Embed Size (px)
Citation preview

Basic Res Cardiol 97; Suppl. 1, I/111 – I/117 (2002) © Steinkopff Verlag 2002
Klara BrixiusPersephone Savvidou-ZarotiUwe MehlhornWilhelm BlochEvangelia G. KraniasRobert H. G. Schwinger
Increased Ca2+-sensitivity of myofibrillar tension in heart failure and its functional implication
� Abstract In human failing myocardium, an increased Ca2+-sensitivity ofmyofilament tension development has been described in Triton X skinnedcardiac myocytes compared to cardiomyocytes obtained from non-failinghuman donor hearts. The present study aimed to investigate whether thereare functional implications of the increased Ca2+-sensitivity in heart failureand whether alterations of myofilament function are already obvious at ear-lier stages of heart failure, such as in cardiac hypertrophy or whether alter-ations of the intracellular Ca2+-homeostasis are able to induce alterations inmyofilament function. Ca2+-activated tension development was measured inTriton X-skinned fibers from human failing and non-failing myocardium.Ca2+-sensitivity of myofilament tension development was significantly shiftedto the left in human failing myocardium. Plots of diastolic free Ca2+ versusdiastolic tension development showed that in a range of similar diastolic Ca2+-concentrations, diastolic tension was significantly enhanced in the failinghearts. The Ca2+/tension relationship was shifted to the right in Triton X-skinned fiber preparations from transgenic renin overexpressing rats(TG(mREN2)27), shown to have concentric hypertrophy. In addition, theCa2+/tension relationship was unchanged in phospholamban knock-out micewith an increased systolic Ca2+ (and enhanced diastolic Ca2+-load).
It is concluded that the increased Ca2+-sensitivity of myofilament tensionobserved in single cardiomyocytes from failing human myocardium may bea phenomenon also present in multicellular preparations and may contributeto the diastolic dysfunction observed in human heart failure. Alterations ofmyofilament function occur at very early stages of heart failure and may bespecies dependent, or dependent on intracellular free Ca2+-levels.
� Key words Cross-bridge interaction – skinned fibers – myosin ATPaseactivity – heart failure
K. Brixius (�) · P. Savvidou-ZarotiR. H. G. SchwingerLaboratory of Muscle Research andMolecular CardiologyClinic III for Internal MedicineUniversity of CologneJoseph-Stelzmann-Str. 950924 Cologne, GermanyTel.: +49-221/4784474Fax: +49-221/4783746E-Mail: [email protected]
U. MehlhornClinic of Cardiothoracic SurgeryUniversity of Cologne
W. BlochInstitute I of AnatomyUniversity of Cologne
E. G. KraniasDepartment of Pharmacology andCell Biophysics, University of Cincinnatti
Introduction
Impaired cardiac contractility results in diminishedorgan perfusion activating a cascade that stimulates theneurohumoral system. Via stimulation of the �-adreno-ceptors or by inducing growth of the cardiac myocytes(e.g., by increase in the endothelin or angiotensin plasma
concentrations), cardiac contractility can be restored, atleast for a short time. However, long-term activation ofthe neurohumoral system leads to profound alterationsin the physiological regulatory mechanisms of cardiacmuscle, such as the diminished inotropic effect of �-agonists in end-stage human heart failure (17), whichhas been attributed to a downregulation of the �1-adrenoceptors (3), as well as to an increase in the

inhibitory Gi-Protein (7) and the �-adrenoceptor kinase(22).
In addition to the �-adrenoceptor downregulation,the frequency-dependent increase in force of contrac-tion is blunted in human failing myocardium (8, 17),which has been suggested to be due to an impaired activ-ity of the Ca2+-ATPase of the sarcoplasmic reticulum(SERCA 2a, 19, 21). There is an ongoing discussion onwhether functional alterations at the level of the myofil-aments in human failing hearts may provide a furtherstress to diseased myocardium. In Triton X-skinned mul-ticellular (18) and single cell preparations (23, 25), anincreased Ca2+-sensitivity of myofilament tension devel-opment in human failing compared to non-failing myo-cardium has been described, which has been attributedto the diminished protein kinase A-dependent phospho-rylation of troponin I due to the downregulation of theleft ventricular �-adrenoceptors. However, there are alsostudies, in which there was no difference in cardiacmyofilament function (10, 11).
