Embed Size (px)
Citation preview

Incontinentia pigmenti: Transmission from father todaughterMichelle M. Emery, MD,a Elaine C. Siegfried, MD,a Mary Seabury Stone, MD,a,bEdwin M. Stone, MD, Phl),? and Shivanand R. Patil, PhOd Iowa City, Iowa
Incontinentia pigmenti (IP) is a well-described genodermatosis that occurs almost exclusivelyin females. IP is characterized by a distinctive skin eruption and a variable presence of multisystem abnormalities. Pedigree analysis is most consistent with an X-linked dominant traitthat is lethal in males. However, 27 reports of IP in males have been published,excluding fourpatients who had Klinefelter's syndrome. It has usually been assumed that these rare affectedmales survive because of genetic mosaicism. Mosaic inheritance of IP is also strongly supported by the characteristic distribution of skin findings along Blaschko's lines. Only one caseof father-to-daughter transmission has been previously reported. We report a second case offather-to-daughter transmission of IP . Chromosomal analysis of blood and fibroblasts fromthe father failed to provide evidence of genetic mosaicism. (J AM ACAD DERMATOL 1993;29 :368-72.)
Incontinentia pigmenti (IP) (Bloch-Sulzbergersyndrome)is characterized by threedistinctivestagesof cutaneous lesions: an initial vesiculobullous stage,a verrucous stage, and a stage of swirled hyperpigmentation. The cutaneous lesions inall stages followBlaschke's lines.\,2 Although the cutaneous findingsare self-limited, IP can be associated with persistentanomalies of the central nervous system, eyes, teeth,nails, and hair. I
IP occurs almost exclusively in females. Geneologic analyses have described a female/ male ratio of37:1. This high ratio may be related in part to a highincidence of spontaneous abortion ofmale fetuses atrisk. I In fact, pedigree analysis of families with IPsupports X-linked dominant transmission with lethality in 46,xY males.': 4 Nonetheless, there arerare reports of surviving males, as summarized byPrendiville et al.5 However, to the best of ourknowledge, there is only one report of father-todaughter transmission of lP.6 We describe an additional case of an affected male who transmitted IPto his daughter. This is also the first case in a male
From the Departments of Dermatology," Pathology,"Ophthalmology,"and Pediatrics," University of Iowa College of Medicine and University of Iowa Hospitals and Clinics.
Reprint requests: Mary S. Stone, MD, Department of Dermatology,200 Hawkins Dr., BT 2052, Iowa City, IA 52245-1090.
Copyright (?J 1993 by the American Academy of Dermatology, Inc .0190-9622/93 $1.00+ .10 16/4/44445
368
in which both lymphocyte and fibroblast chromosome analyses have been performed.
CASE REPORT
A 4-month-old white girl had an eruption on her trunkand extremities. The infant was born at term after an uncomplicatedpregnancy. Theinfant's mother was a healthy22-year-old woman (gravida 1, para I, abortus 0). Themother denied a personal or family history of physicalfeatures of IP.
