Curr Opin Cell Biol 12, 658-665.
Tarricone, C., Dhavan, R., Peng, J., Areces, L. B., Tsai, L. H., and Musacchio, A.
(2001). Structure and regulation of the CDK5-p25(nck5a) complex. Mol Cell 8, 657-669.
Tassan, J. P., Jaquenoud, M., Fry, A. M., Frutiger, S., Hughes, G. J., and Nigg, E. A.
(1995). In vitro assembly of a functional human CDK7-cyclin H complex requires MAT1, a
novel 36 kDa RING finger protein. Embo J 14, 5608-5617.
Tsai, L. H., Delalle, I., Caviness, V. S., Jr., Chae, T., and Harlow, E. (1994). p35 is a
neural-specific regulatory subunit of cyclin-dependent kinase 5. Nature 371, 419-423.
Tsai, L. H., Takahashi, T., Caviness, V. S., Jr., and Harlow, E. (1993). Activity and
expression pattern of cyclin-dependent kinase 5 in the embryonic mouse nervous system.
Development 119, 1029-1040.
Whitmire, E., Khan, B., and Coue, M. (2002). Cdc6 synthesis regulates replication
competence in Xenopus oocytes. Nature 419, 722-725.
Winters, Z. E. (2002). P53 pathways involving G2 checkpoint regulators and the role of
their subcellular localisation. J R Coll Surg Edinb 47, 591-598.
Wu, L., Wells, D., Tay, J., Mendis, D., Abbott, M. A., Barnitt, A., Quinlan, E., Heynen,
A., Fallon, J. R., and Richter, J. D. (1998). CPEB-mediated cytoplasmic polyadenylation and
the regulation of experience-dependent translation of alpha-CaMKII mRNA at synapses.
Neuron 21, 1129-1139.
Yajima, J., Edamatsu, M., Watai-Nishii, J., Tokai-Nishizumi, N., Yamamoto, T., and
Toyoshima, Y. Y. (2003). The human chromokinesin Kid is a plus end-directed microtubule-
based motor. Embo J 22, 1067-1074.
Yang, V. W. (2002). APC as a checkpoint gene: the beginning or the end?
Gastroenterology 123, 935-939.
Yasumoto, H., Kim, S., Zhan, Y., Miyazaki, H., Hoshiga, M., Kaneda, Y., Morishita, R.,
and Iwao, H. (2001). Dominant negative c-jun gene transfer inhibits vascular smooth muscle
cell proliferation and neointimal hyperplasia in rats. Gene Ther 8, 1682-1689.

Bibliography
124
Zarzov, P., Decottignies, A., Baldacci, G., and Nurse, P. (2002). G(1)/S CDK is
inhibited to restrain mitotic onset when DNA replication is blocked in fission yeast. Embo J
21, 3370-3376.
Zerfass-Thome, K., Schulze, A., Zwerschke, W., Vogt, B., Helin, K., Bartek, J.,
Henglein, B., and Jansen-Durr, P. (1997). p27KIP1 blocks cyclin E-dependent transactivation
of cyclin A gene expression. Mol Cell Biol 17, 407-415.
Zhu, Y., Bond, J., and Thomas, P. (2003a). Identification, classification, and partial
characterization of genes in humans and other vertebrates homologous to a fish membrane
progestin receptor. Proc Natl Acad Sci U S A.
Zhu, Y., Rice, C. D., Pang, Y., Pace, M., and Thomas, P. (2003b). Cloning, expression,
and characterization of a membrane progestin receptor and evidence it is an intermediary in
meiotic maturation of fish oocytes. Proc Natl Acad Sci U S A.