The present study investigates the functional implica-tion of an increased Ca2+-sensitivity of myofilament ten-sion development in human failing myocardium. There-fore, simultaneous measurements of the intracellularCa2+-transient and force of contraction were performedin isolated left ventricular trabeculae obtained fromfailing and non-failing human hearts. To investigatewhether alterations of Ca2+-activated myofilament ten-sion development even occur in earlier stages of heartfailure, i.e., in cardiac hypertrophy, the Ca2+/tension rela-tionship was also studied in a transgenic rat model(TG(mREN2)27) of cardiac hypertrophy, since humanmyocardium characterized as NYHA II-III is rarely avail-able for experimental use. To investigate whether alter-ations of the intracellular Ca2+-homeostasis are able toinduce alterations in myofilament function, Ca2+-tensionrelationships were studied in genetically altered micewith an enhanced intracellular Ca2+-homeostasis, i.e., inthe phospholamban knockout mouse (13).
Materials and methods
� Human tissue
Experiments were performed on human left ventricularmyocardium. Tissue was obtained during cardiac trans-plantation (n = 27, 4 female, 23 male; age: 57.5 ± 1.8years). Patients suffered from heart failure clinically clas-sified as NYHA IV on the basis of clinical symptoms andsigns as judged by the attending cardiologist shortlybefore operation. All patients gave written informed con-sent before surgery. Medical therapy consisted of diuret-ics, nitrates, ACE inhibitors and cardiac glycosides.
Patients receiving catecholamines, �-adrenoceptor- orCa2+-antagonists were withdrawn from the study. Non-failing human myocardium was obtained from 12 donors(7 women, 8 men age: 50 ± 3 years). There was no evi-dence of left ventricular dysfunction by echocardiogra-phy. These hearts could not be transplanted for technicalreasons. The study confirms with the declarations ofHelsinki and was approved by the local ethics committee.
� Transgenic animals and controls
Ten male heterozygous TG(mREN2)27 rats aged 12 – 14weeks were compared to 10 normal age-matched maleSprague Dawley rats. TG(mREN2)27 rats exhibited con-centric hypertrophy but no dilation. The homozygousmutant mice (age: 10 – 12 weeks) deficient in phospho-lamban were generated by gene-targeting methodologyin embryonic stem cells as described earlier (13). Bodyweight and heart weight were similar in all groups, i.e.,mice did not show signs of cardiac hypertrophy.
� Ca2+ and force measurements
Intracellular Ca2+ was measured in isolated, electricallydriven (30 to 180 bpm, punctuate stimulation) trabecu-lae by the fluorescence indicator fura-2. The dual excita-tion of fura-2 at 380 nm (Ca2+-free indicator) and 340 nm(Ca2+-bound indicator) allows the determination of theintracellular Ca2+ using the ratiometric technique inde-pendent of the amount of dye present in the preparation(Scientific Instruments, Heidelberg, Germany). Force ofcontraction was continuously recorded by an oscillo-scope. Electrical stimulation was performed by a punc-tuate platinum electrode at one end of the muscle strip.After the muscle strips were pre-stretched with a force of1 mN, they were allowed to equilibrate for 30 – 60 min-utes at a stimulation frequency of 1 Hz and a stimulationvoltage 20% above threshold. Thereafter, the muscleswere gradually stretched along their length-tension curvein 0.05 mm steps until maximum isometric tension wasreached. Under this condition, diastolic tension was sim-ilar in the nonfailing 10.0 ± 0.3 mN and the failing group10.1 ± 0.3 mN. Experiments were performed as describedpreviously (4).
� Calibration of the fura-2 transients
For the calibration of the fura-2 transients, the formulaof Grynkiewicz and coworkers (9) was used:
(R-Rmin)[Ca2+] = KD · · �
(Rmax-R)
I/112 Basic Research in Cardiology, Vol. 97, Suppl. 1 (2002)© Steinkopff Verlag 2002
Background and autofluorescence of the isolated musclestrip and the setup were recorded at the beginning of theexperiment. The autofluorescence did not change whenthe stimulation frequency was increased. Rmin (the fluo-rescence ratio 340/3380 nm in 0 mmol/L Ca2+) and Rmax(the fluorescence ratio 340/380 nm in a saturating con-centration of Ca2+) were determined intracellularly bymetabolic inhibition with the metabolic poison carbonylcyanide m-chloro-phenylhydrazone (5.0 µmol/L; inhibi-tion of oxidative phosphorylation) and rotenone (2.0µmol/L; inhibition of mitochondrial electron transport)in the presence of the Ca2+ ionophore bromo A 23187.Rmin and Rmax could be determined only with an excess of extracellular EGTA or CaCl2. � is the ratio of fluo-rescence in Ca2+-free and Ca2+-containing solution at 380 nm. KD is the effective dissociation constant and wasassumed to be 224 nmol/L (9).