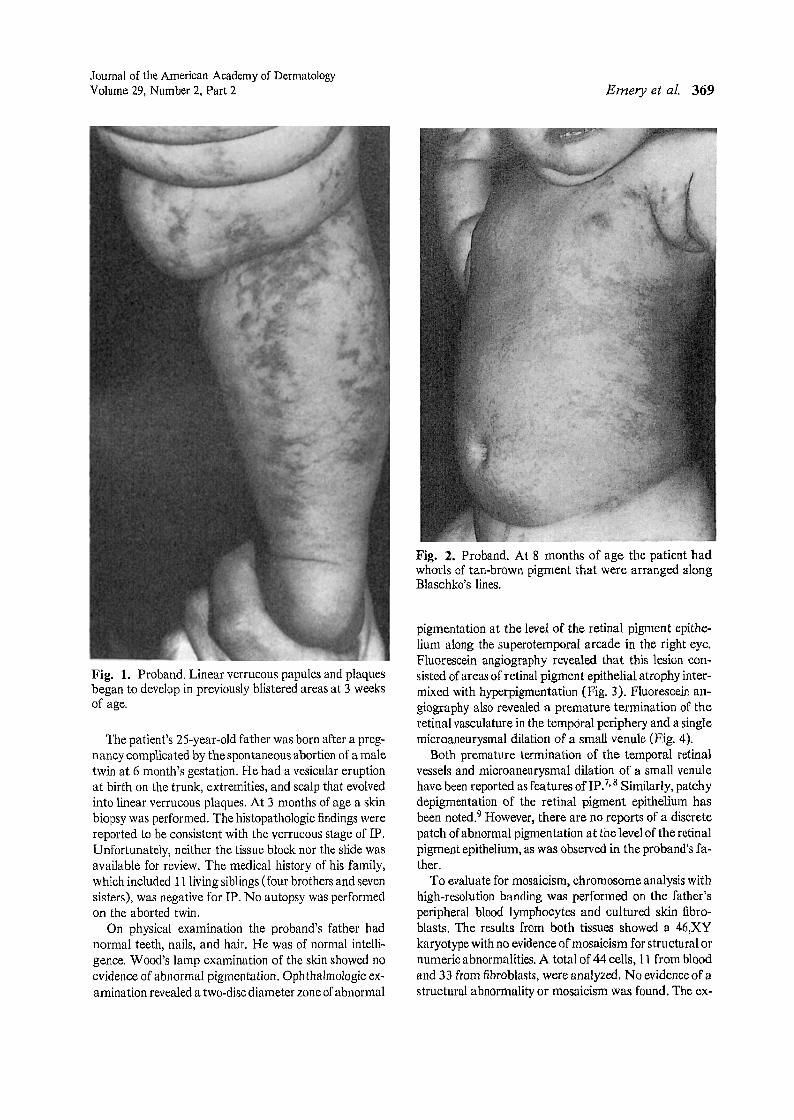
At birth the infant had multiple vesiculobullous lesionson her trunk, extremities. and scalp. Linear verrucousplaques began to develop in previously blistered areas atapproximately 3 weeks of age.
On physical examination the infant was alert and appeared healthy. Her weight was at the 90th percentile,and the head circumference was at the 75th percentile.Muscle tone and motor function were grossly normal. Shehad multiple linear vesicuiobullous lesions on the skin ofthe extremities.She also had linear verrucous papules andplaques located mainly on the extremities (Fig. 1). On thetrunk and extremities she had swirls of tan-brown pigment that were arranged along Blaschke's lines. The hairand nails appeared normal. Findings of an ophthalmologic examination were unremarkable.
By 8 months of age , the infant's mother reported onlyoccasional new vesicles that appeared to be associatedwith "colds." Many of the previous verrucous lesions hadundergone involution, but there were more prominentswirls of hyperpigmentation that followed Blaschke'slines (Fig. 2). Again, muscle tone and motor developmentwere normal.
Journal of the American Academy of DermatologyVolume 29, Number 2, Part 2
Fig. 1. Proband. Linear verrucous papules and plaquesbegan to develop in previously blistered areas at 3 weeksof age.
The patient's 25-year-old father was born after a pregnancy complicated by the spontaneous abortion of a maletwin at 6 month's gestation. He had a vesicular eruptionat birth on the trunk, extremities, and scalp that evolvedinto linear verrucous plaques. At 3 months of age a skinbiopsy was performed. The histopathologic findings werereported to be consistent with the verrucous stage of IP .Unfortunately, neither the tissue block nor the slide wasavailable for review. The medical history of his family,which included 11 living siblings (four brothers and sevensisters) , was negative for IP . No autopsy was performedon the aborted twin.
On physical examination the proband's father hadnormal teeth, nails, and hair. He was of normal intelligence. Wood's lamp examination of the skin showed noevidence of abnormal pigmentation. Oph thalmologic examination revealed a two-discdiameter zone ofabnormal
Emery et at. 369
Fig. 2. Proband. At 8 months of age the patient hadwhorls of tan-brown pigment that were arranged alongBlaschko's lines.