Appendix
125
7 Appendix. 7.1 Nucleotide and deduced amino acid sequence of Human-Ringo1. 1/1 31/11 atg cag aag cat tac aca gtg gcc tgg ttt ctt tac tca gcc cct ggg gta gat ccc agc M Q K H Y T V A W F L Y S A P G V D P S 61/21 91/31 ccc cca tgt agg tcc ctt ggc tgg aaa agg aag agg gag tgg tca gat gaa tct gag gag P P C R S L G W K R K R E W S D E S E E 121/41 151/51 gag ccg gag aag gag ctc gcc cct gag cct gag gag acc tgg gta gtg gag acg ctg tgt E P E K E L A P E P E E T W V V E T L C 181/61 211/71 ggg ctc aag atg aag ctg aag caa cag cga gtg tca ccc atc ctc ctt gag cac cac aag G L K M K L K Q Q R V S P I L L E H H K 241/81 271/91 gac ttc aac agt cag ctt gcc cct ggg gta gat ccc agc ccc ccg cat agg tcc ttt tgc D F N S Q L A P G V D P S P P H R S F C 301/101 331/111 tgg aaa agg aag atg gag tgg tgg gac aaa tct gag gag tcg gag gag gag cca cgg aag W K R K M E W W D K S E E S E E E P R K 361/121 391/131 gtg ctc gcc cct gag cct gag gag atc tgg gtg gcg gag atg ctg tgt ggc ctc aag atg V L A P E P E E I W V A E M L C G L K M 421/141 451/151 aag ctg aag cga cgg cga gtg tcg ctc gtg ctc cct gag cac cac gag gcc ttc aac agg K L K R R R V S L V L P E H H E A F N R 481/161 511/171 ctg ctt gag gat cct gtc att aaa aga ttc ctg gcc tgg gac aaa gat ctg agg gtg tcg L L E D P V I K R F L A W D K D L R V S 541/181 571/191 gac aag tat ctc ctt gct atg gtc ata gcg tat ttc agc cga gcc ggc ttc ccc tcc tgg D K Y L L A M V I A Y F S R A G F P S W 601/201 631/211 caa tac caa cgc ctt cat ttc ttc ctg gct ctc tac ctg gcc aat gac atg gag gag gac Q Y Q R L H F F L A L Y L A N D M E E D 661/221 691/231 gac gag gac tcc aaa caa aac atc ttc cac ttc ctg tat ggg aag aac cgc tct