� Skinned fibers and Ca2+/tension relationship
Left ventricular muscle fibers were prepared according topreviously published procedures (18, 26). Briefly, thefiber bundles (diameter < 0.2 mm) were dissected andpermeabilized at 4 °C for 20h in a solution containing50% (v/v) glycerol, 1% Triton X, and in mM NaN3 10,ATP 5, MgCl2 5, EGTA 4, 1,4-dithioerythritol (DTE) 2,and imidazole 20 (pH 7.0). Afterwards the fibers werestored in a similar solution but without Triton X at–20°C.
The chemically skinned fiber bundles were preparedunder microscopic control and then mounted isometri-cally and connected to a force transducer (AME 801,Senso Nor). In relaxation solution, the fibers length wasadjusted to an extent where resting tension was justthreshold (slack position). Fiber diameter and lengthwere the same in all preparations (125 – 150 µm and 7 –
8 mm, respectively). Sarcomeric length was measured bya-actinin staining (see below). All experiments were per-formed at room temperature as previously described (5).
� Sarcomere length
Measurement of sarcomere length was either obtained bylaser diffraction measurements (rats: 2.00 ± 0.02 µm) orby immunocytochemical labeling of Z-lines by �-actininstaining (murine: 1.95 ± 0.02 µm and human myo-cardium: 2.04 ± 0.03 µm). The �-actinin staining of thefibers and the measurement of sarcomeric length wasperformed as described previously (5).
� Statistics
All values are means ± S.E.M. unless otherwise noted.Student’s t-test, or paired t-test were used to test signifi-cance. P-values of < 0.05 were accepted as significant.
Results
� Human failing myocardium
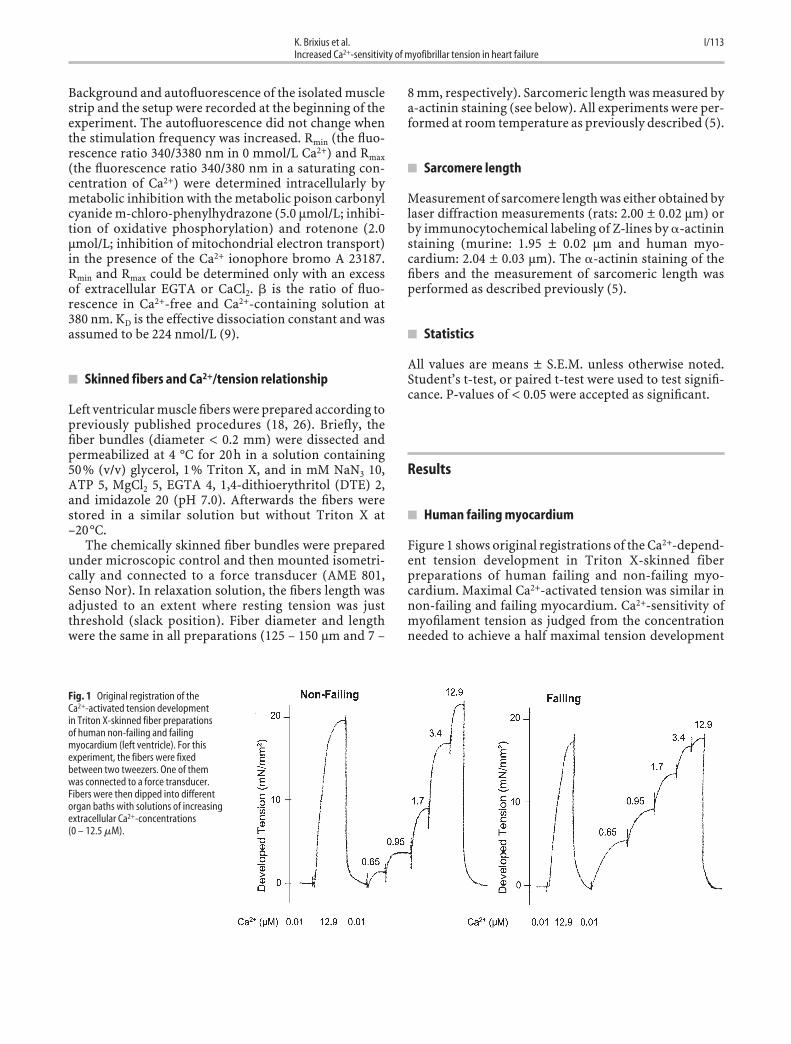
Figure 1 shows original registrations of the Ca2+-depend-ent tension development in Triton X-skinned fiberpreparations of human failing and non-failing myo-cardium. Maximal Ca2+-activated tension was similar innon-failing and failing myocardium. Ca2+-sensitivity ofmyofilament tension as judged from the concentrationneeded to achieve a half maximal tension development
K. Brixius et al. I/113Increased Ca2+-sensitivity of myofibrillar tension in heart failure
Fig. 1 Original registration of the Ca2+-activated tension developmentin Triton X-skinned fiber preparationsof human non-failing and failingmyocardium (left ventricle). For thisexperiment, the fibers were fixedbetween two tweezers. One of themwas connected to a force transducer.Fibers were then dipped into differentorgan baths with solutions of increasingextracellular Ca2+-concentrations (0 – 12.5 �M).