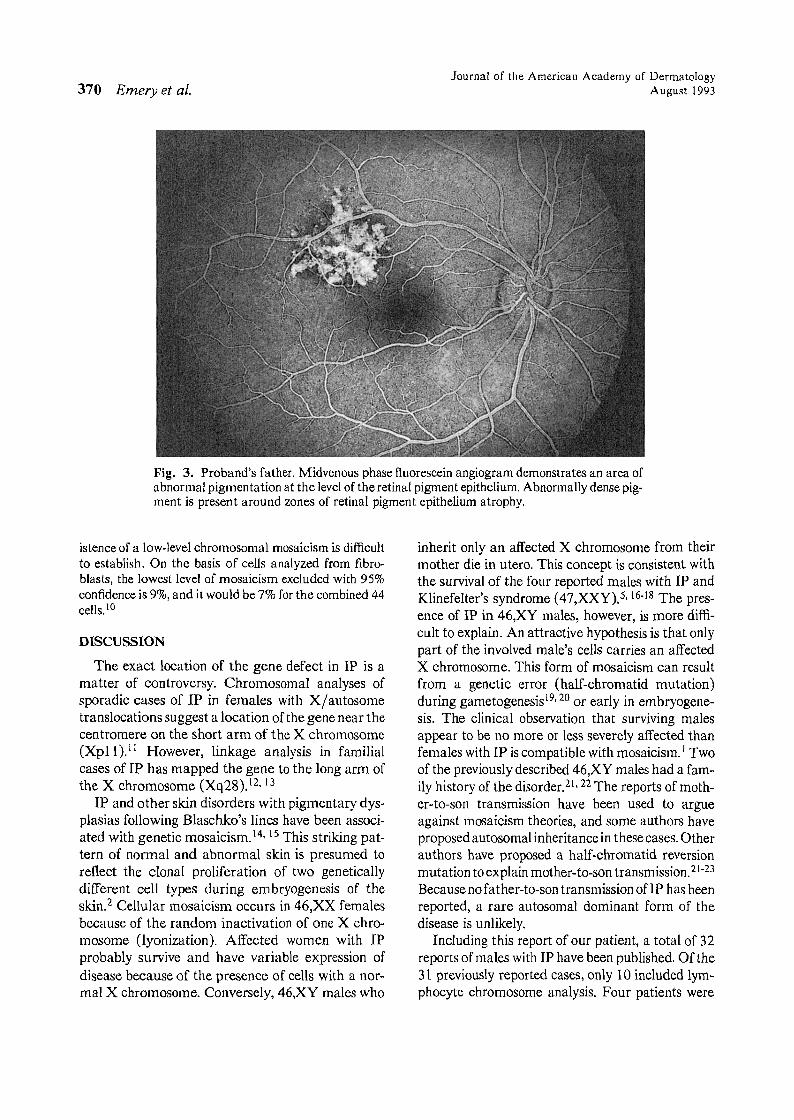
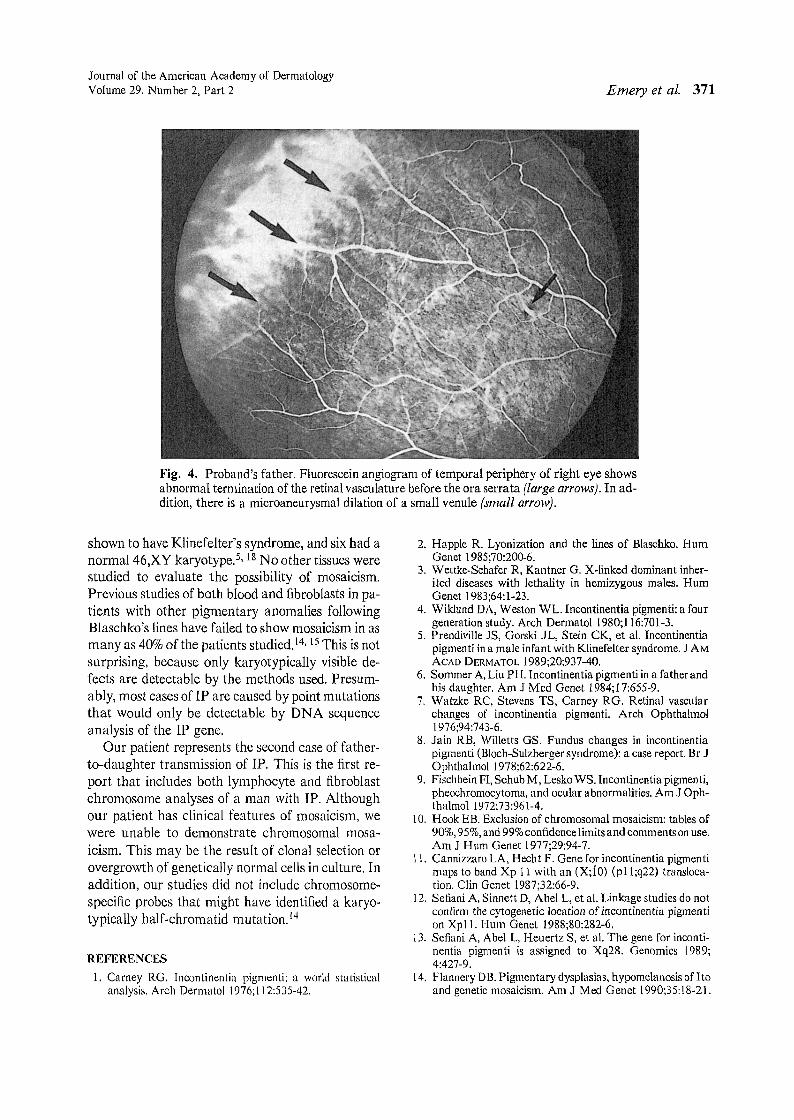
pigmentation at the level of the retinal pigment epithelium along the superotemporal arcade in the right eye.Fluorescein angiography revealed that this lesion consisted of areas of retinal pigment epithelial atrophy intermixed with hyperpigmentation (Fig. 3). Fluorescein angiography also revealed a premature termination of theretinal vasculature in the temporal periphery and a singlemicroaneurysmal dilation of a small venule (Fig. 4).
Both premature termination of the temporal retinalvessels and microaneurysmal dilation of a small venulehave been reported as features oflP'?· 8 Similarly, patchydepigmentation of the retinal pigment epithelium hasbeen noted." However, there are no reports of a discretepatch of abnormal pigmentation at the level of the retinalpigment epithelium, as was observed in the proband's father.
To evaluate for mosaicism, chromosome analysis withhigh-resolution banding was performed on the father'speripheral blood lymphocytes and cultured skin fibroblasts. The results from both tissues showed a 46,XYkaryotype with no evidence of mosaicism for structural ornumeric abnormalities. A total of 44 cells, 11 from bloodand 33 from fibroblasts, were analyzed. No evidence of astructural abnormality or mosaicism was found. The ex-
370 Emery et al.Journal of the American Academy uf Dermatology
August 1993
Fig. 3. Proband's father. Midvenous phase fluorescein angiogram demonstrates an area ofabnormal pigmentation at the level of the retinal pigment epithelium. Abnormally dense pigment is present around zones of retinal pigment epithelium atrophy.
istence of a low-level chromosomal mosaicism is difficultto establish. On the basis of cells analyzed from fibroblasts, the lowest level of mosaicism excluded with 95%confidence is 9%, and it would be 7% for the combined 44cells.!?