cgc ata D E D S K Q N I F H F L Y G K N R S R I 721/241 751/251 ccc ttg ctc cgt aag cgt cgg ttc cag tta tac cgt tcc atg aac ccg agg gcc agg aag P L L R K R R F Q L Y R S M N P R A R K 781/261 811/271 aac cgc tct cac ata ccc ttg gtc cgt aag cgt cgg ttc cag tta cgc cgt tgc atg aac N R S H I P L V R K R R F Q L R R C M N 841/281 871/291 ccg agg gcc agg aag aac cgc tct cag ata gtc ctg ttc cag aaa cgt cgg ttc cac ttc P R A R K N R S Q I V L F Q K R R F H F 901/301 931/311 ttc tgt tcc atg agc tgc agg gct tgg gtt tcc cca gag gag ttg gag gag atc cag gct F C S M S C R A W V S P E E L E E I Q A 961/321 991/331 tat gac cca gag cac tgg gtg tgg gcg cga gat cgc gct cgc ctt tcc tag Y D P E H W V W A R D R A R L S * 1/1 31/11
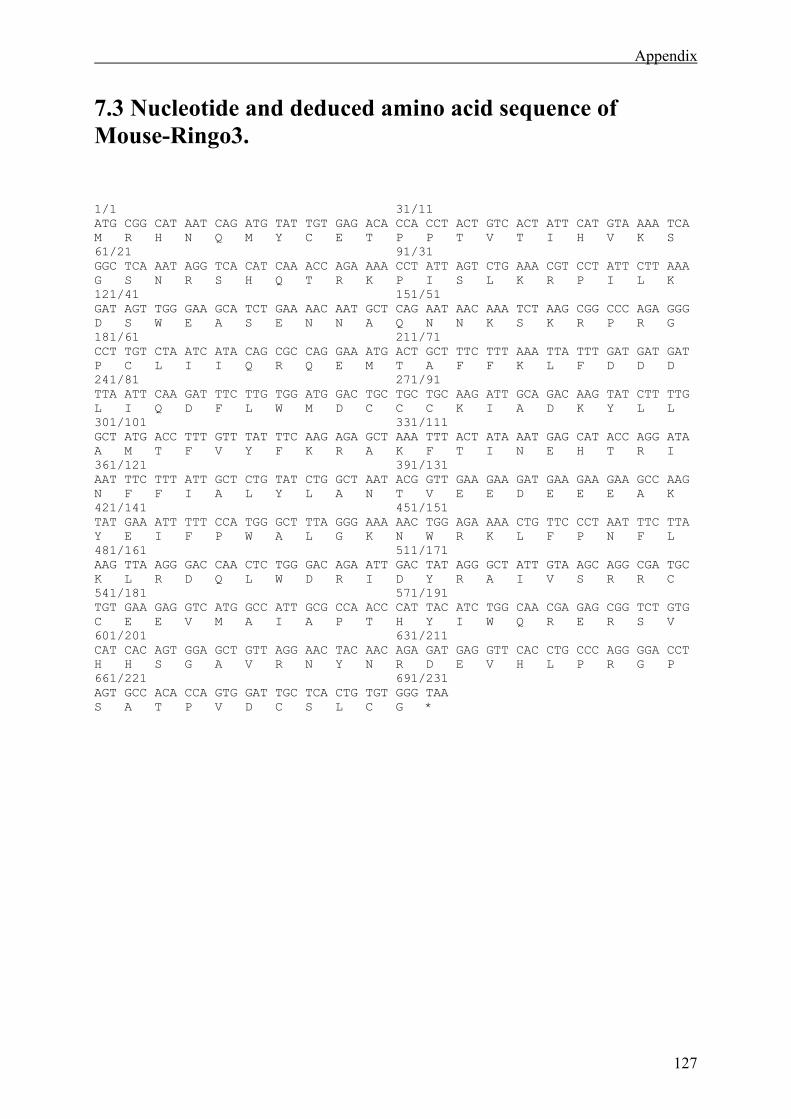
Appendix
127
7.3 Nucleotide and deduced amino acid sequence of Mouse-Ringo3. 1/1 31/11 ATG CGG CAT AAT CAG ATG TAT TGT GAG ACA CCA CCT ACT GTC ACT ATT CAT GTA AAA TCA M R H N Q M Y C E T P P T V T I H V K S 61/21 91/31 GGC TCA AAT AGG TCA CAT CAA ACC AGA AAA CCT ATT AGT CTG AAA CGT CCT ATT CTT AAA G S N R S H Q T R K P I S L K R P I L K 121/41 151/51 GAT AGT TGG GAA GCA TCT GAA AAC AAT GCT CAG AAT AAC AAA TCT AAG CGG CCC AGA GGG D S W E A S E N N A Q N N K S K R P R G 181/61 211/71 CCT TGT CTA ATC ATA CAG CGC CAG GAA ATG ACT GCT TTC TTT AAA TTA TTT GAT GAT GAT P C L I I Q R Q E M T A F F K L F D D D 241/81 271/91 TTA ATT CAA GAT TTC TTG TGG ATG GAC TGC TGC TGC AAG ATT GCA GAC AAG TAT CTT TTG L I Q D F L W M D C C C K I A D K Y L L 301/101 331/111 GCT ATG ACC TTT GTT TAT TTC AAG AGA GCT AAA TTT ACT ATA AAT GAG CAT ACC AGG ATA A M T F V Y F K R A K F T I N E H T R I 361/121 391/131 AAT TTC