(EC50 for Ca2+) was significantly shifted to the left inhuman failing myocardium (Fig. 2).
To investigate whether there is a functional implica-tion of the increased Ca2+-sensitivity of myofilament ten-sion in human failing myocardium, simultaneous meas-urements of the intracellular Ca2+-transient and force ofcontraction were performed in isolated, electrically stim-ulated left ventricular trabeculae of human failing andnon-failing myocardium. The intracellular Ca2+-concen-tration was measured using the Ca2+-indicator fura-2. Ithas been shown by previous studies that the frequency-induced alterations in the amplitude of the isometrictwitch correlates with the Ca2+ released during systole,i.e., with the amplitude of the intracellular Ca2+-tran-sient. To investigate, whether the increased Ca2+-sensi-tivity of myofilament tension may play a role for the dias-
tolic tension development in human myocardium, dias-tolic free Ca2+ was plotted against diastolic tension. At asimilar free diastolic intracellular Ca2+, tension develop-ment is significantly increased in the failing compared tothe non-failing myocardium (Fig. 3). These results indi-cate that the increased Ca2+-sensitivity of myofilamenttension development may contribute to the diastolic dys-function in human failing myocardium, especially atincreased heart rate.
� Hypertrophied myocardium
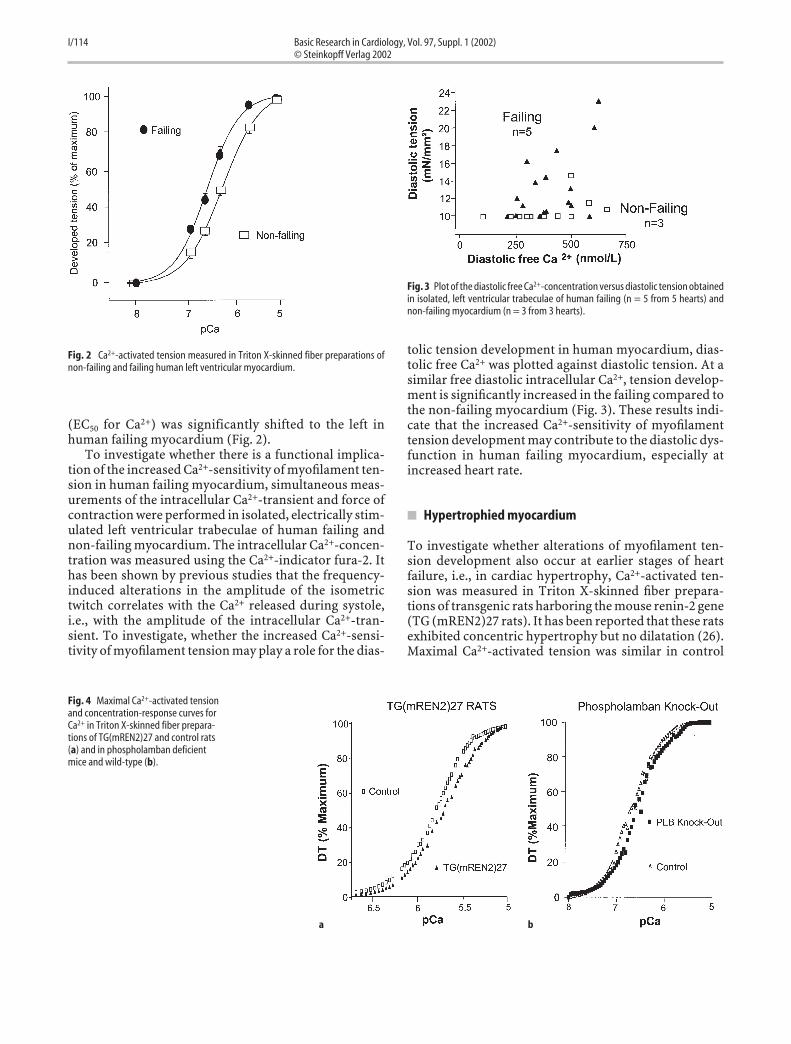
To investigate whether alterations of myofilament ten-sion development also occur at earlier stages of heartfailure, i.e., in cardiac hypertrophy, Ca2+-activated ten-sion was measured in Triton X-skinned fiber prepara-tions of transgenic rats harboring the mouse renin-2 gene(TG (mREN2)27 rats). It has been reported that these ratsexhibited concentric hypertrophy but no dilatation (26).Maximal Ca2+-activated tension was similar in control
I/114 Basic Research in Cardiology, Vol. 97, Suppl. 1 (2002)© Steinkopff Verlag 2002
Fig. 2 Ca2+-activated tension measured in Triton X-skinned fiber preparations ofnon-failing and failing human left ventricular myocardium.
Fig. 3 Plot of the diastolic free Ca2+-concentration versus diastolic tension obtainedin isolated, left ventricular trabeculae of human failing (n = 5 from 5 hearts) andnon-failing myocardium (n = 3 from 3 hearts).