DISCUSSION
The exact location of the gene defect in IP is amatter of controversy. Chromosomal analyses ofsporadic cases of IP in females with X/autosometranslocations suggest a location of the gene near thecentromere on the short arm of the X chromosome(Xpll).ll However, linkage analysis in familialcases of IP has mapped the gene to the long arm ofthe X chromosome (Xq28).l2, 13
IP and other skin disorders with pigmentary dysplasias following Blaschko's lines have been associated with genetic mosaicism.lv 15 This striking pattern of normal and abnormal skin is presumed toreflect the clonal proliferation of two geneticallydifferent cell types during embryogenesis of theskin.? Cellular mosaicism occurs in 46,XX femalesbecause of the random inactivation of one X chromosome (lyonization). Affected women with IPprobably survive and have variable expression ofdisease because of the presence of cells with a normal X chromosome. Conversely, 46,XY males who
inherit only an affected X chromosome from theirmother die in utero. This concept is consistent withthe survival of the four reported males with IP andKlinefelter's syndrome (47,XXY).5, l6-18 The presence of IP in 46,XY males, however, is more difficult to explain. An attractive hypothesis is that onlypart of the involved male's cells carries an affectedX chromosome. This form of mosaicism can resultfrom a genetic error (half-chromatid mutation)during gametogenesis'S 20 or early in embryogenesis. The clinical observation that surviving malesappear to be no more or less severely affected thanfemales with IP iscompatible with mosaicism. I Twoof the previously described 46,XY males had a family history of the disorder.": 22 The reports of mother-to-son transmission have been used to argueagainst mosaicism theories, and some authors haveproposed autosomal inheritance in these cases. Otherauthors have proposed a half-chromatid reversionmutation to explain mother-to-son transmission.U<'Because no father-to-son transmission of IP has beenreported, a rare autosomal dominant form of thedisease is unlikely.
Including this report of our patient, a total of 32reports of males with IP have been published. Of the31 previously reported cases, only 10 included lymphocyte chromosome analysis. Four patients were
Journal of the American Academy of DermatologyVolume 29, Number 2, Part 2 Emery et al. 371
Fig. 4. Proband's father. Fluorescein angiogram of temporal periphery of right eye showsabnormal termination of the retinal vasculature before the ora serrata (large arrows). In addition, there is a microaneurysmal dilation of a small venule (small arrow).
shown to have Klinefelter's syndrome, and six had anormal 46,XY karyotype'- 18 No other tissues werestudied to evaluate the possibility of mosaicism.Previous studies of both blood and fibroblasts in patients with other pigmentary anomalies followingBlaschko's lines have failed to show mosaicism in asmany as 40%of the patients studied. 14, 15This is notsurprising, because only karyotypically visible defects are detectable by the methods used. Presumably, most cases of IP are caused by point mutationsthat would only be detectable by DNA sequenceanalysis of the IP gene.
Our patient represents the second case of fatherto-daughter transmission of IP. This is the first report that includes both lymphocyte and fibroblastchromosome analyses of a man with IP. Althoughour patient has clinical features of mosaicism, wewere unable to demonstrate chromosomal mosaicism. This may be the result of clonal selection orovergrowth of genetically normal cells in culture. Inaddition, our studies did not include chromosomespecific probes that might have identified a karyotypically half-chromatid mutation.l"
REFERENCES
I. Carney RG. lncontinentia pigmenti: a world statisticalanalysis. Arch Dermatol 1976;112:535-42.
2. Happle R. Lyonization and the lines of Blaschko. HumGenet 1985;70:200-6.
3. Wettke-Schafer R, Kantner G. X-linked dominant inherited diseases with lethality in hemizygous males. HumGenet 1983;64:1-23.
4. Wiklund DA, Weston WL. Incontinentia pigmenti: a fourgeneration study. Arch Dermatol 1980;116:701-3.
5. PrendiviIIe JS, Gorski .IL, Stein CK, et al. Incontinentiapigmenti in a male infant with Klinefelter syndrome. J AMACAD DERMATOL 1989;20:937-40.