TTT ATT GCT CTG TAT CTG GCT AAT ACG GTT GAA GAA GAT GAA GAA GAA GCC AAG N F F I A L Y L A N T V E E D E E E A K 421/141 451/151 TAT GAA ATT TTT CCA TGG GCT TTA GGG AAA AAC TGG AGA AAA CTG TTC CCT AAT TTC TTA Y E I F P W A L G K N W R K L F P N F L 481/161 511/171 AAG TTA AGG GAC CAA CTC TGG GAC AGA ATT GAC TAT AGG GCT ATT GTA AGC AGG CGA TGC K L R D Q L W D R I D Y R A I V S R R C 541/181 571/191 TGT GAA GAG GTC ATG GCC ATT GCG CCA ACC CAT TAC ATC TGG CAA CGA GAG CGG TCT GTG C E E V M A I A P T H Y I W Q R E R S V 601/201 631/211 CAT CAC AGT GGA GCT GTT AGG AAC TAC AAC AGA GAT GAG GTT CAC CTG CCC AGG GGA CCT H H S G A V R N Y N R D E V H L P R G P 661/221 691/231 AGT GCC ACA CCA GTG GAT TGC TCA CTG TGT GGG TAA S A T P V D C S L C G *
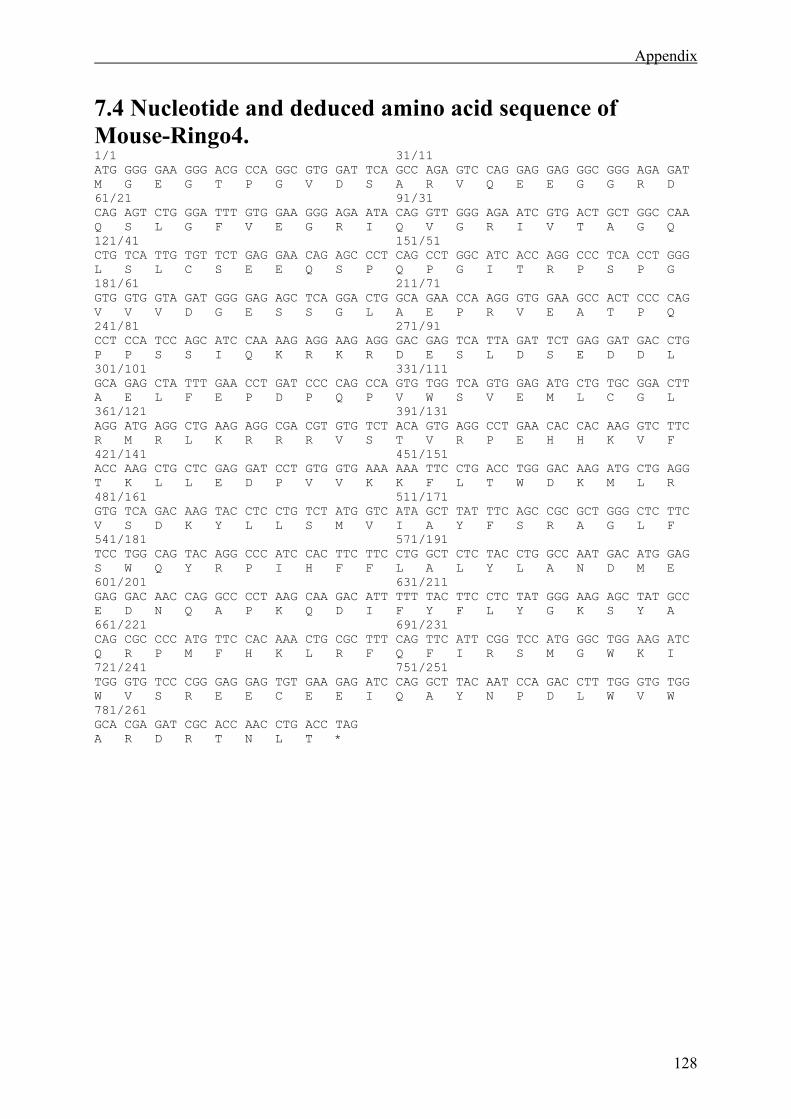
Appendix
128
7.4 Nucleotide and deduced amino acid sequence of Mouse-Ringo4. 1/1 31/11 ATG GGG GAA GGG ACG CCA GGC GTG GAT TCA GCC AGA GTC CAG GAG GAG GGC GGG AGA GAT M G E G T P G V D S A R V Q E E G G R D 61/21 91/31 CAG AGT CTG GGA TTT GTG GAA GGG AGA ATA CAG GTT GGG AGA ATC GTG ACT GCT GGC CAA Q S L G F V E G R I Q V G R I V T A G Q 121/41 151/51 CTG TCA TTG TGT TCT GAG GAA CAG AGC CCT CAG CCT GGC ATC ACC AGG CCC TCA CCT GGG L S L C S E E Q S P Q P G I T R P S P G 181/61 211/71 GTG GTG GTA GAT GGG GAG AGC TCA GGA CTG GCA GAA CCA AGG GTG GAA GCC ACT CCC CAG V V V D G E S S G L A E P R V E A T P Q 241/81 271/91 CCT CCA TCC AGC ATC CAA AAG AGG AAG AGG GAC GAG TCA TTA GAT TCT GAG GAT GAC CTG P P S S I Q K R K R D E S L D S E D D L 301/101 331/111 GCA GAG CTA TTT GAA CCT GAT CCC CAG CCA GTG TGG TCA GTG GAG ATG CTG TGC GGA CTT A E L F E P D P Q P V W S V E M L C