Fig. 4 Maximal Ca2+-activated tensionand concentration-response curves forCa2+ in Triton X-skinned fiber prepara-tions of TG(mREN2)27 and control rats(a) and in phospholamban deficientmice and wild-type (b).
a b

and TG (mREN2)27 rats, but the Ca2+-sensitivity of Ca2+-activated tension was significantly shifted to the right(Fig. 4a). These results indicate that alterations of myofil-ament function also occur at the transition of myocardialhypertrophy into heart failure.
� Altered intracellular Ca2+-homeostasis
To investigate whether a dysregulation of the intracellu-lar Ca2+-homeostasis influences myofilament function,Triton X-skinned fiber preparations from phospholam-ban knock-out mice, which have been shown to have anincreased systolic Ca2+-transient and an enhanced capac-ity to lower diastolic Ca2+ (6, 12), were investigated. Therewas no alteration of the Ca2+-tension relationshipbetween phospholamban knockout mice and their litter-mates (Fig. 4b). Thus, an increase of the intracellular sys-tolic Ca2+-release seems to have no impact on the Ca2+-sensitivity of the myofilaments.
Discussion
The present study investigated the functional implicationof an increased Ca2+-sensitivity of myofilament tensiondevelopment in heart failure and animal models of car-diac hypertrophy and augmented Ca2+-handling. It hasbeen shown by previous studies performed in isolatedTriton X-skinned cardiomyocytes that the Ca2+/tensionrelationship is significantly shifted to the left in humanfailing compared to non-failing myocardium (23, 25).However, there are also studies, in which there was nodifference in cardiac myofilament function (10, 11).These different findings may be due to 1) the use of car-diac tissue obtained from hearts with different etiology(dilated vs. ischemic cardiomyopathy), 2) the use offibers from different cardiac regions (right vs. left ven-tricle) and 3) the use of different skinning procedures(saponine vs. Triton X). In addition, different sarcomerelengths may play a role. In the present study, it wasshown that an increased Ca2+-sensitivity of myofilamenttension development is also present in multicellularskinned preparations and in multicellular intact trabec-ulae during diastole. Thus, the increased Ca2+-sensitivityof myofilament tension may contribute to the diastolicdysfunction observed in human heart failure.