6. Sommer A, Liu PH. Incontinentia pigmenti in a father andhis daughter. Am J Med Genet 1984;17:655-9.
7. Watzke Re, Stevens TS, Carney RG. Retinal vascularchanges of incontinentia pigmenti. Arch Ophthalmol1976;94:743-6.
8. Jain RB, Willetts GS. Fundus changes in incontinentiapigmenti (Bloch-Sulzberger syndrome): a case report. Br JOphthalmol 1978;62:622-6.
9. Fischbein FI, Schub M, Lesko WS. Incontinentia pigrnenti,pheochromocytoma, and ocular abnormalities. Am J Ophthalmol 1972;73:961-4.
10. Hook EB. Exclusion of chromosomal mosaicism: tables of90%,95%, and 99%confidence limits and comments onuse.Am J Hum Genet 1977;29:94-7.
11. Cannizzaro LA, Hech t F. Gene for incontinentia pigmentimaps to band Xp 11 with an (X;iO) (pll;q22) translocation. Clin Genet 1987;32:66-9.
12. Sefiani A, Sinnett D, Abel L, et al. Linkage studies do notconfirm the cytogenetic location ofincontinentia pigmention Xpll. Hum Genet 1988;80:282-6.
13. Sefiani A, Abel L, Heuertz S, et al. The gene for incontinentia pigmenti is assigned to Xq28. Genomics 1989;4:427-9.
14. Flannery DB. Pigmentary dysplasias, hypomelanosis of I toand genetic mosaicism. Am J Med Genet 1990;35:18-21.

372 Emery et al.
15. Sybert VP, Pagon RA, Donlan M, et al. Pigmentaryabnormalities and mosaicism for chromosomal aberration:association with clinicalfeatures similar to hypomelanosisof Ito. J Pediatr 1990;116:581-6.
16. Kunze J, Frenzel HH, Huttig E, et al. Klinefelter'ssyndrome and incontinentia pigmenti bloch-sulzberger.Hum Genet 1977;35:237-40.
17. Ormerod AD, White MI, McKay E, et al. Incontinentiapigmenti in a boywithKlinefelter's syndrome. J Med Genet1987;24:439-41.
18. Garcia-Dorado J, De Unamuno P, Fernandez-Lopez E, etal. Incontinentia pigmenti: XXY male with a familyhistory. Clin Genet 1990;38:128-38.
19. Lenz W. Half chromatid mutations may explain incontinentia pigmenti in males [Letter]. Am J Hum Genet1975;27:690c l.
Journal of the American Academy of DermatologyAugust 1993
20. Hecht F, Hecht BK. The half chromatid mutation modeland bidirectional mutation in incontinentia pigmenti, ClinGenet 1983;24:177-9.
21. Kurczynski TW, Berns JS, Johnson WE. Studies of a family with incontinentia pigmenti variably expressed in bothsexes. J Moo Genet 1982;19:447-51.
22. Hecht F, Hecht BK, Austin WJ. Incontinentia pigmenti inArizona Indians including transmission from mother to soninconsistent with the half chromatid mutation model. ClinGenet 1982;21:293-6.
23. Langenbeck U. Transmission of incontinentia pigmentifrom mother to son is consistent with a half chromatidback-mutation (reversion) model. Clin Genet 1982;22:290-1.






![Tnfa Signaling Through Tnfr2 Protects Skin Against ...eprints.whiterose.ac.uk/81541/1/Tnfa signaling through tnfr2 protects... · genodermatosis incontinentia pigmenti (IP) [17]](https://img.pdfslide.net/doc/110x75/5f3bedf6651a4c137761035c/tnfa-signaling-through-tnfr2-protects-skin-against-signaling-through-tnfr2-protects.jpg)











![First IKBKG Gene Mutation Study in Serbian Incontinentia ... · Incontinentia pigmenti (IP; Bloch-Sulzberg-er syndrome; MIM 308300) is a rare X-linked dominant genodermatosis [5]](https://img.pdfslide.net/doc/110x75/5f3bedf5651a4c1377610355/first-ikbkg-gene-mutation-study-in-serbian-incontinentia-incontinentia-pigmenti.jpg)