G L 361/121 391/131 AGG ATG AGG CTG AAG AGG CGA CGT GTG TCT ACA GTG AGG CCT GAA CAC CAC AAG GTC TTC R M R L K R R R V S T V R P E H H K V F 421/141 451/151 ACC AAG CTG CTC GAG GAT CCT GTG GTG AAA AAA TTC CTG ACC TGG GAC AAG ATG CTG AGG T K L L E D P V V K K F L T W D K M L R 481/161 511/171 GTG TCA GAC AAG TAC CTC CTG TCT ATG GTC ATA GCT TAT TTC AGC CGC GCT GGG CTC TTC V S D K Y L L S M V I A Y F S R A G L F 541/181 571/191 TCC TGG CAG TAC AGG CCC ATC CAC TTC TTC CTG GCT CTC TAC CTG GCC AAT GAC ATG GAG S W Q Y R P I H F F L A L Y L A N D M E 601/201 631/211 GAG GAC AAC CAG GCC CCT AAG CAA GAC ATT TTT TAC TTC CTC TAT GGG AAG AGC TAT GCC E D N Q A P K Q D I F Y F L Y G K S Y A 661/221 691/231 CAG CGC CCC ATG TTC CAC AAA CTG CGC TTT CAG TTC ATT CGG TCC ATG GGC TGG AAG ATC Q R P M F H K L R F Q F I R S M G W K I 721/241 751/251 TGG GTG TCC CGG GAG GAG TGT GAA GAG ATC CAG GCT TAC AAT CCA GAC CTT TGG GTG TGG W V S R E E C E E I Q A Y N P D L W V W 781/261 GCA CGA GAT CGC ACC AAC CTG ACC TAG A R D R T N L T *

7.5 Sequence alignment of Ringo3 alternative splice variants
129Ringo 3a
Ringo 3bRingo 3cRingo 3d
Ringo 3aRingo 3bRingo 3cRingo 3d
Ringo 3aRingo 3bRingo 3c
Ringo 3d
129
7.6 Sequence alignment of Ringo4 alternative splice variants
Ringo 4aRingo 4b
Ringo 4aRingo 4b
130


















![Dissertation thesis [draft title] - MADOC · TellMeWhoYourFriendsAre? DisentanglingtheInterplayofYoung Immigrants’HostCountryIdentification andTheirFriendshipswithNatives Inaugural](https://img.pdfslide.net/doc/110x75/5b8369317f8b9a934f8d31bc/dissertation-thesis-draft-title-madoc-tellmewhoyourfriendsare-disentanglingtheinterplayofyoung.jpg)