It has been shown that in human failing myocardiumcAMP-dependent phosphorylation is decreased due tothe downregulation of the �1-adrenoceptors (1). In arecent study in transgenic mice, it was shown that PKA-dependent alterations of the myofibrillar Ca2+/tensionrelationship may be attributed to the phosphorylation oftroponin I exclusively and that the PKA-dependent phos-phorylation of myosin binding protein C is not involved
in the relaxing effects seen after cAMP-dependent stim-ulation (12). Consequently, the Ca2+/tension relationshipobtained in Triton X-skinned cardiomyocytes wasshifted to the right after protein kinase A-dependentphosphorylation in human failing, but not in humannon-failing myocardium (23, 25). Therefore, it may beconcluded that in human failing myocardium, Ca2+-sen-sitivity of myofilament tension development is involvedin regulation of the diastolic cell function and may beattributed to a decreased PKA-dependent phosphoryla-tion of troponin I. Yet, it cannot be excluded from thepresent study that a re-expression of fetal proteins (e.g.,�-myosin heavy chain) may additionally be involved inthe leftward shift of the Ca2+-dependent tension devel-opment in human failing myocardium.
To investigate whether functional alterations of themyofilament tension development occurs in earlierstages of heart failure, e.g., in cardiac hypertrophy, theCa2+/tension relationship was studied in transgenic ratsoverexpressing the mouse REN-2 gene (TG(mREN2)27rats), since human myocardium at intermediate stages ofNYHA II-III is difficult to obtain. It has been reportedthat the hearts of the TG(mREN2)27 rats exhibited con-centric hypertrophy but no dilation (26). In contrast tothe results in human myocardium, Ca2+-sensitivity oftension development was shifted to the right, whichmeans that Ca2+-sensitivity is decreased in the hyper-trophic compared to the control rats. It has been reportedthat the ratio between �- and �-myosin heavy chainexpression is altered in this rat model of renin overex-pression. In addition, a decreased phosphorylation of themyosin light chain may be responsible for the rightwardshift of the Ca2+/tension relationship in TG(mREN2)27rats, since it has been shown that phosphorylation of themyosin light chain increases Ca2+-sensitivity of myofila-ment tension (14). Consistently, a significant reductionin the phosphorylation of the myosin light chain wasfound in hypertensive rats (14). The reduced Ca2+-sensi-tivity could also result from isoform shifts of troponin T(15). These results indicate that alterations in myofila-ment function may be species dependent (rat vs. human)or may be different in the various studies towards thetransition into heart failure.
Cardiac contraction and relaxation is mainly influ-enced by the alterations of diastolic and systolic intracel-lular free Ca2+-concentrations based upon the Ca2+-induced Ca2+-release of the sarcoplasmic reticulum.Recently, phospholamban-deficient mice were shown toexhibit a significantly enhanced myocardial performancecompared with wild-type controls in working heartpreparations (13). It is feasible that cardiac compensa-tory mechanisms accompanying phospholamban abla-tion occur in these hyperdynamic hearts. Thus, the ryan-odine receptors are down-regulated in the phospholam-ban knock-out mice (6). The Ca2+/tension relationshipwas unchanged in phospholamban knock-out mice in
K. Brixius et al. I/115Increased Ca2+-sensitivity of myofibrillar tension in heart failure

comparison to controls. These results indicate that inaddition to the unchanged protein levels for myosin,actin, troponin I and troponin T (6), regulation of myofil-ament contraction (e.g., by adrenergic stimulation) isunchanged in phospholamban knockout mice.
In summary, the increased Ca2+-sensitivity of myofil-ament tension observed in single cardiomyocytes fromfailing human myocardium is a phenomenon also pres-ent in multicellular preparations and may especially con-tribute to the diastolic dysfunction in human hypertro-phy/heart failure. Alterations of myofilament functionoccur at very early stages of heart failure and may bespecies dependent.
� Limitations of the study
In the present study, non-failing myocardial tissue wasobtained from brain-dead patients, who were treated by
dobutamine. Thus, it cannot be excluded that, due to anincrease of catecholamines, the responsiveness of thecontractile proteins is altered. In the present study, car-diac tissue from different species was investigated, i.e.,rat, mouse and human. Due to the fact that there aredifferences in the regulation of the intracellular Ca2+-transient (2), the cardiac failing phenotype may be dif-ferent between the different species (24). This mayexplain the different alterations described for Ca2+-sen-sitivity between human and rat myocardium.
Acknowledgments We are indebted to all colleagues of the Departmentof the Cardiothoracic Surgery of the University of Cologne and Munichfor providing us with human myocardial samples. The authors thankMs. S. Danneschewski, S. Pfeiffer and K. Rössler for their excellent tech-nical help. We are grateful to K.F. Frank for critical discussion of themanuscript. This study was supported by a grant of Köln Fortune (toK.B., R.H.G.S.) and the Deutsche Forschungsgemeinschaft (toR.H.G.S.).
I/116 Basic Research in Cardiology, Vol. 97, Suppl. 1 (2002)© Steinkopff Verlag 2002
1. Bartel S, Stein B, Eschenhagen T, MendeU, Neumann J, Schmitz W, Krause E-G,Karczewski P, Scholz H (1996) Proteinphosphorylation in isolated trabeculaefrom nonfailing and failing humanhearts. Mol Cell Biochem 157: 171–179
2. Bassani JW, Bassani RA, Bers DM (1994)Relaxation in rabbit and rat cardiac cells:species-dependent differences in cellularmechanisms. J Physiol 476: 279–93
3. Bristow MR,Ginsburg R, Minobe W,Cubiciotti RS; Sagemann WS, Lurie K,Billingham ME, Harrison DC, Stinson EB(1982) Decreased catecholamine sensitiv-ity and �-adrenergic-receptor density infailing human hearts. N Engl J Med 307:205–211
4. Brixius K, Pietsch M, Hoischen S, Müller-Ehmsen J, Schwinger RHG (1997) Effectof inotropic interventions on contractionand Ca2+ transients in the human heart. JAppl Physiol 83: 652–660
5. Brixius K, Mehlhorn U, Bloch W,Schwinger RHG (2000) Different effect ofthe Ca2+ sensitizers EMD 57033 and CGP48506 on cross-bridge cycling in humanmyocardium. J Pharmacol Exp Ther 295:1284–90
6. Chu G, Luo W, Slack JP, Tilgmann C,Sweet WE, Spindler M, Saupe KW, BiovinGP, Moravec CS, Matlib MA, Grupp IL,Ingwall JS, Kranias EG (1996) Compen-satory mechanisms associated with thehyperdynamic function of phospholam-ban deficient mouse hearts. Circ Res 78:1064–1076
7. Feldman AM, Cates AE, Veazey WB,Hershberger RE, Bristow MR, BaughmanKL, Baumgartner WA, Van Dop C(1988A) Increase of the 40,000 mol wtpertussis toxin substrate (G protein) inthe failing human heart. J Clin Invest 82:189–197
8. Feldman MD, Gwathmey JK, Phillips P,Schoen F, Morgan JP (1988B) Reversal of the force-frequency-relationship inworking myocardium from patients withendstage heart failure. J Appl Cardiol 3:273–283
9. Grynkiewicz G, Poenie M, Tsien RY(1985) A new generation of Ca2+ indica-tors with greatly improved fluorescenceproperties. J Biol Chem 260: 3440–3450
10. Hajjar RJ, Gwathmey JK, Briggs GM,Morgan JP (1988) Differential effect ofDPI 201-106 on the sensitivity of themyofilaments to Ca2+ in intact andskinned trabeculae from control andmyopathic human hearts. J Clin Invest 82:1578–84
11. Holubarsch C, Ruf T, Goldstein DJ,Ashton RC, Nickl W, Pieske B, Pioch K,Ludemann J, Wiesner S, Hasenfuss G,Posival H, Just H, Burkhoff D (1996) Exis-tence of the Frank-Starling mechanism inthe failing human heart. Investigations onthe organ, tissue, and sarcomere levels.Circulation 94: 683–9
12. Kentish JC, McCloskey DT, Layland J,Palmer S, Leiden JM, Martin AF, Solaro RJ(2001) Phosphorylation of troponin I byprotein kinase A accelerates relaxationand crossbridge cycle kinetics in mouseventricular muscle. Circ Res 88: 1059–65
13. Luo W, Grupp IL, Harrer J, Ponniah S,Grupp G, Duffy JJ, Doetschman T,Kranias EG (1994) Targeted ablation ofthe phospholamban gene is associatedwith markedly enhanced myocardial con-tractility and loss of �-agonist stimula-tion. Circ Res 75: 401–409
14. Morano I, Lengsfeld M, Ganten U, GantenD, Ruegg JC (1988) Chronic hypertensionchanges myosin isoenzyme pattern anddecreases myosin phosphorylation in therat heart. J Mol Cell Cardiol 20: 875–886
15. Nassar R, Malouf NN, Kelly MB, OakeleyAE, Anderson PA (1991) Force-pCa rela-tion and troponin T isoforms of rabbitmyocardium. Circ Res 69: 1470–75
16. Santana LF, Kranias EG, Lederer WJ(1997) Calcium sparks and excitation-contraction coupling in phospholamban-deficient mouse ventricular myocytes. JPhysiol 503: 21–29
17. Schwinger RHG, Böhm M, Erdmann E(1992) Inotropic and lusitropic dysfunc-tion in myocardium from patients withdilated cardiomyopathy. Am Heart J 123:116–128
18. Schwinger RHG, Böhm M, Koch A,Schmidt U, Morano I, Eissner H, Über-fuhr P, Reichart B, Erdmann E (1994) Thefailing heart is unable to use the Frank-Starling-Mechanism. Circ Res 74: 959–969
References

K. Brixius et al. I/117Increased Ca2+-sensitivity of myofibrillar tension in heart failure
19. Schwinger RHG, Böhm M, Schmidt U,Karczewski P, Bavendiek U, Flesch M,Krause E-G, Erdmann E (1995)Unchanged protein levels of SERCA 2aand phospholamban but reduced Ca2+-uptake and Ca2+-ATPase activity of car-diac sarcoplasmic reticulum frompatients with dilated cardiomyopathycompared to nonfailing patients. Circula-tion 92: 3220–3228
20. Schwinger RHG, Brixius K, Savvidou-Zaroti P, Bölck B, Zobel C, Frank K,Kranias EG, Hoischen S, Erdmann E(2000) The enhanced contractility inphospholamban deficient mouse hearts isnot associated with alterations in Ca2+-sensitivity or myosin ATPase-activity ofthe contractile proteins. Bas Res Cardiol95: 12–18
21. Schwinger RHG, Munch G, Bolck B,Karczewski P, Krause EG, Erdmann E(1999) Reduced Ca(2+)-sensitivity ofSERCA 2a in failing human myocardiumdue to reduced serin-16 phospholambanphosphorylation. J Mol Cell Cardiol 31:479–491
22. Ungerer M, Böhm M, Elce JS, Erdmann E,Lohse MJ (1993) Altered expression ofbeta-adrenergic receptor kinase and beta1-adrenergic receptors in the failinghuman heart. Circulation 87: 454–463
23. Van der Velden J, de Jong JW, Owen VJ,Burton PBJ, Stienen GJM (2000) Effect ofproteinkinase A on calcium sensitivity offorce and its sarcomere length depend-ence in human cardiomyocytes. Cardio-vasc Res 46: 487–95
24. Ventura-Clapier R, Mekhfi H, Oliviero P,Swynghedauw B (1988) Pressure overloadchanges cardiac skinned-fiber mechanicsin rats, not in guinea pigs. Am J Physiol254: H517–24
25. Wolff MR, Buck SH, Stoker SW, GreaserML, Mentzer RM (1996) Myofibrillar cal-cium sensitivity of isometric tension isincreased in human dilated cardiomyo-pathy. J Clin Invest 98: 167–76
26. Zobel C, Brixius K, Pietsch M, Münch G, Bölck K, Schwinger RHG (1998)Unchanged sarcoplasmic reticulum Ca2+-ATPase activity, reduced Ca2+ sensitivity,and negative force-frequency relation-ship in transgenic rats overexpressing themouse renin gene. J Mol Med 76: 533–544
























![Basel 3 & Implication[1]](https://img.pdfslide.net/doc/110x75/577d1e721a28ab4e1e8e9042/basel-3-implication1.jpg)


